
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic and biosynthetic methods for selective cyclisations of 4,5-epoxy alcohols to tetrahydropyrans
James I.
Bowen†
 a,
Luoyi
Wang†
b,
Matthew P.
Crump
a and
Christine L.
Willis
*a
a,
Luoyi
Wang†
b,
Matthew P.
Crump
a and
Christine L.
Willis
*a
aSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: chris.willis@bristol.ac.uk
bCAS Key Laboratory of Microbial Physiological and Metabolic Engineering, State Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China
First published on 14th January 2022
Abstract
Tetrahydropyrans (THPs) are common structural motifs found in natural products and synthetic therapeutic molecules. In Nature these 6-membered oxygen heterocycles are often assembled via intramolecular reactions involving either oxy-Michael additions or ring opening of epoxy-alcohols. Indeed, the polyether natural products have been particularly widely studied due to their fascinating structures and important biological properties; these are commonly formed via endo-selective epoxide-opening cascades. In this review we outline synthetic approaches for endo-selective intramolecular epoxide ring opening (IERO) of 4,5-epoxy-alcohols and their applications in natural product synthesis. In addition, the biosynthesis of THP-containing natural products which utilise IERO reactions are reviewed.
 James I. Bowen | James graduated from the University of Birmingham in 2018 with a MSci in Chemistry with Industrial Experience, having spent one year working at GSK in Stevenage. In his final year he investigated gold catalysed polycyclisation cascades of ynamides under the supervision of Dr Paul Davies, for which he was awarded the Alfred Bader Prize. He then moved to the University of Bristol to join the Chemical Synthesis CDT and is currently in the final year of his PhD studies with Professors Chris Willis and Matt Crump, investigating the biosynthesis of polyketide natural products. |
 Luoyi Wang | Luoyi Wang received his PhD in natural products chemistry from Shanghai Institute of Materia Medica, Chinese Academy of Sciences in 2013. After one year of postdoctoral research at Utah State University and Ohio State University in the US, he joined Professor Chris Willis and her group at the University of Bristol as a PDRA within BrisSynBio working in the field of polyketide biosynthesis. In 2020, he moved to Beijing and established his own research group at the Institute of Microbiology, Chinese Academy of Sciences. His research focuses on mechanistic enzymology in natural products biosynthesis and enzyme engineering for biocatalytic applications. |
 Matthew P. Crump | Matt Crump is Professor of NMR and Structural Biology at the University of Bristol where he established biological NMR across a broad range of chemical and biological research areas. His major interests are on structural and mechanistic aspects of natural product biosynthesis with a focus on NMR and X-ray crystallography of natural product enzymes and their interactions with biosynthetic intermediates. |
 Christine L. Willis | Chris Willis is Professor of Organic Chemistry and Head of Organic and Biological Chemistry at the University of Bristol. Her research focuses on natural product biosynthesis including the application of total synthesis, isotopic labelling, pathway engineering and mechanistic studies to produce biocatalysts and new bioactive molecules. She was the recipient of the Natural Product Chemistry Award of the Royal Society of Chemistry in 2020. |
1. Introduction
Tetrahydropyrans (THPs) are common structural features in many classes of natural products and biologically active molecules such as the potent anti-cancer macrolide lactone bryostatin 1, and various marine polycyclic polyethers including the brevotoxins (Fig. 1). Furthermore, these rings are regularly employed as scaffolds in medicinal chemistry programs and indeed they are the sixth most prevalent ring system amongst all FDA approved small molecule drugs.1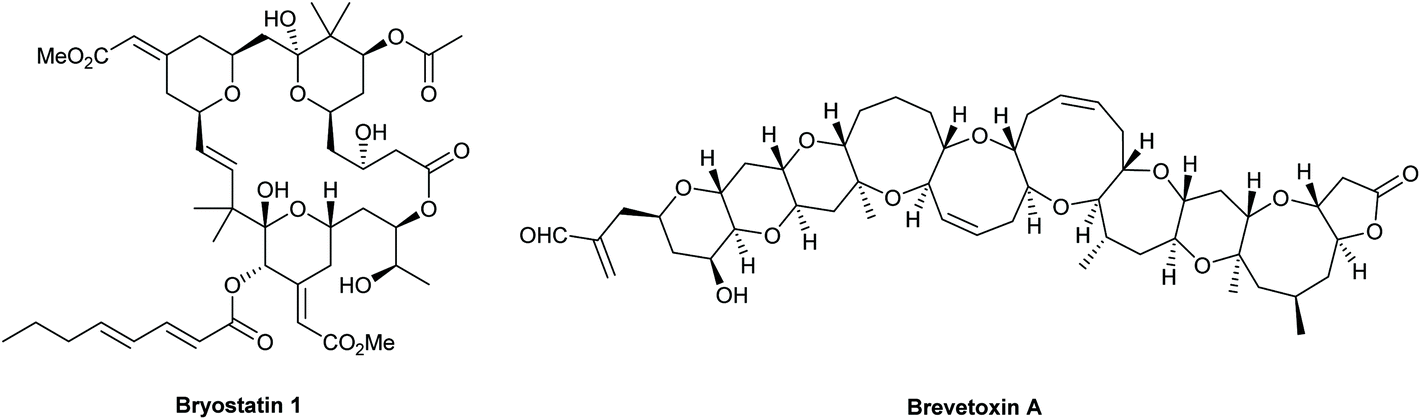 | ||
| Fig. 1 Bryostatin 1 and brevetoxin A. | ||
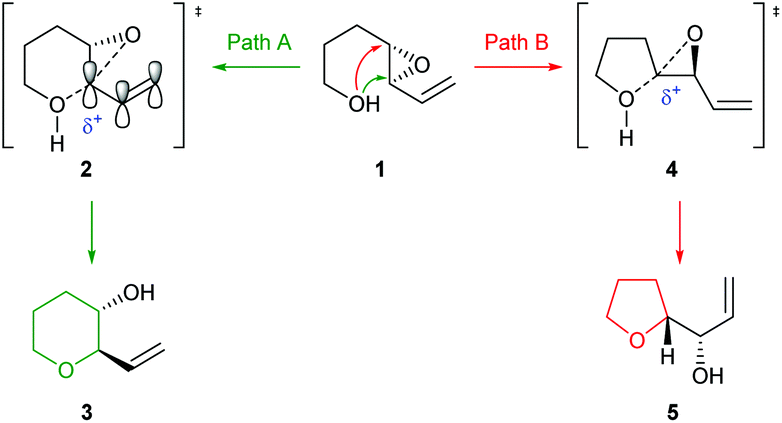
Many methods have been developed for the synthesis of THPs as described in previous informative reviews.2–6 Common approaches include Prins cyclisations, hetero-Diels–Alder reactions and ring closing metatheses (Fig. 2).7–9 In addition, reviews dedicated to the synthesis of natural products with structures incorporating THP rings have been published.10–13 As well as synthetic studies, the biosynthesis of THP-containing natural products is of widespread interest and recent work has revealed three main processes found in Nature: oxa-Michael conjugate addition, modification of hemiacetals and intramolecular nucleophilic opening of epoxides (Fig. 2).14–16 Indeed, intramolecular epoxide ring opening (IERO) of 4,5-epoxy alcohols has also been widely used in organic synthesis to create oxygen heterocycles giving either 5-membered tetrahydrofurans (THFs) or 6-membered THPs depending on the site of attack of the alcohol onto the epoxide (Fig. 3). This review focuses on the synthesis and biosynthesis of THP rings via IERO of 4,5-epoxy alcohols, highlighting how regioselectivity of epoxide ring opening may be achieved.
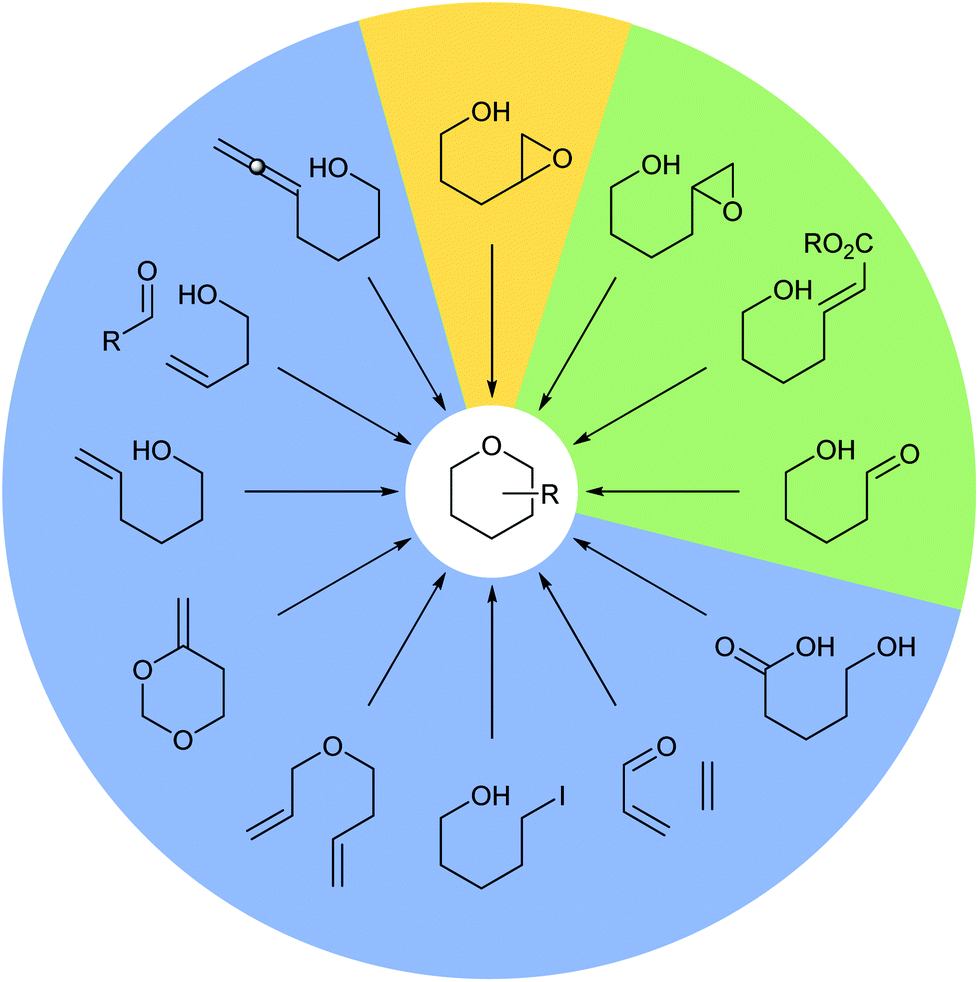 | ||
| Fig. 2 Common methods for the synthesis and biosynthesis of THP rings. Synthetic methods (blue), synthetic and biosynthetic methods (green) and the focus of this review (yellow). | ||
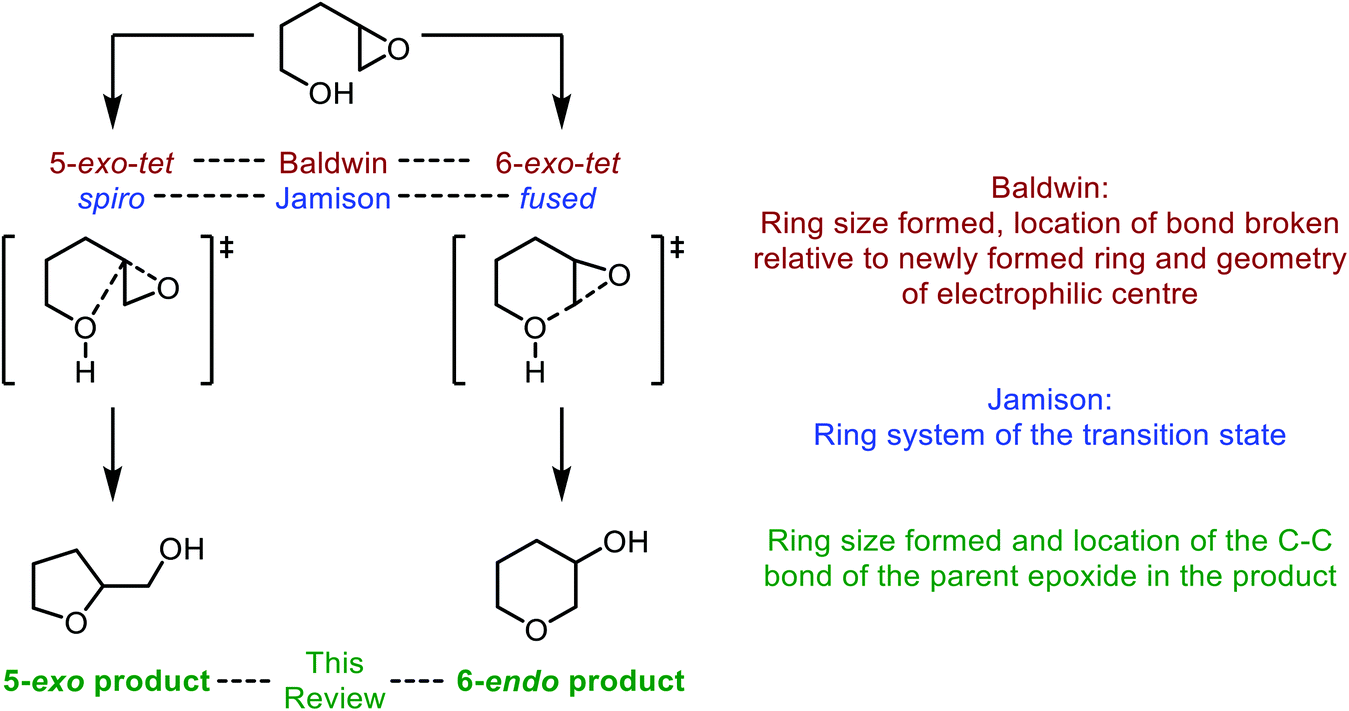 | ||
| Fig. 3 Intramolecular epoxide ring opening of 4,5-epoxy alcohols and corresponding nomenclature as described by Baldwin (red), Jamison (blue) and in this review (green). | ||
In the literature the terms 5-exo and 6-endo have often been used to describe formation of THFs and THPs respectively from 4,5-epoxy alcohols.17 This nomenclature is adopted in this review and refers to the process of intramolecular epoxide ring opening by a numerical prefix, describing the size of the ring formed, followed by exo/endo to indicate whether the C–C bond of the initial epoxide is located outside or inside the newly formed ring respectively. It is important to note that this nomenclature does not relate to Baldwin's rules, a set of guidelines for predicting the outcome of ring-forming reactions whereby the numerical prefix defines the ring size formed, exo and endo refer to whether the bond broken during cyclisation is inside or outside of the newly formed ring, and the suffix indicates the geometry of the electrophile. Using these rules both modes of cyclisation of 4,5-epoxy alcohols are exo processes as the C–O bond broken during ring formation is outside of the newly formed heterocycle (Fig. 3).18,19 According to Baldwin's rules, both 5-exo-tet and 6-exo-tet are favoured processes, whilst 6-endo-tet cyclisations are disfavoured. Jamison et al. have suggested an alternative nomenclature which refers to the transition state of the cyclisation as either spiro or fused for THF and THP formation respectively (Fig. 3).5
Whilst both THF and THP rings can be generated from 4,5-epoxy alcohols, the THF is often the major product as noted by Baldwin.17 This selectivity originates from improved orbital overlap and a lower overall energetic barrier for formation in a 5-exo process.20 Furthermore, formation of 5-membered rings is often preferred kinetically over the corresponding 6-membered rings. Therefore, to form THP rings through intramolecular epoxide ring opening of 4,5-epoxy alcohols, this inherent preference to form the 5-membered ring needs to be overcome. Many synthetic methods have been developed to favour THP formation using substrates and reaction conditions which either stabilise the 6-endo transition state or destabilise the 5-exo transition state. To achieve selectivity in natural product biosynthesis, enzymes are often used. In this review, we will outline synthetic strategies which have been developed to favour THP formation alongside examples of their use in total synthesis. This topic will be divided into substrate controlled and reagent controlled cyclisations with examples of their use in natural product synthesis. Furthermore, examples of THP and THF formation in natural product biosynthesis will be described, highlighting how nature employs enzymes to promote and control IERO.
2. Synthetic methods
Vilotijevic and Jamison have published reviews on the use of epoxide-opening cascades to create ladder polyethers and oxasqualenoid natural products.5,21 In this section of our review, we build upon these publications providing an overview of synthetic approaches to achieve 6-endo-selective epoxide ring opening of 4,5-epoxy alcohols alongside their use in natural product synthesis.2.1 Substrate controlled formation of THPs from 4,5-epoxy alcohols
 | ||
| Scheme 1 Stabilisation of 6-endo transition state by conjugation. | ||
Indeed, acid catalysed cyclisation of unsaturated epoxide 1 proceeded with complete regioselectivity, affording 6-endo product 3 in excellent yield (Scheme 2).23 Various π-systems have been investigated including, vinyl halides and unsaturated esters, where the greater electron withdrawing nature of the conjugated functional group decreases the observed selectivity. Whilst trans-epoxides often give good yields of THPs under these conditions, poor regioselectivity was obtained with the corresponding cis-epoxides. Nicolaou proposed that cis-epoxides are unable to adopt the required planar geometry to achieve stabilisation in the transition state. Indeed, earlier studies by Coxon et al. illustrated that cyclisation of cis-4,5-epoxy alcohols gave preferential 5-exo ring closure compared to the corresponding trans-epoxides.24 The 6-endo transition state for cis-epoxides requires a group to be placed axially, resulting in steric clashes and an overall increased barrier for THP formation (Scheme 2).
