
All-carbon microporous graphitic photocatalyst-promoted reduction of CO2 to CO in the absence of metals or dopant elements†
Ana
Garcia-Mulero
a,
Abdullah M.
Asiri
 b,
Josep
Albero
a,
Ana
Primo
*a and
Hermenegildo
Garcia
*ab
b,
Josep
Albero
a,
Ana
Primo
*a and
Hermenegildo
Garcia
*ab
aInstituto Universitario de Tecnología Química, Consejo Superior de Investigaciones Científicas-Universitat Politecnica de Valencia, Universitat Politecnica de Valencia, 46022 Valencia, Spain. E-mail: aprimoar@itq.upv.es; hgarcia@qim.upv.es
bCenter of Excellence in Advanced Materials Research, King Abdullah University, Jeddah, Saudi Arabia
First published on 15th July 2022
Abstract
Microporous graphitic carbon (mp-C) derived from the pyrolysis of α-, β-, and γ-cyclodextrins exhibited photocatalytic activity in CO2-saturated acetonitrile–water upon irradiation with UV-Vis light and in the presence of triethanolamine, forming H2 (19 μmol h−1) and CO (23 μmol h−1) accompanied by a lesser proportion of CH4 (4 μmol h−1). The most efficient was the mp-C material derived from α-cyclodextrin (mp-Cα) and having a pore dimension of 0.68 nm. The process also occured, although to a much lesser extent, under simulated sunlight or with UV-Vis irradiation in the absence of a sacrificial agent, with H2O being the electron donor. The origin of the CO was proved by isotopic 13C labelling experiments. Photocurrent measurements proved the occurrence of charge separation and the increase in photocurrent intensity in the presence of CO2. Transient absorption spectroscopy was used to detect the charge separate state decay in the microsecond time scale and proved that a fraction of the photogenerated electrons were able to react with CO2.
Introduction
Photocatalysis is gaining interest as a long-term technology to produce materials from abundant feedstock fuels and chemicals by using direct sunlight.1 Since the seminal discovery by Fujishima and Honda of the photoelectrocatalytic activity of TiO2 under UV irradiation,2,3 photocatalysis has been traditionally dominated by transition metal semiconductors.3,4 However, for the sake of sustainability and considering the scarcity of resources, there is much current interest in moving from metal-containing catalysts to metal-free catalysts,5–7 including in the area of photocatalysis.8 Consequently, the number of examples showing that metal-free materials can behave as photocatalysts, even for overall H2O splitting, is continuously increasing. Compared to H2 generation, photocatalytic CO2 reduction is considerably more challenging due to the sluggish kinetics involved in the transfer of several electrons and protons.9 An additional factor to be considered is CO2 adsorption on the photocatalyst surface. For this reason, the number of metal-free photocatalysts for CO2 reduction is comparatively lower, and there is thus a need to expand the number of materials under study, particularly those based predominantly on carbon.10In this context, defective doped graphenes have shown activity for H2 generation.11 More recently, microporous graphitic carbon (mp-C) derived from cyclodextrins has also shown photocatalytic activity for hydrogen generation and oxygen evolution.12 Theoretical calculations indicate that oxidation sites can be associated with the residual population of oxygen atoms in microporous graphitic materials.13 In addition, these calculations also indicate that confinement inside the micropores can facilitate bond cleavage in a kind of preactivation when the size of the molecules fit tight within the graphenic wall of the microporous carbon.13 Photocatalytic hydrogen evolution activity was also found to be increased upon N- or P-doping as a consequence of band alignment, thus the resulting mp-(N)C and mp-(P)G powders were able to even promote overall H2O splitting.
Going further in the study of the photocatalytic activity of this type of metal-free microporous graphitic carbon, it would be of interest to determine if these novel materials can also promote the photocatalytic CO2 reduction, a reaction that is more challenging than hydrogen generation from water due to the need for larger reduction potentials and the large range of products that can be formed. Herein, it is reported that, in the absence of any metal, microporous graphitic carbon materials exhibited photocatalytic activity for selective CO2 reduction to CO in the presence of sacrificial electron donors and that this photocatalytic activity followed the trend observed for overall water splitting, with the most efficient material being the one derived from the smallest α-cyclodextrin.
Results and discussion
The mp-C materials (mp standing for microporous and C corresponding to graphitic carbon) under study were obtained by pyrolysis at 900 °C under an inert atmosphere of α-, β-, and γ-cyclodextrins as previously reported. N- and P-doping was achieved by mixing homogeneously urea or H3PO4 to the cyclodextrin in the proportion indicated before the pyrolysis. The resulting samples were characterized by combustion elemental analysis. The analytical data of the samples under study are presented in Table 1. Importantly, mp-Cα constituted almost exclusively C (>98%) and some residual H. In comparison, oxygen was present in about 9 and 13 wt% in mp-Cβ and mp-Cγ, respectively. This trend in the O content depending on the pore dimension of the resulting mp-C was already observed and it was suggested that oxygen functionalities are better accommodated in larger pores, while their content in mp-Cα is almost negligible. This oxygen content also increased upon doping, in which in addition to the dopant element, the presence of a substantial percentage of oxygen (above 10%) should also be noted. The extent of N- and P-doping was also determined by elemental analysis.The specific surface area of microporous carbons was measured by isothermal CO2 adsorption at 273 K. At this point, it should be noted that in contrast to CO2 mp-Cα did not adsorb N2, a fact that was attributed to its pore size (0.64 nm) being close to the N2 kinetic diameter in comparison with the pore sizes of mp-Cβ and mp-Cγ, which were 0.78 and 0.99 nm, respectively. In the cases of mp-Cβ and mp-Cγ, isothermal N2 adsorption was used to estimate the pore-size distribution (see Fig. S1†), which corresponded in both cases predominantly to dimensions below 20 nm. A remarkable revelation was the significantly much larger area of mp-(P)Cα. It is proposed that the acidity of H3PO4 used as the P precursor was responsible for some additional corrosion of the graphenic walls during the pyrolysis process, leading to a three-fold specific surface area increase.
