
Low-dose exposure to phytosynthesized gold nanoparticles combined with glutamine deprivation enhances cell death in the cancer cell line HeLa via oxidative stress-mediated mitochondrial dysfunction and G0/G1 cell cycle arrest
Abhinav
Prasad
 a,
Ashapurna
Khatua
b,
Yugal Kishore
Mohanta
c,
Muthupandian
Saravanan
*d,
Ramovatar
Meena
*b and
Ilora
Ghosh
*a
a,
Ashapurna
Khatua
b,
Yugal Kishore
Mohanta
c,
Muthupandian
Saravanan
*d,
Ramovatar
Meena
*b and
Ilora
Ghosh
*a
aBiochemistry and Environmental Toxicology Laboratory, Lab#103, School of Environmental Sciences, Jawaharlal Nehru University, New Delhi 110067, India. E-mail: abhi9sesjnu2013@gmail.com; iloraghosh17@gmail.com
bNanotoxicology Laboratory, Lab#312, School of Environmental Sciences, Jawaharlal Nehru University, New Delhi 110067, India. E-mail: ashapurnakhatua9@gmail.com; ramovatarmeena@mail.jnu.ac.in
cDepartment of Applied Biology, School of Biological Sciences University of Science and Technology Meghalaya, Ri-Bhoi-793101, India. E-mail: ykmohanta@gmail.com
dAMR and Nanotherapeutics Laboratory, Department of Pharmacology, Saveetha Dental College, Saveetha Institute of Medical and Technical Sciences (SIMATS), Chennai 600077, India. E-mail: saravananm.sdc@saveetha.com
First published on 30th June 2022
Abstract
Cancer cells use nutrients like D-glucose (Glc) and L-glutamine (Q) more efficiently for their development. This increased nutritional dependency of malignant cells has been commonly employed in various in vitro and in vivo models of anticancer therapies. This study utilized a combination of a low dose (25 μg mL−1) of S2, a phytosynthesized gold nanoparticle (AuNP) that was previously proven to be non-toxic, and deprivation of extracellular glutamine as an anticancer strategy in the human cervical cancer cell line HeLa. We discovered that 24 h Q deprivation led to a less significant decrease in the viability of HeLa cells while a low dose of S2 caused a non-significant reduction in the viability of HeLa cells. However, combining these two treatments resulted in highly significant inhibition of cell growth, as measured by the MTT test and morphological examination. Glutamine starvation in HeLa cells was found to induce cellular uptake of S2 via clathrin-mediated endocytosis, thus facilitating the improved antitumor effects of the combined treatment. Flow cytometry-based assays using fluorescent probes H2DCFDA and MitoSOX Red confirmed that this combination therapy involved the development of oxidative stress conditions owing to a surplus of cytosolic reactive oxygen species (cytoROS) and mitochondrial superoxide (mtSOX) generation. Furthermore, the investigated combinatorial treatment also indicated mitochondrial inactivity and disintegration, as evidenced by the drop in the mitochondrial membrane potential (Δψm) and the decrease in the mitochondrial mass (mtMass) in a flow-cytometric assay utilizing the probes. Tetramethylrhodamine ethyl ester and MitoTracker Green FM, respectively. Cell cycle arrest in the G0/G1 phase, induction of cell death via apoptosis/necrosis, and inhibition of migration capacities of HeLa cells were also seen after the combined treatment. Thus, this research provides insight into a new combinatorial approach for reducing the dose of nanoparticles and increasing their efficacy to better inhibit the growth of human cervical cancer cells by leveraging their extracellular glutamine dependence.
1. Introduction
Nanotechnology, which comprises techniques of material design, synthesis and manipulation at the nanoscale (1–100 nm), has brought about new potential and remarkable progress in several scientific and industrial fields.1 Other than pharmaceutical and biomedical sciences, it finds application in cosmetics, food and feed, environmental remediation, optical devices, electronics (transistors, light emitters), photo-electrochemical technology, space, energy science, catalysis and chemical industries.2 Metal and metal–oxide nanoparticles (NPs) have been employed as medicines, drug–gene delivery systems, diagnostics, and biosensors to identify critical disease biomarkers in the fight against diseases like microbial infections and cancer.3 The effectiveness of these NPs lies in their high surface to volume ratio and high reactivity. However, there are certain drawbacks in the synthesis of these beneficial NPs, such as the employment of toxic reagents, use of sophisticated instruments, undesirable reactions with different biological membranes, generation of harmful by-products, and the lack of biocompatibility.4 These issues need to be addressed before their extensive use. Therefore, green nanotechnology methods that utilize the innovative idea of using living organisms, their biochemicals, biomolecules and biological waste materials as reducing and capping agents to reduce and cap precursor ions to metal/metal–oxide NPs during their intracellular or extracellular synthesis present a strong claim as a potential solution.NPs are traditionally synthesized by physical and chemical methods. Commonly used physical methods are thermal decomposition, gas condensation, chemical vapour deposition/condensation, electro-explosion, mechanical milling, laser ablation and sputtering, whereas chemical methods are chemical reduction, microemulsion, sol–gel synthesis, and the Brust–Schiffrin method.4,5 Chemical methods utilize chemicals such as trisodium citrate, sodium borohydride, hydrazine, formaldehyde, ethanol and ascorbic acid as reducing agents and PPI (polypropylene polybenzyl isocyanate), CTAB (cetrimonium bromide), PEG (polyethylene glycol), PAA (poly(acrylic acid)), TOP (trioctylphosphine), PAMAM (poly(amidoamine)), linoleic acid and oleic acid as capping agents.6 NPs synthesized by these methods are hazardous for human and environmental health. Green-synthesized inorganic NPs fabricated via relatively simple, reproducible, environmentally friendly and cost-effective biological techniques confer considerable enhancements in effectiveness in pharmaceutical and biomedical applications due to enhanced antioxidant, antimicrobial, and anticancer properties, as well as wound healing and biosensing capabilities,7 compared to their traditionally produced physical and chemical counterparts. The enhanced effectiveness of biologically synthesized NPs is due to the improvement in the solubility and bioavailability of phytochemicals and the safety, stability, cytocompatibility, biocompatibility, and pharmacokinetic profiles and systemic clearance of the NPs. Green metallic nanoparticles have shown the combined effect of both biochemicals and biomolecules in the extracts of biological resources and the inherent properties of the metallic counterparts.
Commonly used biological resources in green nanotechnology approaches for NP synthesis are plants, bacteria, fungi and algae.5 Among these, plants (herbs) have advantages over other resources due to the presence of various phytochemicals such as flavonoids, alkaloids, phenols, terpenes, saponins and proteins that not only act as reducing, capping and stabilizing agents but also possess established medicinal properties. Moreover, plant-derived bioactive molecules aid in increasing the reproducibility of size- and shape-controlled NPs.7
There has been steady development in cancer treatment and cure approaches. Despite this, the global incidence and mortality rates of cancer are increasing. Cancer remains the world's second leading cause of death, with 9.96 million deaths expected in 2020 (WHO fact sheets). Cervical cancer is the ninth most often diagnosed cancer, with a 3.1% incidence rate and a mortality rate of 3.4%; it is also the ninth most common cause of cancer death.8
Cancer cells undergo metabolic reprogramming, in which they rearrange their metabolism to maintain high metabolic demands for uncontrolled growth and survival under unfavorable circumstances.9 Cancer cells’ metabolic requirements alter as the disease progresses, and their vulnerabilities shift as well. Tumors in their early stages of growth require increased nutrient uptake and biosynthesis of macromolecules.10L-Glutamine (Q), along with D-glucose, is one of the two major nutritional substrates that meet the increased metabolic needs of many malignancies.11 Glutamine, the most prevalent amino acid in the human bloodstream, is a source of carbon and reduced nitrogen.12 As a source of reduced nitrogen, glutamine catabolizes to synthesize nucleotides, non-essential amino acids, proteins, and hexosamines.13–16 Glutamine, as a carbon source, helps cancer cells meet their bioenergetic and biosynthetic requirements by producing ATP, replenishing TCA cycle intermediates, producing glutathione, thus maintaining redox homeostasis, and synthesizing lipids via reductive carboxylation.13,17–20
Even though most cancer cells have a high rate of aerobic glycolysis (the Warburg effect), some cancer cells are glutamine-dependent for growth and survival, a phenomenon known as “glutamine addiction”.21–23 Extracellular glutamine addiction has been also discovered in the human cervical cancer cell line HeLa.24 In HeLa cells, mitochondrial oxidative phosphorylation (OXPHOS) was also shown to be increased to support their growth.25 Mitochondrial glutaminolysis leads to sequential conversion of Q, first to glutamate by glutaminases, and then to alpha-ketoglutarate (α-KG) by glutamate dehydrogenase 1 or aminotransferases/transaminases. This mitochondrial α-KG then enters the TCA cycle to produce ATP via OXPHOS and provide biosynthetic precursors essential for growth and proliferation.15,26 Thus, by Q deprivation, intervention in glutamine metabolism-related processes that contribute to tumorigenesis could provide novel approaches to improve cancer treatment.
Because of their added advantages, biogenic gold nanoparticles (AuNPs) have been shown to function as antibacterial, antifungal, anticancer, virucidal, and antiparasitic agents, and as sensors, catalysts, and agents in photodynamic and photothermal therapy.27–29 In our recent work, we found that out of the three categories of AuNPs (S1, S2, and S3) synthesized by changing the HAuCl4 concentration and utilizing the leaf extract of Pongamia pinnata, a plant with diverse medicinal properties such as antioxidant, anti-inflammatory, antidiabetic, anticonvulsant, antimicrobial, antiprotozoal, anthelminthic, insecticidal, anticancer and immunomodulatory activity,30 AuNP S2 exhibited the least anticancer activity in HeLa cells with IC50 >200 μg mL−1.31 Higher quantities of drugs or NPs can be harmful to non-targeted cells and tissues.
Therefore, in this work, we devised a strategy that combined a low dose of phytosynthesized AuNP S2 with a Q-depleted growth medium to enhance anticancer effects in HeLa cells by targeting Q addiction. To obtain insight into cellular mechanisms, we also looked at changes in cellular phenomena like cytoROS and mitochondrial superoxide production, status of Δψm, mtMass, and cell cycle profile, all of which are imperative in determining a cell's ability to proliferate and migrate. This research might add to our knowledge of strategizing a potential therapy for many cancer types, particularly those that demonstrate Q addiction.
2. Materials and methods
2.1. Chemicals and reagents
Dulbecco's modified Eagle's medium (DMEM), dimethyl sulfoxide (DMSO), monodansyl cadaverine (MDC), 2′,7′-dichlorofluorescin diacetate (H2DCFDA), carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP), and propidium iodide (PI) were bought from Sigma-Aldrich, USA. SRL, India, provided 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). Fetal bovine serum (FBS), dialyzed FBS, trypsin, tetramethylrhodamine ethyl ester (TMRE), MitoSOX Red, MitoTracker Green FM (MTG), and annexin V-FITC were purchased from Thermo Fisher Scientific.2.2. Phytosynthesized AuNP S2
The current investigation employed spherical AuNP S2 with an average size of 12 nm, produced by green synthesis utilizing 0.5 mM HAuCl4 and the leaf extract of Pongamia pinnata, as described in our earlier study.31 AuNP S2 was shown to have limited anticancer activity in HeLa cells, with IC50 >200 μg mL−1 at 24 h, in comparison with the other two AuNPs, S1 and S3. Relative to S1 and S3, S2 caused less morphological changes, less ROS production, S-phase cell cycle arrest, and extremely limited inhibition of cell migration in HeLa cells.31 As a result, we undertook this investigation to improve the anticancer effectiveness of AuNP S2.2.3. Cell culture and treatments
The HeLa cell line obtained from the National Centre for Cell Science, Pune, India, was maintained at 37 °C, in high glucose (HG) DMEM containing 25.5 mM Glc, 4 mM glutamine, 1 mM pyruvate, 10% (v/v) FBS, 100 U mL−1 penicillin, and 100 μg mL−1 of both streptomycin and amphotericin B in a humified incubator with 5% CO2. PBS-trypsin/EDTA was used to subculture the cells. Following the use of HG DMEM having 10% FBS for the first cycle of cell growth, experimental plates were seeded with an appropriate number of cells (passage number 2 to 4) in fresh HG DMEM having 10% dialyzed FBS and kept overnight to acclimatize the cells to the new serum type. Because FBS is an abundant source of growth factors and nutrients, standard FBS was replaced with dialyzed FBS to allow for a more thorough assessment of the effectiveness of glutamine deprivation and to lessen the FBS effects. There was no significant change in cell viability when HeLa cells were grown for 24 h in HG DMEM with 10% dialyzed FBS versus 10% standard FBS (data not shown).After PBS washing, the cells cultured in HG DMEM containing dialyzed FBS were treated for 24 h with 25 μg mL−1 S2, either in standard LG (low glucose) DMEM, containing the physiological concentration of 5.5 mM Glc, 4 mM glutamine, and 1 mM pyruvate or in Q-depleted LG DMEM, containing only 5.5 mM Glc, and 1 mM pyruvate. The cells grown in LG DMEM were considered as the control.
2.4. Assay for cell viability (MTT test)
The MTT test was used to assess the effect of a non-toxic dose of S2 (25 μg mL−1) and Q deprivation, either alone or in combination, on the viability of HeLa cells. This test was carried out as previously reported with minor changes.32 In a 96-well plate, HeLa cells were seeded at a density of 4 × 103 cells per well in HG DMEM with 10% dialyzed FBS and cultured overnight. The next day, the cells were exposed to S2 for 24 h as described in section 2.3. After the treatment period, the media was removed, and the cells were washed with PBS at room temperature (RT). Then 4 μL of MTT reagent (5 mg mL−1, prepared in PBS), diluted to 200 μL with serum-free medium (SFM), was added to each well and kept for 4 h in an incubator. After that, 100 μL of DMSO per well was added and the absorbance at 570 nm was measured using a SpectraMax M5 microplate reader (Molecular Devices, USA). The following formula was applied for calculating the percentage change in cell viability.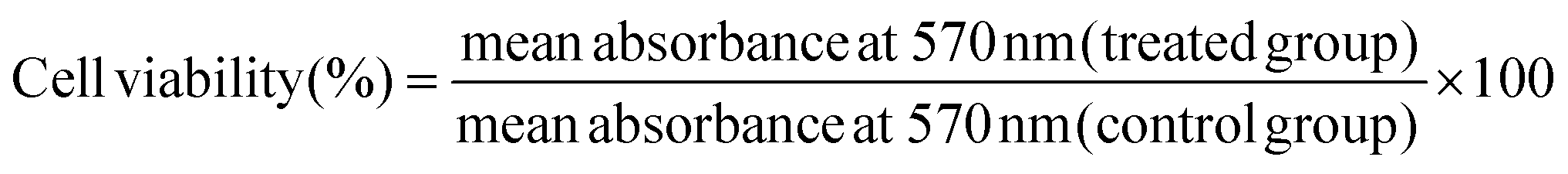
2.5. Morphological investigation
Morphological changes in the HeLa cells in the different exposure groups were investigated by seeding approximately 1 × 105 cells in each well of a 6-well plate and subjecting them to overnight growth. Then the 24 h treatment was carried out as described in section 2.3. An inverted phase-contrast microscope (EA-Prime, LMI Microscopes, UK) was used to capture the images of the cells at 10× magnification. Alterations in cell morphology were studied by comparing images of the treated groups with those of the control.2.6. Flow cytometric label-free assay for intracellular uptake of AuNP
A label-free flow cytometry-based method, which utilizes forward scattering (FSC) and side scattering (SSC) of light to determine the relative uptake of particles by cells, was employed to quantify the uptake of phytosynthesized AuNP S2 by HeLa cells under the experimental conditions.33,34 HeLa cells were seeded at 2 × 105 cells per well in 6-well plates and cultured for 24 h. Then the cells were exposed to AuNP S2 (25 μg mL−1) for 6 h in the presence or absence of glutamine in growth media as described in section 2.3. The inhibitor group included a 1 h pretreatment with an inhibitor of clathrin-dependent endocytosis, MDC (300 μM), before exposure to S2 in Q-deprived media. After that, the cells were washed, harvested by trypsinization, and suspended in ice-cold PBS, and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 events were recorded and analyzed for each sample on a BD FACS Verse™ flow cytometer (Becton, Dickinson and Company, Franklin Lakes, NJ, USA).
000 events were recorded and analyzed for each sample on a BD FACS Verse™ flow cytometer (Becton, Dickinson and Company, Franklin Lakes, NJ, USA).
2.7. Evaluation of cytosolic ROS generation
Flow cytometry was used to examine the effects of a low dose of AuNP S2 and glutamine deficiency on cytoROS production in HeLa cells. The amount of cytoROS is measured using the cationic free radical probe H2DCFDA, which rapidly diffuses into the cell. H2DCFDA is a non-fluorescent dye that turns intensely green fluorescent when it interacts with intracellular ROS in its 2′,7′-dichlorofluorescein (DCF) form after de-esterification. This test was carried out according to a methodology previously described31with certain modifications. 2 × 105 cells were seeded in a 6-well plate, cultured overnight, and then treated for 24 h, as stated in section 2.3. After treatment, the cells were rinsed in PBS at RT before loading for 30 min in the dark with 10 μM H2DCFDA diluted in SFM. After that, the cells were harvested using trypsin and washed with cold PBS, and DCF fluorescence from 10000 cells of each treatment group was captured in triplicate in the FITC (fluorescein-5-isothiocyanate) channel using a flow cytometer (BD FACS Verse). FCS Express™ v7 (De Novo Software, CA, USA) was used to analyze the data.35 The change in cytoROS levels in each treatment group compared to that in the control was assessed by comparing their 2-D histograms of count versus MFIs (median fluorescence intensities), which were plotted as overlays, and then statistically analyzing the change in MFIs of each sample, represented as ratios relative to the control.
2.8. Assay for determination of variation in mitochondrial membrane potential (MMP)
Relative quantification of changes in the MMP upon treatment of HeLa cells with a low dose of S2 in the presence or absence of glutamine was performed by flow cytometry using a standard protocol that included the TMRE probe.36 TMRE is a positively charged, lipophilic, red-orange fluorescent dye that readily permeabilizes and accumulates in active mitochondria with an intact membrane potential. When there is a loss of Δψm, this dye seeps out of the cell and exhibits compromised staining leading to the loss of fluorescence. Therefore, the measurement of TMRE fluorescence produced by cells is utilized for the relative quantification of Δψm. The technique of sample preparation for assessing the status of the Δψm by flow cytometry was similar to the sample preparation for cytoROS assessment as described in section 2.7, except for the probe, which was TMRE used at 100 nM in this case.2.9. Assessment of mitochondrial ROS (mtROS) production
Flow cytometry was used to examine the effect of low-dose AuNP S2 and glutamine depletion on mitochondrial reactive oxygen species production in HeLa cells. MitoSOX Red is a cationic, live-cell permeant mitochondrial superoxide indicator used to measure mtROS.37 MitoSOX can be taken up by actively respiring mitochondria because of the positive charge given by the triphenylphosphonium substituent. MitoSOX Red is a non-fluorescent dye that turns red fluorescent inside the mitochondria when another constituent, dihydroethidium, is oxidized by the mitochondrial superoxide, resulting in the red fluorescent molecule 2-hydroxyethidium, with excitation and emission maxima of 510 and 580 nm, respectively.38 This assay was carried out according to a previously published protocol, with minor modifications.39 2 × 105 cells were seeded in a 6-well plate, cultured overnight, and then treated for 24 h as stated in section 2.3. After treatment, the cells were rinsed in PBS at RT before loading with 2.5 μM MitoSOX Red in SFM for 30 min in the dark. After that, the cells were harvested using trypsin and washed with cold PBS, and fluorescence from 10000 cells of triplicate sample for each group was captured in the PE (phycoerythrin) channel using a flow cytometer (BD FACS Verse™). FCS Express™ v7 (De Novo Software) was used to analyze the data.40 The percentage of MitoSOX Red positive cells (cells with high MitoSOX Red fluorescence and hence enhanced mtROS) and MitoSOX Red negative cells (cells with low MitoSOX Red fluorescence and thus low mtROS) were gated as the P1 and P2 cell populations, respectively. By comparing the P1 (or P2) of each sample, the change in mtROS levels in each treatment group compared to the control was statistically evaluated.
2.10. Determination of mitochondrial mass
Flow cytometry using a Δψm-independent probe, MTG, was used to quantify relative changes in the mtMass (volume) in HeLa cells after treatment with a low dose of S2 combined with Q deprivation. MTG turns green fluorescent when it accumulates in the lipid environment of mitochondria and binds to free thiol groups of cysteine residues in mitochondrial proteins.41 When a mitochondrial breakdown occurs, this dye seeps out of the cells. Thus, the MTG fluorescence generated by a cell's mitochondria has been utilized to represent the mtMass or volume, and a reduction in MTG intensity may indicate mitochondrial damage.41,42 With minor variations, a standard methodology was followed.38 2 × 105 cells were seeded in a 6-well plate, cultured overnight, and then treated for 24 h as stated in section 2.3. Following treatment, the cells were rinsed in PBS at RT before loading 50 nM MTG in SFM for 30 min in the dark. After that, the cells were harvested using trypsin and washed in cold PBS, and fluorescence of 10000 cells from each triplicate was recorded for each treatment group in the FITC channel using a flow cytometer (BD FACS Verse™). FCS Express™ v7 software was used to analyze the data. The percentage of MTG positive cells (cells with high MTG fluorescence and thus increased mtMass) and MTG negative cells (cells with low MTG fluorescence and thus decreased mtMass) in the cell population were represented by gates P1 and P2, respectively. By comparing the P1 (or P2) of each sample, the change in the mtMass content in each treatment group compared to that of the control was statistically evaluated.
2.11. Analysis of cell cycle profile
Flow cytometry was used to analyze the distribution of cell cycle phases in HeLa cells due to different treatments for 24 h. With minor modification in our previously described protocol,43 samples fixed in 70% ethanol (v/v) and stained with a staining solution (prepared in PBS) containing 50 μg mL−1 PI, 100 μg mL−1 RNase A, and 0.1% Triton X-100, were acquired at a low flow rate in a flow cytometer. During analysis, only singlet cells gated on the PI-area versus PI-width dot plots were considered for cell cycle profiling using FCS Express™ v7 (De Novo software).2.12. Annexin V-FITC/PI assay for detection of apoptosis/necrosis
