DOI:
10.1039/D1OB02038B
(Review Article)
Org. Biomol. Chem., 2022,
20, 37-54
Catalytic asymmetric synthesis of α-stereogenic carboxylic acids: recent advances
Received
17th October 2021
, Accepted 12th November 2021
First published on 16th November 2021
Abstract
Chiral carboxylic acids bearing an α-stereogenic center constitute the backbone of many natural products and therapeutic reagents as well as privileged chiral ligands and catalysts. Hence, it is not surprising that a large number of elegant catalytic asymmetric strategies have been developed toward the efficient synthesis of α-chiral carboxylic acids, such as α-hydroxy acids and α-amino acids. In this review, the recent advances in asymmetric synthesis of α-stereogenic free carboxylic acids via organocatalysis and transition metal catalysis are summarized (mainly from 2010 to 2020). The content is organized by the reaction type of the carboxyl source involved, including asymmetric functionalization of substituted carboxylic acids, cyclic anhydrides, α-keto acids, substituted α,β-unsaturated acids and so on. We hope that this review will motivate further interest in catalytic asymmetric synthesis of chiral α-substituted carboxylic acids.
 Rui Niu | Rui Niu was born in Shaanxi Province, China. She received her B.S. degree from Yan'an University in 2019. Currently she is pursuing her M.Sc. degree under the guidance of Dr Jun-Bing Lin at the same university. Her current research interests mainly focus on organocatalytic asymmetric cascade reactions. |
 Yi He | Yi He was born in Shaanxi Province, China. He received his B.S. degree from Yan'an University in 2021. He then worked as a research assistant in Dr Jun-Bing Lin's group at Yan'an University. His current research interests mainly focus on catalytic asymmetric transformations involving prochiral carboxylic acids. |
 Jun-Bing Lin | Jun-Bing Lin received his Ph.D. in 2012 from Lanzhou University under the supervision of Prof. Peng-Fei Xu. After postdoctoral research with Prof. Hong-Yu Li at Lanzhou University and Prof. Shu-Yu Zhang at Shanghai Jiao Tong University, he started his independent research at Yan'an University in 2018. His research interests are the development of new synthetic methods and asymmetric synthesis of functionalized molecules. |
1. Introduction
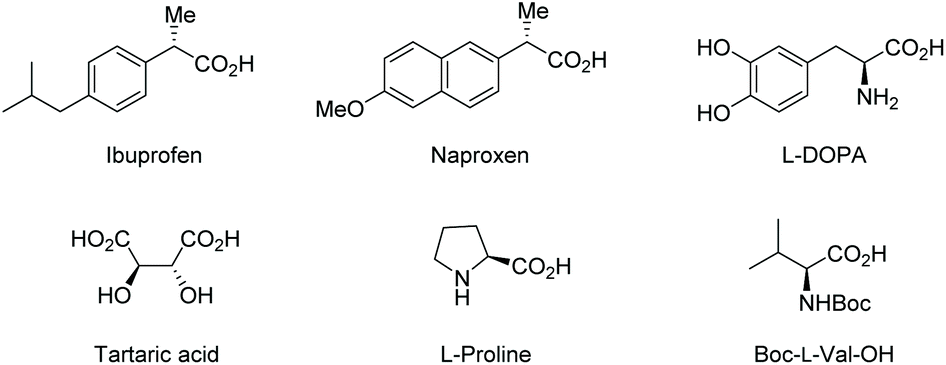
Carboxylic acids bearing an α-stereogenic center constitute the backbone of many natural products and therapeutic reagents1 as well as privileged chiral ligands and catalysts2 (Fig. 1). They also could serve as synthetic intermediates capable of a myriad of transformations owing to the extraordinary versatility of the carboxyl group.3 Therefore, the asymmetric synthesis of this type of intriguing molecule from prochiral or racemic substrates has been a significant domain in asymmetric catalysis, and various catalytic asymmetric strategies have been devised toward this end over the past decades.4
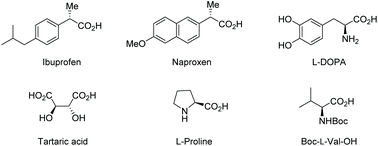 |
| | Fig. 1 Representative examples of α-stereogenic free carboxylic acids. | |
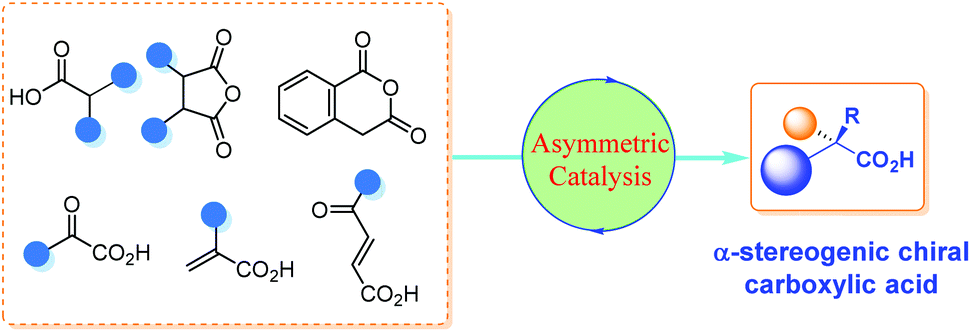
Although there are many reviews on the asymmetric synthesis of carboxylic acid derivatives,5 a timely survey on the catalytic asymmetric synthesis of α-chiral FREE carboxylic acids is highly desirable and potentially beneficial for the advancement of this research area. Herein, the recent advances in catalytic asymmetric synthesis of α-stereogenic carboxylic acids, relying on both organocatalysts and transition metal complexes, are summarized. The content is organized by reaction types of the carboxyl sources involved, including catalytic asymmetric functionalization of substituted carboxylic acids, cyclic anhydrides, α-keto acids, substituted α,β-unsaturated acids and so on (Scheme 1). It should be noted that, some examples involving further transformations of the obtained α-chiral carboxylic acids to esters to facilitate purification procedures are included. However, chiral catalyst-mediated kinetic resolution of racemic substrates6 and enzymatic protocols toward the α-chiral carboxylic acid are not discussed in this review.7
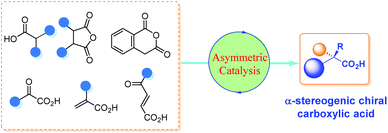 |
| | Scheme 1 Catalytic asymmetric synthesis of α-chiral carboxylic acids. | |
2. Catalytic asymmetric functionalization of substituted carboxylic acids
The direct catalytic asymmetric functionalization of substituted carboxylic acids to access α-chiral free carboxylic acids represents an attractive strategy because of the prevalence of carboxylic acid in nature. In addition, the direct applications of carboxylic acids in such transformations could circumvent the tedious protection/deprotection procedures. However, owing to the inherent Brønsted acidity of the carboxyl functionality, only recently the catalytic asymmetric α- and β-functionalizations of substituted carboxylic acids have been emerging as a powerful method for the synthesis of α-chiral carboxylic acids (Scheme 2).
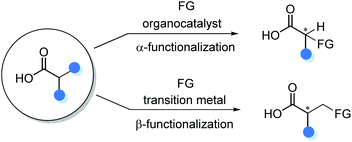 |
| | Scheme 2 Catalytic asymmetric functionalization of substituted carboxylic acids. | |
2.1 Catalytic asymmetric α-functionalization of substituted carboxylic acids
The enediolate-mediated direct addition of a free carboxylic acid to polar electrophiles provides a straightforward method to prepare α-substituted carboxylic acids. However, unlike other carbonyl compounds such as aldehydes and ketones, the catalytic asymmetric addition of α-carbon of free carboxylic acids to electrophilic species to make α-chiral free carboxylic acids was extremely rare, partially because of the relatively low acidity of carboxylic acid α-protons and an undesired decarboxylation process in such transformations. While early enediolate-mediated reactions used stoichiometric amounts of chiral reagents,8 few catalytic asymmetric examples of saturated carboxylic acids leading to α-chiral carboxylic acids have been reported.9
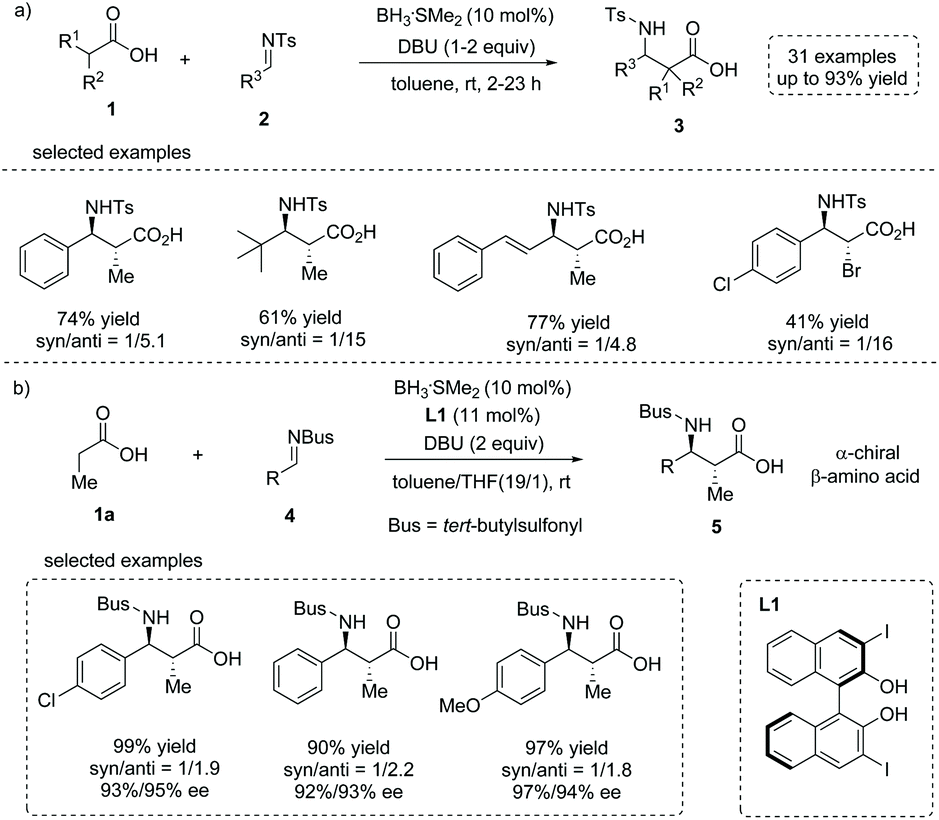
In 2015, Kanai, Shimizu and co-workers reported a boron-catalyzed anti-selective Mannich-type addition of enolizable carboxylic acid 1 to N-Ts imine 2 (Ts = p-toluenesulfonyl).10 This approach combined a borane catalyst (BH3·SMe2) and an organic base (DBU, 1,8-diazabicyclo[5.4.0]undec-7-ene) that enabled the generation of the enediolate species from carboxylic acid as a real nucleophile to react with imines. Using this scenario, a series of β-amino substituted free carboxylic acids 3 were efficiently installed directly from simple carboxylic acids (up to 93% yield, Scheme 3a). The asymmetric variant of the current reaction was also developed by using a chiral boron catalyst (in situ formed from BH3·SMe2 and a substituted BINOL L1) and N-Bus imine 4 (Bus = tert-butylsulfonyl). The α-chiral carboxylic acids 5 with an anti-configuration could be made by this protocol with high yields and ees, albeit with low diastereoselectivities (Scheme 3b). The reaction tolerated multiple functional groups such as chloro and methoxyl groups on the imines. Notably, the Bus-group could be successfully removed with aluminum chloride and anisole, making this chemistry potentially applicable in natural product and peptide synthesis.
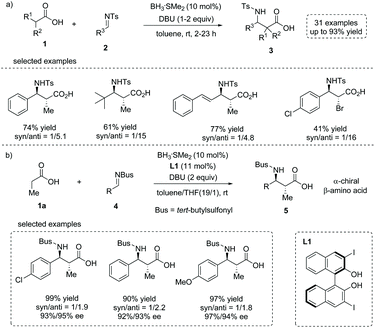 |
| | Scheme 3 Facile synthesis of α-stereogenic carboxylic acids via chemoselective boron-catalyzed Mannich-type addition of carboxylic acids. | |
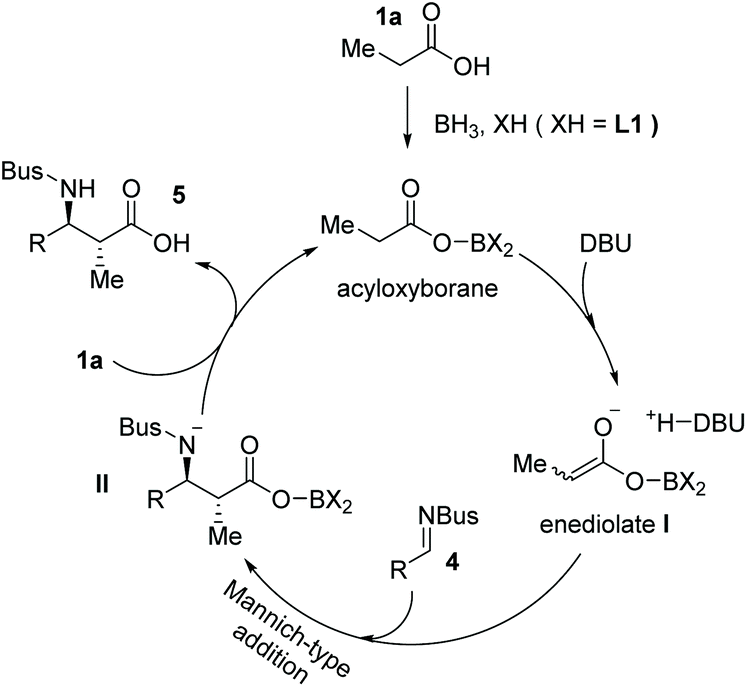
Mechanistically, the activation of carboxylic acids could be achieved with BH3·SMe2 and substituted BINOL L1 to form an acyloxyborane intermediate, which reacts with DBU generating the highly reactive enediolate nucleophile I. The enediolate could take part in subsequent stereoselective Mannich-type anti-addition to imine 4 to form intermediate II, which then reacts with another carboxylic acid 1 delivering the final α-chiral carboxylic acid product 5 and acyloxyborane intermediate for the next catalytic cycle (Scheme 4).
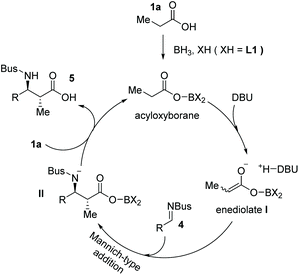 |
| | Scheme 4 Proposed reaction pathway for the asymmetric synthesis of chiral carboxylic acid 5. | |
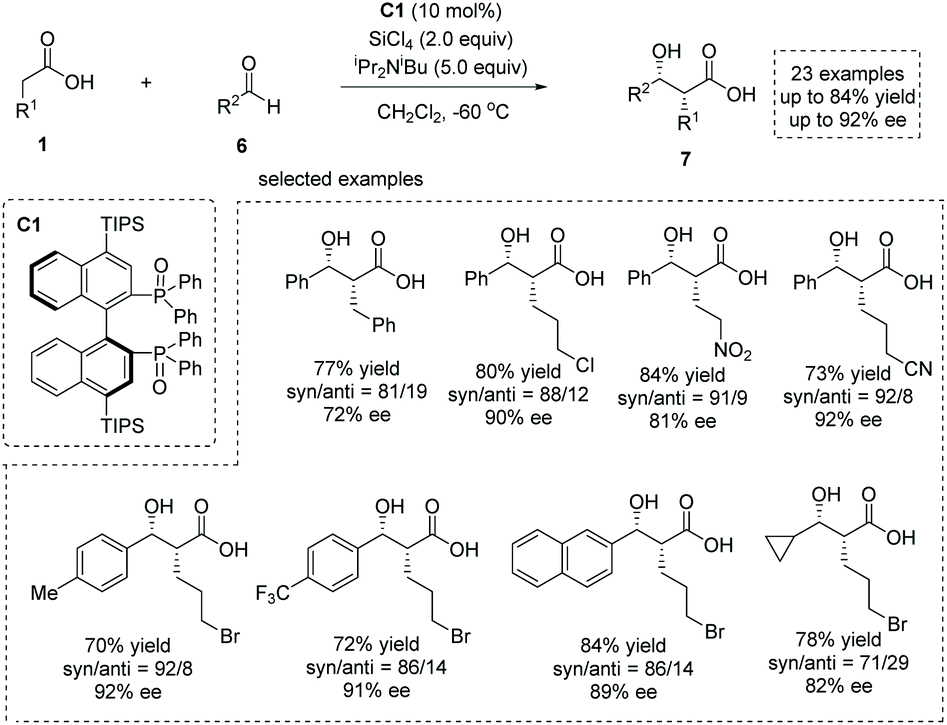
The enediolate-mediated aldol-type addition of carboxylic acids to aldehydes is a straightforward method toward β-hydroxy α-substituted carboxylic acids. On the basis of the successful application of boron-catalyzed nucleophilic activation of carboxylic acids, Kanai, Shimizu and co-workers achieved boron-catalyzed aldol-type reactions of carboxylic acids to simple aldehydes and CF3-activated ketones, respectively.11 Based on a new silicon tetrachloride activation strategy, in 2018, Nakajima, Kotani and co-workers realized an asymmetric aldol-type addition of carboxylic acids to aldehydes leading to chiral α-hydroxy carboxylic acids.12 They accomplished the catalytic generation of enediolate nucleophiles from simple carboxylic acids with silicon tetrachloride in the presence of chiral phosphine oxide C1. This “Lewis base assisted Lewis acid activation”13 mode successfully converted a series of carboxylic acids 1 and aldehydes 6 to the corresponding chiral syn β-hydroxy α-alkyl carboxylic acids 7 with high to excellent enantioselectivities (up to 84% yield and up to 92% ee, Scheme 5).
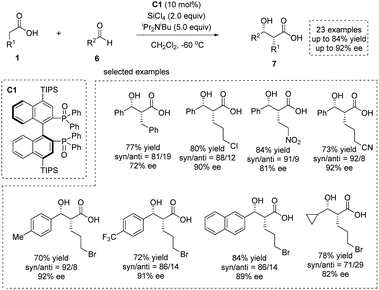 |
| | Scheme 5 Catalytic enantioselective synthesis of α-hydroxy carboxylic acids via silicon tetrachloride and phosphine oxide catalysis. | |
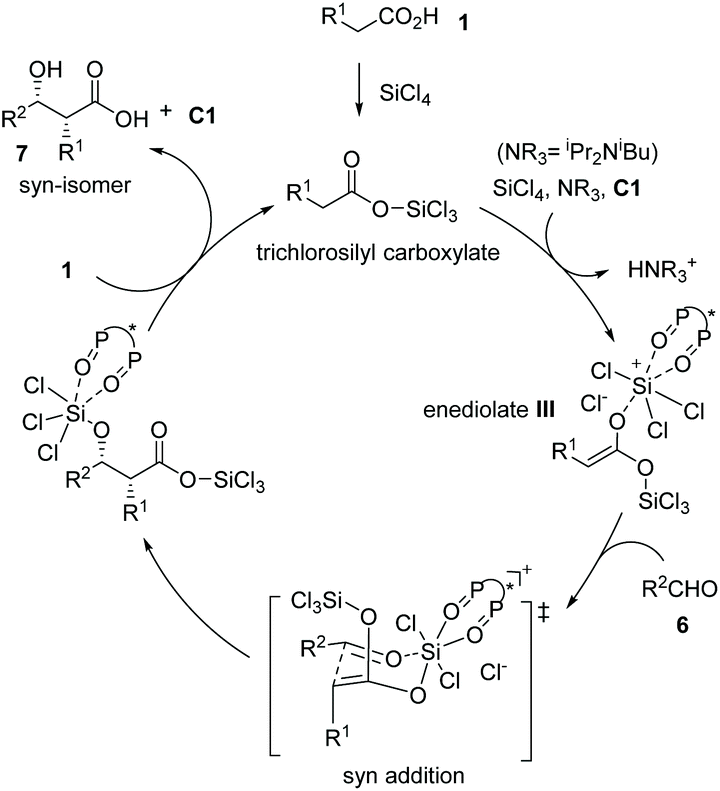
Mechanistically, carboxylic acid 1 was activated by silicon tetrachloride to form trichlorosilyl carboxylate, which underwent deprotonation by a tertiary amine in the presence of the chiral Lewis base (S)-BINAPO (2,2′-bis-(diphenylphosphoryl)-1,1′-binaphthyl) derivative C1 to form bis(trichlorosilyl) enediolate III. The enediolate reacted with aldehydes 6 in a syn addition fashion via a Zimmerman–Traxler chair-like transition-state, furnishing the silicon-protected α-chiral β-hydroxy carboxylic acids. Finally, the reaction between carboxylic acid 1 and the resulting silicon-protected α-chiral β-hydroxy carboxylic acids delivered the syn adduct 7 and regenerated the catalyst for the next cycle (Scheme 6). The coordination of the chiral bis(phosphine oxide) catalyst C1 to the silicon center ensures the enantio-induction of the entire catalytic process.