The Raman spectra of the mp-C samples were almost coincident. As an example, Fig. 1 shows a representative example, while Fig. S2 in the ESI† gathers the Raman spectra of all the samples under study. Three signals corresponding to 2D (broad), G, and D bands appeared at about 2700, 1590, and 1350 cm−1, respectively. These three signatures were characteristic of defective graphenes derived from oligo or polysaccharides. Doping was not reflected in any detectable change in the Raman spectra.
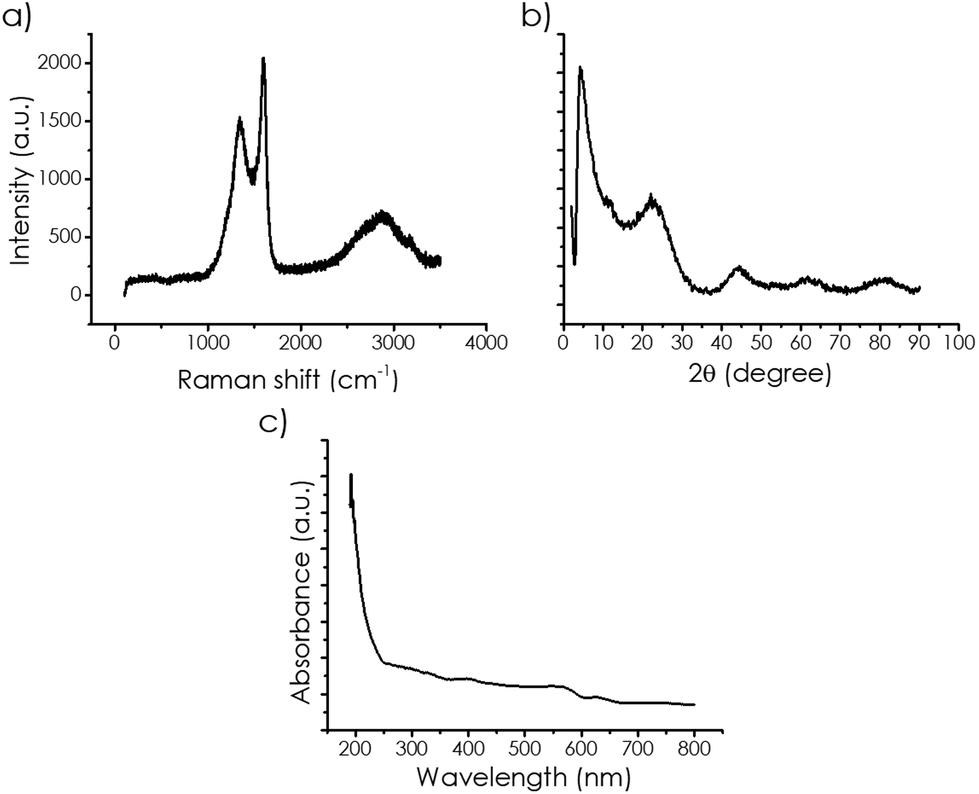 | ||
| Fig. 1 (a) Raman spectra; (b) XRD pattern and (c) UV-Vis spectra of the sample mp-Cα. | ||
The high crystallinity of mp-C was reflected in the XRD patterns of the powders. Fig. 1 also presents a selected example, while the collection of XRD patterns are gathered in Fig. S3 in the ESI.† Broad peaks corresponding to the loose stacking of graphene sheets at about 26°, 45°, and 61°, corresponding to the 002, 004, and 110 face diffractions were recorded. In addition, a new diffraction peak at short angles that was attributed to the presence of the micropores was also recorded for mp-C.
The morphology of the particles was determined by field-emission scanning electron microscopy (Fig. 2 and Fig. S4 in the ESI†). It was observed that upon pyrolysis, the cyclodextrin powders formed a thick crust carbon residue with a smooth surface due to the melting of the cyclodextrin powder and transformation into graphitic carbon. Inspection at a higher magnification of the crust revealed the presence of granules arranged into the form of tubular agglomerates, with most of them longer than one micron length and about 80 nm wide, aligned perpendicular to the surface. Transmission electron microscopy revealed the presence of small particles of about 10–20 nm and microporous channels. Fig. 2 shows a selection of these TEM images. From the contrast between the walls and the voids, the dimensions of the cyclodextrin precursor were used to determine the dimensions of the pores in the resulting graphitic carbon.
 | ||
| Fig. 2 FESEM and TEM images of mp-Cα. | ||
The pore sizes were estimated as 0.68, 0.86, and 0.97 nm for mp-Cα, mp-Cβ and mp-Cγ, respectively. These values were in good accordance with the data previously reported, showing that the diffuse-reflectance-UV-Vis absorption spectra of the samples under study were also very similar, with a continuous absorption over the whole range of UV and visible wavelengths and then decreasing gradually in absorptivity towards longer wavelengths. The conjugated condensed polycyclic aromatic domains present in mp-C should be the chromophores responsible for this absorption in the visible region. Fig. 1 also includes a representative DR-UV-Vis spectrum, while the collection for all the samples is presented in Fig. S5 in the ESI.† Some relative absorption maxima were measured at 280, 400, 550, and 620 nm, coincident for all the mp-C samples. N- and P-doping modified the DR-UV-Vis spectra mostly in the UV region by introducing a more intense absorption band with a tail extending to 350 and 300 nm for mp-(N)Cα and mp-(P)Cα, respectively, while no influence was observed at longer wavelengths. From the optical absorption spectroscopy analysis, the bandgap of the samples could be estimated using Tauc plots. The optical bandgap for each mp-C sample is provided also in Table 1.