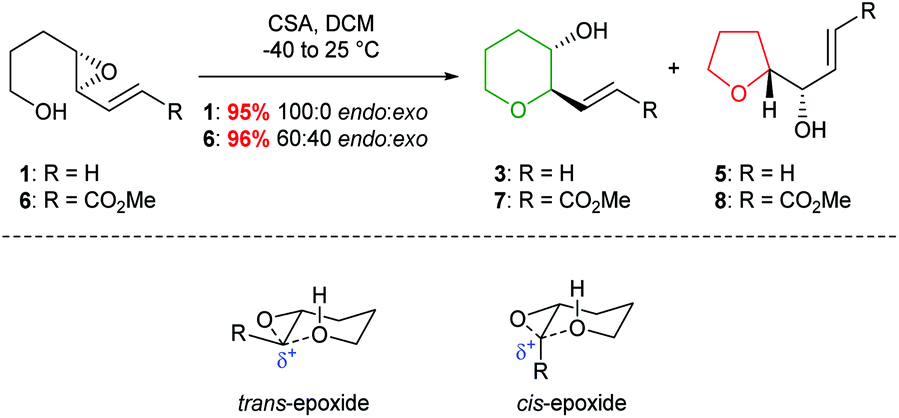 | ||
| Scheme 2 Acid catalysed cyclisation of unsaturated epoxides 1 and 6 (top) and transition states for the cyclisation of cis and trans-epoxides (bottom). | ||
Unsaturated 4,5-epoxy alcohols have been widely used as intermediates in natural product synthesis.25,26 Examples include the preparation of mucocin, amphidinol 3 and the formation of the E ring in maitotoxin, as well as the botcinins, and more recently in the total synthesis of meayamycin B, illustrating the widespread value of the method (Scheme 3).27–32
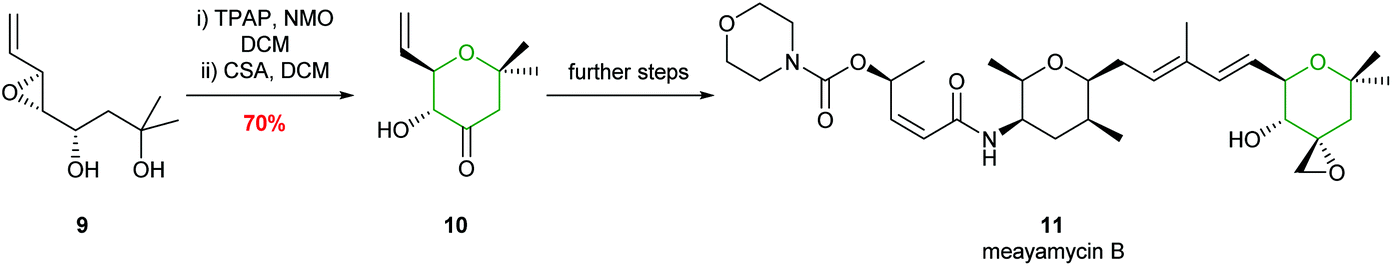 | ||
| Scheme 3 Total synthesis of meayamycin B by Koide et al.32 | ||
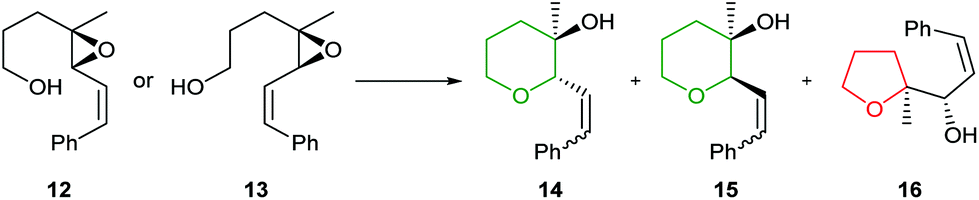
In 1997, Nakata and co-workers reported that introduction of a styryl group adjacent to the epoxide provided greater stabilisation in the 6-endo transition state compared with the alkenic epoxides.33,34 Treatment of trans-epoxides containing a styryl group with either acid or base generated THP products in excellent yield, but improved stereocontrol was achieved using NaH (entries 1 and 2, Table 1). Whilst Nicolaou reported that unsaturated cis-epoxides gave poor 6-endo selectivity under acidic conditions,23 Nakata showed that styryl epoxides may be converted solely to THP products under acidic conditions, with no evidence for the formation of the corresponding THF (entries 3 and 4, Table 1). In contrast, under basic conditions cis-epoxides gave a mixture of 5- and 6-membered rings (entry 5). Isomerisation of the styryl double bond may occur in acid but in general this is not a problem as this directing group is often removed after cyclisation. Hence, although both cis and trans-epoxides with a styryl group can be cleanly cyclised, the need to remove the styryl directing group can be a disadvantage and as such, these substrates are only employed when the stabilisation of alkenic epoxides is insufficient to achieve the desired selectivity. For example, Nakata and co-workers used styryl-containing epoxides in their synthesis of the IJK ring system of brevetoxin B and the total synthesis of hemibrevetoxin B (Scheme 4).35,36
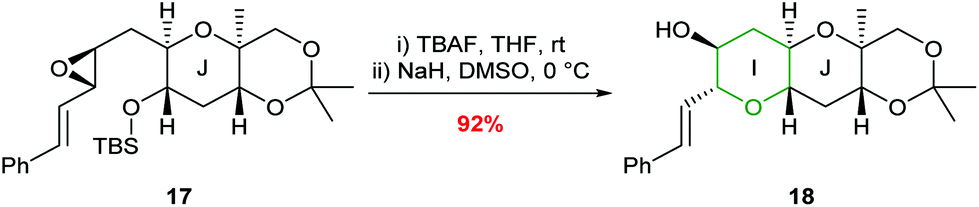 | ||
| Scheme 4 Synthesis of the I ring of brevetoxin B.35 | ||
|
|
||||
|---|---|---|---|---|
| Entry | Epoxide | Conditions | Yield |
14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15:16 :15:16 |
| 1 | 12 | CSA (0.2 eq.), DCM, −78 °C, 1 h | 100% | 90:10:0 |
| 2 | 12 | NaH (10 eq.), DMSO, rt, 2.5 h | 97% | 100:0:0 |
| 3 | 13 | CSA (0.2 eq.), DCM, rt, 10 min | 85% | 30:70:0 |
| 4 | 13 | AcOH–H2O (10:1), rt, 19 h |
62% | 15:85:0 |
| 5 | 13 | NaH (20 eq.), DMSO, rt, 19 h | 77% | 0:66:34 |
Both palladium and rhodium catalysts have been successfully employed in selective 6-endo ring closures of 4,5-epoxy alcohols. Pioneering work by Trost demonstrated that activation of vinyl epoxides could lead to exclusively the 6-endo products, albeit with poor stereoselectivity (Scheme 5).37 The reaction proceeds via initial formation of a π-allylpalladium intermediate, which in turn is trapped by the appended alcohol. The formation of the π-allylpalladium intermediate ensures only the THP is formed.
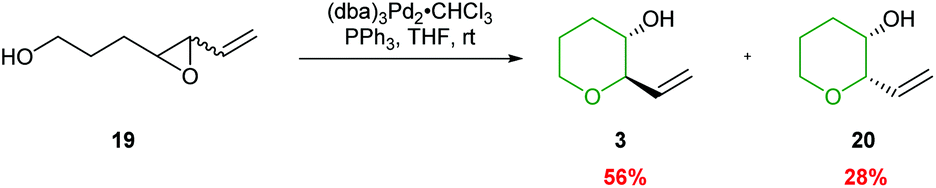 | ||
| Scheme 5 Palladium catalysed cyclisation of 4,5-epoxy alcohols.37 | ||
Through modification of the substrate, Hirama and co-workers converted both cis and trans-epoxides to the corresponding anti and syn-products in excellent yields and stereoselectivities (Table 2).38In situ formation of an ammonium alkoxide nucleophile alongside the use of an α,β-unsaturated ester to replace the alkene were vital for achieving the desired transformation.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Reaction time | Yield |
23:24
|
| 1 | 21 | 5 min | 90% | >99:1 |
| 2 | 22 | 6 min | 86% | 2:98 |

Hirama employed a palladium-mediated cyclisation in the synthesis of the HIJ ring system of ciguatoxin and the AB ring fragment of gambiertoxin 4B (Scheme 6).39,40 The key cyclisations proceeded smoothly in 93% and 74% yields respectively to afford single diastereoisomers.
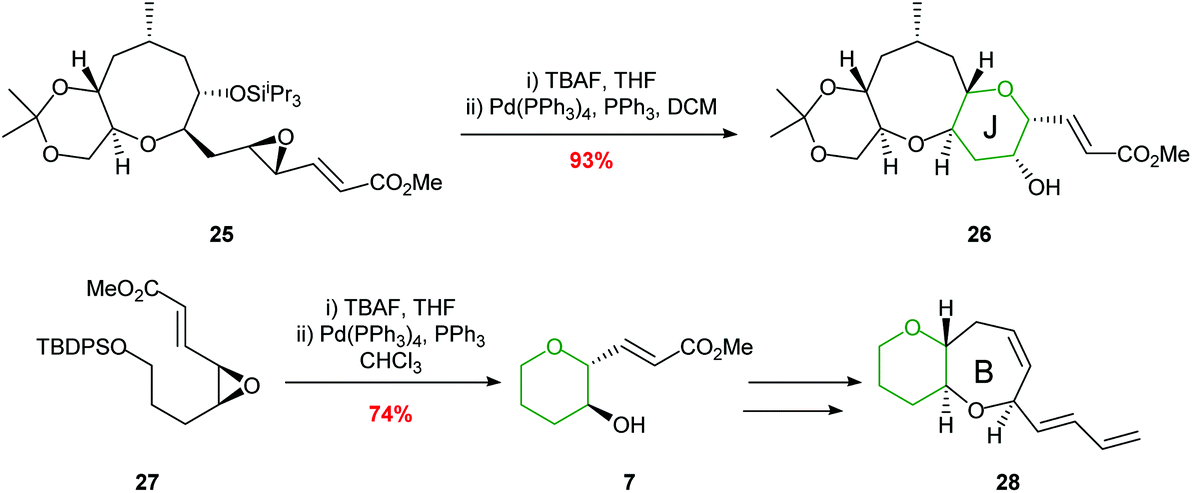 | ||
| Scheme 6 Synthesis of the J ring of ciguatoxin (top) and B ring of gambiertoxin 4B (bottom).39,40 | ||
Ha and co-workers expanded the scope of transition metals by using rhodium catalysis to promote selective THP formation (Scheme 7).41 Stirring trans-epoxide 29 with [Rh(CO)2Cl]2 in THF at room temperature gave anti-THP 24 with overall retention of stereochemistry via a double inversion mechanism. Interestingly, cis-epoxides were unreactive under these conditions. This was proposed to be due to a steric clash preventing the formation of the required π-allylrhodium species.
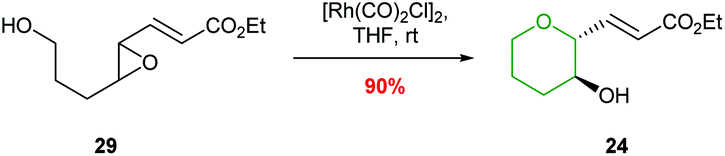 | ||
| Scheme 7 Rhodium catalysed intramolecular epoxide ring opening.41 | ||
The value of this methodology was demonstrated during the synthesis of the ABCDE ring fragment of ciguatoxin C. Hirama reported that cyclisation of acetal 30 under acidic conditions led to some deprotection of the acetal resulting in the required product 31 being isolated in only 41% yield (Scheme 8).42 In contrast using mild rhodium catalysed conditions, the required product 31 was isolated in 84% yield. Furthermore, in 2015 Jamison reported the creation of the B and C rings of (−)-brevisin through a cascade process under rhodium catalysis, whereas acidic conditions gave the required product 33 in low yield (Scheme 8).43
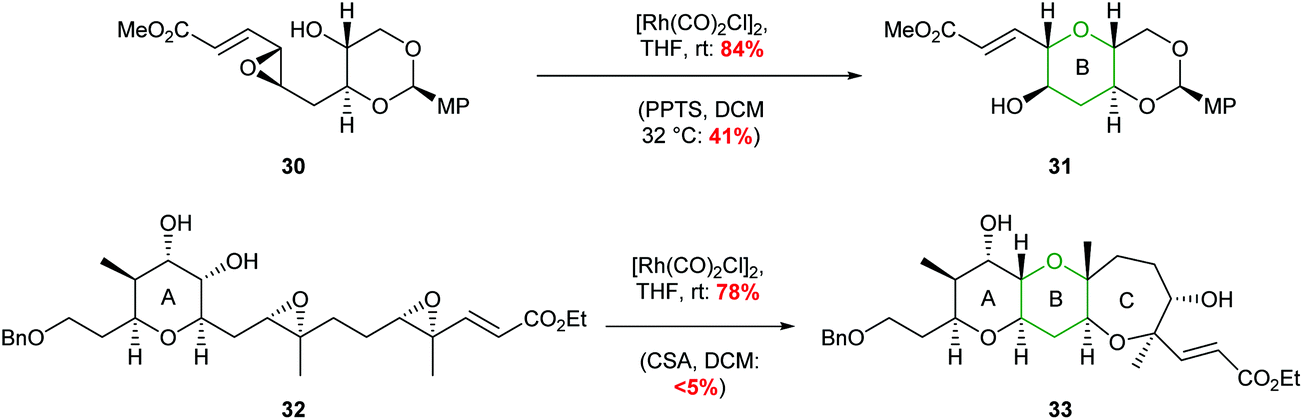 | ||
| Scheme 8 Synthesis of B ring of ciguatoxin C (top) and BC rings of (−)-brevisin (bottom). MP = 4-methoxyphenyl.42,43 | ||
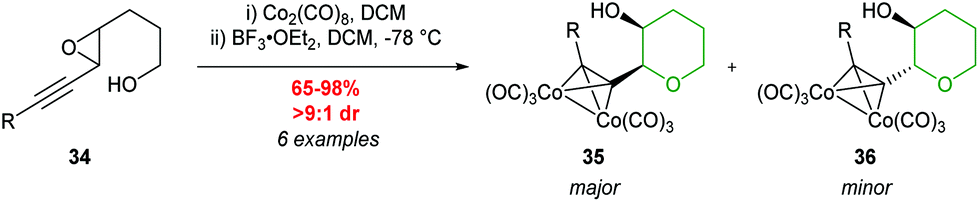 | ||
| Scheme 9 Cyclisation of racemic trans-acetylenic epoxides.44 | ||
 | ||
| Scheme 10 Mechanism for regioselective cyclisation of 4,5-epoxy alcohols 37.45 | ||
Hanaoka and co-workers adopted this chemistry to complete the first total synthesis of (−)-ichthyothereol, where cobalt complexation followed by 6-membered ring formation proceeded in 87% yield and with excellent stereocontrol (Scheme 11).46
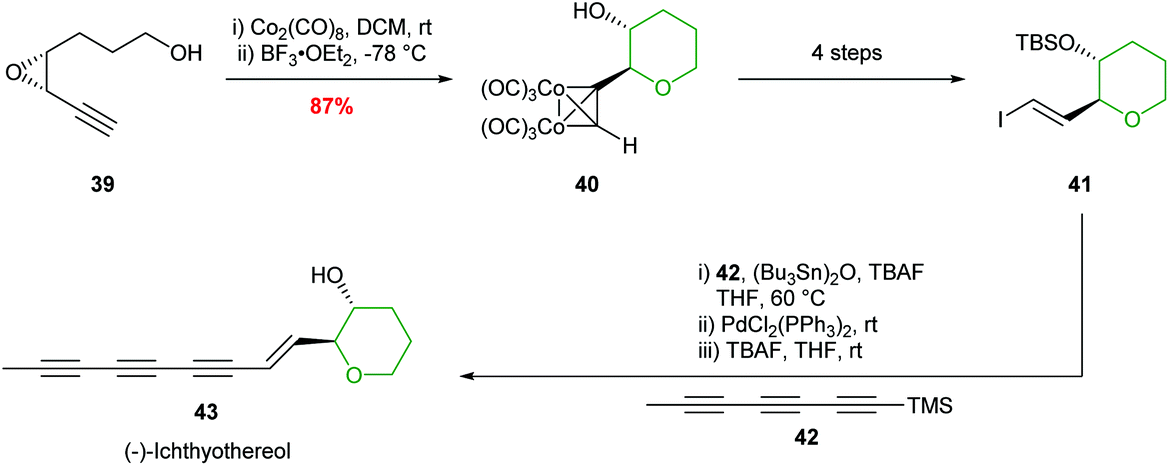 | ||
| Scheme 11 Total synthesis of (−)-ichthyothereol 43.46 | ||
The use of acetylenic epoxides as substrates for the synthesis of oxygen heterocycles was further extended by Hanaoka and co-workers (Table 3).47 Both cis and trans-acetylenic epoxides with electron donating substituents on the alkyne were cleanly converted to the corresponding THPs through activation with BF3·Et2O, in the absence of the cobalt complex (entries C and D, Table 3). This is analogous to the results of Nicolaou (Scheme 2) whereby inversion of stereochemistry at the propargylic position is observed.23 In contrast, substrates containing electron withdrawing or electron neutral groups on the alkyne gave predominantly the 5-exo products.
|
|
|||
|---|---|---|---|
| Entry | R | Yield |
44:45
|
| A | H | 92% | 10:90 |
| B | TMS | 91% | 62:38 |
| C | nBu | 96% | 95:5 |
| D | Ph | 94% | 100:0 |
| E | Bz | 96% | 1:99 |
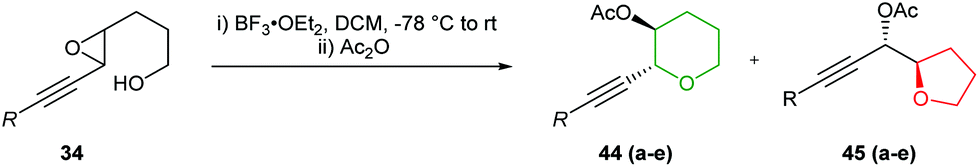
Forsyth showcased the value of this procedure in 2000 during the total synthesis of thyrsiferol (Scheme 12).48 Cyclisation of trisubstituted epoxide 46 with BF3·Et2O gave THP 47 in 91% isolated yield and with complete regiocontrol.
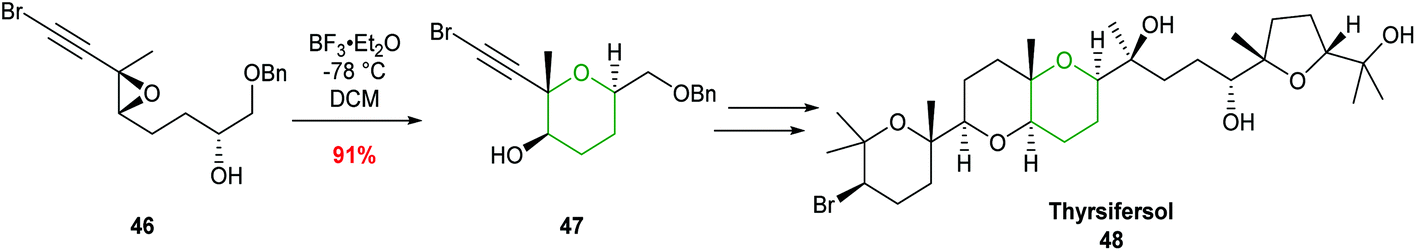 | ||
| Scheme 12 Total synthesis of thyrsiferol by Forsyth.48 | ||
In further studies, Hanaoka et al. illustrated that trisubstituted epoxides with an additional methyl group at either end of the epoxide (i.e. propargylic or homopropargylic positions) also led to 6-endo cyclisation using either the cobalt or Lewis acid mediated protocols.49 However, poor stereoselectivity was often observed for these classes of substrates using either procedure.
In 2004, the synthesis of THPs using an intramolecular Nicholas reaction was reported by Martín et al. (Scheme 13).50 Activation of a dicobalt complexed propargylic alcohol 49 with BF3·Et2O generates carbocation 50 which is trapped by the oxirane oxygen. Although this process does not involve intramolecular epoxide ring opening of an acetylenic epoxide, but instead reversed reactivity of an epoxide attacking the propargylic center, the formal 6-endo product is still formed.
