
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ball milled glyco-graphene oxide conjugates markedly disrupted Pseudomonas aeruginosa biofilms†
Jacopo
Tricomi‡
ab,
Margherita
Cacaci‡
cd,
Giacomo
Biagiotti
ab,
Lucrezia
Caselli
ae,
Lorenzo
Niccoli
fg,
Riccardo
Torelli
d,
Alessio
Gabbani
 h,
Maura
Di Vito
c,
Francesco
Pineider
h,
Mirko
Severi
ab,
Maurizio
Sanguinetti
cd,
Enzo
Menna
ij,
Moreno
Lelli
fg,
Debora
Berti
ae,
Stefano
Cicchi
*ab,
Francesca
Bugli
*cd and
Barbara
Richichi
*ab
h,
Maura
Di Vito
c,
Francesco
Pineider
h,
Mirko
Severi
ab,
Maurizio
Sanguinetti
cd,
Enzo
Menna
ij,
Moreno
Lelli
fg,
Debora
Berti
ae,
Stefano
Cicchi
*ab,
Francesca
Bugli
*cd and
Barbara
Richichi
*ab
aDepartment of Chemistry ‘Ugo Schiff’, University of Firenze, Via della Lastruccia 13, 50019 Sesto Fiorentino, FI, Italy. E-mail: stefano.cicchi@unifi.it; barbara.richichi@unifi.it
bConsorzio Interuniversitario Nazionale per la Scienza e Tecnologia dei Materiali (INSTM), Via G. Giusti, 9, 50121 Firenze, Italy
cDipartimento di Scienze Biotecnologiche di Base, Cliniche Intensivologiche e Perioperatorie, Università Cattolica del Sacro Cuore, Rome, Italy. E-mail: francesca.bugli@unicatt.it
dDipartimento di Scienze di Laboratorio e Infettivologiche, Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome, Italy
eItalian Center for Colloid and Surface Science (CSGI), Via della Lastruccia 3, Sesto Fiorentino, 50019, FI, Italy
fMagnetic Resonance Centre (CERM), Department of Chemistry ‘Ugo Schiff’, University of Firenze, Via Luigi Sacconi 6, 50019 Sesto Fiorentino, FI, Italy
gConsorzio Interuniversitario Risonanze Magnetiche Metalloproteine Paramagnetiche (CIRMMP), Via Luigi Sacconi 6, 50019 Sesto Fiorentino, FI, Italy
hDepartment of Chemistry and Industrial Chemistry, University of Pisa, Via Moruzzi 13, 56132 Pisa, Italy
iDepartment of Chemical Sciences, University of Padova, Via Marzolo 1, 35131 Padova, Italy
jCentre for Mechanics of Biological Materials – CMBM, Via Marzolo 9, 35131 Padova, Italy
First published on 21st June 2022
Abstract
The engineering of the surface of nanomaterials with bioactive molecules allows controlling their biological identity thus accessing functional materials with tuned physicochemical and biological profiles suited for specific applications. Then, the manufacturing process, by which the nanomaterial surface is grafted, has a significant impact on their development and innovation. In this regard, we report herein the grafting of sugar headgroups on a graphene oxide (GO) surface by exploiting a green manufacturing process that relies on the use of vibrational ball mills, a grinding apparatus in which the energy is transferred to the reacting species through collision with agate spheres inside a closed and vibrating vessel. The chemical composition and the morphology of the resulting glyco-graphene oxide conjugates (glyco-GO) are assessed by the combination of a series of complementary advanced techniques (i.e. UV-vis and Raman spectroscopy, transmission electron microscopy, and Magic Angle Spinning (MAS) solid-state NMR (ssNMR) providing in-depth insights into the chemical reactivity of GO in a mechanochemical route. The conjugation of monosaccharide residues on the GO surface significantly improves the antimicrobial activity of pristine GO against P. aeruginosa. Indeed, glyco-GO conjugates, according to the monosaccharide derivatives installed into the GO surface, affect the ability of sessile cells to adhere to a polystyrene surface in a colony forming assay. Scanning electron microscopy images clearly show that glyco-GO conjugates significantly disrupt an already established P. aeruginosa biofilm.
Introduction
Biofilm-associated infections represent the majority of chronic infections including nosocomial ones.1 Specifically, biofilms are composed of bacterial communities that are embedded in a self-produced matrix of extracellular polymeric substances (EPSs).2 Here, the lifestyle of pathogens is clearly distinct from that of free-living (planktonic) bacterial cells. Indeed, while bacteria belonging to the superficial layers can leave the biofilm and colonize new sites, cells of the deeper layer slow down their metabolism. Microorganisms organized in biofilms are more resistant to antimicrobial agents as the results of multifactorial mechanisms3 and, as more recently discovered, of the presence of peculiar phenotypic sub-populations (i.e. persister and viable but non-culturable (VBNC) cells).4–8 Therefore, the discovery of efficient anti-biofilm formulations is a top priority in the current health context.9P. aeruginosa is a multidrug-resistant Gram negative opportunistic bacterium capable of causing lung and bloodstream infections including sepsis and it is now listed as a “priority pathogen” by the World Health Organization.10 The persistence of P. aeruginosa is attributed to its ability to form, under moist conditions, biofilms on almost all inert materials including medical devices (i.e. catheters, ventilator tubes, prostheses) and biotic surfaces, making the biofilm the main virulence factor in most chronic infections.11 The establishment of a chronic infection is determined by the different properties displayed by the P. aeruginosa biofilm.6,12,13 In particular, resistance to antibiotics of P. aeruginosa biofilm-associated cells is related to multifaceted factors, such as the production of the polysaccharide alginate in the matrix and of filamentous phages that might also contribute to slowing the antibiotic diffusion through the biofilm,13 mutations in the regulators of efflux pumps, and the acquisition of resistance determinants through horizontal gene transfer.14
P. aeruginosa produces two small soluble calcium(II)-dependent homotetrameric lectins, the D-galactose-binding LecA and the D-mannose/L-fucose-binding LecB.15 These carbohydrate-binding proteins are key players in the formation of the P. aeruginosa biofilm matrix as they are involved in the specific recognition of glycans expressed on the host cell surface.16,17 In addition, they bind branched side glycan chains of the exopolysaccharides in the matrix thus contributing to the biofilm's structural integrity and increasing the retention of bacterial cells within the biofilm.18 Therefore, the identification of drugs able to interfere with the lectin–sugar interactions is considered an intriguing approach to overcoming the formation of biofilms and the surge of antibiotic resistance.19–21 In particular, the conjugation of D-galactose, L-fucose glycosides and their glycomimetics/analogues to a vast array of multivalent glyco-constructs (i.e. glycopolymers, glycoclusters, glycodendrimers and glyconanoparticles) proved to be a putative therapeutic approach toward the development of antiadhesive agents that by the targeting of either LecA or LecB, interferes with P. aeruginosa adhesion since the early stages of biofilm formation.22–24
In this context, carbon nanomaterials, thanks to their peculiar properties and the high surface area, are suitable platforms for the self-assembly and conjugation of carbohydrates aimed at the creation of versatile nanostructures for applications in nanomedicine.25–27 Among them, pristine (or reduced) graphene and its oxidized form graphene oxide (GO) are attractive nanomaterials for many applications.28 The grafting of simple and complex saccharides,29–31 on the graphene surface through both covalent and non-covalent interactions, provides not just stable aqueous glycographene suspensions, but also enables exploring the biomolecular recognition of glycans by the creation of bioactive glyconanohybrids.27,32,33 However, their combination with carbohydrates is the least explored compared to the other carbon nanomaterials (i.e. fullerenes, carbon nanotubes).32,33 Indeed, glycographene conjugates have been mainly studied in biomedical applications, where the molecular recognition of glycans plays a key role, such as the fabrication of biosensors for the detection of lectins34,35 or carbohydrate binding receptors,36 for bacterial capture and disinfection,37 and also as delivery systems for cancer therapy.38–42 GO proved to be, in some of these applications, a better candidate over pristine graphene due to its potential to be degraded by peroxidase enzymes.43 Notably, the accumulation of GO in specific organs is significantly affected by the thickness of GO sheets and some studies reported the long-term cytocompatibility of highly dispersed GO sheets.43 In addition, the covalent chemical functionalization of GO and the aqueous colloidal stability enhance the rate of biodegradation and thus its biocompatibility.43
The bactericidal activity of GO has been reported44,45 and it was mainly attributed to the combination of physical (i.e. bacteria wrapping and nano-knife action)45 and chemical (i.e. reactive oxygen species production) damages induced by GO nanosheets which cause the collapse of bacterial cells (the ‘nanoscale dewetting’).46 In particular, some of us have recently characterized the antimicrobial properties of GO by evaluating the different mechanisms of action with respect to the environmental conditions adopted to study the bacteria–GO interaction, along with other studies regarding the anti-biofilm properties of GO.46–48
In this work, GO was modified by grafting monosaccharides leading to chemical converted glyco-GO derivatives 1–3 (Fig. 1).
 | ||
| Fig. 1 General structure of glyco-GO conjugates 1–3. | ||
In particular, L-fucose and D-mannose residues have been included for the targeting of LecB whereas D-galactose residues have been included for providing the targeting of LecA. Notably, monosaccharides have been grafted on the GO surface by exploiting an unconventional and innovative approach that we have already employed for the functionalization of structurally different bionanomaterials, and that relies on the use of a vibrational ball milling apparatus.49,50 In the mechanochemical approach the reactions are conducted inside a closed vessel containing the reagents and appropriately sized spheres/balls (i.e. stainless steel, agate or tungsten carbide spheres). Then, the shaking of the container causes the spheres to collide with each other, transferring energy to the reactants.51
In this work, the reactivity of pristine GO with monosaccharides in the mechanochemical process has been investigated by characterizing the glyco-GO conjugates using complementary and advanced techniques. The glyco-GO conjugates, according to the terminal monosaccharide residues, efficiently provide P. aeruginosa biofilm dispersal, by exploiting both the intrinsic GO associated ‘nanoscale dewetting’ effect and by interfering with the lectin–sugar recognition in a synergic way.
Results and discussion
Mechanochemical synthesis and characterization of glyco-GO conjugates 1–3
We have recently reported on the efficient nucleophilic substitution of primary amine groups on the epoxide residues of the GO platform by exploiting the ball milling approach.52 This strategy, which has recently proved to access a wide range of compounds,49,50 allowed us to obtain a functionalized GO conjugate using a green and scalable process under organic solvent-free conditions. In this work, we employed this protocol using vibrational ball mills for the synthesis of glyco-GO conjugates 1–3 (Scheme 1).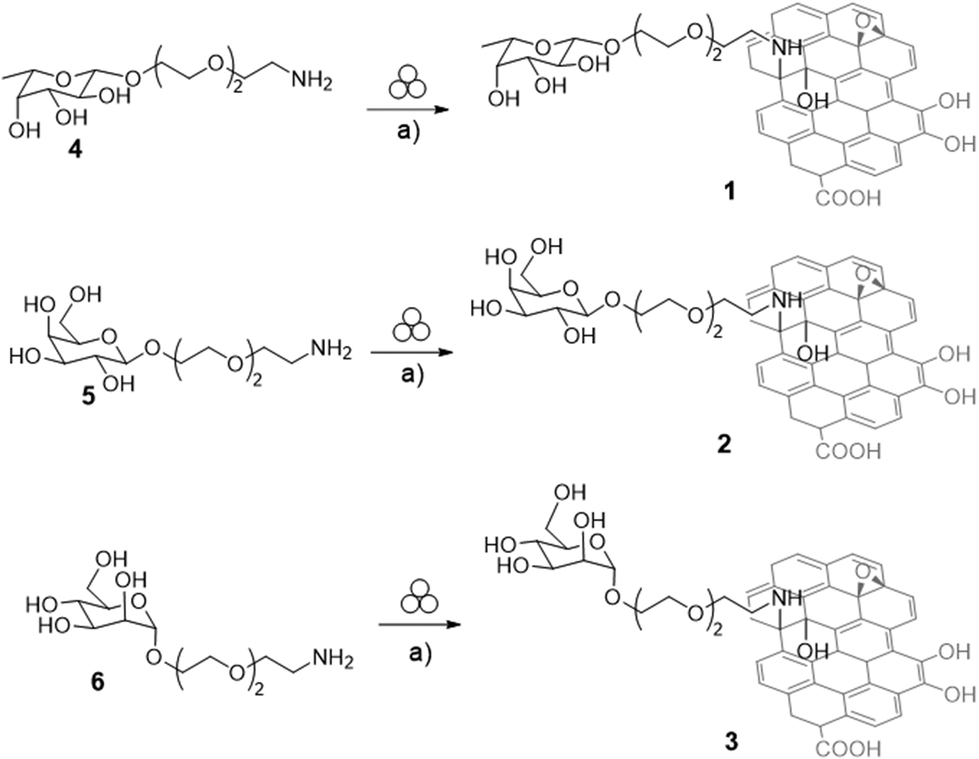 | ||
| Scheme 1 Synthesis of Fuc-GO 1, Gal-GO 2, and Man-GO 3. Reaction conditions: (a) Graphene oxide (GO) powder, 10 mL agate jar, agate spheres Ø1 cm, 25 Hz, 90 min. | ||
At first, glycosides 4–6 (Scheme 1), containing respectively L-fucose (Fuc, compound 4, Scheme 1), D-galactose (Gal, compound 5, Scheme 1), and D-mannose (Man, compound 6, Scheme 1) residues and bearing a PEGylated linker at the anomeric position, have been prepared starting from the corresponding fully acetylated monosaccharides by modifying previously reported protocols (see the ESI, Schemes S1–S3†).53,54
Then, we selected the fucose derivative 4 (Scheme 1) to optimize the milling conditions. The mechanochemical reaction was performed by grinding GO powder and 4 (in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in weight) at 25 Hz for 40 minutes in a 10 mL stainless-steel mixer mill and using two stainless-steel balls (∅ = 1.0 cm). Fucoside 4 is a viscous oil, and thus we performed milling under wet grinding conditions49 by adding water (450 μL) in a solvent-volume/reagent-weight ratio (η)49 of 2.25. The product was easily recovered from the jar using Milli-Q H2O and then, it was centrifuged (8000 rpm, 30 min, 2 cycles). The solid was dispersed in methanol (100 mL) and filtered (0.2 μm, Nylon membrane).
:1 ratio in weight) at 25 Hz for 40 minutes in a 10 mL stainless-steel mixer mill and using two stainless-steel balls (∅ = 1.0 cm). Fucoside 4 is a viscous oil, and thus we performed milling under wet grinding conditions49 by adding water (450 μL) in a solvent-volume/reagent-weight ratio (η)49 of 2.25. The product was easily recovered from the jar using Milli-Q H2O and then, it was centrifuged (8000 rpm, 30 min, 2 cycles). The solid was dispersed in methanol (100 mL) and filtered (0.2 μm, Nylon membrane).