Photocatalytic activity
Photocatalytic CO2 reduction reactions using the mp-C samples as photocatalysts (15 mg) were carried out in a mixture of acetonitrile (12 mL) and H2O (4 mL) adding triethanolamine (TEOA) (4 mL) as a sacrificial electron donor under the output of a 300 W Xe lamp. The course of the reaction was followed by taking aliquots of the head space, which were analyzed in a micro-GC system. The Experimental section provides additional details of the reactor, CO2 pressure, and analysis. The products observed under these conditions were H2 and CO in similar amounts, accompanied by a lesser proportion of CH4. No products in the liquid phase were detected at the end of the reaction by 1H and 13C NMR spectroscopy. Fig. 3 shows the temporal evolution of the three gases using mp-Cα as the photocatalyst, while Fig. 4 presents a complete series of plots for each of the mp-C samples under study.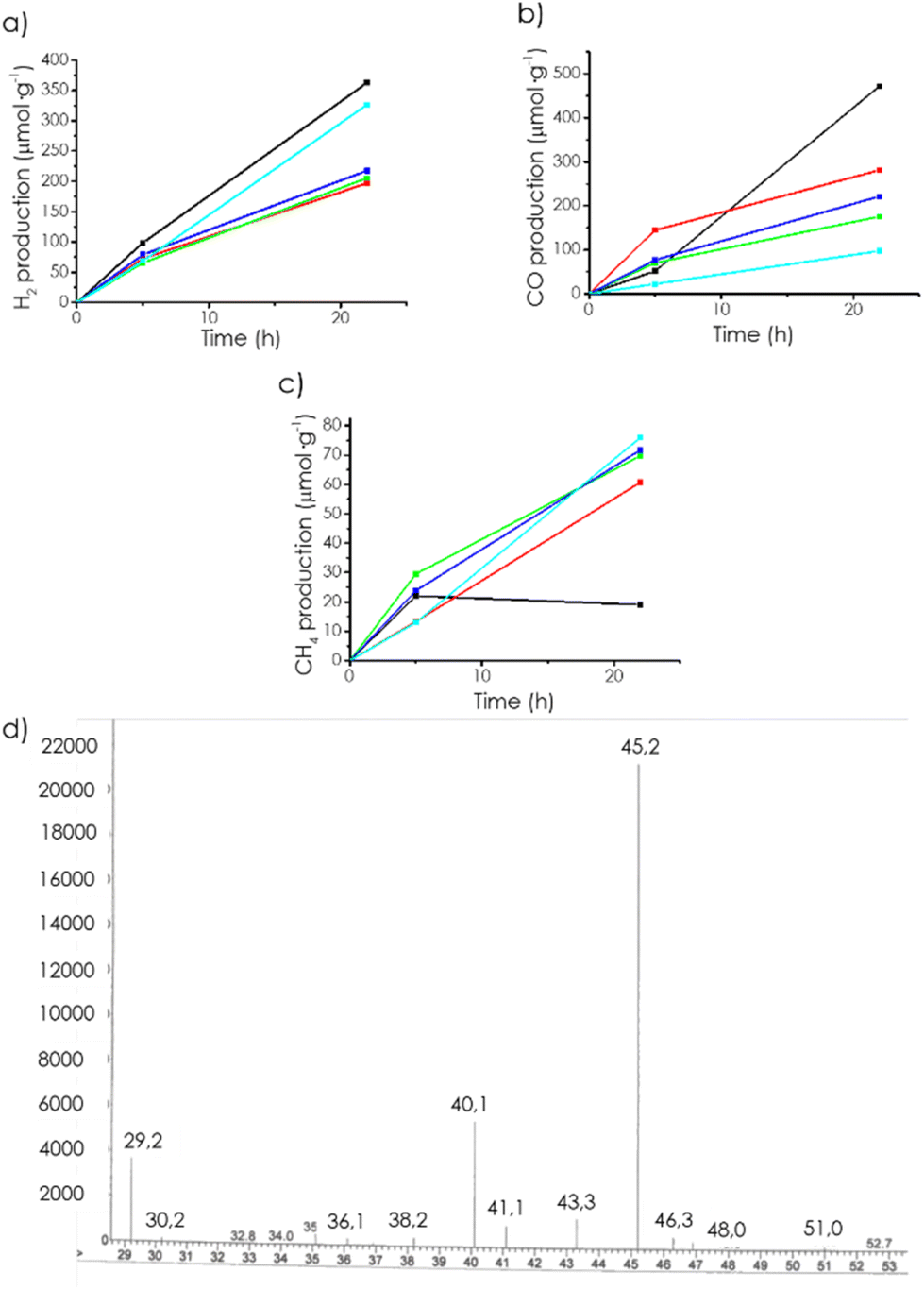 | ||
| Fig. 3 Temporal gas evolution from CO2 dissolved in a CH3CN/H2O mixture containing TEAO using mp-Cα (black), mp-Cβ (red), mp-Cγ (green), mp(N)-Cα (blue), and mp(P)-Cα (cyan) as photocatalysts upon 300 W Xe lamp irradiation: (a) H2, (b) CO, (c) CH4, (d) mass spectra of the test using 13CO2 as the substrate and mp-Cα as the photocatalyst. | ||
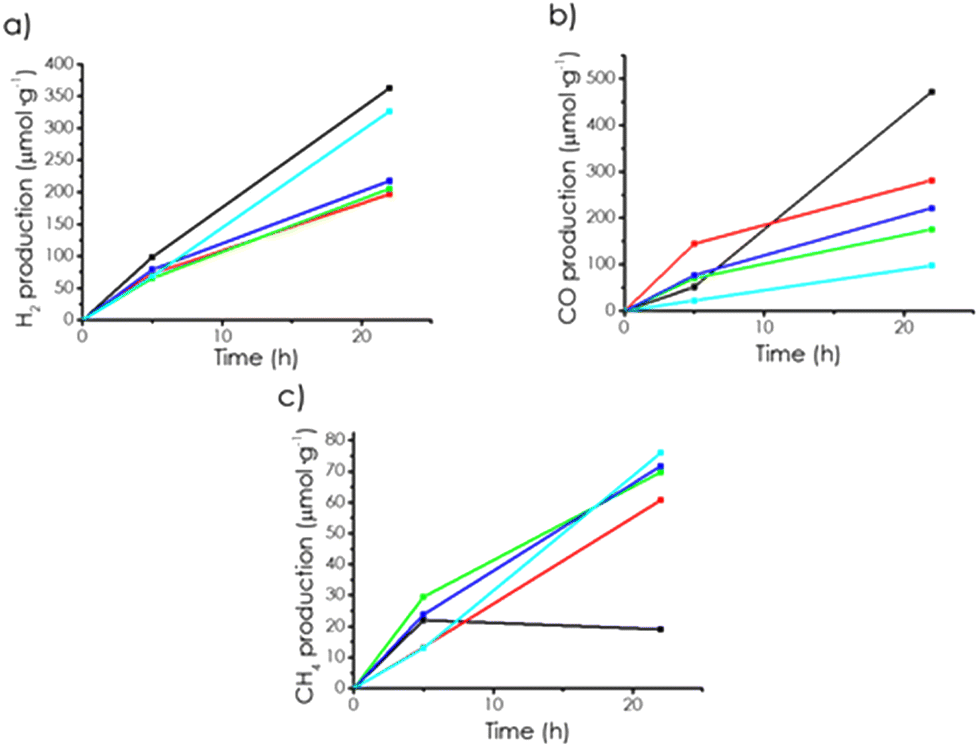 | ||
| Fig. 4 Temporal profiles of (a) H2, (b) CO, and (c) CH4 evolution for mp-Cα (black), mp-Cβ (red), mp-Cγ (green), mp(N)-Cα (blue), and mp(P)-Cα (cyan) upon the irradiation of CO2 dissolved in CH3CN/H2O containing TEAO with the output of a 300 W Xe lamp. | ||
The photocatalytic activity for H2 or CO evolution followed the order mp-Cα > mp-Cβ ≈ mp-Cγ. This order is similar to what has been observed for H2 generation, and it has been interpreted as reflecting the beneficial influence of the spatial confinement and small pore size commensurate with the substrate dimensions to enhance the photocatalytic activity.12 Thus, the sample with the smallest pore size was the most active of the series since the tight fitting of CO2 inside the micropores of mp-Cα should favour electron transfer from the mp-Cα photocatalyst to the substrate. The control experiments, as shown in Fig. S7,† indicated there was negligible gas evolution in the absence of photocatalyst.
No beneficial influence of doping on the photocatalytic CO2 reduction to CO was observed, and the performance of mp-Cα was also higher than those of mp-(N)Cα and mp-(P)Cα (see Fig. 4). This was probably due to the band energy of the various samples, which were in all cases above the thermodynamic potential required for CO2 reduction to CO (see Fig. S8†).
In view of these results, one aspect that is remarkable is the higher efficiency of the materials, such as mp-Cα, constituting greater than 98% C element in the absence of any metal or even dopant element. In the state of the art, it is frequently assumed that doping in the optimal proportion should increase the photocatalytic activity of graphitic carbons, but this assumption did not seem to apply in the present set of samples. It could be that the mp-Cα contained an excessive dopant amount. Also the simultaneous co-doping with two elements could result in an enhancement of the photocatalytic activity. It appears that spatial confinement of the photocatalytic reaction inside small pores was the key factor leading to the higher photocatalytic activity of mp-Cα. The apparent quantum yield of CO formation measured for mp-Cα at 380 nm was determined as 0.02%.
The photocatalyst stability was checked by performing a set of consecutive uses with the same mp-Cα sample, whereby similar temporal evolution profiles were observed for H2, CO, and CH4 in four consecutive uses (Fig. S9†). Furthermore, TEM characterization of the mp-Cα sample after exhaustive use showed that the porosity and morphology of the sample was maintained in the process. These photocatalytic data and characterization support the photocatalyst stability under the irradiation conditions.
Under the same solvent mixture and conditions, but using solar simulated sunlight, mp-Cα was also revealed to be active, although with lesser gas evolution. The results are presented in Fig. S6 in the ESI.† Notably, CH4 (8 μmol h−1) was the major product formed under these conditions, with a significantly lesser evolution of H2 (1.3 μmol h−1) and CO (2 μmol h−1). This product dependence with the irradiation wavelength would indicate the existence of different sites in mp-C. It is proposed that CH4 was the product formed when there was strong CO adsorption as a consequence of the deeper photocatalytic reduction. If this were the case, then CO would be formed in those sites with less CO affinity, while CH4 would be on those other with stronger CO adsorption. It seems that those sites with stronger CO affinity were responsive to longer wavelengths.
To determine the origin of CO and CH4, blank controls experiments under the same conditions by replacing CO2 by Ar were performed, whereby the formation of CO or CH4 was not observed, except in the case of mp-(N)Cα and mp-(P)Cα, for which the presence of CO in minute amounts was observed. Further confirmation of CO2 as the source of CO and CH4 in the photocatalytic reactions was obtained by using 13C-labelled CO2 in the reaction with the mp-Cα photocatalyst and by analyzing the products by mass spectrometry. This technique revealed the presence of a peak at m/z 29 amu and the complete absence of a peak at m/z 28 amu (Fig. 3), providing firm evidence that all the CO was derived from 13CO2. The photocatalytic activity of mp-C to promote artificial photosynthesis, promoting CO2 reduction by H2O was finally also explored. In these measurements, the experimental conditions were similar except that the triethanolamine volume was replaced by additional H2O. In these reactions, CH3CN (12 mL) and H2O (8 mL) were used as the solvent mixture using the same mp-Cα weight (15 mg). In these reactions, the products observed were H2 (30 μmol h−1), CO (2 μmol h−1), and O2. The formation of CH4 could not be detected under these conditions. The temporal product evolution is presented in Fig. S9.† These results were in line with our previous reports showing that mp-Cα was able to photocatalytically generate H2 and O2 from H2O.12 A control experiment, in the absence of CO2 did not show CO evolution. Now if CO2 was also present, then, photocatalytic CO2 reduction also occurred concomitantly with overall water splitting. Again, these results are remarkable and in our knowledge unprecedented, since mp-Cα is composed almost purely of C and no metals or doping elements are present.