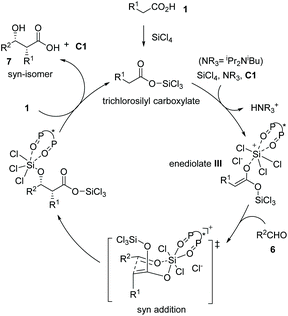 |
| | Scheme 6 Proposed reaction pathway for the asymmetric synthesis of chiral carboxylic acid 7. | |
2.2 Catalytic asymmetric β-functionalization of substituted carboxylic acids
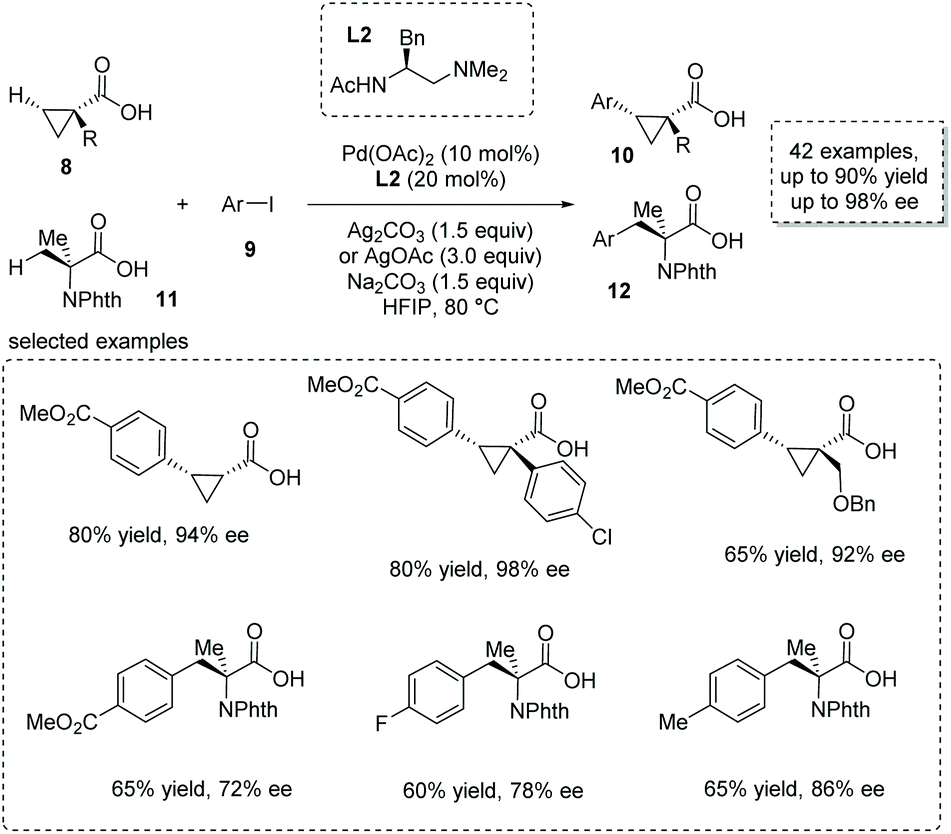
The asymmetric β-functionalization of prochiral carboxylic acids is another powerful method to form α-chiral carboxylic acids. Recently, with the development of C–H activation based on a concerted metalation–deprotonation (CMD) mechanism, the weakly coordinating carboxyl-directing β-functionalization strategy was successfully applied to the catalytic synthesis of α-stereogenic carboxylic acids.14 An elegant piece of work was reported by Yu and co-workers recently based on their previous finding on ligand-accelerated enantioselective methylene C–H bond activation.15 They achieved a Pd(II)-catalyzed challenging enantioselective β-selective C(sp3)–H arylation of cyclopropanecarboxylic acid 8 and 2-aminoisobutyric acid 11, respectively.16 Substituted aryl iodides 9 were used as arylation reagents, and Pd(OAc)2 was the catalyst of choice with monoprotected aminoethyl amine (MPAAM) L2 having an ethylenediamine scaffold as a bidentate chiral ligand. Two stereogenic centers at the α and β positions of the carboxyl group were concomitantly created in this desymmetrization process (Scheme 7). Both cyclopropanecarboxylic and 2-aminoisobutyric acids proved to be competent substrates, giving rise to the corresponding chiral α-stereogenic carboxylic acids 10 and 12 with satisfactory results (53–90% yield, 72–98% ee). Compared to other strategies, a remarkable breakthrough of this methodology is that no exogenous directing groups such as amides were exploited, thus allowing for the direct use of the obtained carboxylic acid for further transformations in a step-economical fashion.
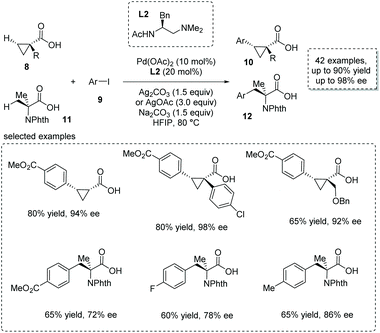 |
| | Scheme 7 Synthesis of chiral cyclopropanecarboxylic and 2-aminoisobutyric acids via Pd(OAc)2 catalyzed enantioselective β-C(sp3)–H arylation of free carboxylic acids. | |
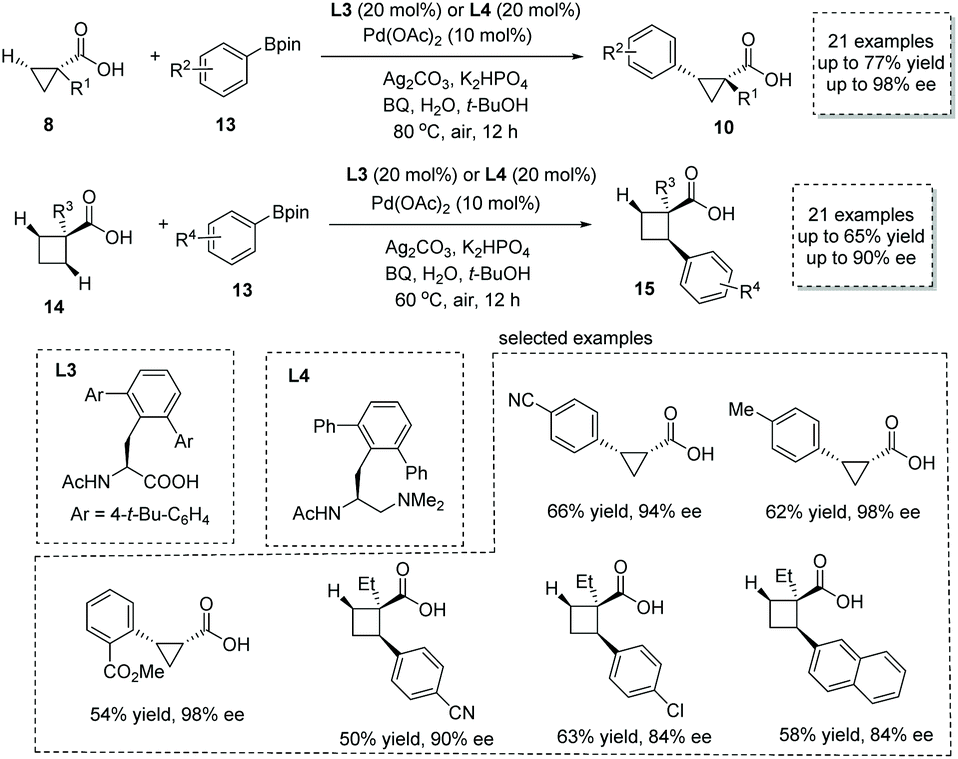
Shortly after, the same group reported a new approach to α-chiral carboxylic acids via the Pd(II)-catalyzed enantioselective β-C(sp3)–H activation/oxidative cross-coupling reaction of free carboxylic acids and aryl-boronic acid pinacol esters 13.17 For most of the cyclopropanecarboxylic acids 8, the combination of Pd(OAc)2 and mono-protected amino acid (MPAA) L3 was the optimal catalytic system, while for cyclobutanecarboxylic acids 14 the combination of Pd(OAc)2 and mono-protected aminoethyl amine (MPAAM) L4 give superior results in terms of efficiency and selectivity (Scheme 8). In both cases, p-benzoquinone (BQ) was used as the external oxidant to regenerate the Pd(II) species. A variety of chiral cyclopropanecarboxylic acids 10 (21 examples, 44–77% yields, 84–98% ee) and cyclobutanecarboxylic acids 15 (21 examples, 42–65% yields, 68–90% ee) with a tertiary or quaternary stereocenter at the α position was readily obtained. Besides aryl-boronic acid pinacol ester, its vinyl counterpart, vinyl boronic ester, also is a competent coupling partner in this reaction, leading to olefin-containing α-chiral carboxylic acids with acceptable efficiency and selectivity. In 2020, van Gemmeren and co-workers reported a direct remote alkynylation of free carboxylic acids,18 and they showed that an asymmetric trial with a chiral bidentate ligand could furnish α-chiral carboxylic acids with an alkyne tether in 39% ee.
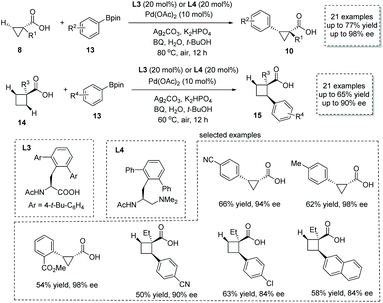 |
| | Scheme 8 Synthesis of chiral cyclopropanecarboxylic and cyclobutanecarboxylic acids via Pd(II) catalyzed enantioselective β-C(sp3)–H activation/oxidative cross-coupling. | |
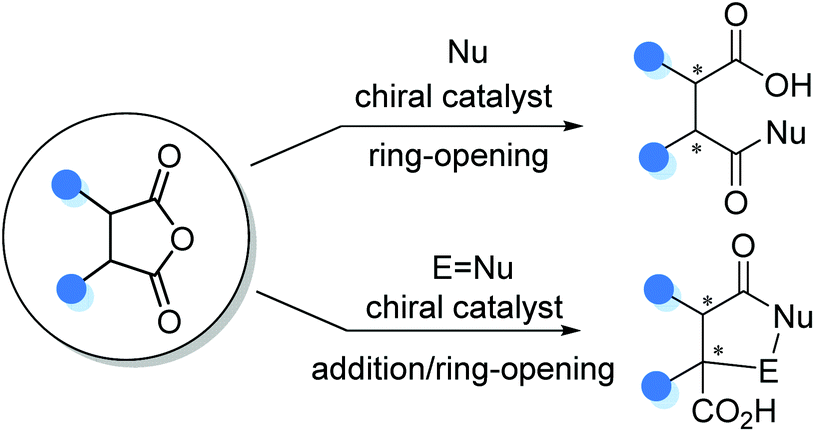
3. Catalytic asymmetric functionalization of cyclic anhydrides
Besides substituted carboxylic acids, the stereoselective functionalization of meso- or prochiral cyclic anhydrides was also a powerful protocol toward asymmetric construction of α-stereogenic free carboxylic acids. Cyclic anhydrides such as succinic and glutaric anhydrides could serve as versatile electrophiles in the carboxylic acid-forming ring-opening reactions, whereas the enolizable ones such as homophthalic anhydrides could act as prochiral nucleophiles to initiate asymmetric addition/ring-opening cascade reaction to install α-chiral carboxylic acids with a cyclic backbone (Scheme 9).
 |
| | Scheme 9 Catalytic asymmetric functionalization of cyclic anhydrides. | |
3.1 Cyclic anhydrides as electrophiles
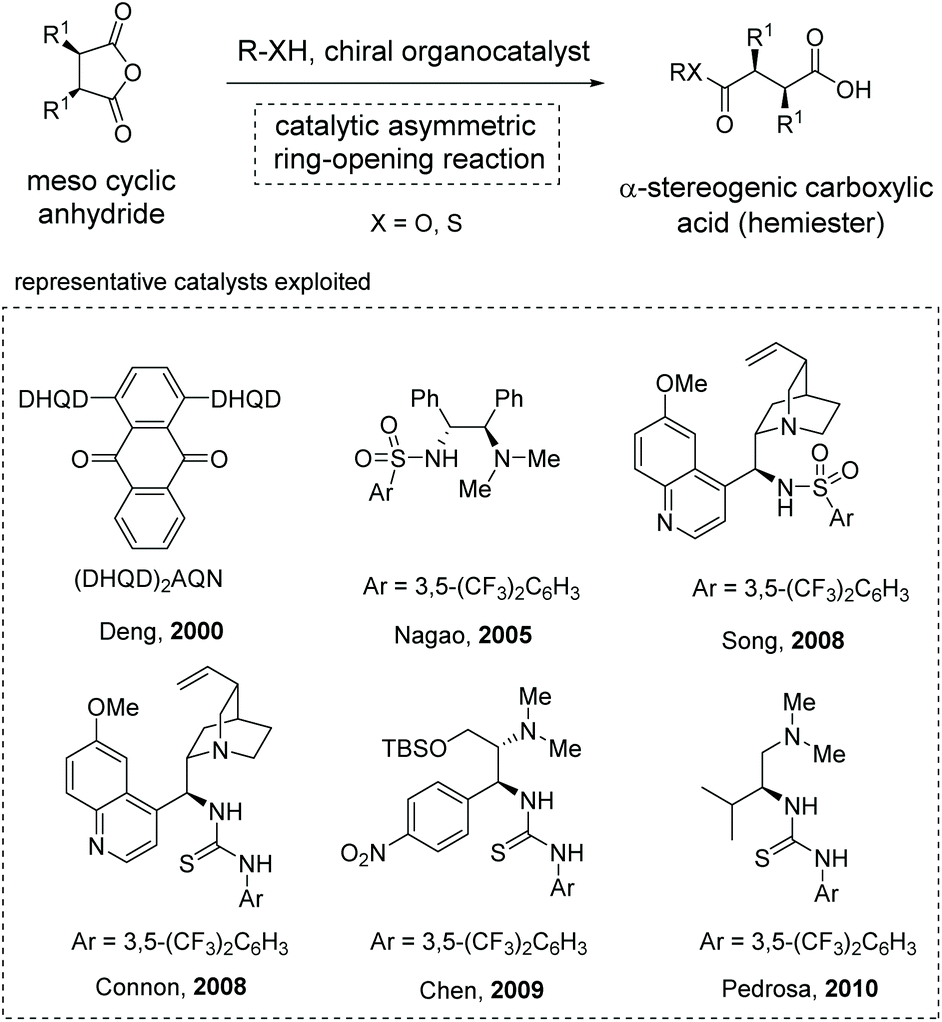
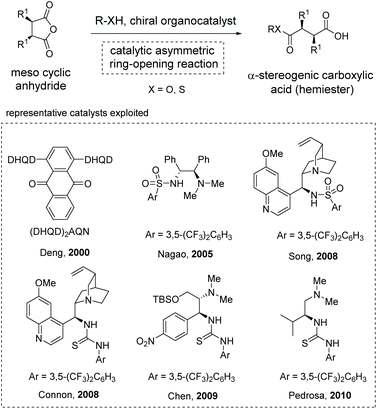
The asymmetric ring-opening of meso-cyclic anhydrides (desymmetrization of meso-anhydrides) with alcohols or alkyl thiols as the nucleophilic species was a reliable strategy for the construction of enantiomerically enriched hemiesters, a type of α-stereogenic carboxylic acid with an ester functionality.19 Owing to the ready availability of anhydride substrates and the increasing significance of chiral hemiesters that contain one or multiple stereogenic centers and two chemically differentiated carbonyl functionalities, substantial progress has been made in this area over the past decades.20 Among the various strategies developed, organocatalytic methods exhibit special robustness in terms of efficiency and selectivity, and various organocatalysts have been successfully applied to this type of transformation by Deng,20a Nagao,20b Song,20d Connon,20e Chen,20f and Pedrosa20g (Scheme 10). Mechanistically, these reactions proceed through a general base catalysis or bifunctional catalysis pathway in most cases.21
 |
| | Scheme 10 Access to hemiester-type α-chiral carboxylic acids via organocatalytic asymmetric ring-opening of meso-cyclic anhydrides. | |
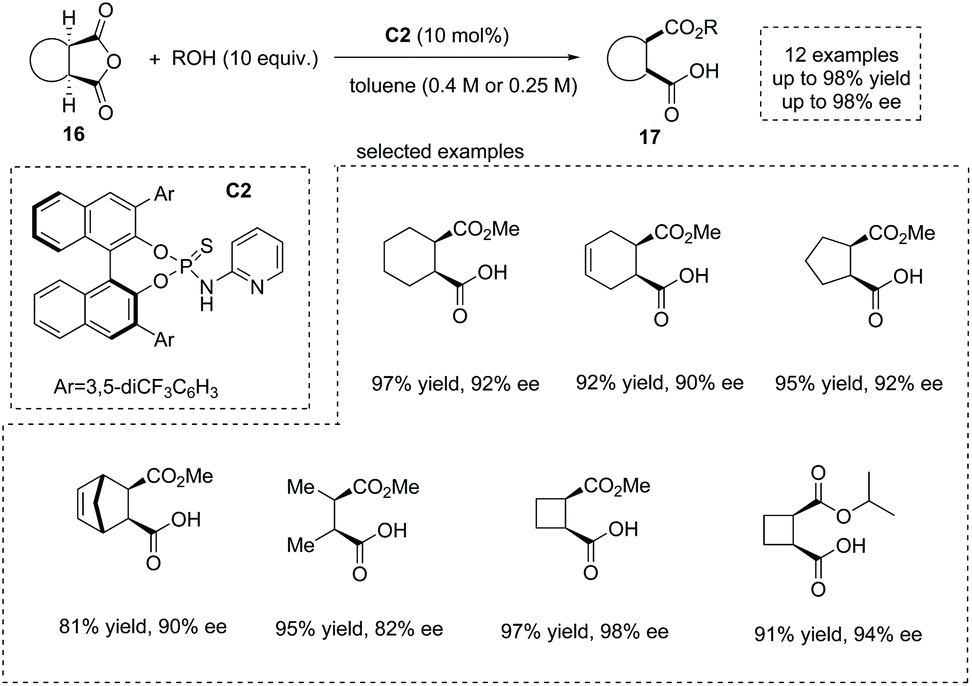
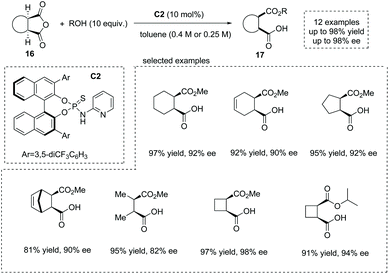
The development of novel and efficient catalysts was a main theme in the field of catalytic asymmetric ring-opening of meso-anhydrides. In 2010, List and co-workers synthesized a series of bifunctional organocatalysts bearing a Brønsted base site and a Brønsted acid site, and evaluated their performance in catalytic asymmetric methanolysis of cis-1,2-cyclohexanedicarboxylic anhydride in toluene. They found that organocatalyst C2 with a basic pyridinyl moiety and an acidic amide moiety was the superior catalyst,22 giving the cyclic α-chiral carboxylic acids 17 with exceedingly good results from the anhydrides 16 (13 examples, 81–98% yields and 82–98% ee) (Scheme 11). Notably, mono-, bi- and tricyclic succinic anhydrides are suitable substrates. Besides methanol, other alcohols such as ethanol, 1-propanol, 2-propanol and propargyl alcohol are also effective nucleophiles in the ring opening of succinic anhydrides. Mechanistically, the synergistic activation of both the alcohol nucleophile and anhydride electrophile by the bifunctional catalyst C2 ensures the efficiency and selectivity of the reaction. The author also demonstrated a short formal synthesis of bioactive (+)-grandisol by this strategy.
 |
| | Scheme 11 Bifunctional organocatalytic asymmetric synthesis of α-chiral carboxylic acids via ring-opening of meso-cyclic anhydrides. | |
In 2013, List et al. reported an elegant strategy for the synthesis of hemiester-type α-chiral carboxylic acids using organotextile catalysis.23 The photochemical immobilization of a cinchona-derived bifunctional sulfonamide catalyst on the textile nylon produced the solid catalyst OrganoTexCat C3, which exhibited impressive stability and catalytic activity in the alcoholytic desymmetrization of meso-anhydrides. Mono-, bi- and tricyclic anhydrides 16 were smoothly converted into α-chiral carboxylic acids 17 with excellent results (up to 94% ee) (Scheme 12). The catalyst also displays excellent recyclability that after two hundred cycling times, the product was still obtained with comparable enantioselectivity to the homogeneous catalysis conditions.
