
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transport among protocells via tunneling nanotubes†
Ingrid Jin
Schanke‡
 ,
Lin
Xue‡
,
Karolina
Spustova
and
Irep
Gözen
*
,
Lin
Xue‡
,
Karolina
Spustova
and
Irep
Gözen
*
Centre for Molecular Medicine Norway, Faculty of Medicine, University of Oslo, 0318 Oslo, Norway. E-mail: irep@uio.no
First published on 13th June 2022
Abstract
We employ model protocell networks for evaluation of molecular transport through lipid nanotubes as potential means of communication among primitive cells on the early Earth. Network formation is initiated by deposition of lipid reservoirs onto a SiO2 surface in an aqueous environment. These reservoirs autonomously develop into surface-adhered protocells interconnected via lipid nanotubes while encapsulating solutes from the ambient buffer. We observe the uptake of DNA and RNA, and their diffusive transport between the lipid compartments via the interconnecting nanotubes. By means of an analytical model we determine key physical parameters affecting the transport, such as nanotube diameter and compartment size. We conclude that nanotube-mediated transport could have been a possible pathway of communication between primitive cells on the early Earth, circumventing the necessity for crossing the membrane barrier. We suggest this transport as a feasible means of RNA and DNA exchange under primitive prebiotic conditions, possibly facilitating early replication.
Introduction
How the first living cell emerged from prebiotic matter on the early Earth is still an unsolved question. Current studies focusing on this problem utilize synthetic model precursors of primitive cells, the ‘protocells’. Protocells carry features of living cells, but are structurally and functionally much simpler.1 A feature in common with a modern cell, which is surrounded by a plasma membrane is a biosurfactant bilayer, which establishes a boundary, an identity, and an interface suitable for chemical exchange.2,3 A bilayer envelope satisfies one of the three conditions of the Chemoton model, the hypothetical chemical entity which features all necessary criteria of ‘living’.4 A membranous protocell is typically prepared under laboratory conditions via self-assembly of bulk amphiphiles in an aqueous solution.2,3In order to cross the boundary between the non-living and living matter, a primitive cell needs to be able to develop, grow and eventually self-replicate, resulting in formation of genetically identical daughter cells. To undergo Darwinian evolution during this process, protocells should attain the ability to sense, and adapt to, relevant changes in the environment. Such changes can include perturbations caused by other protocells. Modern cells, for example, can communicate by secreting chemical signals into their surroundings, which is recognized by nearby cells, that are either in direct contact, or a short distance away.5–9 The response can be manifold, for example tuning the gene expression to change density of the cell population, or induce cell division. Whether bacteria or eukaryotes, cellular communication and division pathways require the coordination of multiple sets of proteins and ligands7 which primitive cells were initially lacking. How protocells could have attained over time the ability to expediently perform communication and division, is a pending question.
Recently, we reported spontaneous formation of protocell-nanotube networks following a set of autonomous shape transformations on solid substrates.10,11 The resulting structure is a population of surface-adhered protocells interconnected with lipid nanotubes. The nanotubular structures within the networks resemble the tunneling nanotubes (TNTs) between mammalian cells, which enable direct communication by transporting signaling molecules and even organelles.12,13 TNTs are also observed in bacterial cells, and likely provide an alternative route of intercellular exchange of cytoplasmic molecules and plasmids.14–17 Whether the nanotubes in protocell networks10 would allow the transport of molecules, e.g. via molecular diffusion,18 was initially not established.
In this work, we have investigated the ability of lipid nanotubes in surface-supported networks to transport prebiologically relevant constituents between the compartments: small water-soluble molecules, RNA and DNA. The protocell networks were formed for that purpose from label-free lipid membranes, allowing the focus of observations to be solely on fluorescently-labeled cargo. Our findings confirm that the nanotubes function as tunneling interconnections between protocells, and are capable of transporting molecules and genetic polymers diffusively among them. We also characterized, supported by a dynamic analytical model, key physical parameters that are influential for the transport process. We hypothesize that nanotubes could have established a feasible means of communication and replication between prebiotic protocells, as in the investigated environment identical RNA and DNA fragments are easily distributed to nearby network nodes.
Results and discussion
Formation of protocell-nanotube networks free of fluorophore
We started our experiments by preparing protocell-nanotube networks (PNNs) according to the protocol by Köksal et al.10 Briefly, a suspension of multilamellar vesicles (MLVs) is brought in contact with a SiO2 surface in an aqueous environment. Upon contact with the surface, each MLV acts as a lipid reservoir and spontaneously spreads as a double bilayer membrane, driven by the surface adhesion. Continuous spreading causes a buildup of membrane tension, and the distal (upper with respect to the surface) bilayer eventually ruptures followed by its transformation to a network of lipid nanotubes. The network resides on the proximal (lower) bilayer. In order to minimize the membrane curvature and overall surface free energy, sections of the nanotubes swell over time and form spherical unilamellar compartments. The resulting structure is a PNN consisting of several lipid compartments connected via lipid nanotubes. The detailed characterization of this process was reported in our earlier work.10,11 A schematic drawing and a fluorescence micrograph of a fluorescently-labeled PNN (control) are shown in Fig. 1a and b respectively. Thereafter we prepared PNNs from label-free lipid membranes in order to exclusively visualize cargo molecules in the network without crosstalk19 with membrane fluorescence. | ||
| Fig. 1 Protocell-nanotube networks (PNNs). (a) Schematic drawing of a protocell-nanotube network. The network and compartments are adhered to a surface supported bilayer. (b) 3D reconstructed confocal fluorescence micrograph of a PNN formed from a fluorescently-labeled membrane (control). The inset shows a xy cross section of the two adjacent vesicles. (c–h) Differential interference contrast (DIC) microscopy images showing PNNs formed from unlabeled lipid membranes. DIC micrographs showing sections from the equator (c and f), and base (d and g) of the PNNs. The magnified version of the regions framed in dashed lines in (d and g) are shown in (e and h), revealing several nanotubes (black arrows). | ||
We used differential interference contrast (DIC) microscopy to visualize the label-free PNNs (Fig. 1c–h), and the lipid nanotubes therein (Fig. 1d, e, g and h). The lack of fluorophore-conjugated lipids appeared to not interfere with the spontaneous formation of phospholipid protocell-nanotube networks. Overall, the findings were consistent with the PNNs formed from lipid preparation containing labeled phospholipids (Fig. 1b).