Photocurrent measurements
To demonstrate the generation of e− and h+ upon illumination and to gain a further understanding of the photocatalytic activity of mp-C, photocurrent measurements were carried out by depositing on a transparent, conductive FTO electrode a well-dispersed suspension of mp-C. Besides mp-C/FTO as the photoelectrode, graphite and Ag/AgCl were used as the counter and reference electrode, respectively, in 1 M aqueous KHCO3 as the electrolyte. The Experimental section provides additional details of the mp-C/electrode area and active mass, as well as the preparation procedure and measurements, while Fig. 4 presents a summary of the photocurrent measurements and the ESI.†Prior characterization of the polarization curves for the mp-Cα/FTO photoelectrodes indicated that they could be used in the range of potential from 1 to −0.5 V vs. Ag/AgCl, since at −1 V vs. Ag/AgCl there was a strong discharge due to the hydrogen evolution reaction (HER). Measurements were carried out under N2- and CO2-saturated conditions (Fig. 5). Large currents were observed at 1 V vs. Ag/AgCl bias potential with no difference in the gas present in the measurements. This indicates that all the charge carriers were mobilized under these extreme conditions. In contrast, at more moderate bias potentials of 0.5 and 0.25 V vs. Ag/AgCl, although the absolute value of the photocurrents were obviously lower depending on the extraction voltage, much large enhancements by light were observed upon CO2 saturation of the electrolyte in comparison to N2 (Fig. 5). At 0 V bias potential vs. Ag/AgCl bias, no charge extraction was clearly observed and a change from positive to negative photocurrent beyond −0.25 V vs. Ag/AgCl that should be close to the conduction band potential of mp-C was measured.
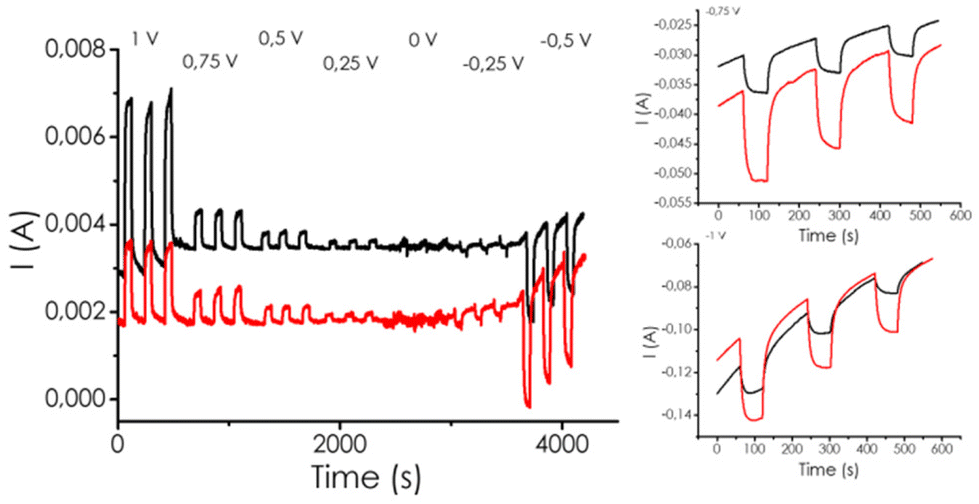 | ||
| Fig. 5 Photocurrent measurements, with bubbling in the electrolyte N2 shown in black and in CO2 in red. The right part corresponds to an expansion of the −0.5 V bias potential. | ||
Beyond this crossover polarization potential, the currents were negative and had a higher intensity in the presence of CO2.
While the photocurrents prove that mp-C undergoes charge separation upon illumination, polarization indicated that it is an n-type semiconductor, whereby the somewhat higher negativity in the presence of CO2 as compared to N2 indicated the occurrence of photocatalytic CO2 reductions, contributing to the process.
Transient absorption spectroscopy
Photoinduced charge separation kinetics and reactivity can be studied by transient absorption spectroscopy (TAS). Upon irradiation with a 355 nm laser pulse of a CH3CN suspension of mp-Cα, the generation of TAS spectra decaying in the microsecond time scale was observed for all the samples. The spectra consisted in all cases of a continuous absorption spanning the whole available monitoring wavelength (300–760 nm), reminiscent to that of the UV-Vis ground state. Fig. 6 shows a representative spectrum recorded 50 μs after the laser pulse. The temporal profile of all the wavelengths were coincident, indicating that they corresponded to a single species or if there were several, they decayed by annihilation and contributed to the absorption similarly. Fitting of the transient signal to first-order kinetics showed low residuals with a lifetime of 60 μs. Identical features, i.e. featureless continuous transient absorption with coincident temporal decays, were similarly recorded for mp-Cβ and mp-Cγ, with a somewhat lower initial signal intensity.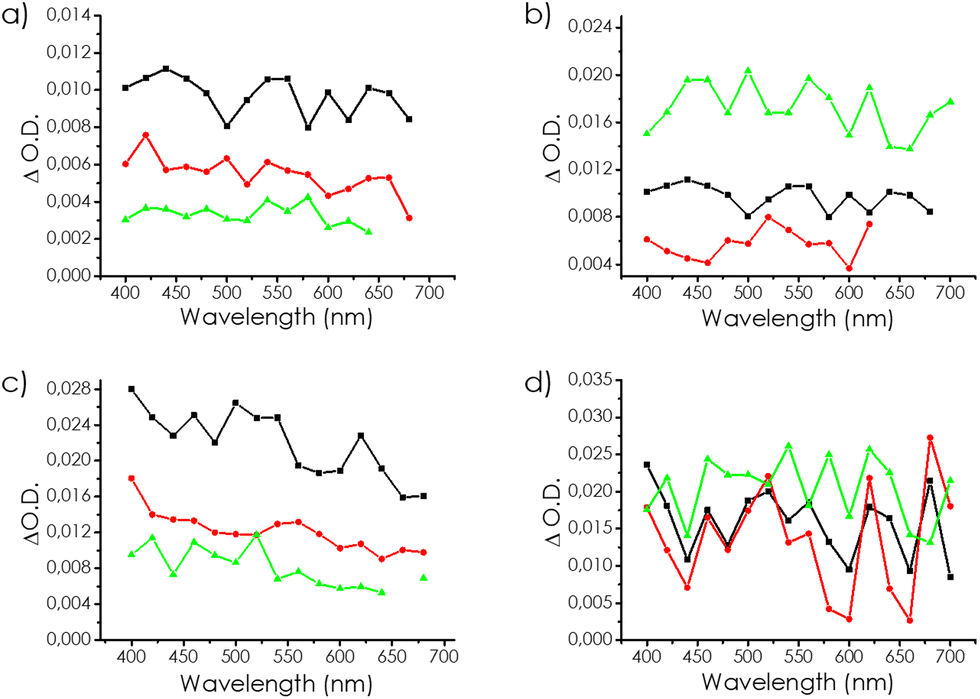 | ||
| Fig. 6 Spectra recorded at 50 μs of the samples mp-Cα (black), mp-Cβ (red), and mp-Cγ (green) in (a) N2 and (c) CO2; and the samples mp-Cα (black), mp(N)-Cα (red), and mp(P)-Cα (green) in (b) N2 and (d) CO2. | ||
To gain information on the nature of the signal, particularly if it corresponds to a charge-separated state, quenching of the signal by O2 was studied. Fig. S10 in the ESI† shows the influence of the quenchers on the transient signal monitored at 400 nm. In the case of mp-Cα, the effect of O2 quenching was marginal with some changes in the intensity and kinetics at 400 and 480 nm, but with no appreciable change in the signal at 640 nm. This trend for O2 quenching was also present, but more remarkable for mp-Cβ. We attributed the increase in the signal intensity in the 400–560 nm region to the prevalence of h+ absorption in this region. In the presence of O2, some photogenerated electrons will be quenched by O2, leading to a lesser prompting of e−/h+ recombination and, therefore, to an excess of h+, which will be reflected in an increase in the intensity of h+ from 400 to 560 nm. The fact that mp-Cβ was more affected than mp-Cα was due to the difference in pore size.
As already commented N2 is not a suitable gas to determine the surface area and N2 and O2 have similar kinetic diameters. To further check this proposal, while providing spectroscopic evidence of the reaction of CO2 with photogenerated e−, TAS measurements were also performed in the presence of CO2. A similar behaviour was observed for O2 and CO2 with a change in the intensity of the temporal profiles and kinetics of the signal in the 480–540 nm region, but no influence at longer wavelengths, which should be mostly due to the unreactive, trapped electrons. In good agreement with our proposal, similar quenching behaviours of O2 and CO2 were observed, as presented in Fig. 7. Notably, the effect of CO2 was even higher than that of O2, in spite of it being a worse electron quencher.
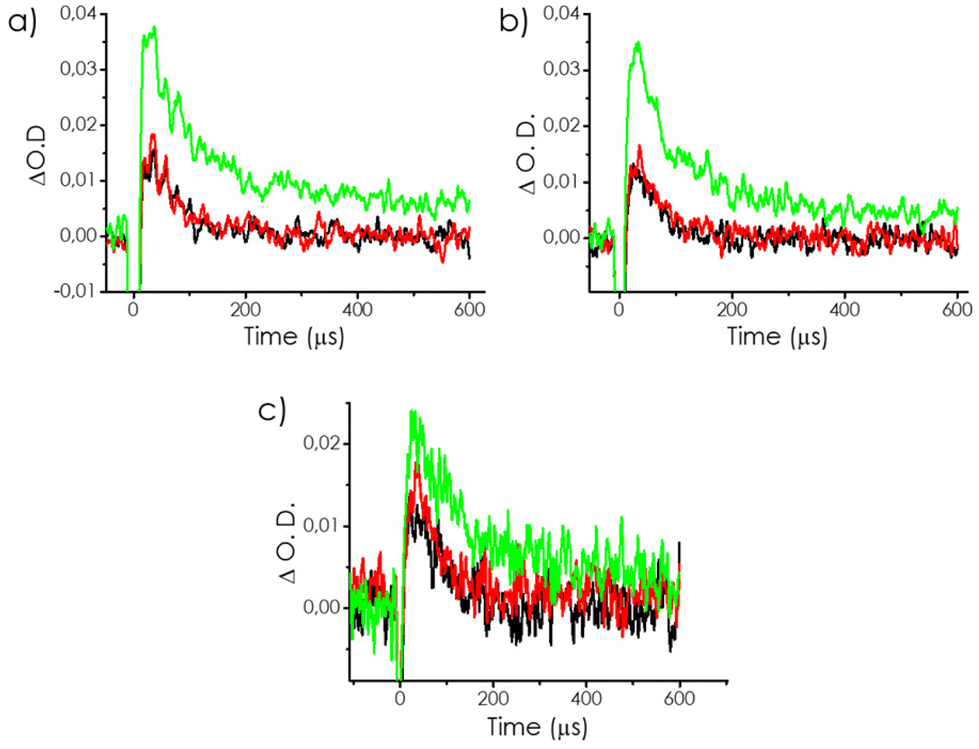 | ||
| Fig. 7 Deactivation kinetics monitored using N2 (black), O2 (red), and CO2 (green) at (a) 400, (b) 500, and (c) 600 nm. | ||
We attribute the higher influence of CO2 to its better ability to diffuse inside the pores of mp-C, as previously commented on when describing the specific surface area measurements by isothermal CO2 adsorption in comparison to N2 adsorption. Therefore, the even higher increase in the intensity of the transient signal at 400 nm was due to the better ability of CO2 to trap internal electrons in mp-Cα and mp-Cβ.
Regarding doped mp-(N)Cα, surprisingly its transient signal was very weak, meaning that not many e−/h+ reached the microsecond time scale and that the N promoted charge recombination. It is well-known in semiconductors that an excessive dopant population can promote charge recombination. Apparently, this should be the case of mp-(N)Cα. In contrast, although the signal for mp-(P)Cα was more intense than for mp-Cα and the quenching behaviour for O2 followed the same trend as for mp-Cα, no influence of the presence of CO2 could be monitored. This means that CO2 was not able to quench the photogenerated electrons.
Overall the results of the TAS study explain the photocatalytic activity results, particularly the lack of a positive influence of the dopant elements on the photocatalytic activity of mp-Cα, but for different reasons. In the case of N-doping this was due to the short lifetime of the charge separation and in the case of P-doping, it was due to the failure to transfer electrons to CO2.
Experimental section
Synthesis of the photocatalyst
First 0.40 mol of the selected precursor (α-cyclodextrin (>98%, Sigma-Aldrich), β-cyclodextrin (>97%, Sigma-Aldrich), or γ-cyclodextrin (>98%, Sigma-Aldrich)) was dissolved in 20 mL of ultrapure water. For the heteroatom doping, 60 mg of urea or 12 μL of H3PO4 was added to the solution too. After 24 h of stirring, it was dried at 60 °C in a silicone bath. The resulting white powder was pyrolyzed in a horizontal electrical oven by heating at 10 °C min−1 up to 900 °C for 2 h under an Ar flux of 200 mL min−1. Finally, it was milled for the following tests and characterizations.Physicochemical characterization
Raman spectra were recorded with 514 nm laser excitation on a Renishaw Raman spectrometer (“Reflex”) equipped with a Leica optical microscope and a charged coupled device camera. The laser power in the sample was 25 mW. Each spectrum was the average of 20 acquisitions at a resolution of 4 cm−1.The XRD pattern was recorded using a Cubix-pro PANalytical diffractometer in the range from 5° to 90° at a scan rate of 1° s−1.
Diffuse reflectance UV–vis spectra in the range of 200–800 were recorded on a Cary 5000 spectrophotometer from Varian.
Solid-state 31P NMR spectra were measured at room temperature using a Bruker AV400WB with π/2 pulse sequences of τ = 5 μs and a relaxation time of 5 s. The experiments were carried out with magic-angle spinning at the rate of 10 kHz. For obtaining the spectra, between 100 and 400 scans were accumulated.
The combustion elemental analyses were measured with a Euro EA 3000 analyzer.
TEM images were recorded on a JEOL JEM 2100F with a voltage of 200 kV coupled with an X-Max energy-dispersive X-ray detector (EDS). Also, FESEM images were obtained on a ZEISS ULTRA 55. Samples were prepared by casting one drop of the suspended material in acetonitrile (ACN) onto a carbon-coated copper TEM grid and allowing it to dry at room temperature.
Photocatalytic tests
The photocatalytic reactions were developed in a 51 mL quartz reactor fitted with a manometer, an inlet valve, and an outlet valve. The irradiation source was a 300 W Xe lamp (Hamamatsu, 1.6 sun power) with UV-Vis light range or a solar simulator (OrielTM, 1 sun power).The mp-C was dispersed in 30 mL of ACN (>99.9%, Sigma-Aldrich) using a sonic tip (FisherbrandTM Model 705 at 40% of 700 W for 1 h using a pulsation of 1 s on and 1 s off) in a concentration of 1.5 mg mL−1. Next, 10 mL of this suspension was introduced in to the reactor with 4 mL of triethanolamine (Sigma-Aldrich) as an electro donor and 4 mL of MilliQ water. To reach the 20 mL of mixture for the reaction, 2 mL more of ACN was added. The system was purged using pure CO2 for 10 min and pressurized until 1.4 bar (absolute pressure).
The gas products were analyzed using a gas chromatograph (Agilent 490 MicroGC) equipped with a molecular sieve 5 Å column with a TC detector and Ar as the carrier gas. Also for CO, a 7890A gas chromatograph was used equipped with a column Carboxen®-1010 PLOT L × I.D. 30 m × 0.53 mm, average thickness 30 μm with a TC detector and He as the carrier gas.
For the stability tests, the material was removed from the reactor between the reactions and washed three times by centrifugation (6000 rpm, 15 min) using MilliQ water before starting a new reaction.
The apparent quantum yield (AQY) was determined under irradiation with a 150 W Xe lamp equipped with a Czerny Turner monochromator. The AQY value was calculated by the equation:
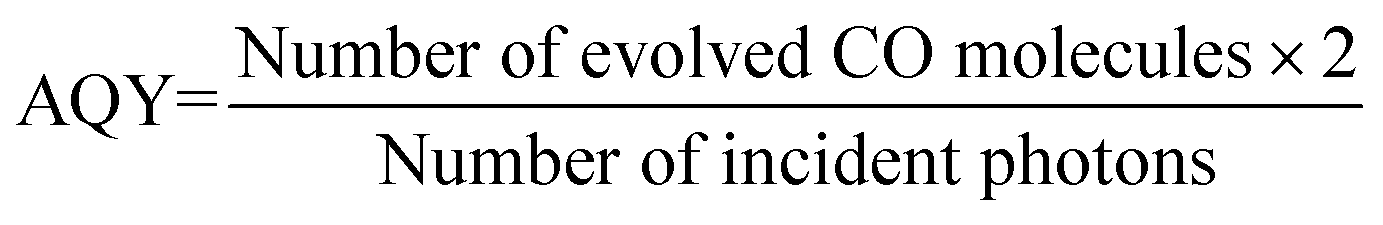
Photoelectrochemical measurements
Photoelectrochemical measurements were developed in a 3-electrode electrochemical cell using Ag/AgCl as the reference electrode, graphite as the counter electrode, and the mp-Cα deposited on fluorine-doped tin oxide (FTO) as the working electrode. A solution 0.5 M of KHCO3 was used as the electrolyte.During the measurements, N2 or CO2 were bubbled in the solution to ensure the electrolyte was completely saturated. Also, each measurement was done twice in two different electrodes to confirm the material behaviour.
Notice that the photocatalyst was deposited on the FTO using a mixture of 15 mg of ultrasonicated photocatalyst (the ultrasonication was developed in 15 mL of ethanol 40%, 1 h, 1 s on, 1 s off and dried at 60 °C) in 300 μL of ethanol and 75 μL of Nafion 5% (w![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :w). The electrode was dried at room temperature overnight. The characteristic of each electrode are presented in Table 2.
:w). The electrode was dried at room temperature overnight. The characteristic of each electrode are presented in Table 2.
| Electrode | Surface area (cm2) | Active mass (mg) |
|---|---|---|
| E1 | 0.7 | 3.4 |
| E2 | 0.65 | 5.3 |
For the chromoamperometric tests, the electrode was allowed to stabilize for 5 min at the selected potential (1 V, 0.75 V, 0.5 V, 0.25 V, 0 V, −0.25 V, −0.5 V, −0.75 V or −1 V vs. the reference electrode). After that, the electrode was irradiated for 1 min using the same Xe lamp that was used in the photocatalytic tests and it was allowed to relax for 2 min. This was repeated five times for each potential to confirm the reproducibility of the tests.
For the cyclic voltammetry tests, the scan speed was 0.02 V s−1, from 1 V to −1 V vs. the reference electrode, for five cycles.
Transient absorption measurements
Transient absorption spectra were recorded using the fourth harmonic of a Q switched Nd:YAG laser (Quantel Brilliant, 266 nm, 15 mJ per pulse, 7 ns FWHM) coupled to mLFP122 Luzchem miniaturized detection equipment. The studied suspension had a concentration of 0.5 mg mL−1 of the mc-Cα in ACN. This transient absorption spectrometer included a 300 W ceramic xenon lamp, 125 mm monochromator, Tektronix TDS-2001C digitizer, compact photomultiplier and power supply, cell holder and fibre-optic connectors, computer interfaces, and a software package developed in the LabVIEW environment from National Instruments. The laser flash generated 5 V trigger pulses with programmable frequency and delay. The rise time of the detector/digitizer was ∼3 ns up to 300 MHz (2.5 GHz sampling). The monitoring beam was provided by a ceramic xenon lamp and delivered through a fibre-optic cable. The laser pulse was probed by a fibre that synchronized the photomultiplier detection system with the digitizer operating in the pre-trigger mode.Conclusions
Microporous graphitic carbons derived from cyclodextrin exhibited activity as photocatalysts for CO2 reduction to CO accompanied by CH4 to some extent. The process took place concomitantly with H2 evolution. The most efficient material in the series was the one having the smallest pore size derived from α-cyclodextrin. This material constituted C at over 99% of its composition and it did not contain any dopant or metal, thus proving the possibility to develop only-carbon photocatalysts. The material exhibited an apparent quantum yield for CO formation of 0.82% and was stable under irradiation conditions. Photocurrent measurements revealed an enhancement of the cathodic photocurrent in the presence of CO2, while transient absorption spectroscopy revealed the quenching of a fraction of the photogenerated e− by CO2 in the submicrosecond time scale. No positive influence of N- or P-doping at the loadings studied was observed, although doping with other elements or co-doping could further enhance the photocatalytic activity for CO2 reduction. Overall the present results are a rare case of an almost all-carbon photocatalyst being able to reduce photocatalytically CO2 without the presence of any metal or even doping element.Author contributions
A. G.-M. prepared the materials and performed the photocatalytic measurements. J. A. recorded and analysed the transient spectra. A. M. A. did materials characterization. A. P. and H. G. conceptualized the work. The manuscript was written through the contribution of all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Spanish Ministry of Science and Innovation (Severo Ochoa, PID-2021-12607OB-C21 and RTI2018-89237-CO2-1) and Generalitat Valenciana (Prometeo 2021/083) are gratefully acknowledged. A. G.-M. thanks the Spanish Ministry of Science and Innovation for a postgraduate scholarship.Notes and references
- P. D. Tran, L. H. Wong, J. Barber and J. S. C. Loo, Energy Environ. Sci., 2012, 5, 5902–5918 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- K. Hashimoto, H. Irie and A. Fujishima, Jpn. J. Appl. Phys., 2005, 44, 8269–8285 CrossRef CAS.
- A. J. Bard, J. Photochem., 1979, 10, 59–75 CrossRef CAS.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro, M. Antonietti and H. García, Chem. Soc. Rev., 2017, 46, 4501–4529 RSC.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Rev., 2014, 114, 6179–6212 CrossRef CAS PubMed.
- D. Su, J. Zhang, B. Frank, A. Thomas, X. Wang, J. Paraknowitsch and R. Schlögl, ChemSusChem, 2010, 3, 169–180 CrossRef CAS PubMed.
- C. Li, Y. Xu, W. Tu, G. Chen and R. Xu, Green Chem., 2017, 19, 882–899 RSC.
- J. Albero, Y. Peng and H. García, ACS Catal., 2020, 10, 5734–5749 CrossRef CAS.
- H. Shen, T. Peppel, J. Stunk and Z. Sun, Sol. RRL, 2020, 4, 1900546 CrossRef CAS.
- J. Albero, D. Mateo and H. García, Molecules, 2019, 24, 906 CrossRef PubMed.
- Y. Peng, A. Rendón-Patiño, A. Franconetti, J. Albero, A. Primo and H. García, ACS Appl. Energy Mater., 2020, 3, 6623–6632 CrossRef CAS.
- A. Rendón-Patiño, A. Santiago Portillo, C. Vallés-García, M. Palomino, S. Navalon, A. Franconetti, A. Primo and H. Garcia, Small Methods, 2020, 4, 1900721 CrossRef.
- A. García-Mulero, A. Rendón-Patiño, A. M. Asiri, A. Primo and H. Garcia, ACS Appl. Mater. Interfaces, 2021, 13, 48753–48763 CrossRef PubMed.
- A. Rendón-Patiño, F. Torres, A. Primo and H. Garcia, Sustainable Energy Fuels, 2022, 6, 2170–2178 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr02655d |
| This journal is © The Royal Society of Chemistry 2022 |