
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral non-stoichiometric ternary silver indium sulfide quantum dots: investigation on the chirality transfer by cysteine†
Lorenzo
Branzi
a,
Finn
Purcell-Milton
 bd,
Chiara
Cressoni
c,
Michele
Back
a,
Elti
Cattaruzza
a,
Adolfo
Speghini
*c,
Yurii K.
Gun'ko
*b and
Alvise
Benedetti
*a
bd,
Chiara
Cressoni
c,
Michele
Back
a,
Elti
Cattaruzza
a,
Adolfo
Speghini
*c,
Yurii K.
Gun'ko
*b and
Alvise
Benedetti
*a
aDepartment of Molecular Science and Nanosystems, Ca’ Foscari University of Venice, Via Torino 155, Venezia Mestre, Italy. E-mail: benedett@unive.it
bSchool of Chemistry, CRANN and AMBER Research Centres, Trinity College Dublin, College Green, Dublin 2, Ireland. E-mail: igounko@tcd.ie
cNanomaterials Research Group, Department of Biotechnology and INSTM, RU of Verona, University of Verona, Strada le Grazie 15, Verona, Italy. E-mail: adolfo.speghini@univr.it
dSchool of Chemical & Pharmaceutical Sciences, Technological University Dublin, Grangegorman, Dublin 2, Ireland
First published on 1st August 2022
Abstract
Chiral semiconductor quantum dots have recently received broad attention due to their promising application in several fields such as sensing and photonics. The extensive work in the last few years was focused on the observation of the chiroptical properties in binary Cd based systems. Herein, we report on the first evidence of ligand-induced chirality in silver indium sulfide semiconductor quantum dots. Ternary disulfide quantum dots are of great interest due to their remarkable optical properties and low toxicity. Non-stoichiometric silver indium sulfide quantum dots were produced via a room temperature coprecipitation in water, in the presence of cysteine as a capping agent. The obtained nanocrystals show a notable photoluminescence quantum yield of 0.24 in water dispersions. Several critical aspects of the nanocrystal growth and chemico-physical characterization, and the optimisation of the surface passivation by the chiral ligand in order to optimize the nanoparticle chirality are thoroughly investigated. Optical spectroscopy methods such as circular dichroism and luminescence as well as nuclear magnetic resonance techniques are exploited to analyze the coordination processes leading to the formation of the ligand–nanocrystal chiral interface. This study highlights the dynamic nature of the interaction between the nanocrystal surface and the chiral ligand and clarifies some fundamental aspects for the transfer and optimization of the chiroptical properties.
Introduction
Chiral semiconductor Quantum Dots (QDs) are among the most investigated chiral nanomaterials due to their unique optical properties, characterized by a distinctive dichroic absorption or circular dichroism (CD) and circular polarized luminescence (CPL). These nanomaterials have been subjected to an extensive investigation during the past decade following the first observation of chiroptical activity in CdS quantum dots prepared by microwave-assisted synthesis in the presence of L- or D-penicillamine by Moloney et al.1 in 2007. The wide potential applications of this novel group of chiral nanomaterials motivated several investigations on other cadmium-based binary QDs in order to modulate the chiroptical properties and to study how the several structural parameters can affect the particle chirality. The investigations on CdSe2–5 and CdTe6–8 nanocrystals have revealed the dependence of the chiroptical properties on the size of the nanocrystals and the ligand type. The influence of 3D nanostructures such as CdS tetrapods,9 as well as the shell systems with type I and type II band alignments CdSe/CdS10–14 and CdS/CdSe10 has been investigated to define the effect of structural features such as nanocrystal shape and the thickness of the shell layers. Investigation on the shape anisotropy of CdSe and CdSe/CdS nanoplates revealed fundamental details on the chiral transfer in anisotropic nanostructures.15 Moreover, several theoretical models for the description of the chiroptical properties observed in different Cd-based QDs have been developed7,10,16,17 and some of the most considerable achievements in this flourishing research field are collected in specific reviews.15,18,19However, despite the wide number of impressive results regarding the chemistry of chiral quantum dots, all these studies are focused only on Cd-based chalcogenides and there is still a lack of investigations on other systems. To the best of our knowledge, this is the first report dealing with ligand-induced chirality in ternary I–III–VI type QDs.
Silver indium sulfide (AIS) QDs are characterized by a broad photoluminescence,20 a giant Stokes shift (>1.0 eV)21 and a broad absorption range.22 The possibility to modify the nanocrystals’ electronic properties (energy gap and photoluminescence quantum yield) according to the I/III metal ratio has stimulated several investigations on non-stoichiometric I–III–VI ternary nanocrystals. Similar to the observations reported on the copper indium sulfide nanocrystals,23,24 a blue-shift was observed for both the absorption and emission bands with the decrease of the I/III metal ratio. In the case of AIS nanocrystals, such an effect on the energy gap in the presence of a sub-stoichiometric Ag content was related to the lowering of the maximum levels of the valence band with the reduction of the contribution from Ag 3d orbitals.25 Moreover, the control on the stoichiometry is fundamental for the optimization of the photoluminescence quantum yield and several reports revealed the highest performances of QDs having In-rich non-stoichiometric compositions.25–29 The luminescence properties of AIS QDs, and especially their broad emission have been subjected to deep investigations; these intrinsic properties that are reported even in single particle measurements have been described by multiple mechanisms, such as the donor–acceptor pair mechanism,30 strong electron–phonon coupling20 and defects localized within the QD's volume.31 Due to these features, AIS QDs have been successfully employed in a broad range of applications such as solar concentrators,22 photoelectrochemical cells32 and as bioimaging probes.29,33
In this investigation we report on the first observation of chirality in I–III–VI QDs prepared via a low temperature approach. The analysis on the interaction between the chiral ligand and the QD's surface by Nuclear Magnetic Resonance (NMR) spectroscopy evidenced the role of dynamic coordination equilibria in the formation of the ligand–nanocrystal chiral interface. Our investigation highlights the critical role of the ligand chemisorption equilibrium in the transfer of chirality in AIS QDs. This observation reveals critical details that must be considered in the optimization of the chiroptical properties for ternary QDs.
Results and discussion
In order to optimize the optical properties of the QDs, in particular the luminescence efficiency, a Ag![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :In atomic ratio of 1:4 was chosen on the basis of the results reported by other authors in the literature.26–29 Cysteine-stabilized non-stoichiometric AIS QDs are directly produced by a facile two-step process at room temperature in water solution depicted in Fig. 1a. In the 1st step, a L- or D-cysteine (Cys) solution is added (see the Materials and methods section) to the metal precursor solution with pH adjusted to 8.5. Then, the QD formation is triggered by the addition of a solution of Na2S which acts as the sulfide source. Consequently, the colour of the reaction mixture turns rapidly from colourless to bright yellow, presumably due to the start of the nucleation process. In fact, for longer reaction times, the solution turns to an orange colour, that remains stable. The UV/Vis spectra presented in Fig. 1b show the rise of an absorption band located at around 350 nm, due to the nanocrystal formation. A constant absorbance of the solution is reached after 40 min from the Na2S addition, indicating the completion of the reaction.