To investigate whether the cell death due to various treatments involved apoptosis and/or necrosis, flow cytometry-based assay utilizing double staining with FITC conjugated annexin V and PI was performed as per the manufacturer's protocol. Briefly, cell pellets obtained by the centrifugation of trypsinized samples from the control and treated groups were washed with cold PBS and resuspended in annexin-binding buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2, pH 7.4) to get ∼1 × 106 cells per mL. To each 100 μL of this cell suspension, 5 μL of annexin V-FITC and 1 μL of the 100 μg mL−1 PI working solution (prepared in annexin-binding buffer) were added and incubated at RT for 15 min. Then 400 μL of annexin-binding buffer was added to this sample, mixed gently, kept on ice, and fluorescence from at least 10000 cells was measured immediately on a BD FACS Verse™ flow cytometer in the FITC and PE channels. Appropriate single-color controls were used for the compensation of fluorescence spillover from FITC into the PE channel. Quadrant gate was applied on the annexin V FITC-area versus PI-area scatter plots of each group to obtain four quadrants, LL (lower-left, annexin V-FITC negative/PI negative), LR (lower-right, annexin V-FITC positive/PI negative), UL (upper-left, annexin V-FITC negative/PI-positive), and UR (upper-right, annexin V-FITC positive/PI-positive) which located the viable cells, cells in early apoptosis, cells in late apoptosis, and necrotic cells, respectively.
2.13. Scratch assay/in vitro cell migration assay
In vitro cell migration/scratch/wound healing test was used to assess cell migration or wound healing capacity.31 In a 6-well plate, 3 × 105 cells per well were grown until 80% confluency. Then, using a sterile 200 μL pipette tip, a scratch/wound was created in each well and treated for 24 h as described in section 2.3. Images obtained using an inverted phase-contrast microscope (EA-Prime, LMI Microscopes, UK) at 10× magnification at 0 h (before treatments) and 24 h (after completion of treatments) were used to measure the wound area. The percentage decrease in the wound area was calculated with respect to the 0 h value of the individual samples.2.14. Statistical and data analysis
Each experiment was carried out three times, mostly in triplicate. The changes were expressed as mean ± standard deviation (SD). One-way ANOVA with Tukey's post hoc test determined the significance of differences in means across groups, using the GraphPad Prism 5 software (San Diego CA, USA). The difference between the experimental and control groups (LG DMEM) was considered significant at p < 0.05. The flow cytometry data were analyzed using the software BD FACSuite™ v1.0 and FCS Express™ v7.10.0007, research use only (RUO) edition (De Novo Software, CA, USA).3. Results
3.1. Assessment of the combined effect of glutamine deprivation and low-dose AuNP S2 exposure on HeLa cell viability
Firstly, to determine how much dependent HeLa cells are on glutamine for growth and viability, we performed the MTT test on cells grown in a Q-depleted medium while maintaining the concentrations of the other two key nutritional substrates, glucose and pyruvate, unchanged. The MTT test indicated that completely removing Q from the growth media for 24 h reduced cell viability by 15 ± 4.07% (p < 0.05). Low-dose (25 μg mL−1 ≪ IC50) S2 exposure had no effect on HeLa cell viability (reduction by 9.57 ± 4.48%). When cells were fed with the same low-dose S2 in a Q-depleted growth medium, however, a highly significant decrease (p < 0.001) in cell viability was found (38.67 ± 4.66%) (Fig. 1).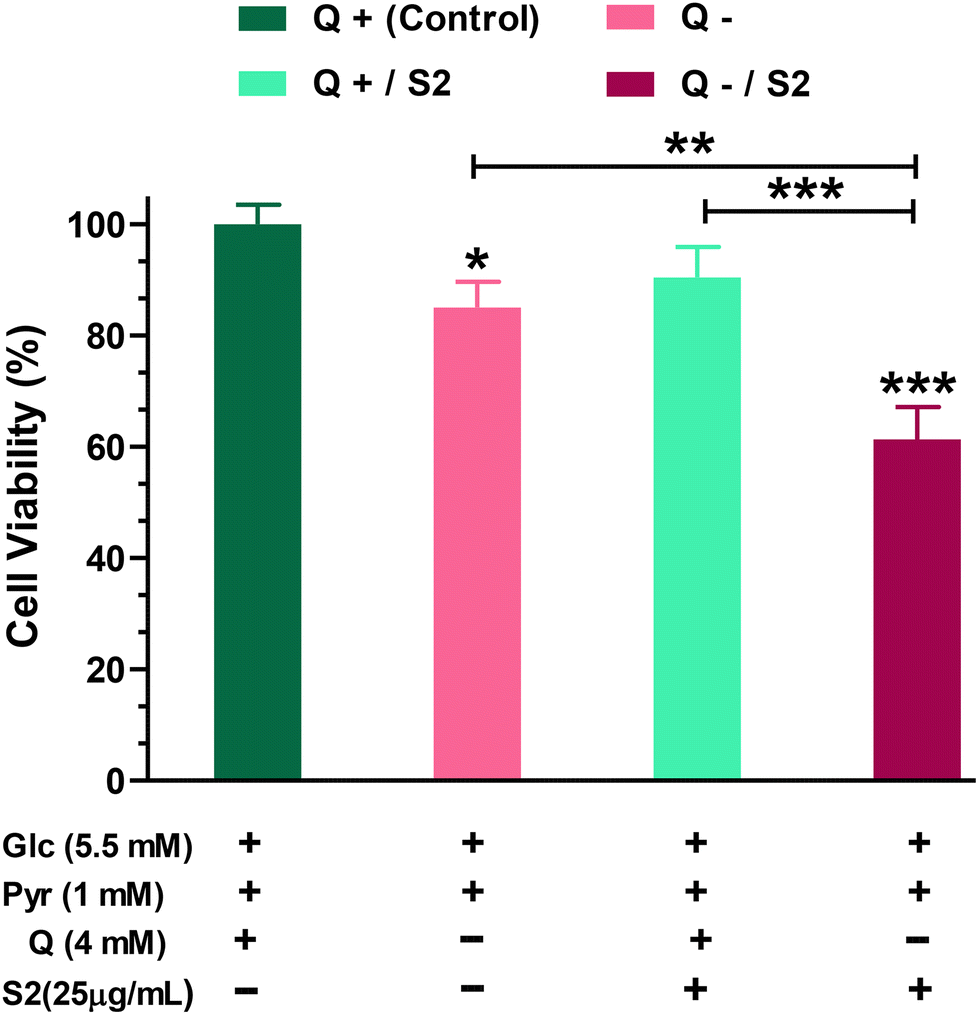 | ||
| Fig. 1 Effect of glutamine deprivation and low-dose AuNP S2 on cell viability in HeLa cancer cells after 24 h, either alone or in combination. Glc = D-glucose, Pyr = Sodium pyruvate, Q = L-glutamine. *p < 0.05, **p < 0.01, ***p < 0.001 compared to the control or between the indicated groups, after applying one-way ANOVA-Tukey's multiple comparison test of significance, on data displayed as mean ± SD (n = 3). | ||
3.2. Examination of morphological changes
The morphological examination showed that compared to the standard size and shape of the HeLa cells (Fig. 2a), Q deprivation for 24 h resulted in cell shrinkage in certain cells, as well as the appearance of some round and dead cells (Fig. 2b). Treatment with 25 μg mL−1 AuNP S2 led to a modest modification in the morphology of these cells in the form of elongated and spiked cells, with a bulging appearance in some of the cells (Fig. 2c). Treatment of HeLa cells with 25 μg mL−1 S2 in Q-deficient growth medium resulted in the greatest variations in size and morphology, with many small, rounded, floating, and dead (apoptotic) cells, as well as some significantly shrunken cells, and very few cells with a normal morphology (Fig. 2d).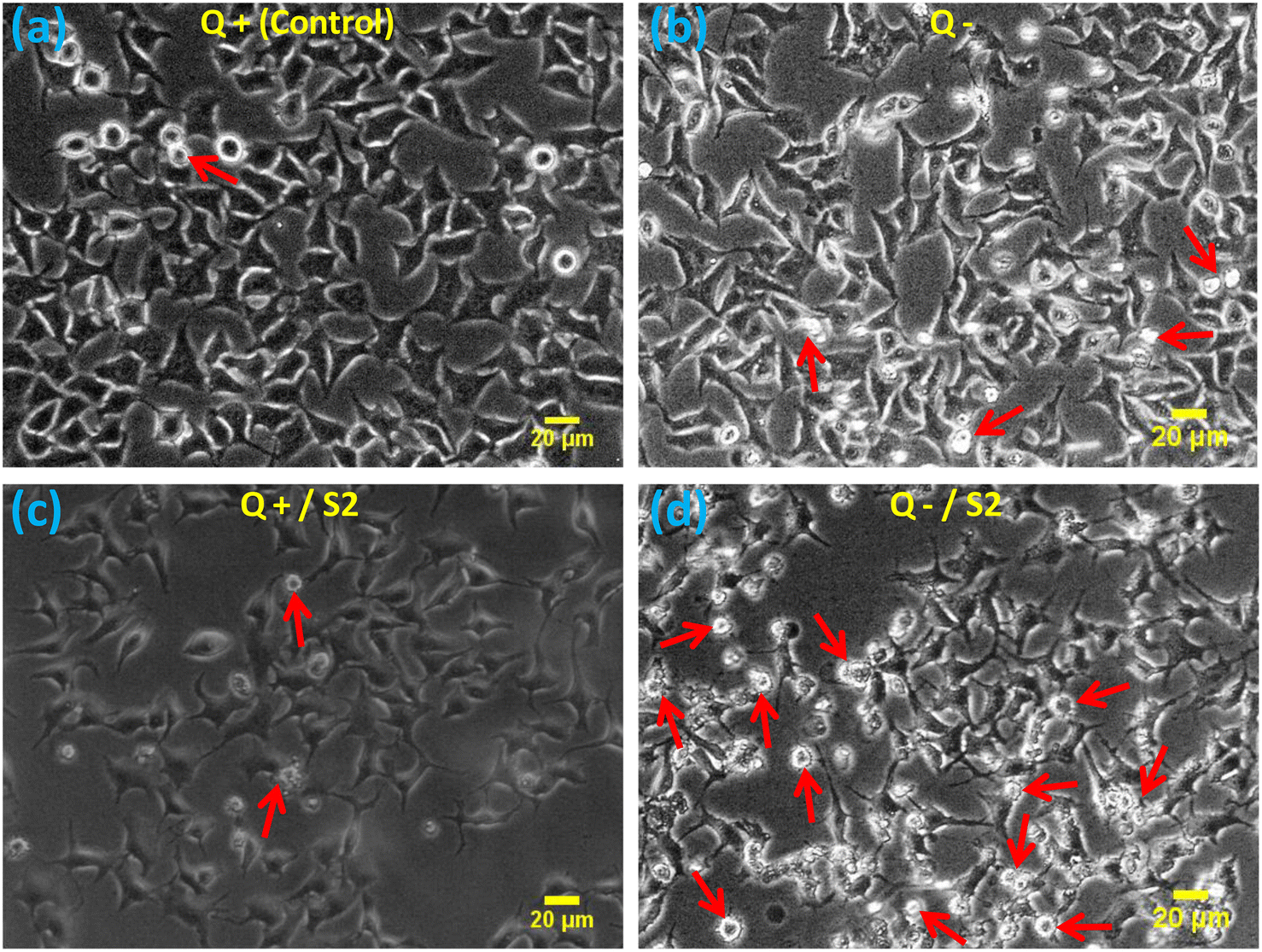 | ||
| Fig. 2 Effect of AuNP S2 on HeLa cell morphology after 24 h, either alone or co-treatment with glutamine deprivation. Red arrows indicate the apoptotic cells. Scale bar = 20 μm at 10× magnification, Q = L-glutamine, + = present, − = absent. | ||
3.3. Determination of cellular uptake of AuNP S2 by label-free flow cytometry
Metallic particles, especially Au, show high scattering properties.44 FSC and SSC provide information on cell size and granularity or internal complexity, respectively. Therefore, the examination of SSC intensity by flow cytometry has been utilized to measure the relative uptake of AuNPs and some other metallic NPs by the cells at varying doses or in different conditions.45–47 Changes in the FSC versus SSC double scatter dot plots in response to the treatment provide additional information on the health of the cell population or individual cells, such as cellular stress and cell damage. Quantification of the uptake of NPs by cells without the utilization of fluorescent dyes offers certain advantages. For example, misinterpretation of the uptake due to fluorescence signals from unbound or labile dyes can be avoided.48 The shift in count versus SSC histograms of different treatment groups relative to the control is shown in Fig. 3a. The extent of rightward shift and increase in the area of these shifted histograms denote relative fold increase in SSC as depicted in Fig. 3b. Compared to the control, the absence of glutamine caused a 1.16 ± 0.03-fold increase (p < 0.01) in SSC, whereas 25 μg mL−1 S2 in Q-supplemented media led to only a 1.10 ± 0.02-fold increase (p < 0.05) in SSC (Fig. 3a and b). However, HeLa cells exposed to S2 under Q deprivation resulted in a 1.3 ± 0.01-fold increase in SSC (p < 0.001), while pretreatment with an inhibitor of clathrin-mediated endocytosis (MDC) in this group showed a 1.07 ± 0.01-fold decrease (p < 0.001) in SSC (Fig. 3a and b). For all the treatment groups, no significant shift in count versus FSC histograms (Fig. 3c) and corresponding fold change in FSC (Fig. 3d) relative to the control was observed. Variability in the response of the HeLa cells to S2 exposure in growth conditions with or without glutamine was also represented as the percentage of cells showing high SSC (cell population P2). In the control, P2 was found to be 7.93% (Fig. 3e). Q deprivation resulted in the increment of P2 to 19.60% (p < 0.05) (Fig. 3f). In contrast, treatment with 25 μg mL−1 S2 in Q-depleted media led to a surge in P2 to 36.88% (p < 0.001) (Fig. 3h) from 11.89% in Q-supplemented media (Fig. 3g), while MDC pretreatment prior to exposure to S2 in media lacking glutamine reduced the percentage of cells with high granularity to 10.45% (Fig. 3i). Non-significant change in FSC and SSC was detected in vehicle control (methanol) for MDC samples (data not shown). These results confirm that Q deprivation for 6 h facilitates induction of uptake of S2 in HeLa cells. The unchanged FSC in all treatment conditions of this study indicated that cell size or cell viability remains unaffected. Thus the change in SSC was primarily due to the uptake of S2 or, to some extent, the development of cellular stress in HeLa cells. Under varied treatment conditions, differential uptake and accumulation of AuNP S2 within the HeLa cell population can be related to cells in different cell cycle phases.49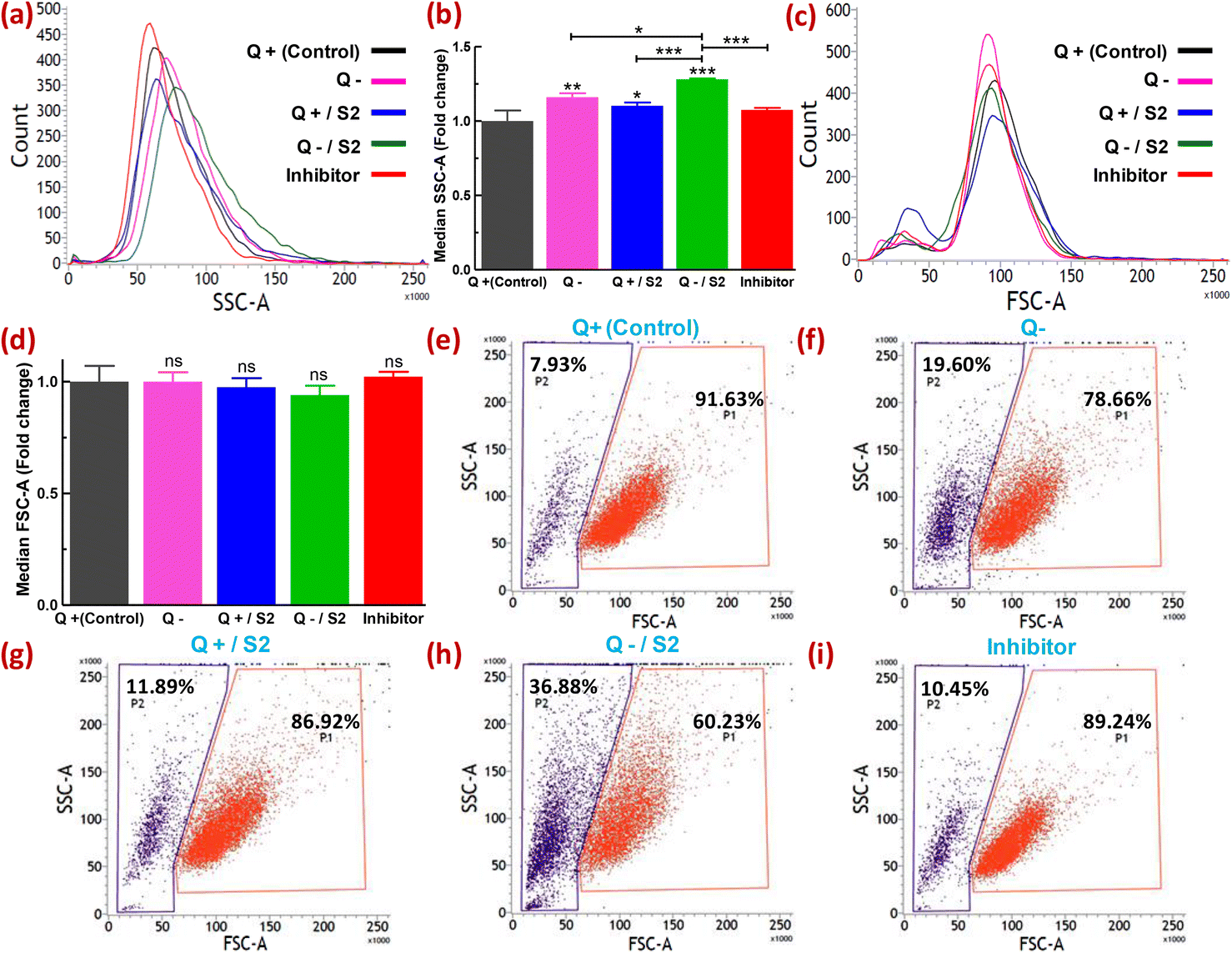 | ||
| Fig. 3 Induction of uptake of S2 in HeLa cells by glutamine deprivation at 6 h, determined by label-free flow cytometry. (a) Representative overlaid histograms showing a shift in count vs median SSC-area histograms of each treatment group from control. (b) The bar graph shows the statistical significance of the changes indicated in (a) as a fold relative to control. (c) Representative and overlaid count vs median FSC-area histograms of each group. (d) Bar diagram for assessment of changes shown in (c) as a fold relative to the control; ns = non-significant. (e−i) Representative dot plots for each group showing P1 and P2 (gated cell populations representing the percentage of cells with high FSC and high SSC, respectively). Inhibitor = Inhibitor of clathrin-mediated endocytosis (monodansyl cadaverine, 300 μM, 1 h). Q = L-glutamine, + = present, − = absent. *p < 0.05, **p < 0.01, ***p < 0.001. Asterisks just over the vertical and horizontal bar in (b) represent the level of significance compared to control and between the indicated groups, respectively. | ||
3.4. Assessment of cytosolic ROS generation
Excess ROS-mediated cell death has been linked to the effect of exposure to a range of biogenic NPs or a nutrient-depleted environment in several cancer cells.50–52 So, we were also curious about the state of cytoROS after treatment with AuNP S2 in glutamine-deprived HeLa cells. We used a flow cytometric technique based on the H2DCFDA probe to assess the state of cytoROS. On observing the DCF fluorescence of each sample, it was clear that both treatments (Q deficiency alone and exposure to 25 μg mL−1 S2 in glutamine-containing medium for 24 h) resulted in less significant changes in cytoROS generation (increments of 1.27 ± 0.14 and 1.42 ± 0.11-fold, respectively, compared to the control) (Fig. 3a and b). The combination of 25 μg mL−1 S2 and 24 h Q deprivation resulted in increased and highly significant cytoROS production (1.96 ± 0.02-fold increase relative to the control, p < 0.001) (Fig. 3a and b). The positive control for this experiment was cells exposed to 200 μM H2O2 for 1 h, which revealed an increase in ROS production similar to the treatment with 25 μg mL−1 S2 in Q-deficient growth medium (1.71 ± 0.06-fold increase compared to the control) (Fig. 3a and b).3.5. Measurement of the mitochondrial membrane potential
Healthy mitochondria have a high trans-membrane potential under normal circumstances.53 Excess ROS causes mitochondrial permeability transition by targeting the mitochondrial membrane's permeability transition pore, resulting in Δψm dissipation.54–56 We used a TMRE probe-based flow cytometric assay to see if the high ROS generated by exposing HeLa cells to a modest dose of AuNP S2 and Q starvation resulted in a collapse in the membrane potential across mitochondria. Assessment of each sample's TMRE fluorescence showed that Q deprivation for 24 h resulted in a non-significant drop in Δψm (1.30 ± 0.05-fold decrease compared to the control, p > 0.05) (Fig. 4c and d). This finding was in line with that of Gwangwa et al., who found that in triple-negative breast cancer cells, MDA-MB-231, Δψm remained unchanged after a less significant (p < 0.05) increase in ROS generation at 24 h.51 Limited exposure to S2 (25 μg mL−1 for 24 h) in glutamine-supplemented medium resulted in no meaningful change in the mitochondrial membrane potential (1.15 ± 0.12-fold reduction relative to the control, p > 0.05) (Fig. 4c and d). A treatment plan that included exposing cells to 25 μg mL−1 S2 and Q depletion for 24 h resulted in a significantly enhanced decrease of the MMP (by 1.74 ± 0.16-fold compared to control, p < 0.05) (Fig. 4c and d). The positive control for this experiment was cells treated to 50 μM FCCP for 30 min, which revealed the largest and most significant drop in Δψm (TMRE MFI ratio = 3.16 ± 0.29-fold compared to the control, p < 0.001) (Fig. 4c and d). | ||
| Fig. 4 Combined effect of glutamine deprivation and low-dose AuNP S2 on cytosolic ROS production and Δψm in HeLa cells at 24 h. (a) Representative overlaid histograms of DCF MFIs from various treatment groups. (b) Bar diagram for comparing changes in DCF MFIs indicated in (a), as a fold increase relative to control and the significance of such changes. (c) Overlapping histograms of TMRE MFIs from various treatments as a measure of Δψm. (d) Bar diagram for a comparative assessment of changes in TMRE MFIs shown in (c), as a fold decrease relative to control and the significance of those changes. Q = L-glutamine, + = present, − = absent. *p < 0.05, **p < 0.01, ***p < 0.001 in comparison with the control or between the indicated groups, following a one-way ANOVA- Tukey's multiple comparison test to evaluate the significance of data reported as mean ± S.D. (n = 3). | ||
3.6. Determination of mitochondrial ROS status
Under physiological conditions, healthy mitochondria produce a basal level of superoxide. So, we used a MitoSOX Red probe-based flow cytometric assay to see if the high ROS generated when HeLa cells were exposed to a low dosage of S2 and Q starvation came solely from the cytosol or if mitochondria-specific ROS also played a role in the generation of overall oxidative stress. By analyzing the fluorescence of MitoSOX Red from each sample, the control sample was found to include only 10.97 ± 0.05% cells with high mtROS (Fig. 5a). In contrast, Q deficiency for 24 h resulted in a less significant increase in the number of cells with abundant mtROS (increase in cells with strong MitoSOX Red fluorescence to 21.79 ± 3.42% from the control) (Fig. 5b and f). Limited exposure to S2 (25 μg mL−1 for 24 h) in Q-containing media did not result in a substantial increase in mitochondrial superoxide generation (increase to only 15.08 ± 1.24% from 10.97 ± 0.05%) (Fig. 5c). Treatment of cells with 25 μg mL−1 S2 in Q-depleted media for 24 h resulted in a highly substantial rise in the mtROS (increase in cells with high MitoSOX Red fluorescence to 58.74 ± 6.08% from the control, p < 0.001) (Fig. 5d and f). The positive control for this experiment was the cells treated with 0.1 μM rotenone for 24 h, which demonstrated the highest and most significant increase in mtROS production (increase in cells with high MitoSOX Red fluorescence to 78.69 ± 2.94% from 10.97 ± 0.05%, p < 0.001) (Fig. 5e and f). | ||
| Fig. 5 Status of mitochondrial ROS after 24 h exposure of HeLa cells to low-dose AuNP S2 under glutamine deprivation. Representative histograms showing variations in MitoSOX Red fluorescence in various treatment groups. (a) Glutamine present (Control). (b) No glutamine. (c) S2 in the presence of glutamine. (d) S2 in the absence of glutamine. (e) Positive control (0.1 μM rotenone, 24 h). (f) Bar graph showing the significance of the changes in data of groups (a–e). Q = L-glutamine, + = present, − = absent. P1 = gate indicating percentage of cells with MitoSOX Redhigh fluorescence (high mtROS), P2 = gate representing percentage of cells with MitoSOX Redlow fluorescence (low mtROS). *p < 0.05, ***p < 0.001 versus control or between the indicated groups, after using one-way ANOVA and Tukey's post hoc test to determine the significance of variation in data shown as mean ± SD (n = 3). | ||
3.7. Assessment of variation in mtMass
We next used an MTG probe-based flow cytometric assay to explore whether the development of oxidative and mitochondrial stress conditions due to elevated cytosolic and mitochondrial ROS, after HeLa cells had been exposed to 25 μg mL−1 S2 combined with Q deprivation for 24 h, affected mitochondria mass. An assessment of MTG fluorescence from each sample resulted in 97.30 ± 1.43% cells with a high mtMass in the control sample (Fig. 6a). Intriguingly, Q deprivation led to a 12.56 ± 3.98% decrease in the percentage of cells with a high mtMass relative to the control, although this decrease was statistically less significant (p < 0.05) (Fig. 6b). S2 exposure alone did not result in a substantial reduction in mitochondrial volume (P1 decreased to 92 ± 0.89% from control) (Fig. 6c and f). The combination of 25 μg mL−1 S2 and Q deprivation for 24 h led to a highly significant loss of mtMass (p < 0.001), as indicated by a reduction in a cell population with strong MTG fluorescence to 65.78 ± 2.20% from 97.30 ± 1.43% in the control (Fig. 6d and f). Cells in the positive control group were treated with 0.1 μM rotenone for 24 h, resulting in the most significant drop in mtMass (decrease in P1 by 32.93 ± 3.75% over the control, p < 0.001) (Fig. 6e and f).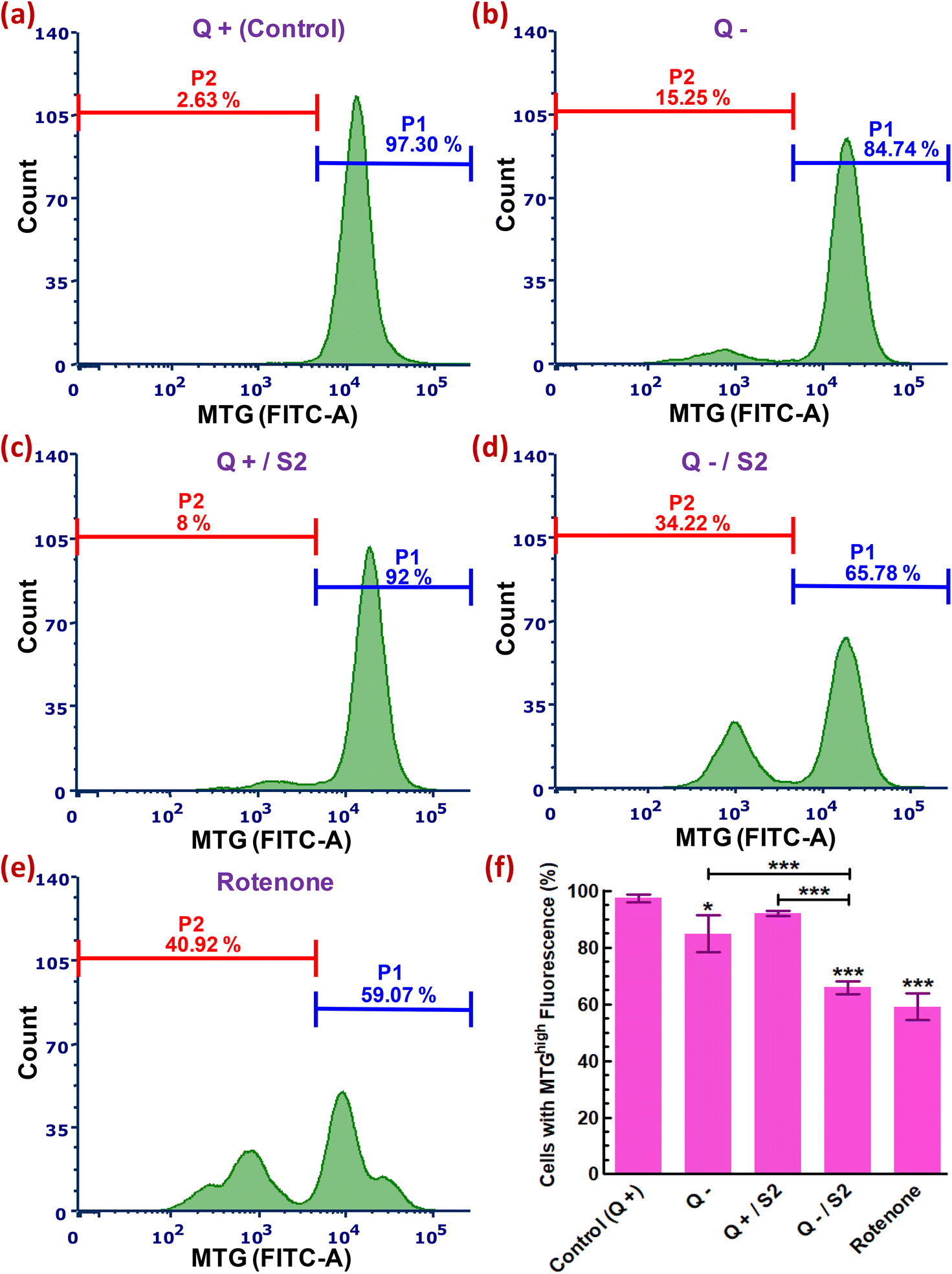 | ||
| Fig. 6 Relative quantification of mitochondrial mass in HeLa cells treated for 24 h with 25 μg mL−1 of AuNP S2 under glutamine deprivation. Representative histograms showing variations in MitoTracker green fluorescence in various treatment groups. (a) Glutamine present (Control). (b) No glutamine. (c) S2 in presence of glutamine. (d) S2 in absence of glutamine. (e) positive control (0.1 μM rotenone, 24 h). (f) Bar graph showing significance of the changes in data of groups (a–e). Q = L-glutamine, + = present, − = absent. P1 = percentage of cells with MTGhigh fluorescence (high mtMass), P2 = percentage of cells with MTGlow fluorescence (low mtMass). *p < 0.05, ***p < 0.001 versus control or between the indicated groups, after performing one-way ANOVA- Tukey's multiple comparison test on data reported as mean ± S.D (n = 3). | ||
3.8. Cell cycle phase distribution profiling
Next, we analyzed the flow cytometry-based distribution profile of cell cycle phases after subjecting the HeLa cells to various treatments. Either Q deprivation for 24 h or treatment with AuNP S2 for the same duration in the presence of glutamine caused S-phase arrest of HeLa cells, as indicated by their highly significant increase (p < 0.001) from 29.5 ± 1.2% (control) to 50.5 ± 3.0% and 45.0 ± 3.0%, respectively (Fig. 7a, b, c and e). Consequently, these two treatment groups also exhibited a reduction in the accumulation of G0/G1-phase cells. But, the effect was more pronounced (p < 0.001) only in the Q depleted conditions. However, both of these exposures resulted in a highly substantial decrease (p < 0.001) in the percentage of G2/M-phase cells. The combination of Q starvation with 25 μg mL−1 S2 led to a highly significant increase (p < 0.001) by 16.1 ± 2.2% in G0/G1-phase cells over the control (59.3 ± 2.1%), and consequently, an exceedingly significant reduction (p < 0.001) in the S-phase cells from 29.5 ± 1.2% to 19.6 ± 3.4% and the G2/M-phase from 11.3 ± 0.9% to 5.0 ± 1.2%, thus inducing G0/G1 arrest (Fig. 7a, d and e). In Fig. 7, the decreased cell count on the Y-axis of the treated groups, relative to the control group, in the order Q−/S2 > Q− > Q+/S2 > Q+ (control) indicates fewer cells that can emit PI fluorescence corresponding to the G0/G1, S, and G2/M phases of the cell cycle, and consequently more cells with sub-G0/G1 DNA content (not shown), that mark DNA damage.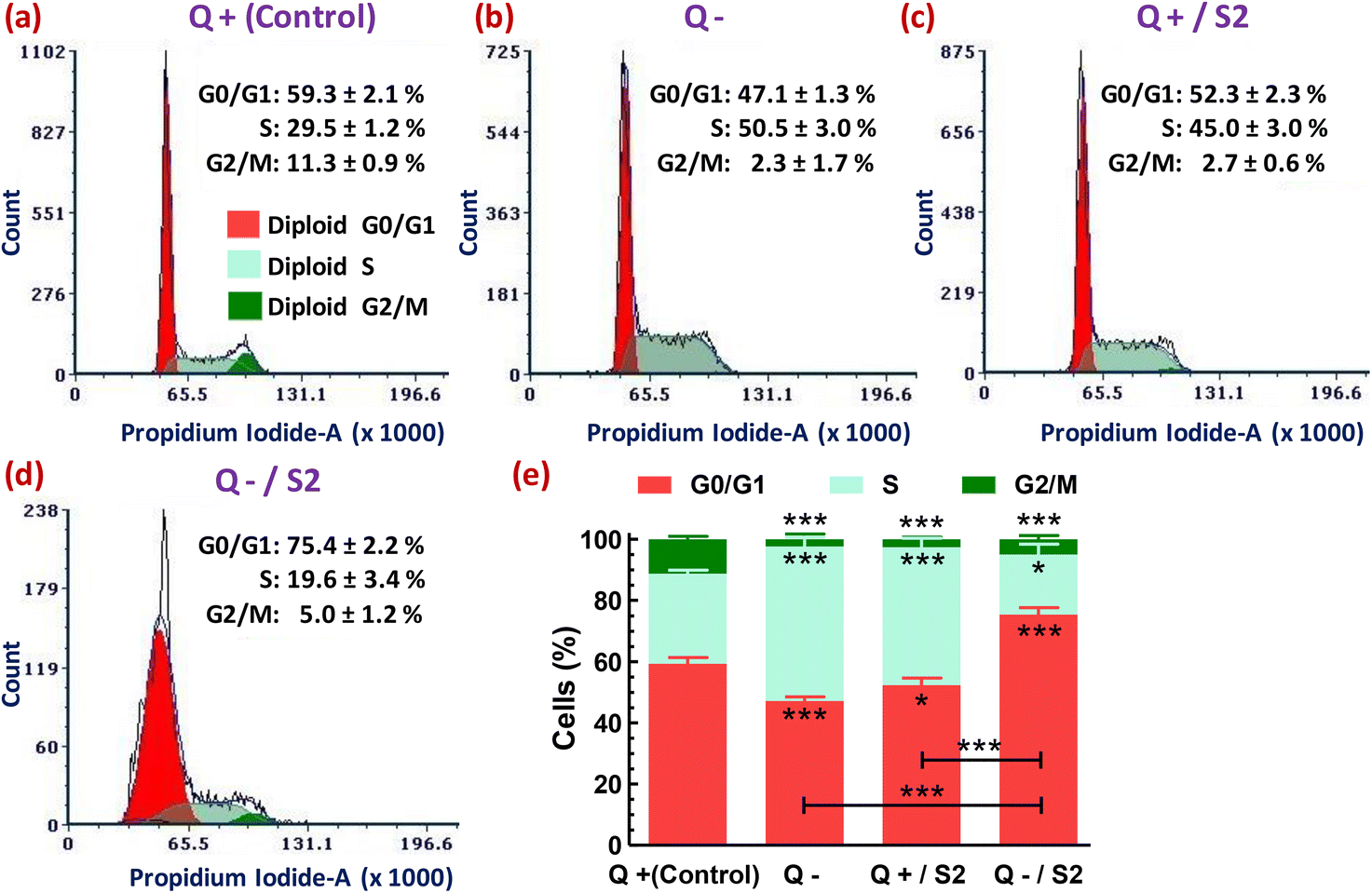 | ||
| Fig. 7 Cell cycle distribution profile in HeLa cells after 24 h of glutamine deprivation and AuNP S2 treatment either alone or in combination. Representative histograms showing cell cycle phase-wise variations in propidium iodide fluorescence in various treatment groups. (a) Glutamine present (Control). (b) No glutamine. (c) S2 in the presence of glutamine. (d) S2 in the absence of glutamine. (e) Stacked bar graph showing the significance of the changes in data of groups (a–d). In (e), asterisks just below (G0/G1 and S-fraction) or above (G2/M fraction) the error bars of red, light green, and dark green-colored vertical bars, and just above the horizontal black-colored bars represent the level of significance compared to respective fractions in the control group and between the indicated groups, respectively. Q = L-glutamine, + = present, − = absent. *p < 0.05, ***p < 0.001 versus control or between the indicated groups. | ||
3.9 Determination of mode of cell death
To determine whether the reduction in cell viability and proliferation brought about by AuNP S2 in Q-starved growth media also led to cell death by apoptosis/necrosis, annexin V-FITC/PI-based flow cytometric assay was performed. The evaluation of the percentage of cells showing variation in FITC and/or PI fluorescence resulted in determining the proportion of viable, early and late apoptotic, and necrotic cells in different treatment groups. The control group contained 98.6 ± 0.6% viable, 0.8 ± 0.3% late apoptotic, and 0.6 ± 0.1% necrotic cells (Fig. 8a). The proportion of viable cells decreased, and late apoptotic/necrotic cells increased in the remaining groups with the most significant percentage reduction (of viable cells) or increase (of late apoptotic/necrotic cells) following the order, rotenone (positive control) > Q−/S2 > Q− > Q+/S2 (Fig. 8e, d, b, c and f). However, the Q+/S2 group displayed the highest number (21.3 ± 1.6%) of early apoptotic cells, followed by the Q−/S2 (17 ± 2.4%), rotenone (16.3 ± 2.5%), Q− (8.4 ± 0.7%) and control (0.3 ± 0.2%) groups.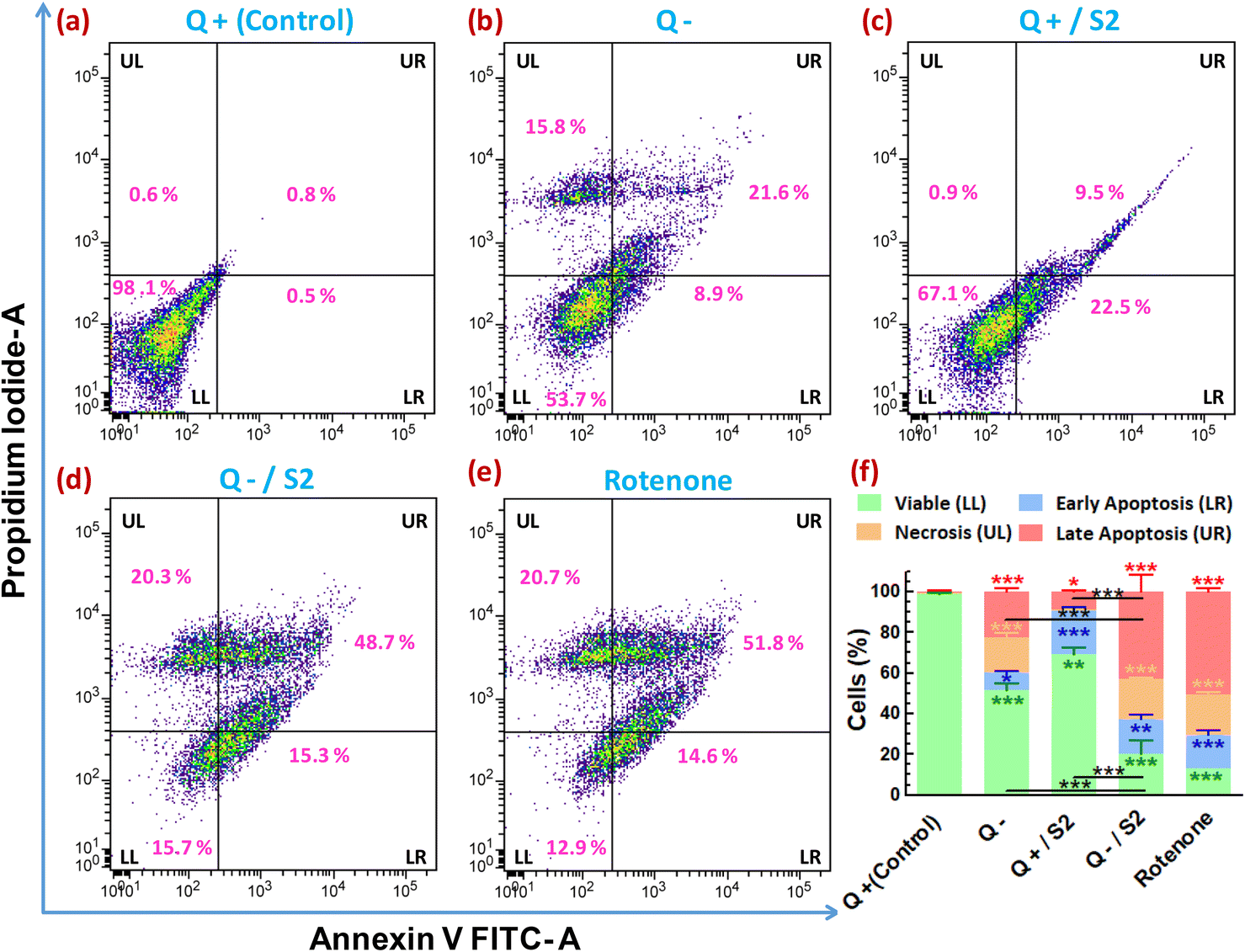 | ||
| Fig. 8 Confirmation of cell death by apoptosis in HeLa cells due to combined treatment of Q-depletion and low-dose AuNP S2 for 24 h. Flow cytometry of Annexin V FITC-PI double-stained cells was used to confirm the apoptotic-necrotic mode of cell death. Representative Annexin V FITC-area versus PI-area density plots of different treatment groups with percentages of cells at different stages of apoptosis shown in the relevant quadrants. Each density plots is divided into four quadrants, namely LL (lower-left), LR (lower-right), UL (upper-left), and UR (upper-right). LL quadrant is Annexin V-FITC negative/PI negative and contains viable cells, whereas LR quadrant is Annexin V-FITC positive/PI negative and contains cells that are in early apoptosis. Cells undergoing necrosis are seen in the UL quadrant, Annexin V-FITC negative/PI-positive. UR quadrant is positive for both Annexin V-FITC and PI and represents cells in late apoptosis. (a) Glutamine present (Control). (b) Glutamine absent. (c) 25 μg mL−1 of S2 in the presence of glutamine. (d) 25 μg mL−1 of S2 in the absence of glutamine. (e) 0.5 μM of rotenone for 24 h, positive control (f) Stacked bar diagram showing percentages of viable cells, cells in different stages of apoptosis and necrotic cells, shown as mean ± SD of three independent experiments, and level of statistical significance of the changes observed in data of groups (a–e). A significant change in UL, LR, UL, and UR fractions (represented by a vertical bar of green, blue, saffron, and red color, respectively) of test groups compared to control, is denoted by asterisks of color similar to the color of respective vertical bars. Non-black colored asterisks just below (LL and LR fractions) or above (UL and UR fractions) the error bars and black-colored asterisks just above the horizontal bars represent the level of significance compared to respective fractions in control and between the indicated groups, respectively. Q = L-glutamine, + = present, − = absent. *p < 0.05, **p < 0.01, ***p < 0.001 versus control or between the indicated groups. | ||
3.10 Effect of AuNP S2 combined with Q deprivation on HeLa cell migration
Finally, the migratory capacity of HeLa cells was assessed to determine the effectiveness of the strategy used in this investigation for inhibiting HeLa cell growth. In vitro wound healing or cell migration assay evaluated the migratory potential of cells subjected to diverse treatments. In the control group, cells migrated from the margins of the wound resulting in a substantial (p < 0.001) decrease of 27.42 ± 4.98% in wound area (width), compared to that at 0 h (Fig. 9a). In contrast to the Q-deprived sample, where a reduction in wound area by 19.91 ± 6.4% (p < 0.01) was observed at 24 h relative to that at 0 h (Fig. 9b), suggesting a comparatively hindered cell migration, a 24 h treatment with S2 in normal growth conditions reduced wound area by 23.68 ± 4.41% relative to 0 h (p < 0.01) (Fig. 9c), suggesting significant cell migration. Exposure of HeLa cells to S2 for 24 h in Q-depleted growth media resulted in a minimal and non-significant reduction in wound area of only 8.60 ± 4.41%, indicating the treatment's effectiveness in suppressing HeLa cell growth and migration.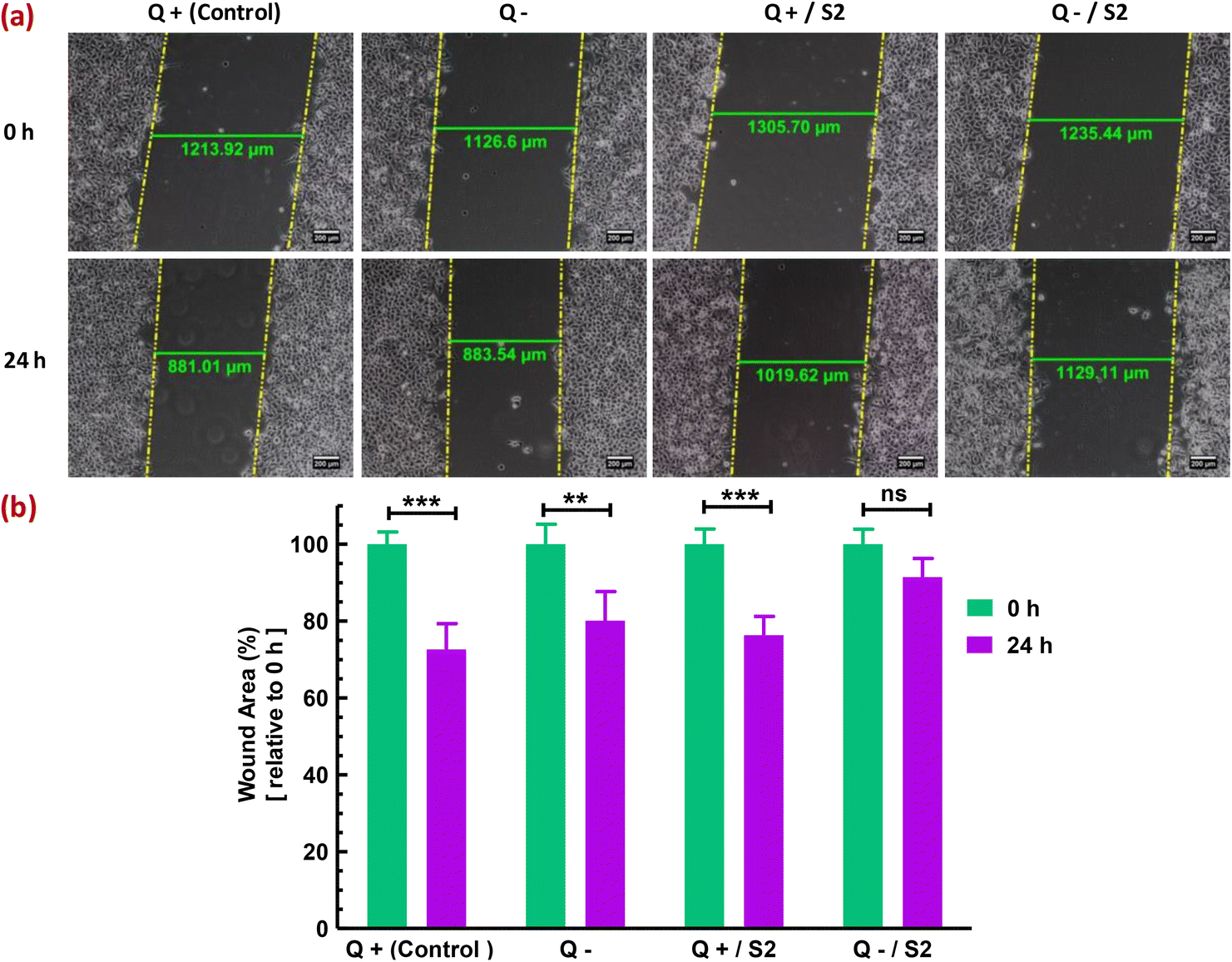 | ||
| Fig. 9 Effect of low-dose AuNP S2 on the in vitro migratory properties of HeLa cells in the presence or absence of glutamine. The wound width (area) was used to assess cell migration and wound healing capacity. Wounds were created with a sterile 200 μL tip, and images were taken with an inverted phase-contrast microscope at 10× magnification at 0 and 24 h. (a) Representative images from three separate studies demonstrated differences in wound area after 24 h of treatment with 25 μg mL−1 of AuNP S2 in the presence or absence of Q, relative to the control. (b) Bar diagram for a comparative analysis of changes in wound width indicated in (a) and the significant level of those changes expressed as mean ± SD (n = 3). + = present; − = absent; Q = L-glutamine, **p < 0.01, ***p < 0.001 signifies level of significance at 24 h versus 0 h. | ||
4. Discussion
For inhibiting cancer cell growth and proliferation, glucose and glutamine metabolism has been targeted by many researchers. Similarly, in recent times, utilization of metallic NPs prepared by green synthesis methods using biological resources as reducing and capping agents has also been proposed and is the focus of many anticancer strategies because of their advantages. Research has shown that biogenic AuNPs synthesized using extracts of biological materials with medicinal features can control the growth and proliferation of cancer cells in a cell-type-dependent manner. However, their regulatory roles and mechanisms in inhibiting cervical cancer cells in a growth environment devoid of extracellular glutamine are still unknown. Therefore, in this study, we explored the potential of a combination therapy that utilized 24 h starvation of glutamine and non-toxic dose (25 μg mL−1) of AuNP S2, a green synthesized nanomaterial from our recent study, possessing anticancer activity with 24 h IC50 >200 μg mL−1 for checking the growth of an in vitro model of cervical cancer, HeLa cells.29 We also aimed to uncover the cellular mechanisms involved in the improved antitumor effects of the studied strategy (Scheme 1).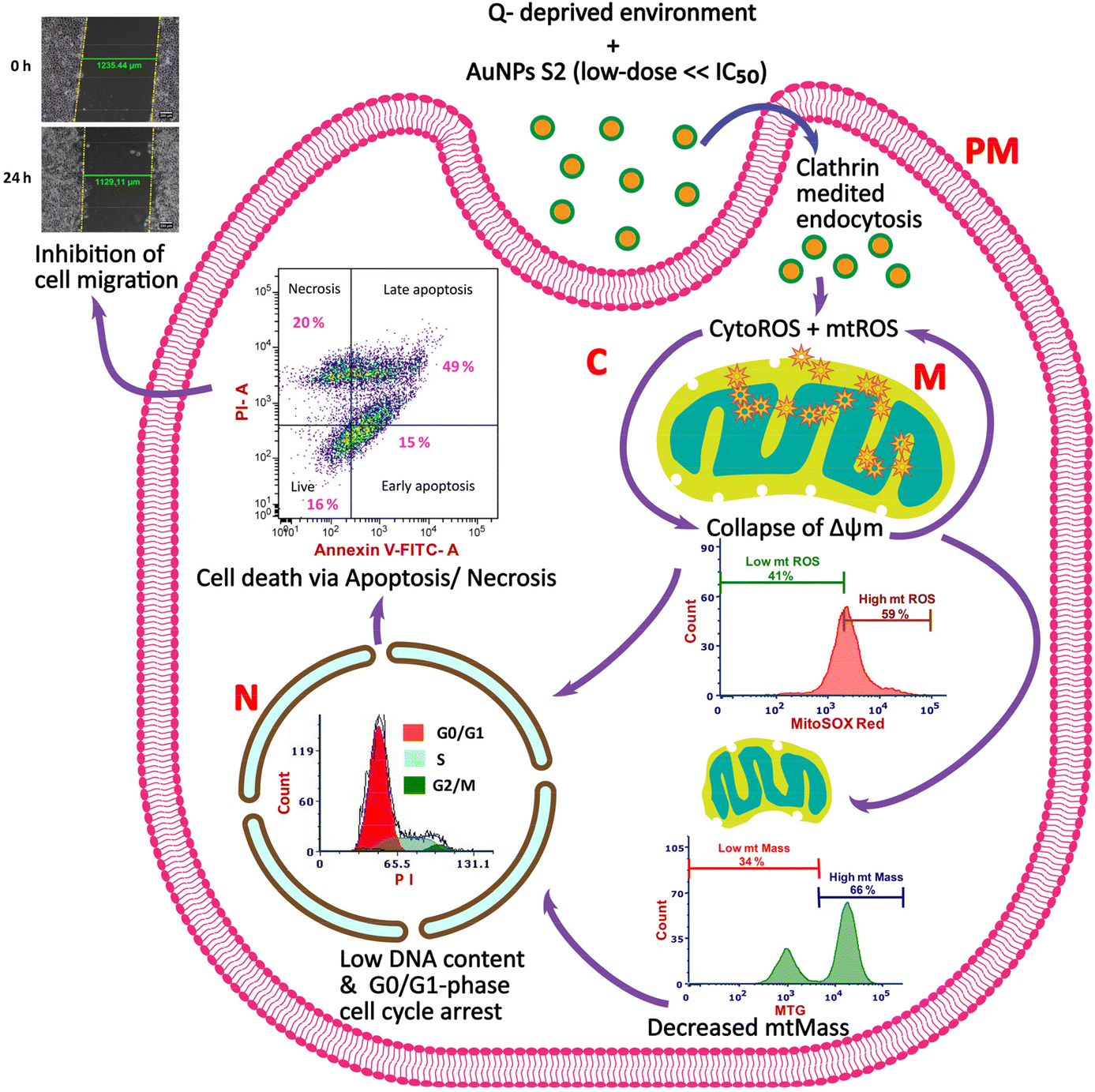 | ||
| Scheme 1 The proposed anticancer mechanisms of non- toxic dose of phytosynthesized AuNPs (S2) combined with glutamine (Q) deprivation in HeLa cells. PM, plasma membrane; C, cytosol; M, mitochondria; N, nucleus; CytoROS, cytosolic reactive oxygen species; mtROS, mitochondrial reactive oxygen species. | ||
Our results from the MTT assay demonstrate that Q withdrawal for a day sensitized the HeLa cells treated with a low dose of AuNP S2 to a significant reduction in cell viability which got reduced by 29% and 24% more than the alone exposure of AuNP S2 and Q depletion, respectively. There are some identifiable morphological characteristics of a cell that is under stress or has undergone the process of cell death due to some treatments, viz., formation of echinoid spikes, membrane blebbing, cell or cytoplasmic shrinkage, nuclear condensation, and formation of the apoptotic body.57,58 So, to support our findings from the cell viability assay, we investigated the morphological alterations using phase-contrast microscopy. The results revealed that 24 h Q deprivation caused cytoplasmic shrinkage and nuclear condensation and the formation of the apoptotic body in only a very small proportion of cells. However, these morphological features, along with the presence of echinoid spikes, got amplified in a substantially larger number of cells when Q depletion was combined with 25 μg mL−1 AuNP S2. Exposure to 25 μg mL−1 AuNP S2 in Q-containing media only led to some cells with an elongated shape and echinoid spikes, primarily characteristic of cells undergoing some form of stress. These observations best explained the inhibitory potential of the proposed combination approach in HeLa cells.
The efficiency of cellular uptake and route of internalization of NPs are among the most crucial factors that determine their toxicity in a cell. Both of these factors themselves are influenced by several other factors, namely particle size, shape, morphology, surface charge, surface chemistry, capping agents, particle hydrophobicity/hydrophilicity, cell type, and extra/intracellular environment that determine biological activity of inorganic NPs.59,60 We also provide evidence that chemopotentiation of a limited dose of AuNP S2 in HeLa cells depended on its intracellular internalization efficiency. Certainly, Q deprivation facilitated a higher degree of cellular uptake of AuNP S2 by HeLa cells, thus conferring a persistent metabolic oxidative stress that culminates in cell death. Furthermore, the utilization of MDC as an inhibitor of clathrin-mediated endocytosis confirmed that internalization of AuNP S2 occurred via clathrin-dependent receptor-mediated endocytosis.
The presence of moderately elevated levels of ROS in cancer cells promotes cell proliferation and differentiation. However, the generation of intracellular ROS in excess amount is expected to cause oxidative damage to nucleotides, lipids, and proteins.61 Therefore, many studies have utilized the modulation of oxidative stress conditions as a therapeutic strategy for cancer inhibition. Glutamine plays an important role in the maintenance of ROS homeostasis by supporting the production of reducing powers, glutathione and nicotinamide adenine dinucleotide phosphate (NADPH), enhancing the activity of ROS scavenger glutathione peroxidase 1 and activating Nrf2 and Wnt3a/β-Catenin antioxidant signalling.12,62,63 Therefore, Q depletion in cancer cells can disrupt the redox homeostasis and sensitize them to oxidative stress-mediated cell death even in the presence of drugs/NPs with less potential of inducing intracellular ROS. In this study, combining low-dose AuNP S2 having reduced capacity to generate cytoROS with Q deprivation significantly enhanced their cytoROS production capability to promote excess ROS-induced inhibition of HeLa cell growth and proliferation.
Mitochondria are of utmost importance for a cancer cell because they provide oncogenic signals, supply ATP, the building block for new cells, and regulate redox homeostasis and apoptosis.64 So, altering mitochondrial health and function provides therapeutic opportunities to potentiate the inhibition of tumor cell proliferation. Together with cytoROS, cancer cells also contain an increased amount of mtROS. Mitochondria-associated ROS are also important signaling molecules that can act as mitogens and contribute to a tumor cell's neoplastic features. However, similar to cytoROS, when the generation of mtROS is too high, it is detrimental to the cell and can induce cell death by apoptosis or necrosis.65 Oxidative stress due to high cytoROS and mtROS can induce the collapse of Δψm and activate the opening of the mitochondrial permeability transition pore that causes the release of mitochondrial cytochrome c into cytosol and initiation of caspase-dependent apoptosis via the activation of caspase-3.66 ROS are also known to cause caspase-independent apoptosis that involves the release of the mitochondrial apoptosis-inducing factor (AIF) into the nucleus. Oxidative stress-induced activation of c-Jun N-terminal kinase could also cause apoptosis by the phosphorylation-mediated inactivation of anti-apoptotic proteins and activation of apoptotic proteins.67 Therefore, we wanted to identify the effects of our strategy on the MMP, mtROS, and mtMass in HeLa cells. Our results indicated that 24 h Q starvation alone could not alter the MMP, whereas it produced the mtROS and reduced the mtMass only to a less significant level. These findings were in line with other reports on similar deprivation strategies in neuroblastoma, colon carcinoma, and hepatocellular carcinoma cells.68,69 Interestingly, the combination of Q deprivation and 25 μg mL−1 of AuNP S2 led to mitochondrial depolarization, a surge in the mtROS, and a reduction in the mtMass to a level that contributed to the mitochondrial dysfunction and significant antitumor activity in HeLa cells. However, when used alone, either of the components of the combinatorial strategy could not bring considerable impact on cell proliferation.
Most proliferating cancer cells tend to arrest in different cell cycle phases when they come across stress conditions, including glucose and glutamine deprivation, or encounter chemical drugs or products, extracts, and NPs from diverse biological sources with cytotoxic potential.51,70–73 Cell cycle arrest provides an opportunity to promote the repair of damaged DNA or trigger the elimination of damaged cells via apoptosis. Investigating cell cycle progression and apoptotic mode of cell death is thus required for the effective screening of prospective therapeutic approaches. We observed S-phase arrest in cervical cancer cells HeLa upon 24 h Q shortage, which was consistent with other researchers’ findings in breast and colon cancer cells at varying time points.74,75 The percentage of cells displaying S-phase cell cycle arrest was higher in treatment consisting of only Q depletion, indicating its higher capacity to inhibit HeLa cell proliferation than AuNP S2 exposure in Q-supplemented growth media. In contrast to S-phase cell cycle arrest shown in HeLa cells by either Q withdrawal or exposure to AuNP S2 in Q-supplemented media, arrest of the cell cycle in the G0/G1-phase by low-dose of AuNP S2 in Q-starved growth conditions can be seen as a more stringent strategy to check uncontrolled division of cells since by arresting in the G0/G1-phase, cells ensure all the requirements for accurate DNA replication and provide more opportunities for DNA repair or to undergo apoptosis if the genomic instability is irreparable. Many studies suggest that biogenic NPs arrest the cell cycle at the G0/G1 phase with a corresponding increase in apoptotic cell death.76,77
Apoptosis is a tightly regulated process and a type of programmed cell death whose vital functions range from normal cell turnover to drug-induced cell death, including the development and functioning of the organs and immune system. Tumor cells have the tendency to acquire resistance to apoptosis by the up- and down-regulation of anti-apoptotic and pro-apoptotic proteins, respectively, and evasion of immune surveillance.78 Apart from characteristic morphological and other biochemical features, it is typically marked by the externalization of phosphatidylserine (PS) to the outer leaflet of the plasma membrane lipid bilayer. Thus, fluorophore-conjugated calcium-dependent recombinant anticoagulant protein annexin V which has a high binding affinity for PS can identify apoptotic cells. Similarly, PI, which binds to the nuclear DNA of a cell with lost plasma membrane integrity, can detect necrotic/late apoptotic cells. Necrosis has been conventionally considered to be a form of accidental or unprogrammed cell death that is a biologically unregulated process occurring due to an overwhelming exposure to chemicals or physical stress inducers, namely temperature, pressure, or osmotic shock. But, based on recent research, a regulated form of necrosis is also known, termed “necroptosis”. Necrosis is characterized by cell and organelle swelling (oncosis) and rupture, induction of mitochondrial permeability transition, relatively rapid loss of plasma membrane integrity, and release of cellular content to cause tissue inflammation.79,80 Early-onset of plasma membrane disintegration in necrosis preferably allows nuclear entry of PI. So, during annexin V/PI dual staining, annexin V negative/PI-positive fluorescence is the characteristic of the necrotic cells. But membrane permeabilization can sometimes also facilitate the binding of fluorochrome-labeled Annexin V to PS, even without their exposure on the surface of the plasma membrane, and emit a positive fluorescence of annexin V. Therefore, annexin V positive/PI-positive fluorescence that typically designates late apoptosis can also represent necrosis/necroptosis,80 and necrosis can be considered to be an extreme form of late apoptosis.78,81 Utilization of annexin V-FITC/PI dual staining indicated that subjecting HeLa cells to Q depletion alone could induce an apoptotic and necrotic mode of cell death in a small proportion of cells, as also observed by Niklison-Chirou et al. in medulloblastoma cells and Sa-Nongdej et al. in glioma stem-like cells.82,83 However, the combination of Q deprivation and 25 μg mL−1 AuNP S2 contributed to a significantly higher rate of apoptotic (early and late combined) cell death compared to Q withdrawal or AuNP S2 alone. The obtained results clearly demonstrated that the potential of AuNP S2 for inducing cell death at a very low dose is greatly enhanced when combined with Q deprivation.
Early steps of metastasis in cancer involve clinically significant aspects like cell migration and tissue invasion.84 Therefore, evaluating tumor cell migration potential is important for developing new therapeutic approaches for cancer inhibition. Our results from the in vitro cell migration assay established that the rate of two-dimensional migration of HeLa cells was significantly lowered by the combination of Q withdrawal and sub-toxic dose of AuNP S2 for 24 h, while either Q depletion or 25 μg mL−1 of AuNP S2 was unable to bring any considerable reduction in their migratory capacity. The obtained results corroborated the antitumor activity of the combination mentioned above, observed in other assays of this study. However, even NPs fabricated via green synthesis may have some possible limitations with regard to their pharmaceutical and biomedical applications for humans, such as NP heterogeneity, and the possibility of conjugation of some toxic components from the biological sources, mainly bacteria and fungi to the synthesized NPs. NP heterogeneity is attributed to the presence of particle sub-populations or surface inhomogeneities and intrinsic to the synthesis method and composition of cell free extracts.85 Effects of the attached toxic components are sometimes not detected in two-dimensional cell culture models due to the lack of cell–cell and cell-extracellulat matrix interactions. Therefore, use of other pre-clinical models such as three-dimensional spheroids/organoids that better mimic the natural cellular environments and in vivo models should be the next step to conduct the research that involves inorganic NPs.
5. Conclusions
Many a time, depending upon the synthesis protocols, biological resources, dose and treatment duration, green synthesized NPs although being non-toxic to normal cells also lack the desired growth and proliferation inhibiting potential toward the target cancer cells. Targeting glucose and glutamine metabolism has shown direct relevance to the alterations in the pathophysiology of cancer. Uncontrolled proliferation and ability to migrate are among the most critical factors responsible for the occurrence of cancer. Overall, this study primarily focused on improving the antitumor effect of a low and non-toxic dose of plant-based gold NPs. We successfully developed a combinatorial strategy involving a sub-toxic dose of the studied AuNPs S2 and targeting the inhibition of glutaminolysis via glutamine deprivation for 24 h in a human cervical cancer cell line, HeLa. Glutamine starvation for 24 h exhibited observable yet less significant anticancer characteristics. But it chemopotentiated the effects of S2 in HeLa cell death. Excess cytosolic and mitochondrial ROS-induced oxidative stress, loss of mitochondrial function owing to mitochondrial membrane depolarization, superoxide-induced mitochondrial stress, and a decrease in mitochondrial mass were all factors that contributed to the enhanced antitumor impact found in this study. Cell cycle arrest in the G0/G1 phase and induction of apoptosis and necrosis due to the treatment strategy adopted in the present study further improved its growth and proliferation inhibitory effects in HeLa cells, as established by the cell viability assay and morphological examination. These effects ultimately halted cancer cell migration. Therefore, this “bench-to-bedside” research might add to our understanding of nutritionally based anticancer signaling in cervical and other malignancies, which might be translated into clinical strategies, after reviewing its molecular mechanisms and efficacy, and addressing the safety concerns through clinical trials.Author contributions
Abhinav Prasad: Conceptualization, data curation, formal analysis, investigation, methodology, software, validation, writing – original draft, writing – reviewing and editing and visualization. Ashapurna Khatua: Investigation and methodology. Yugal Kishore Mohanta: Investigation and writing – reviewing and editing. Muthupandian Saravanan: Conceptualization, formal analysis, writing – reviewing and editing, and validation. Ramovatar Meena: Conceptualization, formal analysis, resources, writing – reviewing and editing and validation. Ilora Ghosh: Conceptualization, data curation, formal analysis, funding acquisition, project administration, resources, supervision, writing – reviewing and editing and validation.Ethical approval
The present research study does not involve any human participants, their data or biological material.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
I. G. acknowledges the Government of India for supporting this study under the JNU DST-PURSE and JNU UPE-II. The Indian Council of Medical Research (ICMR) provided Abhinav Prasad with a Senior Research Fellowship. We appreciate the flow cytometry facility provided by CIF, SES, and Jawaharlal Nehru University. I. G. received funding from the DST-PURSE (Promotion of University Research and Scientific Excellence program of Department of Science and Technology, Government of India) and UGC UPE-II (University with Potential for Excellence, Phase II scheme of University Grant Commission, Government of India) via Jawaharlal Nehru University, New Delhi, India. The funding agencies had no role in the study design, data acquisition, analysis, the decision to publish, and preparation of the manuscript.References
- H. Vahidi, F. Kobarfard, A. Alizadeh, M. Saravananand and H. Barabadi, Green nanotechnology-based tellurium nanoparticles: Exploration of their antioxidant, antibacterial, antifungal and cytotoxic potentials against cancerous and normal cells compared to potassium tellurite, Inorg. Chem. Commun., 2021, 124, 108385, DOI:10.1016/j.inoche.2020.108385.
- S. Iravani, H. Korbekandi, S. V. Mirmohammadi and B. Zolfaghari, Synthesis of silver nanoparticles: chemical, physical and biological methods, Res. Pharm. Sci., 2014, 9, 385–406 CAS.
- D. M. Cruz, E. Mostafavi, A. Vernet-Crua, H. Barabadi, V. Shah, J. L. Cholula-Díaz, G. Guisbiers and T. J. Webster, Green nanotechnology-based zinc oxide (ZnO) nanomaterials for biomedical applications: A review, J. Phys Mater., 2020, 3, 034005 CrossRef CAS.
- I. Ijaz, E. Gilani, A. Nazir and A. Bukhari, Detail review on chemical, physical and green synthesis, classification, characterizations and applications of nanoparticles, Green Chem. Lett. Rev., 2020, 13, 223–245, DOI:10.1080/17518253.2020.1802517.
- D. Chugh, V. S. Viswamalya and B. Das, Green synthesis of silver nanoparticles with algae and the importance of capping agents in the process, J. Genet. Eng. Biotechnol., 2021, 19, 1–21, DOI:10.1186/s43141-021-00228-w.
- H. Duan, D. Wang and Y. Li, Green chemistry for nanoparticle synthesis, Chem. Soc. Rev., 2015, 44, 5778–5792, 10.1039/c4cs00363b.
- M. Saravanan, H. Barabadi and H. Vahidi, Chapter 5 – Green nanotechnology: isolation of bioactive molecules and modified approach of biosynthesis, in Micro and Nano Technologies, Biogenic Nanoparticles for Cancer Theranostics, ed. C. Patra, I. Ahmad, M. Ayaz, A. T. Khalil, S. Mukherjee and M. Ovais, Elsevier, 2021, pp. 101–122. DOI:10.1016/B978-0-12-821467-1.00005-7.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries, Ca-Cancer J. Clin., 2021, 71, 209–249, DOI:10.3322/caac.21660.
- Q. Wei, Y. Qian, J. Yu and C. C. Wong, Metabolic rewiring in the promotion of cancer metastasis: mechanisms and therapeutic implications, Oncogene, 2020, 39, 6139–6156, DOI:10.1038/s41388-020-01432-7.
- B. Faubert, A. Solmonson and R. J. DeBerardinis, Metabolic reprogramming and cancer progression, Science, 2020, 368, eaaw5473, DOI:10.1126/science.aaw5473.
- E. R. Still and M. O. Yuneva, Hopefully devoted to Q: targeting glutamine addiction in cancer, Br. J. Cancer, 2017, 116, 1375–1381, DOI:10.1038/bjc.2017.113.
- J. Zhang, N. N. Pavlova and C. B. Thompson, Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine, EMBO J., 2017, 36, 1302–1315, DOI:10.15252/embj.201696151.
- M. Kodama, K. Oshikawa, H. Shimizu, S. Yoshioka, M. Takahashi, Y. Izumi, T. Bamba, C. Tateishi, T. Tomonaga, M. Matsumoto and K. I. Nakayama, A shift in glutamine nitrogen metabolism contributes to the malignant progression of cancer, Nat. Commun., 2020, 11, 1320, DOI:10.1038/s41467-020-15136-9.
- B. J. Altman, Z. E. Stine and C. V. Dang, From Krebs to clinic: glutamine metabolism to cancer therapy, Nat. Rev. Cancer, 2016, 16, 619–634, DOI:10.1038/nrc.2016.71.
- L. Jin, G. N. Alesi and S. Kang, Glutaminolysis as a target for cancer therapy, Oncogene, 2016, 35, 3619–3625, DOI:10.1038/onc.2015.447.
- J. Son, C. A. Lyssiotis, H. Ying, X. Wang, S. Hua, M. Ligorio, R. M. Perera, C. R. Ferrone, E. Mullarky, N. Shyh-Chang, Y. Kang, J. B. Fleming, N. Bardeesy, J. M. Asara, M. C. Haigis, R. A. DePinho, L. C. Cantley and A. C. Kimmelman, Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway, Nature, 2013, 496, 101–105, DOI:10.1038/nature12040.
- L. Yang, S. Venneti and D. Nagrath, Glutaminolysis: A Hallmark of Cancer Metabolism, Annu. Rev. Biomed. Eng., 2017, 19, 163–194, DOI:10.1146/annurev-bioeng-071516-044546.
- K. P. Michalak, A. Maćkowska-Kędziora, B. Sobolewski and P. Woźniak, Key Roles of Glutamine Pathways in Reprogramming the Cancer Metabolism, Oxid. Med. Cell. Longevity, 2015, 2015, 964321, DOI:10.1155/2015/964321.
- C. T. Hensley, A. T. Wasti and R. J. DeBerardinis, Glutamine and cancer: cell biology, physiology, and clinical opportunities, J. Clin. Invest., 2013, 123, 3678–3684, DOI:10.1172/JCI69600.
- D. Daye and K. E. Wellen, Metabolic reprogramming in cancer: unraveling the role of glutamine in tumorigenesis, Semin. Cell Dev. Biol., 2012, 23, 362–369, DOI:10.1016/j.semcdb.2012.02.002.
- M. V. Liberti and J. W. Locasale, The Warburg Effect: How Does it Benefit Cancer Cells?, Trends Biochem. Sci., 2016, 41, 211–218, DOI:10.1016/j.tibs.2015.12.001.
- D. R. Wise and C. B. Thompson, Glutamine addiction: a new therapeutic target in cancer, Trends Biochem. Sci., 2010, 35, 427–433, DOI:10.1016/j.tibs.2010.05.003.
- K. Vanhove, E. Derveaux, G.-J. Graulus, L. Mesotten, M. Thomeer, J.-P. Noben, W. Guedens and P. Adriaensens, Glutamine Addiction and Therapeutic Strategies in Lung Cancer, Int. J. Mol. Sci., 2019, 20, 252, DOI:10.3390/ijms20020252.
- L. J. Reitzer, B. M. Wice and D. Kennell, Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells, J. Biol. Chem., 1979, 254, 2669–2676 CrossRef CAS PubMed.
- C. Jose, N. Bellance and R. Rossignol, Choosing between glycolysis and oxidative phosphorylation: a tumor's dilemma?, Biochim. Biophys. Acta, 2011, 1807, 552–561, DOI:10.1016/j.bbabio.2010.10.012.
- H. C. Yoo, Y. C. Yu, Y. Sung and J. M. Han, Glutamine reliance in cell metabolism, Exp. Mol. Med., 2020, 52, 1496–1516, DOI:10.1038/s12276-020-00504-8.
- K. X. Lee, K. Shameli, Y. P. Yew, S.-Y. Teow, H. Jahangirian, R. Rafiee-Moghaddam and T. J. Webster, Recent Developments in the Facile Bio-Synthesis of Gold Nanoparticles (AuNPs) and Their Biomedical Applications, Int. J. Nanomed., 2020, 15, 275–300, DOI:10.2147/IJN.S233789.
- M. A. Meléndez-Villanueva, K. Morán-Santibañez, J. J. Martínez-Sanmiguel, R. Rangel-López, M. A. Garza-Navarro, C. Rodríguez-Padilla, D. G. Zarate-Triviño and L. M. Trejo-Ávila, Virucidal Activity of Gold Nanoparticles Synthesized by Green Chemistry Using Garlic Extract, Viruses, 2019, 11, 1111, DOI:10.3390/v11121111.
- L. Rotimi, M. O. Ojemaye, O. O. Okoh, A. Sadimenko and A. I. Okoh, Synthesis, characterization, antimalarial, antitrypanocidal and antimicrobial properties of gold nanoparticle, Green Chem. Lett. Rev., 2019, 12, 61–68, DOI:10.1080/17518253.2019.1569730.
- L. M. R. Al Muqarrabun, N. Ahmat, S. A. S. Ruzaina, N. H. Ismail and I. Sahidin, Medicinal uses, phytochemistry and pharmacology of Pongamia pinnata (L.) Pierre: a review, J. Ethnopharmacol., 2013, 150, 395–420, DOI:10.1016/j.jep.2013.08.041.
- A. Khatua, A. Prasad, E. Priyadarshini, A. K. Patel, A. Naik, M. Saravanan, H. Barabadi, L. Ghosh, B. Paul, R. Paulraj and R. Meena, Emerging Antineoplastic Plant-Based Gold Nanoparticle Synthesis: A Mechanistic Exploration of their Anticancer Activity Toward Cervical Cancer Cells, J. Cluster Sci., 2020, 31, 1329–1340, DOI:10.1007/s10876-019-01742-1.
- T. L. Riss, R. A. Moravec, A. L. Niles, S. Duellman, H. A. Benink, T. J. Worzella and L. Minor, Cell Viability Assays, in S. Markossian, G. Sittampalam, A. Grossman, et al., Eli Lilly & Company and the National Center for Advancing Translational Sciences, Assay Guid. Man., Bethesda (MD), 2013 Search PubMed.
- A. Salvati, I. Nelissen, A. Haase, C. Åberg, S. Moya, A. Jacobs, F. Alnasser, T. Bewersdorff, S. Deville, A. Luch and K. A. Dawson, Quantitative measurement of nanoparticle uptake by flow cytometry illustrated by an interlaboratory comparison of the uptake of labelled polystyrene nanoparticles, NanoImpact, 2018, 9, 42–50, DOI:10.1016/j.impact.2017.10.004.
- H. Klingberg, L. B. Oddershede, K. Loeschner, E. H. Larsen, S. Loft and P. Møller, Uptake of gold nanoparticles in primary human endothelial cells, Toxicol. Res., 2015, 4, 655–666, 10.1039/C4TX00061G.
- S. Persano, F. Vicini, A. Poggi, J. L. C. Fernandez, G. M. R. Rizzo, H. Gavilán, N. Silvestri and T. Pellegrino, Elucidating the Innate Immunological Effects of Mild Magnetic Hyperthermia on U87 Human Glioblastoma Cells: An In Vitro Study, Pharmaceutics, 2021, 13, 1668, DOI:10.3390/pharmaceutics13101668.
- E. Villa, E. Proïcs, C. Rubio-Patiño, S. Obba, B. Zunino, J. P. Bossowski, R. M. Rozier, J. Chiche, L. Mondragón, J. S. Riley, S. Marchetti, E. Verhoeyen, S. W. G. Tait and J.-E. Ricci, Parkin-Independent Mitophagy Controls Chemotherapeutic Response in Cancer Cells, Cell Rep., 2017, 20, 2846–2859, DOI:10.1016/j.celrep.2017.08.087.
- R. Zhou, A. S. Yazdi, P. Menu and J. Tschopp, A role for mitochondria in NLRP3 inflammasome activation, Nature, 2011, 469, 221–225, DOI:10.1038/nature09663.
- L. de B. Monteiro, G. G. Davanzo, C. F. de Aguiar and P. M. M. Moraes-Vieira, Using flow cytometry for mitochondrial assays, MethodsX, 2020, 7, 100938, DOI:10.1016/j.mex.2020.100938.
- M. E. Kauffman, M. K. Kauffman, K. Traore, H. Zhu, M. A. Trush, Z. Jia and Y. R. Li, MitoSOX-Based Flow Cytometry for Detecting Mitochondrial ROS, React. Oxygen Species, 2016, 2, 361–370, DOI:10.20455/ros.2016.865.
- J. Liu, Z. Li, Y. Wang and D. Xing, Overexpression of ALTERNATIVE OXIDASE1a alleviates mitochondria-dependent programmed cell death induced by aluminium phytotoxicity in Arabidopsis, J. Exp. Bot., 2014, 65, 4465–4478, DOI:10.1093/jxb/eru222.
- C. Cottet-Rousselle, X. Ronot, X. Leverve and J.-F. Mayol, Cytometric assessment of mitochondria using fluorescent probes, Cytometry, Part A, 2011, 79, 405–425, DOI:10.1002/cyto.a.21061.
- B. Xiao, X. Deng, W. Zhou and E.-K. Tan, Flow Cytometry-Based Assessment of Mitophagy Using MitoTracker, Front. Cell. Neurosci., 2016, 10, 76, DOI:10.3389/fncel.2016.00076.
- A. Khatua, A. Prasad, H. G. Behuria, A. K. Patel, M. Singh, M. Yasasve, M. Saravanan and R. Meena, Evaluation of antimicrobial, anticancer potential and Flippase induced leakage in model membrane of Centella asiatica fabricated MgONPs, Biomater. Adv., 2022, 212855, DOI:10.1016/j.bioadv.2022.212855.
- M. Faraday and X. The, Bakerian Lecture. —Experimental relations of gold (and other metals) to light, Philos. Trans. R. Soc. London, 1857, 147, 145–181, DOI:10.1098/rstl.1857.0011.
- A. M. Scherbart, J. Langer, A. Bushmelev, D. van Berlo, P. Haberzettl, F.-J. van Schooten, A. M. Schmidt, C. R. Rose, R. P. F. Schins and C. Albrecht, Contrasting macrophage activation by fine and ultrafine titanium dioxide particles is associated with different uptake mechanisms, Part. Fibre Toxicol., 2011, 8, 31, DOI:10.1186/1743-8977-8-31.
- C. Greulich, J. Diendorf, J. Gessmann, T. Simon, T. Habijan, G. Eggeler, T. A. Schildhauer, M. Epple and M. Köller, Cell type-specific responses of peripheral blood mononuclear cells to silver nanoparticles, Acta Biomater., 2011, 7, 3505–3514, DOI:10.1016/j.actbio.2011.05.030.
- C. Greulich, J. Diendorf, T. Simon, G. Eggeler, M. Epple and M. Köller, Uptake and intracellular distribution of silver nanoparticles in human mesenchymal stem cells., Acta Biomater., 2011, 7, 347–354, DOI:10.1016/j.actbio.2010.08.003.
- A. Salvati, C. Aberg, T. dos Santos, J. Varela, P. Pinto, I. Lynch and K. A. Dawson, Experimental and theoretical comparison of intracellular import of polymeric nanoparticles and small molecules: toward models of uptake kinetics, Nanomedicine, 2011, 7, 818–826, DOI:10.1016/j.nano.2011.03.005.
- R. Peters, Z. Herrera-Rivera, A. Undas, M. van der Lee, H. Marvin, H. Bouwmeester and S. Weigel, Single particle ICP-MS combined with a data evaluation tool as a routine technique for the analysis of nanoparticles in complex matrices, J. Anal. At. Spectrom., 2015, 30, 1274–1285, 10.1039/C4JA00357H.
- A. Andleeb, A. Andleeb, S. Asghar, G. Zaman, M. Tariq, A. Mehmood, M. Nadeem, C. Hano, J. M. Lorenzo and B. H. Abbasi, A Systematic Review of Biosynthesized Metallic Nanoparticles as a Promising Anti-Cancer-Strategy, Cancers, 2021, 13, 2818, DOI:10.3390/cancers13112818.
- M. V. Gwangwa, A. M. Joubert and M. H. Visagie, Effects of glutamine deprivation on oxidative stress and cell survival in breast cell lines, Biol. Res., 2019, 52, 15, DOI:10.1186/s40659-019-0224-9.
- Y. Zhao, X. Hu, Y. Liu, S. Dong, Z. Wen, W. He, S. Zhang, Q. Huang and M. Shi, ROS signaling under metabolic stress: cross-talk between AMPK and AKT pathway, Mol. Cancer, 2017, 16, 79, DOI:10.1186/s12943-017-0648-1.
- L. Bouchier-Hayes, C. Muñoz-Pinedo, S. Connell and D. R. Green, Measuring apoptosis at the single cell level, Methods, 2008, 44, 222–228, DOI:10.1016/j.ymeth.2007.11.007.
- S. Marchi, C. Giorgi, J. M. Suski, C. Agnoletto, A. Bononi, M. Bonora, E. De Marchi, S. Missiroli, S. Patergnani, F. Poletti, A. Rimessi, J. Duszynski, M. R. Wieckowski and P. Pinton, Mitochondria-ros crosstalk in the control of cell death and aging, J. Signal Transduction, 2012, 2012, 329635, DOI:10.1155/2012/329635.
- D. M. Hockenbery, Targeting mitochondria for cancer therapy, Environ. Mol. Mutagen., 2010, 51, 476–489, DOI:10.1002/em.20552.
- I. Kim, S. Rodriguez-Enriquez and J. J. Lemasters, Selective degradation of mitochondria by mitophagy, Arch. Biochem. Biophys., 2007, 462, 245–253, DOI:10.1016/j.abb.2007.03.034.
- S. N. Syed Abdul Rahman, N. Abdul Wahab and S. N. Abd Malek, In Vitro Morphological Assessment of Apoptosis Induced by Antiproliferative Constituents from the Rhizomes of Curcuma zedoaria, J. Evidence-Based Complementary Altern. Med., 2013, 2013, 257108, DOI:10.1155/2013/257108.
- A. Saraste and K. Pulkki, Morphologic and biochemical hallmarks of apoptosis, Cardiovasc. Res., 2000, 45, 528–537, DOI:10.1016/S0008-6363(99)00384-3.
- P. Foroozandeh and A. A. Aziz, Insight into Cellular Uptake and Intracellular Trafficking of Nanoparticles, Nanoscale Res. Lett., 2018, 13, 339, DOI:10.1186/s11671-018-2728-6.
- H. Barabadi, H. Vahidi, K. D. Kamali, M. Rashedi and M. Saravanan, Antineoplastic Biogenic Silver Nanomaterials to Combat Cervical Cancer: A Novel Approach in Cancer Therapeutics, J. Cluster Sci., 2020, 31, 659–672, DOI:10.1007/s10876-019-01697-3.
- B. Perillo, M. Di Donato, A. Pezone, E. Di Zazzo, P. Giovannelli, G. Galasso, G. Castoria and A. Migliaccio, ROS in cancer therapy: the bright side of the moon, Exp. Mol. Med., 2020, 52, 192–203, DOI:10.1038/s12276-020-0384-2.
- S. Lee, B. N. Sellers and G. M. DeNicola, The Regulation of NRF2 by Nutrient-Responsive Signaling and Its Role in Anabolic Cancer Metabolism, Antioxid. Redox Signal., 2018, 29, 1774–1791, DOI:10.1089/ars.2017.7356.
- Y. Wang, Q. Wang, J. Li, G. Lu and Z. Liu, Glutamine Improves Oxidative Stress through the Wnt3a/β-Catenin Signaling Pathway in Alzheimer’s Disease In Vitro and In Vivo, BioMed Res. Int., 2019, 2019, 4690280, DOI:10.1155/2019/4690280.
- W.-X. Zong, J. D. Rabinowitz and E. White, Mitochondria and Cancer, Mol. Cell., 2016, 61, 667–676, DOI:10.1016/j.molcel.2016.02.011.
- D. C. Wallace, Mitochondria and cancer, Nat. Rev. Cancer, 2012, 12, 685–698, DOI:10.1038/nrc3365.
- M. Redza-Dutordoir and D. A. Averill-Bates, Activation of apoptosis signalling pathways by reactive oxygen species, Biochim. Biophys. Acta, Mol. Cell Res., 2016, 1863, 2977–2992, DOI:10.1016/j.bbamcr.2016.09.012.
- J. D. West and L. J. Marnett, Endogenous reactive intermediates as modulators of cell signaling and cell death, Chem. Res. Toxicol., 2006, 19, 173–194, DOI:10.1021/tx050321u.
- K. Valter, L. Chen, B. Kruspig, P. Maximchik, H. Cui, B. Zhivotovsky and V. Gogvadze, Contrasting effects of glutamine deprivation on apoptosis induced by conventionally used anticancer drugs, Biochim. Biophys. Acta, Mol. Cell Res., 2017, 1864, 498–506, DOI:10.1016/j.bbamcr.2016.12.016.
- A. Kumar, S. Giri and C. Shaha, Sestrin2 facilitates glutamine-dependent transcription of PGC-1α and survival of liver cancer cells under glucose limitatio., FEBS J., 2018, 285, 1326–1345, DOI:10.1111/febs.14406.
- S. Hubackova, E. Davidova, S. Boukalova, J. Kovarova, M. Bajzikova, A. Coelho, M. G. Terp, H. J. Ditzel, J. Rohlena and J. Neuzil, Replication and ribosomal stress induced by targeting pyrimidine synthesis and cellular checkpoints suppress p53-deficient tumors, Cell Death Dis., 2020, 11, 110, DOI:10.1038/s41419-020-2224-7.
- H. Murad, M. Hawat, A. Ekhtiar, A. AlJapawe, A. Abbas, H. Darwish, O. Sbenati and A. Ghannam, Induction of G1-phase cell cycle arrest and apoptosis pathway in MDA-MB-231 human breast cancer cells by sulfated polysaccharide extracted from Laurencia papillosa, Cancer Cell Int., 2016, 16, 39, DOI:10.1186/s12935-016-0315-4.
- S. Krishnappa, C. M. Naganna, H. K. Rajan, S. Rajashekarappa and H. B. Gowdru, Cytotoxic and Apoptotic Effects of Chemogenic and Biogenic Nano-sulfur on Human Carcinoma Cells: A Comparative Study, ACS Omega, 2021, 6, 32548–32562, DOI:10.1021/acsomega.1c04047.
- M. Morais, V. Machado, F. Dias, C. Palmeira, G. Martins, M. Fonseca, C. S. M. Martins, A. L. Teixeira, J.A. V. Prior and R. Medeiros, Starch-Capped AgNPs’ as Potential Cytotoxic Agents against Prostate Cancer Cells, Nanomaterials , 2021, 11, 256, DOI:10.3390/nano11020256.
- D. Patel, D. Menon, E. Bernfeld, V. Mroz, S. Kalan, D. Loayza and D. A. Foster, Aspartate Rescues S-phase Arrest Caused by Suppression of Glutamine Utilization in KRas-driven Cancer Cells, J. Biol. Chem., 2016, 291, 9322–9329, DOI:10.1074/jbc.M115.710145.
- I. H. Polat, M. Tarrado-Castellarnau, A. Benito, C. Hernandez-Carro, J. Centelles, S. Marin and M. Cascante, Glutamine Modulates Expression and Function of Glucose 6-Phosphate Dehydrogenase via NRF2 in Colon Cancer Cells, Antioxidants, 2021, 10, 1349, DOI:10.3390/antiox10091349.
- O. Oladimeji, J. Akinyelu, A. Daniels and M. Singh, Modified Gold Nanoparticles for Efficient Delivery of Betulinic Acid to Cancer Cell Mitochondria, Int. J. Mol. Sci., 2021, 22, 5072, DOI:10.3390/ijms22105072.
- S. Meenakshisundaram, V. Krishnamoorthy, Y. Jagadeesan, R. Vilwanathan and A. Balaiah, Annona muricata assisted biogenic synthesis of silver nanoparticles regulates cell cycle arrest in NSCLC cell lines, Bioorg. Chem., 2020, 95, 103451, DOI:10.1016/j.bioorg.2019.103451.
- S. Elmore, Apoptosis: a review of programmed cell death, Toxicol. Pathol., 2007, 35, 495–516, DOI:10.1080/01926230701320337.
- D. Tang, R. Kang, T. Vanden Berghe, P. Vandenabeele and G. Kroemer, The molecular machinery of regulated cell death, Cell Res., 2019, 29, 347–364, DOI:10.1038/s41422-019-0164-5.
- X.-M. Hu, Z.-X. Li, R.-H. Lin, J.-Q. Shan, Q.-W. Yu, R.-X. Wang, L.-S. Liao, W.-T. Yan, Z. Wang, L. Shang, Y. Huang, Q. Zhang and K. Xiong, Guidelines for Regulated Cell Death Assays: A Systematic Summary, A Categorical Comparison, A Prospective, Front. Cell Dev. Biol., 2021, 9, 368, DOI:10.3389/fcell.2021.634690.
- H. Malhi, G. J. Gores and J. J. Lemasters, Apoptosis and necrosis in the liver: a tale of two deaths?, Hepatology, 2006, 43, S31–S44, DOI:10.1002/hep.21062.
- M. V. Niklison-Chirou, I. Erngren, M. Engskog, J. Haglöf, D. Picard, M. Remke, P. H. R. McPolin, M. Selby, D. Williamson, S. C. Clifford, D. Michod, M. Hadjiandreou, T. Arvidsson, C. Pettersson, G. Melino and S. Marino, TAp73 is a marker of glutamine addiction in medulloblastoma, Genes Dev., 2017, 31, 1738–1753, DOI:10.1101/gad.302349.117.
- W. Sa-Nongdej, S. Chongthammakun and C. Songthaveesin, Nutrient starvation induces apoptosis and autophagy in C6 glioma stem-like cells, Heliyon, 2021, 7, e06352, DOI:10.1016/j.heliyon.2021.e06352.
- M. F. Leber and T. Efferth, Molecular principles of cancer invasion and metastasis (review), Int. J. Oncol., 2009, 34, 881–895, DOI:10.3892/ijo_00000214.
- J. M. Rabanel, V. Adibnia, S. F. Tehrani, S. Sanche, P. Hildgen, X. Banquy and C. Ramassamy, Nanoparticle heterogeneity: an emerging structural parameter influencing particle fate in biological media?, Nanoscale, 2019, 11, 383–406, 10.1039/c8nr04916e.
| This journal is © The Royal Society of Chemistry 2022 |