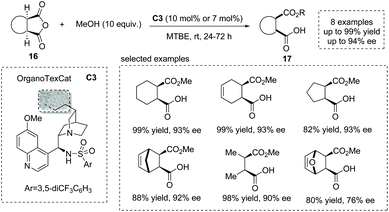 |
| | Scheme 12 Organotextile catalysis strategy for the asymmetric synthesis of α-chiral carboxylic acids. | |
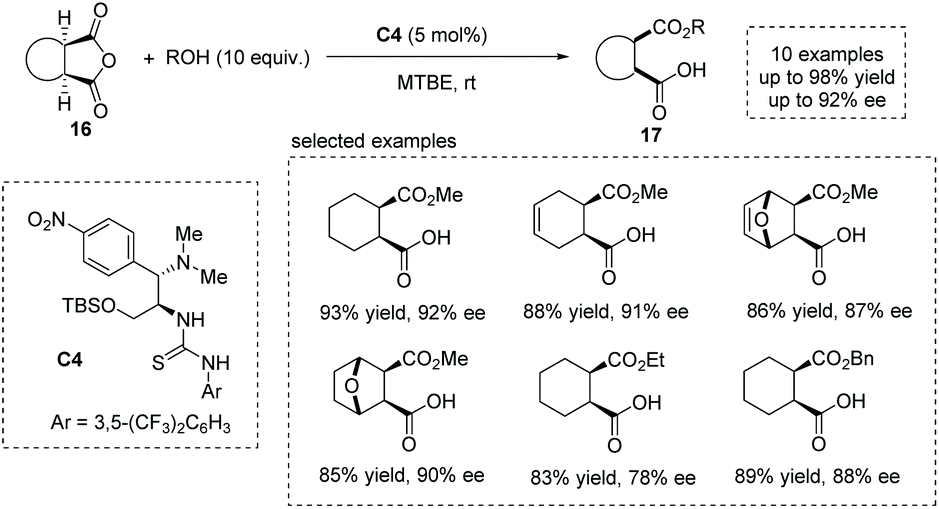
Based on their previous findings,20f in 2016, Chen and co-workers developed a new chloramphenicol-based tertiary amine thiourea bifunctional catalyst C4 for the asymmetric ring-opening of meso-anhydride,24 yielding a series of hemiester-type α-chiral carboxylic acids efficiently (Scheme 13). A bifunctional activation mechanism was proposed to account for the reactivity and selectivity observed.
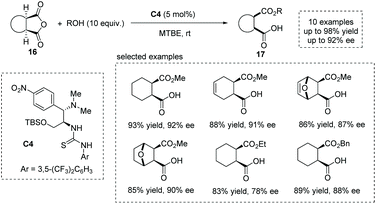 |
| | Scheme 13 Asymmetric synthesis of α-chiral carboxylic acids via chloramphenicol-based tertiary amine thiourea catalyzed ring-opening of meso-cyclic anhydrides. | |
The carbon-centered nucleophiles also could be applicable to the asymmetric ring-opening of cyclic anhydrides to install α-chiral carboxylic acids. Rovis et al. reported that Ni, Pd and Rh complexes were robust catalysts for the asymmetric ring-opening of meso-cyclic anhydrides to construct γ-keto α-chiral carboxylic acids in good yields and ees.25 Recently, Doyle, Rovis and co-workers reported that under irradiation with 34 W blue LEDs, the synergistic catalysis of a Ni salt and a photocatalyst for the asymmetric ring opening of cyclic anhydrides 16 with benzyl trifluoroborates 18 could be achieved,26 delivering γ-keto α-chiral carboxylic acids 19 in good yields and ees (20 examples, up to 90% yield, up to 94% ee) (Scheme 14a). They exploited 4CzIPN, Ni(cod)2 and PhBox L5 as the photocatalyst, Ni catalyst and chiral ligand, respectively. In this process, the benzylic radical could be generated by photo-oxidation of benzyl trifluoroborates. The authors stated that Ni0 oxidative addition to the anhydride, single-electron oxidation of the formed NiII to a NiIII species by the benzylic radical, and subsequent reductive elimination form a likely reaction pathway. The Ni0 oxidative addition to the anhydride was the ee-determining step (Scheme 14b). This chemistry represents a mild catalytic monofunctionalization of cyclic anhydrides and is a significant complement to the anhydride desymmetrization chemistry.
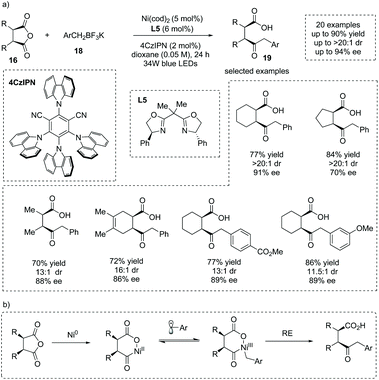 |
| | Scheme 14 Asymmetric synthesis of γ-keto α-chiral carboxylic acids via dual Ni- and photoredox-catalyzed desymmetric benzylation of meso-cyclic anhydrides. | |
3.2 Cyclic anhydrides as nucleophiles
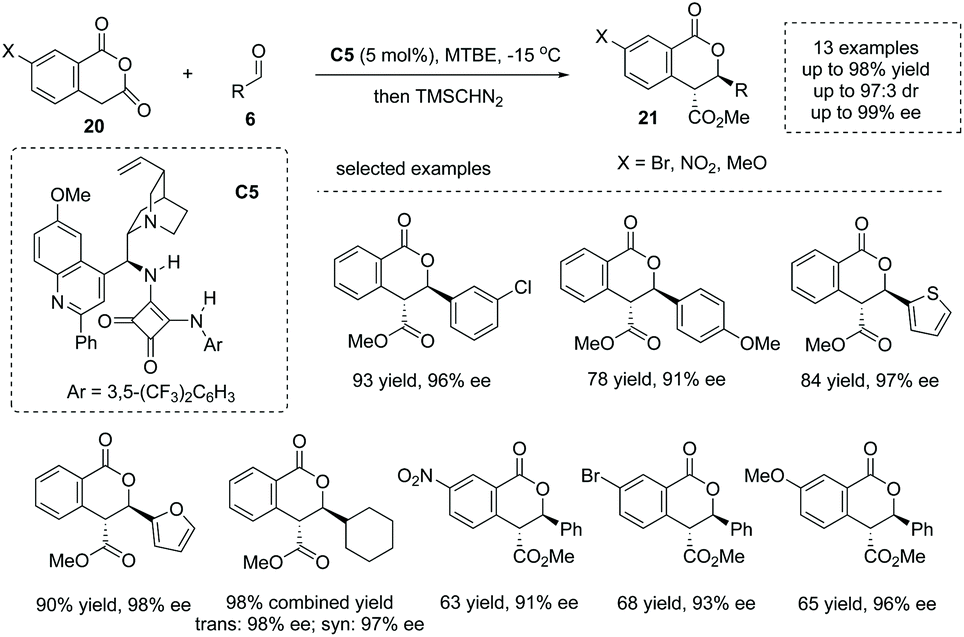
Enolizable anhydrides such as succinic and glutaric anhydrides could also behave as carbon-centered nucleophiles in annulation and multicomponent reactions.27 The addition of enolizable anhydride to certain electrophiles in the presence of a chiral Brønsted base catalyst is another significant strategy to form an α-stereogenic carboxylic acid scaffold. When aldehydes are exploited as electrophiles, an intriguing formal [4 + 2] process could be initiated by the aldol-type addition. Early in 2012, Connon and co-workers reported the first highly enantio- and diastereoselective catalytic asymmetric annulation of homophthalic anhydrides 20 with aryl aldehydes under the influence of the bifunctional tertiary amine squaramide catalyst C5.28 The reaction proceeded through an aldol-type addition of anhydride enolate to aldehyde, followed by an intramolecular lactonization process (Scheme 15). The α-chiral carboxylic acids with a biologically significant dihydro-isocoumarin backbone29 were readily obtained with exceedingly good results in terms of yields and enantioselectivities (13 examples, 78–98% yields, 91–99% ee) (to facilitate the purification and HPLC procedure, the esterification of the carboxylic acid products was performed). Mechanistically, the bifunctional general base–general acid activation mode, where the tertiary amine moiety could activate the homophthalic anhydride nucleophile by deprotonation–enolate formation and the squaramide moiety could activate the aldehyde electrophile by hydrogen bonding interactions, accounts for the reaction outcome observed. Later, the author extended this chemistry via variation of the nucleophiles from homophthalic anhydride to simple aryl-substituted succinic anhydride, which smoothly reacted with aldehydes and activated ketones yielding a special α-chiral carboxylic acid product (paraconic acid) containing a γ-butyrolactone backbone with excellent stereocontrol.30
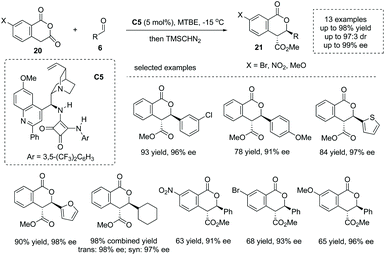 |
| | Scheme 15 Asymmetric synthesis of cyclic carboxylic acids via bifunctional Brønsted base catalyzed addition of homophthalic anhydrides to aldehydes. | |
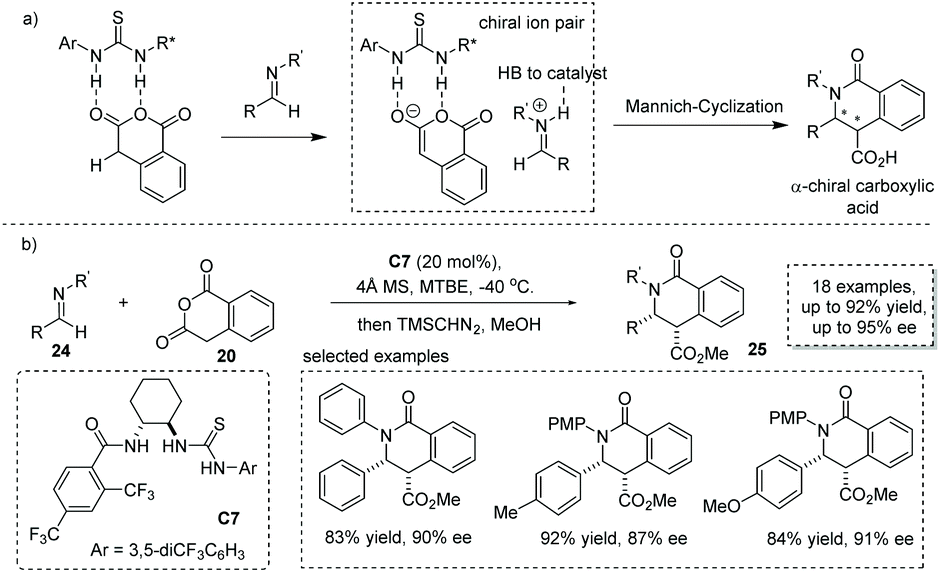
When changing the electrophile from aldehyde and ketone to imine in cycloadditions of enolizable anhydrides, valuable substituted chiral carboxylic acids with a lactam scaffold could be obtained.31 Previously, two possible reaction pathways including (1) the iminolysis of anhydrides–cyclization and (2) iminium enolate-mediated [4 + 2] cycloaddition were proposed for this transformation, which make the catalytic asymmetric variant of this reaction challenging. Connon and co-workers, in 2016, reported that cinchona-derived urea C6 was an efficient catalyst for the asymmetric formal [4 + 2] annulation of homophthalic anhydride 20 and N-mesyl aldimines 22 (Scheme 16),32 and the chiral carboxylic acids with a dihydroisoquinone core were readily produced (to facilitate the purification and HPLC procedure, the esterification of the carboxylic acid products was performed). The key to success lies in the use of N-mesyl aldimines, while more basic imines always undergo rapid uncatalyzed reactions and the imines with larger N-sulphonyl substituents such as Ts imine give the cyclic chiral carboxylic acids with lower ees. Although the diastereocontrol of this reaction is poor, this chemistry demonstrates the first catalytic asymmetric cycloaddition of enolizable anhydride with imines.
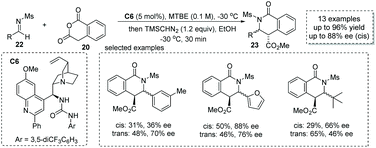 |
| | Scheme 16 Asymmetric synthesis of α-chiral carboxylic acids via organocatalytic addition of homophthalic anhydride to imines. | |
Based on one possible reaction pathway that the reaction proceeds via iminium-enolate intermediates, Seidel and co-workers successfully developed a hydrogen bond donor C7 catalyzed asymmetric formal [4 + 2] cycloaddition of homophthalic anhydride with aryl-protected imine 24.33 The key to success lies in the catalyst bearing two hydrogen-bonding donor sites enabling synergistic activation of both the nucleophile and electrophile via ion pair interactions (Scheme 17a). A series of α-chiral carboxylic acids with a substituted isoquinoline core was readily obtained in satisfactory results with the ee ranging from 66% to 95% (to facilitate the purification and HPLC procedure, the esterification of the carboxylic acid product was performed). DFT calculations revealed that the bifunctional catalyst with two hydrogen bond donor sites exerts a synergistic stabilization effect on the ion pair derived from the homophthalic anhydride and catalyst (Scheme 17b). Recently, Han and co-workers found that under the catalysis of bifunctional cinchona-derived thiourea, homophthalic anhydride reacted with isatin-derived N-Boc imines in a Mannich-type addition,34 and no cycloadduct was observed. This reaction provides a general method for the asymmetric synthesis of oxindole-containing chiral carboxylic acids with a stereogenic center at the α position.
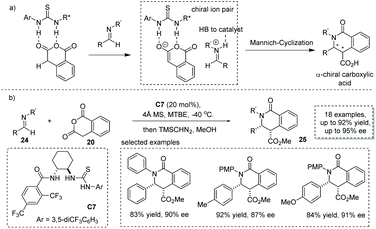 |
| | Scheme 17 Asymmetric synthesis of α-chiral carboxylic acids via the ion-pairing strategy. | |
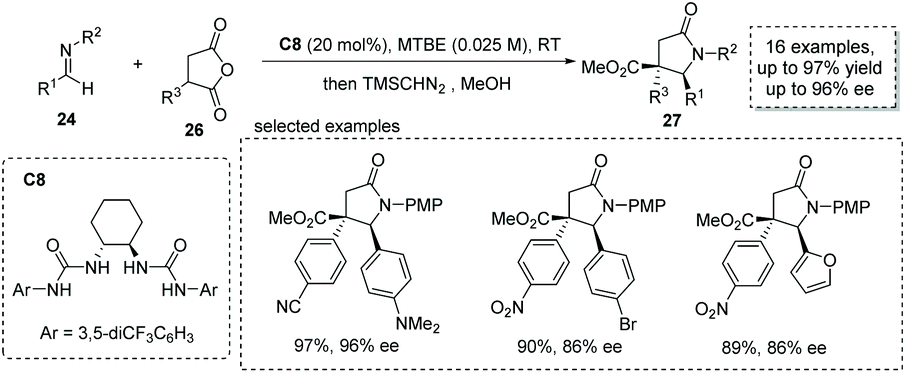
Based on a similar strategy, Connon and co-workers, in 2019, achieved a formal [3 + 2] cycloaddition between α-substituted succinic anhydride 26 and p-methoxyphenyl (PMP)-protected aldimine 24 under the catalysis of chiral bisurea C8, delivering the α-chiral carboxylic acids with a trisubstituted γ-lactam backbone in high yields and ees (16 examples, up to 97% yield, up to 96% ee, Scheme 18).35 The reaction also exhibits broad substrate scope and multiple functional groups are tolerated (to facilitate the purification and HPLC procedure, the esterification of the carboxylic acid products was performed).
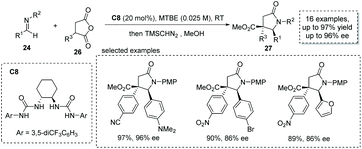 |
| | Scheme 18 Asymmetric synthesis of α-chiral carboxylic acids via bisurea catalyzed formal [3 + 2] cycloaddition. | |
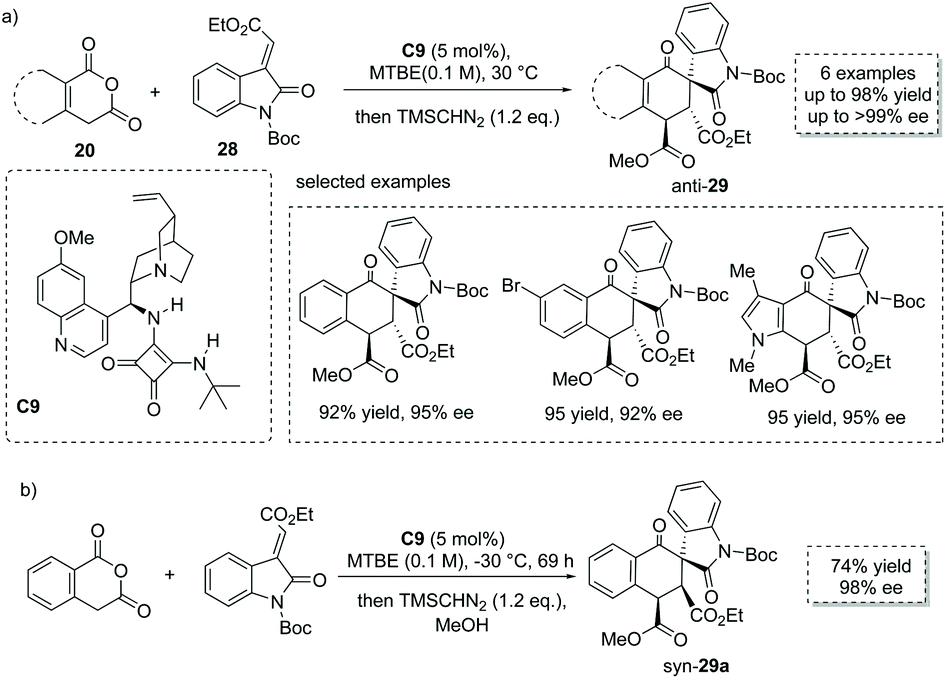
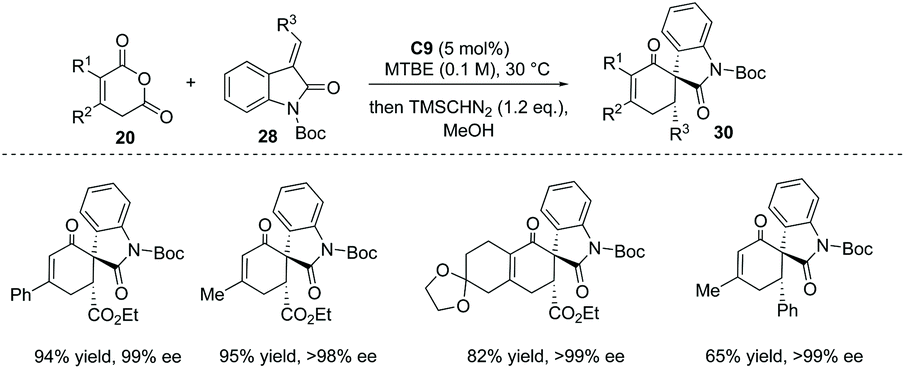
The Tamura cycloaddition refers to the reaction of homophthalic anhydride with activated alkynes and alkenes at high temperature to construct fused ring systems.36 When activated alkenes was utilized, the reaction could potentially produce cyclic carboxylic acids with a quaternary or tertiary stereocenter at the α position with respect to the carboxyl group. With the development of organocatalysis over the past decades, several organocatalytic asymmetric Tamura reactions have been implemented.37 In 2014, Connon and co-workers developed a bifunctional squaramide C9 catalyzed asymmetric Tamura-type reaction between enolizable cyclic anhydride and isatin-derived activated alkenes 28,37a giving rise to spirooxindole cyclohexene structural motifs with a free carboxyl group (Scheme 19a). This chemistry represents the first catalytic asymmetric Tamura reaction and the desired products were obtained in high yields and ees (to facilitate the purification and HPLC procedure, the esterification of the carboxylic acid products was performed). Notably, an interesting temperature-controlled diastereoselectivity switch was observed: at 30 °C products with an anti-configuration were preferentially obtained, whereas the syn diastereomer was selectively formed at −30 °C (Scheme 19b). The authors reason that nucleophile-catalyzed isomerization of the alkylidene oxindole starting materials may be responsible for this observed phenomenon.
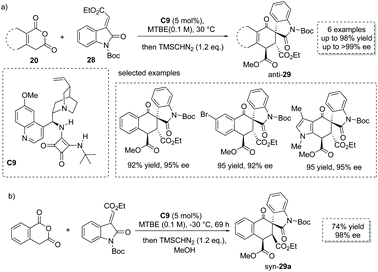 |
| | Scheme 19 Asymmetric synthesis of α-chiral carboxylic acids via bifunctional squaramide catalyzed Tamura reaction. | |
Interestingly, when some enolizable cyclic anhydrides such as phenyl glutaconic anhydrides were used as nucleophiles, the α-chiral carboxylic acids formed underwent immediate decarboxylation, yielding substituted cyclohexanone 30 as the ultimate products in excellent ees (Scheme 20).
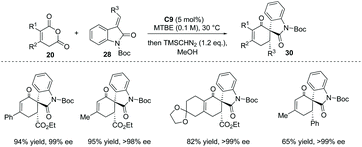 |
| | Scheme 20 Decarboxylation of the formed α-chiral carboxylic acids to afford substituted cyclohexenones. | |
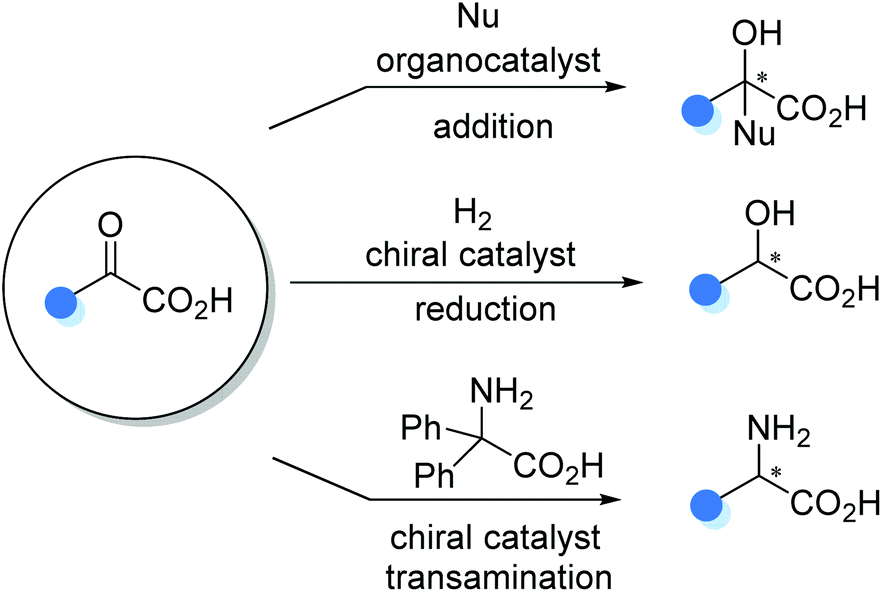
4. Catalytic asymmetric functionalization of α-keto acids
The carboxyl group in α-keto acids significantly improves the electrophilic reactivity of the ketone functionality, making α-keto acids competent prochiral electrophiles in asymmetric transformations. The organocatalytic aldol-type 1,2-addition, transition metal catalyzed asymmetric reduction, and organocatalytic asymmetric transamination are three typical α-keto acid-involved reactions for the preparation of α-substituted chiral carboxylic acids, such as α-hydroxy and α-amino carboxylic acids (Scheme 21).
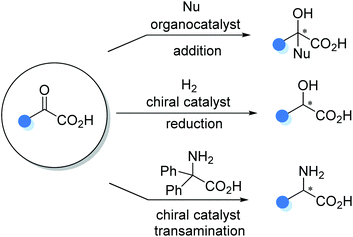 |
| | Scheme 21 Catalytic asymmetric functionalization of α-keto acids. | |
4.1 Catalytic asymmetric addition to α-keto acids
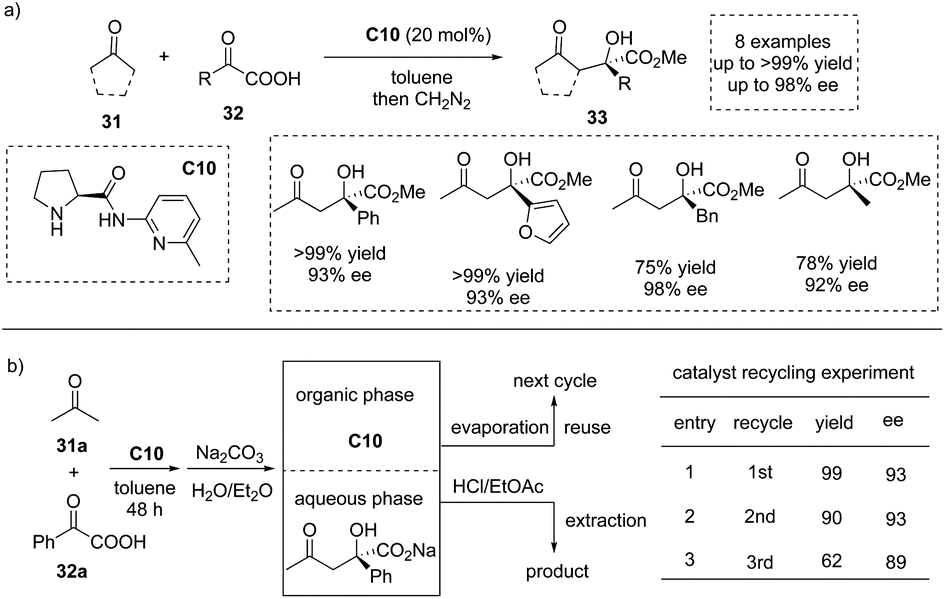
In 2006, Gong and co-workers reported that under the catalysis of an aminopyridine-containing L-prolinamide C10, the enamine-mediated asymmetric aldol-type addition of simple ketones 31 to aryl and alkyl α-keto acid 32 could take place with excellent enantioselectivities (Scheme 22a).38 The reaction proceeded smoothly to give rise to a series of chiral α-hydroxy carboxylic acids with a tetrasubstituted carbon at the α-position (to facilitate the purification and HPLC procedure, the esterification of the carboxylic acid products was performed). Interestingly, the product and the catalyst could be easily separated by a simple aqueous Na2CO3 quenching-phase separation procedure. The product exists in the basic aqueous phase and could be purified by an acidification and extraction work-up, while the catalyst could be recovered by a simple evaporation of the solvent to dryness. The catalyst recycling experiment revealed that after two cycles, the catalyst C10 still exhibited satisfactory catalytic performance (Scheme 22b).
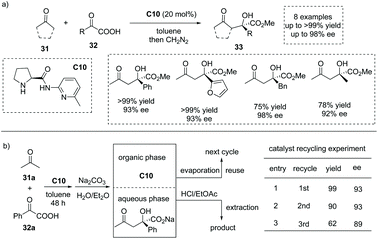 |
| | Scheme 22 Asymmetric synthesis of α-hydroxy carboxylic acids via secondary amine catalyzed aldol-type addition of ketones to α-keto acids. | |
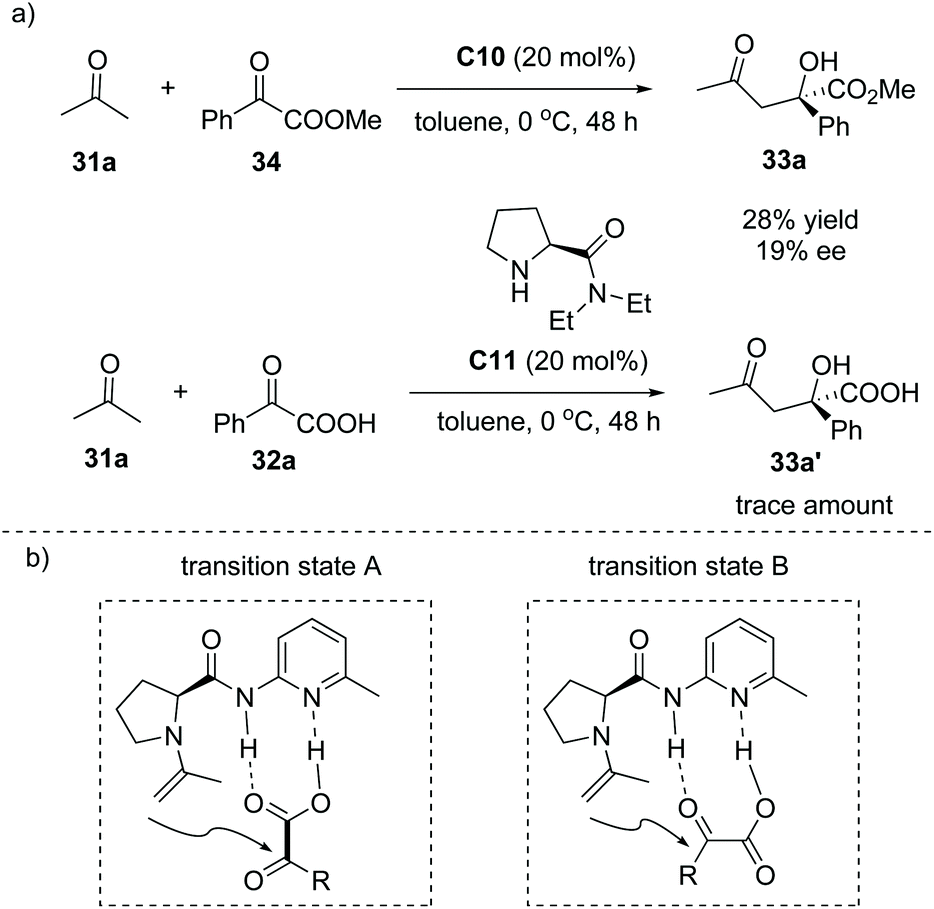
A mechanistic study revealed that when the α-keto ester 34 instead of α-keto acid 32a was used as the aldol acceptor, catalyst C10 gave inferior results (28% yield, 19% ee). The catalyst C11 lacking a hydrogen bond donor site gave only a trace amount of the product (Scheme 23a). In addition, a linear effect for the reaction of α-keto acid 32a and acetone with 20 mol% of C10 in toluene was observed, suggesting that only a single molecule of the catalyst was involved in the reaction. Subsequent DFT calculations implied that the proposed transition state B is much favorable compared to the proposed transition state A during the formation of the product38 (Scheme 23b). These experiments clearly revealed that the enamine activation and the double hydrogen bond interaction between the catalyst and α-keto acid both are responsible for the high efficiency and excellent selectivity of this reaction.
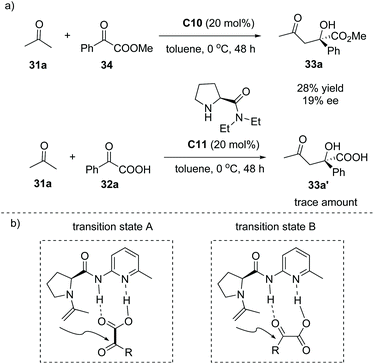 |
| | Scheme 23 Mechanistic study of the formation of 33a. | |
4.2 Catalytic asymmetric hydrogenation of α-keto acids
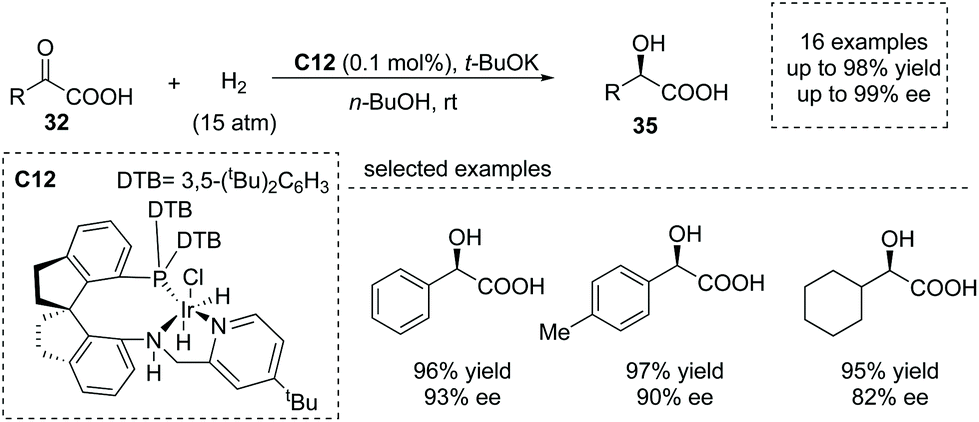
Transition metal complex catalyzed asymmetric hydrogenation of α-keto acids was another significant method to produce α-hydroxy chiral carboxylic acids.39 However, this is a challenging transformation owing to the undesired catalyst deactivation induced by the competitive coordination of the carboxyl group to the center metal. Che, Zhou and co-workers reported that using a highly efficient chiral Ir/SpiroPAP catalyst C12,40 the asymmetric hydrogenation of α-keto acids occurred to give the desired enantioenriched α-hydroxy acids 35 (Scheme 24). The products were made according to this protocol with a high TON (as high as 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and excellent enantioselectivity (up to 99.2% ee) under mild conditions. Interestingly, the fast reduction of the chloro-substituted α-keto acid, 2-chlorobenzoylformic acid, could give rise to chiral 2-chloromandelic acid, a key intermediate of a platelet aggregation inhibitor, Clopidogrel, further highlighting the potential applications of this reaction.
000) and excellent enantioselectivity (up to 99.2% ee) under mild conditions. Interestingly, the fast reduction of the chloro-substituted α-keto acid, 2-chlorobenzoylformic acid, could give rise to chiral 2-chloromandelic acid, a key intermediate of a platelet aggregation inhibitor, Clopidogrel, further highlighting the potential applications of this reaction.
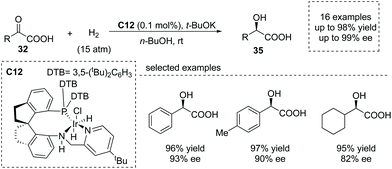 |
| | Scheme 24 Asymmetric synthesis of α-hydroxy carboxylic acids via Ir complex catalyzed hydrogenation of α-keto acids. | |
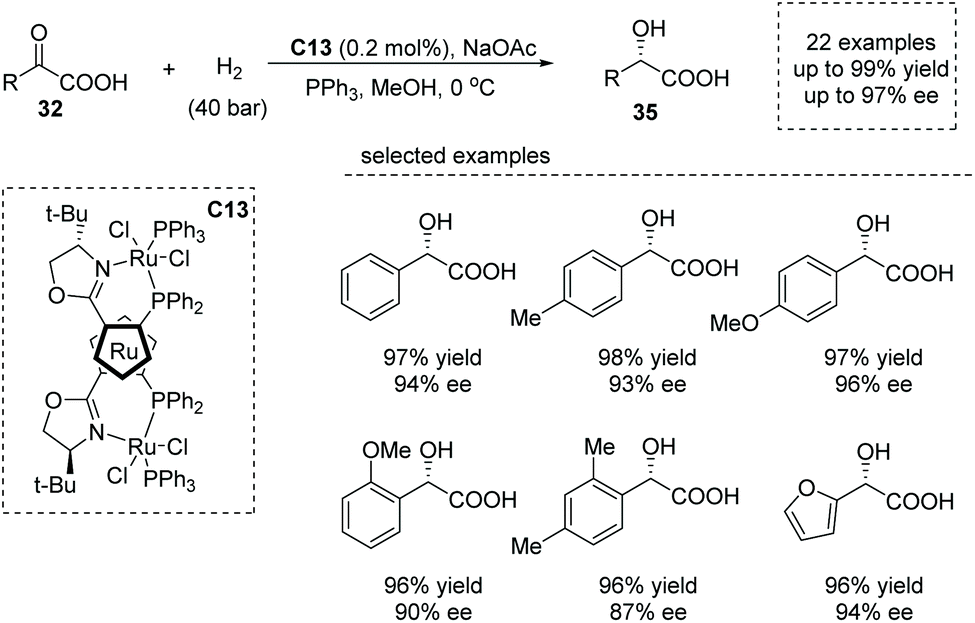
Shortly after, Liu and co-workers reported a similar reaction using 0.2 mol% of a ruthenocenyl phosphino–oxazoline–ruthenium complex (RuPHOX-Ru) catalyst C13,41 also giving rise to chiral α-aryl α-hydroxy carboxylic acids 35 from various substituted benzoylformic acids in high yields and with high enantioselectivities (up to 99% yield, up to 97% ee, Scheme 25). A wide range of functional groups could be tolerated in this transformation, delivering the corresponding products, substituted chiral mandelic acids in a highly efficient manner. The reaction was amenable for gram-scale synthesis with only 0.02 mol% of catalyst dosage, and the obtained chiral carboxylic acids could be directly applied to further synthetically useful transformations. The author further demonstrated the utility of the current reaction in the facile synthesis of the key intermediate of an orally active β3 adrenoceptor agonist, Mirabegron.
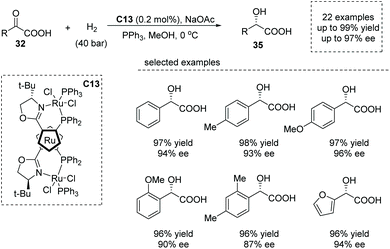 |
| | Scheme 25 Asymmetric synthesis of α-hydroxy carboxylic acids via Ru complex catalyzed hydrogenation of α-keto acids. | |
4.3 Catalytic asymmetric transamination of α-keto acids
The transamination reaction of α-keto acids with substituted primary amines is a biomimetic strategy to make substituted α-amino acids.42 Despite several achievements using activated ketones as substrates, the catalytic asymmetric transamination reaction of α-keto acids is limited. Since pyridoxal/pyridoxamine and their phosphates are the coenzymes of transaminases, in principle, pyridoxal or pyridoxamine derivatives containing a chiral control moiety may potentially act as chiral catalysts for asymmetric transamination processes.
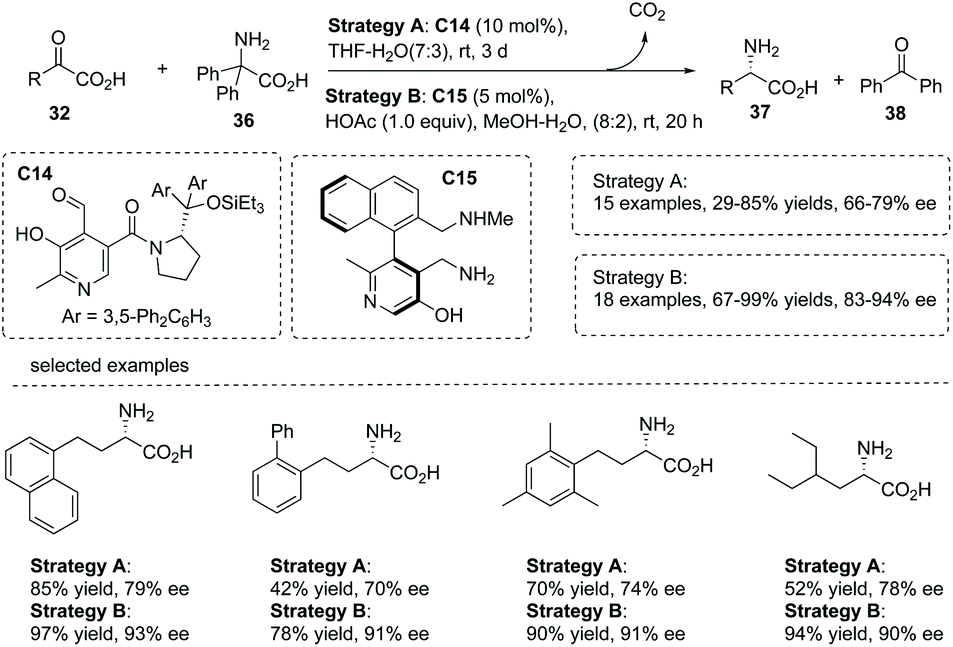
In 2015, an elegant work involving the use of chiral pyridoxal as an organocatalyst in the biomimetic transamination of α-keto acids was reported by Zhao and co-workers.43 This transformation was catalyzed by a chiral pyridoxal C14, which was newly synthesized from commercially available pyridoxine and α,α-diarylprolinols. A series of chiral α-alkyl α-amino acids 37 was readily obtained in 47–90% yields with up to 87% ee under very mild conditions (Scheme 26, strategy A). The current chemistry represents a rare example of exploiting stereochemically defined small molecules as chiral catalysts to mimic the behavior of transaminase in half-transamination. Inspired by the remarkable cooperative effect of pyridoxal/pyridoxamine and the Lys residue in enzymatic transformations, the author further found that under the catalysis of axially chiral pyridoxamine C15 which has a lateral amine chain, the asymmetric transamination of α-keto acids gave rise to chiral α-alkyl α-amino acids 37 with largely improved results in terms of yields and enantioselectivities compared to catalyst C14 (Scheme 26, strategy B).
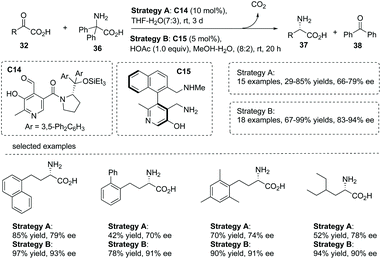 |
| | Scheme 26 Organocatalytic asymmetric synthesis of chiral α-amino carboxylic acids via biomimetic transamination of α-keto acids. | |
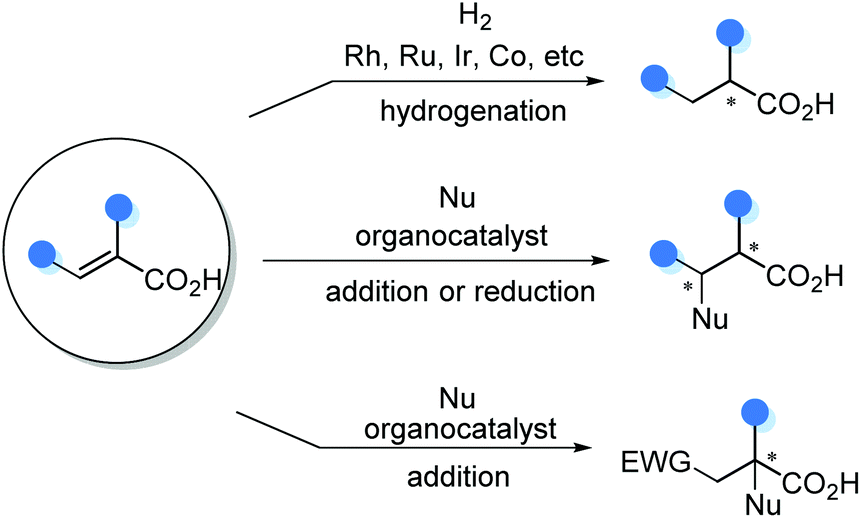
5. Catalytic asymmetric functionalization of substituted α,β-unsaturated acids
The catalytic asymmetric functionalization of substituted α,β-unsaturated acids offers new avenues to make α-chiral carboxylic acids. While chiral Rh, Ru, Ir, Co and other metal complex catalyzed asymmetric hydrogenation of α-substituted acrylic acids has been well-established, recently organocatalytic conjugate addition and reduction of substituted acrylic acids are emerging as a complementary strategy (Scheme 27).
 |
| | Scheme 27 Catalytic asymmetric functionalization of substituted α,β-unsaturated acids. | |
5.1 Transition metal catalyzed asymmetric hydrogenation
The catalytic asymmetric hydrogenation of α-substituted acrylic acid represents a direct, atom-economical and relatively mature approach to enantiopure α-chiral free carboxylic acids.4a,44 Since the pioneering work using the Rh catalyst,45 other metals46 such as Ru,47 Ir48 and recently Co49 complexes have also been applied successfully to the synthesis of α-chiral carboxylic acids with diverse substituents at the α position such as amino, alkyl, aryl and alkoxyl groups.
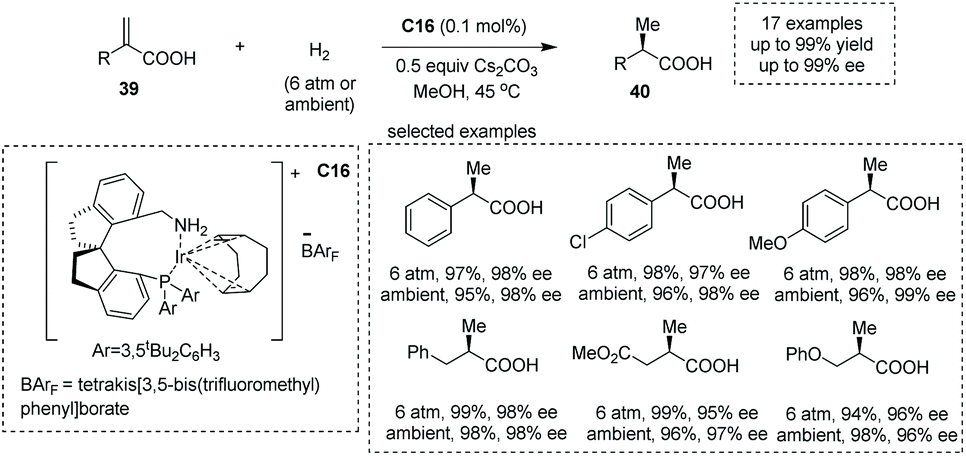
Zhou and co-workers, in 2012, demonstrated that under the catalysis of iridium complexes, an asymmetric hydrogenation of α-alkyl substituted acrylic acids 39 readily occurred to produce α-methyl substituted carboxylic acids 40 in methanol solvent (Scheme 28).48d A new class of chiral P and N ligands with a spirocyclic backbone (chiral spiro aminophosphine ligand) C16 was chosen as the superior chiral ligand for asymmetric induction, and a series of α-methyl carboxylic acids with diverse patterns of substitution were obtained with excellent results (17 examples, 93–99% yields, 94–99% ees). In 2013, the same group reported a chiral spirophosphine oxazoline iridium complex catalyzed asymmetric hydrogenation of unsaturated N- and O-heterocyclic acids, giving rise to chiral heterocyclic carboxylic acids in MeOH in 94–99% yields with excellent ees ranging from 88% to 99%.48f
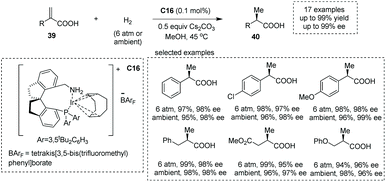 |
| | Scheme 28 Asymmetric synthesis of α-chiral carboxylic acids via chiral Ir complex catalyzed asymmetric hydrogenation. | |
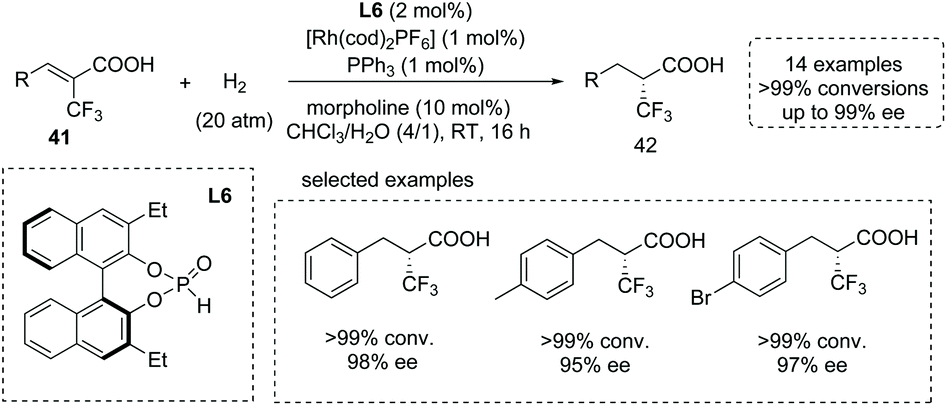
The introduction of trifluoromethyl groups into organic molecules could potentially change the properties of biological molecules, especially with the concurrent formation of a CF3-substituted stereocenter. An impressive protocol for the catalytic synthesis of α-CF3-substituted chiral carboxylic acids 42via a Rh(I)-catalyzed asymmetric hydrogenation of α-CF3 substituted α,β-unsaturated acids 41 was disclosed by Ding and co-workers in 2013. They exploited a chiral secondary phosphine oxide (SPO) L6 and an achiral triarylphosphine as the chiral and achiral ligand,45g respectively (Scheme 29). This mixed-ligand strategy allowed the two ligands to coordinate simultaneously to the central metal and gave the desired α-CF3-substituted propanoic acid derivatives with excellent results (15 examples, >99% conversion in most cases and 93–99% ee). The reactions were performed with 1 mol% Rh(I) catalyst in the mixed solvent of CHCl3 and water at room temperature with wide substrate scope. It is worth noting that while the β-CF3-substituted unsaturated acids were exploited as substrates, β-CF3-substituted chiral carboxylic acids could also be produced efficiently.
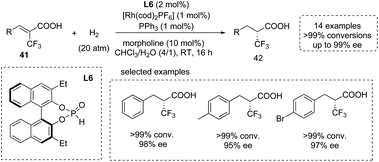 |
| | Scheme 29 Asymmetric synthesis of α-CF3-substituted chiral carboxylic acids via chiral Rh complex catalyzed asymmetric hydrogenation. | |
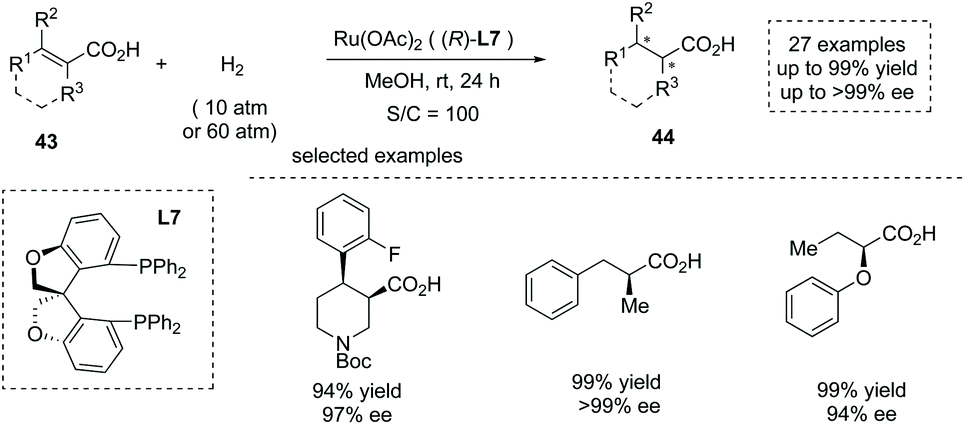
In 2020, Zhang, Chen and co-workers reported a Ru(OAc)2 catalyzed asymmetric hydrogenation of unsaturated acyclic acids 43 to produce α-chiral carboxylic acids 44 (Scheme 30).47e The oxa-spirocyclic diphosphine ligand L7 was synthesized and identified as the optimal ligand, and a series of unsaturated carboxylic acids including unsaturated N-heterocyclic acids, α-alkyl cinnamic acids, and α-aryloxy crotonic acids were smoothly converted into the corresponding chiral carboxylic acids with excellent results (27 examples, up to 99% yield and up to >99% ee). The author also demonstrated the application of the current chemistry to the asymmetric synthesis of several key intermediates of drugs such as Sacubitril and Artemisinin.
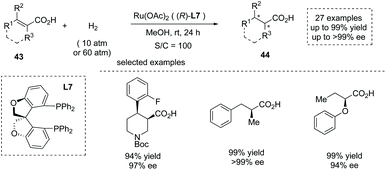 |
| | Scheme 30 Asymmetric synthesis of α-chiral carboxylic acids via chiral Ru complex catalyzed asymmetric hydrogenation. | |
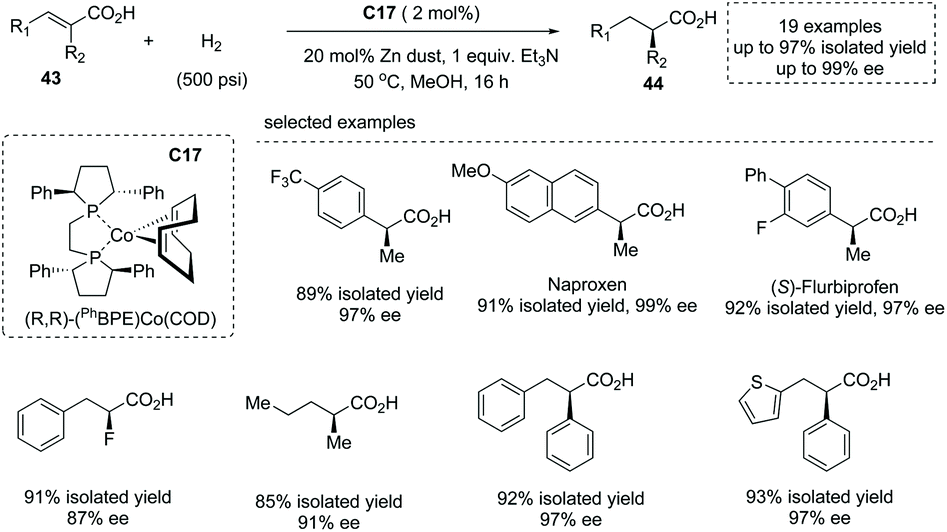
The first-row transition metal Co complex was also a competent catalyst for asymmetric hydrogenation of substituted unsaturated acids to produce α-chiral carboxylic acids. In early 2020, Chirik and co-workers reported a Co complex C17 catalyzed asymmetric hydrogenation of unsaturated acyclic acids49a (Scheme 31). The readily prepared bis(phosphine) cobalt (0) 1,5-cyclooctadiene was exploited as a precatalyst, and under the optimal reaction conditions structurally diverse di-, tri-, and tetra-substituted unsaturated acids 43 underwent enantioselective hydrogenation to afford α-stereogenic carboxylic acids 44 with excellent yields and enantioselectivities (21 examples, up to 97% yields, up to 99% ee). A detailed mechanistic study revealed that this reaction experienced homolytic H2 activation by the cobalt (0) species and cis addition of H2 across alkene double bonds. Using Earth-abundant and environmentally benign cobalt complexes, this chemistry provided an alternative to noble metal catalyzed asymmetric hydrogenation of unsaturated carboxylic acids.
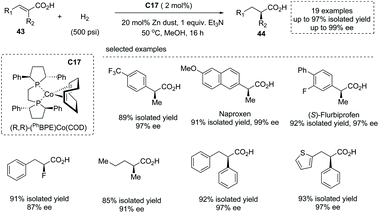 |
| | Scheme 31 Asymmetric synthesis of α-chiral carboxylic acids via chiral Co complex catalyzed asymmetric hydrogenation. | |
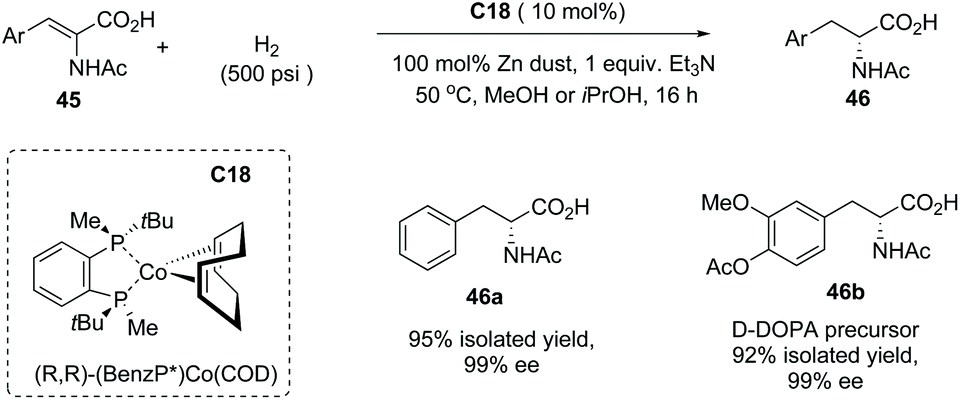
In addition to α-alkyl carboxylic acids, α-amino acid 46 also could be made by this method using the Co complex C18 as the catalyst from dehydro-α-amino acid derivatives 45 with excellent results (Scheme 32). Impressively, the D-DOBA precursor 46b could be efficiently made by this protocol in 92% yield with 99% ee.