:In atomic ratio of 1:4 was chosen on the basis of the results reported by other authors in the literature.26–29 Cysteine-stabilized non-stoichiometric AIS QDs are directly produced by a facile two-step process at room temperature in water solution depicted in Fig. 1a. In the 1st step, a L- or D-cysteine (Cys) solution is added (see the Materials and methods section) to the metal precursor solution with pH adjusted to 8.5. Then, the QD formation is triggered by the addition of a solution of Na2S which acts as the sulfide source. Consequently, the colour of the reaction mixture turns rapidly from colourless to bright yellow, presumably due to the start of the nucleation process. In fact, for longer reaction times, the solution turns to an orange colour, that remains stable. The UV/Vis spectra presented in Fig. 1b show the rise of an absorption band located at around 350 nm, due to the nanocrystal formation. A constant absorbance of the solution is reached after 40 min from the Na2S addition, indicating the completion of the reaction.
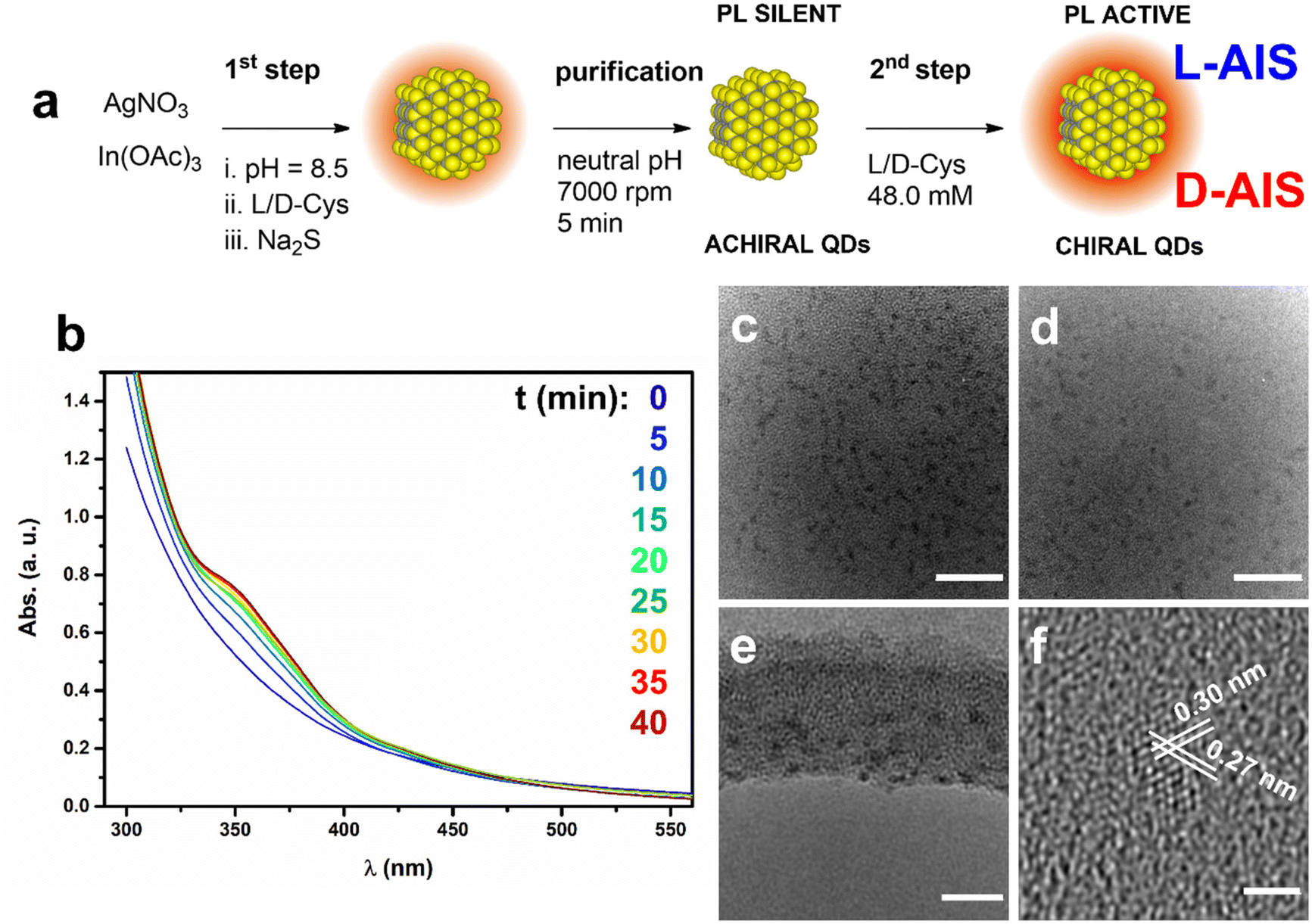 | ||
| Fig. 1 (a) Schematic representation of the synthesis. (b) UV/Vis analysis of the reaction mixture at different reaction times. (c) TEM micrographs of non-stoichiometric AIS QDs with scale bars: (c and d) = 20.0 nm, (e) = 10.0 nm and (f) = 2.0 nm. | ||
After the purification step, the AIS QDs are easily redispersed in water with the addition of an appropriate concentration of L- or D-Cys (see Fig. 1a). During the 2nd step of the synthesis, the nanocrystals are aged for 24 h at room temperature in a solution of the chiral ligand, L- or D-Cys, at a specific concentration. Then, the colloidal solution is diluted to a final concentration of 1.0 mg ml−1 with double distilled water and stored for further characterization. Here we refer to the QD sample prepared using enantiomerically pure L- or D-Cys in both 1st and 2nd steps of the synthesis as L-AIS or D-AIS, respectively.
The TEM images of the QDs after the 2nd step show the presence of nanocrystals with an average size of 2.1 nm ± 0.9 (Fig. 1c–f and Fig. S1†) and the phase contrast images reveal details on the crystallographic structure of the nanocrystals. Indeed, d-spacings of 0.30 and 0.27 nm are estimated by the analysis of the fringes produced by electron diffraction (Fig. 1f and Fig. S2†), related to the 311 and 400 facets of the cubic AIS phase. XPS elemental quantitative analysis is evaluated based on the peak area of the single signals (Fig. S3†) corrected by the elemental sensitivity factor and the relative atomic composition is estimated as follows: C 42.3%, O 18.4%, Ag 3.7%, N 4.6%, In 14.0%, and S 17.0%. The relative Ag/In metal ratio is 1/3.8, thus confirming the expected QD stoichiometry according to the molar ratio of the starting reaction mixture. The detailed comment on the XPS analysis (Fig. S3†) with the peak fit and the relative attributions are reported in the ESI section.†
Crystallographic information on the L- and D-AIS QDs has been gained by analysis of the XRD patterns, shown in Fig. 2a. The XRD patterns show diffractions centred at 2θ values of 27.8°, 44.0°, 47.5° and 53.5°, which can be related to the (311), (330), (440) and (620) crystallographic planes of the cubic AgIn5S8 phase, respectively. This cubic spinel phase is commonly observed in non-stoichiometric indium-rich AgInS2 nanocrystals,25,26 and previous investigations reported on the stabilization of this phase in a wide range of In/Ag stoichiometries.26,34–36 The interpretation of the XRD data is still limited by the large Full Width at Half Maximum (FWHM) of the XRD reflections which is related to the small nanoparticle size.
 | ||
| Fig. 2 Chemico-physical characterization of L-AIS (blue) and D-AIS (red) QDs: (a) XRD patterns; reference pattern: cubic AgIn5S8, ICSD 51619. (b) PLE and PL spectra of the QD solutions. (c and d) FTIR spectra of the QD powders and the reference spectra for zwitterionic L-Cys (black). | ||
The photoluminescence (PL) properties in the UV/visible optical range of colloidal QD dispersions stabilized with the L- or D-Cys ligand with 16.0 mM concentration have been investigated. The emission spectrum upon excitation at 450 nm (Fig. 2b) shows a broad emission peak centred at around 635 nm, with a large FWHM at around 160 nm (0.5 eV). Moreover, a PLQY of 0.24 was obtained for this colloidal dispersion (eqn (S1) and (S2)†). The PL excitation spectrum shows the presence of a shoulder at around 420 nm, and features at 360 nm and at around 322 nm. The PL decay curve of a L-AIS QD dispersion upon pulsed excitation at 453 nm is shown in Fig. S4.† From a fit of this decay curve with a triple exponential function (eqn (S3)†), an average lifetime of 518 ns was evaluated (see eqn (S4) and Table S1†), similar to that reported by other authors for non-stoichiometric AIS QDs.26,28,37 This value arises from different decay components that are a combination of size-dependent and defect-related de-excitation mechanisms.20
The presence of coordinated cysteine on the final AIS QDs has been investigated by the FTIR technique (Fig. 2c and d). The broad absorption band at around 3400 cm−1 can be attributed to the νNH N–H stretching vibration. The broad and intense band centred at 1616 cm−1 is ascribed to the overlap of the aνCO2− carboxylate asymmetric stretching and the aδNH2 asymmetric –NH2 bending indicated by a shoulder at around 1580 cm−1.38 The sharp bands at 1400 and 1340 cm−1 are related to the sνCO2− carboxylate symmetric stretching and sδNH2 –NH2 bending, respectively.38,39 The absorption bands between 1230 and 1020 cm−1 could be related to the δCN and δCH bending modes.40 The absence of a sharp peak at around 940 cm−1 related to the δSH mode indicates that the –SH thiol group is deprotonated and that sulphur is involved in surface metal coordination mainly as an anionic X-type ligand.12,41
The chiroptical properties of the QD colloidal solutions were analysed by UV-visible CD spectroscopy. The CD spectrum of L-AIS QDs (shown in Fig. 3) shows a negative band located at 420 nm and a positive one at 375 nm with an inversion point located at 400 nm. The observation of the chiroptical activity in the exciton region strongly confirms the transfer of chirality to the AIS nanocrystals. The opposite CD signal is observed for D-AIS confirming the isolation of the two enantiomers and the enantioselectivity of the synthesis. The analysis of the origin of the characteristic bisignated peak located in the exciton region presenting the typical shape of the Cotton effect was reported by Ben-Moshe et al.10 on chiral CdSe QDs. The authors related this typical spectroscopic feature to the splitting mechanism induced by the interaction of the chiral ligand with the QD's density of states, leading to the formation of two bands that are preferentially excited by alternating circular polarizations. However, the photoluminescence excitation spectra presented in Fig. 2b reveal the presence of a complex band structure in the proximity of the exciton position and further detailed investigation of the possible contribution of multiple electronic transitions must be considered to achieve a better understanding of the shape of the CD signal observed in ternary QDs. The analysis of the chiroptical properties in the UV region (200–300 nm) reveals the presence of another chiroptically active band located at 222 nm (see Fig. S5†), showing a positive sign for L-AIS and a negative one for D-AIS. The anisotropic g-factors of −0.92 × 10−4 and 0.63 × 10−4 can be calculated from the bands observed at 430 and 375 nm, respectively, for L-AIS. Similarly, for D-AIS, values of +0.93 × 10−4 and −0.58 × 10−4 can be calculated for the bands located at 430 and 375 nm, respectively. These chiroptical properties are in accordance with the observation reported by other authors for similar systems in which the chiroptical properties of isotropic QDs are originated by the induction of a chiral ligand (ligand-induced chirality).5,17,42
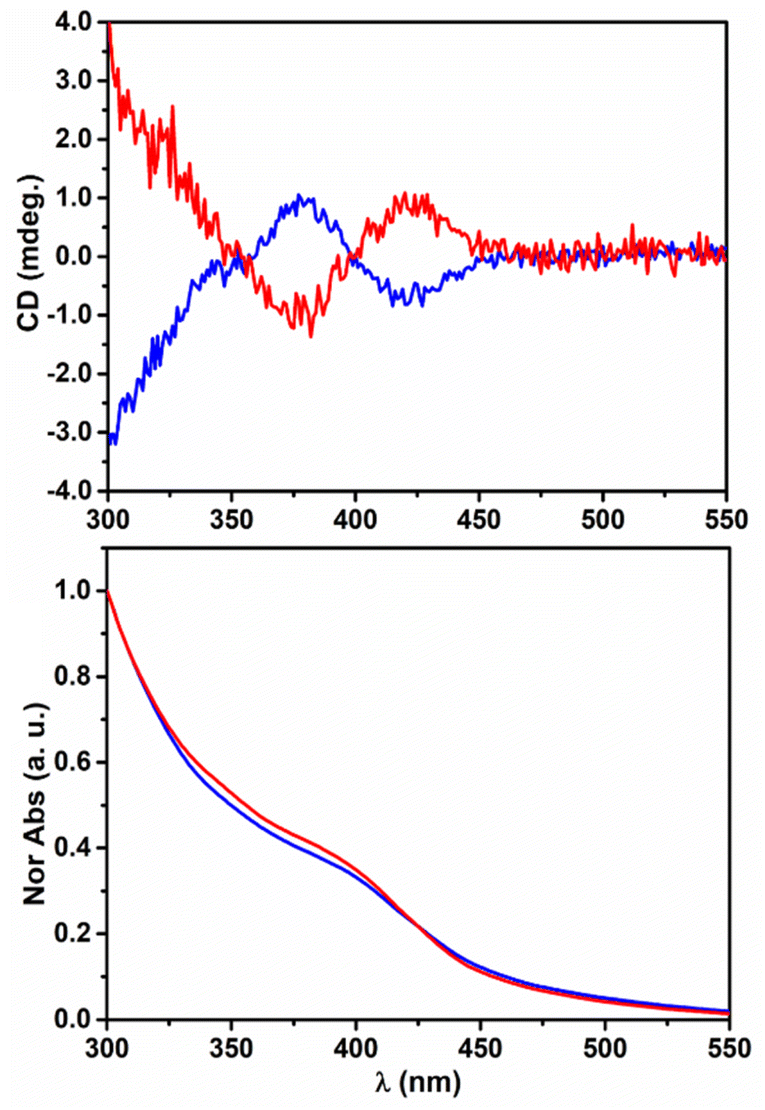 | ||
| Fig. 3 CD (top) and UV/Vis Abs (bottom) spectra of colloidal solutions of L-AIS (blue) and D-AIS (red). | ||
It must be remarked that the 2nd synthetic step significantly affects the spectroscopic properties of the nanocrystals. In particular, an increase of the ligand concentration in the AIS colloids enhances their luminescence efficiency, as shown in Fig. S7.† Moreover, the chiroptical activity of the nanocrystals isolated after the 1st step is readily lost in a few hours after dispersing the nanocrystals in the presence of a low ligand concentration (see Fig. S8†).
The PL kinetic measurements (Fig. S9†) of L-AIS show a high enhancement of the emission after 300 min from the dispersion of the nanocrystals, depending also on the L-Cys concentration (4.0, 8.0 and 16.0 mM). The emission intensity increases up to a plateau value after 24 hours, depending on the ligand concentration. During this period, the maximum of the emission at around 600 nm observed for the nanocrystals isolated after the first step is red shifted to 635 nm. Analogously, an increase in the PLE intensity at around 430 nm is observed (Fig. S10†).
In order to clarify the structural differences of the AIS QDs after the 1st and 2nd steps, an XRD analysis of the two samples was carried out. Fig. S11† shows a comparison of the degree of crystallinity of nanocrystals isolated after the 1st and 2nd reaction steps, and a clear narrowing of the diffraction reflections is observed for both the QD enantiomers. This behaviour can be reasonably related to the increase of the crystallite size during the ageing process. Moreover, this variation of the QDs’ crystal structure is dramatically reflected in their spectroscopic properties, shown in Fig. S12.† In particular, the absorption band centred at around 350 nm, observed during the nucleation step, shifts to 400 nm and the same variation is reflected also in the CD spectra where the intensities of the bands that are related to the inversion point located at 350 nm gradually decrease, resulting in the formation of a new band at around 400 nm. Further details on this process and the study of the kinetics according to different ligand concentrations are reported in the ESI section (Fig. S13†). Similar observations were reported by Soares et al. on the variation of the optical properties of AgInS2/ZnS QDs produced via a microwave-assisted synthesis,37 where the authors suggested the possible formation of small semiconductor nanoclusters in the early steps of the reaction which subsequently grow forming the final nanocrystals for long reaction times. According to this hypothesis the increase of the photoluminescence intensity can be explained by the removal of defects during the second reaction step, due to the particle growth. Moreover, the observed red-shift of the emission band (Fig. S10†) is clearly related to the increase of the nanocrystal size.43
In order to investigate the origin of chirality and to optimize the chiroptical activity of the nanocrystals, the chemisorption equilibrium of the chiral ligand on the nanocrystal surface has been investigated combining CD spectroscopy and NMR titration results, shown in Fig. 4. The analysis covers a range of ligand concentrations (2.0–16.0 mM) in which the highest variation of the chiroptical activity at the exciton position was observed.
 | ||
| Fig. 4 (a) 1H-NMR titration spectra of a 1 mg ml−1 solution of L-AIS QDs with different L-Cys concentrations, the signals that are ascribed to L-cystine are marked with an asterisk. (b) DOSY and (c) CD spectra of L-AIS QD solutions in the presence of different L-Cys concentrations. (d and e) Plots of the intensity of the CD bands and the measured diffusion coefficients respectively. | ||
The 1H-NMR spectrum for the QD dispersion stabilized in a 2.0 mM solution of L-Cys (Fig. 4a) shows broad signals without any well-resolved fine structure for both Hα and Hβ located at 3.94 and 2.94 ppm, respectively. The observed spectral broadening is typically associated with the ligand bonded to the particle surface. The origin of the broadening can be related to several factors: (i) distribution of the chemical shift due to the ligand absorption on different sites on the QD surface,44,45 (ii) distribution of the spin–spin relaxation T2 due to different tumbling dynamics in the absorbed ligands46 and (iii) different packing that can affect the ligand–ligand dynamics.44,47 On increasing the ligand concentration, a significant variation of the resonance frequency can be observed; the position of the peaks shifts from 2.94 to 3.00 ppm and from 3.94 to 3.92 ppm for Hβ and Hα, respectively.
At a higher L-Cys concentration (16.0 mM) it is possible to observe a significant spectral narrowing and the expected fine structure of free zwitterionic cystine can be observed as well (see Fig. S14†). This behaviour suggests that the ligand chemisorption dynamics is in the fast chemical exchange regime kex ≫ 1/τNMR.48 In this case, the chemical exchange between the two (free and bonded) ligand forms exceeds the NMR characteristic time scale and therefore, the exchanging protons cannot be resolved in two distinct sets of frequencies. Instead, for each set of protons, the analysis records a single signal whose observable chemical shift reflects the populations of the free and bonded forms.
The same consideration is also valid for the other NMR quantities such as relaxation times T1 or T2 and diffusion coefficients.49,50 Even if in the presence of a fast chemical exchange, the bands ascribed to the bonded and free forms of the ligand cannot be resolved, further evidence of the ligand–particle interaction can be collected by NOESY analysis (Fig. S15†).51 The spectrum shows the presence of characteristic strong negative crosspeaks for the Hα/Hβ protons of L-Cys. This evidence can be related to ligand–nanoparticle interactions that affect the ligand mobility in solution, causing the ligand coordinated on the particle surface to be characterized by a slow tumbling, responsible for the formation of intense negative crosspeaks.49,51 Under these conditions, we can assume that the resolution of the ligand proton frequencies in different signals according to different binding modes is not feasible and the different contributions are averaged in the single contribution that can be related to the bonded ligand population.