Encapsulation and transport of cargo
Following the formation of PNNs, we introduced fluorescent cargo to the ambient solution in the vicinity of the compartments by means of an open-volume microfluidic device.20,21 A selected sample region was locally superfused with an aqueous buffer containing the cargo molecules (Fig. 2a). The supplied cargo molecules were ATTO 488 (Fig. 2a), a 10-base RNA labeled with fluorescein amidite (FAM), or a 20-base single-stranded DNA (ssDNA), also labeled with FAM. During superfusion, fluorescently-labeled cargo molecules are spontaneously internalized by the protocells. Although the lipid compartments have similar diameter, the fluorescence intensities of their internal volumes vary after superfusion (Fig. S1†). If the uptake solely depended on direct permeation of molecules across the lipid membrane, compartments of similar size would have reached similar internal concentrations after exposure for the same time period.22 Since we observe very different intensities for different vesicles, we think that a transient pore-enhanced uptake route dominates. A similar observation was reported in an earlier study, where the concentration of encapsulated fluorescein inside containers of similar size ranged from 20% to 90% of the concentration of the external medium.22 Finite element simulations of diffusion through membrane pores predicted the experimental findings well.22 According to these simulations, 3–5 pores of nm size in a micrometer-sized container would account for the experimentally observed uptake.22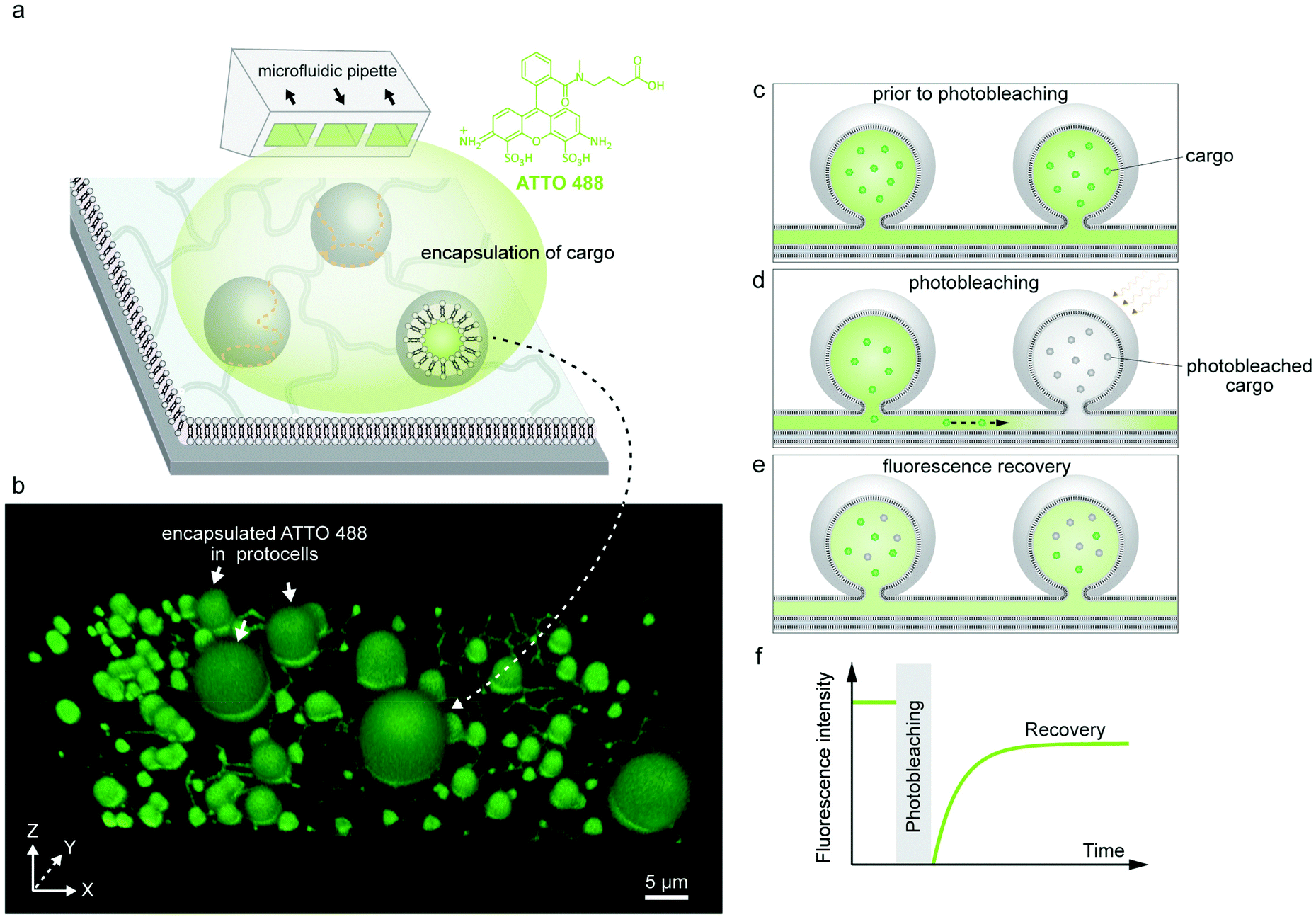 | ||
| Fig. 2 Encapsulation of molecular cargo inside PNN and FRAP experiment. (a) Schematic representation of the encapsulation experiment. An open-volume microfluidic device (microfluidic pipette) creates a confined exposure zone around the protocells, delivering different cargo molecules, e.g. fluorescent dye ATTO 488. (b) 3D confocal micrograph of a label-free PNN upon encapsulating fluorescent dye inside the nanotubes and the protocells. (c–e) Schematic drawing of the FRAP (fluorescence recovery after photobleaching) experiment. (c) The fluorescent cargo is encapsulated inside the protocells and the connecting nanotubes, corresponding to (b). (d) Cargo in one of the protocells in (a) is photobleached using high laser intensity. (e) The fluorescence recovers due to the diffusion of fluorescent cargo from neighboring protocell through the nanotube. (f) Schematic graph depicting typical FRAP curve. | ||
Earlier research has shown that lipid membranes open up transient pores to reduce membrane tension that arises due to factors such as mechanical stress,23 osmotic stress24 or optical illumination.25 The opening of a pore enables the lipids to cover a smaller surface area, lessening the tension on the membrane while simultaneously releasing the excess internal liquid volume. Subsequent closure of the pore is driven by the edge (line) tension of the curved lipids at the edge of the pore.25,26 Determining the position and number of pores in a vesicle can only be performed in very specific environments, e.g. extreme solvent viscosity to stabilize the pores.23
In protocell-nanotube networks, simultaneous collapse and emergence of vesicles on the same membrane region can be observed,10 which indicates variations in local membrane tension. Additionally, during superfusion the network is exposed to a gentle hydrodynamic flow (10–100 nl s−1), which may add to the mechanical stress on the membranous protocells, increasing the membrane tension and facilitating the formation of transient nanopores through which the cargo can diffuse. Our observations on surface-adhered lipid compartment systems11,22,27 show that the transient pores can contract and close, indicated by the prolongation of fluorescence signal of the encapsulated constituents in the sealed compartments which is distinguishable from the ones which immediately leak the constituents (several minutes vs. seconds).22 The latter leads to a sudden decay in fluorescence intensity measured inside the compartments.22