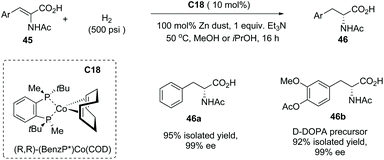 |
| | Scheme 32 Asymmetric synthesis of α-amino carboxylic acids via Co complex C18 catalyzed asymmetric hydrogenation. | |
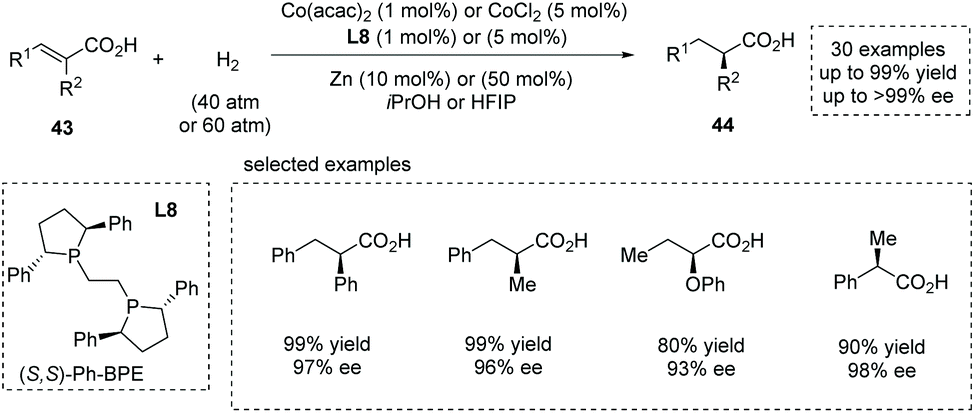
Also in 2020, Zhang, Chen and co-workers reported a novel synthesis of α-chiral carboxylic acids 44via Co(II)-catalyzed asymmetric hydrogenation of α-substituted unsaturated carboxylic acids 43.49b Readily available Co(acac)2 or CoCl2 was used as the Co(II) source. The highly electron-rich and sterically demanding diphosphine ligands proved to be catalytically active, and Ph-BPE L8 was identified as the optimal chiral ligand (Scheme 33). High yields and enantioselectivities are generally achieved for a wide range of substrates such as α-alkyl and α-aryl cinnamic acids, α-alkoxy- and α-aryloxy-substituted unsaturated acids, and α-substituted acrylic acids (30 examples, up to 99% yield and up to 99% ee). The value of the obtained α-chiral carboxylic acids was further demonstrated by the facile (formal) synthesis of several drugs such as (S)-Equol, Rupintrivir, Sacubitril, Naproxen, Ibuprofen and Artemisinin. Notably, a Zn additive acted as a one-electron reductant and was beneficial for achieving full conversion with low catalyst dosage. Mechanistic studies revealed that the carboxyl group played a pivotal role in controlling the reactivity and enantioselectivity via coordination with the center metal. The author later extended this chemistry to the asymmetric hydrogenation of challenging cyclic unsaturated carboxylic acids, providing a facile method for the synthesis of α-chiral cyclic carboxylic acids.49c
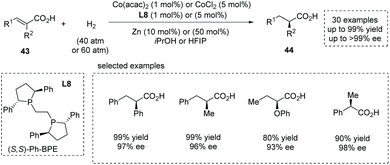 |
| | Scheme 33 Asymmetric synthesis of α-chiral carboxylic acids via Co(II)-catalyzed asymmetric hydrogenation. | |
5.2 Organocatalyzed asymmetric conjugate addition and reduction
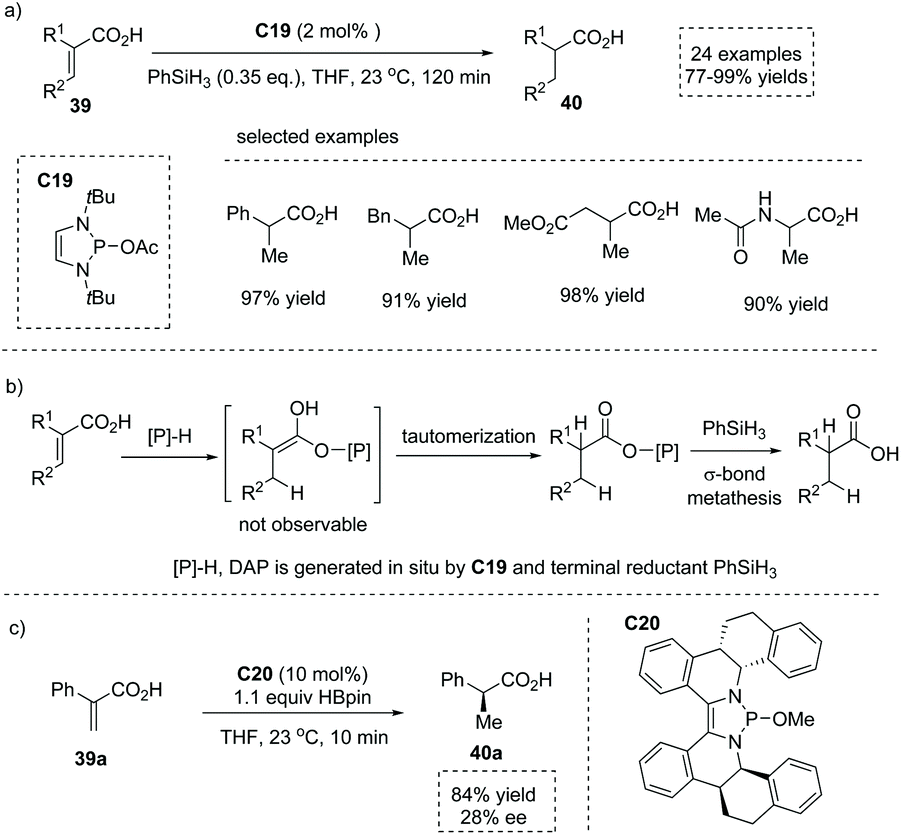
As mentioned above, the chiral transition metal complex catalyzed asymmetric hydrogenation of prochiral substituted unsaturated acids plays a dominant role in the production of α-stereogenic chiral acids. However, despite the great myriad of transformations promoted by various organocatalysts, the metal-free catalytic asymmetric conjugate addition or reduction of unsaturated carboxylic acids to produce α-chiral carboxylic acids still represents an attractive yet challenging goal. As early as 1988, Yamamoto reported a tartaric acid-derived chiral acyloxyborane catalyzed asymmetric Diels–Alder reaction of cyclopentadiene with acrylic acid,50 affording α-chiral carboxylic acid with a bridged bicyclic [2.2.1] scaffold (one example, 93% yield, endo/exo = 96:2, 78% ee). More than thirty years later, in 2020, Cramer and co-workers found that 1,3,2-diazaphospholene (DAP) exhibited potent nucleophilicity and remarkably low basicity, and thus was capable of selectively delivering the hydride faster than it deprotonated the acid in the catalytic conjugate reduction of substituted α,β-unsaturated acids 39 (Scheme 34a).51 The catalytically active DAP was in situ generated from precatalyst C19 and terminal reductant silane PhSiH3. A number of α-stereogenic carboxylic acids 40 including α-alkyl and α-aryl propionic acids, cyclohexanecarboxylic acids, 3-phenylpropionic acids and succinic acids could be made by this methodology. Mechanistic studies revealed that the reaction proceeded via a conjugate reduction involving DAP hydride to an α,β-unsaturated acid, immediate proton tautomerization and a subsequent bond metathesis process (Scheme 34b). Significantly, when a chiral DAP precursor C20 was synthesized and evaluated, asymmetric induction was observed and the enantio-enriched product was obtained in 84% yield and 28% ee (Scheme 34c).
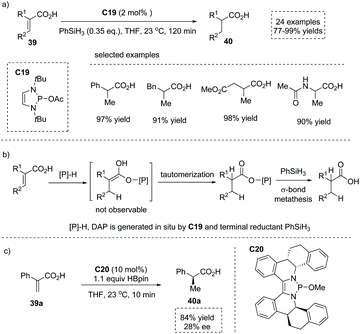 |
| | Scheme 34 Synthesis of α-stereogenic carboxylic acids via DAP catalyzed conjugate reduction of substituted α,β-unsaturated acids. | |
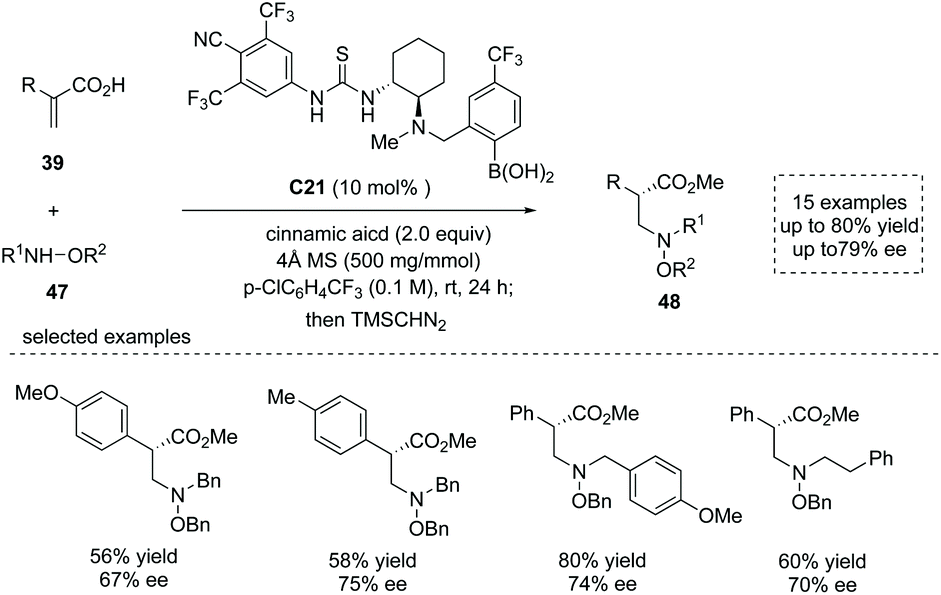
Based on their previous success in the organocatalytic direct conjugate addition of nucleophiles to unsaturated carboxylic acids,52 Takemoto and co-workers recently reported a chiral thiourea-boronic acid hybrid catalyst C21 catalyzed asymmetric aza-Michael addition–protonation to α-substituted acrylic acids 39.53 The substituted hydroxylamines 47 were used as nitrogen-centered nucleophiles and two equivalents of cinnamic acids were used as an additive. The special α-chiral carboxylic acids, β2-amino acid derivatives 48 with diverse α substituents, were readily obtained in moderate yields and ees (15 examples, up to 80% yield and up to 79% ee; to facilitate the purification and HPLC procedure, the esterification of the carboxylic acid products was performed) (Scheme 35). The advantage of α-chiral free carboxylic acids for further transformations was demonstrated by a two-step synthesis of the natural product (−)-nakinadine B from atropic acid in 28% overall yield. Mechanistically, two molecules of cinnamic acid coordinated to the catalyst boron atom forming a resting state of the catalyst, and the reaction proceeded via (1) cinnamic acid/atropic acid ligand exchange; (2) hydrogen bond-directed aza-Michael addition of substituted hydroxylamine, (3) subsequent ee-determining protonation and (4) final cinnamic acid/product ligand exchange to regenerate the catalyst and deliver the β2-amino acid product.
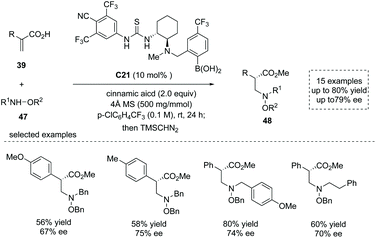 |
| | Scheme 35 Synthesis of α-chiral carboxylic acids via hybrid catalyst C21 catalyzed asymmetric aza-Michael addition/protonation to α-substituted acrylic acids. | |
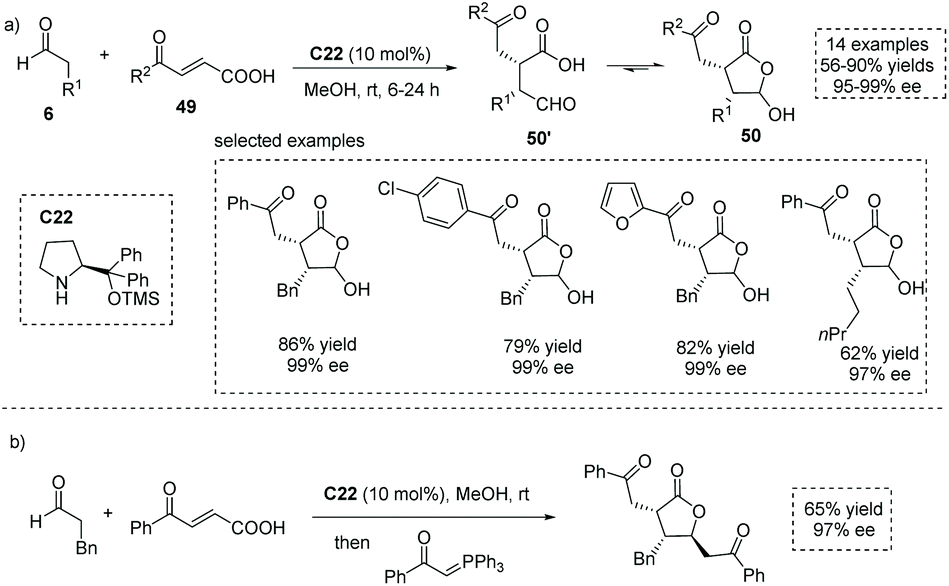
The conjugate addition of carbon-centered nucleophiles to ester activated enones is a commonly exploited strategy to form α-stereogenic centers with respect to the ester functionality.54 In principle, the conjugate addition of certain nucleophiles to carboxylic acid-activated enones (also known as acyl-activated acrylic acid) could produce α-stereogenic chiral carboxylic acids. In 2015, Xu and co-workers reported a chiral secondary amine, Jørgensen–Hayashi catalyst C22 catalyzed asymmetric Michael addition of enolizable aldehydes to carboxylic acid-activated enones 49 (Scheme 36a),55 giving trisubstituted γ-lactone 50 with a hemiacetal moiety in high yields with excellent enantioselectivities (14 examples, up to 90% yield, up to 99% ee). It is worth noting that the carboxylic acid could act as an acidic cocatalyst, thus circumventing the use of additional acidic additives in this enamine-mediated process. Owing to the labile nature of the hemiacetal moiety, the obtained highly functionalized products could be regarded as a type of special α-chiral carboxylic acid 50′ with an aldehyde tether, which could take part in several synthetically valuable transformations (Scheme 36b). On varying the nucleophiles from the enolizable aldehyde to 4-methyl-2-pentenal, a dienamine-mediated γ,γ-coupling of enals with carboxylic acid-activated enones proved feasible.56 Recently, Liu and co-workers found that this chemistry could be applied to the synthesis of complex bicyclic architectures, tetrahydrofuro[2,3-b]furan-2(3H)-one derivatives having four stereogenic centers.57
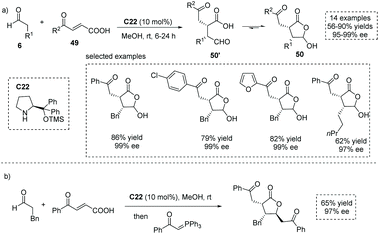 |
| | Scheme 36 Asymmetric synthesis of functionalized hemiacetal-containing lactones as masked α-chiral carboxylic acids via asymmetric conjugate addition of aldehydes to carboxylic acid-activated enones. | |
6. Miscellaneous transformations
The development of new strategies for the catalytic asymmetric synthesis of α-chiral carboxylic acids is a significant task in synthetic organic chemistry. Several strategies involving new starting materials with diverse oxidation levels have been reported recently.
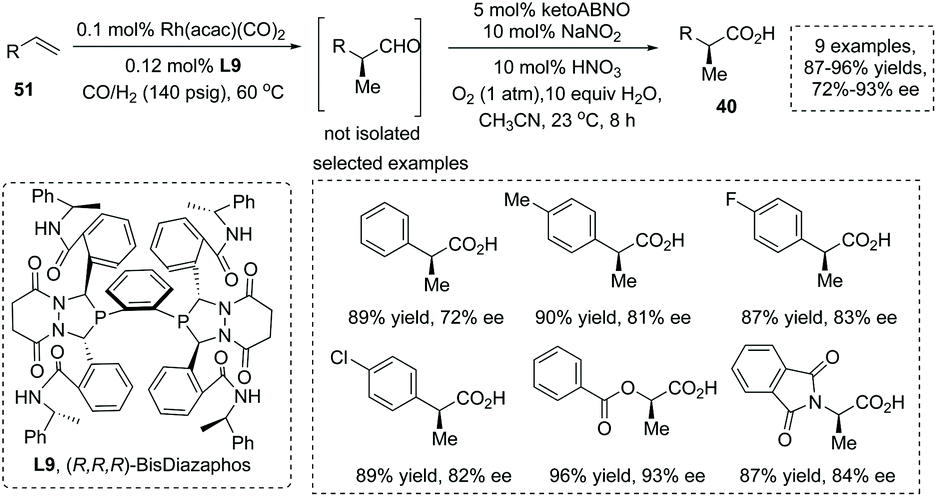
The mild oxidation of α-chiral aldehydes was an efficient strategy to generate α-chiral carboxylic acids. Stahl and Landis et al., in 2016, reported a novel protocol involving nitroxyl/NOx catalytic aerobic oxidation of an aldehyde coupled with Rh catalyzed asymmetric hydroformylation (AHF) of an alkene, providing a practical means for the asymmetric synthesis of α-chiral carboxylic acids from simple alkenes 51.58 The AHF reaction was carried out under known conditions with 0.1 mol% Rh(acac)(CO)2 and 0.12 mol% chiral BisDiazaphos L9 (1 mmol alkene, 1:1 CO/H2, 60 °C) and proceeded with quantitative conversion of the alkenes to α-chiral aldehydes. For the oxidation step, KetoABNO (9-azabicyclo[3.3.1]nonan-3-one N-oxyl) and NaNO2 were identified as the optimal nitroxyl and NOx sources, respectively. It is noteworthy that the utilization of metal-free oxidants under acidic conditions avoided the epimerization of sensitive α-chiral aldehyde intermediates, furnishing the corresponding α-stereogenic carboxylic acids with high overall yield and enantioselectivity (9 examples, 87–96% yields, 72–93% ee) (Scheme 37). Furthermore, the reaction also could be applied to gram-scale synthesis.
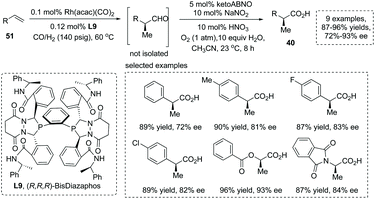 |
| | Scheme 37 Asymmetric synthesis of α-stereogenic carboxylic acids via sequential hydroformylation/oxidation of alkenes. | |
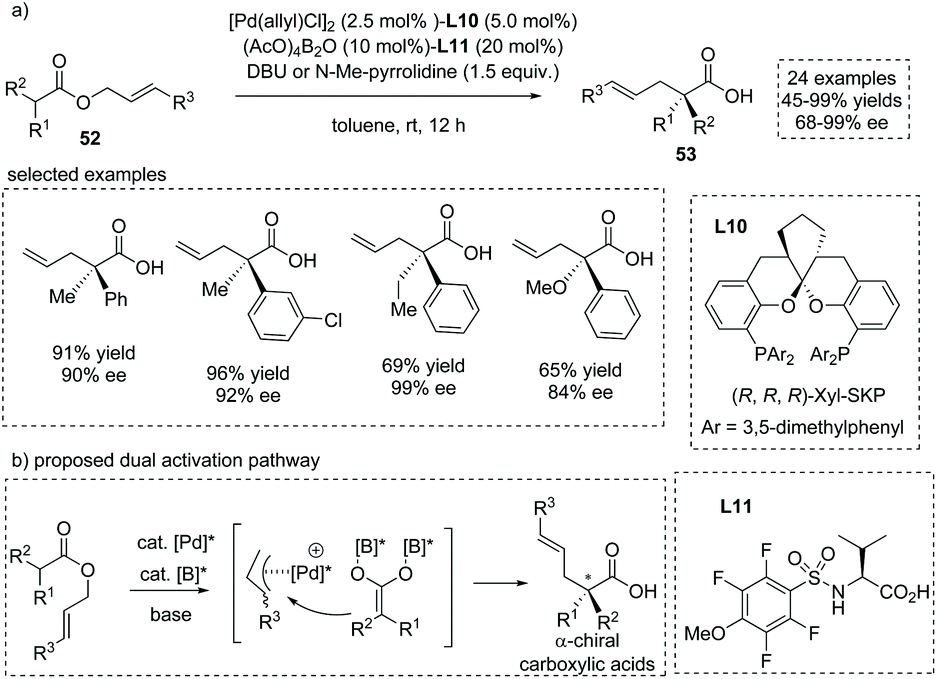
The asymmetric addition of catalytically generated ester-derived enediolate species to metal-bound electrophilic species is another reaction of importance that allows for the facile synthesis of α-substituted carboxylic acids. In 2018, Kanai, Shimizu and co-workers reported an enantioselective synthesis of α-quaternary chiral carboxylic acids via a chiral Pd/B catalyst synergistically catalyzed migratory allylation of α,α-disubstituted O-allyl esters 52.59 Upon exposure to the palladium catalyst, the allyl ester generates a chiral π-allyl palladium complex, wherein boron-catalyzed enolization of the concomitant carboxylate leads to a nucleophilic boryl enediolate species. The two in situ generated intermediates rapidly react and then produce the chiral α-quaternary carboxylic acids 53 in a highly enantiocontrolled fashion. The reaction tolerated a large number of allyl esters with diverse substituents and delivered the chiral carboxylic acids in excellent ees in most cases (Scheme 38). The reaction proceeded via two independent active species and not via a concerted Ireland–Claisen rearrangement pathway; thus the O-allyl product was also observed in some cases. Since independent chiral ligands L10 and L11 were introduced onto palladium and boron atoms, respectively, the match/mismatch effect of the two chiral ligands on the reaction was observed, with the absence of either chiral ligand leading to a pronounced decrease of the ee value.
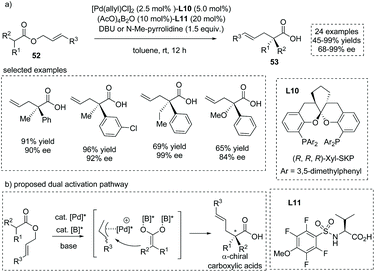 |
| | Scheme 38 Asymmetric synthesis of α-allyl carboxylic acids via Pd/B dual catalyzed migratory allylation. | |
In 2019, the 1,3,2-diazaphospholene (DAP) precursor C23 catalyzed reductive Ireland–Claisen rearrangement of allylic acrylates was reported by Cramer and co-workers,60 leading to α-substituted carboxylic acids in a highly efficient manner. After careful optimization, 1.5 equivalents of pinacol borane were used as the terminal reductant to generate the DAP catalyst. A broad array of substrates 54 bearing various functional groups at the α or β position were all well tolerated, giving rise to structurally diverse α-substituted carboxylic acids 55 efficiently (Scheme 39a). Notably, an asymmetric variant under the catalysis of the chiral DAP precursor C20 produced the highly functionalized α-quaternary chiral carboxylic acids 55a with a tetralin scaffold in 96% yield and 37% ee (Scheme 39b). Mechanistically, once initiated by the conjugate addition of in situ generated DAP hydride across the double bond of acrylate, the reaction could proceed via either the B-[3,3] pathway (with P–C intermediate IV and B–O intermediate VI) or the P-[3,3] pathway (with P–O intermediate V). The author found that the reaction pathway is highly substrate-dependent, and for the substrate 54a, the P-[3,3] pathway occurred exclusively via the chiral catalyst-bound P–O intermediate V. Subsequent σ-bond metathesis between the rearrangement product and HBpin, followed by a alcohol work-up, delivered the enantio-enriched α-stereogenic carboxylic acid 55a (Scheme 39c).
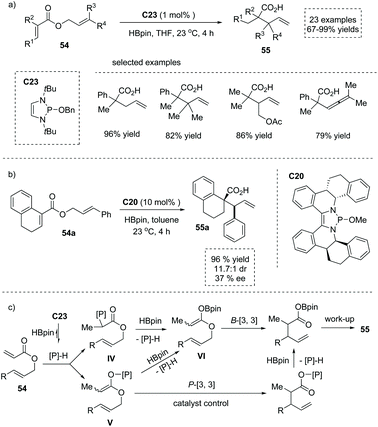 |
| | Scheme 39 Synthesis of α-stereogenic carboxylic acids via DAP catalyzed reduction/Claisen rearrangement of allyl cinnamate. | |
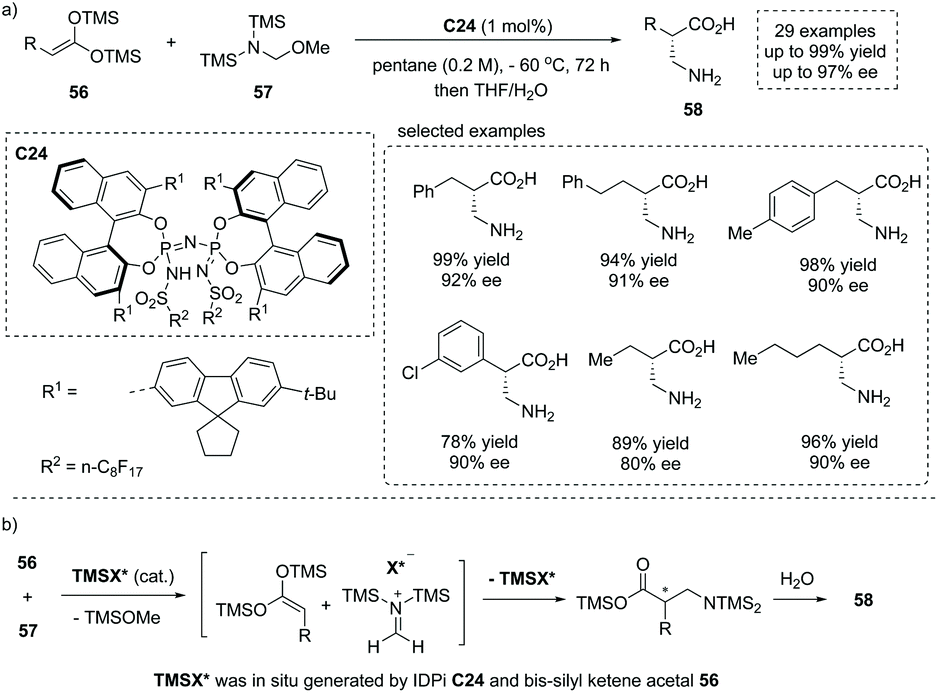
Owing to the significance of chiral β2-amino acids, the direct, catalytic approach to the free, unmodified β2-amino acid represents an extremely appealing yet unreported “dream reaction”. Based on their previous studies on silylium-based asymmetric counteranion-directed catalysis (Si-ACDC), recently List and co-workers reported a chiral imidodiphosphorimidate (IDPi) C24 catalyzed highly enantioselective Mukaiyama Mannich-type reaction of bis-silyl ketene acetal 56 (bis-silyl enediol ether) and silylated aminomethyl ether 57 at a low catalyst loading (1 mol%).61 A broad array of functional groups were well tolerated, and both aromatic and aliphatic free β2-amino acids 58 with a stereogenic center at the α position were readily obtained with high yields and enantioselectivity (29 examples, up to 96% yield, up to 97% ee, Scheme 40a). Besides, the salient features of this chemistry include extremely concise product purification and catalyst recovery. Upon completion of the reaction, the workup of the reaction mixture included a simple extraction with water and washing with dichloromethane without further purification. The pure products could be obtained from the aqueous phase, and the IDPi catalyst C24 could be easily recovered from the organic phase via flash chromatography and acidification. Mechanistically, in situ silylation of the IDPi C24 by bis-silyl ketene acetal 56 generated N-silylated IDPi and/or its diastereomeric O–Si-silatropomers (TMSX*) as the active catalyst, which reacts with silylated aminomethyl ether 57 giving rise to methylene iminium ion–IDPi ion pairs and TMSOMe. Then asymmetric Mannich-type addition and subsequent silyl transfer regenerated the chiral catalyst (TMSX*) for the next catalytic cycle. The final hydrolytic workup of the reaction mixture produced the desired product 58 (Scheme 40b). The Mannich-type addition was the ee-determining step and the facial selectivity was controlled by chiral ion pairing interactions (Scheme 40b).
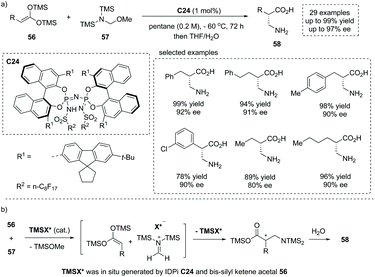 |
| | Scheme 40 Direct synthesis of α-chiral β2-amino acids via chiral IDPi C24 catalyzed asymmetric Mannich-type reaction. | |
7. Conclusions
In summary, asymmetric catalysis is a powerful method for the synthesis of structurally diverse α-chiral carboxylic acids. A variety of catalytic strategies including organocatalysis and transition metal catalysis have been developed for the synthesis of these appealing molecules over the past decades.62 Compared to its ester and amide counterparts, the direct catalytic asymmetric synthesis of α-stereogenic free carboxylic acids features several marked advantages such as enabling the facile synthesis of marketed drugs, direct further synthetic elaboration of the carboxyl group, and in some cases largely simplified procedures for product purification (just need a simple extraction). However, the full potential of this field has not been realized, and new strategies and methods should be implemented: (1) the combination of different technologies such as transition metal catalysis, organocatalysis and photoredox catalysis in a synergistic fashion to achieve unprecedented α-chiral carboxylic acid-forming reactions; (2) the development of new catalytic systems to improve the level of stereo-induction in currently unsatisfactory reactions such as DAP–catalyzed conjugate reduction of unsaturated carboxylic acids; and (3) the development of new α-chiral carboxylic acid forming reactions using new readily available substrates with different oxidation levels. We hope that this review will provide some insights into this stimulating research area and motivate further interest in new efficient and practical means leading to α-stereogenic carboxylic acids in a highly stereocontrolled fashion.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
We are grateful for the financial support from the Natural Science Basic Research Plan in Shaanxi Province of China (2021JQ-613), the Education Department of Shaanxi Province (19JK0965), and Yan'an University (YDBK2018-19).
Notes and references
-
(a) S. Noguchi, S. Kishimoto, I. Minamida, M. Obayashi and K. Kawakita, Chem. Pharm. Bull., 1971, 19, 646–648 CrossRef CAS;
(b) T. Y. Shen, Angew. Chem., Int. Ed. Engl., 1972, 11, 460–472 CrossRef CAS;
(c) N. Bodor, R. Woods, C. Raper, P. Kearney and J. J. Kaminski, J. Med. Chem., 1980, 23, 474–480 CrossRef CAS PubMed;
(d) F. von Nussbaum, M. Brands, B. Hinzen, S. Weigand and D. Häbich, Angew. Chem., Int. Ed., 2006, 45, 5072–5129 CrossRef CAS.
-
(a) C. Min and D. Seidel, Chem. Soc. Rev., 2017, 46, 5889–5902 RSC;
(b) B. List, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS;
(c) B.-F. Shi, N. Maugel, Y.-H. Zhang and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882–4886 CrossRef CAS PubMed;
(d) T. Kodama, P. N. Moquist and S. E. Schaus, Org. Lett., 2011, 13, 6316–6319 CrossRef CAS;
(e) M. Wasa, K. M. Engle, D. W. Lin, E. J. Yoo and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 19598–19601 CrossRef CAS PubMed;
(f) K. S. L. Chan, H.-Y. Fu and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 2042–2046 CrossRef CAS PubMed;
(g) Y. Luan, K. S. Barbato, P. N. Moquist, T. Kodama and S. E. Schaus, J. Am. Chem. Soc., 2015, 137, 3233–3236 CrossRef CAS;
(h) F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252–256 CrossRef CAS.
-
(a) L. J. Gooßen, N. Rodríguez and K. Gooßen, Angew. Chem., Int. Ed., 2008, 47, 3100–3120 CrossRef;
(b) N. Rodríguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC;
(c) L. C. Morrill and A. D. Smith, Chem. Soc. Rev., 2014, 43, 6214–6226 RSC;
(d) M. T. Knowe, M. W. Danneman, S. Sun, M. Pink and J. N. Johnston, J. Am. Chem. Soc., 2018, 140, 1998–2001 CrossRef CAS;
(e) L. Vicens, M. Bietti and M. Costas, Angew. Chem., Int. Ed., 2021, 60, 4740–4746 CrossRef CAS PubMed.
-
(a) S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2017, 50, 988–1001 CrossRef CAS;
(b) A. Das and B. Maji, Chem. – Asian J., 2021, 16, 397–408 CrossRef CAS.
-
(a) K. Bera and I. N. N. Namboothiri, Asian J. Org. Chem., 2014, 3, 1234–1260 CrossRef CAS;
(b) J. Moschner, V. Stulberg, R. Fernandes, S. Huhmann, J. Leppkes and B. Koksch, Chem. Rev., 2019, 119, 10718–10801 CrossRef CAS;
(c) V. A. Larionov, N. V. Stoletova and V. I. Maleev, Adv. Synth. Catal., 2020, 362, 4325–4367 CrossRef CAS;
(d) X.-X. Zhang, Y. Gao, X.-S. Hu, C.-B. Ji, Y.-L. Liu and J.-S. Yu, Adv. Synth. Catal., 2020, 362, 4763–4793 CrossRef CAS;
(e) F. J. Aguilar Troyano, K. Merkens, K. Anwar and A. Gómez-Suárez, Angew. Chem., Int. Ed., 2021, 60, 1098–1115 CrossRef CAS PubMed;
(f) H.-Q. Cao, J.-K. Li, F.-G. Zhang, D. Cahard and J.-A. Ma, Adv. Synth. Catal., 2021, 363, 688–729 CrossRef CAS.
-
(a) Y. Chen and L. Deng, J. Am. Chem. Soc., 2001, 123, 11302–11303 CrossRef CAS;
(b) L. Tang and L. Deng, J. Am. Chem. Soc., 2002, 124, 2870–2871 CrossRef CAS PubMed;
(c) K. Ishihara, Y. Kosugi, S. Umemura and A. Sakakura, Org. Lett., 2008, 10, 3191–3194 CrossRef CAS PubMed;
(d) I. Shiina, K. Nakata and Y.-s. Onda, Eur. J. Org. Chem., 2008, 5887–5890 CrossRef CAS;
(e) X. Yang and V. B. Birman, Adv. Synth. Catal., 2009, 351, 2301–2304 CrossRef CAS PubMed;
(f) I. Shiina, K. Nakata, K. Ono, Y.-s. Onda and M. Itagaki, J. Am. Chem. Soc., 2010, 132, 11629–11641 CrossRef CAS.
-
(a) P.-Y. Wang, Y.-J. Chen, A.-C. Wu, Y.-S. Lin, M.-F. Kao, J.-R. Chen, J.-F. Ciou and S.-W. Tsai, Adv. Synth. Catal., 2009, 351, 2333–2341 CrossRef CAS;
(b) K. Fesko, M. Uhl, J. Steinreiber, K. Gruber and H. Griengl, Angew. Chem., Int. Ed., 2010, 49, 121–124 CrossRef CAS PubMed;
(c) Y.-P. Xue, C.-H. Cao and Y.-G. Zheng, Chem. Soc. Rev., 2018, 47, 1516–1561 RSC.
-
(a) D. A. Evans, J. V. Nelson, E. Vogel and T. R. Taber, J. Am. Chem. Soc., 1981, 103, 3099–3111 CrossRef CAS;
(b) C. E. Stivala and A. Zakarian, J. Am. Chem. Soc., 2011, 133, 11936–11939 CrossRef CAS;
(c) Y. Ma, C. E. Stivala, A. M. Wright, T. Hayton, J. Liang, I. Keresztes, E. Lobkovsky, D. B. Collum and A. Zakarian, J. Am. Chem. Soc., 2013, 135, 16853–16864 CrossRef CAS PubMed;
(d) P. Lu, J. J. Jackson, J. A. Eickhoff and A. Zakarian, J. Am. Chem. Soc., 2015, 137, 656–659 CrossRef CAS;
(e) Y. Ma, K. A. Mack, J. Liang, I. Keresztes, D. B. Collum and A. Zakarian, Angew. Chem., Int. Ed., 2016, 55, 10093–10097 CrossRef CAS;
(f) K. Yu, P. Lu, J. J. Jackson, T.-A. D. Nguyen, J. Alvarado, C. E. Stivala, Y. Ma, K. A. Mack, T. W. Hayton, D. B. Collum and A. Zakarian, J. Am. Chem. Soc., 2017, 139, 527–533 CrossRef CAS;
(g) K. Yu, B. Miao, W. Wang and A. Zakarian, Org. Lett., 2019, 21, 1930–1934 CrossRef CAS.
-
(a) C. R. Hauser and W. J. Chambers, J. Am. Chem. Soc., 1956, 78, 4942–4944 CrossRef CAS;
(b) T. Morisawa, M. Sawamura and Y. Shimizu, Org. Lett., 2019, 21, 7466–7469 CrossRef CAS PubMed;
(c) T. Tanaka, R. Yazaki and T. Ohshima, J. Am. Chem. Soc., 2020, 142, 4517–4524 CrossRef CAS PubMed;
(d) K. Sun, M. Ueno, K. Imaeda, K. Ueno, M. Sawamura and Y. Shimizu, ACS Catal., 2021, 11, 9722–9728 CrossRef CAS.
- Y. Morita, T. Yamamoto, H. Nagai, Y. Shimizu and M. Kanai, J. Am. Chem. Soc., 2015, 137, 7075–7078 CrossRef CAS.
-
(a) H. Nagai, Y. Morita, Y. Shimizu and M. Kanai, Org. Lett., 2016, 18, 2276–2279 CrossRef CAS;
(b) K. Ishizawa, H. Nagai, Y. Shimizu and M. Kanai, Chem. Pharm. Bull., 2018, 66, 231–234 CrossRef CAS.
- S. Kotani, Y. Yoshiwara, M. Ogasawara, M. Sugiura and M. Nakajima, Angew. Chem., Int. Ed., 2018, 57, 15877–15881 CrossRef CAS PubMed.
- S. E. Denmark and R. A. Stavenger, Acc. Chem. Res., 2000, 33, 432–440 CrossRef CAS.
-
(a) R. Giri, N. Maugel, J.-J. Li, D.-H. Wang, S. P. Breazzano, L. B. Saunders and J.-Q. Yu, J. Am. Chem. Soc., 2007, 129, 3510–3511 CrossRef CAS;
(b) P. Dolui, J. Das, H. B. Chandrashekar, S. S. Anjana and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 13773–13777 CrossRef CAS;
(c) K. K. Ghosh, A. Uttry, A. Mondal, F. Ghiringhelli, P. Wedi and M. van Gemmeren, Angew. Chem., Int. Ed., 2020, 59, 12848–12852 CrossRef CAS;
(d) J. Das, D. K. Mal, S. Maji and D. Maiti, ACS Catal., 2021, 11, 4205–4229 CrossRef CAS.
- G. Chen, W. Gong, Z. Zhuang, M. S. Andrä, Y.-Q. Chen, X. Hong, Y.-F. Yang, T. Liu, K. N. Houk and J.-Q. Yu, Science, 2016, 353, 1023–1027 CrossRef CAS.
- P.-X. Shen, L. Hu, Q. Shao, K. Hong and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 6545–6549 CrossRef CAS PubMed.
- L. Hu, P.-X. Shen, Q. Shao, K. Hong, J. X. Qiao and J.-Q. Yu, Angew. Chem., Int. Ed., 2019, 58, 2134–2138 CrossRef CAS.
- F. Ghiringhelli, A. Uttry, K. K. Ghosh and M. van Gemmeren, Angew. Chem., Int. Ed., 2020, 59, 23127–23131 CrossRef CAS PubMed.
-
(a) Y. Chen, P. McDaid and L. Deng, Chem. Rev., 2003, 103, 2965–2984 CrossRef CAS;
(b) I. Atodiresei, I. Schiffers and C. Bolm, Chem. Rev., 2007, 107, 5683–5712 CrossRef CAS;
(c) M. D. Díaz de Villegas, J. A. Gálvez, P. Etayo, R. Badorrey and P. López-Ram-de-Víu, Chem. Soc. Rev., 2011, 40, 5564–5587 RSC;
(d) Z. Rodriguez-Docampo and S. J. Connon, ChemCatChem, 2012, 4, 151–168 CrossRef CAS;
(e) A. Borissov, T. Q. Davies, S. R. Ellis, T. A. Fleming, M. S. W. Richardson and D. J. Dixon, Chem. Soc. Rev., 2016, 45, 5474–5540 RSC.
-
(a) Y. Chen, S.-K. Tian and L. Deng, J. Am. Chem. Soc., 2000, 122, 9542–9543 CrossRef CAS;
(b) T. Honjo, S. Sano, M. Shiro and Y. Nagao, Angew. Chem., Int. Ed., 2005, 44, 5838–5841 CrossRef CAS;
(c) B. Rodríguez, T. Rantanen and C. Bolm, Angew. Chem., Int. Ed., 2006, 45, 6924–6926 CrossRef PubMed;
(d) S. H. Oh, H. S. Rho, J. W. Lee, J. E. Lee, S. H. Youk, J. Chin and C. E. Song, Angew. Chem., Int. Ed., 2008, 47, 7872–7875 CrossRef CAS;
(e) A. Peschiulli, Y. Gun'k and S. J. Connon, J. Org. Chem., 2008, 73, 2454–2457 CrossRef CAS;
(f) S.-X. Wang and F.-E. Chen, Adv. Synth. Catal., 2009, 351, 547–552 CrossRef CAS;
(g) R. Manzano, J. M. Andrés, M.-D. Muruzábal and R. Pedrosa, J. Org. Chem., 2010, 75, 5417–5420 CrossRef CAS;
(h) A. Peschiulli, B. Procuranti, C. J. O′ Connor and S. J. Connon, Nat. Chem., 2010, 2, 380–384 CrossRef CAS PubMed.
-
(a) M. Baidya, M. Horn, H. Zipse and H. Mayr, J. Org. Chem., 2009, 74, 7157–7164 CrossRef CAS;
(b) H. Li, X. Liu, F. Wu, L. Tang and L. Deng, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20625–20629 CrossRef CAS;
(c) H. Yang and M. W. Wong, J. Am. Chem. Soc., 2013, 135, 5808–5818 CrossRef CAS PubMed;
(d) K. Blise, M. W. Cvitkovic, N. J. Gibbs, S. F. Roberts, R. M. Whitaker, G. E. Hofmeister and D. Kohen, J. Org. Chem., 2017, 82, 1347–1355 CrossRef CAS PubMed.
- V. N. Wakchaure and B. List, Angew. Chem., Int. Ed., 2010, 49, 4136–4139 CrossRef CAS.
- J.-W. Lee, T. Mayer-Gall, K. Opwis, C. E. Song, J. S. Gutmann and B. List, Science, 2013, 341, 1225–1229 CrossRef CAS.
- L.-J. Yan, H.-F. Wang, W.-X. Chen, Y. Tao, K.-J. Jin and F.-E. Chen, ChemCatChem, 2016, 8, 2249–2253 CrossRef CAS.
-
(a) E. A. Bercot and T. Rovis, J. Am. Chem. Soc., 2002, 124, 174–175 CrossRef CAS;
(b) E. A. Bercot and T. Rovis, J. Am. Chem. Soc., 2004, 126, 10248–10249 CrossRef CAS;
(c) M. J. Cook and T. Rovis, J. Am. Chem. Soc., 2007, 129, 9302–9303 CrossRef CAS;
(d) J. B. Johnson, E. A. Bercot, C. M. Williams and T. Rovis, Angew. Chem., Int. Ed., 2007, 46, 4514–4518 CrossRef CAS.
- E. E. Stache, T. Rovis and A. G. Doyle, Angew. Chem., Int. Ed., 2017, 56, 3679–3683 CrossRef CAS PubMed.
- M. González-López and J. T. Shaw, Chem. Rev., 2009, 109, 164–189 CrossRef.
- C. Cornaggia, F. Manoni, E. Torrente, S. Tallon and S. J. Connon, Org. Lett., 2012, 14, 1850–1853 CrossRef CAS PubMed.
-
(a) P. Kongsaeree, S. Prabpai, N. Sriubolmas, C. Vongvein and S. Wiyakrutta, J. Nat. Prod., 2003, 66, 709–711 CrossRef CAS PubMed;
(b) R. H. Cichewicz, F. A. Valeriote and P. Crews, Org. Lett., 2004, 6, 1951–1954 CrossRef CAS;
(c) M. Enomoto and S. Kuwahara, Angew. Chem., Int. Ed., 2009, 48, 1144–1148 CrossRef CAS;
(d) N. Nazir, S. Koul, M. A. Qurishi, M. H. Najar and M. I. Zargar, Eur. J. Med. Chem., 2011, 46, 2415–2420 CrossRef CAS;
(e) S. Wan, F. Wu, J. C. Rech, M. E. Green, R. Balachandran, W. S. Horne, B. W. Day and P. E. Floreancig, J. Am. Chem. Soc., 2011, 133, 16668–16679 CrossRef CAS.
-
(a) F. Manoni, C. Cornaggia, J. Murray, S. Tallon and S. J. Connon, Chem. Commun., 2012, 48, 6502–6504 RSC;
(b) C. Cornaggia, S. Gundala, F. Manoni, N. Gopalasetty and S. J. Connon, Org. Biomol. Chem., 2016, 14, 3040–3046 RSC;
(c) R. Claveau, B. Twamley and S. J. Connon, Chem. Commun., 2018, 54, 3231–3234 RSC;
(d) U. Farid, M. L. Aiello and S. J. Connon, Chem. – Eur. J., 2019, 25, 10074–10079 CrossRef CAS PubMed.
- N. Castagnoli, J. Org. Chem., 1969, 34, 3187–3189 CrossRef CAS.
- S. A. Cronin, A. G. Collar, S. Gundala, C. Cornaggia, E. Torrente, F. Manoni, A. Botte, B. Twamley and S. J. Connon, Org. Biomol. Chem., 2016, 14, 6955–6959 RSC.
- C. L. Jarvis, J. S. Hirschi, M. J. Vetticatt and D. Seidel, Angew. Chem., Int. Ed., 2017, 56, 2670–2674 CrossRef CAS PubMed.
- Z. Chang, C. Ye, J. Fu, P. Chigumbu, X. Zeng, Y. Wang, C. Jiang and X. Han, Adv. Synth. Catal., 2019, 361, 5516–5520 CrossRef CAS.
- A. G. Collar, C. Trujillo, B. Lockett-Walters, B. Twamley and S. J. Connon, Chem. – Eur. J., 2019, 25, 7275–7279 CrossRef CAS.
- Y. Tamura, A. Wada, M. Sasho and Y. Kita, Tetrahedron Lett., 1981, 22, 4283–4286 CrossRef CAS.
-
(a) F. Manoni and S. J. Connon, Angew. Chem., Int. Ed., 2014, 53, 2628–2632 CrossRef CAS;
(b) U. Nath and S. C. Pan, J. Org. Chem., 2017, 82, 3262–3269 CrossRef CAS PubMed;
(c) H. Xu, F. Sha, Q. Li and X.-Y. Wu, Org. Biomol. Chem., 2018, 16, 7214–7222 RSC;
(d) A. G. Collar, C. Trujillo and S. J. Connon, Chem. – Eur. J., 2019, 25, 7270–7274 CrossRef CAS PubMed;
(e) B. Lockett-Walters, C. Trujillo, B. Twamley and S. Connon, Chem. Commun., 2019, 55, 11283–11286 RSC.
-
(a) Z. Tang, L.-F. Cun, X. Cui, A.-Q. Mi, Y.-Z. Jiang and L.-Z. Gong, Org. Lett., 2006, 8, 1263–1266 CrossRef CAS;
(b) X.-Y. Xu, Z. Tang, Y.-Z. Wang, S.-W. Luo, L.-F. Cun and L.-Z. Gong, J. Org. Chem., 2007, 72, 9905–9913 CrossRef CAS PubMed.
-
(a) L. Zhu, Q. Meng, W. Fan, X. Xie and Z. Zhang, J. Org. Chem., 2010, 75, 6027–6030 CrossRef CAS;
(b) L. Zhu, H. Chen, Q. Meng, W. Fan, X. Xie and Z. Zhang, Tetrahedron, 2011, 67, 6186–6190 CrossRef CAS.
- P.-C. Yan, J.-H. Xie, X.-D. Zhang, K. Chen, Y.-Q. Li, Q.-L. Zhou and D.-Q. Che, Chem. Commun., 2014, 50, 15987–15990 RSC.
- H. Guo, J. Li, D. Liu and W. Zhang, Adv. Synth. Catal., 2017, 359, 3665–3673 CrossRef CAS.
-
(a) K. Bernauer, R. Deschenaux and T. Taura, Helv. Chim. Acta, 1983, 66, 2049–2058 CrossRef CAS;
(b) R. Deschenaux and K. Bernauer, Helv. Chim. Acta, 1984, 67, 373–377 CrossRef CAS;
(c) L. Liu, W. Zhou, J. Chruma and R. Breslow, J. Am. Chem. Soc., 2004, 126, 8136–8137 CrossRef CAS.
-
(a) L. Shi, C. Tao, Q. Yang, Y. E. Liu, J. Chen, J. Chen, J. Tian, F. Liu, B. Li, Y. Du and B. Zhao, Org. Lett., 2015, 17, 5784–5787 CrossRef CAS PubMed;
(b) X. Lan, C. Tao, X. Liu, A. Zhang and B. Zhao, Org. Lett., 2016, 18, 3658–3661 CrossRef CAS PubMed;
(c) Y. E. Liu, Z. Lu, B. Li, J. Tian, F. Liu, J. Zhao, C. Hou, Y. Li, L. Niu and B. Zhao, J. Am. Chem. Soc., 2016, 138, 10730–10733 CrossRef CAS PubMed.
-
(a) W. S. Knowles, Angew. Chem., Int. Ed., 2002, 41, 1998–2007 CrossRef CAS;
(b) R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008–2022 CrossRef CAS;
(c) S. Khumsubdee and K. Burgess, ACS Catal., 2013, 3, 237–249 CrossRef CAS.
-
(a) W. S. Knowles, M. J. Sabacky, B. D. Vineyard and D. J. Weinkauff, J. Am. Chem. Soc., 1975, 97, 2567–2568 CrossRef CAS;
(b) A. Miyashita, A. Yasuda, H. Takaya, K. Toriumi, T. Ito, T. Souchi and R. Noyori, J. Am. Chem. Soc., 1980, 102, 7932–7934 CrossRef CAS;
(c) W. S. Knowles, Acc. Chem. Res., 1983, 16, 106–112 CrossRef CAS;
(d) M. van den Berg, A. J. Minnaard, E. P. Schudde, J. van Esch, A. H. M. de Vries, J. G. de Vries and B. L. Feringa, J. Am. Chem. Soc., 2000, 122, 11539–11540 CrossRef CAS;
(e) R. Hoen, J. A. F. Boogers, H. Bernsmann, A. J. Minnaard, A. Meetsma, T. D. Tiemersma-Wegman, A. H. M. de Vries, J. G. de Vries and B. L. Feringa, Angew. Chem., Int. Ed., 2005, 44, 4209–4212 CrossRef CAS PubMed;
(f) W. Chen, P. J. McCormack, K. Mohammed, W. Mbafor, S. M. Roberts and J. Whittall, Angew. Chem., Int. Ed., 2007, 46, 4141–4144 CrossRef CAS PubMed;
(g) K. Dong, Y. Li, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2013, 52, 14191–14195 CrossRef CAS.
- G. Szöllősi, T.-a. Hanaoka, S.-i. Niwa, F. Mizukami and M. Bartók, J. Catal., 2005, 231, 480–483 CrossRef.
-
(a) T. Ohta, H. Takaya, M. Kitamura, K. Nagai and R. Noyori, J. Org. Chem., 1987, 52, 3174–3176 CrossRef CAS;
(b) T. Sturm, W. Weissensteiner and F. Spindler, Adv. Synth. Catal., 2003, 345, 160–164 CrossRef CAS;
(c) X. Cheng, Q. Zhang, J.-H. Xie, L.-X. Wang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2005, 44, 1118–1121 CrossRef CAS PubMed;
(d) J. Li, J. Shen, C. Xia, Y. Wang, D. Liu and W. Zhang, Org. Lett., 2016, 18, 2122–2125 CrossRef CAS PubMed;
(e) G.-Q. Chen, J.-M. Huang, B.-J. Lin, C. Shi, L.-Y. Zhao, B.-D. Ma, X.-B. Ding, Q. Yin and X. Zhang, CCS Chem., 2020, 2, 468–477 CrossRef CAS.
-
(a) S. Li, S.-F. Zhu, C.-M. Zhang, S. Song and Q.-L. Zhou, J. Am. Chem. Soc., 2008, 130, 8584–8585 CrossRef CAS PubMed;
(b) S. Li, S.-F. Zhu, J.-H. Xie, S. Song, C.-M. Zhang and Q.-L. Zhou, J. Am. Chem. Soc., 2010, 132, 1172–1179 CrossRef CAS PubMed;
(c) S. Yang, S.-F. Zhu, C.-M. Zhang, S. Song, Y.-B. Yu, S. Li and Q.-L. Zhou, Tetrahedron, 2012, 68, 5172–5178 CrossRef CAS;
(d) S.-F. Zhu, Y.-B. Yu, S. Li, L.-X. Wang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2012, 51, 8872–8875 CrossRef CAS PubMed;
(e) S. Song, S.-F. Zhu, Y. Li and Q.-L. Zhou, Org. Lett., 2013, 15, 3722–3725 CrossRef CAS PubMed;
(f) S. Song, S.-F. Zhu, L.-Y. Pu and Q.-L. Zhou, Angew. Chem., Int. Ed., 2013, 52, 6072–6075 CrossRef CAS PubMed;
(g) M.-L. Li, S. Yang, X.-C. Su, H.-L. Wu, L.-L. Yang, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2017, 139, 541–547 CrossRef CAS PubMed.
-
(a) H. Zhong, M. Shevlin and P. J. Chirik, J. Am. Chem. Soc., 2020, 142, 5272–5281 CrossRef CAS PubMed;
(b) X. Du, Y. Xiao, J.-M. Huang, Y. Zhang, Y.-N. Duan, H. Wang, C. Shi, G.-Q. Chen and X. Zhang, Nat. Commun., 2020, 11, 3239 CrossRef CAS PubMed;
(c) X. Du, Y. Xiao, Y. Yang, Y.-N. Duan, F. Li, Q. Hu, L. W. Chung, G.-Q. Chen and X. Zhang, Angew. Chem., Int. Ed., 2021, 60, 11384–11390 CrossRef CAS PubMed.
- K. Furuta, Y. Miwa, K. Iwanaga and H. Yamamoto, J. Am. Chem. Soc., 1988, 110, 6254–6255 CrossRef CAS PubMed.
-
(a) J. H. Reed and N. Cramer, ChemCatChem, 2020, 12, 4262–4266 CrossRef CAS;
(b) J. H. Reed, J. Klett, C. Steven and N. Cramer, Org. Chem. Front., 2020, 7, 3521–3529 RSC.
-
(a) T. Azuma, A. Murata, Y. Kobayashi, T. Inokuma and Y. Takemoto, Org. Lett., 2014, 16, 4256–4259 CrossRef CAS PubMed;
(b) N. Hayama, T. Azuma, Y. Kobayashi and Y. Takemoto, Chem. Pharm. Bull., 2016, 64, 704–717 CrossRef CAS PubMed;
(c) N. Hayama, R. Kuramoto, T. Földes, K. Nishibayashi, Y. Kobayashi, I. Pápai and Y. Takemoto, J. Am. Chem. Soc., 2018, 140, 12216–12225 CrossRef CAS PubMed;
(d) K. Michigami, H. Murakami, T. Nakamura, N. Hayama and Y. Takemoto, Org. Biomol. Chem., 2019, 17, 2331–2335 RSC.
- H. Murakami, A. Yamada, K. Michigami and Y. Takemoto, Asian J. Org. Chem., 2021, 10, 1097–1101 CrossRef CAS.
-
(a) R. Shintani, K. Ueyama, I. Yamada and T. Hayashi, Org. Lett., 2004, 6, 3425–3427 CrossRef CAS PubMed;
(b) Z. Wang, Z. Yang, D. Chen, X. Liu, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2011, 50, 4928–4932 CrossRef CAS PubMed.
- J.-B. Lin, S.-M. Xu, J.-K. Xie, H.-Y. Li and P.-F. Xu, Chem. Commun., 2015, 51, 3596–3599 RSC.
- W. Jiang, J. Zhou, A.-J. Ma, D. Li, Y.-Y. Ma, D.-G. Zhao, S.-H. Hou, J.-B. Lin and S.-Y. Zhang, Org. Chem. Front., 2020, 7, 571–577 RSC.
- W.-W. Zhao and Y.-K. Liu, Org. Chem. Front., 2017, 4, 2358–2363 RSC.
- K. C. Miles, M. L. Abrams, C. R. Landis and S. S. Stahl, Org. Lett., 2016, 18, 3590–3593 CrossRef CAS.
- T. Fujita, T. Yamamoto, Y. Morita, H. Chen, Y. Shimizu and M. Kanai, J. Am. Chem. Soc., 2018, 140, 5899–5903 CrossRef CAS PubMed.
- J. H. Reed, P. A. Donets, S. Miaskiewicz and N. Cramer, Angew. Chem., Int. Ed., 2019, 58, 8893–8897 CrossRef CAS PubMed.
- C. Zhu, F. Mandrelli, H. Zhou, R. Maji and B. List, J. Am. Chem. Soc., 2021, 143, 3312–3317 CrossRef CAS PubMed.
-
(a) S. Feng and S. L. Buchwald, J. Am. Chem. Soc., 2021, 143, 4935–4941 CrossRef CAS PubMed;
(b) Y.-H. Yao, X.-J. Zou, Y. Wang, H.-Y. Yang, Z.-H. Ren and Z.-H. Guan, Angew. Chem., Int. Ed., 2021, 60, 23117–23122 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*