A Diffusion Ordered Spectroscopy (DOSY) analysis of the colloidal solutions in the presence of an increasing ligand concentration, shown in Fig. 4b, evidences the increase of the diffusion coefficient from 5.25 to 10.23 × 10−10 m2 s−1 for the Hβ. The variation of the diffusion coefficient for the Hα is even more evident, and the following diffusion coefficients can be estimated for the colloidal solutions with different L-Cys concentrations: 4.36 (2.0 mM), 6.92 (4.0 mM), 7.41 (6.0 mM), 8.32 (8.0 mM) and 9.12 (16.0 mM) × 10−10 m2 s−1. It must be remarked that the signal for the free L-Cys reference shows a much higher diffusion coefficient, which is estimated around 12.0 × 10−10 m2 s−1 for both Hβ and Hα. As expected, the presence of a fast exchange between the ligand coordinated on the particle surface and the free ligand in solution prevents the measurement of distinct diffusion coefficients for the two species (Db and Df for the bonded and free ligand, respectively). However, since the observed diffusion coefficient is the average of the two populations, it is possible to observe the effect of the increasing content of the free ligand population that shifts the observed value for the diffusion coefficient toward Df. It is worth noting that the variations of the observed diffusion coefficients can be related to the intensity variations of the CD bands located at 375 and 415 nm (Fig. 4c). In fact, the intensity of the CD signals increases on increasing the ligand concentration until a certain saturation value at around 8.0 mM is reached and no further increase is observed for higher L-Cys ligand concentrations. The UV/Vis absorption spectra of the sample at different L-Cys concentrations are shown in Fig. S16.† Since no significant variation is observed in the exciton region, we exclude that the variation of the ligand concentration in the investigated range affects the structure or composition of the L-AIS nanocrystals. The variations of the CD intensity and diffusion coefficients, shown in Fig. 4d and e, confirm this trend. It must be remarked that this comparison is possible thanks to the enhancement of the resolution in the diffusion dimension with respect to the small variations which are observed in the chemical shift.
An estimation of the bonded (xb) and free (xf) ligand population in the different colloidal solutions can be calculated according to eqn (S5) and (S6)† assuming the diffusion coefficient for the sample with the lowest total ligand concentration (2.0 mM) as a reference for the bonded ligand (Db). Even though this approximation gives an excess in the estimation of the bonded population, it was successfully employed by Copenhaver et al. for the estimation of the ligand coverage of QDs.44 By means of this rough estimation we observe a large increase in the bonded population for concentrations below 8.0 mM (Fig. S17†). Instead, for higher L-Cys concentrations, the free form is favoured, minimizing the effects on the composition of the chiral ligand shell and thus on the chiral transfer, due to the possible saturation of the ligand sites on the particle surface.
A similar behaviour can be observed also for the disulfide dimer L-cystine, as this species is present as an impurity in commercial cysteine (Fig. S14†) and it is in equilibrium with its monomeric form.52 The negative NOESY cross-peaks show that even this species is involved in the coordination equilibria with the particle surface (Fig. S15†). Furthermore, as observed with L-Cys, the diffusion coefficient for both the Hα and Hβ in L-cystine are shifted towards higher values with the increase of the ligand concentration (Fig. S18†). However, due to the low content of dimers in solution, the analysis shows a huge broadening in the diffusion dimension and for this reason our considerations are mainly focused only on L-Cys as the principal chiral ligand present in solution.
Based on our observations, chiral ternary AIS QDs produced via coprecipitation reaction in the presence of a chiral ligand present similar chiroptical properties to the more investigated Cd-based QDs. The particle morphology, crystalline structure and entity of the chiral induction suggest that the chirality is originated at the ligand–nanocrystal interface. As successfully employed for Cd-based chiral QDs, chiral AIS QDs can be described as a hybrid core–shell system composed of an achiral inorganic core surrounded by a chiral organic ligand shell (Fig. 5).5,8,10,16 In such nanosystems, the chiroptical activity can be originated by different processes that can be mainly described as electronic or structural. The electronic mechanism arises from the ligand chemisorbed on the particle surface which can affect the electronic state of the inorganic achiral core via dipolar induction.10,53,54 On the other hand, structural chirality can be introduced at the ligand–nanocrystal interface by distortions of the nanocrystal surface by the chiral ligand.6,16,55 Further analysis would be needed to clearly address the entity of the different contributions to the QD chirality.
 | ||
| Fig. 5 Schematic representation on the ligand–nanoparticle chiral hybrid structure. | ||
The main difference in the Cd-based chiral QDs is the fast exchange of the ligand molecules on the particle surface observed by NMR spectroscopy. The analysis reveals that in the presence of a low ligand concentration (2.0 mM) sub-coordinated particles are present, and under these conditions the chiroptical activity can be increased by increasing the concentration of the ligand in solution (Fig. 4d and e). In this way it is possible to drive the chemisorption equilibrium toward the saturation of the particle surface sites increasing the surface coverage which consequently increases the chiroptical activity. The organic chiral shell can be populated until a certain value corresponding to the surface saturation is reached for a ligand concentration above 8.0 mM, evidenced by both CD and DOSY analyses. A further increase of the ligand concentration in solution mainly affects the free ligand population with only a minor variation of the bonded population, that does not affect further the particle chirality. This behaviour is different from the concentration-dependent study reported by Kuznetsova et al.12 for CdSe/CdS core–shell QDs produced via post-synthetic phase-transfer ligand exchange. In that case, the intensity of the CD signal grew on increasing the ligand concentration until a critical point, and for a higher ligand concentration a strong reduction of the CD signal was observed. This behaviour was ascribed to the variation of the cysteine binding mode that is preferentially tridentate under the low ligand concentration conditions, while a bidentate coordination is favoured at higher ligand concentrations, causing a loss of the QD chirality. It is reasonable to conclude that the stability of the chiroptical properties for the present AIS QDs at high ligand concentrations is related to the lower affinity of the cysteine to the Ag+ ad In3+ ions exposed on the particle surface. As shown in Fig. S17,† the excess of ligand produced in the concentration above the saturation conditions (above 8.0 mM) contributes largely to the free ligand population without significantly affecting the ligand–nanocrystal interface and consequently the chiroptical activity. In fact, Fig. S19† shows that the CD signals of L- and D-AIS colloidal solutions are observed even in the presence of a ligand concentration of 100.0 mM. This conclusion is supported by the evidence of a slow exchange regime observed by Kuznetsova et al.12 in the NMR study of the interaction between cystine and CdSe/CdS QDs, clearly evidencing a higher ligand affinity for the Cd2+ cations exposed on the QD surface under the investigated conditions. Similar dynamics was reported in the NMR analysis of interface chemistry of water soluble CdTe QDs stabilized with thioglycolic acid which shows close (3.39 and 3.36 ppm) but distinct resonances for the α protons related to the free and bonded ligands, whose contributions can be resolved by the deconvolutions of the peaks.56 However, it must be considered that the effect on the ligand dynamics can also be affected by the different pH values as the stability of the thiolate ligand on the surface of Cd-based quantum dots strongly increases in alkaline pH57 and further studies on the effect of pH and solvent on the dynamics of the chiral ligand shell seem to be fundamental to understand the optimization of the chiroptical activity of chiral QDs.
Conclusions
Ligand-induced chirality in ternary non-stoichiometric silver indium sulfide QDs is observed for the first time with the isolation of L-AIS and D-AIS QDs stabilized with L- or D-cysteine respectively. We presented a two-step synthesis that allows the preparation of water soluble non-stoichiometric AIS chiral QDs with a PLQY of 0.24. The observed chiroptical activities of L-AIS and D-AIS QDs are in agreement with the literature data regarding chiral induction in Cd-based QDs induced by ligands suggesting that similar processes may be responsible for the chiral induction in the nanocrystals’ achiral inorganic matrixes.The investigation of the ligand–nanocrystal interface exploiting different NMR techniques allowed the collection of information on the interaction between the chiral ligand and the nanoparticle surface. In particular, we observed that under the investigated conditions, the chiral ligand is characterized by a fast exchange dynamics between the L-Cys molecules bonded to the nanocrystal surface and those free in solution. Finally, the combination of the NMR evidence with the CD data allowed the optimization of the chiroptical properties of AIS QDs.
Materials and methods
Starting materials
L-Cysteine (97%), AgNO3 (99%), In(OAc)3 (99%), Na2S anhydrous (99%), rhodamine 6G, deuterium oxide (99.9%), sodium hydroxide and hydrochloric acid (37%) were purchased from Sigma-Aldrich (Merck). D-Cysteine (95%) was purchased from Fluorochem. All the chemicals were employed without further purification.Cysteine-stabilized non-stoichiometric AIS QD synthesis, 1st step
In 90.0 ml of deionized water, 0.01 M In(AcO)3 (24.0 ml, 0.24 mmol) and 0.01 M AgNO3 (6.0 ml, 0.06 mmol) were added under constant stirring. The pH of the colourless solution was adjusted to 8.5 with the addition of 1.0 M NaOH, and the solution turned opaque with a dark-yellow colour. 0.16 M of optically pure solution of L- or D-cysteine was added (6.0 ml, 0.96 mmol) and the solution turned colourless. An aqueous solution of 1.0 M Na2S was added (0.480 ml, 0.48 mmol) and the colour of the resulting solution turned rapidly to orange. This solution was kept under constant stirring at room temperature for 1 h. The QDs were precipitated by the addition of 0.1 M HCl, adjusting the pH to approximately 6. The QDs were collected by centrifugation at 7000 rpm for 5 min and washed three times with deionized water.Cysteine-stabilized non-stoichiometric AIS QD synthesis, 2nd step
The freshly-prepared QD pellet was dispersed in 10.0 ml of enantiomerically pure L- or D-cysteine (48.0 mM) solution and a solution of 0.1 M NaOH was used to adjust the pH to 6.3. The QD dispersion was stored in air, monitoring the UV/Vis and CD properties for at least 24 h to ensure the complete growth of the nanocrystals. Then, the QD dispersions were diluted to a final QD concentration of 1.0 mg ml−1 and a ligand concentration of 16.0 mM.Photoluminescence life-time determination
The luminescence decay measurements were performed on a Nanolog/Fluorolog-3-2iHR320 (Horiba) spectrofluorimeter. The luminescence decay curve was acquired on the QD aqueous dispersion at 1.0 mg ml−1 concentration and at a cysteine concentration of 16.0 mM. The QD dispersion was excited with a 453 nm NanoLED with a pulse width of 1.2 ns. The emission decay curve was collected at 615 nm, centred on the maximum emission of QDs.Photoluminescence quantum yield determination
The quantum yield was estimated by comparison with a known fluorescence standard according to eqn (S1) and (S2).† Rhodamine 6G was chosen as the standard considering a quantum yield of 0.90 ± 0.02 in a water solution.58 The QDs and standard solution in deionized water were prepared adjusting the dilution in order to obtain an absorbance of 0.05 at the excitation wavelength (485 nm). The emission spectra were collected exciting with radiation at 485 nm and measuring the emission in the 500–850 nm range. A spectral resolution of 2.0 nm was employed in both excitation and emission.NMR titrations
30.0 mg of L-AIS QDs were dispersed in 30.0 ml of D2O, in the presence of 1.5 mM acetonitrile as a reference for the diffusion order spectroscopy experiments. Samples of 0.700 ml with different L-cysteine concentrations (2.0, 4.0, 6.0, 8.0, 16.0 mM) were collected. The pH of the solutions was adjusted to 8.0 ± 0.1 with the addition of a few microliters of 0.1 M NaOH in D2O. The solutions were stored in the dark for 24 h to ensure equilibration, and then, the QD dispersions were centrifuged at 9000 rpm for 5 min to remove possible aggregates and the samples were transferred in 0.5 mm NMR tubes for the analysis.Absorption spectra
The UV/Vis analysis was carried out with a Cary 60 (Agilent) UV/Vis spectrophotometer.Morphological characterization
High resolution transmission electron microscopy (HRTEM) and scanning transmission electron microscopy (STEM) high-angle annular dark-field imaging (HAADF) were carried out using an FEI Titan operating at a beam voltage of 300 kV, at the advanced microscopy laboratory in Trinity College Dublin.Structural characterization
The X-ray powder diffraction measurement was recorded with a Panalytical Advanced Powder Diffractometer.Steady state luminescence spectra
Luminescence excitation and emission spectra were collected using a FluoroMax-4 (Horiba).FTIR spectra
FTIR spectra were recorded using a NEXUS-FTIR instrument implementing a Nicolet Diffuse Reflectance accessory (DRIFT).XPS spectra
XPS spectra were acquired with nonmonochromatic Al Kα radiation (1486.6 eV) using a PerkinElmer Φ 5600ci spectrometer, at a working pressure in the 10−7 Pa range. The BE values were referred to the Fermi level. Uncertainties in the BE values were around 0.2 eV. Fits of raw spectra were done adopting a Shirley-type background and Gaussian–Lorentzian peak shapes using a non-linear least-square fitting program (XPSPEAK41 software). Atomic fractions were evaluated using sensitivity factors as provided by Θ V5.4A software. The relative uncertainty of the calculated atomic fraction of the different elements was around 5–10%. The circular dichroism spectra were recorded with Jasco spectropolarimeters J1500 and J815.NMR
A Bruker Avance III 400 MHz (proton frequency of 400.23 MHz) equipped with a 5.0 mm BBOF probe was employed for the 1H titration experiment and the NOESY spectra (mixing time 500 ms). The DOSY investigation was carried out on a Bruker Avance II 600 MHz (proton frequency 600.13 MHz) equipped with a 5.0 mm TLC cryo probe. The DOSY data have been processed with TopSpin Dynamics Center software.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Science Foundation Ireland and Biorbic, Bioeconomy Research Centre (grant number SFI 16/RC/3889) for the financial support. The authors acknowledge the “Centro Piattaforme Tecnologiche” University of Verona for the circular dichroism measurement with the Jasco J1500 CD spectrophotometer. A. S. and C. C. would like to thank the University of Verona for financial support in the framework of “Joint Projects 2018”.References
- M. P. Moloney, Y. K. Gun'ko and J. M. Kelly, Chem. Commun., 2007, 3900–3902 RSC.
- S. A. Gallagher, M. P. Moloney, M. Wojdyla, S. J. Quinn, J. M. Kelly and Y. K. Gun'ko, J. Mater. Chem., 2010, 20, 8350–8355 RSC.
- K. Varga, S. Tannir, B. E. Haynie, B. M. Leonard, S. V. Dzyuba, J. Kubelka and M. Balaz, ACS Nano, 2017, 11, 9846–9853 CrossRef CAS PubMed.
- J. K. Choi, B. E. Haynie, U. Tohgha, L. Pap, K. W. Elliott, B. M. Leonard, S. V. Dzyuba, K. Varga, J. Kubelka and M. Balaz, ACS Nano, 2016, 10, 3809–3815 CrossRef CAS PubMed.
- U. Tohgha, K. K. Deol, A. G. Porter, S. G. Bartko, J. K. Choi, B. M. Leonard, K. Varga, J. Kubelka, G. Muller and M. Balaz, ACS Nano, 2013, 7, 11094–11102 CrossRef CAS PubMed.
- T. Nakashima, Y. Kobayashi and T. Kawai, J. Am. Chem. Soc., 2009, 131, 10342–10343 CrossRef CAS PubMed.
- Y. Zhou, M. Yang, K. Sun, Z. Tang and N. A. Kotov, J. Am. Chem. Soc., 2010, 132, 6006–6013 CrossRef CAS PubMed.
- G. Li, X. Fei, H. Liu, J. Gao, J. Nie, Y. Wang, Z. Tian, C. He, J.-L. Wang, C. Ji, D. Oron and G. Yang, ACS Nano, 2020, 14, 4196–4205 CrossRef CAS PubMed.
- J. E. Govan, E. Jan, A. Querejeta, A. Kotov and Y. K. Gun'ko, Chem. Commun., 2010, 46, 6072–6074 RSC.
- A. Ben-Moshe, A. Teitelboim, D. Oron and G. Markovich, Nano Lett., 2016, 16, 7467–7473 CrossRef CAS PubMed.
- A. K. Visheratina, A. O. Orlova, F. Purcell-Milton, V. A. Kuznetsova, A. A. Visheratin, E. V. Kundelev, V. G. Maslov, A. V. Baranov, A. V. Fedorov and Y. K. Gun'ko, J. Mater. Chem. C, 2018, 6, 1759–1766 Search PubMed.
- V. A. Kuznetsova, E. Mates-torres, N. Prochukhan, M. Marcastel, F. Purcell-milton, J. O. Brien, A. K. Visheratina, M. Martinez-carmona, Y. Gromova, M. Garcia-melchor and Y. K. Gun'ko, ACS Nano, 2019, 13, 13560–13572 CrossRef CAS PubMed.
- J. Cheng, J. Hao, H. Liu, J. Li, J. Li, X. Zhu, X. Lin, K. Wang and T. He, ACS Nano, 2018, 12, 5341–5350 CrossRef CAS PubMed.
- J. Hao, Y. Li, J. Miao, R. Liu, J. Li, H. Liu, Q. Wang and H. Liu, ACS Nano, 2020, 14, 10346–10358 CrossRef CAS PubMed.
- V. Kuznetsova, Y. Gromova, M. Martinez-Marmona, F. Purcell-Milton, E. Ushakova, S. Cherevkov, V. Maslov and Y. K. Gun'ko, Nanophotonics, 2020, 10, 797–824 CrossRef.
- S. D. Elliott, M. P. Moloney and Y. K. Gun'ko, Nano Lett., 2008, 8, 2452–2457 CrossRef CAS PubMed.
- X. Gao, X. Zhang, K. Deng, B. Han, L. Zhao, M. Wu, L. Shi, J. Lv and Z. Tang, J. Am. Chem. Soc., 2017, 139, 8734–8739 CrossRef CAS PubMed.
- J. Kumar, K. T. George and L. M. Liz-Marzán, Chem. Commun., 2016, 52, 12555–12569 RSC.
- A. O. Govorov, Y. K. Gun'ko, J. M. Slocik, V. A. Gérard, Z. Fan and R. R. Naik, J. Mater. Chem., 2011, 21, 16806–16818 RSC.
- O. Stroyuk, F. Weigert, A. Raevskaya, F. Spranger, C. Würth, U. Resch-Genger, N. Gaponik and D. R. T. Zahn, J. Phys. Chem. C, 2019, 123, 2632–2641 CrossRef CAS.
- A. S. Baimuratov, I. V. Martynenko, A. V. Baranov, A. V. Fedorov, I. D. Rukhlenko and S. Y. Kruchinin, J. Phys. Chem. C, 2019, 123, 16430–16438 CrossRef CAS.
- M. L. Zaffalon, V. Pinchetti, A. Camellini, S. Vikulov, C. Capitani, B. Bai, M. Xu, F. Meinardi, J. Zhang, L. Manna, M. Zavelani-Rossi, S. A. Crooker and S. Brovelli, Energy Mater. Adv., 2021, 2021, 20–22 Search PubMed.
- M. Uehara, K. Watanabe, Y. Tajiri, H. Nakamura and H. Maeda, J. Chem. Phys., 2012, 129, 134709 CrossRef PubMed.
- D. Nam, W. Song and H. Yang, J. Colloid Interface Sci., 2011, 361, 491–496 CrossRef CAS PubMed.
- M. Dai, S. Orawa, T. Kameyama, K.-I. Okazaki, A. Kudo, S. Kuwabata, Y. Tsuboi and T. Torimoto, J. Mater. Chem., 2012, 22, 12851–12858 Search PubMed.
- S. P. Hong, H. K. Park, H. J. Oh, H. Yang and R. Y. Do, J. Mater. Chem., 2012, 22, 18939–18949 RSC.
- W. Xiang, C. Xie, J. Wang, J. Zhong, X. Liang, H. Yang, L. Luo and Z. Chen, J. Alloys Compd., 2014, 588, 114–121 CrossRef CAS.
- X. Hu, T. Chen, Y. Xu, M. Wang, W. Jiang and W. Jiang, J. Lumin., 2018, 200, 189–195 CrossRef CAS.
- M. D. Regulacio, K. Y. Win, S. L. Lo, S.-Y. Zhang, X. Zhang, M.-Y. Han and Y. Zheng, Nanoscale, 2013, 5, 2322–2327 RSC.
- Y. Hamanaka, T. Ogawa and M. Tsuzuki, J. Phys. Chem. C, 2011, 115, 1786–1792 CrossRef CAS.
- I. V. Martynenko, A. S. Baimuratov, F. Weigert, J. X. Soares, L. Dhamo, P. Nickl, I. Doerfel, J. Pauli, I. D. Rukhlenko, A. V. Baranov and U. Resch-Genger, Nano Res., 2019, 12, 1595–1603 CrossRef CAS.
- X. Tong, A. I. Channa, Y. You, P. Wei, X. Li, F. Lin, J. Wu, A. Vomiero and Z. M. Wang, Nano Energy, 2020, 76, 105062 CrossRef CAS.
- J.-Y. Chang, G.-Q. Wang, C.-Y. Cheng, W.-X. Lin and J.-H. Hsu, J. Mater. Chem., 2012, 22, 10609–10618 RSC.
- H. Guo, J. Liu, B. Luo, X. Huang, J. Yang, H. Chen, L. Shi, X. Liu, D. Benetti, Y. Zhou, G. S. Selopal, F. Rosei, Z. Wang and X. Niu, J. Mater. Chem. C, 2021, 9, 9610–9618 RSC.
- L. Lin, C. Wu, C. Lai and T. Lee, Chem. Mater., 2008, 20, 4475–4483 CrossRef CAS.
- L.-H. Lin, C.-C. Wu and T.-C. Lee, Cryst. Growth Des., 2007, 12, 4–11 Search PubMed.
- J. X. Soares, K. D. Wegner, D. S. M. Ribeiro, A. Melo, I. Häusler and J. L. M. Santos, Nano Res., 2020, 13, 2438–2450 CrossRef CAS.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies Contents, John Wiley & Sons Ltd, Baffins Lane, 3rd edn, 2001 Search PubMed.
- N. Kazuo, Infrared and Raman Spectra of Inorganic and Coordination Compounds. Part B: Applications in Coordination, Organometallic and Bioinorganic Chemistry, 6th edn, 2009 Search PubMed.
- A. Pawlukojć, J. Leciejewicz, A. J. Ramirez-Cuesta and J. Nowicka-Scheibe, Spectrochim. Acta, Part A, 2005, 61, 2474–2481 CrossRef PubMed.
- N. Kojima, Y. Sogiura and H. Tanaka, Bull. Chem. Soc. Jpn., 1976, 49, 3023–3028 CrossRef CAS.
- M. Puri and V. E. Ferry, ACS Nano, 2017, 11, 12240–12246 CrossRef CAS PubMed.
- Y. Hamanaka, T. Ogawa, M. Tsuzuki, K. Ozawa and T. Kuzuya, J. Lumin., 2013, 133, 121–124 CrossRef CAS.
- X. Ji, D. Copenhaver, C. Sichmeller and X. Peng, J. Am. Chem. Soc., 2008, 130, 7012–7021 Search PubMed.
- M. Malicki, J. M. Hales, M. Rumi, S. Barlow, L. McClary, S. R. Marder and J. W. Perry, Phys. Chem. Chem. Phys., 2010, 12, 6267–6277 RSC.
- R. Grisorio, D. Quarta, A. Fiore, L. Carbone, G. P. Suranna and C. Giansante, Nanoscale Adv., 2019, 1, 3639–3646 RSC.
- M. J. Hostetler, J. E. Wingate, C.-J. Zhong, J. E. Harris, R. W. Vachet, M. R. Clark, J. D. Londono, S. J. Green, J. J. Stokes, G. D. Wignall, G. L. Glish, M. D. Porter, N. D. Evans and R. W. Murray, Langmuir, 1998, 14, 17–30 CrossRef CAS.
- D. A. Brain, Prog. Nucl. Magn. Reson. Spectrosc., 2003, 43, 63–103 CrossRef.
- Z. Hens and J. C. Martins, Chem. Mater., 2013, 25, 1211–1221 CrossRef CAS.
- C. S. Johnson, J. Magn. Reson., Ser. A, 1993, 102, 214–218 CrossRef CAS.
- B. Fritzinger, I. Moreels, P. Lommens, R. Koole, Z. Hens and J. C. Martins, J. Am. Chem. Soc., 2009, 131, 3024–3032 CrossRef CAS PubMed.
- A. Rigo, A. Corazza, M. Luisa, M. Rossetto, R. Ugolini and M. Scarpa, J. Inorg. Biochem., 2004, 98, 1495–1501 CrossRef CAS PubMed.
- A. O. Govorov, Z. Fan, P. Hernandez, J. M. Slocik and R. R. Naik, Nano Lett., 2010, 10, 1374–1382 CrossRef CAS PubMed.
- Y. Zhou, Z. Zhu, W. Huang, W. Liu, S. Wu, X. Liu, Y. Gao, W. Zhang and Z. Tang, Angew. Chem., Int. Ed., 2011, 50, 11456–11459 CrossRef CAS PubMed.
- R. Zhou, K. Wei, J. Zhao and Y. Jiang, Chem. Commun., 2011, 47, 6362–6364 RSC.
- C. A. Martínez Bonilla, M.-H. Torres Flórez, D. R. Molina Velasco and V. V. Kouznetsov, New J. Chem., 2019, 43, 8452–8458 RSC.
- J. Aldana, N. Lavelle, Y. Wang and X. Peng, J. Am. Chem. Soc., 2018, 127, 3901–3909 Search PubMed.
- D. Magde, R. Wong and P. G. Seybold, Photochem. Photobiol., 2002, 75, 327–334 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr03330e |
| This journal is © The Royal Society of Chemistry 2022 |