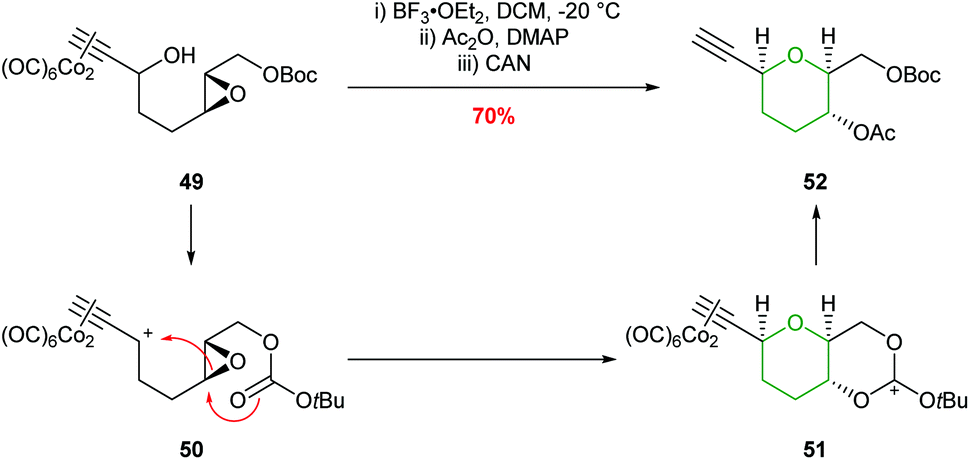 | ||
| Scheme 13 Intramolecular Nicholas reaction developed by Martín.50 | ||
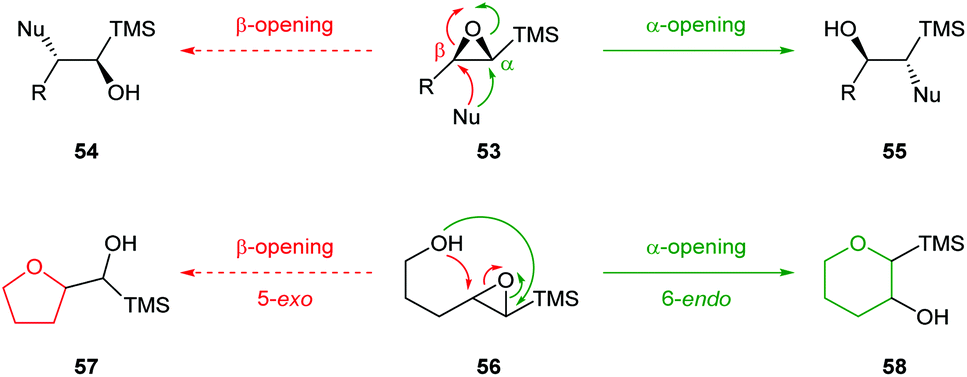 | ||
| Scheme 14 Inter and intramolecular nucleophilic ring opening of epoxy silanes 53 and 56.51 | ||
Initially, Schaumann et al. conducted acid catalysed cyclisations of trans-4,5-epoxy alcohols which showed that 1,4-anti diastereoisomers 59 afforded the 6-endo products 61 whereas 1,4-syn diastereoisomers 62 yielded two epimeric 5-exo products 66 (Scheme 15).54 These results were rationalised by the ability of the 1,4-anti diastereoisomers to adopt a chair-like conformation 60 in the transition state whilst cyclisation of 1,4-syn diastereoisomers would require a boat-like transition state 63. To avoid this high energy pathway, the reaction instead could proceed via an SN1 mechanism, facilitated by the β-cation stabilisation effect of silicon.
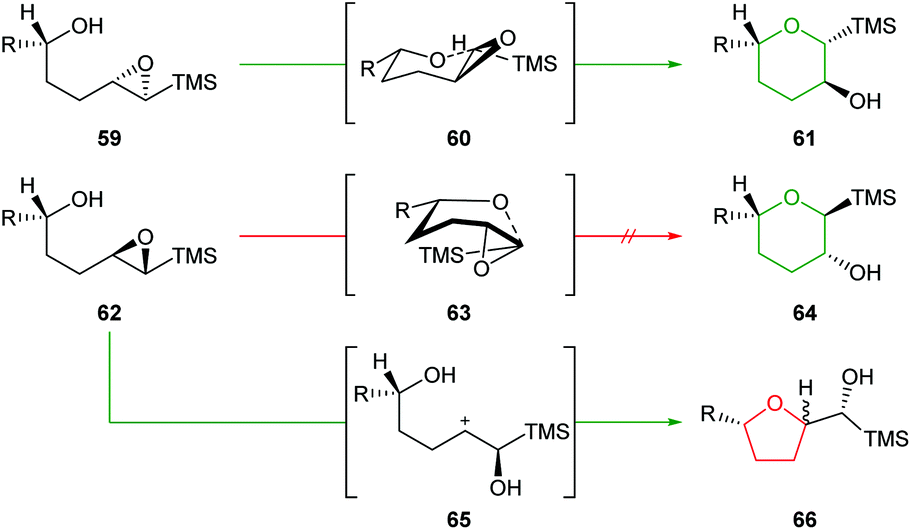 | ||
| Scheme 15 Intramolecular ring opening of 1,4-anti (top) and 1,4-syn (bottom) epoxy alcohols.54 | ||
Jamison further investigated cyclisations of epoxysilanes as a strategy for THP synthesis in ladder polyethers.55 In this case, trisubstituted epoxysilane 67, which places silicon in the axial position during cyclisation, gave THP 69 as the major product, whereas the diastereomer 71 produced a complex mixture of products (Scheme 16). The use of epoxysilanes as substrates is of particular value for the selective synthesis of THPs as the silyl directing group may be readily removed via a TBAF promoted Brook rearrangement.
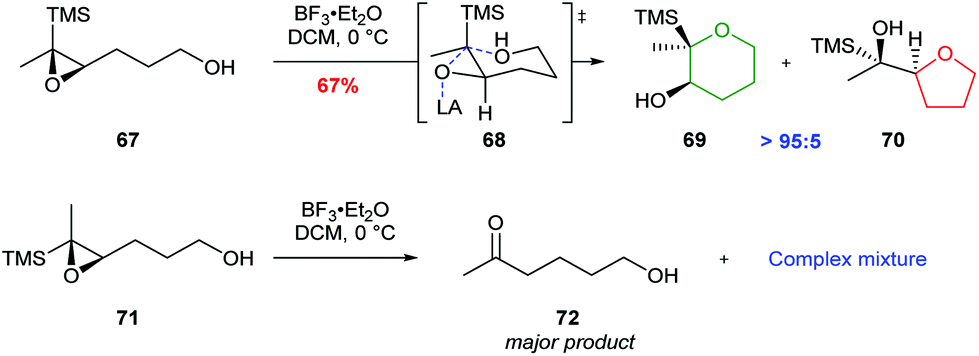 | ||
| Scheme 16 Cyclisation of cis-epoxysilane 67 and trans-epoxysilane 71 by Jamison.55 | ||
Jamison used this chemistry to good effect in iterative cascades, creating fused THP rings in excellent stereoselectivities and yields (Scheme 17).56,57 It was found that while BF3·Et2O was effective in the synthesis of isolated tetrahydropyrans, poor selectivity was obtained in cascade reactions. In contrast, the use of mildly basic conditions (Cs2CO3 and CsF in MeOH) facilitated the formation of fused THPs with concomitant deprotection of the silyl directing groups via a homo-Brook rearrangement (Scheme 17).
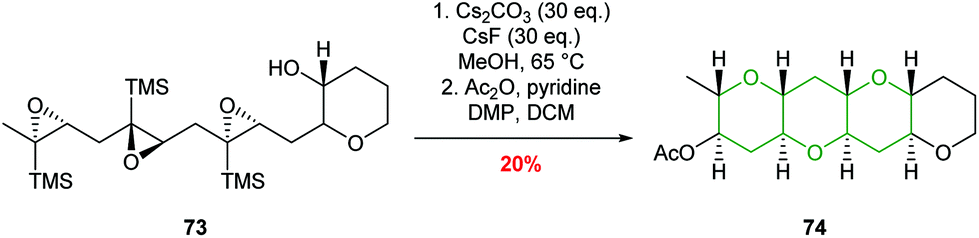 | ||
| Scheme 17 Epoxide-opening cascade directed by silicon.56,57 | ||
Jauch and co-workers used BF3·Et2O in the conversion of epoxysilane 75 to create the 6-endo product in 81% yield (Scheme 18).58 The silyl directing group was subsequently removed with TBAF to furnish THP 76 required for the construction of the jerangolids.
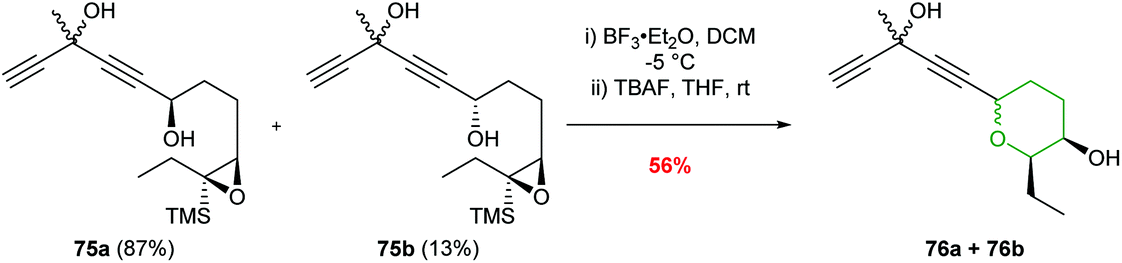 | ||
| Scheme 18 Synthesis of eastern THP ring of the jerangolids by Jauch.58 | ||
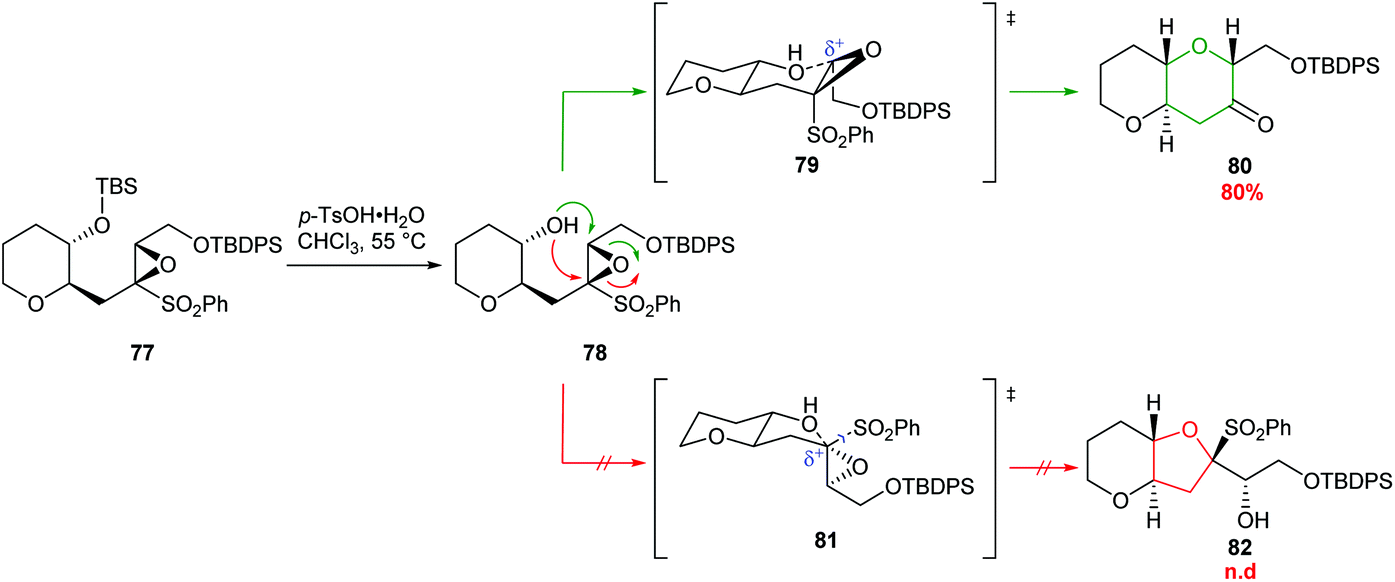 | ||
| Scheme 19 6-endo cyclisation of epoxysulfone 77.59 | ||
A common structural feature of polyether marine toxins is the presence of methyl groups on trans-fused polytetrahydropyran ring systems. Mori constructed these scaffolds in excellent yields from epoxysulfones (Scheme 20).61 For example, treatment of epoxysulfone 77 with p-TsOH at 55 °C gave THP 80 in 80% yield (Scheme 19). Alternative reaction conditions were required when the substrate possessed either a tetrasubstituted epoxide (83) or a silyl protect tertiary alcohol (85 and 87) rather than a secondary alcohol (Scheme 20). Treatment of epoxide 83 with TsOH at 0 °C gave 84 in 90% yield whereas at higher temperatures 1,2-sulfonyl migration occurred leading to ketone 89 as the major product. For tertiary silyl ether 85, Lewis acidic conditions were required to facilitate clean conversion to the THP 86. Finally, for the formation of dimethyl-substituted THP 88 from 87, BF3·Et2O was used to promote cyclisation, and Tl(TFA)3 was added to suppress the problematic 1,2-sulfonyl rearrangement.
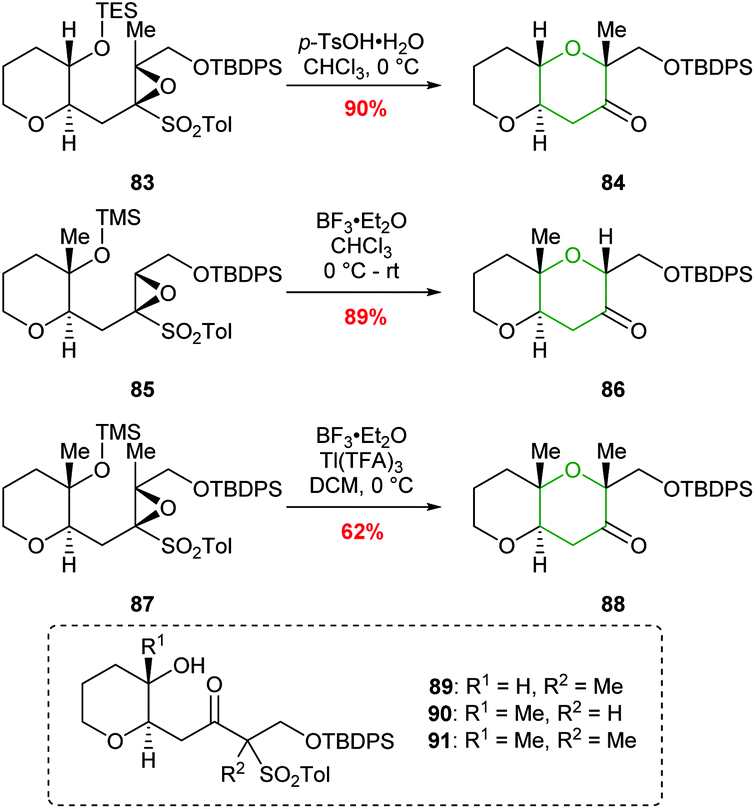 | ||
| Scheme 20 Conversion of methyl substituted epoxy sulfones 83, 85 and 87 to the corresponding THP products.61 | ||
The relative stereochemistry of the epoxysulfone substrate proved to be important in the outcome of reactions with Brønsted acids with cyclisation only occurring with substrates such as 78, whilst 92, 93 and 94 were unreactive (Fig. 4). These differences in reactivity were proposed to be due to steric interactions preventing the unreactive isomers adopting the 6-endo transition states.62 To expand the substrate scope, Mori used alternative reaction conditions to effect cyclisation and deliver THPs (Scheme 21). Thus, the silyl ether of the epoxysulfone (95 or 96) was first deprotected with TBAF and the resultant alcohols treated with MgBr2·Et2O and finally addition of DBU gave bicyclic ketone 99 in good yield and excellent stereocontrol.62 Although this is not an intramolecular epoxide ring opening process, the formal 6-endo product is obtained.
 | ||
| Fig. 4 Proposed transition states and corresponding steric clashes for the intramolecular epoxide ring opening of epoxysulfones 78, 92, 93 and 94.62 | ||
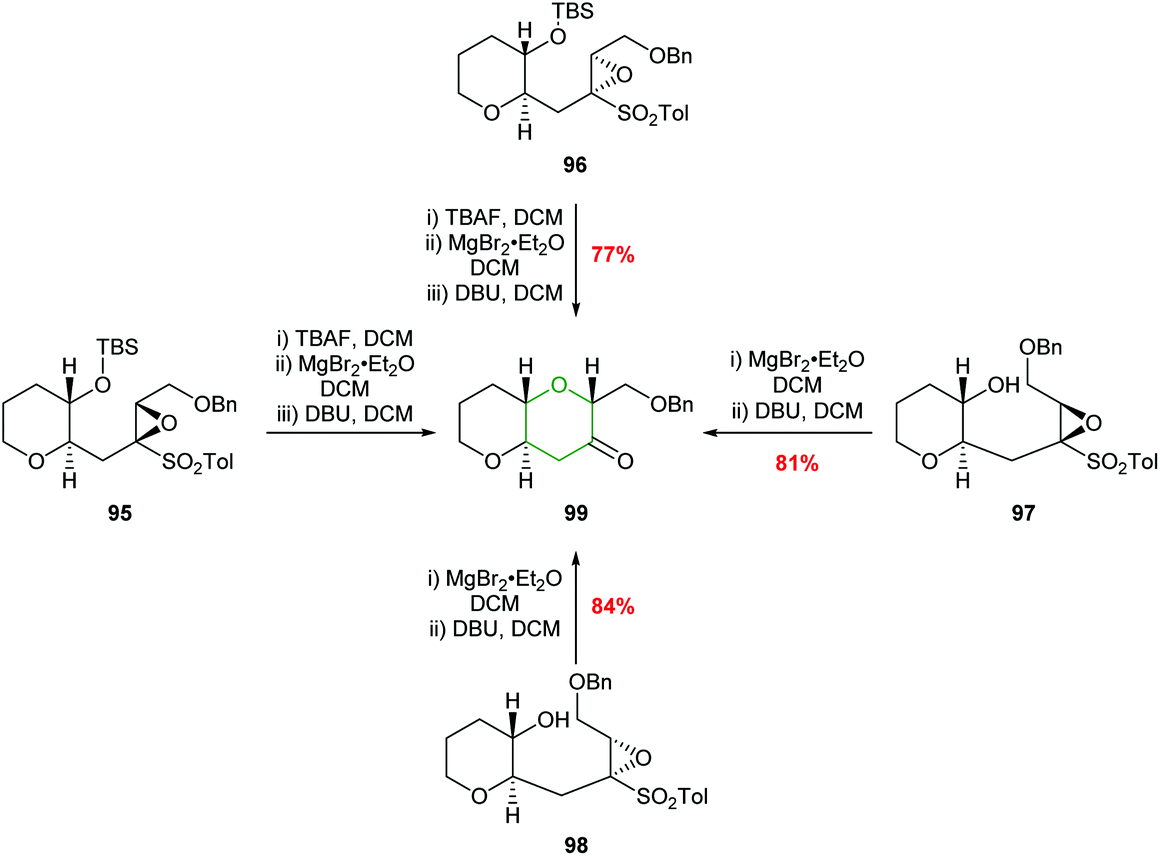 | ||
| Scheme 21 Alternative procedure for the synthesis of THP 99.62 | ||
Sulfonyl directed intramolecular epoxide ring opening has been widely used by Mori and others to prepare THP containing natural products and some examples are shown in Scheme 22.63–68
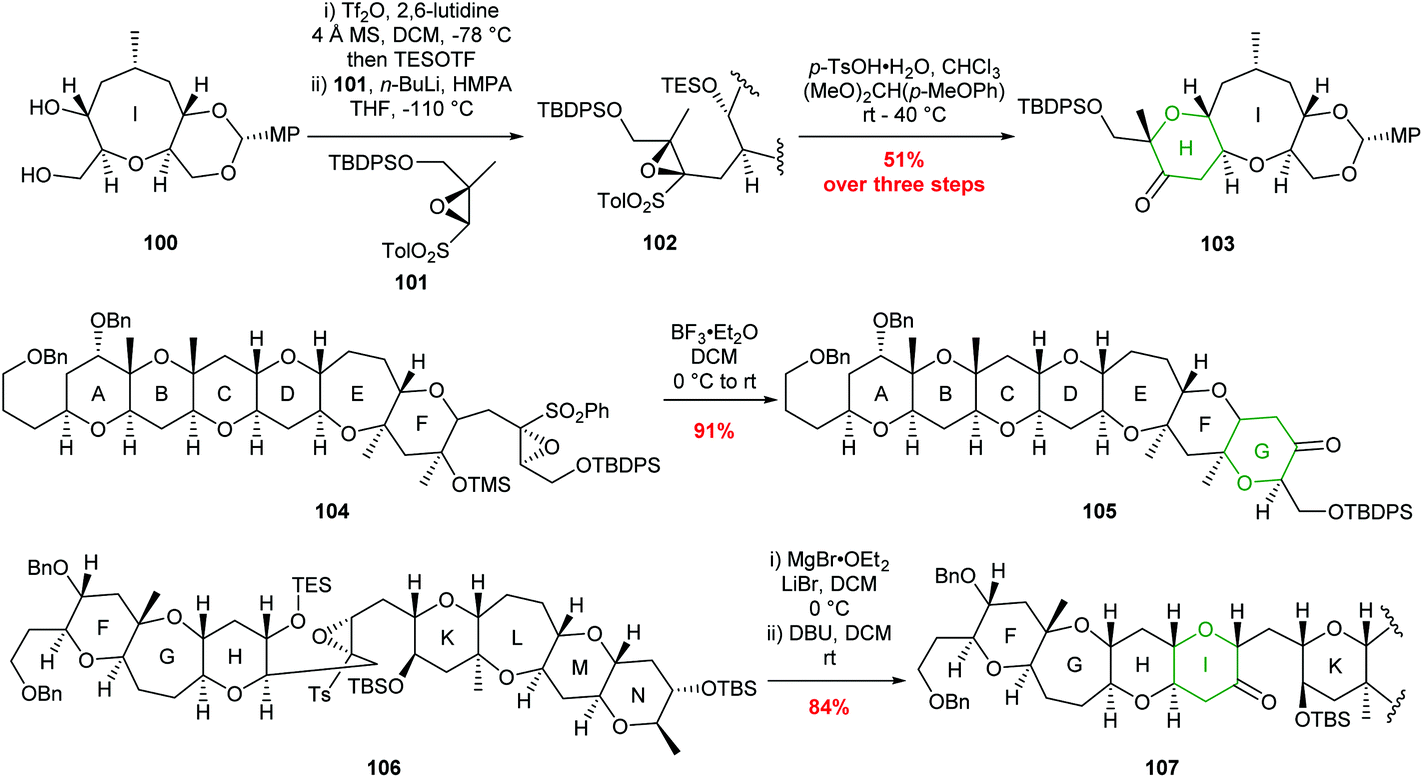 | ||
| Scheme 22 Examples of sulfonyl directed intramolecular epoxide ring opening in total synthesis. H ring of ciguatoxin (top), G ring of gambierol (middle) and I ring of gymnocin-A (bottom). MP = 4-methoxyphenyl.65,66,68 | ||
 | ||
| Scheme 23 Gold-catalysed cyclisation of allenic epoxides 108 and 110.72 | ||
A further example of this regiocontrol was demonstrated by Holton in the total synthesis of hemibrevetoxin B reported in 2002 (Scheme 24).73 Reaction of 112 with N-(phenylseleno)phthalimide promotes a cascade process leading to formation of 6- and 7-membered oxygen heterocycles in 83% yield. The use of hexafluoroisopropanol (HFIP) as a solvent was proposed to be important to increase the charge separation in the reaction and therefore improve selectivity.
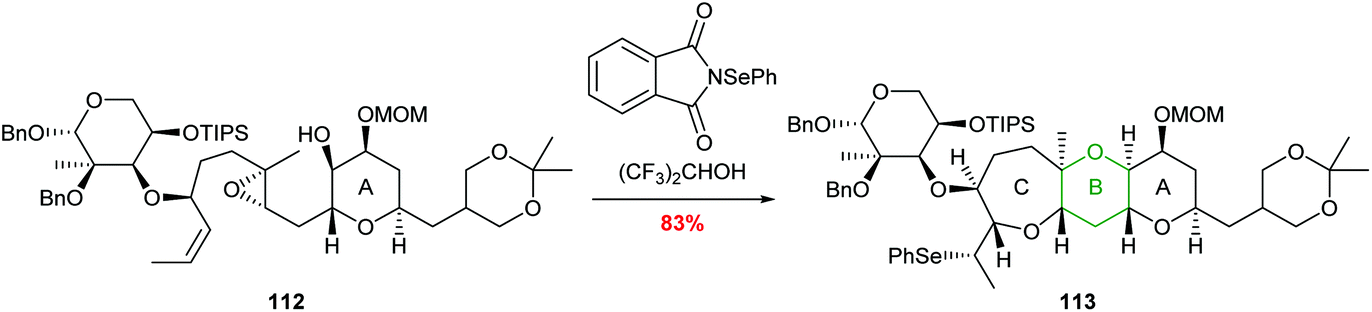 | ||
| Scheme 24 Synthesis of the B and C rings of hemibrevetoxin B.73 | ||
More recently, Qu and co-workers demonstrated how trialkyl substituted epoxides are converted to the corresponding THP products through reaction with 1-ethyl-3-methylimidazolium tetrafluoroborate ([EMIM]BF4) in perfluoro-tert-butanol (PFTB).74 In contrast, disubstituted epoxides formed solely the THF products. These contrasting results indicate the important role that an extra substituent on the epoxide can play in controlling 6-endo cyclisation. Many selective cyclisation cascades have been used to good effect to generate the core structures of marine polycyclic polyether natural products.
2.2 Reagent controlled formation of THPs from 4,5-epoxy alcohols
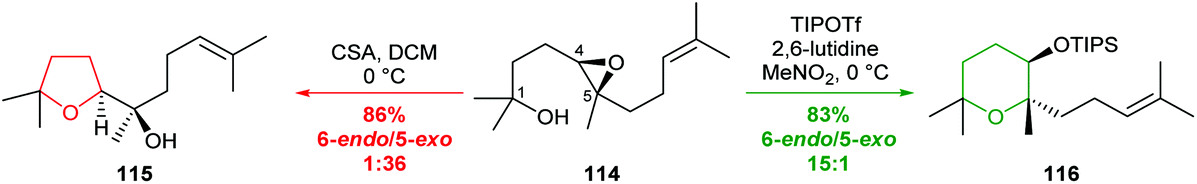 | ||
| Scheme 25 Reagent-controlled selectivity in cyclisations of 4,5-epoxy alcohol 114.75 | ||
In 2007, Morimoto demonstrated the value of these reagent-controlled cyclisations to create both 5 and 6 membered rings with excellent selectivity in the total synthesis of (+)-enshuol 121 (Scheme 26).77 Whilst this methodology still imposes constraints on the substrate structure, it demonstrates the power of using different reagents to control selectivity.
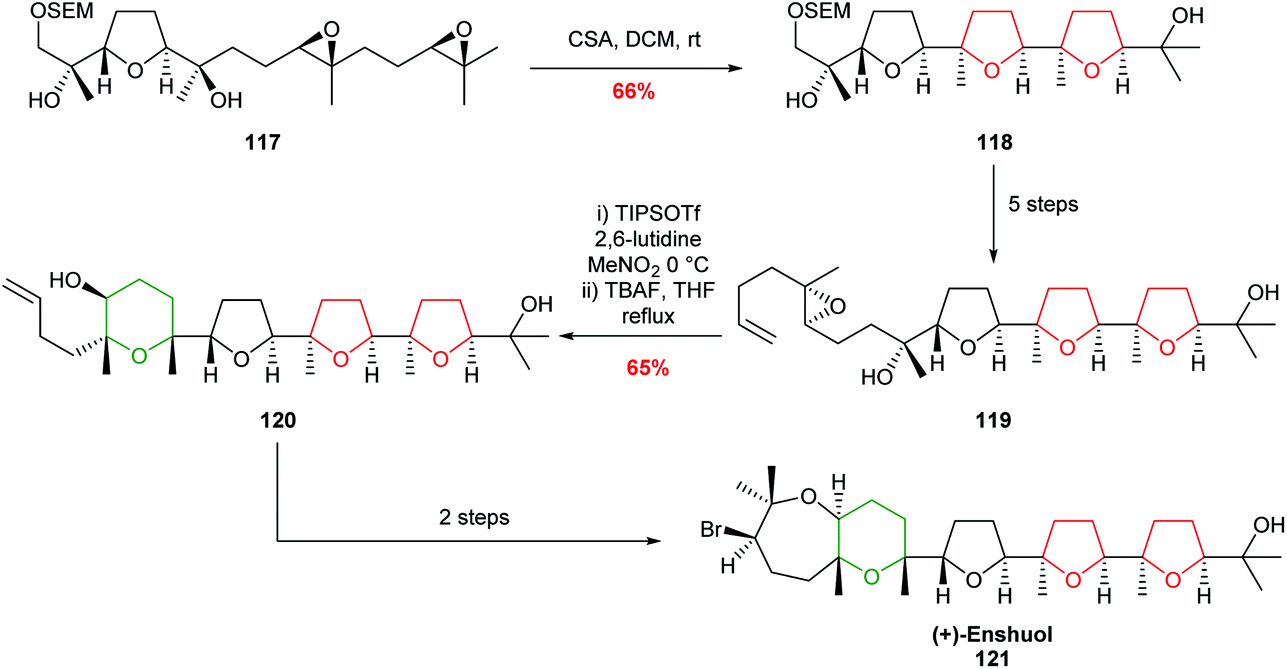 | ||
| Scheme 26 Total synthesis of (+)-enshuol by TIPSOTf promoted 6-endo cyclisation.77 | ||
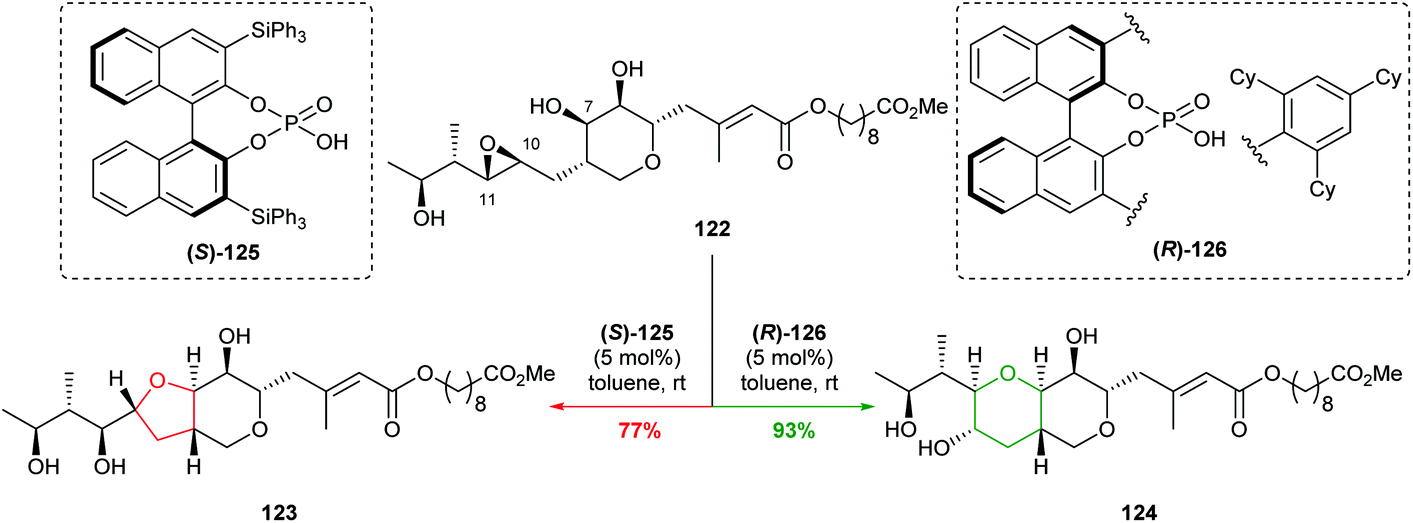 | ||
| Scheme 27 Cyclisations of epoxide 122 using chiral phosphoric acid catalysts.78 | ||
Through a combination of mechanistic and computational studies, Nagorny proposed the selectivity originated from steric clashes between the catalyst and substrate set up by a hydrogen bond network. In particular, steric interactions between (R)-126 and 122 increase the energy of the 5-exo transition state, whilst no such interactions occur between (S)-125 and 122. Although this is a substrate specific case, it elegantly illustrates the use of non-covalent interactions with a chiral catalyst in intramolecular attack on epoxides. With the aid of computational methods and experimental design, this work may pave the way forward for developing new catalysts for controlling cyclisation of a variety of 4,5-epoxy alcohols.
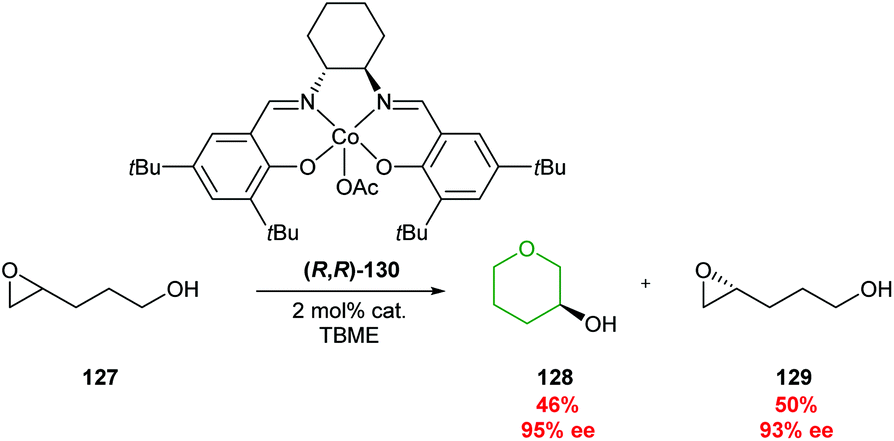 | ||
| Scheme 28 Kinetic resolution of 4,5-epoxy alcohols through cobalt catalysis.79 | ||
Later in 2006, Toste developed a vanadium catalysed kinetic resolution starting from unsaturated alcohols (Scheme 29).81 The approach proceeded via an initial resolution of α-hydroxy ester 131, followed by subsequent diastereoselective epoxidation of 132 and finally regioselective intramolecular epoxide ring opening to afford THP products 135. Cyclisation is proposed to proceed via coordination of the vanadium complex to the hydroxy ester 133via a chair like transition state 134.4 To the best of our knowledge, neither the cobalt or vanadium catalysed processes have been used in the total synthesis of natural products.
 | ||
| Scheme 29 Vanadium catalysed synthesis of 2,5-trans-tetrahydropyrans.81 | ||
|
|
||
|---|---|---|
| Entry | Conditions |
138:139
|
| 1 | Cs2CO3, MeOH | 1:2.7 |
| 2 | BF3·Et2O, DCM | 1.4:1 |
| 3 | AcOH, toluene | 1.6:1 |
| 4 | H2O | >10:1 |
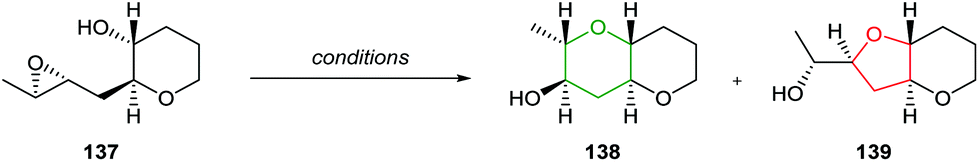
Whilst a methyl substituent on a trisubstituted epoxide can direct nucleophilic attack as described in section 2.1.5, interestingly Jamison demonstrated that cyclisation of THP templated substrates proceeded to give endo products such as 141 under aqueous conditions (Scheme 30).70
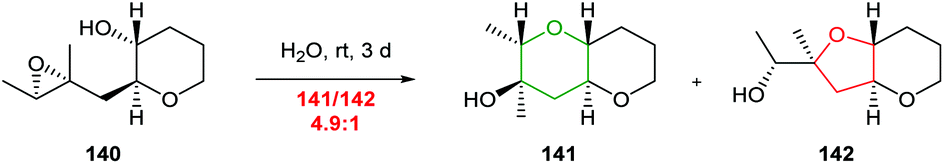 | ||
| Scheme 30 Water promoted 6-endo cyclisation of 4,5-epoxy alcohol 140.70 | ||
Building on the use of the THP ring template, Jamison developed a benzylidene acetal template to achieve selective 6-endo cyclisation which has the advantage that following reaction, the template was readily removed (Scheme 31).87 Both water and silica gel were used to promote the desired cyclisation.
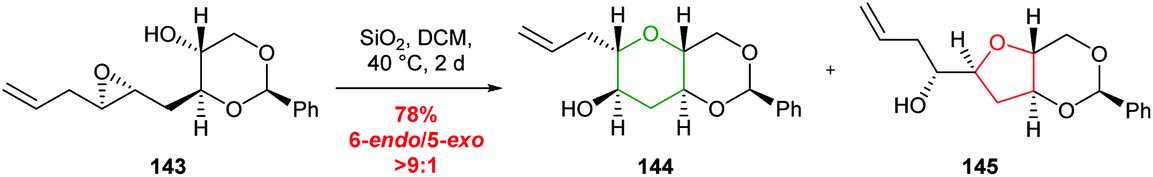 | ||
| Scheme 31 Silica gel promoted 6-endo cyclisation of benzylidene acetal templated epoxide 143.87 | ||
This methodology was used to good effect in the synthesis of the HIJK ring system of gymnocin A through a polycyclisation cascade in water, and the J ring of yessotoxin (Scheme 32).87,88
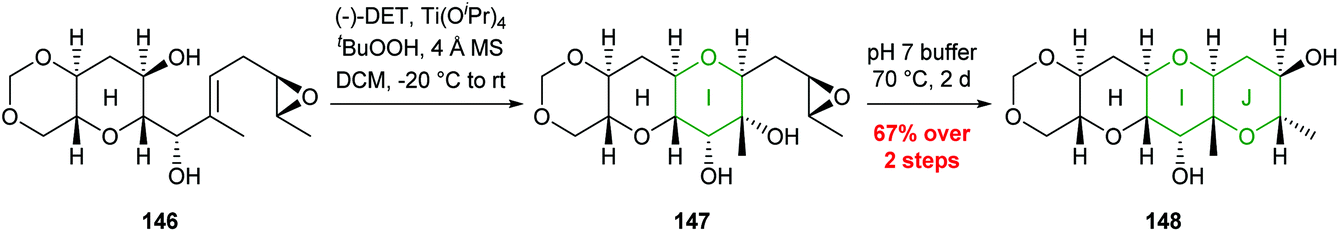 | ||
| Scheme 32 Synthesis of J ring of yessotoxin by Jamison.88 | ||
 | ||
| Scheme 33 Cyclisation of epoxy alcohol 149 under acidic conditions and antibody catalysis (top) and transition state mimic 154 used to produce antibody catalyst 26D9.89,91 | ||
A similar approach was investigated by Chiosis, but poor selectivity was achieved.92 Nevertheless, both these studies illustrate the power of antibody catalysis to overcome the inherent preference for 5-exo cyclisation of 4,5-epoxy alcohols.
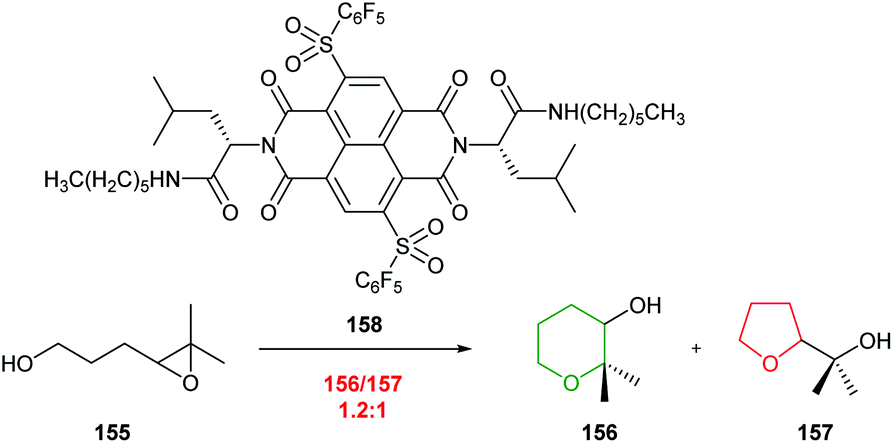 | ||
| Scheme 34 Cyclisation of 4,5-epoxy alcohol 155 catalysed by NDI 158.93 | ||
2.3 Further methods
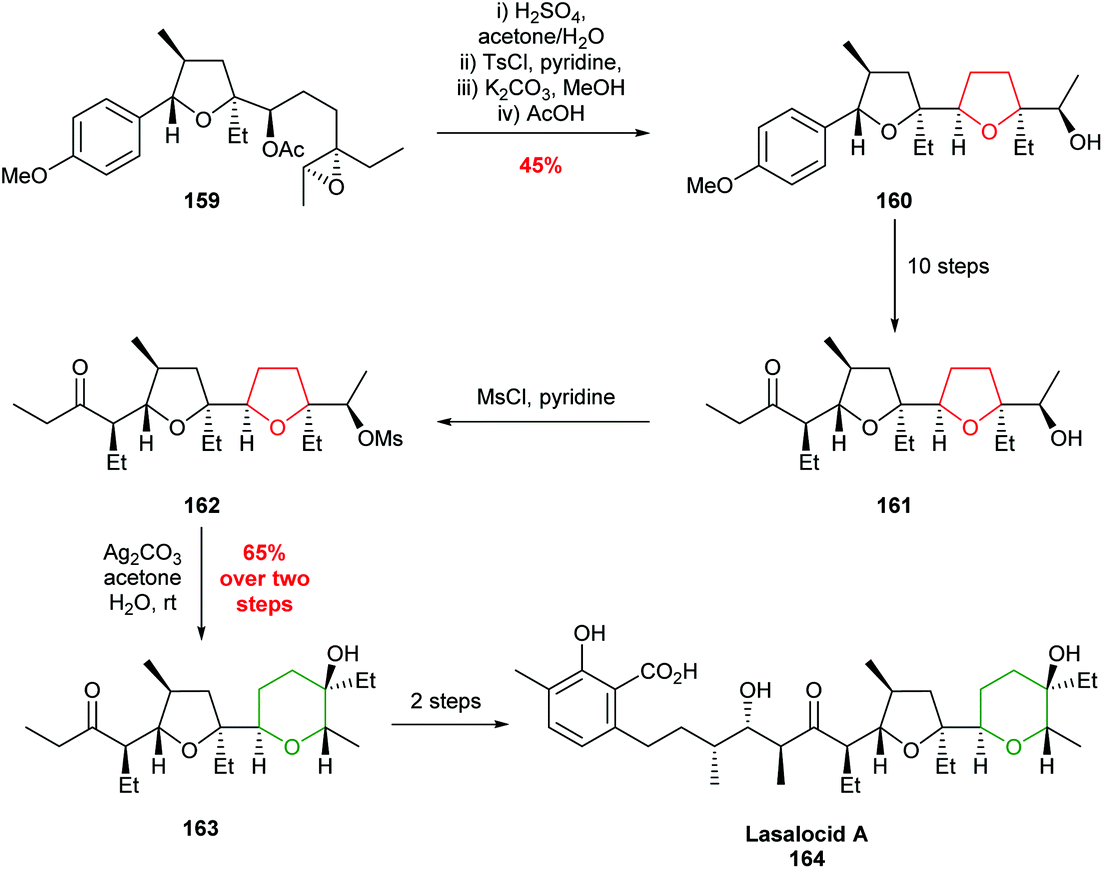 | ||
| Scheme 35 Total synthesis of lasalocid A.95 | ||
Nakata and co-workers reported further investigations into this ring expansion and first converted unsaturated alcohol 165 to an epoxide which cyclised to the expected THF on treatment with camphorsulfonic acid in DCM (Scheme 36).96 The key ring expansion step was performed by heating mesylate 166 in AcOH and water in the presence of Zn(OAc)2 giving, after acetylation, THP 167 in 75% yield.
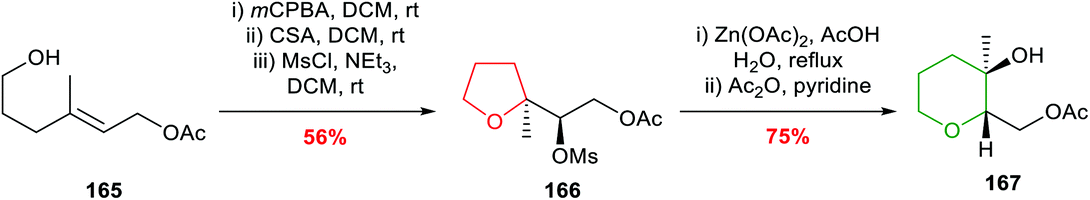 | ||
| Scheme 36 Preparation of THP 167 by ring expansion of 5-exo cyclisation product.96 | ||
This methodology was used in the synthesis of a model compound 170 with the ST rings of maitotoxin (Scheme 37).34,97
 | ||
| Scheme 37 Synthesis of model ST rings of maitotoxin.34,97 | ||
Zinc acetate in refluxing acetic acid was required for the ring expansion of mesylates 166 and 169. However, Nakata showed that by using chloromethylsulfonate (Mc) as the leaving group, rather than mesylate, ring expansion was achieved without the need for acetic acid (Scheme 38).98,99 These modified conditions were used in the efficient conversion of THF 171 to THP 167. Furthermore, it was shown that ring expansion may be performed in the absence of a Lewis acid, albeit giving 6-membered rings in lower yield.
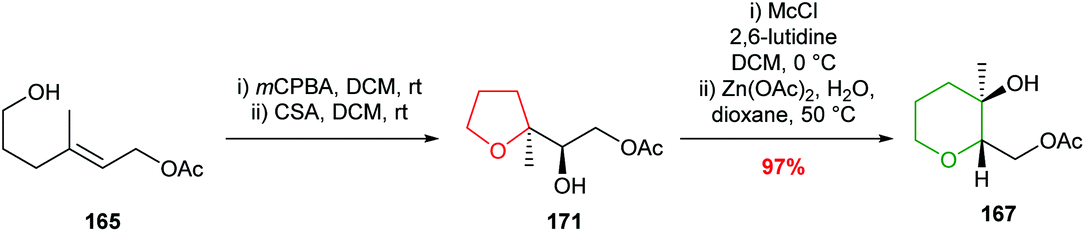 | ||
| Scheme 38 Alternative conditions for the rearrangement-ring expansion of 5-exo product 171. Mc = chloromethylsulfonate.99 | ||
McDonald used this ring expansion strategy to create the B ring of 15,28-dideoxy-15,28-didehydrothyrsenol in 31% yield over 2 steps from alcohol 172 (Scheme 39).100
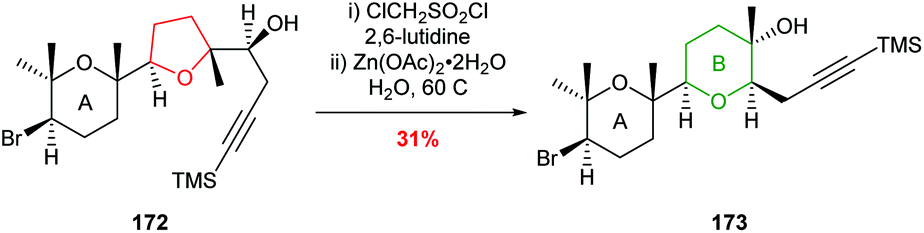 | ||
| Scheme 39 Synthesis of B ring in 15,28-dideoxy-15,28-didehydrothyrsenol.100 | ||
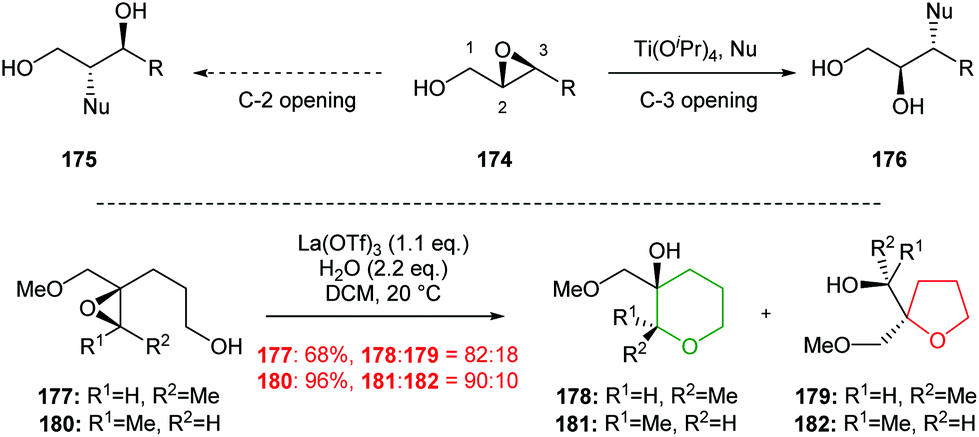 | ||
| Scheme 40 Inter- and intramolecular Lewis acid catalysed regioselective epoxide ring opening.101,102 | ||
Two examples of the use of La(OTf)3 mediated cascade reactions have been reported leading to bicyclic and tricyclic products 184 and 186 (Scheme 41).103 There remains opportunities for exploitation of this methodology in natural product synthesis.
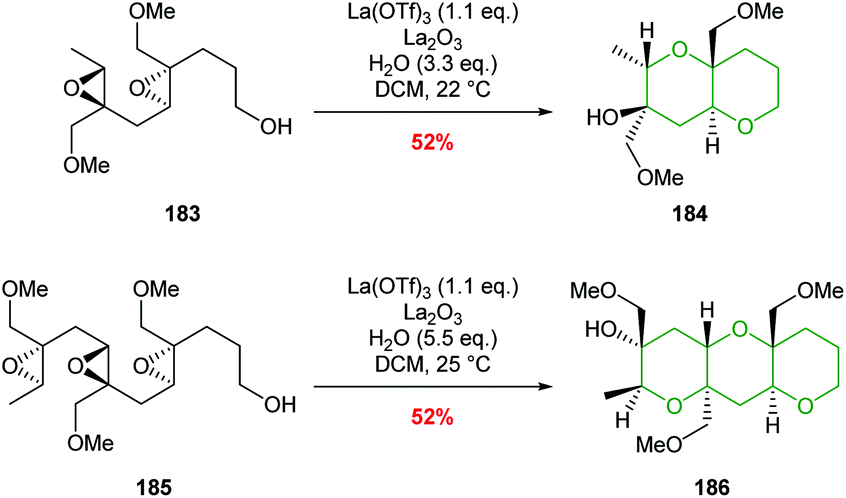 | ||
| Scheme 41 La(OTf)3 catalysed polyepoxide ring opening cascades.103 | ||
2.4 Summary of synthetic methods
In summary, many methods have been developed for the preparation of oxygen heterocycles from 4,5-epoxy alcohols. Structural features may be incorporated into the substrate, such as double and triple bonds, epoxysilanes and epoxysulfones, which favour formation of tetrahydropyrans via either stabilisation of a 6-endo transition state or destabilization of a 5-exo transition state, to afford tetrahydropyrans in high yields. These strategies have been used in natural product synthesis. There are exciting prospects for the use of reagents and catalysts in IERO to control 5-exo and 6-endo cyclisation of 4,5-epoxy alcohols. Both Lewis acids and chiral phosphoric acids have been used in regioselective cyclisations but to date these methods have not been widely used in the total synthesis of natural products. However, taking inspiration from nature might facilitate the optimisation of catalysts and development of novel methods for reagent controlled 6-endo cyclisation for THP formation. In the next section the biosynthesis of THPs via cyclisations of 4,5-epoxy alcohols are reviewed.3. Biosynthetic methods
Compared with the diversity of synthetic methods for the preparation of saturated oxygen heterocycles, a more limited set of reactions are used in natural product biosynthesis. However, epoxide formation/intramolecular epoxide-opening cascades are common processes used in the biosynthesis of many natural products. In some cases, epoxide hydrolases (EHs) have been identified which control the stereochemical outcome of cyclisation. When EHs are absent, labile epoxide biosynthetic intermediates 127 often spontaneously cyclise to generate 5-exo THF products (Scheme 42).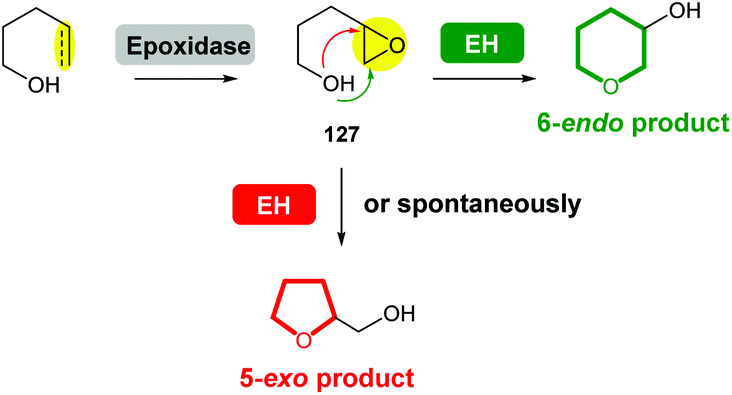 | ||
| Scheme 42 Biosynthesis of THPs and THFs via epoxide formation/epoxide-opening cascade reactions. EH: epoxide hydrolase. | ||
Interestingly, the biosynthesis of some natural products with structures containing THF rings has also been reported to be mediated by EHs, or in some cases, the 5-exo cyclisation is significantly accelerated by the presence of an EH. While this review focuses on THP formation, the biosynthesis of THF rings via a similar epoxide formation/epoxide-opening cascade reaction is topical and therefore is also discussed.
3.1 Formation of THPs from 4,5-epoxy alcohols catalysed by epoxide hydrolases (EHs)
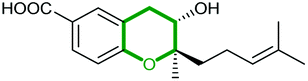
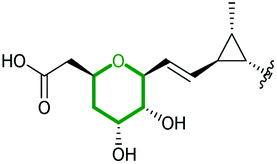
Using in vivo and in vitro studies of the flavin-dependent monooxygenase (FMO) Lsd18, Minami et al. demonstrated that Lsd18 is responsible for oxidising the diene precursor 187 to afford bisepoxyprelasalocid A 188 (Scheme 43).105 In search for the enzyme responsible for selective epoxide-opening, Oikawa and co-workers identified Lsd19 as the putative epoxide hydrolase from the lasalocid biosynthetic gene cluster. In vitro turnover assays of bisepoxyprelasalocid A 188 with purified Lsd19 showed conversion of the synthetic substrate into lasalocid A (Scheme 43).104 In the absence of Lsd19, the synthetic bisepoxyprelasalocid A 188, which was obtained in only 3% yield, spontaneously cyclises to generate the THF ring of isolasalocid A 189.104 As discussed in section 2.3.1, Kishi synthesised the THP ring of lasalocid A by initial 5-exo cyclisation of an analogous epoxide 159 followed by ring expansion (Scheme 35). Whilst an elegant approach, this contrast between the synthesis and biosynthesis of the lasalocid THP ring highlights how nature can elegantly employ EH enzymes to readily overcome inherent selectivity for 5-exo cyclisation of 4,5-epoxy alcohols.
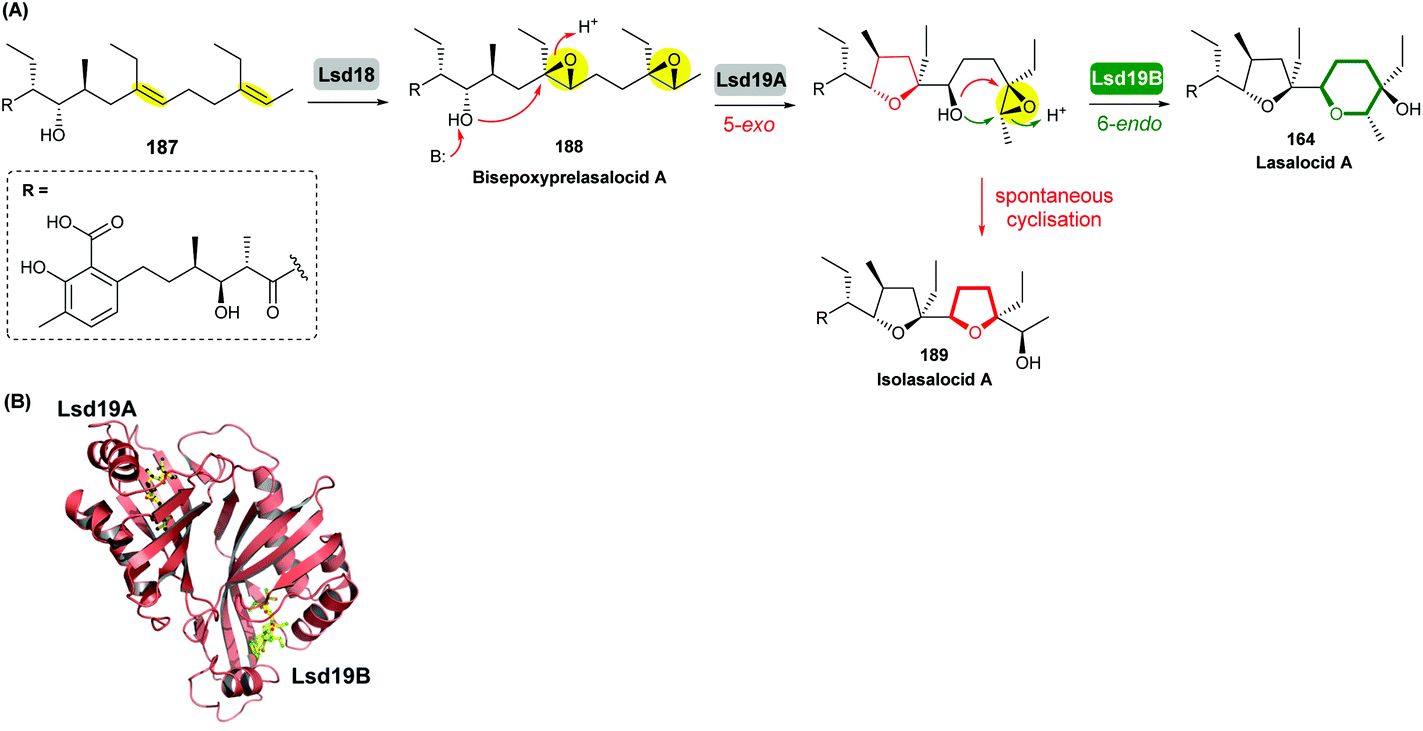 | ||
| Scheme 43 (A) THP ring formation in lasalocid A biosynthesis; (B) Crystal structure of Lsd19 with bound substrate (left half) and product (right half) analogues shown by stick models.104–107 | ||
Concurrent in vivo studies by the Leadlay group on the targeted deletion of the lsd19 gene (previously identified as lasB) in the lasalocid producing strain further supported the role of Lsd19 in directing biosynthesis towards lasalocid A rather than towards isolasalocid A.106 In the ΔlasB mutant of Streptomyces lasaliensis, production of lasalocid A was no longer detected, and isolasalocid A was the exclusive product.106
To gain a better understanding of how Lsd19 catalyses THP formation, Hotta et al. determined the X-ray crystal structure of Lsd19 in complex with its substrate and a product analogue to 1.59 Å resolution (Scheme 43).107 The epoxide hydrolase consists of two catalytic domains including the N-terminal region Lsd19A and the C-terminal region Lsd19B. The 5-exo and 6-endo epoxide-opening steps are catalysed by Lsd19A and Lsd19B, respectively. On the basis of structural analysis and quantum mechanical calculations, a mechanism of general acid–base catalysis has been proposed in which an aspartate residue acts as a base that activates the hydroxyl group on the substrate for a nucleophilic attack on the epoxide carbon, while a tyrosine residue acts as an acid to stabilise the negative charge that develops on the epoxide oxygen during the transition state of the reaction.107
The terminal triene portion 190 of the polyene precursor firstly undergoes bisepoxidation catalysed by the flavin-dependent monooxygenase AurC to form a proposed bisepoxide 191, which is then transformed into an isolable THF intermediate 192 through a 5-exo epoxide-opening step catalysed by the epoxide hydrolase AurD (Scheme 44). This stable THF intermediate then undergoes another round of epoxidation (by AurC) to give 193 and in this case cyclisation occurs via 6-endo ring opening (by AurD) to yield the dioxabicyclo-octane ring product 194.108
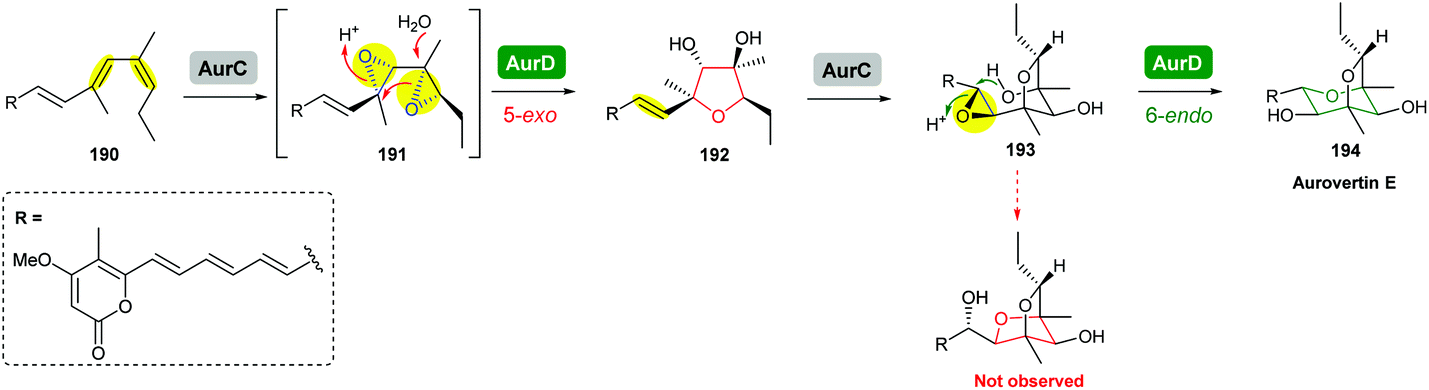 | ||
| Scheme 44 Proposed late-stage biosynthesis of aurovertins.108 | ||
 | ||
| Scheme 45 (A) Rearrangement products of pseudomonic acid A (195) formed under either acidic or basic conditions; (B) THP ring formation in mupirocin/thiomarinol biosynthesis; (C) crystal structure of MupZ; (D) proposed mechanism of MupZ-catalysed THP ring formation; (E) structure of thiomarinol A (202).109,110 | ||
Using whole-cell biotransformations, Crump, Willis and co-workers showed that the C8–C16 single bond in the acyclic precursor 198 is oxidised by the Rieske non-haem oxygenase MupW to give the corresponding 8,16-epoxide 199 (presumably through an 8,16-alkene intermediate), which spontaneously cyclises giving a five-membered tetrahydrofuran ring product 201 (Scheme 45).110 When both MupW and the epoxide hydrolase MupZ are included in the biotransformation system, the hydroxylated THP ring product 200 is then generated. Deletion of the mupZ gene in the mupirocin producing strain abolished production of the THP ring metabolites and the THF ring compounds were produced instead. Based on X-ray crystallographic studies, molecular modelling and mutagenesis experiments of MupZ, the 6-endo epoxide-opening has been proposed to proceed through an acid–base mechanism, in which the Glu54 residue initially deprotonates the 5-hydroxyl group of the substrate (Scheme 45D), then the Tyr41 residue protonates the epoxide oxygen and subsequently stabilises the developing transition state, leading to the 6-endo THP ring product 200.110 This mechanism of general acid–base catalysis was found to be similar to that of the THP ring formation in lasalocid A biosynthesis.107
The thiomarinols, produced by marine bacteria such as Pseudoalternomonas sp. SANK 73390, are a group of natural products that closely resemble mupirocin and also show significant antibiotic activities against MRSA.111 Their structures are assembled on a highly equivalent polyketide backbone to mupirocin but are esterified by an 8-hydroxynonanoic acid chain and bear an additional non-ribosomal peptide synthase (NRPS)-derived pyrrothine moiety (Scheme 45E). TmlW/TmlZ are the MupW/MupZ equivalents in thiomarinol biosynthesis and each share over 55% sequence identity to their counterparts in the mupirocin pathway. Using the same whole-cell biotransformation approach, Wang et al. recently demonstrated that thiomarinol biosynthesis employs the same THP ring forming mechanism that requires the dual action of the Rieske non-haem oxygenase TmlW and the epoxide hydrolase TmlZ.111 It was also shown that these two pairs of enzymes are cross compatible between the mupirocin and thiomarinol systems.111
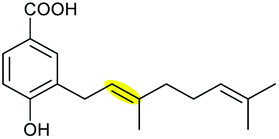
Using in vitro enzymatic assays, He et al. demonstrated that the 3-geranyl-4-hydroxybenzoate precursor 203 is converted by XimD to an unstable epoxide intermediate 204, which spontaneously transforms to a benzofuran product 205. Although the extra methyl substituent on the epoxide intermediate may have a directing effect that has been applied in synthetic efforts to favour the THP ring formation (see section 2.1.5 for examples), no THP ring product was reported in this case. However, when XimE was added in the assay, the 6-membered ring benzopyran xiamenmycin B 206 was formed as the major product (Scheme 46).112 Crystallographic structures of XimE, with and without product, suggested a synergistic mechanism in which residues E136, Y46, and H102 are functionally important to catalysis, substrate binding, and structural stabilisation (Scheme 46). Notably, in studies on substrate scope, these two enzymes exhibited high promiscuity capable of producing 14 products with 6 different benzoheterocyclic scaffolds.112 In a follow-up study, this pair of substrate-promiscuous enzymes were further employed for combinatorial biosynthesis of a series of benzoheterocyclic derivatives with greater structural diversity.113
 | ||
| Scheme 46 (A) THP ring formation in xiamenmycin biosynthesis; (B) crystal structure of XimE; (C) proposed mechanism of XimE-catalysed pyran ring formation.112 | ||
3.2 Formation of THPs from 4,5-epoxy alcohols in which no EH has been identified
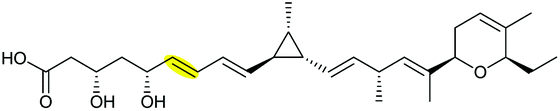
The biosynthetic pathway was proposed by Reeves and co-workers through analysis of the biosynthetic gene cluster and characterisation of compounds produced by gene knockout strains.114 Disruption of the ambJ gene that encodes for a flavin-dependent epoxidase yielded ambruticin J lacking the THP ring. Thus, the THP ring formation was proposed to occur through epoxidation of ambruticin J 207 by AmbJ, followed by selective epoxide-opening of 208 at C-7 to give ambruticin F (Scheme 47). It is not known whether this cyclisation is under enzymatic or substrate control, but to-date no epoxide hydrolase has been identified in the ambruticin gene cluster.114 Nevertheless, as demonstrated by the work of Nicolaou (section 2.1), alkenic epoxides such as epoxy-ambruticin J 208 readily under IERO to form THP products due to stabilisation of the 6-endo transition state by the adjacent π-system.22 Our more recent studies involving cyclisation of model 4,5-epoxy alcohols are in accord with the proposal that both the 8,9-alkene and 5-alcohol of epoxy-ambruticin J 208 may play a role in controlling 6-endo cyclisation.115 In 2021 the total synthesis of ambruticin J was reported enabling further biosynthetic studies to be carried out to elucidate the role of AmbJ.115,116
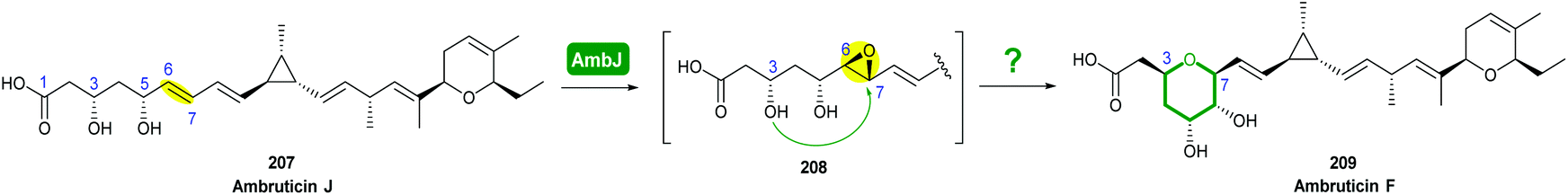 | ||
| Scheme 47 Proposed formation of the hydroxylated THP in ambruticin biosynthesis.114 | ||
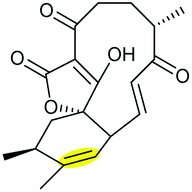
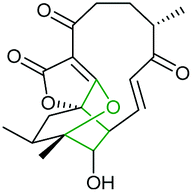
Combining in vivo gene inactivation with in vitro biochemical studies, Li et al. showed that the ether ring formation and hydroxylation in abyssomicin 2 biosynthesis is catalysed by the cytochrome P450 AbmV.118 This was proposed to result from a domino reaction sequence involving, (i) epoxidation of the 11,12-alkene in abyssomicin 6 to generate an epoxide intermediate 210 followed by, (ii) epoxide ring-opening via nucleophilic attack of the tetronate OH upon C12 to afford abyssomicin 2 (Scheme 48).
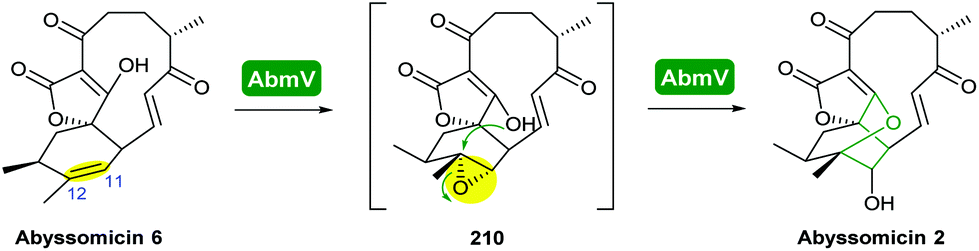 | ||
| Scheme 48 Proposed late stage pathway for abyssomicin biosynthesis.118 | ||
3.3 Formation of THFs catalysed by EHs
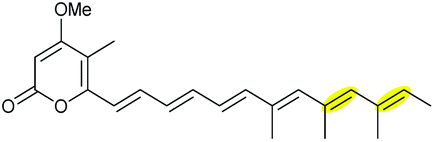
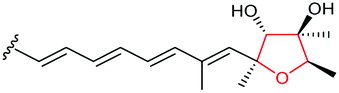
Using Aspergillus nidulans as a heterologous expression host, Wang and co-workers reconstituted the pathway and demonstrated that four genes, ctvA, ctvB, ctvC, and ctvD, are sufficient to produce citreoviridin.119 Overexpression of the two genes ctvA and ctvB produced citreomontanin. In order to form the THF ring with the correct stereochemistry, the terminal alkene of citreomontanin with an E-16,17-alkene needs to undergo isomerisation to yield the Z-16,17 isomer, a step that could be catalysed by the flavin-dependent monooxygenase CtvC (Scheme 49). Bisepoxidation by CtvC then forms (17R,16R,15S,14R)-bisepoxide 211. Addition of ctvC to the overexpression system generated a mixture of new products, with citreoviridin being the major one among other unidentified products (possibly due to spontaneous hydrolysis and degradation of the unstable bisepoxide intermediate). Finally, when ctvD was added to generate the ctvABCD overexpression strain, the only observed product was citreoviridin (Scheme 49).119
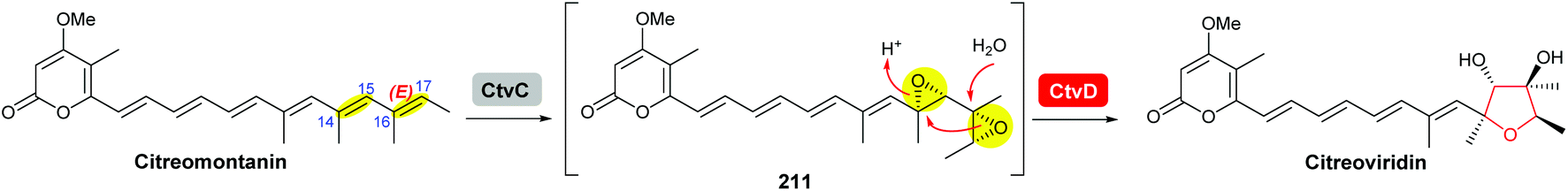 | ||
| Scheme 49 Proposed late stages biosynthesis of citreoviridin.119 | ||
Using gene knockout experiments and in vitro enzymatic turnover assays, Araki et al. demonstrated that AscE is a P450 monooxygenase that catalyses stereoselective epoxidation of the terminal olefin of ilicicolin A to produce ilicicolin A epoxide 213, which is then hydroxylated by another P450 monooxygenase AscH to yield the 16-hydroxy-ilicicolin A epoxide intermediate 214 (Scheme 50). This epoxy alcohol 214 is then cyclised by AscI to form the THF ring in ascofuranol 215, and finally oxidation of the secondary alcohol to a ketone by AscJ delivers the product ascofuranone 216. It is interesting to note that the 16-hydroxy-ilicicolin A epoxide intermediate 214 can also be converted into ascofuranol nonenzymatically under acidic conditions. AscI shares 29% and 27% identities, respectively, to the epoxide hydrolases AurD and CtvD in aurovertin and citreoviridin biosynthesis (see previous sections).120
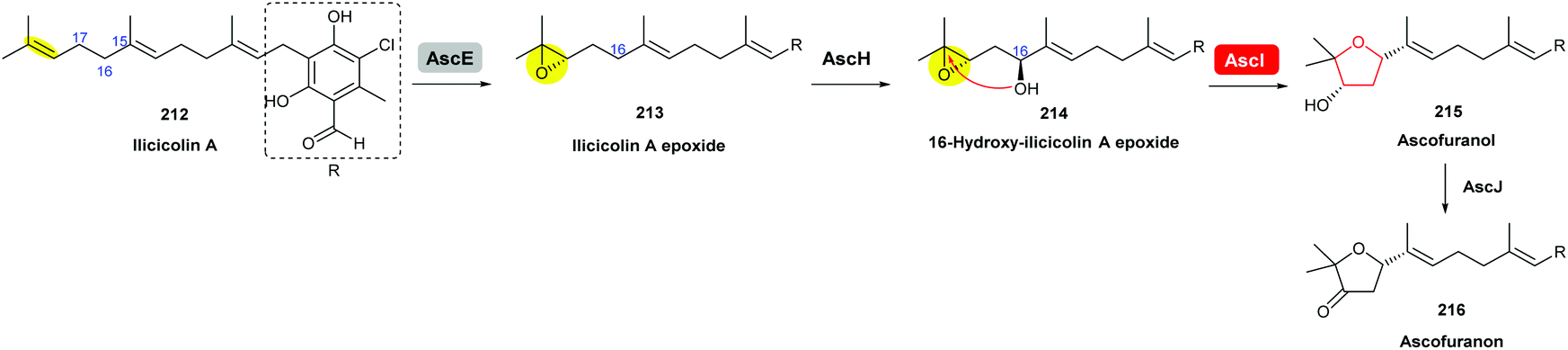 | ||
| Scheme 50 Proposed late-stage biosynthesis of ascofuranone.120 | ||
Based on isotopic feeding studies, gene inactivation experiments and bioinformatic analysis, a biosynthetic sequence has been proposed for the late stage transformation of aurachin B to aurachin A (Scheme 51).122,123 The FAD-dependent monooxygenase AuaJ is believed to catalyse epoxidation of the 2′,3′-alkene on the farnesyl chain to give 217. Intramolecular attack of the epoxide intermediate 217 by the 3-hydroxyl would then result in the epoxide-opening and cyclisation to afford the THF ring. The proposed epoxide intermediate 217 has not been detected from the cell extracts, which may be due to rapid turnover. Although this process was proposed to be catalysed by an epoxide hydrolase, the function of AuaI remains to be confirmed by further studies.
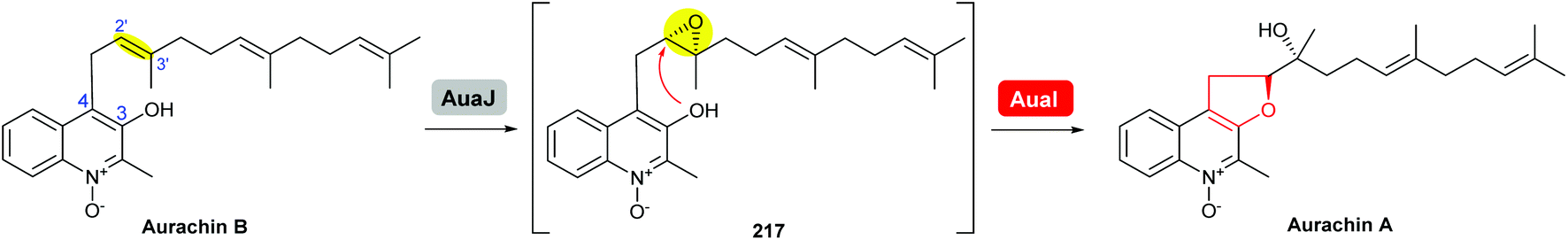 | ||
| Scheme 51 Proposed late-stage biosynthesis of aurachin A.122,123 | ||
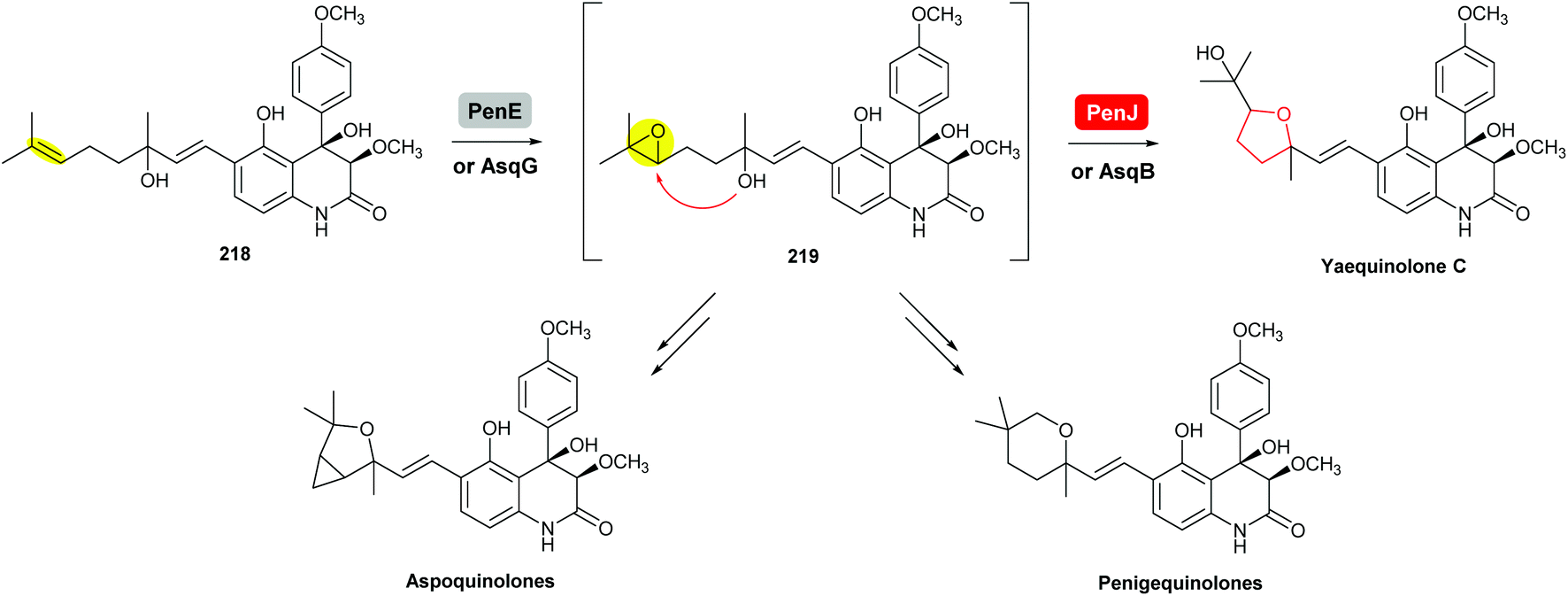 | ||
| Scheme 52 THF ring formation in yaequinolone C biosynthesis.125,126 | ||
Yaequinolone C has been proven to be an off-pathway shunt product found during studies on penigequinolone biosynthesis.125 Formation of the THF ring in yaequinolone C has been shown by Zou et al. to result from epoxidation of the terminal olefin of the precursor 218 catalysed by the flavin-dependent monooxygenase PenE, followed by the PenJ-catalysed epoxide-opening of 219 to afford yaequinolone C (Scheme 52).126 It was found that yaequinolone C could also be formed when the alkene precursor 218 was incubated with only PenE, suggesting the epoxide-opening step could happen spontaneously. When the biotransformation was performed with both PenE and PenJ, the conversion of the precursor 218 to yaequinolone C was significantly elevated (∼10-fold), indicating the role of PenJ as an epoxide hydrolase in enhancing the rate of the 5-exo cyclisation step.126
The corresponding flavin-dependent monooxygenase/epoxide hydrolase pair AsqG/AsqB in the aspoquinolone pathway have been shown to be functionally equivalent in their ability to catalyse the same transformations.125 These two pairs of enzymes were also found to be cross compatible in mix-and-match experiments,125 which is similar to the mupirocin/thiomarinol case.111
In the presence of further enzymes, epoxide intermediate 219 undergoes further modifications to yield the final metabolites penigequinolones and aspoquinolones.125
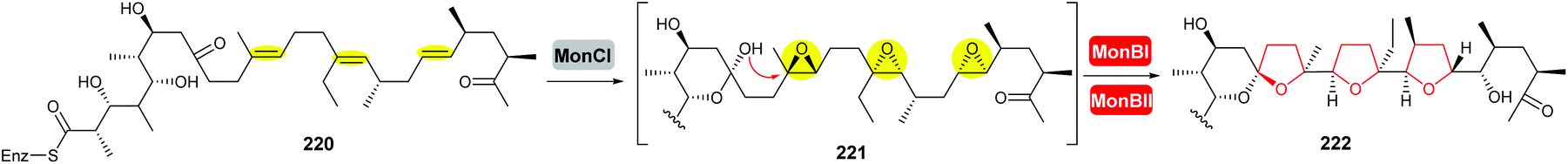 | ||
| Scheme 53 THF ring formation in monensin biosynthesis.127,128 | ||
Deletion of the monCI gene, which encodes a putative flavin-dependent epoxidase from the monensin biosynthetic gene cluster, led to accumulation of an E,E,E-triene precursor 220 in mutant strains of Streptomyces cinnamonensis, suggesting MonCI is responsible for formation of the triepoxide intermediate 221.127 Subsequent epoxide-opening of the triepoxide intermediate was originally thought to be catalysed by MonCII but was later found to be controlled by the MonBI and MonBII epoxide hydrolases.128 Using structurally simple substrate analogues, Oikawa and co-workers demonstrated remarkable synergistic effect between MonBI and MonBII in catalysing epoxide-opening cascade reactions.129 When used alone in turnover reactions, MonBI was inactive and MonBII was only weakly active. However, the epoxide opening activity was dramatically enhanced with the addition of MonBI to the MonBII reaction mixture.129
Other epoxide hydrolases that have been proposed to be involved in THF ring biosynthesis include SalBI/SalBII and from salinomycin pathway,130 NigBI/NigBII from the nigericin pathway,131 NanI from the nanchangmycin,132 MadI from the maduramicin pathway,133 and Pak24/Pak25 from the K-41A pathway.134
3.4 Summary of biosynthetic methods
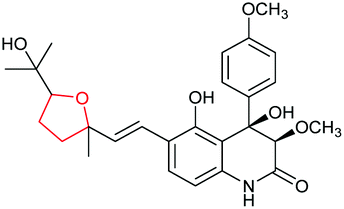
THP and THF rings are often essential structural moieties for bioactivities of many natural products and the formation of these oxygen heterocycles via epoxide formation/epoxide-opening cascade reactions has been demonstrated in a variety of bacterial and fungal natural product biosynthetic pathways. The epoxidation step is commonly catalysed by a flavin-dependent monooxygenase on an alkene substrate. However, interestingly in the mupirocin/thiomarinol systems, a Rieske non-haem oxygenase catalyses oxidation of a non-activated alkane precursor to an epoxide via an alkene. For regioselective intramolecular opening of epoxide biosynthetic intermediates, an epoxide hydrolase (EH) is often employed to yield the 6-endo cyclisation THP product. Only three protein structures (Lsd19, MupZ and XimE) have been reported for these EHs so far and X-ray crystallographic studies have suggested a similar mechanism of general acid–base catalysis. In the cases of ambruticins and abyssomicins where EHs have not been identified, further studies would be required to determine whether their THP ring formation steps are under enzymatic or substrate control. Cyclisation of 4,5-epoxy alcohols to generate THFs has been shown to either occur spontaneously or to be mediated and significantly accelerated by EHs, although no protein structure has been reported for this type of EHs yet. These epoxidation/epoxide-opening cascade reactions are summarised in Table 5.| Natural product | Strain | Substrate | Epoxidase | Epoxide hydralase | Product | Ref. |
|---|---|---|---|---|---|---|
| Lasalocid A | Streptomyces lasaliensis |
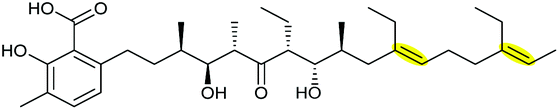
|
Lsd18 (FMO) | Lsd19 (PDB ID: 3RGA) |

|
104–107 |
| Aurovertin | Calcarisporium arbuscula |

|
AurC (FMO) | AurD |
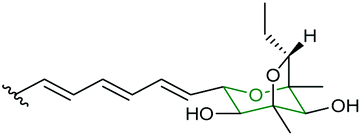
|
108 |
| Mupirocin | Pseudomonas fluorescens |
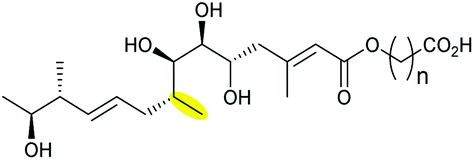
|
MupW (Rieske non-haem oxygenase) | MupZ (PDB ID: 6FXD) |
|
110 |
| Thiomarinol | Pseudoalternomonas sp. | TmlW (Rieske non-haem oxygenase) | TmlZ | 111 | ||
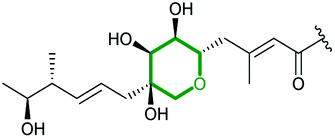
| Xiamenmycin | Streptomyces xiamenensis |
|
XimD (FMO) | XimE (PDB ID: 6ISK) |
|
112 |
| Ambruticin | Sorangium cellulosum |
|
AmbJ (FMO) | Not reported |
|
114 |
| Abyssomicin | Streptomyces koyangensis |
|
AbmV (P450) | Not reported |
|
118 |
| Citreoviridin | Penicillium sp. |
|
CtvC (FMO) | CtvD |
|
119 |
| Ascofuranone | Acremonium egyptiacum |
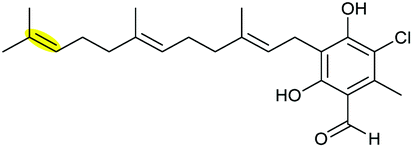
|
AscE (P450) | AscI |
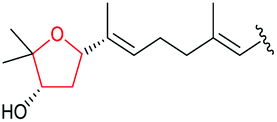
|
120 |
| Aurachin | Stigmatella aurantiaca |
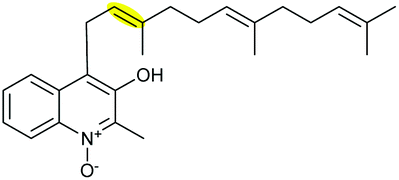
|
AuaJ (FMO) | AuaI |

|
122 |
| Penigequinolone | Penicillium & Aspergillus sp. |
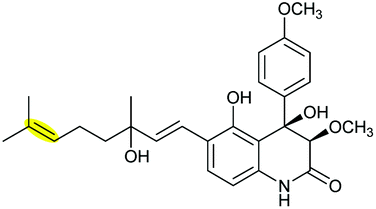
|
PenE (FMO) | PenJ |
|
125 and 126 |
| Aspoquinolone | Aspergillus Nidulans | AsqG (FMO) | AsqB | 126 | ||
| Monensin | Streptomyces cinnamonensis |
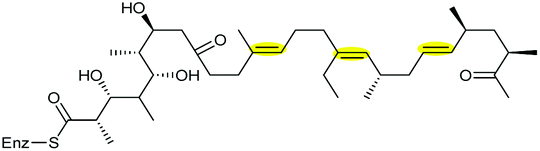
|
MonCI (FMO) | MonBI, MonBII |

|
127–129 |
4. Future outlook
4.1 Combining chemical synthesis and biosynthesis
Despite advances in chemical methodology aided by computational studies, the total synthesis of complex natural products remains a challenging endeavour. Modular approaches to the total synthesis of target compounds often provide flexibility for the assembly of libraries of natural product analogues but may rely on the use of protecting groups, precious metals, toxic reagents and solvents whilst generating substantial amounts of unwanted by-products. On the other hand, enzyme-catalysed transformations offer clean and selective methods to generate target compounds but there can be issues to overcome in terms of narrow substrate specificities and scale-up. Hybrid strategies combining chemical synthesis and biotransformations continue to provide exciting new opportunities to gain efficient access to supplies of complex natural products and their analogues that are not accessible by individual synthetic or biotechnological approaches.1354.2 Enzyme engineering
In a world with rising demands for clean, efficient, and selective catalysts, biosynthetic enzymes will continue to provide a repertoire of powerful tools to generate complex molecules and their derivatives. However, the catalytic potential of these enzymes is still far from fully explored.136In a recent study, Xu and co-workers engineered XimE from xiamenmycin biosynthesis to improve its catalytic activity for the preparation of angular pyranocoumarins by site-directed mutagenesis (Scheme 54).113 Guided by the crystallographic structure of XimE and molecular docking, the Y119A mutant of XimE was generated. The proportion of THP product 226 to THF product 225 increased from 51.7% in wild-type XimE to 79.2% in the Y119A mutant.113
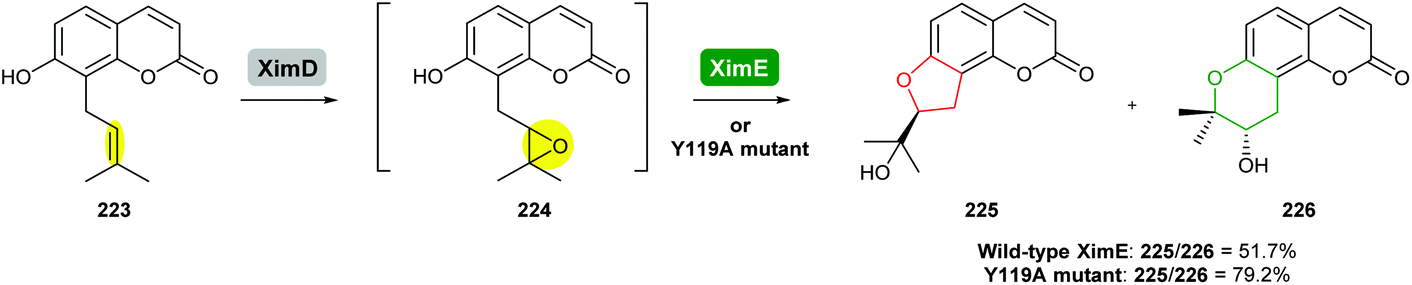 | ||
| Scheme 54 Formation of pyranocoumarins by engineered XimE.113 | ||
As further epoxide hydrolases are discovered, rational engineering of these enzymes to expand their substrate scope, enhance their catalytic activity and to possibly switch their regioselectivity in epoxide-opening process will be a fascinating area of research. For enzymes to be utilised in combination with organic synthesis, it will be necessary to optimise their stability, substrate scope and performance under a variety of conditions such as in the presence of organic solvents. We anticipate such efforts will tune and diversify the functions of these enzymes to provide biocatalysts that bring the benefits of nature's biosynthetic machinery to chemical synthesis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC Bristol Chemical Synthesis Centre for Doctoral Training (EP/L015366/1) for a studentship to JIB and the BBSRC/EPSRC for funding through the Bristol Centre for Synthetic Biology (BB/L01386X/1) to LW.References
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845 CrossRef CAS PubMed.
- M. A. Perry, S. D. Rychnovsky and N. Sizemore, Synthesis of Saturated Oxygenated Heterocycles I, in Topics in Heterocyclic Chemistry, ed. J. Cossy, Springer, Berlin, Heidelberg, 2014, vol. 35, pp. 43–95 Search PubMed.
- O. V. Salomatina, O. I. Yarovaya and V. A. Barkhash, Russ. J. Org. Chem., 2005, 41, 155 CrossRef CAS.
- I. Larrosa, P. Romea and F. Urpí, Tetrahedron, 2008, 64, 2683 CrossRef CAS.
- I. Vilotijevic and T. Jamison, Mar. Drugs, 2010, 8, 763 CrossRef CAS PubMed.
- F. Vetica, P. Chauhan, S. Dochain and D. Enders, Chem. Soc. Rev., 2017, 46, 1661 RSC.
- C. Olier, M. Kaafarani, S. Gastaldi and M. P. Bertrand, Tetrahedron, 2010, 66, 413 CrossRef CAS.
- X. Han, G. Peh and P. E. Floreancig, Eur. J. Org. Chem., 2013, 2013, 1193 CrossRef CAS.
- A. Deiters and S. F. Martin, Chem. Rev., 2004, 104, 2199 CrossRef CAS PubMed.
- P. A. Clarke and S. Santos, Eur. J. Org. Chem., 2006, 2006, 2045 CrossRef.
- N. M. Nasir, K. Ermanis and P. A. Clarke, Org. Biomol. Chem., 2014, 12, 3323 RSC.
- K. Fujiwara and A. Murai, Bull. Chem. Soc. Jpn., 2004, 77, 2129 CrossRef CAS.
- H. Fuwa, Mar. Drugs, 2016, 14, 65 CrossRef PubMed.
- F. Hemmerling and F. Hahn, Beilstein J. Org. Chem., 2016, 12, 1512 CrossRef CAS PubMed.
- S. Meng, G.-L. Tang and H.-X. Pan, ChemBioChem, 2018, 19, 2002 CrossRef CAS PubMed.
- M.-C. Tang, Y. Zou, K. Watanabe, C. T. Walsh and Y. Tang, Chem. Rev., 2017, 117, 5226 CrossRef CAS PubMed.
- J. E. Baldwin, J. Chem. Soc., Chem. Commun., 1976, 734 RSC.
- I. V. Alabugin and K. Gilmore, Chem. Commun., 2013, 49, 11246 RSC.
- K. Gilmore, R. K. Mohamed and I. V. Alabugin, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2016, 6, 487 CAS.
- J. Na, K. N. Houk, C. G. Shevlin, K. D. Janda and R. A. Lerner, J. Am. Chem. Soc., 1993, 115, 8453 CrossRef CAS.
- I. Vilotijevic and T. F. Jamison, Angew. Chem., Int. Ed., 2009, 48, 5250 CrossRef CAS PubMed.
- K. C. Nicolaou, M. E. Duggan, C.-K. Hwang and P. K. Somers, J. Chem. Soc., Chem. Commun., 1985, 111, 1359 RSC.
- K. C. Nicolaou, C. V. C. Prasad, P. K. Somers and C. K. Hwang, J. Am. Chem. Soc., 1989, 111, 5330 CrossRef CAS.
- J. Coxon, M. Hartshorn and W. Swallow, Aust. J. Chem., 1973, 26, 2521 CrossRef CAS.
- I. Kadota, H. Ueno and Y. Yamamoto, Tetrahedron Lett., 2003, 44, 8935 CrossRef CAS.
- K. Sato and M. Sasaki, Angew. Chem., Int. Ed., 2007, 46, 2518 CrossRef CAS PubMed.
- P. A. Evans and V. S. Murthy, Tetrahedron Lett., 1999, 40, 1253 CrossRef CAS.
- P. Neogi, T. Doundoulakis, A. Yazbak, S. C. Sinha, S. C. Sinha and E. Keinan, J. Am. Chem. Soc., 1998, 120, 11279 CrossRef CAS.
- M. Kanemoto, M. Murata and T. Oishi, J. Org. Chem., 2009, 74, 8810 CrossRef CAS PubMed.
- Y. Sakamoto, G. Matsuo, H. Matsukura and T. Nakata, Org. Lett., 2001, 3, 2749 CrossRef CAS PubMed.
- H. Fukui and I. Shiina, Org. Lett., 2008, 10, 3153 CrossRef CAS PubMed.
- R. K. Bressin, S. Osman, I. Pohorilets, U. Basu and K. Koide, J. Org. Chem., 2020, 85, 4637 CrossRef CAS PubMed.
- H. Matsukura, M. Morimoto, H. Koshino and T. Nakata, Tetrahedron Lett., 1997, 38, 5545 CrossRef CAS.
- T. Nakata, J. Synth. Org. Chem., Jpn., 1998, 56, 940 CrossRef CAS.
- H. Matsukura, N. Hori, G. Matsuo and T. Nakata, Tetrahedron Lett., 2000, 41, 7681 CrossRef CAS.
- M. Morimoto, H. Matsukura and T. Nakata, Tetrahedron Lett., 1996, 37, 6365 CrossRef CAS.
- B. M. Trost and A. Tenaglia, Tetrahedron Lett., 1988, 29, 2927 CrossRef CAS.
- T. Suzuki, O. Sato and M. Hirama, Tetrahedron Lett., 1990, 31, 4747 CrossRef CAS.
- T. Oishi, K. Maeda and M. Hirama, Chem. Commun., 1997, 1289 RSC.
- T. Suzuki, O. Sato, M. Hirama, Y. Yamamoto, M. Murata, T. Yasumoto and N. Harada, Tetrahedron Lett., 1991, 32, 4505 CrossRef CAS.
- J. D. Ha, E. Y. Shin, S. K. Kang, J. H. Ahn and J.-K. Choi, Tetrahedron Lett., 2004, 45, 4193 CrossRef CAS.
- M. Inoue, F. Saito, M. Iwatsu, Y. Ishihara and M. Hirama, Tetrahedron Lett., 2007, 48, 2171 CrossRef CAS.
- K. W. Armbrust, M. G. Beaver and T. F. Jamison, J. Am. Chem. Soc., 2015, 137, 6941 CrossRef CAS PubMed.
- C. Mukai, Y. Ikeda, Y. Sugimoto and M. Hanaoka, Tetrahedron Lett., 1994, 35, 2179 CrossRef CAS.
- B. J. Teobald, Tetrahedron, 2002, 58, 4133 CrossRef CAS.
- C. Mukai, N. Miyakoshi and M. Hanaoka, J. Org. Chem., 2001, 66, 5875 CrossRef CAS PubMed.
- C. Mukai, Y. Sugimoto, Y. Ikeda and M. Hanaoka, Tetrahedron Lett., 1994, 35, 2183 CrossRef CAS.
- I. C. González and C. J. Forsyth, J. Am. Chem. Soc., 2000, 122, 9099 CrossRef.
- C. Mukai, S. Yamaguchi, I. J. Kim and M. Hanaoka, Chem. Pharm. Bull., 2001, 49, 61 CrossRef PubMed.
- F. R. P. Crisóstomo, T. Martín and V. S. Martín, Org. Lett., 2004, 6, 565 CrossRef PubMed.
- P. F. Hudrlik, J. P. Arcoleo, R. H. Schwartz, R. N. Misra and R. J. Rona, Tetrahedron Lett., 1977, 18, 591 CrossRef.
- W. E. Fristad, T. R. Bailey, L. A. Paquette, R. Gleiter and M. C. Boehm, J. Am. Chem. Soc., 1979, 101, 4420 CrossRef CAS.
- T. Ikeno, J. Harada and S. Tomoda, Chem. Lett., 1999, 28, 409 CrossRef.
- G. Adiwidjaja, H. Flörke, A. Kirschning and E. Schaumann, Tetrahedron Lett., 1995, 36, 8771 CrossRef CAS.
- T. P. Heffron and T. F. Jamison, Org. Lett., 2003, 5, 2339 CrossRef CAS PubMed.
- G. L. Simpson, T. P. Heffron, E. Merino and T. F. Jamison, J. Am. Chem. Soc., 2006, 128, 1056 CrossRef CAS PubMed.
- T. P. Heffron, G. L. Simpson, E. Merino and T. F. Jamison, J. Org. Chem., 2010, 75, 2681 CrossRef CAS PubMed.
- J. Lenhof, M. Hutter, V. Huch and J. Jauch, Eur. J. Org. Chem., 2020, 2020, 5833 CrossRef CAS.
- Y. Mori, K. Yaegashi and H. Furukawa, J. Am. Chem. Soc., 1996, 118, 8158 CrossRef CAS.
- Y. Mori, Chem. Pharm. Bull., 2019, 67, 1 CrossRef CAS PubMed.
- Y. Mori, H. Furuta, T. Takase, S. Mitsuoka and H. Furukawa, Tetrahedron Lett., 1999, 40, 8019 CrossRef CAS.
- Y. Mori, K. Yaegashi and H. Furukawa, Tetrahedron Lett., 1999, 40, 7239 CrossRef CAS.
- Y. Mori, K. Yaegashi and H. Furukawa, J. Am. Chem. Soc., 1997, 119, 4557 CrossRef CAS.
- Y. Mori, K. Nogami, H. Hayashi and R. Noyori, J. Org. Chem., 2003, 68, 9050 CrossRef CAS PubMed.
- M. Inoue, S. Yamashita, A. Tatami, K. Miyazaki and M. Hirama, J. Org. Chem., 2004, 69, 2797 CrossRef CAS PubMed.
- H. Furuta, Y. Hasegawa and Y. Mori, Org. Lett., 2009, 11, 4382 CrossRef CAS PubMed.
- T. Sakai, A. Ishihara and Y. Mori, J. Org. Chem., 2017, 82, 3976 CrossRef CAS PubMed.
- T. Sakai, S. Matsushita, S. Arakawa, K. Mori, M. Tanimoto, A. Tokumasu, T. Yoshida and Y. Mori, J. Am. Chem. Soc., 2015, 137, 14513 CrossRef CAS PubMed.
- F. E. McDonald, X. Wang, B. Do and K. I. Hardcastle, Org. Lett., 2000, 2, 2917 CrossRef CAS PubMed.
- C. J. Morten and T. F. Jamison, J. Am. Chem. Soc., 2009, 131, 6678 CrossRef CAS PubMed.
- F. Bravo, F. E. McDonald, W. A. Neiwert, B. Do and K. I. Hardcastle, Org. Lett., 2003, 5, 2123 CrossRef CAS PubMed.
- M. A. Tarselli, J. L. Zuccarello, S. J. Lee and M. R. Gagné, Org. Lett., 2009, 11, 3490 CrossRef CAS PubMed.
- A. Zakarian, A. Batch and R. A. Holton, J. Am. Chem. Soc., 2003, 125, 7822 CrossRef CAS PubMed.
- F. Li, S. Ren, P. Li, P. Yang and J. Qu, Angew. Chem., Int. Ed., 2020, 59, 18473 CrossRef CAS PubMed.
- Y. Morimoto, Y. Nishikawa, C. Ueba and T. Tanaka, Angew. Chem., Int. Ed., 2006, 45, 810 CrossRef CAS PubMed.
- Y. Morimoto, Y. Nishikawa and M. Takaishi, J. Am. Chem. Soc., 2005, 127, 5806 CrossRef CAS PubMed.
- Y. Morimoto, H. Yata and Y. Nishikawa, Angew. Chem., Int. Ed., 2007, 46, 6481 CrossRef CAS PubMed.
- S. Wang, A. J. Arguelles, J. Tay, M. Hotta, P. M. Zimmerman and P. Nagorny, Chem. – Eur. J., 2020, 26, 4583 CrossRef CAS PubMed.
- M. H. Wu, K. B. Hansen and E. N. Jacobsen, Angew. Chem., Int. Ed., 1999, 38, 2012 CrossRef CAS PubMed.
- K. B. Hansen, J. L. Leighton and E. N. Jacobsen, J. Am. Chem. Soc., 1996, 118, 10924 CrossRef CAS.
- A. Blanc and F. D. Toste, Angew. Chem., Int. Ed., 2006, 45, 2096 CrossRef CAS PubMed.
- I. Vilotijevic and T. F. Jamison, Science, 2007, 317, 1189 CrossRef CAS PubMed.
- C. J. Morten, J. A. Byers, A. R. Van Dyke, I. Vilotijevic and T. F. Jamison, Chem. Soc. Rev., 2009, 38, 3175 RSC.
- Z. Wang, Y.-T. Cui, Z.-B. Xu and J. Qu, J. Org. Chem., 2008, 73, 2270 CrossRef CAS PubMed.
- J. A. Byers and T. F. Jamison, J. Am. Chem. Soc., 2009, 131, 6383 CrossRef CAS PubMed.
- J. A. Byers and T. F. Jamison, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16724 CrossRef CAS PubMed.
- A. R. Van Dyke and T. F. Jamison, Angew. Chem., Int. Ed., 2009, 48, 4430 CrossRef CAS PubMed.
- L. C. Czabaniuk and T. F. Jamison, Org. Lett., 2015, 17, 774 CrossRef CAS PubMed.
- K. Janda, C. Shevlin and R. Lerner, Science, 1993, 259, 490 CrossRef CAS PubMed.
- J. Na, K. N. Houk, C. G. Shevlin, K. D. Janda and R. A. Lerner, J. Am. Chem. Soc., 1993, 115, 8453 CrossRef CAS.
- K. Gruber, B. Zhou, K. N. Houk, R. A. Lerner, C. G. Shevlin and I. A. Wilson, Biochemistry, 1999, 38, 7062 CrossRef CAS PubMed.
- G. Chiosis, Tetrahedron Lett., 2000, 41, 801 CrossRef CAS.
- X. Zhang, X. Hao, L. Liu, A.-T. Pham, J. López-Andarias, A. Frontera, N. Sakai and S. Matile, J. Am. Chem. Soc., 2018, 140, 17867 CrossRef CAS PubMed.
- M. Paraja and S. Matile, Angew. Chem., Int. Ed., 2020, 59, 6273 CrossRef CAS PubMed.
- T. Nakata, G. Schmid, B. Vranesic, M. Okigawa, T. Smith-Palmer and Y. Kishi, J. Am. Chem. Soc., 1978, 100, 2933 CrossRef CAS.
- T. Nakata, S. Nomura and H. Matsukura, Tetrahedron Lett., 1996, 37, 213 CrossRef CAS.
- K. Nagasawa, N. Hori, R. Shiba and T. Nakata, Heterocycles, 1997, 44, 105 CrossRef CAS.
- T. Nakata, S. Nomura, H. Matsukura and M. Morimoto, Tetrahedron Lett., 1996, 37, 217 CrossRef CAS.
- N. Hori, K. Nagasawa, T. Shimizu and T. Nakata, Tetrahedron Lett., 1999, 40, 2145 CrossRef CAS.
- F. E. McDonald and M. A. Smart, Heterocycles, 2012, 86, 535 CrossRef.
- M. Caron and K. B. Sharpless, J. Org. Chem., 1985, 50, 1557 CrossRef CAS.
- K. Fujiwara, T. Tokiwano and A. Murai, Tetrahedron Lett., 1995, 36, 8063 CrossRef CAS.
- T. Tokiwano, K. Fujiwara and A. Murai, Synlett, 2000, 335 CAS.
- Y. Shichijo, A. Migita, H. Oguri, M. Watanabe, T. Tokiwano, K. Watanabe and H. Oikawa, J. Am. Chem. Soc., 2008, 130, 12230 CrossRef CAS PubMed.
- A. Minami, M. Shimaya, G. Suzuki, A. Migita, S. S. Shinde, K. Sato, K. Watanabe, T. Tamura, H. Oguri and H. Oikawa, J. Am. Chem. Soc., 2012, 134, 7246 CrossRef CAS PubMed.
- L. Smith, H. Hong, J. B. Spencer and P. F. Leadlay, ChemBioChem, 2008, 9, 2967 CrossRef CAS PubMed.
- K. Hotta, X. Chen, R. S. Paton, A. Minami, H. Li, K. Swaminathan, I. I. Mathews, K. Watanabe, H. Oikawa, K. N. Houk and C.-Y. Kim, Nature, 2012, 483, 355 CrossRef CAS PubMed.
- X.-M. Mao, Z.-J. Zhan, M. N. Grayson, M.-C. Tang, W. Xu, Y.-Q. Li, W.-B. Yin, H.-C. Lin, Y.-H. Chooi, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2015, 137, 11904 CrossRef CAS PubMed.
- C. M. Thomas, J. Hothersall, C. L. Willis and T. J. Simpson, Nat. Rev. Microbiol., 2010, 8, 281 CrossRef CAS PubMed.
- L. Wang, A. Parnell, C. Williams, N. A. Bakar, M. R. Challand, M. W. van der Kamp, T. J. Simpson, P. R. Race, M. P. Crump and C. L. Willis, Nat. Catal., 2018, 1, 968 CrossRef CAS.
- L. Wang, Z. Song, P. R. Race, J. Spencer, T. J. Simpson, M. P. Crump and C. L. Willis, Chem. Sci., 2020, 11, 5221 RSC.
- B. B. He, T. Zhou, X. L. Bu, J. Y. Weng, J. Xu, S. Lin, J. T. Zheng, Y. L. Zhao and M. J. Xu, ACS Catal., 2019, 5391 CrossRef CAS.
- X. L. Bu, B. B. He, J. Y. Weng, C. C. Jiang, Y. L. Zhao, S. M. Li, J. Xu and M. J. Xu, ACS Synth. Biol., 2020, 9, 2282 CrossRef CAS PubMed.
- B. Julien, Z.-Q. Tian, R. Reid and C. D. Reeves, Chem. Biol., 2006, 13, 1277 CrossRef CAS PubMed.
- J. I. Bowen, L. Wang, M. P. Crump and C. L. Willis, Org. Biomol. Chem., 2021, 19, 6210 RSC.
- K. Trentadue, C. Chang, A. Nalin and R. E. Taylor, Chem. – Eur. J., 2021, 27, 11126 CrossRef CAS PubMed.
- C. Sadaka, E. Ellsworth, P. Hansen, R. Ewin, P. Damborg and J. Watts, Molecules, 2018, 23, 1371 CrossRef PubMed.
- Q. Li, W. Ding, Z. Yao, J. Tu, L. Wang, H. Huang, S. Li and J. Ju, Org. Lett., 2018, 20, 4854 CrossRef CAS PubMed.
- T. S. Lin, Y. M. Chiang and C. C. C. Wang, Org. Lett., 2016, 18, 1366 CrossRef CAS PubMed.
- Y. Araki, T. Awakawa, M. Matsuzaki, R. Cho, Y. Matsuda, S. Hoshino, Y. Shinohara, M. Yamamoto, Y. Kido, D. K. Inaoka, K. Nagamune, K. Ito, I. Abe and K. Kita, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8269 CrossRef CAS PubMed.
- Y. Katsuyama, K. Harmrolfs, D. Pistorius, Y. Li and R. Müller, Angew. Chem., Int. Ed., 2012, 51, 9437 CrossRef CAS PubMed.
- D. Pistorius, Y. Li, A. Sandmann and R. Müller, Mol. Biosyst., 2011, 7, 3308 RSC.
- G. Höfle and B. Kunze, J. Nat. Prod., 2008, 71, 1843 CrossRef PubMed.
- Y. Matsuda and I. Abe, Nat. Prod. Rep., 2016, 33, 26 RSC.
- Y. Zou, M. Garcia-Borràs, M. C. Tang, Y. Hirayama, D. H. Li, L. Li, K. Watanabe, K. N. Houk and Y. Tang, Nat. Chem. Biol., 2017, 13, 325 CrossRef CAS PubMed.
- Y. Zou, Z. Zhan, D. Li, M. Tang, R. A. Cacho, K. Watanabe and Y. Tang, J. Am. Chem. Soc., 2015, 137, 4980 CrossRef CAS PubMed.
- A. Bhatt, C. B. W. Stark, B. M. Harvey, A. R. Gallimore, Y. A. Demydchuk, J. B. Spencer, J. Staunton and P. F. Leadlay, Angew. Chem., Int. Ed., 2005, 44, 7075 CrossRef CAS PubMed.
- A. R. Gallimore, C. B. W. Stark, A. Bhatt, B. M. Harvey, Y. Demydchuk, V. Bolanos-Garcia, D. J. Fowler, J. Staunton, P. F. Leadlay and J. B. Spencer, Chem. Biol., 2006, 13, 453 CrossRef CAS PubMed.
- K. Sato, A. Minami, T. Ose, H. Oguri and H. Oikawa, Tetrahedron Lett., 2011, 52, 5277 CrossRef CAS.
- M. E. Yurkovich, P. A. Tyrakis, H. Hong, Y. Sun, M. Samborskyy, K. Kamiya and P. F. Leadlay, ChemBioChem, 2012, 13, 66 CrossRef CAS PubMed.
- B. M. Harvey, T. Mironenko, Y. Sun, H. Hong, Z. Deng, P. F. Leadlay, K. J. Weissman and S. F. Haydock, Chem. Biol., 2007, 14, 703 CrossRef CAS PubMed.
- Y. Sun, X. Zhou, H. Dong, G. Tu, M. Wang, B. Wang and Z. Deng, Chem. Biol., 2003, 10, 431 CrossRef CAS PubMed.
- R. Liu, F. Fang, Z. An, R. Huang, Y. Wang, X. Sun, S. Fu, A. Fu, Z. Deng and T. Liu, J. Ind. Microbiol. Biotechnol., 2020, 47, 275 CrossRef CAS PubMed.
- J. Chen, J. Chen, C. Gui, C. Gui, Q. Wei, J. Liu, L. Ye, X. Tian, Y. C. Gu, Q. Li, J. Ju and J. Ju, Org. Lett., 2020, 22, 4627 CrossRef CAS PubMed.
- F. Hahn and F. M. Guth, Nat. Prod. Rep., 2020, 37, 1300 RSC.
- K. Chen and F. H. Arnold, Nat. Catal., 2020, 3, 203 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |