
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly of colloidal single-layer carbon nitride†
Oleksandr
Stroyuk
 *a,
Oleksandra
Raievska
a,
Christoph J.
Brabec
ab,
Volodymyr
Dzhagan
cd,
Yevhenii
Havryliuk
ef and
Dietrich R. T.
Zahn
ef
*a,
Oleksandra
Raievska
a,
Christoph J.
Brabec
ab,
Volodymyr
Dzhagan
cd,
Yevhenii
Havryliuk
ef and
Dietrich R. T.
Zahn
ef
aForschungszentrum Jülich GmbH, Helmholtz-Institut Erlangen Nürnberg für Erneuerbare Energien (HI ERN), 91058 Erlangen, Germany. E-mail: o.stroyuk@fz-juelich.de
bFriedrich-Alexander-Universität Erlangen-Nürnberg, Materials for Electronics and Energy Technology (i-MEET), Martensstrasse 7, 91058 Erlangen, Germany
cV. Lashkaryov Institute of Semiconductors Physics, National Academy of Sciences of Ukraine, 41 Nauky Av., 03028 Kyiv, Ukraine
dTaras Shevchenko National University of Kyiv, 64 Volodymyrs'ka St., 01601 Kyiv, Ukraine
eSemiconductor Physics, Chemnitz University of Technology, D-09107 Chemnitz, Germany
fCenter for Materials, Architectures, and Integration of Nanomembranes (MAIN), Chemnitz University of Technology, D-09107 Chemnitz, Germany
First published on 11th August 2022
Abstract
We introduce a new concept of a “bottom-to-top” design of intercalate carbon nitride compounds based on the effects of self-assembly of colloidal single-layer carbon nitride (SLCN) sheets stabilized by tetraethylammonium hydroxide NEt4OH upon ambient drying of the water solvent. These effects include (i) formation of stage-1 intercalates of NEt4OH during the ambient drying of SLCN colloids on glass substrates and (ii) the spontaneous formation of layered hexagonally-shaped networks of SLCN sheets on freshly-cleaved mica surfaces. The dynamics of the intercalate formation was followed by in situ X-ray diffraction allowing different stages to be identified, including the deposition of a primary “wet” intercalate of hydrated NEt4OH and the gradual elimination of excessive water during its ambient drying. The intercalated NEt4+ cations show a specific “flattened” conformation allowing the dynamics of formation and structure of the intercalate to be probed by vibrational spectroscopies. The two-dimensional self-assembly on mica is assumed to be driven both by the internal hexagonal symmetry of heptazine units and by a templating effect of the mica surface.
Introduction
Intercalation chemistry of graphite is a well-established and blossoming area yielding a variety of materials with unique adsorption, catalytic, and electrochemical characteristics.1–3 The recent research boom aiming at graphene4–7 provided an additional impetus for the chemistry of graphite intercalates as intermediates for the graphite exfoliation and production of graphene suspensions.2,3,8 The graphene-stimulated renaissance of interest in graphite has also inspired studies of its analogs, in particular, graphitic carbon nitride (GCN).9–11 Due to a combination of high photochemical activity, stability, and sensitivity to visible light GCN is considered as one of the most promising and “green” semiconductor materials for artificial photosynthesis and environmental photocatalysis.9,12–16 Introduction of ionic and molecular species between the carbon nitride sheets due to intercalation is expected to strongly affect the interaction of GCN with light, the dynamics of the photogenerated charge carriers, the adsorption of reactants, etc. and, therefore, progress in the intercalation chemistry of GCN can boost its applications in photocatalysis and photoelectrochemistry.In the case of GCN the intercalation compounds are typically produced spontaneously during the polycondensation or prepared after the synthesis as a intermediate step for the exfoliation of GCN into single and a-few-layer species. The first approach is exemplified by intercalation of GCN compounds with alkali metals formed spontaneously when GCN is produced via the polycondensation in molten eutectic mixtures of alkali halides.17–19 GCN/alkali metal intercalates showed superior photocatalytic properties in water splitting due to the involvement of the intercalated species in the separation of photogenerated charge carriers.17 Li-intercalated GCN was found to be active as an electrochemical humidity sensor.18
The GCN intercalation as a preparatory step for exfoliation was reported for several intercalating species, including H2SO4,20 ethylenediamine,21 HCl,22 NH4Cl,23 and water.24 The exfoliation of intercalates is facilitated due to a large increase of the interlayer distance (H2SO4, ethylenediamine),20,21 decomposition of the intercalated compound to gaseous products (NH4Cl),23 or a reaction with the GCN sheets releasing large volumes of gases (H2O).24
All these examples refer to a “classic” “top-to-bottom” route to the intercalate compounds produced by forcing intercalating species into the interlayer galleries by the synthesis conditions or external stimuli. At the same time, no reports on an alternative “bottom-to-top” strategy for the design of GCN intercalates starting from single-layer or a-few-layer polyheptazine or polytriazine imide sheets can be found, despite the current progress in the exfoliation of GCN and the synthesis of such two-dimensional (2D) species.25–27
In the present work we introduce a new concept of a “bottom-to-top” design of intercalate carbon nitride compounds using self-assembly effects shown by colloidal single-layer polyheptazine sheets upon ambient drying of the water solvent. The proposed methodology potentially allows the morphology and chemistry of the polyheptazine sheets to be tuned and the nature of intercalated species to be varied independently opening routes to a broad variety of new spatially ordered supramolecular functional assemblies.
Results and discussion
Earlier we reported27,28 a new approach to the exfoliation of bulk GCN to single-layer sheets in boiling aqueous solutions of several tetraalkylammonium hydroxides, from which tetraethylammonium hydroxide NEt4OH was selected for detailed investigations. We found that the exfoliation results in the formation of stable aqueous colloidal solutions containing single-layer polyheptazine species with a thickness of 3.5–4.0 Å and lateral size depending on the concentration and decreasing down to 40–50 nm for the most diluted colloids.27,28 We also showed the applicability of this exfoliation technique for different GCN samples produced at different temperature/duration of the melamine polycondensation and found clear effect of the history of the preparation of GCN precursor on the optical properties of the final SLCN colloids.27,29 In the latter work, however, we did not explore the details of composition and structure of colloidal SLCN produced from different GCN precursors.Here, we report for the first time two phenomena of self-assembly of SLCN sheets observed at solvent evaporation from SLCN colloids. The first self-assembly phenomenon observed mainly by X-ray diffraction (XRD) is the formation of a layered material composed of SLCN sheets with NEt4OH intercalated in the interlayer galleries. The second self-assembly effect observed by atomic force microscopy (AFM) results in the formation of regular hexagonal patterns on the surface of mica.
Both self-assembly effects were tracked for a series of colloidal SLCN samples produced by the exfoliation of bulk GCN powders synthesized at different temperatures of melamine polycondensation (Tpc). We found that the self-assembly results in very similar products for all studied SLCN samples indicating the feasibility of a broad tailoring of the properties of self-assemblies by tuning the properties of the original “building blocks” – the SLCN sheets. Due to this limited effect of the “history” of the GCN and SLCN precursors on the geometry of the self-assembled products we provide here only a concise report on the characteristics of both the bulk GCN precursors and exfoliated SLCN colloids with most of the data supplied in the ESI.†
Characterization of bulk GCN samples
The variation of Tpc as well as of the polycondensation duration tpc at a constant Tpc are the two most efficient and simple methods of tailoring the structure and optical properties of bulk GCN, which can be expected to be later translated to the single-layer exfoliation products. The polycondensation of melamine was performed in air at 400–700 °C with 1 h duration and at 700 °C for various periods of time (0.5–2 h). In the further discussion the samples of bulk GCN will be referred to as “GCN-Tpc” according to the polycondensation temperature used in the synthesis and implying tpc = 1 h. More details on the synthesis and exfoliation of bulk GCN are provided in ESI.†The XRD inspection of the polycondensation products showed that the melamine conversion at 400 °C does not yield GCN but results in an intermediate under-condensed heptazine species (ESI, Fig. S1a,† curve 2). By increasing Tpc to 500 °C a mixture of GCN with under-condensed products is produced (Fig. S1a,† curve 3), while at higher Tpc GCN appears to be sole product (Fig. S1a,† curves 4–6). At these higher Tpc a characteristic (002) peak at 27–28° as well as a low-intensity feature at ca. 13° corresponding to the intra-layer periodicity are observed.12,25,30 The increase of Tpc from 500 to 700 °C results in a gradual shift of the (002) peak to higher angles (lower interplanar distances) indicating an increased ordering and packing of the polyheptazine sheets. A similar, but smaller shift is also observed for an increase of the polycondensation duration (tpc) at a constant Tpc (ESI, Fig. S1b†).
A Fourier-transform infrared (FTIR) spectroscopy inspection of the polycondensation products shows a characteristic triazine/heptazine “fingerprint” vibration at 800–810 cm−1 as well as a set of skeletal vibration bands of polyheptazine species at 1200–1700 cm−1 (ESI, Fig. S2a†). As Tpc is increased from 400 to 500 °C an additional peak appears at 890 cm−1 assigned to hydroperoxide species.31 The intensity of this peak increases almost linearly with an increase in Tpc (ESI, Fig. S2b†). The peroxidation of bulk GCNs is expected to provide exfoliated SLCN species with oxygen-containing functionalities, their density easily controlled by varying Tpc. No distinct differences were found in the FTIR spectra of GCNs produced at 700 °C for different tpc (ESI, Fig. S2c†).
The bulk GCN products exhibited relatively bright photoluminescence (PL) emitted in a broad band centered at ca. 2.3–2.5 eV (500–550 nm) depending on the polycondensation conditions (ESI, Fig. S3a and c†). The PL maximum energy was found to shift from 2.53 eV (490 nm) to 2.31 eV (540 nm) as Tpc was increased from 400 to 700 °C (ESI, Fig. S3b†). This shift originates from both a denser structure of the GCNs produced at higher Tpc and from a higher density of oxygen functionalities.12,25,30
An X-ray photoemission spectroscopy (XPS) study of bulk GCNs revealed a distinct dependence of their structure on the polycondensation conditions. The high-resolution XP spectra in the ranges of C 1s and N 1s electron binding energies show the presence of aliphatic sp3-hybridized and aromatic sp2-hybridized carbon and nitrogen,12,25,30 with their ratios depending on Tpc (ESI, Fig. S4a†). In particular, the GCN-650 sample shows a much higher relative population of aromatic C (ca. 287.6–288.0 eV) and N (397.8–398.2 eV) as compared to GCN-400. The fractions of sp2-C and sp2-N were found to increase proportionally to Tpc (ESI, Fig. S4b and c†), but decrease for longer tpc, indicating a partial thermal destruction of the samples.
Characterization of exfoliated SLCN samples
The set of bulk GCN samples produced at varied Tpc/tpc was subjected to exfoliation in boiling aqueous 1.36 M (20 wt%) solution of NEt4OH yielding quantitatively in each case stable and transparent colloidal solutions with a nominal C3N4 concentration of 0.54 M (50 g L−1). In the further discussion we will refer to the samples of colloidal SLCNs as “SLCN-Tpc” according to the polycondensation temperature used in the synthesis of the corresponding bulk GCN precursor and implying tpc = 1 h.Inspection of the solutions by dynamic light scattering showed that the average hydrodynamic size of SLCN sheets decreases from 80–90 nm for Tpc = 400 °C to ca. 35 nm for Tpc = 700 °C with a simultaneous strong narrowing of the size distribution (ESI, Fig. S5†). The decrease of the lateral size of SLCN sheets originates, most probably, from intra-sheet defects and oxygen functionalities introduced at higher Tpc, which can be attacked during exfoliation resulting in the disintegration of polyheptazine sheets to smaller species. The presence of oxygen functionalities can be readily observed for SLCN colloids produced from bulk GCN-650 and GCN-700 samples by complex absorption bands at λ > 400 nm, which overlap with the fundamental absorption band of polyheptazine and originate from nπ* transitions in the oxygen functionalities (ESI, Fig. S6a and b†). However, despite the edge defects and oxygen functionalities, the SLCN sheets preserve a high aromaticity, ca. 90% of nitrogen atoms present in the sp2-hybridized form for Tpc ≥ 500 °C as shown by XPS (ESI, Fig. S7†). We note that colloidal SLCN, both before and after purification, retains the high PL efficiency of the original GCN samples with a broadband “blue” emission and spectral parameters close to those of the corresponding bulk precursors (ESI, Fig. S6c and d†).
The thickness of SLCN particles was probed by AFM on freshly-cleaved atomically-smooth mica substrates. The roughness of the mica substrates was below 1 Å (ESI, Fig. S8†), while a layer of pure NEt4OH after drying was found to be 2–3 Å thick (ESI, Fig. S9† and Fig. 1c, bars 1), thus not interfering with the AFM characterization of SLCN sheets.
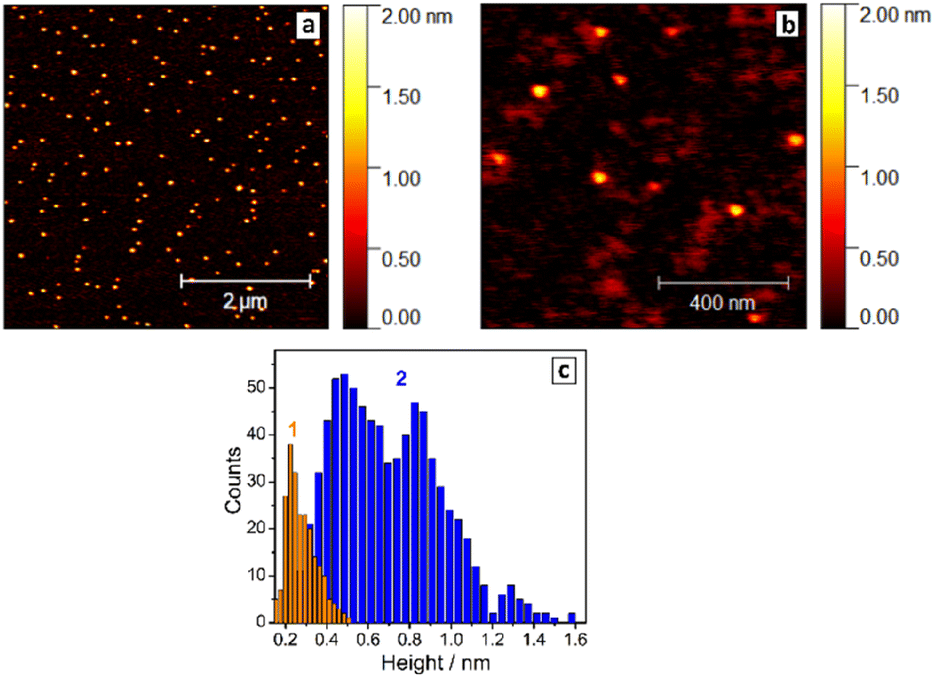 | ||
| Fig. 1 (a and b) AFM images of the SLCN-700 sample produced by exfoliation of bulk GCN-700. (c) Height distribution based on the AFM measurements plotted for pure NEt4OH solution (1) and SLCN-700 sample (2), both produced under the same conditions. | ||
Exemplary AFM images of SLCN-700 particles on mica (Fig. 1a and b) show multiple discrete species of different thickness allowing a reliable statistical analysis to be performed for many hundreds of particles using a recently reported approach.32 The analysis reveals a multimodal distribution of SLCN-700 sheet heights with distinct maxima at 4.5–5.0 Å, 8.5–9.0 Å, and 13.0–13.5 Å (Fig. 1c, bars 2), which we assign to single-, double-, and triple-layer sheets, respectively.
Similar multimodal distributions were also plotted (ESI, Fig. S10†) for the SLCN samples produced from GCN-400, GCN-600, and GCN-700 (tpc = 2 h). In all cases, the height distributions peak at around 4.5 Å, 9.0 Å, and 13–13.5 Å indicating the presence of SLCN as well as double- and triple-layer sheets with a height increment of ca. 4.5 Å per layer. This value is larger than expected for single-layer polyheptazine, 3.5–4 Å,27 most probably because of the adsorption of the NEt4OH stabilizer on the sheet surface and its presence between the single layers. This assumption is supported later by the XRD data.
Generally, the height distribution of SLCN on mica was found to shift to larger values for higher concentrations of the colloidal solutions used to prepare the samples (ESI, Fig. S11†) indicating the tendency of SLCN sheets for lateral stacking. These observations illustrate an inherent capability of SLCN sheets to stacking self-assembly, which will be addressed in detail in the next sections. At the same time, no clear evidence for the self-assembly could be gathered by AFM, XRD, and other methods for “raw” colloidal SLCN solutions without subjecting them to a purification and elimination of excessive NEt4OH present in the systems.
Purification of SLCN colloids
As mentioned, the raw exfoliation products contain 0.54 M carbon nitride (in terms of a molecular C3N4 unit) and 1.36 M NEt4OH as a stabilizer. Such colloids were subjected to purification via addition of a “poor” solvent, in this case 2-propanol, and centrifugation resulting in the formation of a precipitate (see more details in ESI). Dissolution of the precipitate in DI water produced stable colloidal solutions with spectral properties identical to those of non-purified solutions, but with a much smaller NEt4OH amount as evidenced by the almost neutral pH (pH 8) of the final colloids. We denote further the purified colloids by an additional letter “p”, for example, SLCN-600-p.A decrease in the NEt4OH concentration was also evidenced by XPS showing a drastic decrease of the aliphatic amine nitrogen peak at 401 eV after the purification (Fig. 2a). Still, the purified SLCN colloids were found to contain residuals of NEt4OH even after several precipitation/redispersion cycles, indicating that these residuals are strongly adsorbed on the SLCN sheets.
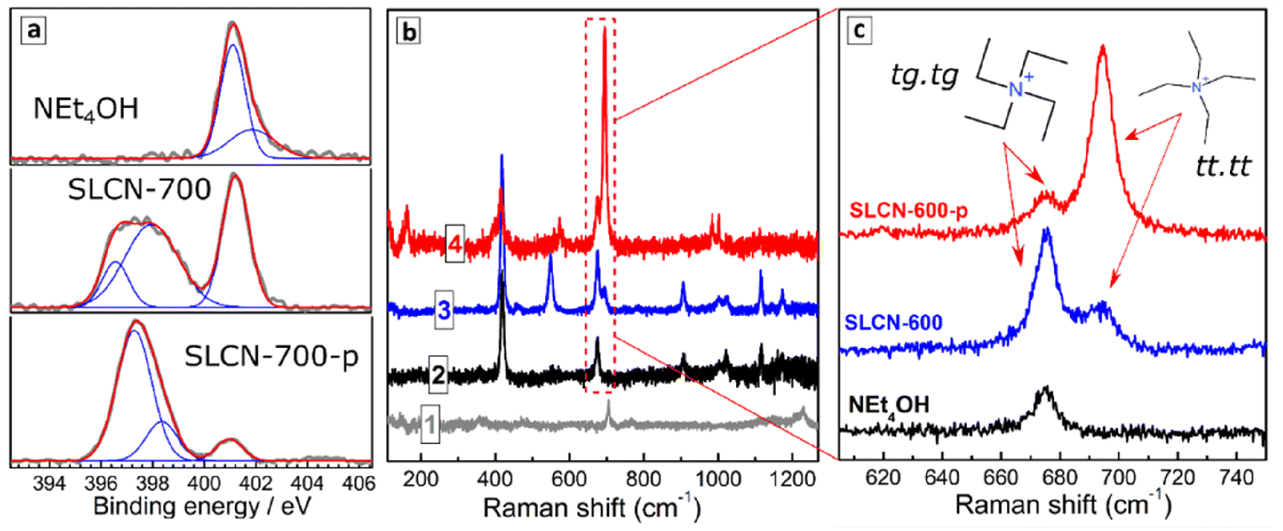 | ||
| Fig. 2 (a) High-resolution XPS spectra of NEt4OH, as well as non-purified and purified SLCN-700 colloidal solution in the range of N 1s electron binding energies. (b) Raman spectra of bulk CN-600 (curve 1), NEt4OH (2), non-purified SLCN-600 (3), and purified SLCN-600-p (4). (c) A close-up on the 620–740 cm−1 range of Raman spectra from (b). | ||
Raman spectra of dried non-purified SLCN colloids are dominated by peaks typical for pure NEt4OH (compare curves 2 and 3, Fig. 2b), which is present in a high excess. The purification resulted in marked elimination of the NEt4OH signals as well as in a considerable redistribution of relative intensities of residual peaks (compare curves 3 and 4, Fig. 2b). The peaks in the range of 660–710 cm−1 were scrutinized in more detail (Fig. 2c) because the relative intensities of the Raman features in this range can provide information on the predominant conformation of NEt4+ cations present in the system.
Only a single peak at 675 cm−1 is observed in this range for NEt4OH, while non-purified SLCN colloids feature two peaks at 675 and 695 cm−1. These peaks can be assigned to two predominant conformations of NEt4+ cation reported earlier in detail.33,34 The tetraethyl-ammonium cation was reported to exist both in crystals and in solutions in the form of all-trans (tt.tt) conformation (Greek cross, tt.tt) and a trans–gauche conformation of each Et–N–Et fragment (Nordic cross, tg.tg), both shown as insets in Fig. 1c. The tg.tg and tt.tt conformations were reported to have Raman peaks at 662–665 cm−1 and 675–685 cm−1, respectively, with the exact positions depending on the counterion and aggregation state.33–36 As we observe no other peaks in this range for colloidal SLCN and bulk CN does not reveal any Raman features in this range as well, we can assume that the peaks at 675 and 695 cm−1 correspond to the tg.tg and tt.tt conformations of NEt4+ cations present in the studied system. The shifts from the reported ranges are likely to originate from interactions with the solvent.
Only the tg.tg conformation is present in pure aqueous solutions of NEt4OH, while in the presence of SLCN sheets two conformations exist in an equilibrium (Fig. 1c). As the tt.tt conformation is more planar than the tg.tg form, we assume that this conformation is stabilized due to the adsorption of the NEt4+ cation on the SLCN sheets, while the rest of free NEt4OH in solution remains in the tg.tg conformation. Similar transitions between the tg.tg and tt.tt conformations of the NEt4+ cation were observed for zeolites depending on the strength of interaction between the cation and the host materials.35–37
After purification, the relative intensity of 695 cm−1 peak increases strongly indicating that the tt.tt form of NEt4+, which we assigned to adsorbed tetraethyl-ammonium cations, prevails in such a system. A residual peak at 675 cm−1 is also observed after purification indicating an equilibrium between adsorbed and free NEt4+ cations.
Tracking of the SLCN self-assembly by XRD
Spontaneous drying of purified SLCN colloids results in solid products showing XRD patterns differing markedly from those of the bulk GCN precursors. Fig. 3a shows an exemplary XRD pattern of the dried SLCN-600-p colloid, which was aged for 1 day before the inspection. The diffractogram reveals two major peaks corresponding to interplanar distances of 7.50 and 3.75 Å, and a minor peak at 5.00 Å, while the original bulk GCN-600 shows a major peak at 3.20 Å and a minor feature at 6.95 Å.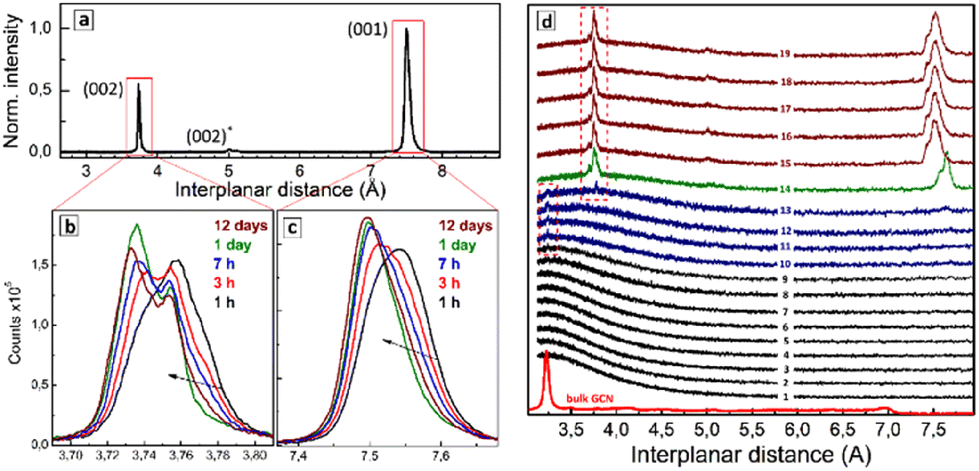 | ||
| Fig. 3 (a) XRD pattern of a dried residue from purified SLCN-600-p colloid after 1-day ageing of the dried sample. Panels (b) and (c) show close-ups of the two major peaks observed in (a) for different periods of sample ageing. (d) Curves 1–19 – XRD patterns detected from a drying drop of purified SLCN-600-p colloid with a 2 min time step between each curve. Red curve – XRD pattern of original bulk GCN-600 presented for comparison. | ||
These observations indicate that drying of the purified SLCN colloids results in the self-assembly of single colloidal sheets into layered structures with an interplanar distance much larger than that of the original bulk material. In this view, we assign the peaks at 7.50 Å and 3.75 Å to (001) and (002) interplanar distances of the stage-1 intercalate of NEt4OH stabilizer.
The gallery height of the intercalate, calculated from the position of (001) peak and the well-reported thickness of single-layer polyheptazine carbon nitride (3.23 Å) was found to be 7.50 − 3.23 = 4.27 Å, which is in the reported range of the size of tetraalkylammonium cations38 and linear aliphatic amines39 intercalated into graphite. In particular, linear aliphatic diamines were reported to intercalate graphite in two forms, a more flattened one with a size of 3.7–4.5 Å and a more bulky elongated conformation with a size of 4.9–7.2 Å.39 Two forms of tetrabutyl-ammonium cation intercalates in graphite were reported, corresponding to a bulkier conformation (gallery height of 8.9 Å) and a flattened form (gallery height of 4.7 Å).38 The intercalation of tetraethylammonium cations into graphite was reported to result in a gallery height of 4.7 Å.40 Similar sizes of respectively 4.3 Å and 4.8 Å were reported for tetrapropyl- and tetrabutylammonium cations intercalated into graphite.41 A decrease in the efficiency of electrochemical intercalation from tetramethyl- over tetraethyl- to tetrabutylammonium cations into graphite was interpreted42 as a result of the spatial restriction of the intercalate and the formation of flattened (tt.tt) conformations of both tetraethylammonium and tetrabutylammonium cations in the interlayer galleries.
The Raman spectroscopic data discussed above indicate that the NEt4+ cation is present on the surface of SLCN sheets in a flattened tt.tt conformation, which can be assumed to be preserved also in the final intercalate. The Raman data also show the presence of the more bulky tg.tg conformation of the NEt4+ cation, which can also enter the intercalate and be responsible for the (002) peak at 5.00 Å. The gallery height calculated for this peak as 2 × 5.00 − 3.23 = 6.77 Å, is in the range of the reported size of the bulkier forms of elongated diamine39 and tetrabutyl-ammonium cation38 intercalates in graphite, supporting our assumption.
All samples of purified SLCN produced at different temperatures and durations of polycondensation of the original bulk precursors showed the same behavior yielding stage-1 intercalates with the same structure (ESI, Fig. S12†). Drying of non-purified SLCN colloids results in a much more complex XRD patterns with many admixtures introducing large uncertainties in the analysis of their structure. So, the exfoliation/purification procedure reported here for SLCN sheets allows performing a “bottom-to-top” synthesis of the intercalated carbon nitride starting from the very building blocks and finishing with the bulk-like layered products. We expect that a large variety of intercalates can be produced by this bottom-to-top approach by introducing potential intercalating agents into SLCN colloids and initiating the self-assembly by solvent evaporation.
A periodic XRD inspection of the drying solid SLCN residuals showed that NEt4OH is not the sole intercalate possible in the present system. In particular, ageing of the freshly dried sample under ambient environment was found to result in a gradual shift of the main (001) peak from 7.54 Å in 1 h after the sample drying to 7.52 Å after 3 h, and finally to 7.50 Å after 1 day, not changing anymore at more prolonged ageing (Fig. 3c). The (002) peak showed a complex structure with at least three separate components contributing to the peak shape (Fig. 3b). Similar to the (001) peak, the sample ageing resulted in a general shift of the (002) peak to lower interplanar distances complicated by a redistribution of intensities of the constituting components. Such evolutions of the (001) and (002) peaks were observed for all tested samples and accelerated under heating and under a nitrogen flow directed to the sample surface. This indicates that the most probable reason for the structural evolution of the samples was gradual evaporation of water intercalated between layers together with NEt4OH.
A very similar dynamic behavior was reported for graphite oxide, which showed an evolution of the interplanar distance upon drying and re-intercalation of water,42–44 as well as other molecules, such as CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 45 or aliphatic alcohols.42 A number of tetraalkylammonium cations were reported to be intercalated into graphite oxide in a hydrated form, with the size of hydrated NEt4+ cation estimated to be 6.5–7.5 Å.46 Graphite oxide typically shows continuous shifts of the (002) peak with no separate components distinguishable, most probably due to a high structural inhomogeneity of this material.42,43 Intercalation of water was also reported for poly(triazineimide) resulting in an expansion of the interplanar (002) distance from 3.23 Å to 3.29 Å,22,47 which matches the range of changes observed in the present work for the (002) peak of the drying SLCN-NEt4OH intercalate. When heated, the poly(triazineimide) lost water and the gallery height was restored to the original value indicating the total reversibility of water intercalation.22,47
45 or aliphatic alcohols.42 A number of tetraalkylammonium cations were reported to be intercalated into graphite oxide in a hydrated form, with the size of hydrated NEt4+ cation estimated to be 6.5–7.5 Å.46 Graphite oxide typically shows continuous shifts of the (002) peak with no separate components distinguishable, most probably due to a high structural inhomogeneity of this material.42,43 Intercalation of water was also reported for poly(triazineimide) resulting in an expansion of the interplanar (002) distance from 3.23 Å to 3.29 Å,22,47 which matches the range of changes observed in the present work for the (002) peak of the drying SLCN-NEt4OH intercalate. When heated, the poly(triazineimide) lost water and the gallery height was restored to the original value indicating the total reversibility of water intercalation.22,47
The complex structure of the (002) peak found here for the intercalated restacked SLCN was not reported before for this kind of materials and requires a separate investigation. Similarly, complex XRD patterns were reported for the electrochemical intercalation of tetraoctylammonium cations into graphite,48 where an increase in the applied voltage resulted in the conversion of the graphite (002) peak into a series of multiple peaks separated by ca. 0.3 Å corresponding to multinary stage intercalates.48
The dynamics of the intercalate formation from colloidal SLCN solutions can be tracked by monitoring the XRD pattern of a drying drop of the colloidal solution periodically. Fig. 3d shows a collection of XRD patterns recorded each 2 min from a drying drop of the SLCN-600-p colloid. No changes were observed during the first tens of minutes (curves 1–8) indicating that an over-saturation of the solution should be reached prior to the intercalate starting to form. Then a small and broadened peak appears at 3.23–3.24 Å (curves 9–12) corresponding to the interplanar (002) distance of the original bulk GCN. We assign this peak to the formation of a primary non-intercalated product from SLCN sheets, which are non-stabilized or under-stabilized by NEt4OH. The Raman data discussed above showed that both adsorbed and free NEt4+ cations are present in the purified SLCN colloid supporting that the presence of non-stabilized or under-stabilized SLCN sheets can be expected due to NEt4+ adsorption/desorption equilibria. This non- (under-)stabilized species are expected to be the least stable and precipitate first due to the drying-induced oversaturation forming a primary non-intercalated deposit.
In the following course of drying an additional feature at 3.78 Å appears along with the peak at 3.23–3.24 Å, which can be assigned to the interplanar (002) distance of a SLCN–NEt4OH intercalate (Fig. 3d, curve 13). Then, a rather abrupt and pronounced change of the XRD pattern occurs and distinct and relatively intense (001) and (002) peaks of the intercalate start to dominate the diffractogram (curve 14). From this moment on, the shape of the XRD pattern is not changing considerably any more indicating that the formation of stage-1 intercalate is the dominating process in the system.
The contours of freshly-formed (001) and (002) peaks (Fig. 3d, curves 14–19) differ from those observed for aged intercalates (Fig. 3b and c), most probably due to differences in the amount of intercalated water as well as due to unequal conditions of the acquisition of XRD patterns (high acquisition rate for monitoring of the drop drying, high integration time for the inspection of the aged intercalates). We note also that the primary intercalate (curve 14) is characterized by larger (001) and (002) distances than those of the intercalate formed later (curves 15–19). This fact can be accounted for by the loose character of the primary intercalate with much more water present in the interlayer galleries. As the total mass of the intercalated deposit increases this excess water is, most probably, “squeezed out” from the galleries leaving only water belonging to the solvation shell of NEt4+, which is then gradually eliminated during the sample drying as discussed above for Fig. 3b and c.
The self-assembly into a layered intercalate observed in the present system can open new general synthetic approaches to complex materials by introducing other intercalating species and inducing spontaneous self-assembly, thus forming intercalates in a “bottom-to-top” approach. As discussed above, this bottom-to-top self-assembly was observed for all NEt4OH-stabilized SLCN samples exfoliated from GCN precursors, which were, in turn, synthesized at different temperatures and durations of polycondensation. The general character of the self-assembly means that simultaneously with the variation of the intercalate molecules, we can tailor the composition and structure of the layered polyheptazine host leading to a very broad variability of the functional properties of final products. Similar approaches are currently developed for other few-layer GCN-based materials. For example, the self-assembly of GCN sheets modified with iron-polyoxometalates allowed membranes for water purification to be designed.49
Tracking of the SLCN self-assembly by FTIR
Similar to Raman spectra, an analysis of FTIR spectra in the range of NEt4+ vibrations allows the most probable conformation of the cation to be identified. In this way, NEt4+ can be used as a marker providing information on the self-assembly processes in SLCN colloids. For this analysis we focus on a section of the FTIR spectra at 900–1250 cm−1 (Fig. 4a), where distinct CH3 rocking vibrations of the NEt4+ cation at about 1000 cm−1 and 1175 cm−1 can be observed,31,50,51 while neither GCN nor SLCN show own vibrations in this range. As discussed above for Raman spectral data, the NEt4+ cation has two distinct conformations, a more planar tt.tt conformation with a tetragonal symmetry and a more bulky tg.tg conformation with a cubic symmetry. The shape of the CH3 rocking vibrations in the IR spectra of NEt4+ salts was reported to depend on the cation conformation.50,51 In particular, for the cubic form the CH3 rocking vibrations at ca. 1000 cm−1 and at 1175 cm−1 were reported to be present as single peaks, while for the more planar tetragonal form these features were reported to split into two bands.50,51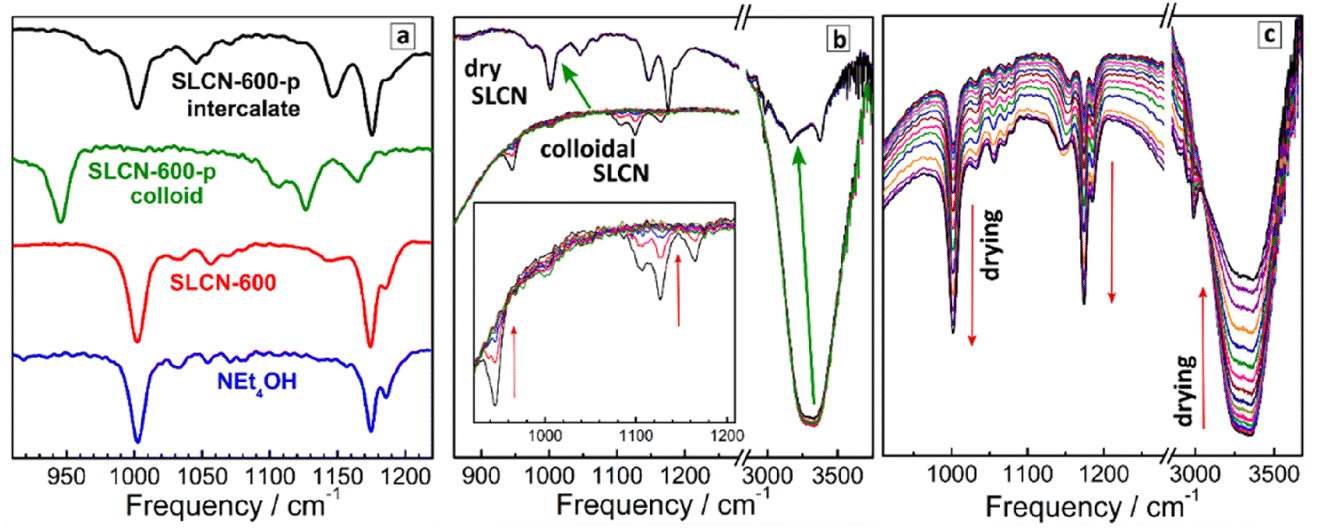 | ||
| Fig. 4 (a) ATR FTIR spectra of an aqueous solution of NEt4OH, non-purified colloidal SLCN-600 (identical spectra for colloidal solution and dry residue), purified SLCN-600-p colloid, and intercalate produced by drying of colloidal SLCN-600-p. (b and c) ATR FTIR spectra of aqueous colloidal SLCN-600-p solution (b) and aqueous solution of NEt4OH (c) measured in 15 s intervals between each spectrum in the course of drying of a solution drop (3 μL) on the diamond surface of the ATR setup. Inset in (b) shows an enlarged 900–1200 cm−1 section of the spectra. | ||
Fig. 4a shows that both CH3 rocking bands of pure NEt4OH appear as single peaks at 1002 cm−1 and 1175 cm−1 with a small satellite at 1185 cm−1. The FTIR spectrum of non-purified SLCN-600, both in the form of the original colloidal solution and after solvent evaporation, is very similar to that of pure NEt4OH. At the same time, the FTIR spectrum of purified SLCN-600 colloid is distinctly different from both pure NEt4OH and non-purified SLCN-600 (Fig. 4a). First, both CH3 rocking modes are shifted to lower frequencies, most probably indicating a strong adsorption of residual NEt4+ cations in the purified colloid to the SLCN sheets affecting the geometry of the cation and the energy of vibrations. Secondly, the original feature at 1175 cm−1 appears splitted into two bands at 1125 cm−1 and 1165 cm−1. From this fact we can conclude that the adsorbed NEt4+ cation is predominantly present in the more planar tetragonal conformation, which is in agreement with the above-discussed Raman data.
The FTIR spectrum of a dried SLCN-600-p sample (Fig. 4a), in turn, appears to have an intermediate shape between free NEt4+ in NEt4OH and adsorbed NEt4+ cation in the original purified colloid. Both CH3 rocking bands are split into two components (1175/1145 cm−1 and 1002/1046 cm−1), however, the bands are shifted to higher frequencies and come closer to the frequencies of pure NEt4OH. From these observations it can be assumed that the NEt4+ cations intercalated between layers remain mostly in the planar tetragonal conformation but, being placed between two SLCN layers, experience a lower influence from each of the layers and show vibration frequencies closer to those of the free tetraethylammonium cation.
Similar to the above-discussed XRD data, we followed the formation of the SLCN–NEt4OH intercalate by drying a drop of purified colloidal SLCN-600-p on the surface of a diamond ATR accessory and periodically (each 15 s) taking FTIR spectra of the drying droplet (Fig. 4b). As a reference we performed a similar procedure with a droplet of pure aqueous NEt4OH solution, for which drying is accompanied by a constant growth of all NEt4+-related bands and a decrease in the intensity of water features at 3000–3500 cm−1 (Fig. 4c).
In contrast, almost no changes can be observed during the drying of purified SLCN-600-p colloid till a critical point, at which the FTIR spectrum changes abruptly and in a pronounced way (the transition shown by green arrows in Fig. 4b). In this critical point the FTIR spectrum typical for colloidal SLCN converts into the FTIR spectrum of the dried intercalate and no further changes can be observed at longer drying times. Simultaneously with this spectral transition the intensity and shape of water-related features change abruptly as well, most probably, due to shielding of residual water from the diamond surface by the deposited intercalate. The abrupt character of the observed changes indicates that the formation of the intercalate requires a certain threshold over-saturation of the solution and is similar to a phase transition. A gradual and small decrease of the intensity of the vibration bands of both strongly adsorbed NEt4+ and water observed before the critical point can be explained by the deposition of non-intercalated layered polyheptazine, as revealed by XRD tracking, which can also shield the ATR diamond from the colloidal solution.
Tracking of the SLCN self-assembly by AFM
We found that the character of interaction of SLCN sheets with the mica substrates observed for purified colloidal samples differs drastically from that discussed above for non-purified SLCN colloids. In particular, drying of the purified SLCN colloids on mica results in the formation of Voronoi tessellation-like periodic networks with a hexagonal symmetry (exemplified in Fig. 5a and b for SLCN-600-p and SLCN-700-p). The self-assembly of SLCN sheets into hexagonally shaped networks occurs in relatively large sample areas of hundreds of μm2 showing roughly the same shape and size of elemental hexagonal units for different samples, but different thicknesses of “walls” depending on the history of the original SLCN colloids (Fig. 5c). This self-assembly effects have a general character for colloidal SLCN samples produced at different conditions of the synthesis of bulk GCN and different exfoliation conditions (ESI, Fig. S13†) allowing the chemical composition of the single layers of the intercalate to be tailored at different steps of the production of final intercalates. | ||
| Fig. 5 AFM images (a and b) of SLCN-600-p (a) and SLCN-700-p (b) samples on mica. (c) Particle height distributions calculated from panoramic AFM images for SLCN samples produced from bulk GCN synthesized at 400 °C, 1 h calcination (bars 1), 600 °C, 1 h (2), 700 °C, 1 h (3), and 700 °C, 2 h (4). (d) Exemplary height profiles for the SLCN-700-p sample plotted along the lines shown in (b). | ||
When compared to conventional Voronoi patterns showing a distribution of various polygons, the SLCN self-assemblies reveal a distinct tendency to hexagonal symmetry, which can have multiple origins, including (i) inherent symmetry of polyheptazine sheets and (ii) directing influence of the mica substrates. In the first case, one can expect translation of the inner hexagonal symmetry of the heptazine building blocks of SLCN sheets to a larger scale due to specific interlayer interaction. This explanation can be supported by examining the structure of the “walls” of the hexagonal SLCN self-assembly. Profiling through the “fingers” forming “walls” of the patterns (profile 1 in Fig. 5b) shows them to be of equal height of ca. 9 Å corresponding to double-layer sheets. At the same time, profiling through the nodes of the hexagons (profile 2 in Fig. 5b) shows that the middle part of the wall between two nodes is thinner than the nodes themselves, in the present case by a single period of 4.5 Å. This geometry implies that the walls are formed by crossed ribbons formed from SLCN sheets resulting in the nodes being thicker than the ribbons. Then, the hexagonal symmetry of the observed self-assembled structures can be naturally explained if a specific angle of 120° is assumed to be the most beneficial for the interaction between SLCN layers.
Similar self-directing effects are typically observed for GCN nano-/micro-tubes formed by the polycondensation of supramolecular precursor assemblies.52,53 It is assumed that such GCN tubes are formed from hexagonally shaped rods produced by stacking of the hexagonally shaped supramolecular precursor complexes.53 In this way, the hexagonal symmetry of the precursor molecules forming the supramolecular complexes is translated to the hexagonal shape of the final GCN tubes at the nano-/micro-level.52 Similar structure-directing effects can also be assumed to take place in the present system.
Alternatively, a directing influence of mica substrate can be inferred to explain the formation of periodic hexagonal patterns. For example, some DNA molecules were reported to self-assemble on the mica surface into hexagonal networks with no specific interactions between single DNA molecules, which could explain such geometry.54 Many other examples of hexagonal self-assemblies were reported and attributed to specific interactions between the adsorbate and mica, the latter having hexagonal lattice symmetry. The scope of such adsorbates includes Pd nanowires,55 ribbon-like 1D coordination polymers,56 and peptide nanorods,57–60 and collagen fibrils.61 Similar effects were also observed by AFM for other atomically-smooth surfaces, such as highly-oriented pyrolitic graphite, which induced hexagonal self-assembly of heptanoic acid.62 In the latter case the hexagonal patterns were formed both due to oriented pre-assembly of the solute and the templating effect of the substrate surface.
The validity of the assumption on the templating role of mica surface could be verified by an AFM observation of the shape of SLCN formations on other substrates. However, the substrates tested by us so far, in particular, glass, polished silicon, as well as thermally evaporated gold and platinum films, did not provide sufficient smoothness for measurements comparable with those made on the freshly-cleaved mica.
As discussed above, the thickness of the walls in the hexagonal self-assembly of SLCN on mica depends both on the history of samples (type of bulk precursor) and on the concentration of the colloidal solution deposited on the mica substrate. In particular, for the same SLCN sample the thickness of the walls forming hexagonal patterns was found to be directly proportional to the SLCN concentration (Fig. 6; ESI, Fig. S14†). At that, the patterns retain the characteristic hexagonal symmetry.
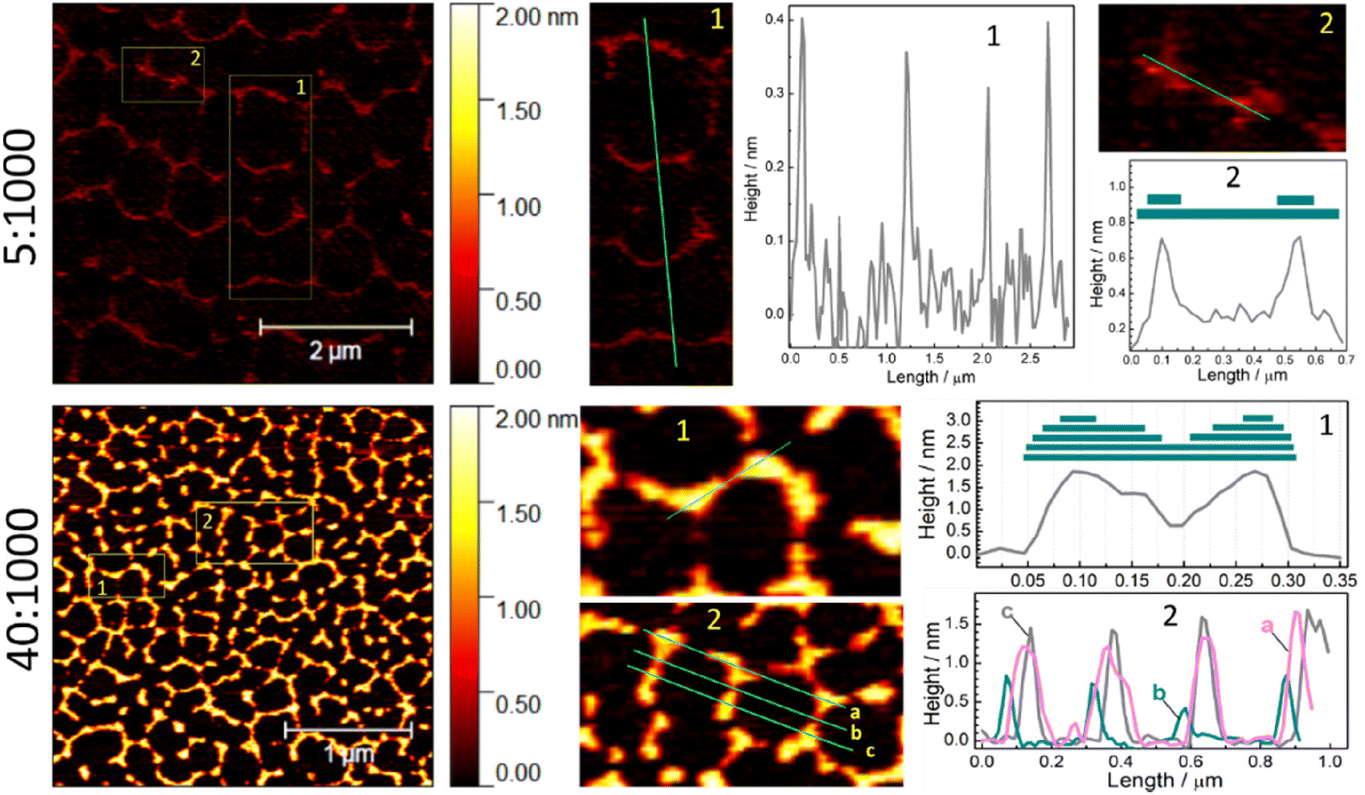 | ||
| Fig. 6 AFM images and roughness profiles for two samples of SLCN-700-p on mica produced at two different dilutions of the original colloid. Areas 1 and 2 are presented as enlarged images, lines show the direction of profiles presented in the graphs. | ||
In the case of the lowest tested SLCN concentration (dilution of 5:1000, Fig. 6), very thin periodic formations can be observed in the AFM images, which already show a hexagonal motif with some of the walls and sections missing due to the lack of the “building” material. Profiling through the walls of such self-assemblies (see section 1 and profile 1 for the case of 5:1000 in Fig. 6) shows them to be single-layer polyheptazine sheets with a thickness of 3.5–4 Å. Profiling through two nodes (section 2 and profile 2 for the case of 5:1000, Fig. 6) shows two-layer formations, which is in accordance with the above-proposed mechanism of the formation of self-assembly via overlapping of two (several) SLCN ribbons at a certain angle.
Qualitatively the same situation is observed for the samples produced from more concentrated samples (dilution of 40:1000, Fig. 6). Here we also observe that the nodes are thicker than walls between them (section 1 and profile 1 for the case of 40:1000, Fig. 6) indicating that the oriented overlapping of SLCN ribbons can be the driving force for the self-assembly. Profiling through several periodicities of the hexagonal network (section 2 and profiles 2 for the case of 40:1000, Fig. 6) shows that nodes and walls are of the same thickness throughout the entire network. Similar conclusions can also be drawn from the profiling of a set of samples with 5 different dilutions (10:1000–70:1000, ESI, Fig. S14†). Overall, the wall thickness was found to increase from ca. 4 Å for the minimal dilution tested (5:1000) to ca. 18 Å for 70:1000, the latter corresponding to four-layer walls, while the hexagonal symmetry of the self-assembled network is preserved in all cases.
The dependence of the geometry of SLCN patterns on the concentration can be regarded as a combination of two self-assembly effects found and discussed in the present work. On one side, the SLCN sheets self-assemble on mica into regular hexagonal patterns. On the other side, the walls of the hexagonal network are formed due to the stacking self-assembly of SLCN sheets and the formation of intercalates with NEt4OH discussed in the previous sections. Therefore, by exploiting both self-assembly effects we potentially can deposit SLCN sheets onto mica in a sequential manner and grow an array of closely-packed vertically-aligned hexagonal wells and even tubes, their depth tailored with a precision of a single polyheptazine layer.
Conclusions
We report on two new effects of self-assembly of single-layer polyheptazine carbon nitride (SLCN) sheets, which open perspectives for bottom-to-top design of novel layered functional materials. First, the SLCN sheets spontaneously form layered stage-1 intercalates of tetraethylammonium hydroxide when an aqueous colloidal SLCN solution stabilized by NEt4OH is left drying in ambient conditions. The dynamics of the intercalate formation was followed by in situ XRD allowing several steps of this process to be discriminated, including the abrupt start of the self-assembly due to over-saturation of the colloidal SLCN solution, deposition of a primary “wet” intercalate of hydrated NEt4OH and the gradual elimination of excessive water during the ambient drying of the freshly deposited intercalate. At that, the NEt4+ cations are intercalated in a preferred flattened conformation having specific vibrational properties, allowing the formation of intercalate to be tracked by FTIR and Raman spectroscopy using the tetraethyl-ammonium cation as a spectral probe to get insights in the dynamics of self-assembly and the structure of final intercalates.These self-assembly effects have a general character for colloidal SLCN samples produced using NEt4OH as an exfoliating agent at different conditions of the synthesis of the original bulk graphitic carbon nitride (GCN) and different exfoliation conditions, allowing the chemical composition of the single layers of the intercalate to be tailored at different steps of the production of final intercalates. We expect that other molecules and ionic species can also be incorporated into intercalates along with NEt4OH, if introduced into SLCN colloids prior to the self-assembly. Such co-intercalation can potentially be used to further functionalize the SLCN self-assemblies, however, the feasibility of this approach is still to be proven.
Secondly, when dried on freshly-cleaved mica, the SLCN sheets form layered hexagonally-shaped networks, which can be observed in relatively large areas of the substrate (hundreds of μm2). These self-assembly effects can be driven both by the translation of internal hexagonal symmetry of heptazine units to a larger micro-scale and by a templating effect specific for mica surfaces. Similar to the formation of intercalates, the 2D self-assembly on mica has a general character for SLCN sheets produced by the NEt4OH-driven exfoliation from different GCNs allowing the chemistry of the “walls” of the hexagonal self-assemblies to be tuned via changing the properties of the bulk precursors. The thickness of the “walls” can be easily controlled by the SLCN content in the solution deposited on mica. The scope of substrates for the SLCN self-assembly reported in the present work is limited to glass and mica, but we expect these self-assembly phenomena to be of a general character for other polar surfaces.
By combining both self-assembly effects we can potentially design new ordered coatings showing a long-range geometrical regularity and a broad tunability of the chemical properties of layered “walls” forming the hexagonal patterns. The colloidal SLCN species stabilized by NEt4OH reveal highly intense broad-band photoluminescence,28 photochemical activity,63 and the capability of charge transfer when combined with semiconductor27,64 and metal27,65 nanocrystals. These functional properties are expected to be preserved in the final intercalates and 2D self-assemblies making such materials promising for light-conversion applications.
Author contributions
O. Stroyuk: conceptualization (lead), investigation (equal), writing – original draft preparation (lead); O. Raievska: investigation (lead), methodology (lead); C. J. Brabec: conceptualization (equal), funding acquisition (equal), writing – review & editing (equal); V. Dzhagan: investigation (equal), writing – review & editing (equal); Y. Havryliuk: investigation (equal), writing – review & editing (equal); D. R. T. Zahn: conceptualization (equal), funding acquisition (lead), writing – review & editing (equal).Conflicts of interest
The authors declare no conflicts of interests.Acknowledgements
Prof. D. R. T. Zahn and O. Raievska acknowledge financial support by the Deutsche Forschungsgemeinschaft (DFG project ZA-146/45-1). V. Dzhagan thanks the Visiting Scholar Program for funding his research stay at TU Chemnitz. Ye. Havryliuk is grateful to the Alexandr von Humboldt Foundation for the financial support of his research stay at the Chemnitz University of Technology.Notes and references
- Y. Li, Y. Lu, P. Adelhelm, M.-M. Titirici and Y.-S. Hu, Chem. Soc. Rev., 2019, 48, 4655 RSC.
- M. S. Stark, K. L. Kuntz, S. J. Martens and S. C. Warren, Adv. Mater., 2019, 31, 1808213 CrossRef PubMed.
- M. Laipan, L. Xiang, J. Yu, B. R. Martin, R. Zhu, J. Zhu, H. He, A. Clearfield and L. Sun, Prog. Mater. Sci., 2020, 109, 100631 CrossRef CAS.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906 CrossRef CAS PubMed.
- J. M. Allen, T. C. Vincent and K. B. Richard, Chem. Rev., 2010, 110, 132 CrossRef PubMed.
- A. Ambrosi, C. K. Chua, A. Bonanni and M. Pumera, Chem. Rev., 2014, 114, 7150 CrossRef CAS PubMed.
- A. C. Ferrari, F. Bonaccorso, V. Fal'ko, K. S. Novoselov, S. Roche, P. Bøggild, S. Borini, F. H. L. Koppens, V. Palermo and N. Pugno, et al. , Nanoscale, 2015, 7, 4598 RSC.
- A. M. Abdelkader, A. J. Cooper, R. A. W. Dryfe and I. A. Kinloch, Nanoscale, 2015, 7, 6944 RSC.
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596 CrossRef CAS.
- J. Zhu, P. Xiao, H. Li and S. A. C. Carabineiro, ACS Appl. Mater. Interfaces, 2014, 6, 16449 CrossRef CAS PubMed.
- J. Liu, H. Wang and M. Antonietti, Chem. Soc. Rev., 2016, 45, 2308 RSC.
- T. S. Miller, A. B. Jorge, T. M. Suter, A. Sella, F. Corà and P. F. McMillan, Phys. Chem. Chem. Phys., 2017, 19, 15613 RSC.
- A. Savateev, I. Ghosh, B. König and M. Antonietti, Angew. Chem., Int. Ed., 2018, 57, 15936 CrossRef CAS PubMed.
- M. Volokh, G. Peng, J. Barrio and M. Shalom, Angew. Chem., Int. Ed., 2019, 58, 6138 CrossRef CAS PubMed.
- L. Lin, Z. Yu and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 6164 CrossRef CAS PubMed.
- Y. Shen, A. J. Dos santos-Garcia and M. J. Martín de Vidales, Processes, 2020, 9, 66 CrossRef.
- H. Gao, S. Yan, J. Wang, Y. A. Huang, P. Wang, Z. Li and Z. Zou, Phys. Chem. Chem. Phys., 2013, 15, 18077 RSC.
- Z. Zhang, J. Huang, Q. Yuan and B. Dong, Nanoscale, 2014, 6, 9250 RSC.
- S. Y. Chong, J. T. A. Jones, Y. Z. Khimyak, A. I. Cooper, A. Thomas, M. Antonietti and M. J. Bojdys, J. Mater. Chem. A, 2013, 1, 1102 RSC.
- Z. Teng, H. Lv, C. Wang, H. Xue, H. Pang and G. Wang, Carbon, 2017, 113, 63 CrossRef CAS.
- H. Zhang, S. O. Park, J. H. Seo, S. H. Joo, M. Huang, C. Wang, M. Wang, S. K. Kwak and R. S. Ruoff, J. Phys. Chem. C, 2021, 125, 9947 CrossRef CAS.
- T. M. Suter, T. S. Miller, J. K. Cockcroft, A. E. Aliev, M. C. Wilding, A. Sella, F. Corà, C. A. Howard and P. F. McMillan, Chem. Sci., 2019, 10, 2519 RSC.
- H. Xu, J. Yan, X. She, L. Xu, J. Xia, Y. Xu, Y. Song, L. Huang and H. Li, Nanoscale, 2014, 6, 1406 RSC.
- P. Yang, H. Ou, Y. Fang and X. Wang, Angew. Chem., Int. Ed., 2017, 56, 3992 CrossRef CAS PubMed.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150 CrossRef CAS PubMed.
- J. Zhang, Y. Chen and X. Wang, Energy Environ. Sci., 2015, 8, 3092 RSC.
- O. Stroyuk, O. Raievska and D. R. T. Zahn, Phys. Chem. Chem. Phys., 2021, 23, 20745 RSC.
- Ya. V. Panasiuk, A. E. Raevskaya, O. L. Stroyuk, P. M. Lytvyn and S. Ya. Kuchmiy, RSC Adv., 2015, 5, 46843 RSC.
- O. L. Stroyuk, Ya. V. Panasiuk, A. E. Raevskaya and S. Ya. Kuchmiy, Theor. Exp. Chem., 2015, 51, 243, DOI:10.1007/s11237-015-9423-9.
- Z. Zhou, Y. Zhang, Y. Shen, S. Liu and Y. Zhang, Chem. Soc. Rev., 2018, 47, 2298 RSC.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies (Tables and Charts), John Wiley & Sons, 3rd edn, 2001 Search PubMed.
- O. Stroyuk, O. Raievska, D. Solonenko, C. Kupfer, A. Osvet, M. Batentschuk, C. J. Brabec and D. R. T. Zahn, RSC Adv., 2021, 11, 21145 RSC.
- C. Naudin, F. Bonhomme, J. L. Bruneel, L. Ducasse, J. Grondin, J. C. Lassegues and L. Servant, J. Raman Spectrosc., 2000, 31, 979 CrossRef CAS.
- T. Takekiyo and Y. Yoshimura, Chem. Phys. Lett., 2006, 420, 8 CrossRef CAS.
- J. E. Schmidt, D. Fu, M. W. Deem and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 16044 CrossRef CAS PubMed.
- T. Ikuno, W. Chaikittisilp, Z. Liu, T. Iida, Y. Yanaba, T. Yoshikawa, S. Kohara, T. Wakihara and T. Okubo, J. Am. Chem. Soc., 2015, 137, 14533 CrossRef CAS PubMed.
- H. V. Brand, L. A. Curtiss, L. E. Iton, F. R. Trouw and T. O. Brun, J. Phys. Chem., 1994, 98, 1293 CrossRef CAS.
- W. Sirisaksoontorn, A. A. Adenuga, V. T. Remcho and M. M. Lerner, J. Am. Chem. Soc., 2011, 133, 12436 CrossRef CAS PubMed.
- T. Maluangnont, M. M. Lerner and K. Gotoh, Inorg. Chem., 2011, 50, 11676 CrossRef CAS PubMed.
- W. Sirisaksoontorn and M. M. Lerner, Carbon, 2014, 69, 582 CrossRef CAS.
- W. Sirisaksoontorn and M. M. Lerner, Inorg. Chem., 2013, 52, 7139 CrossRef CAS PubMed.
- A. J. Cooper, M. Velický, I. A. Kinloch and R. A. W. Dryfe, J. Electroanal. Chem., 2014, 730, 34 CrossRef CAS.
- A. V. Talyzin, S. M. Luzan, T. Szabó, D. Chernyshev and V. Dmitriev, Carbon, 2011, 49, 1894 CrossRef CAS.
- A. Lerf, A. Buchsteiner, J. Pieper, S. Schöttl, I. Dekany, T. Szabo and H. P. Boehm, J. Phys. Chem. Solids, 2006, 67, 1106 CrossRef CAS.
- D. Kim, D. W. Kim, H. K. Lim, J. Jeon, H. Kim, H. T. Jung and H. Lee, J. Phys. Chem. C, 2014, 118, 11142 CrossRef CAS.
- Z. H. Liu, Z. M. Wang, X. Yang and K. Ooi, Langmuir, 2002, 18, 4926 CrossRef CAS.
- F. Foglia, A. J. Clancy, J. Berry-Gair, K. Lisowska, M. C. Wilding, T. M. Suter, T. S. Miller, K. Smith, F. Demmel, M. Appel, V. G. Sakai, A. Sella, C. A. Howard, M. Tyagi, F. Corà and P. F. McMillan, Sci. Adv., 2020, 6, eabb6011 CrossRef CAS PubMed.
- Z. Chen, D. Guo, L. Si and G. Xie, Scanning, 2017, 9438573 Search PubMed.
- H. Lan, F. Wang, M. Lan, X. An, H. Liu and J. Qu, Environ. Sci. Technol., 2019, 53, 6981 CrossRef CAS PubMed.
- W. H. J. de Beer and A. M. Heyns, Spectrochim. Acta, Part A, 1981, 37, 1099, DOI:10.1016/0584-8539(81)80156-0.
- W. H. J. De Beer, A. M. Heyns, P. W. Richter and J. B. Clark, J. Solid State Chem., 1981, 36, 171 CrossRef CAS.
- O. Stroyuk, O. Raievska and D. R. T. Zahn, RSC Adv., 2020, 20, 34059 RSC.
- J. Barrio and M. Shalom, ChemCatChem, 2018, 10, 5573 CrossRef CAS.
- S. Hamada and S. Murata, Angew. Chem., 2009, 121, 6952 CrossRef.
- Z. Wang, T. Kong, K. Zhang, H. Hu, X. Wang, J. Hou and J. Chen, Mater. Lett., 2007, 61, 251 CrossRef CAS.
- Q. Evrard, G. Cucinotta, F. Houard, G. Calvez, Y. Suffren, C. Daiguebonne, O. Guillou, A. Caneschi, M. Mannini and K. Bernot, Beilstein J. Nanotechnol., 2019, 10, 2440, DOI:10.3762/bjnano.10.234.
- B. Dai, S. G. Kang, T. Huynh, H. Lei, M. Castelli, J. Hu, Y. Zhang and R. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8543 CrossRef CAS PubMed.
- F. Zhang, H. N. Du, Z. X. Zhang, L. N. Ji, H. T. Li, L. Tang, H. Bin Wang, C. H. Fan, H. J. Xu, Y. Zhang, J. Hu, H. Y. Hu and J. H. He, Angew. Chem., Int. Ed., 2006, 45, 3611 CrossRef CAS PubMed.
- H. Li, F. Zhang, Y. Zhang, M. Ye, B. Zhou, Y. Z. Tang, H. J. Yang, M. Y. Xie, S. F. Chen, J. H. He, H. P. Fang and J. Hu, J. Phys. Chem. B, 2009, 113, 8795 CrossRef CAS PubMed.
- S. Kang, H. Li, T. Huynh, F. Zhang, Z. Xia, Y. Zhang and R. Zhou, ACS Nano, 2012, 6, 9276 CrossRef CAS PubMed.
- W. W. Leow and W. Hwang, Langmuir, 2011, 27, 10907 CrossRef CAS PubMed.
- G. Velpula, C. Martin, B. Daelemans, G. Hennrich, M. Van der Auweraer, K. S. Mali and S. De Feyter, Chem. Sci., 2021, 12, 13167 RSC.
- O. Stroyuk, Y. Panasiuk, A. Raevskaya and S. Kuchmiy, Phys. Status Solidi B, 2019, 256, 1800279 CrossRef.
- O. L. Stroyuk, A. E. Raevskaya, Y. V. Panasiuk, V. F. Plyusnin, V. M. Dzhagan, S. Schulze and D. R. T. Zahn, FlatChem, 2017, 2, 38 CrossRef CAS.
- O. Stroyuk, A. Raevskaya, G. Grodzyuk, N. Andriushina, M. Skoryk, V. Yefanov, S. Schulze and D. R. T. Zahn, RSC Adv., 2019, 9, 444, 10.1039/C8RA09454C.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the synthesis of bulk GCNs and colloidal SLCNs; details of spectral and structural characterization of the samples; XRD, XPS, and FTIR data as well as PL spectra for GCN samples produced in different conditions; distributions of hydrodynamic size, XPS data, absorption and PL spectra for different colloidal SLCN samples; AFM images of clean mica and a NEt4OH layer on mica; AFM images and height distributions for SLCN samples produces from different GCNs; XRD of a set of intercalates produced by restacking of different samples of SLCN; AFM images of self-assemblies produced from different SLCN purified samples as well as of SLCN-700-p sample at different dilution. See DOI: https://doi.org/10.1039/d2nr03477h |
| This journal is © The Royal Society of Chemistry 2022 |