The concentration of FAM-RNA and FAM-ssDNA inside the compartments after 4 min of superfusion was observed to be lower compared to ATTO 488 (Fig. S1†). This is likely due to the higher molecular weight of the RNA and DNA, slowing down their diffusion through the pores (6.8 kDa for FAM-ssDNA, 3.4 kDa for FAM-RNA vs. 0.8 kDa for ATTO 488). The cargo molecules that are not encapsulated by the PNN during superfusion, are aspirated by the microfluidic pipette due to continuous recirculation.21 Upon termination of superfusion, the ambient buffer instantly becomes free of cargo.
The encapsulated cargo could be observed within the model protocells and nanotubes of the network via confocal microscopy. The confocal micrograph presented in Fig. 2b shows encapsulated ATTO 488 inside a surface-supported PNN. It confirms that the cargo molecules can enter the nanotubes, but cannot alone affirm their ability to transport the cargo. There remains still the possibility that the nanotubes are not conducting, but contain membrane defects which could block the tubes and prevent the exchange of molecules between the compartments.
After encapsulating the cargo, we investigated the transport of the molecules within the network by means of fluorescence recovery after photobleaching (FRAP) experiments (Fig. 2c–f). FRAP is a commonly used technique in cell biology and biomaterials science to determine dynamic processes e.g. membrane fluidity,13 protein localization and mobility,28 protein trafficking in intercellular nanotubes29,30 and intracellular protein transport in ER or Golgi.31,32 When an isolated lipid vesicle encapsulating a fluorescent solution suspended in a non-fluorescent aqueous environment is photobleached, its intensity does not recover as there is no access to a new source of fluorophores for replenishment (Fig. S2†).33 If the vesicles are physically connected through the tunneling nanotubes (Fig. 2c and d), and the molecules are able to diffuse through the tubes, the fluorescence of the photobleached vesicles recovers (Fig. 2e and f).
We verified this hypothesis by encapsulating ATTO 488, FAM-RNA and FAM-ssDNA in several nodes of the protocell networks, photobleaching selected compartments in the network and subsequently measuring the recovery of the initially photobleached compartments. The results from multiple experiments are shown in Fig. 3. Each plot in graphs Fig. 3a, h and l, labeled with a capital letter, shows the fluorescence recovery of a single compartment within a network. Fig. 3a–g are associated with the experiments using ATTO 488, Fig. 3h–k RNA, and Fig. 3l–o DNA as encapsulated molecular cargo. Fig. 3b–d and e–g show confocal microscopy time series from two different experiments. For each experiment, the recovery of the compartments encircled in dashed lines was monitored and plotted in Fig. 3a (I and B). Plot E in Fig. 3h is obtained from the lipid compartment shown in Fig. 3i–k, and plot C in Fig. 3l from the compartment in Fig. 3m–o. Confocal microscopy time series corresponding to all other plots shown in Fig. 3 are presented in Fig. S3–5.†
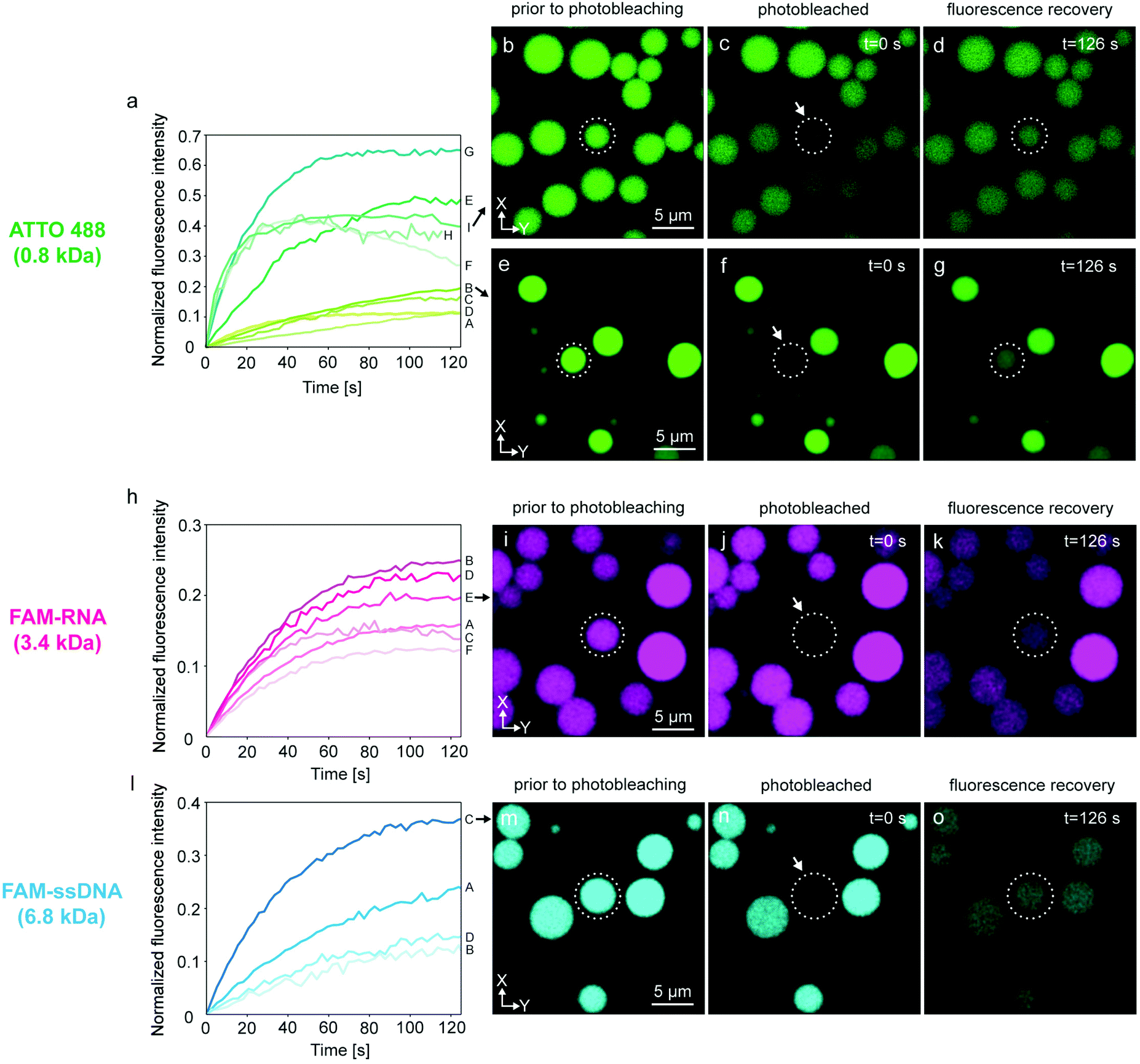 | ||
| Fig. 3 FRAP of selected compartments within PNNs. Three different cargo molecules, ATTO 488 (green color), FAM-RNA (magenta color) and FAM-ssDNA (cyan color) were loaded into protocell networks. (a) Plots show 9 FRAP experiments of ATTO 488 (A–I), each plot representing one experiment. (b–d) Confocal micrographs corresponding to plot I in panel (a): (b) ATTO 488-containing model protocell prior to, (c) during, (d) after, photobleaching. (e–g) Micrographs of the experiment corresponding to plot B in panel (a). (h) FRAP curves for RNA (plots A to F). (i–k) Micrographs of the FRAP experiment corresponding to plot E in panel (h). (l) Plots showing 4 FRAP experiments for DNA (plots A to D). Micrographs of experiment C are shown in (m–o). The compartments monitored for recovery are encircled in white dashed lines. | ||
Plots A–D in Fig. 3a show a final recovery of ∼10–20% of the initial fluorescence intensity. Plots E–I show more rapid recovery up to 65% (plot G) of initial intensity. The compartment represented in plot H could only be monitored until 115 s and further data collection could not be achieved. Plots F and I show a decline after reaching 40% of the initial intensity. Decrease in fluorescence intensity can be due to inherent photobleaching caused by continuous imaging,34 or leakage from the compartments into the ambient solution via transient pores or defects in the membrane.25,35 Sott et al. characterized diffusive transport of fluorescein within vesicle-nanotube networks.18 These networks were manually generated by a microneedle technique. The authors concluded that dissipation factors such as leakage and photobleaching led to a minor reduction of fluorescent contents in the network. We provided a detailed discussion in ESI section 4† based on the experimental results depicted in Fig. S6.† It is plausible that up to a 20% fluorescence loss over time could be due to leakage through the membrane, but we also take into account content loss from photobleaching. The latter is not a physical loss, but a reduction of signal from fluorescent species that are still within the container.
Another possibility is transport of content from the recovered vesicle to adjacent compartments through other established nanotubular connections. It is challenging to determine to which exact compartments transfer of material would occur, as protocell-nanotube networks are extending out of the field of view for tens to hundreds of micrometers. RNA-containing compartments (Fig. 3h–k and Fig. S4†) recover ∼12–26% of the original intensity over a time period of 100–120 s. It was more challenging to internalize DNA inside the network compared to ATTO 488 and RNA, due to its comparatively larger size. We conducted a total of four FRAP experiments with DNA (Fig. 3l–o and Fig. S5†). The amount of recovery varied between ∼12% and 36%. We observed the transfer of all cargo molecules between the compartments, and established thus proof of principle.
Geometric parameters influencing transport
We employed a simple analytical model to determine the impact of certain geometrical parameters for the molecular transport within the protocell-nanotube networks (Fig. 4). Although other transport mechanisms between vesicles via nanotubes have been reported, e.g. Marangoni transport,36 for the model we mainly focus on molecular diffusion as means of transport. Rate equations describing the equilibration of particles between two37,38 or more38 chambers connected with a capillary (Fig. 4a) were previously established. Volume of the compartments, diffusion coefficient of the molecules traveling between the compartments, length and radius of the connective nanoconduits were taken into account to calculate the diffusion rate37,38 and relaxation time.37 | ||
| Fig. 4 Geometric parameters influencing molecular diffusion in a two-vesicle system. (a) The analytical model is based upon two vesicles with diameters d1 and d2, connected through nanotube with a radius of r and distance L. ATTO 488 transports to the photobleached vesicle. Plot depicted in a black continuous line in (b-e) is experiment shown in Fig. S6.† Dashed lines in (b-e) are obtained with different variables applied to eqn (3). One of the curves in each graph fits to the recovery curve of the photobleached vesicle (solid line) while changing the (b) tube radius (r), (c) diffusion coefficient of the cargo molecule (D), (d) compartment diameter (d), and (e) nanotube length (L). | ||
The fluorescence recovery, F, over time, t, takes the form of an exponential function (Fig. 2f) and a FRAP curve can be fitted to:39
 | (1) |
For a simple system with two spherical vesicles, the relaxation time, τrelax, is given by:37
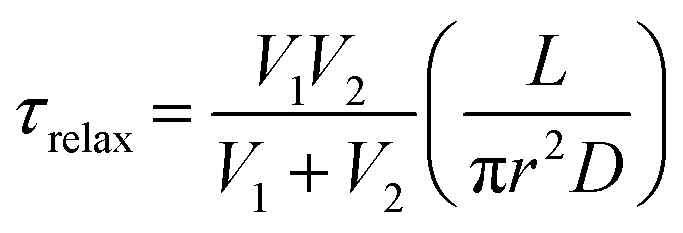 | (2) |
 | (3) |
The above-mentioned models are based on simple, closed systems (dead ends) containing straight tubular connections37,38,40 (Fig. 4a), where the networks in our experiments typically consists of multiple protocells with a branched, complex network of nanotubes extending outwards with many inter-connections and junctions (Fig. 1). Another feature that is different in our experiments is that the receiving (photobleached) lipid compartments are not free of cargo but contain the cargo with quenched fluorophores (Fig. 2d). Despite these differences, the models appear to provide a good approximation to the experimental system we have.
We compared the fluorescence recovery time of the compartment shown in Fig. S6† to the relaxation time (eqn (3)) predicted by the model37 (Fig. 4b–e). The two adjacent compartments in the experiment contain ATTO 488, which has a diffusion coefficient (D) of 400 μm2 s−1.41 We assume that the protocell compartments are spherical and connected with a straight tube of length L between the centers of their base. Each of the two compartments has a diameter of 4.2 μm and we take L = 4.2 μm since the compartments are located very close to each other (Fig. S6†). Despite their proximity, it is not likely that these two compartments are fused, based on the mathematical model characterizing the fusion within the PNNs11 (Fig. S7†). The radius of the nanotube, r, is the unknown variable.
Recovery time based on nanotubular connections with varying nanotube radii were plotted while keeping d1 = 4.2 μm, d2 = 4.2 μm, D = 400 μm2 s−1, L = 4.2 μm (cf.eqn (1) and (3)) (Fig. 4b). The best match of the experimental curve to the theoretical curve was achieved for the tube with a radius of 50 nm (Fig. 4b). This is within the range of tunneling nanotubes observed in cellular12 and artificial systems.42
Next, we focused on diffusion coefficient of the cargo molecule. Fig. 4c shows how diffusion coefficients varying from 200–800 μm2 s−1 would change the recovery time of the photobleached molecules, i.e. the time for the fluorescently-labeled molecules to travel from one compartment to the photobleached one in the two-vesicle system our model is based on (Fig. 4a and c). The recovery curve for ATTO 488 shown in ESI section S4,† follows the trend predicted for D = 400 μm2 s−1 in Fig. 4c. This agrees with the reported diffusion coefficient of ATTO 488.41 Since we do not have any control on the locations of vesicle nucleation sites, which later transform into protocells, we could not identify an isolated two-vesicle network in experiments involving RNA and DNA. Here we would expect a slower transport as the diffusion coefficients of the RNA and DNA are smaller than of ATTO 488. For a 20-base single-stranded DNA in aqueous solution, D was determined to be 152 μm2 s−1 (ref. 43) and for 10-base RNA, 186 μm2 s−1.44 Therefore in a closed two-vesicle setting, the recovery, i.e., transport, of RNA/DNA would be similar to the one shown with red dashed line in Fig. 4c.
Fig. 4d shows multiple plots corresponding to compartments with varying sizes. As anticipated, the larger the diameter of the compartments, the more time it takes to reach equilibrium. For the receiving (photobleached) vesicle this means more volume needs to be filled, while for the donating vesicle it means a lower probability for the cargo to reach the entrance to the nanotube.
Finally we investigated the length of the nanotube (Fig. 4e). With increasing nanotube length the travel time of the cargo in the nanotube increases, resulting in slower fluorescence recovery (Fig. 4e). The nanotube length, related to the distance between the protocells, is directly proportional to the relaxation time (eqn (3)). The exact length of the connecting nanotubes in the experiments is difficult to predict, as they are almost never straight due to pinning and branching.11,45
The recovery curve of the experiment shown in Fig. S6† (plot shown with a continuous black line in Fig. 4b–e) declines gradually over time. This can be due to inherent photobleaching caused by continuous imaging,34 or leakage from the compartments25,35 as discussed above.
Conclusion
Our findings confirm that the lipid nanotubes in surface-adhered protocell networks are open, and allow molecular transport between the interconnected bilayer-encapsulated compartments. The rate of the diffusive transport is highly dependent on the structure of the network and influenced by the parameters such as the radius and length of the nanotubes, size of the compartments and the diffusion coefficient of the molecule that is transported. There is a physical limit to how small the radius of the lipid nanotubes can be without the presence of curvature stabilizing proteins,46 rendering the other parameters deciding factors for the diffusion rate in a prebiotically relevant context. We conclude that it appears feasible to increase complexity by encapsulating reactants for prebiotic reactions in PNNs, in order to gain a deeper understanding of possible chemical communication processes within primitive cell populations at the origin of life.Materials and methods
Lipid preparation
Lipid suspensions were prepared with soybean polar extract and E. coli polar extract (Avanti Polar Lipids, USA) (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 wt%), using the dehydration-rehydration method.47 Briefly, lipids were dissolved in chloroform in a 10 mL pear shaped bottom flask leading to a final concentration of 10 mg mL−1. Chloroform is toxic and highly volatile and should always be handled under a fume hood with associated personal protective equipment. For the sample shown in Fig. 1b, 1 wt% of lipid-conjugated fluorophore 16:0 Rhod Liss PE (Avanti Polar Lipids, USA) was added into the lipid mixture, and for the sample shown in Fig. S2a,† 1 wt% ATTO 655 DOPE (Atto-Tech GmbH, Germany). 300 μL of the dissolved lipid mixture was placed in a rotary evaporator and the solvent was removed at 24 rpm and reduced pressure (20 kPa) for 6 hours to form a dry lipid film. The dry lipid film was rehydrated with 3 mL phosphate-buffered saline (PBS) followed by addition of 30 μL glycerol. The PBS contained 5 mM Trizma base, 30 mM K3PO4, 30 mM KH2PO4, 3 mM MgSO4·7H2O, and 0.5 mM Na2-EDTA (pH = 7.4, adjusted with H3PO4). The lipid suspension was kept at 4 °C overnight to allow swelling of the lipid cake. The following day, the suspension was sonicated for 5–10 s at room temperature, leading to formation of a giant vesicle suspension. The suspension was aliquoted and stored at −18 °C.
:50 wt%), using the dehydration-rehydration method.47 Briefly, lipids were dissolved in chloroform in a 10 mL pear shaped bottom flask leading to a final concentration of 10 mg mL−1. Chloroform is toxic and highly volatile and should always be handled under a fume hood with associated personal protective equipment. For the sample shown in Fig. 1b, 1 wt% of lipid-conjugated fluorophore 16:0 Rhod Liss PE (Avanti Polar Lipids, USA) was added into the lipid mixture, and for the sample shown in Fig. S2a,† 1 wt% ATTO 655 DOPE (Atto-Tech GmbH, Germany). 300 μL of the dissolved lipid mixture was placed in a rotary evaporator and the solvent was removed at 24 rpm and reduced pressure (20 kPa) for 6 hours to form a dry lipid film. The dry lipid film was rehydrated with 3 mL phosphate-buffered saline (PBS) followed by addition of 30 μL glycerol. The PBS contained 5 mM Trizma base, 30 mM K3PO4, 30 mM KH2PO4, 3 mM MgSO4·7H2O, and 0.5 mM Na2-EDTA (pH = 7.4, adjusted with H3PO4). The lipid suspension was kept at 4 °C overnight to allow swelling of the lipid cake. The following day, the suspension was sonicated for 5–10 s at room temperature, leading to formation of a giant vesicle suspension. The suspension was aliquoted and stored at −18 °C.
Surface preparation
SiO2 surfaces were fabricated at the Norwegian Micro- and Nano-Fabrication Facility at the University of Oslo (MiNaLab). SiO2 films were deposited onto glass cover slips (Menzel Gläser #1, 100–150 μm thickness; WillCo Wells B.V., Amsterdam, NL) by E-beam physical vapor deposition using an EvoVac instrument (Ångstrom Engineering, Canada), to a final film thickness of 84 nm.Sample preparation
For sample preparation, two 4 μL droplets of lipid suspension were placed on a clean glass and dehydrated in desiccator for 20–25 min under low pressure. The dry lipid film was rehydrated for 10 min with 0.5–1 mL of Na-HEPES buffer containing 10 mM HEPES and 100 mM NaCl (pH = 7.8, adjusted with NaOH). The rehydrated suspension was later transferred into an observational chamber containing Ca-HEPES buffer (10 mM HEPES, 100 mM NaCl and 4 mM CaCl2, pH = 7.8, adjusted with NaOH). The sample was kept at room temperature for 2–3 days for PNN formation and protocell growth. Alternatively, to speed up the protocell growth, the sample was incubated at 35 °C until the following day to promote protocell growth.11Encapsulation of cargo molecules
The cargo molecules were delivered with an open-volume microfluidic pipette20,21 (Fluicell AB, Sweden) positioned using 3-axis hydraulic micromanipulator (Narishige, Japan) to the vicinity of the protocell-nanotube structures. The protocells were superfused with solutions of Ca-HEPES buffer containing 500 μM of ATTO 488 carboxyl (Atto-Tech GmbH, Germany). DNA solution was prepared and delivered in nuclease-free water (Thermo Fisher Scientific, USA) by dissolving 200 μM of 20-base ssDNA (5′-/56-FAM/TGT ACG TCA CAA CTA CCC CC-3′, Integrated DNA Technologies, USA). 10-base RNA oligomers (5′-FAM-AAA AAA AAA A-3′, Dharmacon, USA) were dissolved in nuclease-free water to a final concentration of 100 μM. Exposing the nanotube networks rapidly to nuclease-free deionized water -a hypotonic environment- facilitated the rapid swelling and growth of membranous compartments (Movie S1†), reducing the time period of growth from hours10 to minutes.Microscopy imaging and analysis
Imaging has been performed using Laser scanning confocal microscope DMi8 (Leica Microsystems, Germany) equipped with a 40× oil objective (NA: 1.3). The 3D fluorescence micrographs in Fig. 1b and 2b were reconstructed using the Leica Application Suite X Software (Leica Microsystems, Germany). Image enhancement (brightness/contrast) of fluorescence micrographs in the figures was performed with the Adobe Photoshop CS4 (Adobe Systems, USA). Schematic drawings were created with Adobe Illustrator CS4 (Adobe Systems, USA).FRAP
FRAP experiments were performed using the FRAP module in the Leica Application Suite X Software. UV diode laser (405 nm, 50 mW) with 50% intensity was used for the photobleaching. A circular region of interest (ROI) on each protocell was defined. The ROIs were monitored during pre-photobleaching (∼10 s), photobleaching (∼10 s) and post-photobleaching (>115 s), consecutively.The FRAP curves in Fig. 3, 4 and S6† show the recovery period after photobleaching. The FRAP curve in Fig. S2† includes the period prior to photobleaching. The normalized fluorescence intensity (Inormalized) of the plots in Fig. 3, 4 and S2† was determined as:  , where I(t) is the fluorescence intensity at time t, I0 is the fluorescence intensity right after the photobleaching/at the beginning of the recovery, and Iinitial is the fluorescence intensity prior to the photobleaching. In Fig. S6,† the normalized fluorescence intensity of the donor (Inormalized,donor) and acceptor (Inormalized,acceptor) protocell after photobleaching was determined as:
, where I(t) is the fluorescence intensity at time t, I0 is the fluorescence intensity right after the photobleaching/at the beginning of the recovery, and Iinitial is the fluorescence intensity prior to the photobleaching. In Fig. S6,† the normalized fluorescence intensity of the donor (Inormalized,donor) and acceptor (Inormalized,acceptor) protocell after photobleaching was determined as:  and
and 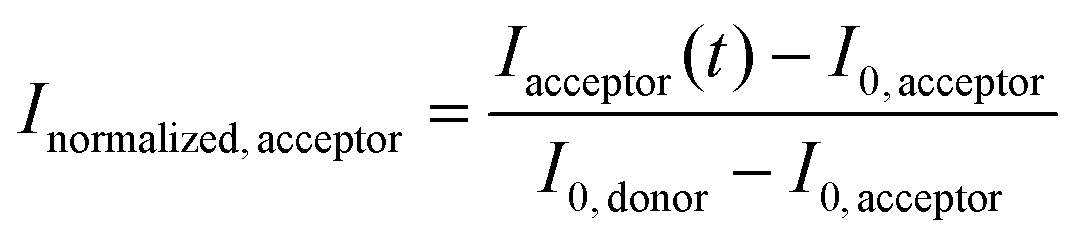 , respectively. Idonor(t) and Iacceptor(t) are the fluorescence intensity of donor and acceptor protocell, respectively, at time t. I0, donor and I0, acceptor are the fluorescence intensities when t = 0, i.e. the beginning of the fluorescence recovery, for the donor and acceptor protocell, respectively. All graphs in Fig. 3, 4, S2 and S6† were plotted using MATLAB R2020b.
, respectively. Idonor(t) and Iacceptor(t) are the fluorescence intensity of donor and acceptor protocell, respectively, at time t. I0, donor and I0, acceptor are the fluorescence intensities when t = 0, i.e. the beginning of the fluorescence recovery, for the donor and acceptor protocell, respectively. All graphs in Fig. 3, 4, S2 and S6† were plotted using MATLAB R2020b.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was made possible through financial support obtained from the Research Council of Norway (Norges Forskningsråd) Project Grant 274433, UiO: Life Sciences Convergence Environment, as well as the startup funding provided by the Centre for Molecular Medicine Norway (RCN 187615) at the University of Oslo.References
- P. Walde, Building artificial cells and protocell models: Experimental approaches with lipid vesicles, BioEssays, 2010, 32(4), 296–303 CrossRef CAS PubMed.
- A. J. Dzieciol and S. Mann, Designs for life: protocell models in the laboratory, Chem. Soc. Rev., 2011, 41(1), 79–85 RSC.
- S. F. Jordan, H. Rammu, I. N. Zheludev, A. M. Hartley, A. Maréchal and N. Lane, Promotion of protocell self-assembly from mixed amphiphiles at the origin of life, Nat. Ecol. Evol., 2019, 3(12), 1705–1714 CrossRef PubMed.
- T. Gánti, The principles of life, Oxford University Press, 2003 Search PubMed.
- Y. Huang, B. Zucker, S. Zhang, S. Elias, Y. Zhu, H. Chen, T. Ding, Y. Li, Y. Sun, J. Lou, M. M. Kozlov and L. Yu, Migrasome formation is mediated by assembly of micron-scale tetraspanin macrodomains, Nat. Cell Biol., 2019, 21(8), 991–1002 CrossRef CAS PubMed.
- M. Liang, L. Ying, P. Junya, W. Danni, Z. Xiaoxin, C. Yitong, C. Lilian, Y. Xiaojun, D. Yanan and Y. Li, Discovery of the migrasome, an organelle mediates release of cytoplasmic contents during cell migration, Cell Res., 2015, 25, 24–38 CrossRef PubMed.
- B. Alberts, J. H. Wilson and T. Hunt, Molecular biology of the cell, Garland Science, New York, 6th edn, 2015 Search PubMed.
- P. Albuquerque and A. Casadevall, Quorum sensing in fungi – a review, Med. Mycol., 2012, 50(4), 337–345 CrossRef CAS PubMed.
- K. Papenfort and B. L. Bassler, Quorum sensing signal-response systems in Gram-negative bacteria, Nat. Rev. Microbiol., 2016, 14(9), 576–588 CrossRef CAS PubMed.
- E. S. Köksal, S. Liese, I. Kantarci, R. Olsson, A. Carlson and I. Gözen, Nanotube-Mediated Path to Protocell Formation, ACS Nano, 2019, 13, 6867–6878 CrossRef.
- E. S. Köksal, S. Liese, L. Xue, R. Ryskulov, L. Viitala, A. Carlson and I. Gözen, Rapid Growth and Fusion of Protocells in Surface–Adhered Membrane Networks, Small, 2020, 16(38), 2002529 CrossRef PubMed.
- A. Rustom, R. Saffrich, I. Markovic, P. Walther and H.-H. Gerdes, Nanotubular Highways for Intercellular Organelle Transport, Science, 2004, 303(5660), 1007–1010 CrossRef CAS PubMed.
- X. Wang and H. H. Gerdes, Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells, Cell Death Differ., 2015, 22(7), 1181–1191 CrossRef CAS.
- G. P. Dubey and S. Ben-Yehuda, Intercellular Nanotubes Mediate Bacterial Communication, Cell, 2011, 144(4), 590–600 CrossRef CAS PubMed.
- A. K. Baidya, S. Bhattacharya, G. P. Dubey, G. Mamou and S. Ben-Yehuda, Bacterial nanotubes: a conduit for intercellular molecular trade, Curr. Opin. Microbiol., 2018, 42, 1–6 CrossRef CAS PubMed.
- G. P. Dubey, G. B. Malli Mohan, A. Dubrovsky, T. Amen, S. Tsipshtein, A. Rouvinski, A. Rosenberg, D. Kaganovich, E. Sherman, O. Medalia and S. Ben-Yehuda, Architecture and Characteristics of Bacterial Nanotubes, Dev. Cell, 2016, 36(4), 453–461 CrossRef CAS PubMed.
- J. Matko and E. A. Toth, Membrane nanotubes are ancient machinery for cell-to-cell communication and transport. Their interference with the immune system, Biol. Futura, 2021, 72(1), 25–36 CrossRef PubMed.
- K. Sott, T. Lobovkina, L. Lizana, M. Tokarz, B. Bauer, Z. Konkoli and O. Orwar, Controlling Enzymatic Reactions by Geometry in a Biomimetic Nanoscale Network, Nano Lett., 2006, 6(2), 209–214 CrossRef CAS PubMed.
- J. W. Lichtman and J.-A. Conchello, Fluorescence microscopy, Nat. Methods, 2005, 2(12), 910–919 CrossRef CAS PubMed.
- A. Ainla, E. T. Jansson, N. Stepanyants, O. Orwar and A. Jesorka, A Microfluidic Pipette for Single-Cell Pharmacology, Anal. Chem., 2010, 82(11), 4529–4536 CrossRef CAS PubMed.
- A. Ainla, G. D. M. Jeffries, R. Brune, O. Orwar and A. Jesorka, A multifunctional pipette, Lab Chip, 2012, 12(7), 1255–1261 RSC.
- K. Spustova, E. S. Köksal, A. Ainla and I. Gözen, Subcompartmentalization and Pseudo–Division of Model Protocells, Small, 2021, 17(2), e2005320 CrossRef PubMed.
- O. Sandre, L. Moreaux and F. Brochard-Wyart, Dynamics of Transient Pores in Stretched Vesicles, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(19), 10591–10596 CrossRef CAS.
- Y. Levin and M. A. Idiart, Pore dynamics of osmotically stressed vesicles, Phys. A, 2004, 331(3), 571–578 CrossRef CAS.
- E. Karatekin, O. Sandre and F. Brochard-Wyart, Transient pores in vesicles, Polym. Int., 2003, 52(4), 486–493 CrossRef CAS.
- I. Gozen and P. Dommersnes, Pore dynamics in lipid membranes, Eur. Phys. J.: Spec. Top., 2014, 223(9), 1813–1829 CAS.
- E. S. Köksal, I. Põldsalu, H. Friis, S. J. Mojzsis, M. Bizzarro and I. Gözen, Spontaneous Formation of Prebiotic Compartment Colonies on Hadean Earth and Pre-NoachianMars**, ChemSystemsChem, 2022, 4, e20210004 Search PubMed.
- E. A. Reits and J. J. Neefjes, From fixed to FRAP: measuring protein mobility and activity in living cells, Nat. Cell Biol., 2001, 3(6), E145–E147 CrossRef CAS PubMed.
- S. Desir, P. Wong, T. Turbyville, D. Chen, M. Shetty, C. Clark, E. Zhai, Y. Romin, K. Manova-Todorova, T. K. Starr, D. V. Nissley, C. J. Steer, S. Subramanian and E. Lou, Intercellular transfer of oncogenic KRAS via tunneling nanotubes introduces intracellular mutational heterogeneity in colon cancer cells, Cancers, 2019, 11(7), 892 CrossRef CAS PubMed.
- N. Rainy, D. Chetrit, V. Rouger, H. Vernitsky, O. Rechavi, D. Marguet, I. Goldstein, M. Ehrlich and Y. Kloog, H-Ras, transfers from B to T cells via tunneling nanotubes, Cell Death Dis., 2013, 4(7), e726 CrossRef CAS PubMed.
- A. S. Verkman, Solute and macromolecule diffusion in cellular aqueous compartments, Trends Biochem. Sci., 2002, 27(1), 27–33 CrossRef CAS PubMed.
- J. F. Presley, K. Hirschberg, K. J. M. Zaal, J. Lippincott-Schwartz, N. B. Cole and T. A. Schroer, ER-to-Golgi transport visualized in living cells, Nature, 1997, 389(6646), 81–85 CrossRef CAS PubMed.
- A. Fragasso, N. De Franceschi, P. Stömmer, E. O. van der Sluis, H. Dietz and C. Dekker, Reconstitution of Ultrawide DNA Origami Pores in Liposomes for Transmembrane Transport of Macromolecules, ACS Nano, 2021, 15(8), 12768–12779 CrossRef CAS PubMed.
- M. Kang, M. Andreani and A. K. Kenworthy, Validation of normalizations, scaling, and photofading corrections for FRAP data analysis, PLoS One, 2015, 10(5), e0127966 CrossRef.
- A. S. Ladokhin, W. C. Wimley and S. H. White, Leakage of membrane vesicle contents: determination of mechanism using fluorescence requenching, Biophys. J., 1995, 69(5), 1964–1971 CrossRef CAS.
- J. Hurtig and O. Orwar, Injection and transport of bacteria in nanotube-vesicle networks, Soft Matter, 2008, 4(7), 1515–1520 RSC.
- L. Dagdug, A. M. Berezhkovskii, S. Y. Shvartsman and G. H. Weiss, Equilibration in two chambers connected by a capillary, J. Chem. Phys., 2003, 119(23), 12473–12478 CrossRef CAS.
- L. Lizana and Z. Konkoli, Diffusive transport in networks built of containers and tubes, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 72(2 Pt 2), 026305–026305 CrossRef CAS PubMed.
- C. Kappel and R. Eils, Fluorescence recovery after photobleaching with the Leica TCS SP2, Confocal Appl. Lett., 2004, 18, 1–12 Search PubMed.
- H. Zhang, Artificial and Intercellular Nanotubes, PhD, Chalmers University of Technology, 2013 Search PubMed.
- P. Kapusta, Absolute Diffusion Coefficients: Comilation of Reference Data for FCS Calibration, 2010 Search PubMed.
- T. Lobovkina, P. Dommersnes, J.-F. Joanny, J. Hurtig and O. Orwar, Zipper dynamics of surfactant nanotube Y junctions, Phys. Rev. Lett., 2006, 97(18), 188105–188105 CrossRef PubMed.
- E. Stellwagen and N. C. Stellwagen, Determining the electrophoretic mobility and translational diffusion coefficients of DNA molecules in free solution, Electrophoresis, 2002, 23(16), 2794–2803 CrossRef CAS PubMed.
- A. Werner, Predicting translational diffusion of evolutionary conserved RNA structures by the nucleotide number, Nucleic Acids Res., 2011, 39(3), e17 CrossRef PubMed.
- T. Bilal and I. Gzen, Formation and dynamics of endoplasmic reticulum-like lipid nanotube networks, Biomater. Sci., 2017, 5(7), 1256–1264 RSC.
- Y. F. Barooji, A. Rørvig-Lund, S. Semsey, S. N. S. Reihani and P. M. Bendix, Dynamics of membrane nanotubes coated with I-BAR, Sci. Rep., 2016, 6(1), 30054–30054 CrossRef CAS PubMed.
- M. Karlsson, K. Nolkrantz, M. J. Davidson, A. Strömberg, F. Ryttsén, B. Åkerman and O. Orwar, Electroinjection of Colloid Particles and Biopolymers into Single Unilamellar Liposomes and Cells for Bioanalytical Applications, Anal. Chem., 2000, 72(23), 5857–5862 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr02290g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |