
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pd–In intermetallic nanoparticles with high catalytic selectivity for liquid-phase semi-hydrogenation of diphenylacetylene†‡
Si
Chen
a,
Xiaohui
Huang
bc,
Dieter
Schild
 d,
Di
Wang
be,
Christian
Kübel
bce and
Silke
Behrens
*a
d,
Di
Wang
be,
Christian
Kübel
bce and
Silke
Behrens
*a
aInstitute of Catalysis Research and Technology (IKFT), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: Silke.Behrens@kit.eu
bInstitute of Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
cJoint Research Laboratory Nanomaterials, Technische Universität Darmstadt, Jovanka-Bontschits-Straße 2, 64287, Darmstadt, Germany
dInstitute for Nuclear Waste Disposal (INE), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
eKarlsruhe Nano Micro Facility, Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344, Eggenstein-Leopoldshafen, Germany
First published on 23rd November 2022
Abstract
Intermetallic nanoparticles (NPs) are highly interesting materials in catalysis due to their geometrically ordered structures and altered electronic properties, but the synthesis of defined intermetallic NPs remains a challenge. Here, we report a novel and facile approach for the synthesis of intermetallic Pd–In NPs in ionic liquids (ILs) at moderate temperatures. Depending on the molar ratio of the metal precursors and the reaction temperature, single-phase Pd3In, PdIn and Pd3In7 NPs were obtained, which was confirmed, e.g. by powder X-ray diffraction, electron microscopy, and optical emission spectroscopy with inductively coupled plasma. The Pd–In NPs stabilized in ILs were used as catalysts in the liquid-phase semi-hydrogenation of diphenylacetylene (DPA). Highly ordered PdIn NPs with a CsCl type structure revealed both high activity and selectivity to cis-stilbene even at full DPA conversion. Intermetallic compounds such as PdIn can be used to isolate contiguous Pd atoms with another base metal into single Pd sites, thereby increasing the catalytic selectivity of Pd while stabilizing the individual sites in the intermetallic structures. This work may provide new pathways for the synthesis of single-phase intermetallic NPs and future insights into a more rational design of bimetallic catalysts with specific catalytic properties.
Introduction
The semi-hydrogenation of alkynes to their corresponding alkenes is one of the most important chemical reactions in diverse industrial and synthetic applications.1,2 The purification of ethylene from steam cracking, for example, is a large-scale, industrial process in which impurities in acetylene are removed by selective hydrogenation.3 Optimal design of catalyst materials and operating conditions are decisive to the selective hydrogenations. Currently, Pd-based heterogeneous catalysts, such as Lindlar4,5 and Pd–Ag alloys,6 are widely used in this reaction because of their physico–chemical properties and high activity, but the limited selectivity to the desired alkenes remains a challenge, especially at high conversion.7,8 Hence, the development of effective strategies for improving the selectivity of Pd catalysts without compromising the activity has attracted broad interest.It is well-known that bimetallic nanoparticles (NPs), which can be present as alloys,9,10 core–shell,8 or (segregated) hetero structures, generally exhibit unique physico–chemical properties and enhanced catalytic performance compared to their monometallic counterparts due to synergistic effects between the two metals.11–13 Many efforts have been devoted to synthesize and optimize Pd catalysts by bimetallic Pd–M alloys (e.g. with M = Cu,14 Ag,15–17 Au,18,19 Zn,20 Ga,21 In,22–24 Sn,25,26 or Bi)27 or even ternary Pd-based phases (such as Pd–Bi–Se28 or Pd–Ga–Sn),29,30 improving catalytic selectivity for the selective hydrogenation of alkynes. In addition, non-precious, binary alloys (Ni/Fe),31 intermetallic compounds (Al13Fe4),32 transition metal-free Zintl phases (BaGa2), or non-precious, ternary phases (Cu–Ni–Fe)33 have been reported as interesting catalytic materials for alkyne semi-hydrogenation in the gas or liquid phase. In general, alloys with random-type structures are prone to changes in response to different reaction conditions, including reconstructions, segregation and oxidative/reductive evolution, thus resulting in reduced catalytic activity and stability.34 In this direction, nanoparticles of intermetallic compounds have attracted extensive attention, because they combine specific crystal structures with long-range atomic ordering and altered electronic structures caused by chemical bonding.35–39 For example, Armbrüster et al. reported that unsupported intermetallic PdGa and Pd2Ga NPs show high selectivity (77% and 60%) and long-term stability for the semi-hydrogenation of acetylene.40 As an electronic analog of Ga in group 13 of the periodic table, alloying In with Pd-based catalysts was reported as a promising alternative to the Pd–Ga systems. Similar to Pd–Ga systems, Pd–In catalysts have isolated Pd sites on the catalyst surface, but have higher stability and oxidation resistance (oxidative potentials ∼−0.3 V (In) and ∼−0.5 V (Ga)). Even at the molecular level, heterobimetallic Pd–In complexes represent interesting homogeneous catalysts in the hydrogenation of alkynes.41 For example, Pd(μ-O2C(CH3))4In(O2C(CH3)) was demonstrated to homogeneously catalyze the liquid-phase hydrogenation of phenylacetylene and styrene in acetic acid, dimethylformamide, and ethyl acetate. Pd metal was not formed until complete hydrogenation to the alkene, with phenylacetylene and styrene acting as stabilizing π ligands for the active form of the complex. The defined 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry of the Pd–In complex was further exploited in incipient wetness impregnation for generation of PdIn particles on various supports.22,42 The impregnated complexes were treated in several steps by calcination at high temperatures (550 °C), followed by H2 reduction upon 400 °C to obtain particles of the desired intermetallic PdIn phase with high selectivity in gas- and liquid-phase semi-hydrogenation of alkynes.22,43,44 The stoichiometry was relevant with respect to alkyne semi-hydrogenation. For gas-phase acetylene semi-hydrogenation, Feng et al. demonstrated that intermetallic PdIn NPs with isolated, single-atom Pd sites exhibited higher selectivity than Pd3In NPs, which was further supported by density functional theory (DFT) calculations.45 To the best of our knowledge, however, so far there have been no studies on defined particles of other Pd–In phase compositions in the liquid-phase semi-hydrogenation of diphenylacetylene.
:1 stoichiometry of the Pd–In complex was further exploited in incipient wetness impregnation for generation of PdIn particles on various supports.22,42 The impregnated complexes were treated in several steps by calcination at high temperatures (550 °C), followed by H2 reduction upon 400 °C to obtain particles of the desired intermetallic PdIn phase with high selectivity in gas- and liquid-phase semi-hydrogenation of alkynes.22,43,44 The stoichiometry was relevant with respect to alkyne semi-hydrogenation. For gas-phase acetylene semi-hydrogenation, Feng et al. demonstrated that intermetallic PdIn NPs with isolated, single-atom Pd sites exhibited higher selectivity than Pd3In NPs, which was further supported by density functional theory (DFT) calculations.45 To the best of our knowledge, however, so far there have been no studies on defined particles of other Pd–In phase compositions in the liquid-phase semi-hydrogenation of diphenylacetylene.
In general, the Pd–In system can form several intermetallic compounds with different Pd/In stoichiometry, including Pd3In, Pd2In, PdIn, Pd3In7, Pd2In3, and Pd5In3, and it is challenging to achieve single-phase particles.46 In addition, thermal annealing (typically over 500 °C) of dry NP powders is typically required to obtain compounds with the ordered intermetallic structure, while such high temperatures may easily cause NP aggregation/sintering with an increase of particle size.47,48 In this context, we make use of the unique physico–chemical properties of room temperature ionic liquids (ILs) as tunable and neoteric reaction medium (with low-volatility, thermal stability, high polarity, high conductivity and solubility) for the synthesis of intermetallic Pd–In NPs at moderate temperatures.49–51 A series of Pd–In bimetallic NPs with different structures and well-defined phase compositions was obtained, which were characterized by powder X-ray diffraction (XRD), scanning and transmission electron microscopy with energy-dispersive X-ray analysis (SEM-EDX, TEM, high-angle, annular dark-field scanning transmission electron microscopy (HAADF-STEM)), optical emission spectroscopy with inductively coupled plasma (ICP-OES), and X-ray photoelectron spectroscopy (XPS). The single-phase PdIn, Pd3In and Pd3In7 NP sols in the ionic liquids were employed as catalysts in the liquid-phase semi-hydrogenation of diphenyl acetylene (DPA) to investigate the influence of catalyst composition and structure on the catalytic performance. In particular, intermetallic PdIn NPs with a molar Pd/In ratio of 1:1 revealed a high activity and excellent selectivity towards stilbene.
Experimental
Materials and chemicals
Disodium tetrachloridopalladate(II) (Na2PdCl4, 99.99% trace metal basis), indium(III) chloride (InCl3, 99%), potassium triethylhydridoborate (K[BEt3H]) (1 M in THF, Superhydride®) and methyltrioctylammonium bromide ([OMA]Br), cis-stilbene (96%), trans-stilbene (96%), diphenylacetylene (98%) were purchased from Sigma Aldrich, while 1,2-diphenylethane (>99%) was received from VWR Chemicals. Methyltrioctylammonium bis(trifluoromethylsulfonyl)imide ([OMA][NTf2]) was obtained from IoLiTec (H2O content <100 ppm; halide content <100 ppm). The ILs were dried in vacuum (10−3 mbar, 6 h at 70 °C; then 16 h at room temperature (RT)) prior to use. Anhydrous tetrahydrofuran (THF) and acetonitrile were purchased from Sigma Aldrich. All chemicals were used as received without further purification. All procedures of Pd–In NPs synthesis and catalytic testing were carried out in anhydrous solvents using Schlenk techniques or the anoxic glove box under argon atmosphere (H2O <0.1 ppm, O2 <0.1 ppm).Synthesis of PdIn and Pd3In7 NPs in [OMA][NTf2]
For the synthesis of intermetallic PdIn NPs, Na2PdCl4 (36.7 mg, 0.125 mmol) and InCl3 (27.6 mg, 0.125 mmol) (molar Pd/In precursor ratio of 1:1) were dissolved in 4 ml [OMA][NTf2]. After the solution was heated to 60 °C, [OMA][BEt3H] (3 mL, 1.5 M in THF) was rapidly injected into the solution while stirring, resulting in a black suspension instantly. Then, the reaction was kept at 95 °C for 1.5 h before it was cooled down to RT. THF and BEt3 were removed in vacuum (30 min, 10−3 mbar) to yield intermetallic PdIn NPs in [OMA][NTf2]. Pd3In7 NPs were synthesized in the IL by a similar procedure employing a molar Pd/In precursor ratio of 3:7 (Na2PdCl4 (22.5 mg, 0.075 mmol) and InCl3 (38.7 mg, 0.175 mmol)).
Synthesis of Pd3In NPs in [OMA][NTf2]
For the synthesis of Pd3In NPs, Na2PdCl4 (55.2 mg, 0.1875 mmol) and InCl3 (13.8 mg, 0.0625 mmol) were dissolved in 4 ml [OMA][NTf2] with a molar Pd/In ratio of 3:1 ratio. After the solution was heated to 60 °C, [OMA][BEt3H] (3 mL, 1.5 M in THF) was rapidly injected into the solution under stirring, resulting in a black suspension instantly. Then, the reaction was kept at 200 °C for 3 h before it was cooled down to RT. THF and BEt3 were removed in vacuum (30 min, 10−3 mbar) to yield Pd3In NPs dispersed in [OMA][NTf2].
Synthesis of Pd NPs in [OMA][NTf2]
Na2PdCl4 (73.5 mg, 0.25 mmol) was dissolved in 4 ml [OMA][NTf2] to get a dark brown solution. [OMA][BEt3H] (3 mL, 1.5 M in THF) was rapidly injected at room temperature while stirring, resulting in a black suspension instantly. Then, the reaction mixture was further heated to 60 °C and stirred for 3 h. After cooling to RT, THF and BEt3 were removed in vacuum (30 min, 10−3 mbar).Characterization
XRD patterns of the NP powders were obtained on a PANalytical X'Pert Pro X-ray diffractometer employing a Bragg–Brentano geometry with Cu-Kα radiation (λ = 0.154060 nm) and a Ni filter. Bragg angles ranging from 5° to 120° (2θ) were recorded over a period of 16 h at RT. The reflections were analyzed by X'Pert Highscore Plus (PANalytical) and compared to reference data reported in the International Centre for Diffraction Data (ICDD) database. The crystallite sizes of NPs were determined by the Scherrer equation using LaB6 as a standard to account for instrumental broadening. After synthesis, Pd and Pd–In NPs were precipitated from the IL by adding acetonitrile and centrifuging (5600 rpm, 5 min, 3 times) and dried under vacuum for 30 min. Then the dry powder was deposited onto the XRD sample holder for XRD measurements. Quantitative elemental analysis of the composition of Pd–In NPs was carried out with ICP-OES (Agilent 725 ICP-OES spectrometer). For ICP-OES analysis, the NPs were precipitated and dissolved in aqua regia. EDX analysis was performed on a Gemini SEM 500 from Zeiss GmbH equipped with a Schottky field emission cathode. Particle size, morphology, and composition were characterized by TEM on a Tecnai F20 ST TEM (200 kV) (Thermofisher Scientific) equipped with an EDAX EDS X-ray spectrometer. High-angle annular dark-field (HAADF) scanning transmission electron microscopy (STEM) imaging and energy dispersive X-ray spectroscopy (EDS) analysis were performed on a Themis 300 transmission electron microscope (Thermofisher Scientific), equipped with a probe aberration corrector and a Super-X EDX detector operated at 300 kV. More than 100 particles were measured in the TEM to calculate mean particle sizes and to establish size histograms. XPS measurements were carried out on an XPS system PHI 5000 VersaProbe Probe II (ULVAC-PHI Inc.) with a scanning microprobe X-ray source [monochromatic Al-Kα (1486.7 eV) X-rays]. Samples were prepared on pieces of silicon wafer inside the anoxic glove box and moved into the XPS without air contact by use of a transfer vessel. Binding energies (BE) of elemental lines were charge referenced by setting In 3d5/2 assigned to In(0) to 443.9 eV. BE of C 1s of all spectra is at 285.3 eV.Catalytic performance test
The liquid-phase hydrogenation of DPA was carried out in an autoclave reactor (200 mL, Premex) which was equipped with a Teflon liner, a mechanical blowing stirrer (Teflon) and baffles (stainless steel), a thermocouple and a heating bath. Typically, 5.6 mmol DPA in 30 mL THF was loaded in the autoclave reactor in the glove box. Then 1 mL Pd–In NPs@IL catalyst (nmetal 56 μmol) was added to the DPA solution. After the reaction temperature of 80 °C was reached and stable for at least 20 min, hydrogen was introduced into the reactor to start the reaction at a stirring speed of 1200 rpm. Throughout the catalytic experiment, the hydrogen pressure in the reactor was maintained constantly at 10 bar. Meanwhile, the consumption of hydrogen was recorded automatically as pressure drop in the gas burette. The reactants and products were periodically sampled and then analyzed by gas chromatography (GC-FID, Agilent) equipped with a J&W SCIENTIFIC DB5 column (Agilent Technology, 30 m × 0.25 mm, 0.25 μm film thickness). Typically, the catalytic tests were performed twice over each Pd–In catalyst and the relative error range of CST selectivity and yield values was approximately ±8% for two tests under the same reaction condition.Conversion (X), selectivity (S) and yield (Y) were calculated according to the following equations:
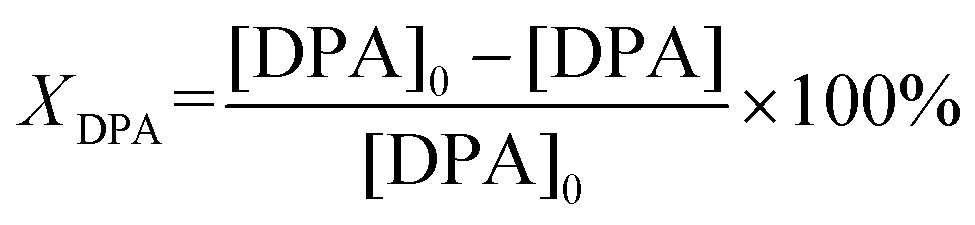
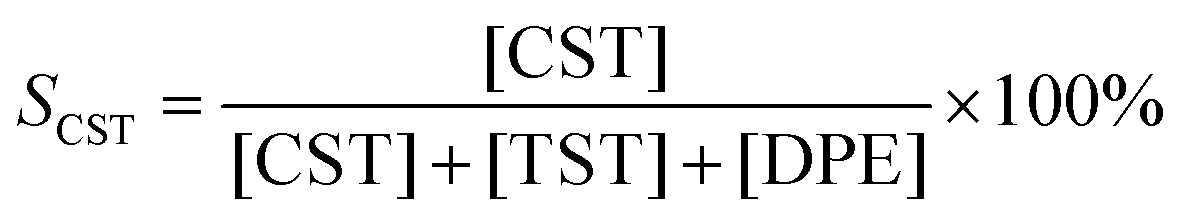
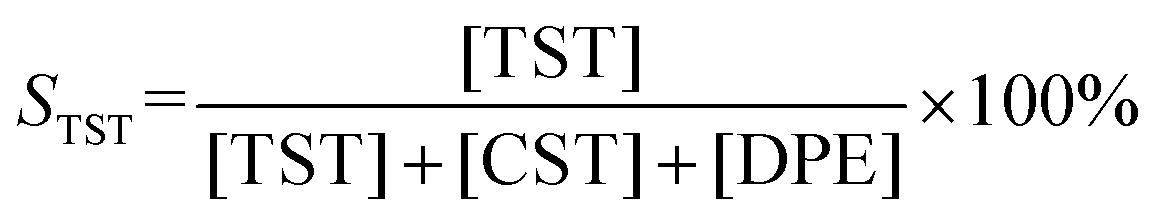
| YCST = SCSTXDPA10−2, YTST = STSTXDPA10−2 |
Results and discussion
Recently, we have reported on the one-step synthesis of Pd2Sn, PdSn, Pt3Sn and PtSn IMNPs in [OMA][NTf2] at 200 °C.25,52 By weakly coordinating anions and cations, ILs are capable to control both particle nucleation and growth processes during NP synthesis. In addition, semi-organized clusters of IL anions and cations can stabilize the NPs by forming protective layers. In this study, bimetallic Pd–In NPs with different compositions and structures were prepared via the co-reduction of the metal precursors in [OMA][NTf2], while no additional ligands or surfactants were employed. In a typical synthesis, the Na2PdCl4 and InCl3 precursors were dissolved in the IL and an excess of the reducing agent was rapidly injected into the solution at 60 °C while vigorously stirring. The initially dark brown color turned to black, which indicated the formation of NPs. After increase of the reaction temperature from 60 °C to 95 °C or 200 °C, accordingly, a series of Pd–In NPs were received as black dispersion in the IL. By varying the precursor ratios of Pd and In, we were able to obtain Pd–In NPs with tunable compositions and structures. Reference Pd NPs were also prepared using a similar procedure. The Pd–In NPs were isolated as a powder after successively washing and centrifuging and used for further characterization by TEM, SEM-EDX, XRD, XPS, and ICP-OES analysis. Thus, to avoid contamination of the IL-stabilized NPs with alkali metal ions, we employed [OMA][BEt3H] where the reducing agent [BEt3H]− is coupled to the respective IL cation [OMA]+ by salt metathesis. Superhydrides (potassium or lithium triethyl borohydride) are well-known as strong reducing agents for use in NP synthesis.53 Alkali metal halides (e.g. KCl, KBr or LiCl) are formed as by-products which are typically insoluble in non-polar reaction media, precipitate and can then be separated from the NP sol, e.g. by subsequent cooling. However, these alkali metal halides are typically highly soluble in ILs and difficult to separate, which may contaminate the NP sol. Bönnemann et al. first combined the tetraalkylammonium stabilizers (e.g. N(octyl)4+) with [BEt3H]−, which they used for the synthesis of various transition metal colloids.54,55XRD analysis was employed to analyze the structure and phase composition of the Pd–In NPs. The XRD patterns of Pd–In bimetallic NPs with different compositions and crystal structures are shown in Fig. 1 (for corresponding calculated XRD patterns see Fig. S1‡). The XRD pattern of the Pd reference reveals reflections at 40.1°, 46.5°, 67.9°, 81.8° and 86.3° (2θ) which are characteristic for the face-centered cubic (fcc) Pd phase. The reflections of the Pd3In NPs (molar Pd/In ratio 3:1) were at 39.2°, 45°, 66°, 79.2°, and 83.5° (2θ) and thus, shifted towards lower diffraction angels as compared to their monometallic Pd NP counterparts. This is in good agreement with Pd3In L10 phase with AuCu3 type structure (ICDD 98-024-7193) instead of tetragonal ZrAl3 and TiAl3 type structures mainly reported at high temperature.56 However, it was not possible to determine whether the cubic Pd3In NPs reveal an ordered structure as superlattice reflections were not observed due to the closely similar X-ray scattering factors of Pd and In.57 The molar Pd/In ratio as determined by ICP-OES and SEM-EDX analysis is 3:1 and corresponds to the molar precursor ratio used for NP synthesis (Table 1 and Fig. S6‡). For PdIn NPs, we observed a different set of reflections at 39.4° (110), 56.8° (200), 71.3° (211), and 84.5° (220) (2θ) which were consistent with the ordered intermetallic PdIn B2 phase with CsCl type structure (ICDD 98-005-9473). The molar Pd/In ratio as determined by ICP-OES and SEM-EDX is 1:1 (Table 1). Rietveld analysis further supported the single-phase composition of the intermetallic PdIn NPs (Fig. S2‡). For the Pd–In NPs (molar ratio 3:7), the XRD diagrams exhibited sharper reflections matching the reflections of the intermetallic Pd3In7 reference (ICDD 98-040-8314). Metallic In or In2O3 was not observed by XRD and Rietveld analysis (Table 1 and Fig. S4‡). For all NPs, the crystallite sizes were calculated according to the Scherrer equation. For Pd3In, PdIn and Pd NPs, the reflections were rather broad and of low intensity due to the small NP size. The crystallite sizes were 4 nm (Pd NPs), 5 nm (Pd3In NPs), and 9 nm (PdIn NPs) with a slight increase with increasing In content. The crystallite sizes were in a similar range as the mean particle sizes determined by TEM images (Fig. 2). However, the crystallite size was calculated to be 60 nm for intermetallic Pd3In7 NPs and thus, much larger than the ones for Pd, PdIn and Pd3In NPs.
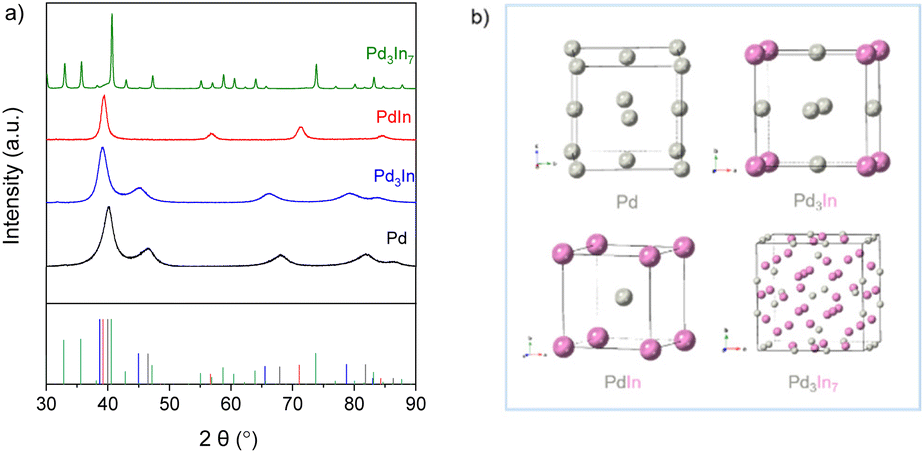 | ||
| Fig. 1 (a) X-ray diffraction patterns and (b) illustration of the unit cell and crystal structures of the L10 Pd3In, B2 PdIn and Pd3In7 phase (references: Pd3In7 (green, ICDD 98-040-8314), PdIn (red, ICDD 98-005-9473), Pd3In (blue, ICDD 98-024-7193), Pd (black, ICDD 98-004-1517)). | ||
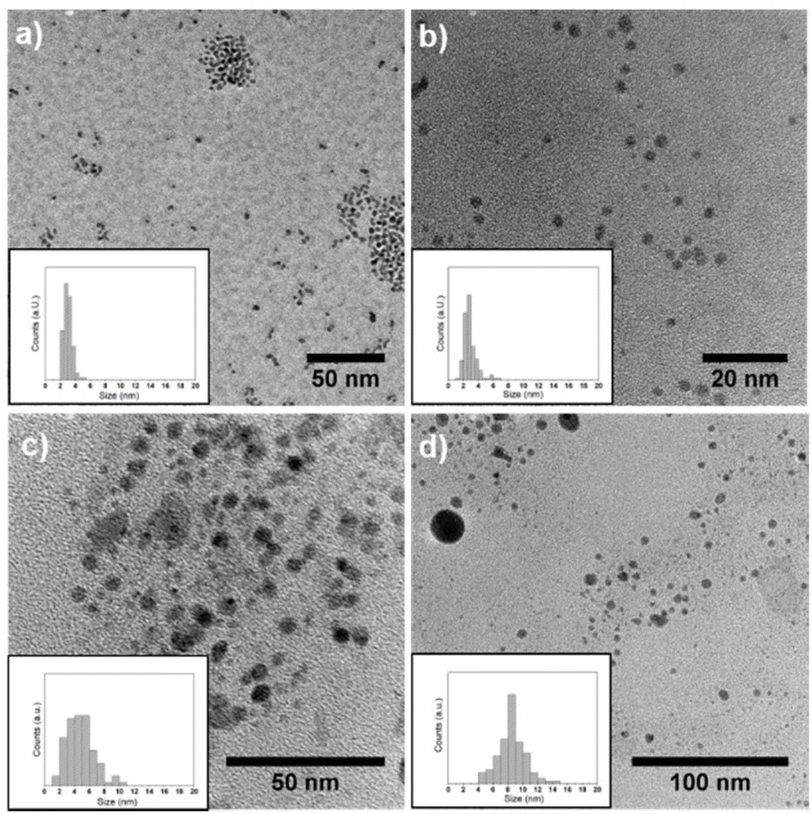 | ||
| Fig. 2 TEM images with particle size distribution of (a) Pd NPs, (b) Pd3In NPs, (c) PdIn NPs, and (d) Pd3In7 NPs. | ||
| Entry | Catalyst | Molar Pd:In ratio |
Mean NP sizeb (nm) | Crystallite sizec (nm) | Bragg angle (hkl) | |
|---|---|---|---|---|---|---|
| Precursor | NPsa | |||||
| a Determined by ICP-OES and SEM-EDX. b Determined by statistical measurement from a number n of NPs (n > 100) based on TEM images. c The crystallite size was calculated according to the Scherrer equation. | ||||||
| 1 | Pd | 1:0 |
1:0 |
3.0 ± 0.5 | 4 | 40.1° (111) |
| 2 | Pd3In | 3:1 |
3:1 |
2.9 ± 0.9 | 5 | 39.2° (111) |
| 3 | PdIn | 1:1 |
1:1 |
4.8 ± 1.8 | 9 | 39.4° (110) |
| 4 | Pd3In7 | 3:7 |
1:2.3 |
8.5 ± 1.8 | 60 | 40.7° (330) |
The Pd–In NPs were isolated from the IL by precipitation and their morphology was examined by transmission electron microscopy (TEM). The mean particle size and size distribution of the Pd–In NPs were determined by statistical measurement from a number n of NPs (n > 100) based on the TEM images (Table 1 and Fig. 2). The monometallic Pd (Fig. 2a) and Pd3In NPs (Fig. 2b) had a nearly spherical shape and a small size with an average diameter about 3 nm, which was in good agreement with crystallite sizes calculated by the Scherrer equation (Table 1). Similarly, the intermetallic PdIn NPs (Fig. 2c) also revealed a spherical shape with a mean diameter of 4.8 ± 1.8 nm, which was slightly larger compared to the Pd and Pd3In NPs. It should be noted that a few particles with larger diameters of around 10 nm were also present. The Pd3In7 NPs exhibited the largest diameter (8.5 ± 1.8 nm) with a contribution of relatively large NPs as observed in Fig. 2d. This is in good agreement with the much larger crystallite size calculated based on the Scherrer equation by XRD analysis (Table 1). It should be noted that the Scherrer formula does not consider the particle size distribution. If a few large and small NPs are present in the powder sample, the diffraction profile represents a convolution of narrow and broad reflections, whereby the narrow reflections of larger NPs dominate the profile. Overall, the particle sizes increased with increasing amount of In used for the synthesis of bimetallic Pd–In NPs.
Fig. 3a displays a high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of intermetallic PdIn NPs. Fig. 3b shows a high-resolution HRTEM of a single PdIn NP with a lattice spacing of 0.23 nm, which can be assigned to the (110) planes of the intermetallic B2 PdIn phase (space group: Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m).45 To further investigate the distribution of Pd and In, EDS elemental maps and EDS line profiles of PdIn nanoparticles were obtained (Fig. 3c–f and Fig. S10‡), which indicated an overall homogeneous distribution of Pd and In within each particle without phase segregation. These results clearly support the successful synthesis of intermetallic PdIn NPs. In addition, Pd3In and Pd3In7 NPs were analyzed by HAADF-STEM imaging, EDS elemental mapping and EDS line scan profiles (Fig. S8, S9, S11 and S12‡). They also showed a uniform Pd and In distribution over the particle core, but some surface enrichment of the more dominant element (i.e. Pd for Pd3In and In for Pd3In7) was observed in this case. For the Pd3In7 NPs, a thin amorphous In oxide coating was further observed besides the crystalline Pd3In7 core by HAADF-STEM imaging (Fig. S9‡), which may be assigned to the partial oxidation of the In atoms on the surface upon exposure to air during sample preparation. For Pd3In NPs, some minor (statistical) variation in the Pd/In ratio for few NPs can also not be completely excluded (Fig. S8d‡).
m).45 To further investigate the distribution of Pd and In, EDS elemental maps and EDS line profiles of PdIn nanoparticles were obtained (Fig. 3c–f and Fig. S10‡), which indicated an overall homogeneous distribution of Pd and In within each particle without phase segregation. These results clearly support the successful synthesis of intermetallic PdIn NPs. In addition, Pd3In and Pd3In7 NPs were analyzed by HAADF-STEM imaging, EDS elemental mapping and EDS line scan profiles (Fig. S8, S9, S11 and S12‡). They also showed a uniform Pd and In distribution over the particle core, but some surface enrichment of the more dominant element (i.e. Pd for Pd3In and In for Pd3In7) was observed in this case. For the Pd3In7 NPs, a thin amorphous In oxide coating was further observed besides the crystalline Pd3In7 core by HAADF-STEM imaging (Fig. S9‡), which may be assigned to the partial oxidation of the In atoms on the surface upon exposure to air during sample preparation. For Pd3In NPs, some minor (statistical) variation in the Pd/In ratio for few NPs can also not be completely excluded (Fig. S8d‡).
 | ||
| Fig. 3 HAADF-STEM analysis of intermetallic PdIn NPs and elemental distribution: (a) representative HAADF-STEM image, (b) atomic-resolution HRTEM image of an individual PdIn NP. (c) HAADF-STEM image of PdIn NPs and corresponding EDS elemental mapping of (d) Pd, (e) In, and (f) Pd + In. | ||
Additionally, X-ray photoelectron spectroscopy (XPS) was performed on PdIn, Pd3In and Pd3In7 NPs to further investigate the electronic interaction between Pd and In. The XPS spectra of the Pd 3d and In 3d region are shown in Fig. 4. In the Pd 3d region (Fig. 4a), the Pd 3d5/2 peaks were located at 335.9, 336.1 and 336.4 eV, respectively for Pd3In, PdIn and Pd3In7 NPs, gradually shifted to higher binding energies (BE) compared to Pd(0) (335.1 eV). Upon addition of In, the electronic states of Pd are gradually filled for the In-rich phases, resulting in a partial negative charge on the Pd atoms. This was previously attributed to a simple charge transfer from In to Pd atoms according to Pauling's electronegativity values (1.78 (In), 2.20 (Pd)) and in agreement with the formation of intermetallic Pd–In NPs.58 Similar results have also been reported for intermetallic Pd–Ga59 and Pd–Sn60 NPs. However, it should be noted that such a simple charge transfer model, based on (minor) electronegativity differences, cannot account, for example, for the strong covalent bonding character in the 1:1 Pd1In1 intermetallic compound influencing the electronic VB structure of the bulk phase. For all Pd–In NPs, the In 3d5/2 elemental line at 443.9 eV dominates and corresponds to indium in the metallic state as reported for intermetallic Pd–In compounds.61,62 Shoulders at higher binding energy of In 3d5/2 (444.9 eV) are characteristic for In3+ and decreased for In-poor intermetallic NPs (Table S1‡). In addition, the valence band (VB) spectra revealed the density of states at the Fermi energy (Ef) caused by Pd 4d states (Fig. S13‡). With increasing In content the maximum of the VB shifted to higher BE between 2 and 4 eV below Ef attributed to localized Pd 4d states.63 It is well known that the position of d-band center with respect to the Fermi level has a significant effect on the catalytic adsorption characteristics.64
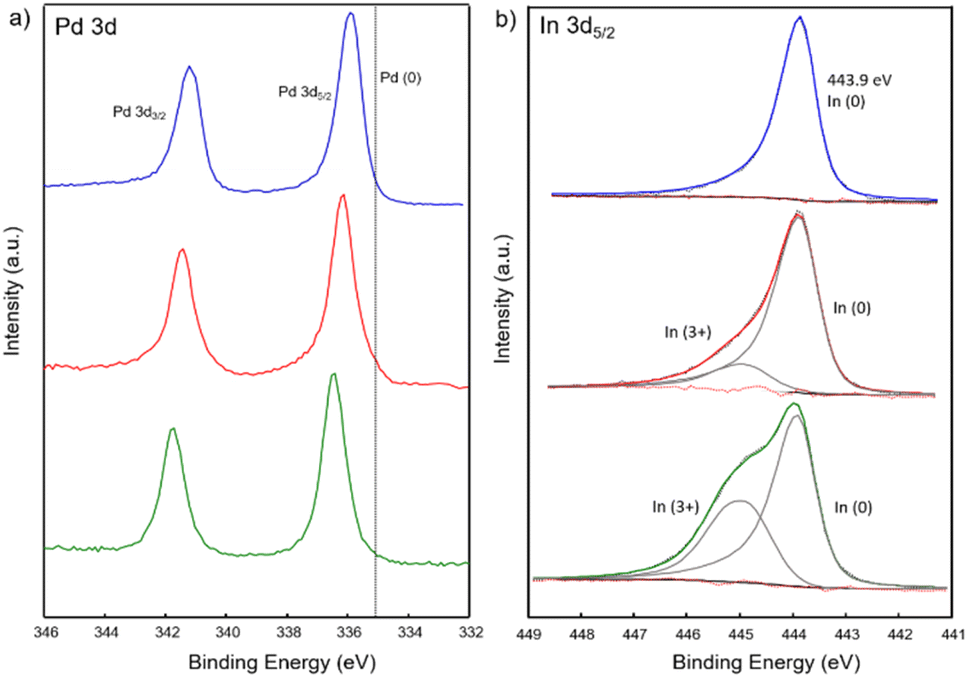 | ||
| Fig. 4 (a) Pd 3d and (b) In 3d5/2 XPS spectra of PdIn (red), Pd3In (blue) and Pd3In7 (green) NPs. | ||
The catalytic properties of the as-prepared PdIn, Pd3In and Pd3In7 NPs dispersed in the IL along with the monometallic Pd NPs were tested in the liquid-phase, semi-hydrogenation of DPA. Fig. 5 shows the possible reaction pathways, intermediates and products in the hydrogenation of DPA. DPA hydrogenation involves the semi-hydrogenation to (cis)-stilbene (CST) or (trans)-stilbene (TST) and their hydrogen-mediated isomerization. The catalytic hydrogenation of inner alkynes reveals an intrinsic stereoselectivity to (Z)-alkenes, because of their syn addition style and the catalytic hydrogenation of alkynes to (E)-alkenes, in principle, barely occurs.27 Recently, a tandem catalytic system was reported comprising Pd3Pb/SiO2 or Pd3Bi/SiO2 and RhSb/SiO2 for alkyne semi-hydrogenation and for alkene isomerization, respectively, which allowed also the one-pot TST synthesis from DPA.27,65 Generally, DPA is hydrogenated to stilbene and subsequently to undesired DPE. CST and TST derivates are interesting for use as dyes, liquid crystals, optical brighteners, OLEDs, or in the production of food additives. However, the selective hydrogenation of substituted carbon–carbon triple bonds can be challenging because the heats of adsorption of the reactants and intermediates are often similar. DPA hydrogenation is also associated with irreversible over-hydrogenation to 1,2-diphenylethane (DPE), which reduces the yield of cis- and trans-alkenes.
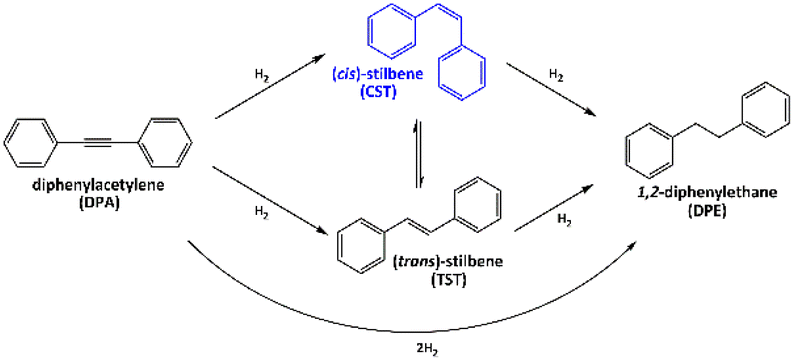 | ||
| Fig. 5 Reaction pathways for the hydrogenation of diphenylacetylene (DPA). | ||
The catalytic performance of the Pd–In NPs with different compositions and crystal structures is compared to the Pd reference NPs in Fig. 6 and summarized in Table 2. For the monometallic Pd reference NPs, the Smax towards the desired CST and DPA conversion were 30% and 100% after 10 min of reaction, respectively, with DPE as the only product. Alloying Pd with In increased both CST selectivity and yield for all Pd–In NP catalysts. In general, the addition of In influences the surface geometric and electronic structures as well as the PdHx hydride formation of Pd catalysts.66 The Pd–In based catalysts revealed a significantly enhanced Smax (CST) in the range of 83 to 85% which was maintained even for higher DPA conversions (Fig. 6d). For example, intermetallic PdIn NPs showed 100% DPA conversion after 15 min which was comparable to the Pd NPs, while the CST selectivity significantly increased to 83%. Thus, the maximum CST yield increased from 30% for the Pd reference NPs to 83% for the PdIn NPs (Fig. 6c, Fig. S14‡ and Table 2). The Pd3In NP catalyst also revealed an enhanced maximum CST yield (70%) which was reached after 30 min of reaction. The volcano-type curve was comparable to earlier findings of Shaun K. Johnston et al., where TiO2-supported Pd3In NPs revealed a higher selectivity than their Pd counterpart but they were also less selective at full conversion (Fig. S15‡).67 Interestingly, the CST selectivity of PdIn NPs only slightly decreased over reaction time while the selectivity considerably decreased to 29.3% over Pd3In NP catalyst as a function of the reaction time (Fig. 6b): In case of intermetallic PdIn NP catalysts, the over hydrogenation to DPE seemed to be inhibited. In this case, a CST and TST yield of 75% and 5%, respectively, were reached after 15 min of reaction and remained nearly constant over 3 h of reaction (Table S2‡). For the intermetallic Pd3In7 (In-rich) catalyst, also a high maximum CST selectivity of 85% was observed but at the same time the DPA conversion decreased to 45%, affording an overall low Ymax(CST) of 40%.
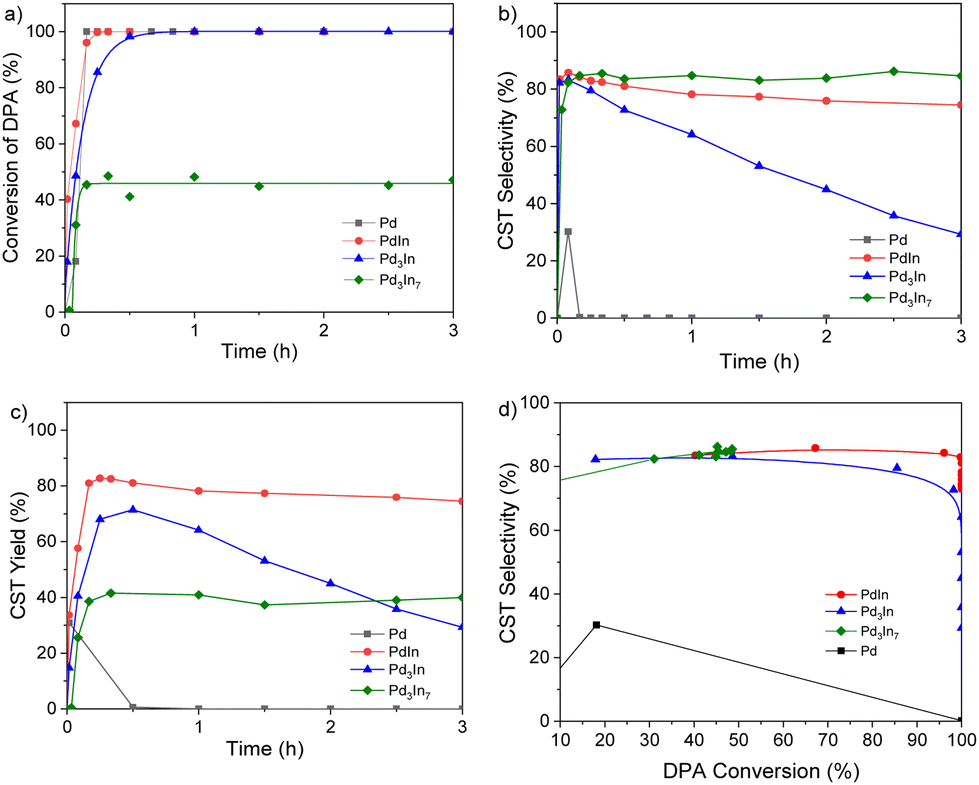 | ||
| Fig. 6 Catalytic performance of Pd@IL, Pd3In@IL, PdIn@IL, and Pd3In7@IL in the selective hydrogenation of diphenylacetylene (reaction conditions: 80 °C, 10 bar H2, Pd–In NPs@IL, 30 ml THF, 5.6 mmol DPA): (a) conversion, (b) CST selectivity, (c) CST yield over reaction time and (d) CST selectivity over DPA conversion. | ||
| Entry | Catalysts | Y(CST) [%] | S(CST) [%] | ||
|---|---|---|---|---|---|
| Y max | Y DPA50 | S max | S DPA50 | ||
| 1 | Pd | 30 (5 min) | 4 | 30 (5 min) | 17 |
| 2 | PdIn | 83 (15 min) | 43 | 86 (5 min) | 82 |
| 3 | Pd3In | 70 (30 min) | 38 | 83 (5 min) | 82 |
| 4 | Pd3In7 | 47 (20 min) | N/A | 85(10 min) | N/A |
The rather low activity over Pd3In7 NPs could be a result of the much higher In content of the NPs and/or the In surface segregation and the consequently lower number of active Pd centers on the NP surface (Fig. S16‡). In addition, the Pd3In7 NPs were also larger in size than the other Pd–In NPs and the Pd reference catalyst. The DPA conversion did not exceed 45% over time of reaction, probably due to catalyst deactivation. However, it should be noted that the catalyst structure did not change after reaction. The XRD patterns of the PdIn, Pd3In and Pd3In7 NPs after catalytic reaction are displayed in Fig. S17–19.‡Fig. 6d shows the CST selectivity as a function of DPA conversion. It is apparent that all Pd–In catalysts reveal a higher CST selectivity compared to the Pd reference over the total course of the hydrogenation reaction. Especially, the intermetallic PdIn NPs showed a much higher CST selectivity compared to Pd3In and Pd3In7 at high DPA conversion (>95%), yet remained around 80%. Overall, the intermetallic PdIn NPs synthesized in ILs combined high activity and selectivity and exhibited the best catalytic properties of the catalysts studied in this work, as maintaining excellent cis-stilbene selectivity at full conversion of DPA is a major challenge. The catalytic performance of other catalysts in the semi-hydrogenation of DPA is summarized in Table S3.‡ It should be noted that the catalytic activity and selectivity are not only influenced by the reaction conditions but also by the presence of different support materials.
The enhanced catalytic performance of Pd-based, bimetallic catalysts is usually ascribed to the isolation of active Pd sites (geometric effect) and/or the modified electronic structure (electronic effect). Based on DFT calculations, Pd single atoms were shown to be surrounded by In atoms in the most exposed (110) plane of PdIn, resulting in complete isolation of Pd atoms.45 Besides inhibition of Pd hydride formation and electronic effects, isolated Pd sites offered by the intermetallic structure of PdIn NPs could have also contributed to the enhanced catalytic performance observed in this study.68 For Pd3In NP catalysts, Pd surface enrichment and the high Pd content could be one reason for the lower CST selectivity at 100% DPA conversion, e.g., favoring the consecutive hydrogenation to DPE or affecting adsorption energies.
According to a stepwise hydrogenation mechanism, the route and selectivity of the reaction are determined by two factors, a kinetic (i.e. the reaction rate ratio for alkyne vs. alkene hydrogenation) and a thermodynamic factor (i.e. the alkyne and alkene adsorption energies). The overall decrease of the reaction rates and, in particular, the decrease in CST hydrogenation rates, was previously reported for alloying Pd with In in supported Pd/In catalysts.68 However, the Pd modification with In did not reveal a direct correlation between the change in the kinetic parameters of the reaction and the selectivity suggesting the importance of a thermodynamic factor. Negative adsorption energies for acetylene (−107 kJ mol−1) and ethylene (−49 kJ mol−1) adsorption at monometallic Pd(111) surfaces indicate relatively slow alkene desorption from Pd, causing overhydrogenation to the alkane.27 In general, the adsorption energy of alkenes is typically reduced in the presence of secondary metal atoms adjacent to Pd atoms, accelerating also alkene desorption.27 Recently, also high CST selectivities were reported for unsupported nanoporous Pd (Table S3‡) where the addition of a base further enhanced the CST selectivity.69 Heterolytic H–H bond cleavage was suggested to contribute to the enhanced catalytic performance in this case. We have previously demonstrated the synthesis of intermetallic PdSn NPs with orthorhombic structure (Pnma) in ILs using a similar approach. These PdSn NPs exhibited a comparably high CST selectivity in the semi-hydrogenation of DPA under these conditions while DPA conversion and CST yield were ∼30% and ∼24%, respectively, and thus much lower than for the PdIn NP catalysts.25
Conclusions
We have developed single-phase intermetallic Pd–In NPs at temperatures below 200 °C using ILs as reaction media. Depending on the reaction conditions (in particular the molar ratio of the metal precursors), single-phase Pd–In NPs with different crystal structures were received in the IL while no additional capping agents were added. The impact of the structure and composition of PdIn, Pd3In and Pd3In7 NPs on the catalytic performance was studied in the liquid-phase semi-hydrogenation of DPA and compared to Pd reference NPs. The catalytic properties of the Pd–In catalysts in the semi-hydrogenation of DPA were significantly governed by the elemental composition and crystal structure, where the intermetallic PdIn NPs with a CsCl-type structure revealed both a very high activity and excellent selectivity towards CST. Notably, the CST selectivity and yield at 100% DPA conversion was only slightly decreased over PdIn catalysts, which were superior to the Pd3In and Pd3In7 NP catalysts as well as monometallic Pd reference NPs. The enhanced catalytic performance of intermetallic PdIn NPs could be a consequence of the isolation of active Pd sites on the surface by surrounding In atoms, a modified electronic structure caused by charge transfer from In to Pd and/or the suppression of the Pd hydride formation for PdIn NPs. However, this will be subject of further experimental and theoretical studies. These findings may open new avenues for design of single-phase intermetallic NPs of other compositions to develop model catalysts for more in-depth studies of the material descriptors in this reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the China Scholarship Council. We are grateful to Karlsruhe Nano Micro Facility (KNMFi) for supporting the TEM work. We would like to thank Dr. Zimmermann (IKFT) for the measurement of SEM-EDX. We also would like to acknowledge Birgit Rolli (IKFT) for the support with GC measurements and Armin Lautenbach (IKFT) for ICP-OES analysis. S.B. acknowledges support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – SFB 1441 – Project-ID 426888090 (projects B2 and C2).References
- G. Vilé, D. Albani, N. Almora-Barrios, N. López and J. Pérez-Ramírez, ChemCatChem, 2016, 8, 21–33 CrossRef.
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 683–733 CrossRef CAS PubMed.
- M. T. Ravanchi, S. Sahebdelfar and S. Komeili, Rev. Chem. Eng., 2018, 34, 215–237 CrossRef.
- J. M. Slocik and R. R. Naik, Adv. Mater., 2006, 18, 1988–1992 CrossRef CAS.
- G. Vile, N. Almora-Barrios, S. Mitchell, N. Lopez and J. Perez-Ramirez, Chemistry, 2014, 20, 5926–5937 CrossRef CAS PubMed.
- R. Li, W. Yao, Y. Jin, W. Jia, X. Chen, J. Chen, J. Zheng, Y. Hu, D. Han and J. Zhao, Chem. Eng. J., 2018, 351, 995–1005 CrossRef CAS.
- Q. Luo, Z. Wang, Y. Chen, S. Mao, K. Wu, K. Zhang, Q. Li, G. Lv, G. Huang, H. Li and Y. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 31775–31784 CrossRef CAS PubMed.
- T. Mitsudome, T. Urayama, K. Yamazaki, Y. Maehara, J. Yamasaki, K. Gohara, Z. Maeno, T. Mizugaki, K. Jitsukawa and K. Kaneda, ACS Catal., 2015, 6, 666–670 CrossRef.
- D. Chatterjee, S. Shetty, K. Muller-Caspary, T. Grieb, F. F. Krause, M. Schowalter, A. Rosenauer and N. Ravishankar, Nano Lett., 2018, 18, 1903–1907 CrossRef CAS PubMed.
- G. H. Wang, J. Hilgert, F. H. Richter, F. Wang, H. J. Bongard, B. Spliethoff, C. Weidenthaler and F. Schuth, Nat. Mater., 2014, 13, 293–300 CrossRef CAS PubMed.
- L. Wang, A. Holewinski and C. Wang, ACS Catal., 2018, 8, 9388–9398 CrossRef CAS.
- J. E. S. van der Hoeven, J. Jelic, L. A. Olthof, G. Totarella, R. J. A. van Dijk-Moes, J. M. Krafft, C. Louis, F. Studt, A. van Blaaderen and P. E. de Jongh, Nat. Mater., 2021, 20, 1216–1220 CrossRef CAS PubMed.
- X.-M. Song, J.-M. Wu and M. Yan, Thin Solid Films, 2009, 517, 4341–4347 CrossRef CAS.
- Z. Wang, L. Yang, R. Zhang, L. Li, Z. Cheng and Z. Zhou, Catal. Today, 2016, 264, 37–43 CrossRef CAS.
- A. V. Rassolov, G. O. Bragina, G. N. Baeva, I. S. Mashkovsky and A. Y. Stakheev, Kinet. Catal., 2020, 61, 869–878 CrossRef CAS.
- V. V. Chesnokov, A. S. Chichkan and Z. R. Ismagilov, Kinet. Catal., 2017, 58, 649–654 CrossRef CAS.
- K. Wang, G. Li, C. Wu, X. Sui, Q. Wang and J. He, J. Cluster Sci., 2015, 27, 55–62 CrossRef.
- P. Concepción, S. García, J. C. Hernández-Garrido, J. J. Calvino and A. Corma, Catal. Today, 2016, 259, 213–221 CrossRef.
- P. Tegeder, M. Freitag, K. M. Chepiga, S. Muratsugu, N. Moller, S. Lamping, M. Tada, F. Glorius and B. J. Ravoo, Chemistry, 2018, 24, 18682–18688 CrossRef CAS PubMed.
- D. V. Glyzdova, N. S. Smirnova, N. N. Leont'eva, E. Y. Gerasimov, I. P. Prosvirin, V. I. Vershinin, D. A. Shlyapin and P. G. Tsyrul'nikov, Kinet. Catal., 2017, 58, 140–146 CrossRef CAS.
- M. Armbrüster, K. Kovnir, M. Behrens, D. Teschner, Y. Grin and R. Schlögl, J. Am. Chem. Soc., 2010, 132, 14745–14747 CrossRef PubMed.
- I. S. Mashkovsky, P. V. Markov, G. O. Bragina, G. N. Baeva, A. V. Rassolov, I. A. Yakushev, M. N. Vargaftik and A. Y. Stakheev, Nanomaterials, 2018, 8, 769–784 CrossRef PubMed.
- M. Neumann, D. Teschner, A. Knop-Gericke, W. Reschetilowski and M. Armbrüster, J. Catal., 2016, 340, 49–59 CrossRef CAS.
- Y. Luo, S. A. Villaseca, M. Friedrich, D. Teschner, A. Knop-Gericke and M. Armbrüster, J. Catal., 2016, 338, 265–272 CrossRef CAS.
- C. Dietrich, S. Chen, G. Uzunidis, M. Hahsler, Y. Trautlein and S. Behrens, ChemistryOpen, 2021, 10, 296–304 CrossRef PubMed.
- S. K. Johnston, N. Cherkasov, E. Pérez-Barrado, A. Aho, D. Y. Murzin, A. O. Ibhadon and M. G. Francesconi, Appl. Catal., A, 2017, 544, 40–45 CrossRef CAS.
- S. Furukawa and T. Komatsu, ACS Catal., 2016, 6, 2121–2125 CrossRef CAS.
- I. G. Aviziotis, A. Götze, F. Göhler, H. Kohlmann and M. Armbrüster, Z. Anorg. Allg. Chem., 2018, 644, 1777–1781 CrossRef CAS.
- O. Matselko, R. R. Zimmermann, A. Ormeci, U. Burkhardt, R. Gladyshevskii, Y. Grin and M. Armbrüster, J. Phys. Chem. C, 2018, 122, 21891–21896 CrossRef CAS.
- O. Matselko, U. Burkhardt, Y. Prots, R. R. Zimmermann, M. Armbrüster, R. Gladyshevskii and Y. Grin, Eur. J. Inorg. Chem., 2017, 2017, 3542–3550 CrossRef CAS.
- R. K. Rai, M. K. Awasthi, V. K. Singh, S. R. Barman, S. Behrens and S. K. Singh, Catal. Sci. Technol., 2020, 10, 4968–4980 RSC.
- M. Armbruster, K. Kovnir, M. Friedrich, D. Teschner, G. Wowsnick, M. Hahne, P. Gille, L. Szentmiklosi, M. Feuerbacher, M. Heggen, F. Girgsdies, D. Rosenthal, R. Schlogl and Y. Grin, Nat. Mater., 2012, 11, 690–693 CrossRef CAS PubMed.
- B. Bridier and J. Perez-Ramirez, J. Am. Chem. Soc., 2010, 132, 4321–4327 CrossRef CAS PubMed.
- C. H. Wu, C. Liu, D. Su, H. L. Xin, H.-T. Fang, B. Eren, S. Zhang, C. B. Murray and M. B. Salmeron, Nat. Catal., 2018, 2, 78–85 CrossRef.
- M. Zhou, C. Li and J. Fang, Chem. Rev., 2021, 121, 736–795 CrossRef CAS.
- S. Furukawa, K. Ochi, H. Luo, M. Miyazaki and T. Komatsu, ChemCatChem, 2015, 7, 3472–3479 CrossRef CAS.
- M. Armbruster, Sci. Technol. Adv. Mater., 2020, 21, 303–322 CrossRef PubMed.
- V. R. Naina, S. Wang, D. I. Sharapa, M. Zimmermann, M. Hähsler, L. Niebl-Eibenstein, J. Wang, C. Wöll, Y. Wang, S. K. Singh, F. Studt and S. Behrens, ACS Catal., 2021, 11, 2288–2301 CrossRef CAS.
- S. Han, C. He, Q. Yun, M. Li, W. Chen, W. Cao and Q. Lu, Coord. Chem. Rev., 2021, 445, 214085 CrossRef CAS.
- M. Armbrüster, G. Wowsnick, M. Friedrich, M. Heggen and R. Cardoso-Gil, J. Am. Chem. Soc., 2011, 133, 9112–9118 CrossRef PubMed.
- I. P. Stolarov, I. A. Yakushev, A. V. Churakov, N. V. Cherkashina, N. S. Smirnova, E. V. Khramov, Y. V. Zubavichus, V. N. Khrustalev, A. A. Markov, A. P. Klyagina, A. B. Kornev, V. M. Martynenko, A. E. Gekhman, M. N. Vargaftik and I. I. Moiseev, Inorg. Chem., 2018, 57, 11482–11491 CrossRef CAS PubMed.
- P. V. Markov, G. O. Bragina, G. N. Baeva, O. P. Tkachenko, I. S. Mashkovskii, I. A. Yakushev, M. N. Vargaftik and A. Y. Stakheev, Kinet. Catal., 2016, 57, 617–624 CrossRef CAS.
- P. V. Markov, A. V. Bukhtiyarov, I. S. Mashkovsky, N. S. Smirnova, I. P. Prosvirin, Z. S. Vinokurov, M. A. Panafidin, G. N. Baeva, Y. V. Zubavichus, V. I. Bukhtiyarov and A. Y. Stakheev, Kinet. Catal., 2020, 60, 842–850 CrossRef.
- P. V. Markov, G. O. Bragina, G. N. Baeva, I. S. Mashkovskii, A. V. Rassolov, I. A. Yakushev, M. N. Vargaftik and A. Y. Stakheev, Kinet. Catal., 2016, 57, 625–631 CrossRef CAS.
- Q. Feng, S. Zhao, Y. Wang, J. Dong, W. Chen, D. He, D. Wang, J. Yang, Y. Zhu, H. Zhu, L. Gu, Z. Li, Y. Liu, R. Yu, J. Li and Y. Li, J. Am. Chem. Soc., 2017, 139, 7294–7301 CrossRef CAS PubMed.
- H. Okamoto, J. Phase Equilib., 2003, 24, 481–481 CrossRef CAS.
- Q. Wang, Z. L. Zhao, Z. Zhang, T. Feng, R. Zhong, H. Xu, S. T. Pantelides and M. Gu, Adv. Sci., 2020, 7, 1901279 CrossRef CAS PubMed.
- T. Wang, J. Liang, Z. Zhao, S. Li, G. Lu, Z. Xia, C. Wang, J. Luo, J. Han, C. Ma, Y. Huang and Q. Li, Adv. Energy Mater., 2019, 9, 1803771 CrossRef.
- K. Ding, H. Yang, Y. Cao, C. Zheng, S. B. Rapole and Z. Guo, Mater. Chem. Phys., 2013, 142, 403–411 CrossRef CAS.
- Y. Qiao, W. Ma, N. Theyssen, C. Chen and Z. Hou, Chem. Rev., 2017, 117, 6881–6928 CrossRef CAS PubMed.
- C. Vollmer and C. Janiak, Coord. Chem. Rev., 2011, 255, 2039–2057 CrossRef CAS.
- C. Dietrich, M. Hähsler, W. Wang, C. Kübel and S. Behrens, ChemNanoMat, 2020, 6, 1854–1862 CrossRef CAS.
- H. Bönnemann, W. Brijoux, R. Brinkmann, R. Fretzen, T. Joussen, R. Köppler, B. Korall, P. Neiteler and J. Richter, J. Mol. Catal., 1994, 86, 129–177 CrossRef.
- H. Bönnemann, R. Brinkmann and P. Neiteler, Appl. Organomet. Chem., 1994, 8, 361–378 CrossRef.
- H. Bönnemann and R. M. Richards, Eur. J. Inorg. Chem., 2001, 2001, 2455–2480 CrossRef.
- H. Kohlmann, J. Solid State Chem., 2010, 183, 367–372 CrossRef CAS.
- I. Harris, M. Norman and A. Bryant, J. Less-Common Met., 1968, 16, 427–440 CrossRef CAS.
- C. Rameshan, H. Lorenz, L. Mayr, S. Penner, D. Zemlyanov, R. Arrigo, M. Haevecker, R. Blume, A. Knop-Gericke and R. Schlögl, J. Catal., 2012, 295, 186–194 CrossRef CAS PubMed.
- M. Armbrüster, R. Schlögl and Y. Grin, Sci. Technol. Adv. Mater., 2014, 15, 034803 CrossRef PubMed.
- S. Mondal, V. S. K. Choutipalli, B. K. Jena, V. Subramanian and C. R. Raj, J. Phys. Chem. C, 2020, 124, 9631–9643 CrossRef CAS.
- A. García-Trenco, A. Regoutz, E. R. White, D. J. Payne, M. S. P. Shaffer and C. K. Williams, Appl. Catal., B, 2018, 220, 9–18 CrossRef.
- Y.-J. Chen, Y.-R. Chen, C.-H. Chiang, K.-L. Tung, T.-K. Yeh and H.-Y. Tuan, Nanoscale, 2019, 11, 3336–3343 RSC.
- B. Verbeek, P. Larsen and W. Gerits, Vacuum, 1983, 33, 813–814 CrossRef CAS.
- P. Markov, A. Bukhtiyarov, I. Mashkovsky, N. Smirnova, I. Prosvirin, Z. Vinokurov, M. Panafidin, G. Baeva, Y. V. Zubavichus and V. Bukhtiyarov, Kinet. Catal., 2019, 60, 842–850 CrossRef CAS.
- S. Furukawa, A. Yokoyama and T. Komatsu, ACS Catal., 2014, 4, 3581–3585 CrossRef CAS.
- Y. Cao, Z. Sui, Y. Zhu, X. Zhou and D. Chen, ACS Catal., 2017, 7, 7835–7846 CrossRef CAS.
- S. K. Johnston, T. A. Bryant, J. Strong, L. Lazzarini, A. O. Ibhadon and M. G. Francesconi, ChemCatChem, 2019, 11, 2909–2918 CrossRef CAS.
- P. Markov, G. Bragina, G. Baeva, I. Mashkovskii, A. Rassolov, I. Yakushev, M. Vargaftik and A. Y. Stakheev, Kinet. Catal., 2016, 57, 625–631 CrossRef CAS.
- Y. Lu, X. Feng, B. S. Takale, Y. Yamamoto, W. Zhang and M. Bao, ACS Catal., 2017, 7, 8296–8303 CrossRef CAS.
Footnotes |
| † Dedicated to Prof. Dr Dieter Fenske on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr03674f |
| This journal is © The Royal Society of Chemistry 2022 |