Thermogravimetric analysis (TGA) under an air atmosphere (see the ESI, Fig. S1† Fuc-GO 1a) showed a relevant residual weight at 500 °C and above, not observed with pristine GO, whereas both the organic and GO components are expected to decompose at lower temperatures. In particular, the pyrolysis of the oxygenated groups on the basal GO plane and of the sugar is supposed to occur up to about 350 °C, followed by complete sample combustion.55 Accordingly, inductively coupled plasma atomic emission spectroscopy (ICP-AES) analyses of Fuc-GO 1a (see the ESI, Table S1†) showed the presence of metallic residues (i.e. iron and chromium), incorporated during the milling process.56 In order to avoid the acidic work-up to remove the metallic debris,56 that were non-compatible with the presence of glycosides, we decided to modify the grinding conditions. Indeed, despite the use of wet conditions, the viscosity of the monosaccharide derivative resulted in sticky reaction mixtures, which could have hindered the adequate motion of the balls during the milling process. Therefore, we decided to include NaCl as an inert milling auxiliary49 and to work under liquid-assisted grinding (LAG)49 conditions by reducing the η value to 0.46 (200 μL of water), aiming at improving the homogeneity of the reaction mixture and the overall mixing. In doing so, GO powder and 4 (Scheme 1) were milled in a ratio of 1:1 in the presence of NaCl (230 mg) and water (200 μL) using the same milling conditions described before (25 Hz, 40 minutes, 10 mL mixer mill, two stainless-steel balls ∅ = 1.0 cm). In this case, only a slightly lower residual weight was detected by TGA (see the ESI, Fig. S1,† Fuc-GO 1b). Accordingly, ICP-AES analysis (see the ESI, Table S1†) showed that the amount of metal debris was slightly reduced under these conditions (% Fe: 13.11 of 1bvs. 16.10 of 1a; % Cr: 2.04 of 1bvs. 3.70 of 1a).
On the basis of these findings, agate milling media (both jars and balls) were used aimed at avoiding the leaching of metallic debris and at maintaining the mechanochemical efficiency of the process. In addition, the amount of inert milling auxiliary was increased (430 mg vs. 230 mg) whereas the η value was reduced to 0.25 (150 μL of water). Thus, GO powder and 4 (Scheme 1) were milled in a ratio of 1:1 at 25 Hz for 40 minutes in a 10 mL mixer mill and using two agate balls (∅ = 1.0 cm). In turn, the product was recovered from the jar using Milli-Q H2O and then centrifuged (8000 rpm, 30 min, 2 cycles). The solid was dispersed in methanol (100 mL) and filtered (0.2 μm, Nylon membrane). As expected, no trace of metallic residues was detected by ICP analysis (see the ESI, Table S1,† Fuc-GO 1). Accordingly, TGA shows the complete combustion of the sample with a residual weight close to that of pristine GO (Fig. 2, Fuc-GO 1), excluding contamination from the milling process.
 | ||
| Fig. 2 (A) Thermogravimetric analysis (TGA) under an air atmosphere; (B) Raman spectra; and (C) TEM images of Fuc-GO 1, Gal-GO 2, Man-GO 3, GObm (pristine GO milled without monosaccharide derivatives) and pristine GO. | ||
On this basis, we decided to extend the protocol for the grafting of the galactose (compound 5, Scheme 1) and mannose (compound 6, Scheme 1) glycosides on the pristine GO surface. TGA analyses in air (Fig. 2A and see the ESI Fig. S2†) and ICP-AES data (see the ESI, Table S1†) of the resulting Gal-GO 2 and Man-GO 3 conjugates confirmed that contamination was avoided, as for the case of Fuc-GO 1. Thermograms from the glyco-GO conjugates 1–3 (Fig. 2A) show a lower combustion temperature (centered at about 420 °C) than pristine GO (about 460 °C). This effect can be ascribed to the mechanical action of ball milling, inducing fragmentation of GO as also evidenced by TEM (vide infra), and making oxygenated groups more exposed. In fact, the same temperature lowering is observed with a GO sample subjected to the milling procedure, but without sugar (Fig. 2A, named GObm derivative). The weight loss ascribed to the organic moiety is supposed to occur in a range of temperatures partly superimposed to the weight losses due to the pyrolysis and combustion of oxygenated groups and the basal plane of GO, centered at 180 and 400 degrees respectively. By comparing the weight losses of the three glyco-GO conjugates with that of GObm in the intermediate region (230–330 °C), less affected by the two main decompositions, a relative (underestimated) evaluation of sugar loadings is obtained for Fuc-GO 1 (3.1%), Gal-GO 2 (5.1%), and Man-GO 3 (3.2%). TGA data are in agreement with the trend obtained by elemental analysis (see the ESI, Table S2†).
Then, all the conjugates were further characterized by means of a series of complementary techniques such as UV-vis spectroscopy, Fourier-transform infrared (FTIR) spectroscopy, transmission electron microscopy (TEM), Raman spectroscopy and solid-state NMR aiming at an in-depth evaluation of the chemical reactivity of pristine GO in the milling process.
FT-IR spectroscopy has been reported as a useful methodology for defining the chemistry involved in the reaction of GO.43 In particular, the change in the intensity of the epoxide peak at 1230 cm−1 was monitored in order to evaluate GO functionalization by epoxide ring opening. However, as previously reported,43 such a band is usually small and can be overlapped by other bands, thus making the comparative analysis of the spectra difficult. This is the case of IR spectra of the glyco-GO conjugates 1–3 which were compared with the IR spectrum of pristine GO (see the ESI, Fig. S3†). Therefore, FT-IR spectroscopy cannot be used to confirm the outcome of the reaction. The UV-Vis absorption spectra of GO glycoconjugates 1–3 (see the ESI, Fig. S4†) showed a broadening in the GO absorption peak at 230 nm and as expected no other absorption bands are introduced upon functionalization with the monosaccharide moieties.
Then, Raman spectroscopy was employed to acquire the Raman spectra of all the conjugates, as well as of the pristine GO and GObm. Fig. 2B shows the Raman spectra acquired at a laser excitation wavelength λmax = 532 nm in the frequency range 102–3203 cm−1, while Table 1 summarizes the most significant spectral parameters for the different samples.
| Sample | GO | GObm | Fuc-GO 1 | Gal-GO 2 | Man-GO 3 |
|---|---|---|---|---|---|
| D (cm−1) | 1347 | 1352 | 1367 | 1350 | 1354 |
| G (cm−1) | 1594 | 1590 | 1590 | 1586 | 1591 |
| I D/IG | 0.93 | 0.91 | 0.88 | 0.92 | 0.94 |
The Raman spectra of all compounds (Fig. 2B) show three main diagnostic bands, marked as D, G, and 2D, which are typical of graphene-based materials.57 Specifically, the high intensity D and G bands result from the presence of defects in the material, and the in-plane vibration of the sp2 hybridized carbon atoms, respectively. The 2D band, of lower intensity, is related to the number of graphene layers. The positions of the D and G band maxima are related to the hybridization of carbon atoms, while the ratio between their intensities (ID/IG) accounts for the degree of order within the material.58 From Table 1, no major variations in the ID/IG ratio emerge between different samples, indicating that both the ball milling process and the conjugation with monosaccharides do not significantly alter the degree of order present in the original GO. In contrast, significant shifts in the positions of the G and D bands with respect to the pristine GO are observed for GObm, Fuc-GO 1, Gal-GO 2, and Man-GO 3, i.e., a 3–8 cm−1 blue shift and a 3–20 cm−1 red shift for the G and D bands, respectively. The shifts of G and D peaks recorded for GObm suggest the possible presence of additional effects, likely related to the modifications of pristine GO occurring during the ball milling process. Indeed, TEM analyses (Fig. 2C) of both ball-milled glyco-GO conjugates 1–3 and GObm provided insights into the effect of milling on the morphology of the GO. As expected, pristine GO is highly dispersible in water, and only few layer GOs have been identified in the TEM images. In particular, GO sheets are characterized by crinkles and folded regions, due to the overlay of multiple sheets, with a lateral size of a few microns. The TEM analysis of the GObm (Fig. 2C) showed that the milling caused fragmentation of the pristine GO sheets reducing the lateral size. However, their dispersibility in water seems to be not affected, as proved by the complete absence of aggregates in the sample. After functionalization with the monosaccharide derivatives the sheets became more contrasted (Fig. 2C, Fuc-GO 1, Gal-GO 2, and Man-GO 3). The increase of contrast might be due to an increase in the surface defects of the material. In addition, the TEM images of ball milled glyco-GO conjugates 1–3 also confirmed the absence of contamination of the samples due to the leaching from the jar surface.
The ball-milled glyco-GO conjugates 1–3 and GObm were investigated also through Magic Angle Spinning (MAS) solid-state NMR (ssNMR) (Fig. 3).
 | ||
| Fig. 3
13C CP MAS spectra of the glyco-GO conjugates 1–3, and GObm. Spectra are acquired at 20.0 T (850 MHz 1H Larmor frequency, 213.8 MHz 13C Larmor frequency) and 20.0 kHz MAS frequency. The glyco-GO conjugates 1–3 show a clear shoulder at around 55–70 ppm (light blue arrow) that corresponds to the resonances of the saccharide units and it is not present in the GObm spectrum, and other saccharide resonances fall at 70–85 ppm but overlap with the GO resonances. Additional resonances of the saccharide linkers are visible from 20 to 45 ppm (orange arrows). Spectra were acquired with 40960 scans and with a CP contact time of 100 μs. Further experimental details are reported in the ESI.† | ||
13C MAS ssNMR spectra allow us to monitor the different functionalities present on the GO surface, monitoring either the effective presence of the monosaccharides on the GO surface or the transformation in GO upon the ball milling process.59–62 In Fig. 3, we report the comparison of the 13C 1D Cross-Polarization MAS spectra (CP-MAS) for glyco-GO conjugates 1–3 and GObm. Ball milled GO (GObm) shows a characteristic band at 65–80 ppm, which is usually attributed to the epoxide (C–O–C) and C–OH groups present on the GO surface.63,64 In Fig. 3 the CP-MAS spectrum is recorded with a very short contact time of 100 μs, in order to selectively detect the resonances of protonated carbon atoms, and in particular of the sugar functionalities. For this reason, the resonances of the epoxide carbons and the aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C quaternary carbons at around 59 and 129 ppm, respectively, are weak and essentially broadened below detection.61,64
C quaternary carbons at around 59 and 129 ppm, respectively, are weak and essentially broadened below detection.61,64
Another band at around 30 ppm can be attributed to aliphatic carbons already present in the material before functionalization, and probably formed during the ball milling procedure; similar resonances are observed in highly oxidized GO.65 We report in Fig. S5 (see the ESI†) the spectrum of Gal-GO 2 acquired with direct 13C excitation (without CP) where all the carbon resonances are observed. Nevertheless, this spectrum does not profit from the enhanced sensitivity of CP and the resonances of the monosaccharide and aliphatic functionalities are barely observed. In CP-MAS the 13C magnetization is increased by transferring polarization from the neighboring protons, not necessarily protons bound to 13C carbons but in close proximity to them (coupled with a dipolar interaction). With the short contact time used in spectra in Fig. 3, the weak intensity of carbon signals can reflect the distance of these aromatic carbon atoms from functionalities bearing protons and only carbons a few Å from protons are essentially observed. The lack of resolution in the carbon resonances reflects the heterogeneous structure of the GO surface, which has no well defined structure, and is typical of these nanomaterials. Upon functionalization, glyco-GO conjugates 1–3 show additional resonances, corresponding to the methylene groups of the short PEG chain and the monosaccharide resonances. These are responsible for the new resonance observed in the range of 20–45 ppm (orange arrows in Fig. 3) and from 55 to 70 ppm (light blue arrow), these correspond to the saccharide-linker and the saccharide unit, respectively. The resonances of the saccharide unit are clearly observed in all glyco-GO conjugates 1–3 and not observed on GObm, and the additional saccharide resonances fall from 70 to 85 ppm but overlap with the C–OH GObm resonances and cannot be distinguished.
To further confirm the presence of monosaccharide and PEG functionalities on the GO surface, we also performed a 1H–13C 2D FSLG (Frequency-Switched Lee–Goldberg) experiment. In this ssNMR correlation experiment the resonances of the 13C spins are correlated with protons present in close proximity, either bound protons or the C–OH proton of the sugar or eventually strongly adsorbed water in contact with the surface. The proton resolution in the indirect dimension is enhanced by the simultaneous use of the Lee–Goldberg sequence (at 100 kHz radio-frequency power) and the high magnetic field (20.0 T, 850 MHz 1H Larmor frequency). To ensure the selectivity in the correlation, the CP contact time in the 1H to 13C magnetization transfer was set to 100 μs. The 2D spectrum of Gal-GO 2 is reported in Fig. S6:† it clearly shows sugar resonances at 60–70 ppm that are partially overlapped with the unresolved GO C–OH resonances. These sugar resonances that are visible as a shoulder in the 1D spectrum in the 2D are more clearly distinguished. Additionally, the resonances of the saccharide linker can be clearly recognized as peaks from 40 to 20 ppm in carbon and around 3–4 ppm in the proton dimension. This spectrum confirms the effective functionalization of the GO surface.
Antimicrobial and antibiofilm activity of glyco-GO conjugates 1–3
The bactericidal effect of glyco-GO conjugates 1–3 was assessed by measuring the minimal bactericidal concentration (MBC, the lowest concentration of drug that kills more than 99% of the bacterial population) values against P. aeruginosa (see the ESI, Table S3†). All the MBC values against planktonic cells were in the μg mL−1 range. In particular, Fuc-GO 1 and Gal-GO 2 conjugates showed a lower MBC (32 μg mL−1) compared to pristine GO (64 μg mL−1). Then, Fuc-GO 1 was selected as a sample conjugate and planktonic cells were treated with a sub-MBC concentration (16 μg mL−1). Scanning electron microscopy (SEM) revealed an evident effect on the cell morphology (Fig. 4A and B). After overnight incubation, the SEM images showed P. aeruginosa deflated cells (Fig. 4B), with an evident alteration of the cell wall. Although the bacteria are not lysed and still alive, cellular suffering appeared evident.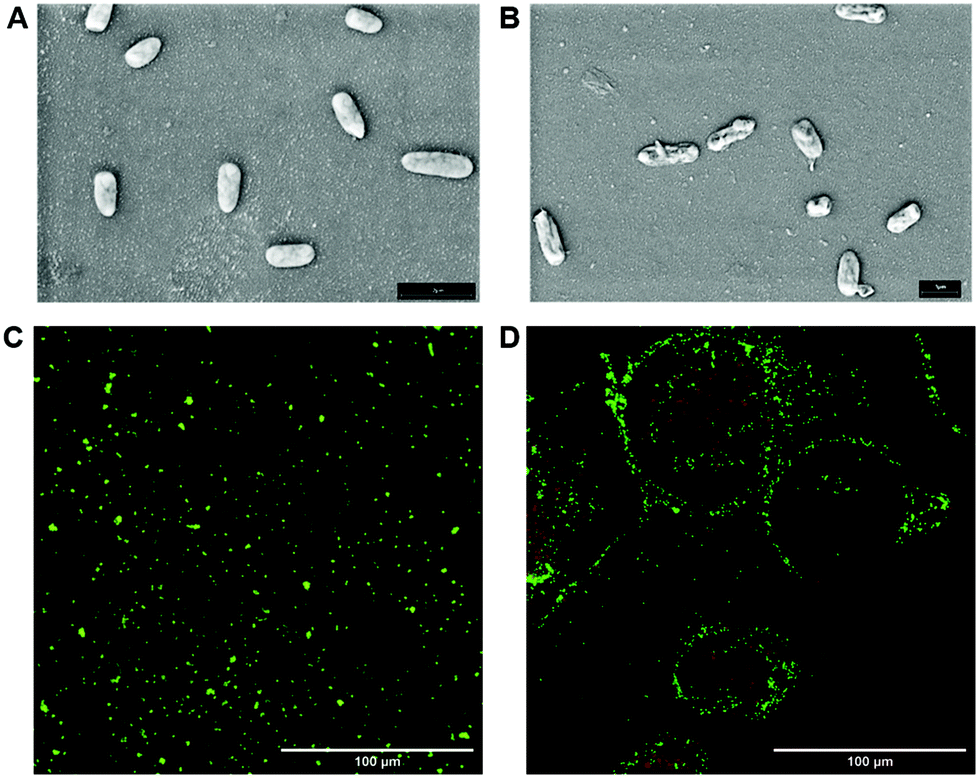 | ||
| Fig. 4 SEM representative images of P. aeruginosa cells: untreated cells (A) and after treatment with Fuc-GO 1 (B). Magnification: 3000×; scale bar 1 μm. Representative fluorescence images of untreated cells (C) and after treatment with Fuc-GO 1 (D) P. aeruginosa cells obtained with a Cytation 5 cell imaging multi-mode reader. Green indicates live cells, and red indicates dead cells. Yellow bar: 100 μm. | ||
To confirm our hypothesis, a live/dead assay was performed using a LIVE/DEAD® BacLight™ bacterial viability kit. This assay discriminates between viable (alive) colored in green and non-viable (dead) colored in red bacterial cells.66 Specifically, two nucleic acid binding fluorescent dyes, SYTO 9 and propidium iodide, stain bacteria with intact cell membranes in green and bacteria with damaged cell walls in red. Fig. 4C and D show that planktonic P. aeruginosa treated with Fuc-GO 1 maintains high viability compared to untreated cells (Fig. 4C), in the presence of a low percentage of dead cells.
Then, the ability of the glyco-GO conjugates 1–3 to disrupt the P. aeruginosa biofilm was evaluated. Specifically, a colony forming unit (CFU) is a parameter that shows the ability of a compound to detach sessile cells from a specific surface. Thus, P. aeruginosa cells were treated with selected glyco-GO conjugates 1–3 and then were allowed to adhere to a polystyrene surface. CFUs per mL of bacterial cells adhered to the polystyrene surface were calculated (Fig. 5).
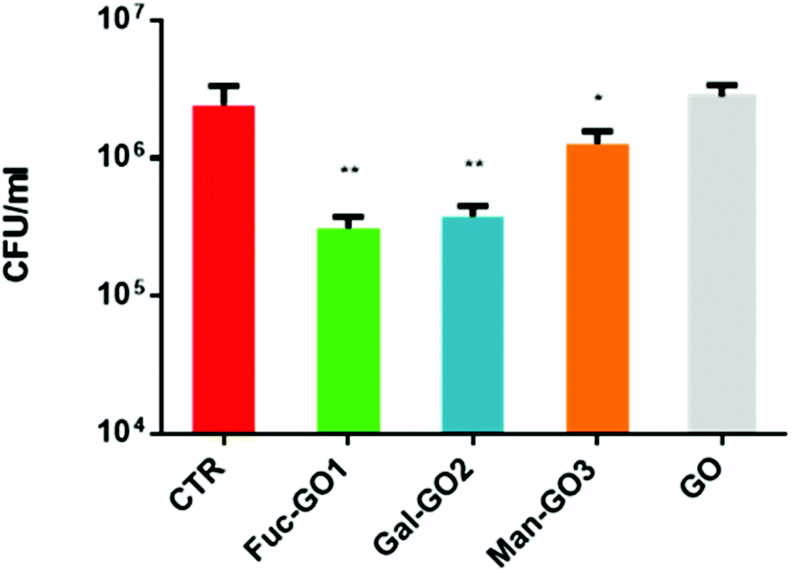 | ||
| Fig. 5 Mean colony-forming unit (CFU) values obtained after treatment of a mature P. aeruginosa biofilm with different glyco-GO conjugates 1–3. The CFU from an untreated biofilm is reported as a control (CTR). ** = 0.002; * = 0.048. | ||
Data obtained showed a significant reduction in CFUs per mL of the P. aeruginosa biofilm treated with all glyco-GO conjugates compared with the untreated biofilm (CRT) and the biofilm treated with pristine GO (Fig. 5). In particular, the fucose bearing conjugate 1, and to a slightly lesser extent the galactose bearing conjugate 2, turned out to be most effective in disrupting cell adhesion. The glyco-GO conjugate 3 showed significantly lower activity according to the known lower-affinity of D-mannose vs. LecB.67,68 Then, the morphological and structural changes on the microbial biofilm were analyzed by SEM. Accordingly, SEM images showed that the treatment with Fuc-GO 1 and Gal-GO 2 heavily affected the P. aeruginosa biofilm ultrastructure (Fig. 6).
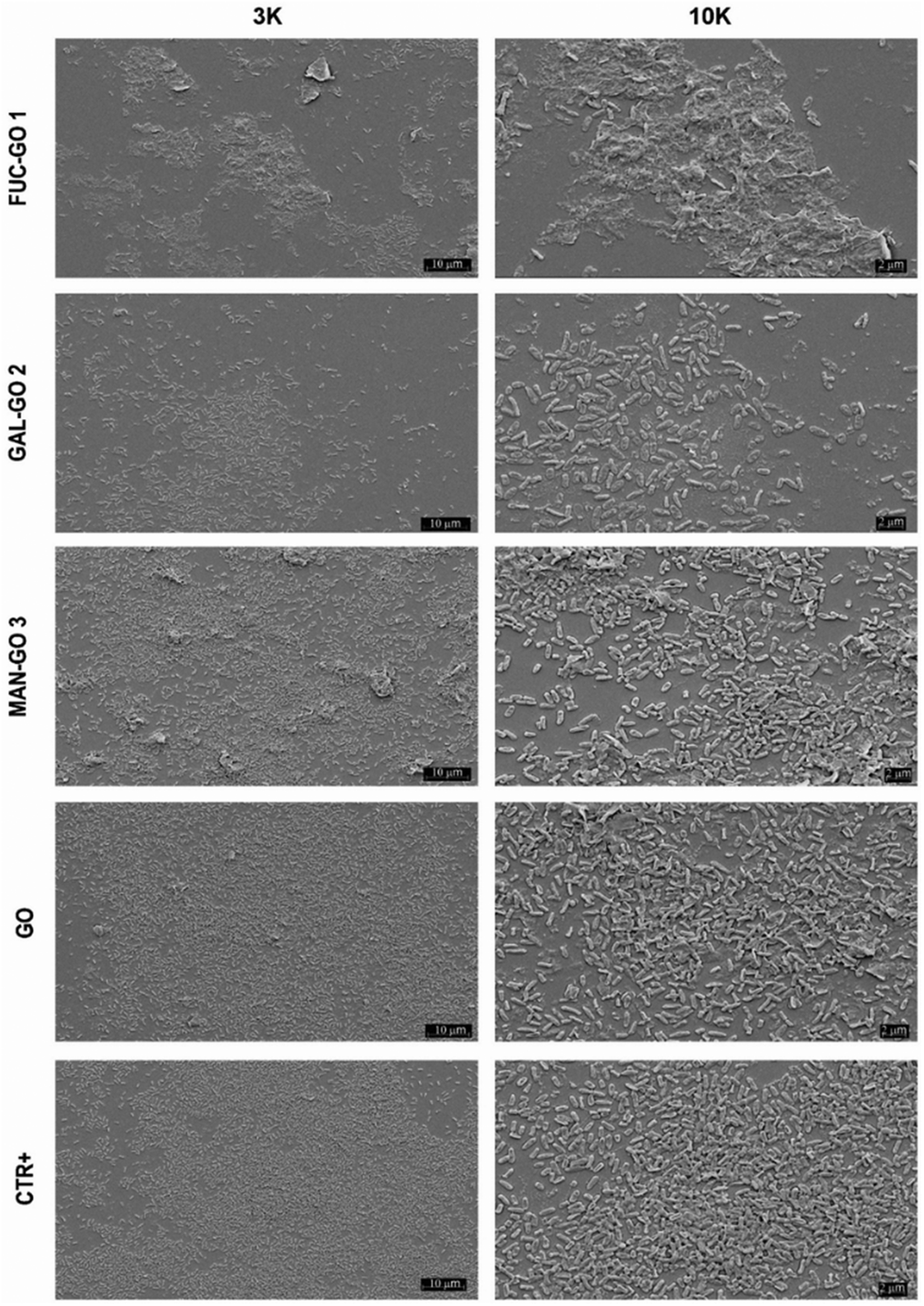 | ||
| Fig. 6 SEM images of the P. aeruginosa biofilm treated (72 h) with different glyco-GO conjugates 1–3 and pristine GO at 256 μg ml−1. Magnification: 3 K and 10 K. | ||
Both the untreated biofilm (CRT) and biofilm treated with pristine GO maintained a cellular density and organization typical of a mature biofilm. Biofilm demolition by glyco-GO conjugates appeared clearly based on the reduction of bacterial cells with a decrease in their three-dimensional organization (Fig. 6).
The specific recognition of carbohydrates by bacterial lectins governs critical host–microbe interactions. Lectins further mediate the colonization of bacteria helping to develop biofilms. Our findings support the hypothesis that the monosaccharide headgroups on the graphene surface are able to target P. aeruginosa lectins by interfering with the lectin–sugar recognition events that regulate biofilm formation.
Experimental
Materials and methods
All reagents, whose synthesis is not described, were commercially available and were used without any further purification, if not specified otherwise. GO powder was purchased from Nanesa. Ball milling was carried out with a Powtech GT 300 mixer mill, using a 10 mL agate mixing mill and agate balls (∅ 1 cm). Filtration was carried out using a glass vacuum filter with a 0.2 μm Nylon membrane (Sartorius). UV-vis spectra were recorded with a Varian Cary 4000 UV-vis spectrophotometer using a 1 cm cell. Potassium bromide (KBr) pellets were prepared using 100 mg of potassium bromide and 1.0 mg of sample. The spectra were recorded under an inert atmosphere and a pure potassium bromide pellet was used as the background. TEM micrographs were acquired using a JEOL 100 SX, operating at 100 kV. Samples were prepared by drop drying a dilute suspension of GO in water (after 10 minutes of sonication) onto 200 mesh carbon-coated copper grids. The NMR spectra of glycoside derivatives were recorded using an Inova 400 instrument.Bacterial strain, media and culture conditions
P. aeruginosa clinical strain was obtained from positive blood culture of patients hospitalized at the Fondazione Policlinico Universitario A. Gemelli IRCCS in Rome. The isolate was rapidly identified by matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS) and tested for purity on MacConkey agar plates. For in vitro biofilm formation, P. aeruginosa was cultured at a density of 108 colony-forming units (CFUs) per ml in Brain Heart Infusion (BHI) broth with 0.25% glucose for 4 days in a 24 well plate with 13 mm diameter glass coverslips.MBC evaluation
Minimal bactericidal concentrations (MBCs) were determined by broth microdilution susceptibility tests, according to the European Committee on Antimicrobial Susceptibility Testing (EUCAST) international guidelines. To evaluate MBCs, 5 μL of the content of each well were seeded on MacConkey medium agar plates and incubated for 24 h at 37 °C. The MBC was defined as the lowest concentration determining the death of 99.9% or more of the initial inoculum.Biofilm demolition assay
Once the biofilms were formed, after 4 days of incubation, the culture medium was removed, the wells were washed three times with PBS and the samples were treated with the glyco-GO conjugates in a final volume of 200 μl of ultrapure water for 24 h at 37 °C. The demolition activity was measured with a CFU valuation assay and observed by scanning electron microscopy (SEM).SEM evaluation
P. aeruginosa biofilms grown on a 13 mm glass disk and treated with different glyco-GO derivatives 1–3 were investigated by SEM. Biofilms were fixed with 2.5% glutaraldehyde and dried using increasing concentrations (from 10 to 100%) of ethanol. The samples were mounted onto an aluminum stab using double-sided carbon tape and coated with a gold/palladium film (80:20) by using a high-resolution sputter coater (Agar Scientific B7234). A microbial suspension of P. aeruginosa equal to 5 × 107 cfu ml−1 was incubated overnight at 37° C with a sub-MBC concentration of Fuc-GO 1 corresponding to 16 μg ml−1. The bacteria were then centrifuged, washed 3 times with PBS and deposited on a polyurethane coverslip (Nunc, Thermofisher, USA). For SEM evaluation, the samples were dehydrated and metallized as described above. For the live and dead assay, cells were stained using a BacLight™ bacterial viability kit (Themo Fisher, USA) according to the manufacturer's instructions. Images were then observed using a Cytation 5 cell imaging multi-mode reader (Biotek, Agilent Technologies USA), with excitation at 480 nm and emission at 525 nm for green channel detection and at 586 nm and 525nm for red channel detection, respectively. Untreated control bacteria were processed in parallel.
Conclusions
The widespread occurrence of P. aeruginosa biofilms has a significant clinical and economic impact.In this regard, the identification of P. aeruginosa biofilm dispersal agents able to disrupt an already established matrix provides an attractive antibacterial strategy against a Gram negative bacterium, which can be resistant to multiple antibiotic agents. In this context, we report the preparation and comprehensive characterization of glyco-GO nanomaterials which proved capable of disrupting already established P. aeruginosa biofilms. Notably, the effect observed with the glyco-GO conjugates 1 and 2 was much stronger than the partial effect observed with pristine GO, supporting the key role of sugar grafting on the GO surface in the antibiofilm profile of the graphene-based nanomaterials. The control of surface functionalization of the graphene-based materials is essential for the design of innovative custom nanomaterials able to interfere with biological processes. In this regard, we demonstrated that ball milling is a valuable approach to preparing functionalized GO conjugates and we provided relevant insights into the chemistry involved in the milling process by exploiting complementary techniques. Therefore, the outcome of this work can pave the way for the controlled functionalization of the GO surface by using a green and reliable process.
Author contributions
B.R., F.B. and S.C. are responsible for conceptualization, funding, acquisition, resources and writing – original draft & review & editing. J.T., M.C., G.B., L.C., L.N., R.T., A.G., and E.M. are responsible for data curation, and investigation. R.T., M.V., F.P., M.S., M.S., E.M., M.L. and D.B. are responsible for formal analysis, methodology, resources and writing – original draft. J.T., M.C., L.C., R.T., and M.V. are responsible for validation and visualization. B.R., F.B. and S.C. are responsible for supervision and B.R. is responsible for project administration. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. R., S. C., G. B., J. T., M. L. and L. N. thank MIUR-Italy (“Progetto Dipartimenti di Eccellenza 2018–2022” allocated to Department of Chemistry “Ugo Schiff”). B. R. and G. B. thank Fondazione Umberto Veronesi for supporting the post-doctoral fellowship of G. B. Funding from the University of Padova (P-DiSC#05BIRD2021-UNIPD) is acknowledged by E. M. M. L. thanks Fondazione CR Firenze for funding. This work benefited from access to CERM/CIRMMP, an Instruct-ERIC centre, and specifically to Instruct-ITALIA (ITA008). M. C. thanks Ministero della Salute, Ricerca finalizzata, Starting Grant, Project Code: SG-2018-12366369.References
- R. Patel, Clin. Orthop. Relat. Res., 2005, 437, 41–47 CrossRef PubMed.
- H.-C. Flemming, J. Wingender, U. Szewzyk, P. Steinberg, S. A. Rice and S. Kjelleberg, Nat. Rev. Microbiol., 2016, 14, 563–575 CrossRef CAS PubMed.
- O. Ciofu, C. Moser, P. Ø. Jensen and N. Høiby, Nat. Rev. Microbiol., 2022 DOI:10.1038/s41579-022-00682-4.
- S. Helaine, A. M. Cheverton, K. G. Watson, L. M. Faure, S. A. Matthews and D. W. Holden, Science, 2014, 343, 204–208 CrossRef CAS PubMed.
- R. A. Bamford, A. Smith, J. Metz, G. Glover, R. W. Titball and S. Pagliara, BMC Biol., 2017, 15, 121 CrossRef PubMed.
- L. R. Mulcahy, J. L. Burns, S. Lory and K. Lewis, J. Bacteriol., 2010, 192, 6191–6199 CrossRef CAS PubMed.
- Y. Zhang, X. Pan, Y. Tian, H. Liu, X. Chen, B. Ge, Z. Wang, X. Tang, S. Lei, W. Yao, Y. Ren, Y. Tian, J. Li, P. Fu, J. Xin, Y. Sun, J. Cao and Z. Wang, Adv. Atmos. Sci., 2022, 1–15 Search PubMed.
- O. Goode, A. Smith, A. Zarkan, J. Cama, B. M. Invergo, D. Belgami, S. Caño-Muñiz, J. Metz, P. O'Neill, A. Jeffries, I. H. Norville, J. David, D. Summers and S. Pagliara, mBio, 2021, 12, e00909–e00921 CrossRef CAS PubMed.
- N. Venkatesan, G. Perumal and M. Doble, Future Microbiol., 2015, 10, 1743–1750 CrossRef CAS PubMed.
- J. Botelho, F. Grosso and L. Peixe, Drug Resistance Updates, 2019, 44, 100640 CrossRef PubMed.
- J. J. Harrison, H. Almblad, Y. Irie, D. J. Wolter, H. C. Eggleston, T. E. Randall, J. O. Kitzman, B. Stackhouse, J. C. Emerson, S. Mcnamara, T. J. Larsen, J. Shendure, L. R. Hoffman, D. J. Wozniak and M. R. Parsek, PLoS Genet., 2020, 16, e1008848 CrossRef CAS PubMed.
- M. Rybtke, P. Ø. Jensen, C. H. Nielsen and T. Tolker-Nielsen, Infect. Immun., 2020, 89, e00631–e00620 CrossRef PubMed.
- O. Ciofu and T. Tolker-Nielsen, Front. Microbiol., 2019, 10, 913 CrossRef PubMed.
- G. Zaborskyte, J. B. Andersen, K. N. Kragh and O. Ciofu, Antimicrob. Agents Chemother., 2017, 61, e02292–e02216 CrossRef CAS PubMed.
- N. Gilboa-Garber, Methods Enzymol., 1982, 83, 378–385 CAS.
- D. Tielker, S. Hacker, R. Loris, M. Strathmann, J. Wingender, S. Wilhelm, F. Rosenau and K.-E. Jaeger, Microbiology, 2005, 151, 1313–1323 CrossRef CAS PubMed.
- S. P. Diggle, R. E. Stacey, C. Dodd, M. Cámara, P. Williams and K. Winzer, Environ. Microbiol., 2006, 8, 1095–1104 CrossRef CAS PubMed.
- D. Passos da Silva, M. L. Matwichuk, D. O. Townsend, C. Reichhardt, D. Lamba, D. J. Wozniak and M. R. Parsek, Nat. Commun., 2019, 10, 2183 CrossRef PubMed.
- J. Meiers, E. Siebs, E. Zahorska and A. Titz, Curr. Opin. Chem. Biol., 2019, 53, 51–67 CrossRef CAS PubMed.
- M. B. Calvert, V. R. Jumde and A. Titz, Beilstein J. Org. Chem., 2018, 14, 2607–2617 CrossRef CAS PubMed.
- S. Wagner, R. Sommer, S. Hinsberger, C. Lu, R. W. Hartmann, M. Empting and A. Titz, J. Med. Chem., 2016, 59, 5929–5969 CrossRef CAS PubMed.
- G. Michaud, R. Visini, M. Bergmann, G. Salerno, R. Bosco, E. Gillon, B. Richichi, C. Nativi, A. Imberty, A. Stocker, T. Darbre and J.-L. Reymond, Chem. Sci., 2016, 7, 166–182 RSC.
- A. Bernardi, J. Jiménez-Barbero, A. Casnati, C. De Castro, T. Darbre, F. Fieschi, J. Finne, H. Funken, K.-E. Jaeger, M. Lahmann, T. K. Lindhorst, M. Marradi, P. Messner, A. Molinaro, P. V. Murphy, C. Nativi, S. Oscarson, S. Penadés, F. Peri, R. J. Pieters, O. Renaudet, J.-L. Reymond, B. Richichi, J. Rojo, F. Sansone, C. Schäffer, W. B. Turnbull, T. Velasco-Torrijos, S. Vidal, S. Vincent, T. Wennekes, H. Zuilhof and A. Imberty, Chem. Soc. Rev., 2013, 42, 4709–4727 RSC.
- E. Siebs, E. Shanina, S. Kuhaudomlarp, P. Silva Figueiredo Celestino Gomes, C. Fortin, P. H. Seeberger, D. Rognan, C. Rademacher, A. Imberty and A. Titz, ChemBioChem, 2022, 23, e2021005 CrossRef PubMed.
- P. Arosio, G. Comito, F. Orsini, A. Lascialfari, P. Chiarugi, C. Ménard-Moyon, C. Nativi and B. Richichi, Org. Biomol. Chem., 2018, 16, 6086–6095 RSC.
- M. A. Dos Santos Ramos, P. Da Silva, L. Spósito, L. De Toledo, B. Bonifácio, C. F. Rodero, K. Dos Santos, M. Chorilli and T. M. Bauab, Int. J. Nanomed., 2018, 13, 1179–1213 CrossRef CAS PubMed.
- Y. Chen, A. Star and S. Vidal, Chem. Soc. Rev., 2013, 42, 4532 RSC.
- X. Huang, Z. Yin, S. Wu, X. Qi, Q. He, Q. Zhang, Q. Yan, F. Boey and H. Zhang, Small, 2011, 7, 1876–1902 CrossRef CAS PubMed.
- J. Liu, G. Chen and M. Jiang, Macromolecules, 2011, 44, 7682–7691 CrossRef CAS.
- B. K. Chegeni, A. D. Tehrani and M. Adeli, J. Taiwan Inst. Chem. Eng., 2019, 96, 176–184 CrossRef.
- Q. Yang, X. Pan, K. Clarke and K. Li, Ind. Eng. Chem. Res., 2012, 51, 310–317 CrossRef CAS.
- N. Losada-Garcia, I. Rodriguez-Oliva, M. Simovic, D. I. Bezbradica and J. M. Palomo, ACS Omega, 2020, 5, 4362–4369 CrossRef CAS PubMed.
- Z. P. Michael, A. Star and S. Vidal, in Carbohydrate Nanotechnology, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2015, pp. 123–135 Search PubMed.
- Q. Chen, W. Wei and J. M. Lin, Biosens. Bioelectron., 2011, 26, 4497–4502 CrossRef CAS PubMed.
- Y. Chen, H. Vedala, G. P. Kotchey, A. Audfray, S. Cecioni, A. Imberty, S. Vidal and A. Star, ACS Nano, 2012, 6, 760–770 CrossRef CAS PubMed.
- Z. Li, S.-S. Deng, Y. Zang, Z. Gu, X.-P. He, G.-R. Chen, K. Chen, T. D. James, J. Li and Y.-T. Long, Sci. Rep., 2013, 3, 2293 CrossRef PubMed.
- Z. Qi, P. Bharate, C. H. Lai, B. Ziem, C. Böttcher, A. Schulz, F. Beckert, B. Hatting, R. Mülhaupt, P. H. Seeberger and R. Haag, Nano Lett., 2015, 15, 6051–6057 CrossRef CAS PubMed.
- N. Gupta, A. K. Jangid, M. Singh, D. Pooja and H. Kulhari, ChemMedChem, 2018, 13, 2644–2652 CrossRef CAS PubMed.
- W. Liang, Y. Huang, D. Lu, X. Ma, T. Gong, X. Cui, B. Yu, C. Yang, C. Dong and S. Shuang, Polymers, 2019, 11, 133 CrossRef PubMed.
- S. Kang, Y. L. Hong, B. C. Ku, S. Lee, S. Ryu, D. H. Min, H. Jang and Y. K. Kim, Nanotechnology, 2018, 29, 475604 CrossRef PubMed.
- T. Yin, J. Liu, Z. Zhao, Y. Zhao, L. Dong, M. Yang, J. Zhou and M. Huo, Adv. Funct. Mater., 2017, 27, 1604620 CrossRef.
- R. Lima-Sousa, D. de Melo-Diogo, C. G. Alves, E. C. Costa, P. Ferreira, R. O. Louro and I. J. Correia, Carbohydr. Polym., 2018, 200, 93–99 CrossRef CAS PubMed.
- R. Kurapati, F. Bonachera, J. Russier, A. R. Sureshbabu, C. Ménard-Moyon, K. Kostarelos and A. Bianco, 2D Mater., 2017, 5, 015020 CrossRef.
- X. Zou, L. Zhang, Z. Wang and Y. Luo, J. Am. Chem. Soc., 2016, 138, 2064–2077 CrossRef CAS PubMed.
- H. Mohammed, A. Kumar, E. Bekyarova, Y. Al-Hadeethi, X. Zhang, M. Chen, M. S. Ansari, A. Cochis and L. Rimondini, Front. Bioeng. Biotechnol., 2020, 8, 465 CrossRef PubMed.
- V. Palmieri, F. Bugli, M. C. Lauriola, M. Cacaci, R. Torelli, G. Ciasca, C. Conti, M. Sanguinetti, M. Papi and M. De Spirito, ACS Biomater. Sci. Eng., 2017, 3, 619–627 CrossRef CAS PubMed.
- C. Martini, F. Longo, R. Castagnola, L. Marigo, N. M. Grande, M. Cordaro, M. Cacaci, M. Papi, V. Palmieri, F. Bugli and M. Sanguinetti, Antibiotics, 2020, 9, 692 CrossRef CAS PubMed.
- V. Palmieri, F. Bugli, M. Cacaci, G. Perini, F. De Maio, G. Delogu, R. Torelli, C. Conti, M. Sanguinetti, M. De Spirito, R. Zanoni and M. Papi, Nanomedicine, 2018, 13, 2867–2879 CrossRef CAS PubMed.
- D. Tan and F. García, Chem. Soc. Rev., 2019, 48, 2274 RSC.
- P. Andreozzi, L. Tamberi, E. Tasca, G. E. Giacomazzo, M. Martinez, M. Severi, M. Marradi, S. Cicchi, S. Moya, G. Biagiotti and B. Richichi, Beilstein J. Org. Chem., 2020, 16, 2272–2281 CrossRef CAS PubMed.
- E. Colacino, G. Ennas, I. Halasz, A. Porcheddu and A. Scano, Mechanochemistry, De Gruyter, Berlin, Boston, 2020 Search PubMed.
- G. Biagiotti, A. Salvatore, G. Toniolo, L. Caselli, M. Di Vito, M. Cacaci, L. Contiero, T. Gori, M. Maggini, M. Sanguinetti, D. Berti, F. Bugli, B. Richichi and S. Cicchi, ACS Appl. Mater. Interfaces, 2021, 13, 26288–26298 CrossRef CAS PubMed.
- N. Kong, M. R. Shimpi, J. H. Park, O. Ramström and M. Yan, Carbohydr. Res., 2015, 405, 33–38 CrossRef CAS PubMed.
- A. K. Sanki and L. K. Mahal, Synlett, 2006, 455–459 CAS.
- M. M. Velásquez-Rojas, F. F. Contreras-Torres, V. Meza-Laguna, E. Álvarez-Zauco, M. H. Farías, V. A. Basiuk and E. V. Basiuk, Mater. Chem. Phys., 2021, 260, 124127 CrossRef.
- I. Y. Jeon, S. Y. Bae, J. M. Seo and J. B. Baek, Adv. Funct. Mater., 2015, 25, 6961–6975 CrossRef CAS.
- D. López-Díaz, M. López Holgado, J. L. García-Fierro and M. M. Velázquez, J. Phys. Chem. C, 2017, 121, 20489–20497 CrossRef.
- R. Muzyka, S. Drewniak, T. Pustelny, M. Sajdak and Ł. Drewniak, Materials, 2021, 14, 769 CrossRef CAS PubMed.
- M. A. Vieira, G. R. Gonçalves, D. F. Cipriano, M. A. Schettino, E. A. Silva Filho, A. G. Cunha, F. G. Emmerich and J. C. C. Freitas, Carbon, 2016, 98, 496–503 CrossRef CAS.
- W. Cai, R. D. Piner, F. J. Stadermann, S. Park, M. A. Shaibat, Y. Ishii, D. Yang, A. Velamakanni, S. J. An, M. Stoller, J. An, D. Chen and R. S. Ruoff, Science, 2008, 321, 1815–1817 CrossRef CAS PubMed.
- W. Gao, L. B. Alemany, L. Ci and P. M. Ajayan, Nat. Chem., 2009, 1, 403–408 CrossRef CAS PubMed.
- S. Caputo, A. Kovtun, F. Bruno, E. Ravera, C. Lambruschini, M. Melucci and L. Moni, RSC Adv., 2022, 12, 15834–15847 RSC.
- I. A. Vacchi, C. Spinato, J. Raya, A. Bianco and C. Ménard-Moyon, Nanoscale, 2016, 8, 13714–13721 RSC.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- A. Rawal, S. H. Che Man, V. Agarwal, Y. Yao, S. C. Thickett and P. B. Zetterlund, ACS Appl. Mater. Interfaces, 2021, 13, 18255–18263 CrossRef CAS PubMed.
- O. Goode, A. Smith, U. Łapińska, R. Bamford, Z. Kahveci, G. Glover, E. Attrill, A. Carr, J. Metz and S. Pagliara, ACS Infect. Dis., 2021, 7, 1848–1858 CrossRef CAS PubMed.
- R. Sommer, D. Hauck, A. Varrot, S. Wagner, A. Audfray, A. Prestel, H. M. Möller, A. Imberty and A. Titz, ChemistryOpen, 2015, 4, 756–767 CrossRef CAS PubMed.
- R. Loris, D. Tielker, K. E. Jaeger and L. Wyns, J. Mol. Biol., 2003, 331, 861–870 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Protocols for the preparation of compounds 4–6, 12, 13, 15, 16, 18, and 19; methods for TGA, ICP-AES, and IR, UV-vis, Raman and NMR spectroscopy of glyco-GO conjugates 1–3. See DOI: https://doi.org/10.1039/d2nr02027k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |