
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of amphiphilic fluorinated peptides: evaluation of self-assembly properties and hydrogel formation†
Suvrat
Chowdhary
 a,
Robert Franz
Schmidt
b,
Anil Kumar
Sahoo
cd,
Tiemo
tom Dieck
a,
Thomas
Hohmann
a,
Boris
Schade
e,
Kerstin
Brademann-Jock
f,
Andreas F.
Thünemann
f,
Roland R.
Netz
c,
Michael
Gradzielski
b and
Beate
Koksch
*a
a,
Robert Franz
Schmidt
b,
Anil Kumar
Sahoo
cd,
Tiemo
tom Dieck
a,
Thomas
Hohmann
a,
Boris
Schade
e,
Kerstin
Brademann-Jock
f,
Andreas F.
Thünemann
f,
Roland R.
Netz
c,
Michael
Gradzielski
b and
Beate
Koksch
*a
aInstitute of Chemistry and Biochemistry, Freie Universität Berlin, Arnimallee 20, 14195 Berlin, Germany. E-mail: beate.koksch@fu-berlin.de
bInstitute of Chemistry, Technische Universität Berlin, Straße des 17. Juni 124, 10623 Berlin, Germany
cDepartment of Physics, Freie Universität Berlin, Arnimallee 14, 14195 Berlin, Germany
dMax Planck Institute of Colloids and Interfaces, Am Mühlenberg 1, 14476 Potsdam, Germany
eInstitute of Chemistry and Biochemistry and Core Facility BioSupraMol, Freie Universität Berlin, Fabeckstraße 36a, 14195 Berlin, Germany
fFederal Institute for Materials Research and Testing (BAM), Unter den Eichen 87, 12205 Berlin, Germany
First published on 22nd June 2022
Abstract
Advanced peptide-based nanomaterials composed of self-assembling peptides (SAPs) are of emerging interest in pharmaceutical and biomedical applications. The introduction of fluorine into peptides, in fact, offers unique opportunities to tune their biophysical properties and intermolecular interactions. In particular, the degree of fluorination plays a crucial role in peptide engineering as it can be used to control the characteristics of fluorine-specific interactions and, thus, peptide conformation and self-assembly. Here, we designed and explored a series of amphipathic peptides by incorporating the fluorinated amino acids (2S)-4-monofluoroethylglycine (MfeGly), (2S)-4,4-difluoroethylglycine (DfeGly) and (2S)-4,4,4-trifluoroethylglycine (TfeGly) as hydrophobic components. This approach enabled studying the impact of fluorination on secondary structure formation and peptide self-assembly on a systematic basis. We show that the interplay between polarity and hydrophobicity, both induced differentially by varying degrees of side chain fluorination, does affect peptide folding significantly. A greater degree of fluorination promotes peptide fibrillation and subsequent formation of physical hydrogels in physiological conditions. Molecular simulations revealed the key role played by electrostatically driven intra-chain and inter-chain contact pairs that are modulated by side chain fluorination and give insights into the different self-organization behaviour of selected peptides. Our study provides a systematic report about the distinct features of fluorinated oligomeric peptides with potential applications as peptide-based biomaterials.
Introduction
Self-assembling peptides (SAPs) are often composed of an amphiphilic structure motif based on alternating arrangements of hydrophobic and hydrophilic residues.1–4 More than two decades ago the first and most prominent variants EAK16-II H2N-(Ala–Glu–Ala–Glu–Ala–Lys–Ala–Lys)2-OH and RADA16-II H2N-(Arg–Ala–Arg–Ala–Asp–Ala–Asp–Ala)2-OH were discovered by Zhang et al.5 These peptides served as essential motifs to study the hierarchical construction of β-sheet-based macroassemblies.2,5–8 Since then, SAPs have attracted paramount interest in biomedical research for their biocompatibility, biodegradability, and biofunctionality. They were utilized in the field of tissue engineering by functioning as extracellular matrix mimics for cell proliferation and wound healing.9–14 Their self-assembly is driven by non-covalent interactions such as hydrogen bonding, electrostatic interactions, hydrophobic interactions, aromatic interactions (π–π stacking) and van der Waals forces.15–18 A promising approach to produce novel functional peptide-based biomaterials consists of the systematic incorporation of fluorinated amino acids. These non-natural building blocks turned out to be a powerful tool to fine-tune biophysical and chemical properties of peptides and proteins.19 Fluorine possesses unique properties including a strong inductive effect combined with high electronegativity. The replacement of a single C–H bond with C–F is generally considered to be isosteric.20,21 Investigations of the effects of fluorinated amino acids on hydrophobicity,22,23 secondary structure formation,24,25 protein–protein interactions,26,27 amyloid folding kinetics,28,29 proteolytic stability,30 the chemical and biological properties of fluorinated peptide-based materials,31 and the integration of fluorine into bacteria32,33 have been reported by our group. The vast majority of previous studies including our own efforts examining fluorinated amino acids in the context of peptide and protein chemistry were limited to the incorporation of one or only a few of these building blocks. Moreover, the chemical nature and biological features of polyfluorinated peptides with a large proportion of fluorinated aliphatic amino acids have not yet been studied. Thus, we were motivated to address the question of the impact that several of such building blocks would have on fluorine-specific interactions in peptide self-assembly.The fabrication of polyfluorinated peptides obviously requires generous amounts of fluorinated amino acids.34 Thus, we have recently reported an improved synthetic strategy to obtain the fluorinated amino acid (2S)-4-monofluoroethylglycine (MfeGly) at the gram scale.35 Moreover, Soloshonok et al. developed a general and practical synthetic process to obtain enantiomerically pure Fmoc-protected fluorinated amino acid through an asymmetric and Ni(II)-complex mediated stereoselective synthesis (see Scheme S1 in the ESI†).36–39 In current attempts, we have extended this strategy to the synthesis of a diverse range of aliphatic fluorinated amino acids with different side chain patterns.40
In this work, we designed an amphipathic motif including lysine and the well-studied non-proteogenic amino acid α-aminobutyric acid (Abu), which has been reported as a suitable hydrophobic building block for SAPs.41,42 The varying degree of fluorination was adjusted by the iterative incorporation of its derivatives (2S)-4-monofluoroethylglycine (MfeGly), (2S)-4,4-difluoroethylglycine (DfeGly) and (2S)-4,4,4-trifluoroethylglycine (TfeGly). With this peptide library we assessed the impact of fluorine-specific interactions on the intrinsic hydrophobicity, secondary structure formation, self-assembling properties, the morphology of amyloid-like aggregates and the formation of peptide hydrogels. Molecular simulations of the different fluorinated peptides demonstrate that the peptide–peptide interactions are finely tuned by the ability of fluorine atoms to form electrostatic contact pairs with positively partially charged atoms on the backbone and side chains. This ability in turn depends critically on the number of fluorinated substituents, based on which we explain how the fluorination degree controls peptide structure formation.
Results and discussion
Peptide design: estimation of sequence length, hydrophobicity, and fluorine-induced polarity
Our rational design is based on a cationic Abu–Lys repeating unit. A π-system derived from 4-aminobenzoic acid ([4]Abz, PABA) was placed on the C-terminus; this building block is a widely used fluorescent probe and enables precise control over peptide stock concentrations.43 To determine a chain length sufficient for β-sheet formation, we characterized a series of AbuK-derived peptides with repeating units ranging from five to eight alternating residues and studied their ability to form secondary structures under physiological conditions via CD spectroscopy (Fig. 1a). High peptide concentrations (2 wt%) were chosen to induce peptide self-assembly. As can be seen from the CD spectra, only AbuK14 (Ac-(Abu–Lys)7-[4]Abz-NH2) and AbuK16 (Ac-(Abu–Lys)8-[4]Abz-NH2) formed β-sheet structures (λmin = 214–220 nm), whereas the remaining variants (AbuK10–13, AbuK15) tend to form polyproline type II helices (PPII). This is proven by the characteristic positive and negative maxima at λmax = 218–228 nm and λmin = 198–205 nm. The PPII helix comprises an extended left-handed helical structure and was also found for similar Ala–Lys derived SAPs.44,45 A further minimum at λmin = 228–230 nm could hint for a minor population of β-turn like conformations through intramolecular hydrophobic interactions between the Abu residues.46,47 As the 16-meric AbuK16 was shown to form β-sheets, we synthesized the polyfluorinated amphipathic peptides MfeGlyK16 (Ac-(MfeGly-Lys)8-[4]Abz-NH2), DfeGlyK16 (Ac-(DfeGly-Lys)8-[4]Abz-NH2) and TfeGlyK16 (Ac-(TfeGly-Lys)8-[4]Abz-NH2) by substitution of Abu with each fluorinated derivative according to the eight repeating unit pattern (Fig. 1b).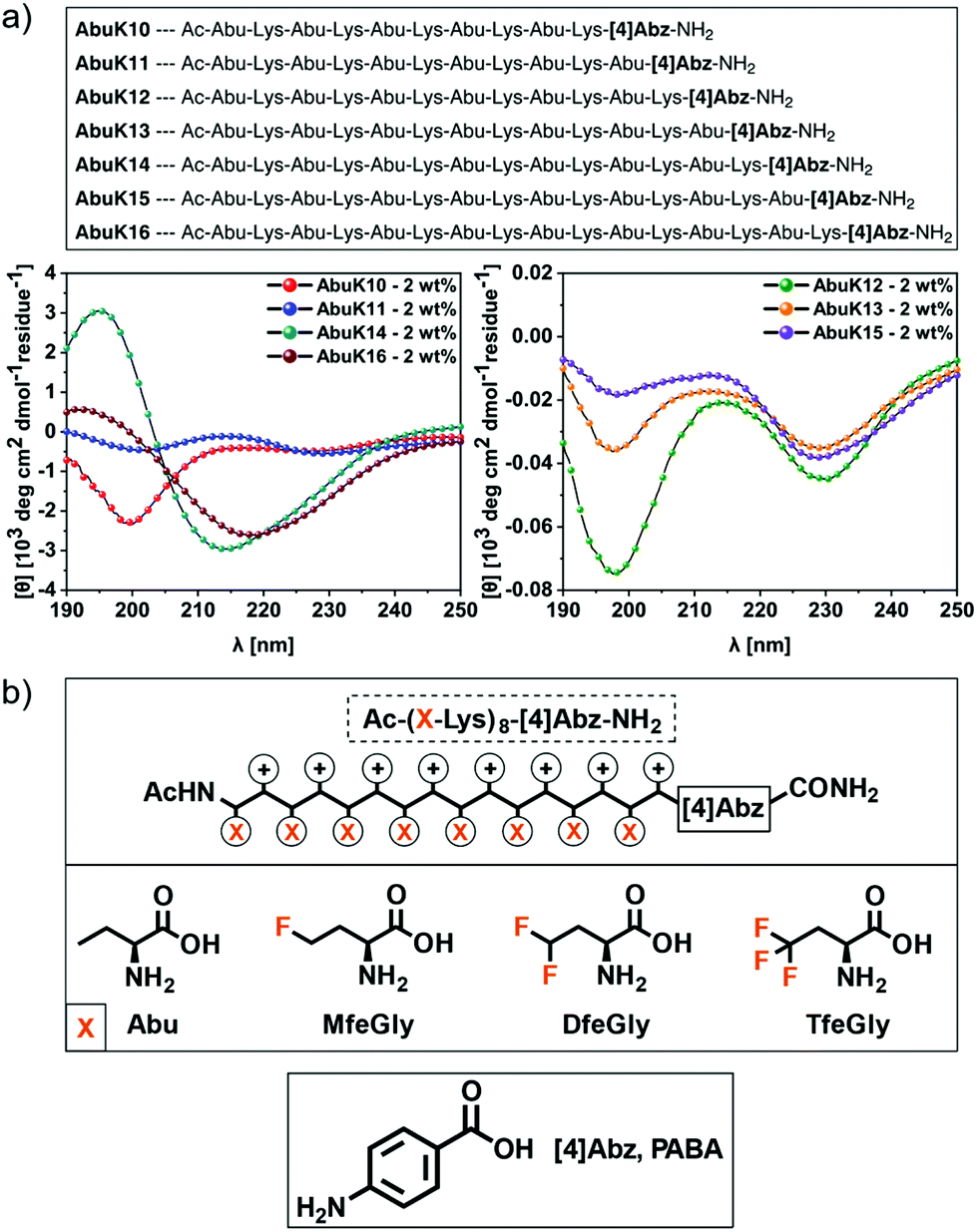 | ||
| Fig. 1 (a) CD spectra of 2 wt% AbuK10–AbuK16 in 50 mM Bis-tris propane + 150 mM NaCl, pH 7.4 recorded at 37 °C. (b) Peptide motif (AbuK16, MfeGlyK16, DfeGlyK16, TfeGlyK16) and chemical structures of Abu and its derivatives MfeGly, DfeGly and TfeGly as well was [4]Abz. | ||
Rational design applying fluorinated amino acids in peptide scaffolds crucially depends on a reliable determination of their hydrophobic nature. Estimation of the intrinsic hydrophobicity is indispensable to discuss fluorine-specific interactions for each oligopeptide. In this work, the hydrophobic properties of peptides were determined through a RP-HPLC based assay (Fig. 2a). In general, the hydrophobicity of the peptides increases with the number of fluorine substituents on the individual amino acids. However, MfeGlyK16 is more polar than AbuK14 and AbuK16, which is in accordance with prior observed trends in fluorine-induced hydrophobicity with this particular amino acid series.28,48 We thus conclude that the origin of the decrease in non-polar character of MfeGlyK16 lies rather in the physicochemical properties of its side chain than in its overall fluorine content.
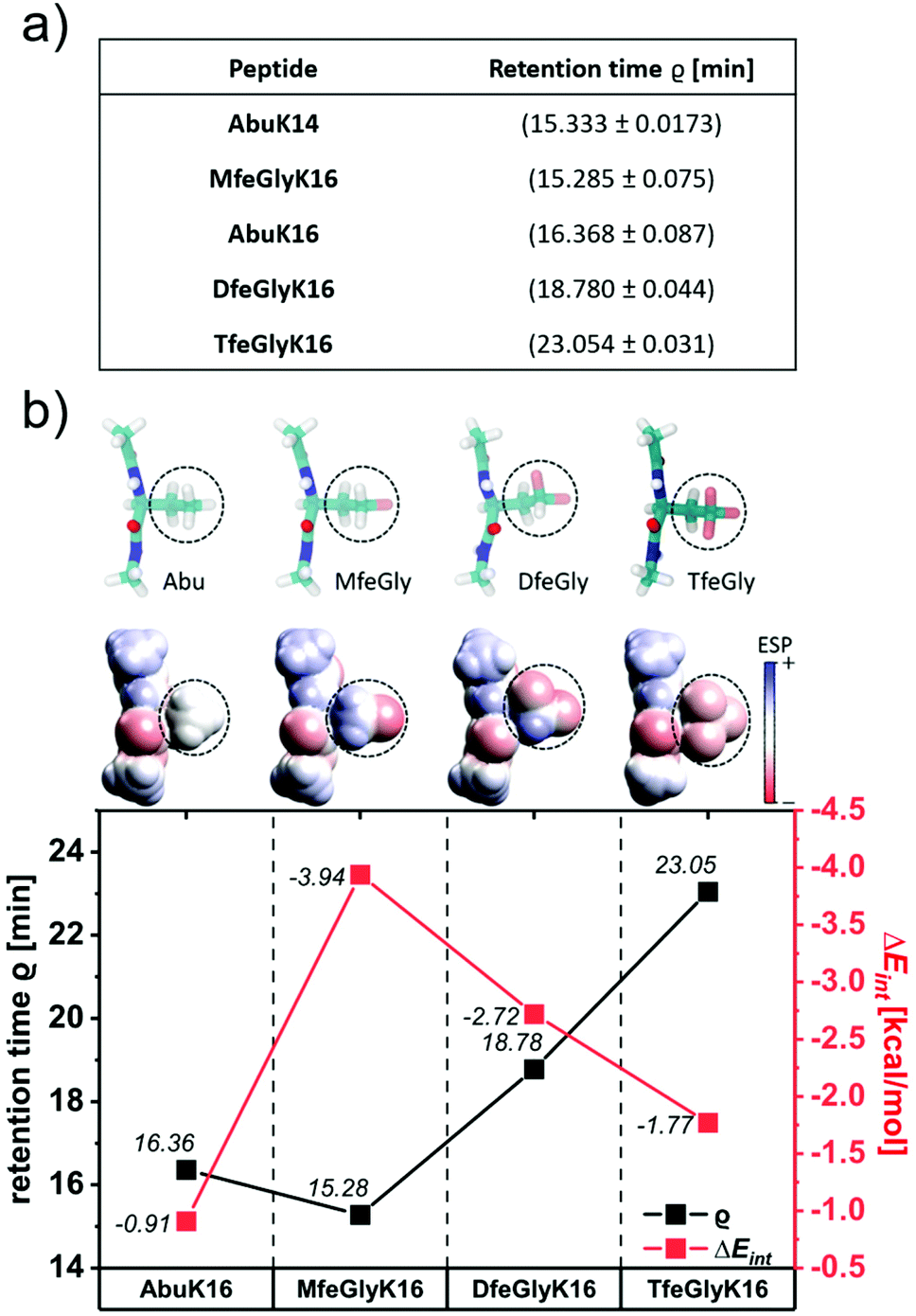 | ||
| Fig. 2 (a) Retention times ϱ of AbuK14, AbuK16, MfeGlyK16, DfeGlyK16 and TfeGlyK16 as experimental index of intrinsic hydrophobicity. An increase in ϱ corresponds to an enhanced non-polar character of respective peptide. The eluents were (A) H2O + 0.1% (v/v) TFA and (B) ACN + 0.1% (v/v) TFA by applying a gradient of 5% → 40% (B) over 30 min. (b) Geometry optimized structures of different amino acid residues marked with dashed circles with the N-terminal acetyl cap and the C-terminal N-methylamide cap obtained from quantum mechanical (MP2/6-31G*) calculations are shown in the ball-stick representation (top row). Atoms are colored according to atom types: carbon (cyan), nitrogen (blue), oxygen (red), hydrogen (white), and fluorine (pink). The corresponding space-filling models are colored according to the calculated electrostatic potential (ESP) showing the varying polarities for the different side chains. The calculated interaction energies ΔEint (HF/6-31G*) of water with an Abu, MfeGly, DfeGly and TfeGly side chain are plotted against retention times ϱ of AbuK16, MfeGlyK16, DfeGlyK16 and TfeGlyK16. For comparison, we also calculated the change in ΔEint for water–water interactions (−5.85 kcal mol−1). | ||
Theoretical approaches to determining the hydrophobic nature of fluorinated amino acids emphasized a change in apparent non-polar character through side chain-based interactions in aqueous conditions triggered by fluorination.49 To gain deeper insights into the impact of fluorine-specific interactions, we used quantum mechanical (QM) calculations for the residues of Abu, MfeGly, DfeGly and TfeGly-derived motifs. The QM geometry optimized structures, their electrostatic potential (ESP) maps and water interaction energies ΔEint for the different types of amino acids are shown in Fig. 2b. The ESP maps reveal different degrees of side chain polarities, the lowest for Abu and the highest for MfeGly. Interestingly, with further increase in fluorination (DfeGly, TfeGly), the polarity decreases again. To quantify the hydrophobicity of the side chains, we calculated ΔEint for the different types of amino acids by many initial configurations of an amino acid–water complex for each amino acid type. ΔEint is found to be the smallest for Abu (−0.91 kcal mol−1) and the largest for MfeGly (−3.94 kcal mol−1). Like the side chain polarity, ΔEint decreases with further increase of fluorination: DfeGly (−2.72 kcal mol−1) and TfeGly (−1.77 kcal mol−1). It should be noted that these ΔEint values are smaller than the water–water interaction energy of −5.85 kcal mol−1, which implies that all these amino acids are hydrophobic. The theoretical values corroborate the experimental trends of peptide hydrophobicity, emphasizing the impact of fluorine-induced polarity changes as seen for MfeGlyK16.
Secondary structure formation of amphipathic peptides
All oligopeptides were studied over a wide concentration range (0.1–1 wt%) by means of CD spectroscopy (Fig. 3). Secondary structure formation was investigated under physiologically buffered (pH 7.4) and basic (pH 9.0) conditions. The results at pH 7.4 show for AbuK16 and MfeGlyK16 at concentrations of 0.1–1 wt% (Fig. 3a–d) the typical course of absorption of a PPII-like conformation as discussed for AbuK-derived peptides above. Interestingly, the formation of β-sheets was not observed for MfeGlyK16 at the highest concentration of 2 wt% (Fig. 3d), very different from the observations for AbuK14 and AbuK16 (see Fig. 1a). Increasing the degree of fluorination triggers β-sheet formation (≥0.25 wt%) for DfeGlyK16; for lower concentrations (0.1 wt%) a similar conformation as given for AbuK16 and MfeGlyK16 was observed (Fig. 3a–d). A growth in fluorine-content on each individual amino acid increases the hydrophobic nature while, simultaneously, decreasing the polarity and, therefore, promotes β-sheet formation. In this manner, the most fluorinated peptide TfeGlyK16 was found to form β-sheets at all selected concentrations (Fig. 3a–d). | ||
| Fig. 3 CD spectra of amphipathic oligopeptides AbuK16 (green), MfeGlyK16 (red), DfeGlyK16 (blue) and TfeGlyK16 (violet) at (a) 0.1 wt%, (b) 0.25 wt%, (c) 0.5 wt% & (d) 1 wt% (also 2 wt% for MfeGlyK16) concentration in 50 mM Bis-tris propane + 150 mM NaCl, pH 7.4 recorded at 37 °C. CD spectra of (e) 0.25 wt% amphipathic oligopeptides AbuK14, AbuK16, MfeGlyK16, DfeGlyK16 and TfeGlyK16 and (f) 0.1–1 wt% MfeGlyK16 in 50 mM Bis-tris propane + 150 mM NaCl, pH 9.0 recorded at 37 °C. | ||
We further studied these peptides under basic conditions (pH 9.0) (Fig. 3e and f) that promote neutralization of the formal positive charges and, thus, induce β-sheet formation. All peptides undergo β-sheet formation at a concentration of 0.25 wt%. Peptides AbuK14 and AbuK16 form β-sheets at indicated concentrations at pH 9.0, whereas significantly higher concentrations are necessary under physiological conditions (2 wt%). The only exception in this regard is the variant MfeGlyK16, which does not form β-sheets even at higher concentrations of 1 wt% (see Fig. 3f). Comparison of MfeGlyK16 with AbuK14, both possessing similar values of peptide hydrophobicity, underlines the lack of β-sheet assembly for MfeGlyK16 due to its side chain properties. Likewise, calculated ΔEint values suggests the residue of MfeGly to be better accommodated in a water-exposed environment. We propose this circumstance, phenomenologically, as a notable driving force maintaining MfeGlyK16 in a PPII-like conformation. Beside the polar effects of each fluorinated side chain, their intrinsic secondary structure propensities are also crucial factors to understand fluorine-driven peptide folding. As reported by Gerling et al. the MfeGly side chain possesses the highest helical propensity among its fluorinated derivatives DfeGly and TfeGly.28 In correlation with the CD data, we suggest a synergistic effect of intrinsic folding propensity and fluorine-induced polarity by multiple incorporation of MfeGly causing the folding pattern of MfeGlyK16. For this purpose, we executed MD simulations to further elucidate this experimental finding. These theoretical results are discussed below.
Peptide self-assembly and characterization of fluorinated peptide-based hydrogels
Earlier reports demonstrated the supramolecular assemblies of amphipathic peptides to possess similar properties as the cross-β-sheet structure of amyloid fibrils.50,51 In order to study structural features of these peptides we used thioflavin T (ThT) fluorescence spectroscopy (Fig. 4a). This dye displays a strong fluorescence upon binding to amyloid-like morphologies caused by rotational immobilization, leading to an increase in fluorescence emission with a maximum at 485 nm. All samples were analyzed after 24 h of incubation time. We additionally studied all solutions by cryo-EM to determine the morphology of the formed aggregates (Fig. 4b–e). For AbuK16 and MfeGlyK16 we did not observe any increase in fluorescence intensity (FL) (range: 0.01–1 wt%). As discussed above, CD spectroscopy at given concentrations confirms our finding that these peptides do not form β-sheets but instead PPII helices. In contrast, DfeGlyK16 shows a 9.5-fold enhanced fluorescence emission (1 wt%) compared to the control sample (ThT-dye in buffer without peptide, fluorescence intensity FL485nm = 1.0), indicating the formation of amyloid-like aggregates. A small increase in FL intensity was observed for DfeGlyK16 at a minimal concentration of 0.25 wt%, which is also the lowest concentration revealing β-sheet structures in the respective CD spectra. TfeGlyK16 samples show a dramatic enhancement in fluorescence intensity of up to 180-fold at 1 wt%. Furthermore, the presence of amyloid-like fibrils was confirmed by Congo red (CR) staining experiments (see ESI, Fig. S97†). Our results indicate a correlation between the degree of fluorination and the ability to form amyloid-like structures. Cryo-EM studies were performed with those solutions for which a secondary structure pattern or amyloid-like behavior was detected by ThT staining. DfeGlyK16 (0.25 wt%) (Fig. 4d) and TfeGlyK16 (0.1 wt%) (Fig. 4e) both form amyloid-like structures. As expected, through CD and ThT staining experiments, AbuK16 (0.25 wt%) and MfeGlyK16 (0.25 wt%) did not form any fibrillary structures (see ESI, Fig. S98–S102†). At elevated concentrations of 2 wt%, however, we detected β-sheet formation for AbuK14 and AbuK16 by CD measurements (see prior data in Fig. 1a). Cryo-EM of these AbuK-derived sequences at 2 wt% concentration revealed narrow ribbons composed of fibrillar strands in a highly regular line pattern (Fig. 4b and c). A similar morphology from assembled fibrils based on a de novo designed coiled coil-based amyloidogenic peptide was studied in prior work.52 CD data of MfeGlyK16 at similar concentrations provide a PPII-like structural pattern, and no similar β-sheet assemblies were found in this case. We also applied small-angle X-ray scattering (SAXS) experiments on supposedly amyloid-like fibrils containing solutions of the peptides AbuK16, MfeGlyK16, DfeGlyK16 and TfeGlyK16 over a wide range of concentration (see ESI† for detailed SAXS interpretation). SAXS data from peptides DfeGlyK16 and TfeGlyK16 (both 1 wt% and 2 wt%) scale approximately with q−1 (TfeGlyK16) and q−2 (DfeGlyK16) at low q-values (see Fig. S111 and S112 in the ESI†). Such scaling behavior indicates a nearly circular cross-section for the fibrils of TfeGlyK16 (interpreted as circular cylinder) and a flat cross-section for DfeGlyK16 (interpreted as extended parallelepiped).29,53 Data evaluation with both theoretical models in terms of time-resolved experiments [with measurement frames of n = 1 (recorded ca. 120 s after sample preparation) and n = 50 (recorded 3000 s after sample preparation)] revealed an increase of the major semi axis from a = 2.7 nm to 11.0 nm for DfeGlyK16 (1 wt%) and from 2.9 nm to 4.4 nm (2 wt%). In contrast, the short semi-axis of the cross section is constant at b = 0.85 nm. For TfeGlyK16 an increase of the a-axis from 2.8 nm to 3.2 nm and from 2.8 nm to 3.1 nm at 1 wt% and 2 wt%, respectively, was determined. Here, the short semi-axis of the cross section is constant at b = 0.95 nm. An overview on the differences of cross-sections found for both polyfluorinated systems between data frame n = 1 and n = 50 is given in Fig. 4f. | ||
| Fig. 4 (a) Thioflavin T assays of AbuK16 (green), MfeGlyK16 (red), DfeGlyK16 (blue) and TfeGlyK16 (violet) incubated for 24 h at 37 °C in 50 mM Bis-tris propane + 150 mM NaCl containing 20 μM ThT dye, pH 7.4. Fluorescence emission was measured at 485 nm and normalized to a negative control (solely buffer) with a FL intensity of 1.0. Cryo-EM micrographs of (b) AbuK14 (2 wt%, diluted to 0.2 wt%), (c) AbuK16 (2 wt%, diluted to 0.2 wt%), (d) DfeGlyK16 (0.25 wt%) and (e) TfeGlyK16 (0.1 wt%) + image of a TfeGlyK16-based hydrogel at pH 7.4 (0.5 wt%). All samples were dissolved in 50 mM Bis-tris propane + 150 mM NaCl, pH 7.4. Insets display magnified areas of the micrographs indicated by white arrow heads. The scale bar denotes 200 nm for each micrograph. (f) Fibril cross sections corresponding to the SAXS model curves for samples of DfeGlyK16 and TfeGlyK16 at concentrations of 1 wt% and 2 wt% obtained through SAXS experiments. The cross-sections are of elliptical shape defined by major and minor semi-axis a and b, respectively. Plotted are the cross-sections derived from SAXS measurement frames n = 1 (after 120 seconds (grey ellipses)) and 50 (after 3000 seconds (colored ellipses)). Fibril growth is illustrated by arrows. | ||
After having characterized the mesoscopic structure of the different systems, we then turned to studying their macroscopic viscoelastic properties. Here, we performed strain-controlled oscillatory shear rheology measurements with the aim of evaluating the influence of fluorination on the mechanical properties of these gel matrices at pH 7.4 and pH 9.0. Amplitude γ (maximum deformation) sweeps at 1 Hz oscillation frequency were performed before the frequency-dependent measurements to ensure that the value γ of the deformation was always chosen such that the experimental conditions remained in the linear viscoelastic (LVE) regime. As a result, the amplitude γ for the oscillatory measurements was fixed at a value of 0.1%. The frequency sweeps were conducted in an angular frequency range of 0.314 to 314 rad s−1 to determine the storage and loss moduli G′ and G′′ (Fig. 5). First, we investigated the peptides AbuK16, MfeGlyK16, DfeGlyK16 and TfeGlyK16 at physiological conditions of pH 7.4 (Fig. 5a and b). As the non-fluorinated AbuK16 (0.5 wt%) formed only a low viscous solution at physiological conditions, we established a further reference sample comprising a Leu–Lys repeating unit (LeuK16, Ac-(Leu–Lys)8-[4]Abz-NH2) to distinguish between the impact of hydrophobicity and fluorine substituents. The Leu–Lys unit was utilized before by Schneider and co-workers for the development of peptide-based hydrogels.54 Through our RP-HPLC assay, we found LeuK16 (ϱ = 28.84 ± 0.025 min) to possess greater hydrophobicity than TfeGlyK16 (see ESI, Fig. S96†). In the case of TfeGlyK16 measurements were done also at lower concentrations of 0.25 and 0.1 wt%. These data are given in Fig. 5b and show a very strong reduction of the viscoelastic properties upon dilution, the elastic properties being reduced by a factor of around 80. Finally, for all 0.5 wt% samples we also studied their rheological behavior at pH 9.0 to determine how the change in pH affects the viscoelastic properties of the systems. Looking at Fig. 5c one observes that especially the elastic properties described by G′ are now much closer than at pH 7.4. For all samples at pH 7.4 and pH 9.0, G′ is at about one order of magnitude larger than G′′, indicating that these are gel-like systems, for which the elastic properties dominate.17 Both moduli increase somewhat with increasing frequency, thereby showing power law behavior, but with a rather small exponent. The plateau storage modulus G0, is of particular interest for gel-like systems. According to classical rubber elasticity theory,55G0 can be related to the crosslinking density ν of the gel. The crosslinking density can in turn be used to estimate an average mesh size ξ in the system, given as:
| ν = G0/kT = ξ−3 |
 | ||
| Fig. 5 Storage modulus G′ and loss modulus G′′ as measured in frequency sweeps (γ = 0.1%) at T = 37 °C for: (a) the peptides AbuK16, MfeGlyK16, DfeGlyK16, TfeGlyK16 and LeuK16 (all 0.5 wt%) at pH 7.4; (b) the peptide TfeGlyK16 at concentrations of 0.5 wt%, 0.25 wt%, and 0.1 wt% at pH 7.4; (c) the peptides AbuK16, MfeGlyK16, DfeGlyK16, TfeGlyK16 and LeuK16 (all 0.5 wt%) at pH 9.0. For sample preparation, freeze-dried peptides were dissolved in 50 mM Bis-tris propane + 150 mM NaCl, either pH 7.4 or pH 9.0. | ||
| Name | c (wt%) | pH | G 0 (Pa) | ξ (nm) |
|---|---|---|---|---|
| LeuK16 | 0.5 | 7.4 | 4869 ± 124 | 9.83 ± 0.08 |
| 9 | 564.4 ± 7.2 | 20.17 ± 0.09 | ||
| TfeGlyK16 | 0.5 | 7.4 | 670 ± 19 | 19.05 ± 0.18 |
| 9 | 70.24 ± 1.4 | 40.4 ± 0.3 | ||
| 0.25 | 7.4 | 8.63 ± 0.79 | 81.3 ± 2.5 | |
| 0.1 | 7.4 | 1.09 ± 0.12 | 161.9 ± 5.7 | |
| DfeGlyK16 | 0.5 | 7.4 | 15.30 ± 1.04 | 67.14 ± 1.5 |
| 9 | 554 ± 15 | 20.29 ± 0.18 | ||
| AbuK16 | 0.5 | 7.4 | 4.81 ± 0.22 | 98.72 ± 1.54 |
| 9 | 264.1 ± 5.1 | 25.98 ± 0.17 | ||
| MfeGlyK16 | 0.5 | 7.4 | 0.53 ± 0.02 | 205.37 ± 2.53 |
| 9 | 208.8 ± 0.83 | 28.10 ± 0.04 |
An interesting behavior is observed upon increasing the pH from 7.4 to 9.0, which lowers the overall charge of the peptide originating from the Lys residues (Fig. 5c). Upon this change, G0 increases tremendously for MfeGlyK16 (209 Pa), AbuK16 (264 Pa) and DfeGlyK16 (554 Pa) but drops by almost one order of magnitude for TfeGlyK16 (70 Pa) and LeuK16 (564 Pa), thereby bringing all the values closer together. This corresponds to an increase of G0 by a factor of 394 (MfeGlyK16), 55 (AbuK16) and 36 (DfeGlyK16), but also a reduction of G0 by a factor of 9.6 (TfeGlyK16) and 8.6 (LeuK16), respectively. The greatest change in viscoelastic stability is observed for the peptide with the lowest hydrophobicity (MfeGlyK16) within this work and becomes weaker with increasing non-polar properties (AbuK16, DfeGlyK16). This surprising loss in G0 of TfeGlyK16-derived hydrogels was confirmed by further measurements at both pH values with independently prepared samples (see ESI, Fig. S104–106†). As an explanation, we suggest a major leverage of side chain-neutralization on the viscoelastic stability of these supramolecular matrices depending rather on peptide hydrophobicity than on fluorine-specific interactions. An almost equal loss in G0 value in context of pH change found for LeuK16 in correlation to TfeGlyK16 serves as further confirmation of this experimental finding. Hence, the divergence in rheological properties between DfeGlyK16 and TfeGlyK16-based hydrogels is, in particular, an interesting phenomenon as it seems to be triggered by only a single H to F substitution of each amino acid residue.
MD simulations of amphipathic peptides
Finally, we have performed MD simulations of AbuK16, MfeGlyK16, DfeGlyK16, and TfeGlyK16 in explicit solvent at two different pH values: pH 7 (where for simplicity we assume all Lys residues to be charged) and pH 11 (where we assume all Lys residues to be charge neutral). The aim was to understand how fluorine-specific interactions may modify inter-peptide interactions and thereby control the formation of higher-ordered structures as observed in the above-discussed experiments. Interestingly, we find intra-strand contact pair formations between the fluorine atom of each MfeGly residue (MfeGlyK16) and backbone hydrogen atoms located on the peptide's amide bonds (Fig. 6a). Such contact pairs are absent in the cases of DfeGlyK16 and TfeGlyK16, whose side chains are randomly oriented as found for AbuK16 (Fig. 6a). The intra-strand contact pair formation observed for MfeGlyK16 can be rationalized by the strongly polar MfeGly side chain as described before. The free energy profiles of inter-strand interaction as a function of the inter-strand separation for the different peptide types at pH 7 are shown in Fig. 6b, which reveal two distinct minima for each peptide type but MfeGlyK16, for which there is only one minimum. The shallow minimum at a larger inter-peptide separation observed for AbuK16, DfeGlyK16, and TfeGlyK16 is due to the partially hydrophobic, non-standard (synonymous for Abu, MfeGly, DfeGly and TfeGly) amino acid (AA) side chain–side chain interaction, whereas the global minimum at a smaller inter-peptide separation is due to the non-standard AA side chain–Lys side chain and non-standard AA side chain–backbone interactions (see snapshots in Fig. 6c). In contrast, the minimum free-energy structure of MfeGlyK16 is due to the polar, non-standard AA side chain–backbone and non-standard AA side chain–side chain interactions, and the charged Lys residues protrude out to minimize the electrostatic repulsion. Although negative and comparable free energy values for all cases imply that every peptide type can form strongly bounded dimers, the possibility of peptide fibril formation depends on whether two such dimers can in turn form favorable contacts with each other. As depicted in Fig. 6d for AbuK16, DfeGlyK16, and TfeGlyK16, two dimers placed parallel to each other can form favorable contacts between the non-standard AA and Lys side chains (which are found to be important for the stability of a dimer as well). In contrast, two MfeGlyK16 dimers placed parallel to each other face charged end groups of the Lys residue (–NH3+), and thus repel each other. Therefore, higher-order structure formation or fibrillization at pH 7 is predicted to be possible for AbuK16, DfeGlyK16, and TfeGlyK16, but not for MfeGlyK16, explaining our experimental findings at physiological conditions. The free energy profiles at pH 11, shown in Fig. S109† (ESI), reveal only one free-energy minimum for each peptide type, the depth of which is more than that of the corresponding peptide type at pH 7. The increased interaction strength is due to the reduced electrostatic repulsion between the two strands, as Lys residues are charge neutral at pH 11. For MfeGlyK16, the intra-strand contact pair formations (Fig. 6a) expose the backbone atoms of MfeGly amino acids that can form H-bonds with the solvent-exposed backbone atoms of another MfeGlyK16 strand, resulting in a very compact MfeGlyK16 dimer with enhanced side chain–side chain interactions (see snapshots in Fig. S109 within the ESI†). These strong cooperative interactions lead to a 2–3 times deeper free energy minimum for MfeGlyK16 than other peptide types, for which the inter-peptide interactions are dominated by weak backbone-side chain and side chain–side chain contacts. Like at pH 7, for AbuK16, DfeGlyK16, and TfeGlyK16, two dimers placed parallel to each other can form the same favorable contacts as found in the stable structure of a single dimer, and hence the dimer structure can be periodically extended from either side by adding more dimers to produce large-scale peptide fibrils. For MfeGlyK16, the dimer structure is the most stable among the different peptide types and the inter-dimer interaction strength is expected to be lower due to the only possible side chain–side chain interaction as backbone atoms in a dimer complex are shielded by MfeGly side chains given by intra-strand contact pair formation. Hence, higher-order structures are expected to be less stable for MfeGlyK16 and would provide an explanation for its structural properties. | ||
| Fig. 6 MD simulation results for amphipathic peptides; water is included in the simulations, but not shown for clarity. (a) Snapshots for representative parts of periodic AbuK16, MfeGlyK16, and TfeGlyK16 single strands (taken from umbrella sampling simulations when two strands are far apart) are shown in the space-filling representation and atoms are colored as: H (white), C (cyan), N (blue), O (red), F (pink). Fluorine atoms from MfeGly residues and the amide-backbone derived hydrogen atoms form strong intra-strand contact pairs which are marked by red, dashed ellipses. Such contact pairs are not observed for Abu and TfeGly. (b) The potential of mean force (PMF) per amino acid (AA) as a function of inter-strand separation dxy at pH 7 (charged Lys), depicting the free energy profile of interaction between two peptide strands for side chains with different degree of fluorinations. The global minimum for each peptide type is marked by a triangle, whereas a secondary shallow minimum at a farther distance (when present) is marked by a star symbol. (c) Structures corresponding to free energy minima for each peptide type are shown in ball-stick representation; the terminal group of each side chain is highlighted in the space-filling representation. Atom colors are the same as in (a) and colors of enclosing boxes are the same as colors of the PMF profiles in (b). Dominant binding modes are given at the top of each snapshot. (d) Schematic depicting the possibility of fibrillization of the dimer complex for the different peptide types. One strand of a dimer complex (shown in the ball-stick representation) is colored as light-gray, whereas the other strand is colored as dark-gray. The end groups of side chains from Lys (–NH3+), Abu (–CH3), MfeGly (–CFH2), DfeGly (–CF2H), and TfeGly (–CF3) are highlighted in space-filling representation. | ||
Conclusions
In this work, we systematically designed and characterized a library of oligopeptides with high numbers of fluorinated amino acids. The peptides AbuK16, MfeGlyK16, DfeGlyK16 and TfeGlyK16 served as models to evaluate the impact of fluorine-specific interactions in the context of secondary structure formation, peptide self-assembly and hydrogel formation. The fluorination degree of the aliphatic side chain plays a crucial role in determining the peptide intrinsic hydrophobic properties. This led not only to the observation of different secondary structures such as PPII helices or β-sheets, but also to fluorine-driven self-assembly into ordered nanostructures. On the other hand, we found for MfeGlyK16 at physiological conditions no evidence of β-sheet assembly, explained by MD simulations that find strong dimer formation preventing peptide fibrillization. Rheological characterization revealed a correlation between the hydrophobic nature of each fluorinated amino acid and an enhanced viscoelastic stability of resulting hydrogel matrices as shown for MfeGlyK16, DfeGlyK16 and TfeGlyK16 in physiological conditions, but also a loss in mechanical stiffness for the latter variant at pH 9.0. This study firstly established and studied a library of distinctive aliphatic and polyfluorinated SAPs for which fluorine-specific interactions were evinced by significant alterations of intra- and intermolecular interactions. The underlined design principle, the unique properties of the peptides and resulting hydrogel matrices will contribute to the future development of de novo designed fluorinated biomaterials.Experimental section
General methods
1H-, 13C- and 19F-NMR spectra (see ESI†) were recorded at room temperature using a JEOL ECX 400 (JEOL, Tokyo, Japan), a JEOL ECP 500 (JEOL, Tokyo, Japan) or a Bruker AVANCE III 700 (700 MHz, BRUKER, Billerica, MA, USA). Chemical shifts δ are reported in ppm with the solvent resonance (MeOH-d4) as the internal standard. HRMS were determined on an Agilent 6220 ESI-TOF MS instrument (Agilent Technologies, Santa Clara, CA, USA). For analysis, the MassHunter Workstation Software Version B.02.00 (Agilent Technologies, Santa Clara, CA, USA) was used. IR Spectra were recorded on an ALPHA II FT-IR spectrometer (Bruker, USA). All NMR and IR spectra were evaluated by using Mnova/Mestrenova (Mestrelab Research, CA, USA). Elemental analysis was proceeded by use of an VARIO EL elemental analyzer (Elementar Analysensysteme GmbH, Langenselbold, Germany). All essential data for compound characterization is placed within the ESI.† All chemicals were purchased from commercial sources (Merck, Sigma-Aldrich, VWR, Fluorochem) and used without further purification. The Fmoc-protected fluorinated amino acid TfeGly was synthesised according to literature (see ESI,† chapter “Gram scale synthesis and characterization of fluorinated amino acid Fmoc-TfeGly-OH”). MfeGly and DfeGly were synthesised by Suvrat Chowdhary and Thomas Hohmann.Synthesis and purification of peptides
All peptides were synthesized with a microwave-equipped Liberty Blue™ peptide synthesizer (CEM, Matthews, NC, USA). A Rink Amide ProTide™ resin (CEM, Matthews, NC, USA) was utilized and the synthesis was performed either in 0.05 mmol or 0.1 mmol scale using oxyma/DIC as activating reagents (0.05 mmol scale: 0.5 M oxyma in DMF and 0.25 M DIC in DMF/0.1 mmol scale: 1 M oxyma in DMF and 0.5 M DIC in DMF). Coupling of native Fmoc-protected amino acids occurred in DMF using 5 eq. of substance (for fluorinated amino acids only 1.5 eq. were used) with 5 eq. of activating reagents and double couplings of 4 min coupling time (for fluorinated amino acid: mono coupling of 10 min) at 90 °C. For deprotection of the N-terminus, a 10% piperazine (w/v) solution in EtOH/NMP (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) with 0.1 M HOBt was used. Acetylation was done manually in three batches using acetic anhydride (10% v/v) and DIPEA (10% v/v) in DMF (6 mL). All peptides were cleaved from the resin by treatment with TFA/TIPS/H2O (90/5/5) for three hours using sonication at room temperature. Then the resins were washed with TFA and DCM, and excess of solvents were removed by evaporation. Peptides were dried by lyophilization before purification with preparative reversed phase HPLC. Purification of synthesized peptides was performed on a Knauer low-pressure HPLC system (Knauer GmbH, Berlin, Germany) sold by VWR (Darmstadt, Germany), comprising a LaPrep Sigma preparative pump (LP1200), a ternary low-pressure gradient, a dynamic mixing chamber, a 6-port-3-channel injection valve with an automated preparative 10 mL sample loop, a LaPrep Sigma standard 1-channel-UV-detector (LP3101), a flow cell with 0.5 mm thickness and a 16-port LaPrep Sigma fractionation valve (LP2016). A Kinetex RPC18 endcapped (5 μM, 100 Å, 250 × 21.2 mm, Phenomenex®, USA) HPLC-column was used. A Security GuardTM PREP Cartridge Holder Kit (21.20 mm, ID, Phenomenex®, USA) served as pre-column. As eluents water and ACN, both containing 0.1% (v/v) TFA were applied. HPLC runs were performed with a flow rate of 15.0 mL min−1, UV-detection occurred at 220 nm for respective peptides. Data analysis occurred with an EZChrom Elite-Software (Version 3.3.2 SP2, Agilent). After separation, the purity of the collected fractions was determined by analytical HPLC. Analytical HPLC was carried out on a Chromaster 600 bar DAD-System with CSM software or a Hitachi Primaide™ UV-HPLC system (both from VWR/Hitachi, Darmstadt, Germany). A Kinetex® RP-C18 (5 μM, 100 Å, 250 × 4.6 mm, Phenomenex®, USA) column and a SecurityGuard™ Cartridge Kit equipped with a C18 cartridge (4 × 3.0 mm, Phenomenex®, USA) as pre-column was used. Otherwise, a Luna® RP-C8 (5 μm, 100 Å, 150 × 3 mm, Phenomenex®, USA) column was used. As eluents water and ACN, both containing 0.1% (v/v) TFA were applied. A flow rate of 1 mL min−1 was used and UV-detection occurred at 220 nm or 280 nm for respective peptides. Data analysis was done with EZ Chrom ELITE software (version 3.3.2, Agilent). The resulting pure peptides (>95%) were obtained after lyophilization of the collected fractions. All essential data for the quantification of purified peptides (HPLC data, HRMS spectra) can be found in the ESI (Fig. S37–S91 and Tables S1–S22†).
:9) with 0.1 M HOBt was used. Acetylation was done manually in three batches using acetic anhydride (10% v/v) and DIPEA (10% v/v) in DMF (6 mL). All peptides were cleaved from the resin by treatment with TFA/TIPS/H2O (90/5/5) for three hours using sonication at room temperature. Then the resins were washed with TFA and DCM, and excess of solvents were removed by evaporation. Peptides were dried by lyophilization before purification with preparative reversed phase HPLC. Purification of synthesized peptides was performed on a Knauer low-pressure HPLC system (Knauer GmbH, Berlin, Germany) sold by VWR (Darmstadt, Germany), comprising a LaPrep Sigma preparative pump (LP1200), a ternary low-pressure gradient, a dynamic mixing chamber, a 6-port-3-channel injection valve with an automated preparative 10 mL sample loop, a LaPrep Sigma standard 1-channel-UV-detector (LP3101), a flow cell with 0.5 mm thickness and a 16-port LaPrep Sigma fractionation valve (LP2016). A Kinetex RPC18 endcapped (5 μM, 100 Å, 250 × 21.2 mm, Phenomenex®, USA) HPLC-column was used. A Security GuardTM PREP Cartridge Holder Kit (21.20 mm, ID, Phenomenex®, USA) served as pre-column. As eluents water and ACN, both containing 0.1% (v/v) TFA were applied. HPLC runs were performed with a flow rate of 15.0 mL min−1, UV-detection occurred at 220 nm for respective peptides. Data analysis occurred with an EZChrom Elite-Software (Version 3.3.2 SP2, Agilent). After separation, the purity of the collected fractions was determined by analytical HPLC. Analytical HPLC was carried out on a Chromaster 600 bar DAD-System with CSM software or a Hitachi Primaide™ UV-HPLC system (both from VWR/Hitachi, Darmstadt, Germany). A Kinetex® RP-C18 (5 μM, 100 Å, 250 × 4.6 mm, Phenomenex®, USA) column and a SecurityGuard™ Cartridge Kit equipped with a C18 cartridge (4 × 3.0 mm, Phenomenex®, USA) as pre-column was used. Otherwise, a Luna® RP-C8 (5 μm, 100 Å, 150 × 3 mm, Phenomenex®, USA) column was used. As eluents water and ACN, both containing 0.1% (v/v) TFA were applied. A flow rate of 1 mL min−1 was used and UV-detection occurred at 220 nm or 280 nm for respective peptides. Data analysis was done with EZ Chrom ELITE software (version 3.3.2, Agilent). The resulting pure peptides (>95%) were obtained after lyophilization of the collected fractions. All essential data for the quantification of purified peptides (HPLC data, HRMS spectra) can be found in the ESI (Fig. S37–S91 and Tables S1–S22†).
Lyophilization
To lyophilize the synthesized peptides a laboratory freeze dryer ALPHA 1-2 LD (Christ Gefriertrocknungsanlagen GmbH, Osterode am Harz, Germany) was used.Sample preparation – exchange of TFA salts
All purified peptides were inevitably obtained as corresponding TFA salts during resin cleavage and subsequent RP-HPLC purification using eluents containing 0.1% TFA. Peptide samples (about 13 mg each) were dissolved in 800 μL Milli-Q-H2O and transferred on a VariPure IPE exchange column (100 mg, 3 mL) (Agilent Technologies, Santa Clara, CA, USA). These columns were previously washed and pre-conditioned with MeOH (3 × 3 mL) and Milli-Q-H2O (3 × 3 mL). The resin was additionally washed with Milli-Q-H2O (500 μL) and the collected peptide fractions were combined. Afterwards, desired samples were lyophilized to obtain the peptide with bicarbonates as counter-ions.Preparation of peptide stock solutions and self-assembly
Peptide stock solutions were prepared by dissolving lyophilized peptide powder (10–15 mg) in 1,1,1,3,3,3-hexafluoropropan-2-ol [HFIP] (2 mL) and treatment for 15 min in an ultrasound-bath to dissolve preformed aggregates. An aliquot of 10 μL was evaporated and the dried peptide film dissolved in a 6 M guanidine hydrochloride (GndHCl) solution (pH 7.4), resulting into a dilution factor (DL) of 100. These samples were measured via UV detection at 280 nm by use of an Eppendorf BioPhotometer plus with semi-micro-VIS Cuvettes (PMMA) 10 × 100 (Eppendorf, Hamburg, Germany). All UV spectra were baseline corrected with a reference spectrum of a sample containing solely buffer solution. The UV absorbance given through the fluorophore p-aminobenzoic acid (PABA) at the C-terminus of the peptides at 280 nm was evaluated in triplicates. By use of a calibration curve derived from p-aminohippuric acid (PAH) (see ESI, Fig. S92†), the concentration of each stock solution was calculated. If not otherwise stated, all peptide samples were treated following this protocol before each measurement: an aliquot from the HFIP peptide stock solution was taken and evaporated. The dried peptide was then dissolved in respective buffer and vortexed (1 min), sonicated (5 min) and finally ultracentrifuged (1 min) at room temperature.RP-HPLC assay for estimation of hydrophobicity
The protocol for the RP-HPLC assay was previously established by our group.28,29 Peptide samples were dissolved in 250 μL of a mixture of 5% (v/v) ACN in 95% (v/v) Milli-Q-water containing 0.1% TFA and filtered over a syringe filter with 0.2 μm pore size. The overall concentration of each sample was 0.2 mM. The retention times of all samples were determined on a C18 column (Capcell C18, 5 μm) using a LaChrom-ELITE-HPLC-System (VWR International) with UV detection at 280 nm. A linear gradient from 5 to 40% ACN + 0.1% TFA in 30 min was applied at room temperature and all experiments were performed in triplicates.QM calculations
All QM calculations were performed using Gaussian 16.1.56Abu, MfeGly, DfeGly, and TfeGly-derived motifs, as shown in Fig. 2b, were taken for QM calculations. Geometry optimizations of these structures were done at the MP2/6-31G* level of theory. Water interaction energies were obtained from the HF/6-31G* single point calculations of the geometry optimized structures. Electrostatic potential maps for the geometry optimized structures were rendered using the Avogadro software.57 Dihedral energy scans were performed at the MP2/6-31G* level of theory. Force field parameters and partial atomic charges for the amino acid Abu and its fluorinated variants (MfeGly, DfeGly, and TfeGly) were initially obtained from CHARMM36m58 and CGenFF59 parameters, using the CGenFF program.60,61 As there were large penalties for dihedral angles associated with side chain rotations (Cα–Cβ–Cγ–F and Cα–Cβ–Cγ–H) and partial atomic charges, new parameters were derived from QM energy scans and water interaction energies, respectively using the FFParam package.62 Optimized partial atomic charges and dihedral parameters for the different amino acids are given in the ESI† (Chapter 12).Equilibrium MD simulations
To study interpeptide interactions, two periodic polypeptide chains, each with the long-axis oriented along the z-direction, with an interaxial distance dxy of 2.5 nm, arranged antiparallel (ap) to each other were considered. Each system was solvated in a rectangular box of size 5 × 5 × 5.832 nm3. If needed, enough counterions (Cl− ions) were added to charge neutralize the whole system. The simulation box is shown in Fig. S108† (ESI). CHARMM-compatible TIP3P water63,64 and ion parameters65 were used. The solvated system was subjected to energy minimization using the steepest descent algorithm, for removing any unfavourable contacts. The simulation for each case was performed for 500 ns in the NpxyLzT ensemble, with Lz per amino acid = 3.6 Å, at T = 300 K and pxy = 1 bar with periodic boundary condition in xyz directions, using the GROMACS 2020.1 package.66 The stochastic velocity rescaling thermostat67 with a time constant of τT = 0.1 ps was used to control the temperature, while for the pressure control a semi-isotropic Parrinello–Rahman barostat68 was used with a time constant of τp = 1 ps and a compressibility of κ = 4.5 × 10−5 bar−1. The LINCS algorithm69 was used to convert the bonds with H-atoms to constraints, allowing a timestep of Δt = 2 fs. Electrostatics interactions were computed using the particle mesh Ewald (PME) method70 with a real-space cut-off distance of 1.2 nm, while van der Waals (VDW) interactions were modelled using Lennard-Jones potentials with a cut-off distance of 1.2 nm where the resulting forces smoothly switch to zero between 1 nm to 1.2 nm.Umbrella sampling simulations
To calculate the free energy landscape or the potential of mean force (PMF) between two periodic polypeptide chains, the final configuration obtained from the equilibrium MD simulation was first pulled in either direction to generate initial conformations for two polypeptide chains at different interaxial separations. Total 40–50 umbrella windows, with an inter-window spacing of 0.35 Å, were simulated in the NpxyLzT for 30 ns each. During these simulations, an additional umbrella potential with a spring constant of 10000 kJ mol−1 nm−2 was used to restrain the interaxial separation to a given distance. Every 100 fs data was collected, and the last 20 ns simulation data for each window was used to obtain the PMF using the weighted histogram analysis method (WHAM).71,72 The g_wham module73 of GROMACS was used for performing the WHAM analysis and calculating error bars using the bootstrap method.
CD spectroscopy
Circular dichroism experiments were performed using a Jasco J-810 spectropolarimeter fitted with a recirculating chiller (D-76227, Karlsruhe). Data were recorded using 0.1 mm Quartz Suprasil® cuvettes (Hellma) equipped with a stopper. Spectra were recorded at 37 °C from 190 to 250 nm at 0.2 nm intervals, 1 nm bandwidth, 4 s response time and a scan speed of 100 nm min−1. Baselines were recorded and were subtracted from the data. Each reported CD value represents the average of minimum three measurements. Further CD spectra can be found in the ESI (Fig. S93–S95†)Congo red (CR) assay for fibril detection
Aliquots of peptide HFIP-stock solutions (0.5 wt%) were dried and then redissolved in 50 mM Bis-tris propane + 150 mM NaCl with addition of 50 μM Congo red (overall pH 7.4). Negative controls were prepared by dissolving corresponding samples in buffer without dye. After dissolution, the standard self-assembly protocol was applied, and all samples were incubated overnight at 37 °C. UV spectra (300–700 nm) were recorded for all samples using a Varian Cary 50 UV-VIS Spectrophotometer (Agilent, USA) and 0.5 mm Quartz Suprasil® cuvettes (Hellma). Experimental data can be found in the ESI (Fig. S97†).Thioflavin T (ThT) fluorescence assay for fibril detection
A suitable protocol for this assay was recently published by our group.29 Aliquots of peptide HFIP-stock solutions were dried and then redissolved in 50 mM Bis-tris propane + 150 mM NaCl with addition of 20 μM Thioflavin T (overall pH 7.4). The buffer containing ThT was previously filtered over a nylon syringe filter with 0.2 μm pore size. After dissolution, the sample was sonicated for 30 s, transferred on a BRAND® microplate (size: 96 wells, color: black; Sigma-Aldrich), sealed to prevent evaporation and placed in an Infinite® M Nano+ plate reader (Tecan Deutschland GmbH, Crailsheim, Germany). ThT fluorescence (λex = 420 nm, λem = 485 nm, Z-position: 15173 nm [manual], gain: 80 [manual], lag time: 0 μs, integration time: 20 μs) was measured after 24 h incubation at 37 °C. The fluorescence intensity at 485 nm was normalized with respect to the negative control solely containing buffer (set as FLint 1.0).
Cryogenic electron microscopy (cryo-EM)
Perforated carbon film-covered microscopical 200 mesh grids (R1/4 batch of Quantifoil, MicroTools GmbH, Jena, Germany) were cleaned with chloroform and hydrophilized by 60 s glow discharging at 10 μA in a EMSCOPE SC500 before 4 μl aliquots of the peptide solution were applied to the grids. The samples were vitrified by automatic blotting and plunge freezing with a FEI Vitrobot Mark IV (Thermo Fisher Scientific Inc., Waltham, Massachusetts, USA) using liquid ethane as cryogen. The vitrified specimens were transferred to the autoloader of a FEI TALOS ARCTICA electron microscope (Thermo Fisher Scientific Inc., Waltham, Massachusetts, USA). This microscope is equipped with a high-brightness field-emission gun (XFEG) operated at an acceleration voltage of 200 kV. Micrographs were acquired on a FEI Falcon 3 direct electron detector (Thermo Fisher Scientific Inc., Waltham, Massachusetts, USA) using the 70 μm objective aperture at a nominal magnification of 28000, corresponding to a calibrated pixel size of 3.75 Å per pixel, respectively.
Small-angle-X-ray scattering (SAXS)
SAXS measurement were performed in a flow-through capillary with a Kratky-type instrument (SAXSess from Anton Paar, Austria) at 37 ± 1 °C. The SAXSess has a low sample-to-detector distance of 0.309 m, which is appropriate for investigations of liquid samples with low scattering intensities. The measured intensity was converted to absolute scale according to Orthaber et al.74 The scattering vector q is defined in terms of the scattering angle θ and the wavelength λ of the radiation (λ = 0.154 nm): thus, q = 4πn/λsinθ. Deconvolution (slit length desmearing) of the SAXS curves was performed with the SAXS-Quant software. Samples analyzed with SAXS were used as prepared, i.e. samples were mixed with buffer solution, vortexed for 20 s and filled in the capillary. Curve fitting was conducted with SASfit.75
Rheological characterization of amphipathic peptide hydrogels
Before each measurement, peptide samples of AbuK16, MfeGlyK16, DfeGlyK16, TfeGlyK16 and LeuK16 were dissolved in 50 mM Bis-tris propane + 150 mM NaCl (either pH 7.4 or pH 9.0), treated as mentioned above and incubated at 37 °C for 24 h. All rheological measurements were performed on an Anton Paar MCR 502 WESP temperature-controlled rheometer in strain-imposed mode at physiological temperature (37 °C). For all measurements, a parallel plate geometry with chromium oxide coating was used, with a diameter of 50 mm for the upper rotating plate. The gap size between the plates was set to 175 μm. The sample and geometry were surrounded by a solvent trap to reduce effects of solvent evaporation. Further experimental data can be found in the ESI (Fig. S97†).Author contributions
B. K. and S. C. conceived the overall project. B. K. provided guidance on data analysis, interpretation, and manuscript preparation. S. C. designed and developed concepts and experiments, synthesized, and purified all peptides, performed CD, UV and FL experiments, established and proceeded a RP-HPLC-based hydrophobicity plot for peptides, prepared samples for further experiments and wrote the manuscript. R. F. S. performed rheology experiments and wrote the manuscript. A. K. S. performed simulations. A. K. S. and R. R. N. analyzed and interpreted data and wrote the manuscript. T. t. D. assisted S. C. in synthesis and purification of peptides and circular dichroism experiments. K. B.-J. and A. F. T. performed SAXS experiments and SAXS data analysis. T. H. provided fluorinated amino acids. B. S. performed cryoEM experiments, analyzed, and interpreted data. R. R. N. and M. G. provided expertise and feedback.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. C., T. H., B. S., R. R. N. and B. K. gratefully acknowledges financial support by the Deutsche Forschungsgemeinschaft (DFG) through the collaborative research center CRC 1349 “Fluorine-Specific Interactions”, project no. 387284271. R. F. S., M. G., and R. R. N. were funded by the DFG grant CRC 1449 “Dynamic Hydrogels at Biointerfaces”, project no. 431232613. We thank Dr Johann Moschner, Dr Katharina Hellmund, Dr Dorian Jamal Mikolajczak, Dr Ana Rita de Lima Fernandes, Dr Allison Berger, and Dr Chaitanya Kumar Thota for fruitful scientific discussions and expertise. We thank Dr Jakob Leppkes for his support during the synthesis of fluorinated amino acids. We would like to acknowledge the assistance of the Core Facility BioSupraMol supported by the DFG. We thank Benedikt Kirmayer for assistance in the preparation of cryo-EM samples and micrographs. A. K. S. and R. R. N. acknowledge support from the Max-Planck MaxWater initiative.Notes and references
- A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Nat. Rev. Chem., 2020, 4, 615–634 CrossRef CAS.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- J. P. Schneider, D. J. Pochan, B. Ozbas, K. Rajagopal, L. Pakstis and J. Kretsinger, J. Am. Chem. Soc., 2002, 124, 15030–15037 CrossRef CAS PubMed.
- B. Ozbas, J. Kretsinger, K. Rajagopal, J. P. Schneider and D. J. Pochan, Macromolecules, 2004, 37, 7331–7337 CrossRef CAS.
- S. Zhang, T. Holmes, C. Lockshin and A. Rich, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 3334–3338 CrossRef CAS PubMed.
- S. Zhang, T. C. Holmes, C. M. DiPersio, R. O. Hynes, X. Su and A. Rich, Biomaterials, 1995, 16, 1385–1393 CrossRef CAS PubMed.
- F. Gelain, Z. Luo and S. Zhang, Chem. Rev., 2020, 120, 13434–13460 CrossRef CAS PubMed.
- F. Gelain, Z. Luo, M. Rioult and S. Zhang, npj Regener. Med., 2021, 6, 9 CrossRef CAS PubMed.
- C. J. C. Edwards-Gayle and I. W. Hamley, Org. Biomol. Chem., 2017, 15, 5867–5876 RSC.
- V. Castelletto, C. J. C. Edwards-Gayle, F. Greco, I. W. Hamley, J. Seitsonen and J. Ruokolainen, ACS Appl. Mater. Interfaces, 2019, 11, 33573–33580 CrossRef CAS PubMed.
- S. Motamed, M. P. Del Borgo, K. Kulkarni, N. Habila, K. Zhou, P. Perlmutter, J. S. Forsythe and M. I. Aguilar, Soft Matter, 2016, 12, 2243–2246 RSC.
- P.-X. Zhang, N. Han, Y.-H. Kou, Q.-T. Zhu, X.-L. Liu, D.-P. Quan, J.-G. Chen and B.-G. Jiang, Neural Regener. Res., 2019, 14, 51–58 CrossRef PubMed.
- K. S. Hellmund, B. von Lospichl, C. Böttcher, K. Ludwig, U. Keiderling, L. Noirez, A. Weiß, D. J. Mikolajczak, M. Gradzielski and B. Koksch, Pept. Sci., 2021, 113, e24201 CAS.
- S. Lee, T. H. T. Trinh, M. Yoo, J. Shin, H. Lee, J. Kim, E. Hwang, Y.-B. Lim and C. Ryou, Int. J. Mol. Sci., 2019, 20, 5850 CrossRef CAS PubMed.
- J. Y. C. Lim, Q. Lin, K. Xue and X. J. Loh, Mater. Today Adv., 2019, 3, 100021 CrossRef.
- X. Zhao and S. Zhang, Chem. Soc. Rev., 2006, 35, 1105–1110 RSC.
- L. M. De Leon Rodriguez, Y. Hemar, J. Cornish and M. A. Brimble, Chem. Soc. Rev., 2016, 45, 4797–4824 RSC.
- J. Kopeček and J. Yang, Acta Biomater., 2009, 5, 805–816 CrossRef PubMed.
- J. N. Sloand, M. A. Miller and S. H. Medina, Pept. Sci., 2021, 113, e24184 CAS.
- A. A. Berger, J.-S. Völler, N. Budisa and B. Koksch, Acc. Chem. Res., 2017, 50, 2093–2103 CrossRef CAS PubMed.
- E. N. G. Marsh, Acc. Chem. Res., 2014, 47, 2878–2886 CrossRef CAS PubMed.
- S. A. Samsonov, M. Salwiczek, G. Anders, B. Koksch and M. T. Pisabarro, J. Phys. Chem. B, 2009, 113, 16400–16408 CrossRef CAS PubMed.
- M. Salwiczek, S. Samsonov, T. Vagt, E. Nyakatura, E. Fleige, J. Numata, H. Cölfen, M. T. Pisabarro and B. Koksch, Chem. – Eur. J., 2009, 15, 7628–7636 CrossRef CAS PubMed.
- S. Huhmann, E. K. Nyakatura, H. Erdbrink, U. I. M. Gerling, C. Czekelius and B. Koksch, J. Fluorine Chem., 2015, 175, 32–35 CrossRef CAS.
- T. Vagt, E. Nyakatura, M. Salwiczek, C. Jäckel and B. Koksch, Org. Biomol. Chem., 2010, 8, 1382–1386 RSC.
- S. Ye, B. Loll, A. A. Berger, U. Mülow, C. Alings, M. C. Wahl and B. Koksch, Chem. Sci., 2015, 6, 5246–5254 RSC.
- J. Leppkes, N. Dimos, B. Loll, T. Hohmann, M. Dyrks, A. R. Wieseke, B. G. Keller and B. Koksch, RSC Chem. Biol., 2022, 3(6), 773–782 RSC.
- U. I. M. Gerling, M. Salwiczek, C. D. Cadicamo, H. Erdbrink, C. Czekelius, S. L. Grage, P. Wadhwani, A. S. Ulrich, M. Behrends, G. Haufe and B. Koksch, Chem. Sci., 2014, 5, 819–830 RSC.
- S. Chowdhary, J. Moschner, D. J. Mikolajczak, M. Becker, A. F. Thünemann, C. Kästner, D. Klemczak, A.-K. Stegemann, C. Böttcher, P. Metrangolo, R. R. Netz and B. Koksch, ChemBioChem, 2020, 21, 3544–3554 CrossRef CAS PubMed.
- S. Huhmann and B. Koksch, Eur. J. Org. Chem., 2018, 2018, 3667–3679 CrossRef CAS.
- C. K. Thota, A. A. Berger, B. Harms, M. Seidel, C. Böttcher, H. von Berlepsch, C. Xie, R. Süssmuth, C. Roth and B. Koksch, Pept. Sci., 2020, 112, e24130 CAS.
- F. Agostini, L. Sinn, D. Petras, C. J. Schipp, V. Kubyshkin, A. A. Berger, P. C. Dorrestein, J. Rappsilber, N. Budisa and B. Koksch, ACS Cent. Sci., 2021, 7, 81–92 CrossRef CAS PubMed.
- J.-S. Völler, M. Dulic, U. I. M. Gerling-Driessen, H. Biava, T. Baumann, N. Budisa, I. Gruic-Sovulj and B. Koksch, ACS Cent. Sci., 2017, 3, 73–80 CrossRef PubMed.
- J. Moschner, V. Stulberg, R. Fernandes, S. Huhmann, J. Leppkes and B. Koksch, Chem. Rev., 2019, 119, 10718–10801 CrossRef CAS PubMed.
- J. Leppkes, T. Hohmann and B. Koksch, J. Fluorine Chem., 2020, 232, 109453 CrossRef CAS.
- H. Mei, T. Hiramatsu, R. Takeda, H. Moriwaki, H. Abe, J. Han and V. A. Soloshonok, Org. Process Res. Dev., 2019, 23, 629–634 CrossRef CAS.
- J. Han, R. Takeda, X. Liu, H. Konno, H. Abe, T. Hiramatsu, H. Moriwaki and V. A. Soloshonok, Molecules, 2019, 24, 4521 CrossRef CAS PubMed.
- Y. Nian, J. Wang, H. Moriwaki, V. A. Soloshonok and H. Liu, Dalton Trans., 2017, 46, 4191–4198 RSC.
- T. T. Romoff, A. B. Palmer, N. Mansour, C. J. Creighton, T. Miwa, Y. Ejima, H. Moriwaki and V. A. Soloshonok, Org. Process Res. Dev., 2017, 21, 732–739 CrossRef CAS.
- T. Hohmann, M. Dyrks, S. Chowdhary, M. Weber, D. Nguyen, J. Moschner and B. Koksch, ChemRxiv, 2022, this content is a preprint and has not been peer-reviewed.
- R. Gambaretto, L. Tonin, C. Di Bello and M. Dettin, Biopolymers, 2008, 89, 906–915 CrossRef CAS PubMed.
- G. D'Auria, M. Vacatello, L. Falcigno, L. Paduano, G. Mangiapia, L. Calvanese, R. Gambaretto, M. Dettin and L. Paolillo, J. Pept. Sci., 2009, 15, 210–219 CrossRef PubMed.
- K. Güçlü, G. Kıbrıslıoğlu, M. Özyürek and R. Apak, J. Agric. Food Chem., 2014, 62, 1839–1845 CrossRef PubMed.
- A. K. Pandey, K. M. Thomas, C. R. Forbes and N. J. Zondlo, Biochemistry, 2014, 53, 5307–5314 CrossRef CAS PubMed.
- T. J. Measey, R. Schweitzer-Stenner, V. Sa and K. Kornev, Macromolecules, 2010, 43, 7800–7806 CrossRef CAS.
- Y. Cho, L. B. Sagle, S. Iimura, Y. Zhang, J. Kherb, A. Chilkoti, J. M. Scholtz and P. S. Cremer, J. Am. Chem. Soc., 2009, 131, 15188–15193 CrossRef CAS PubMed.
- T. J. Measey and R. Schweitzer-Stenner, J. Am. Chem. Soc., 2006, 128, 13324–13325 CrossRef CAS PubMed.
- M. Salwiczek, E. K. Nyakatura, U. I. M. Gerling, S. Ye and B. Koksch, Chem. Soc. Rev., 2012, 41, 2135–2171 RSC.
- J. R. Robalo and A. Vila Verde, Phys. Chem. Chem. Phys., 2019, 21, 2029–2038 RSC.
- Y. Chen, Y. Hua, W. Zhang, C. Tang, Y. Wang, Y. Zhang and F. Qiu, Int. J. Nanomed., 2018, 13, 2477–2489 CrossRef CAS PubMed.
- A. Bertolani, L. Pirrie, L. Stefan, N. Houbenov, J. S. Haataja, L. Catalano, G. Terraneo, G. Giancane, L. Valli, R. Milani, O. Ikkala, G. Resnati and P. Metrangolo, Nat. Commun., 2015, 6, 7574 CrossRef PubMed.
- M. S. de Freitas, R. Rezaei Araghi, E. Brandenburg, J. Leiterer, F. Emmerling, K. Folmert, U. I. M. Gerling-Driessen, B. Bardiaux, C. Böttcher, K. Pagel, A. Diehl, H. v. Berlepsch, H. Oschkinat and B. Koksch, J. Struct. Biol., 2018, 203, 263–272 CrossRef CAS PubMed.
- J. Schmitt, V. Calabrese, M. A. da Silva, S. Lindhoud, V. Alfredsson, J. L. Scott and K. J. Edler, Phys. Chem. Chem. Phys., 2018, 20, 16012–16020 RSC.
- I. M. Geisler and J. P. Schneider, Adv. Funct. Mater., 2012, 22, 529–537 CrossRef CAS.
- P. J. Flory and J. Rehner Jr., J. Chem. Phys., 1943, 11, 521–526 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, 2016 Search PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. de Groot, H. Grubmüller and A. D. MacKerell, Nat. Methods, 2017, 14, 71–73 CrossRef CAS PubMed.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. Mackerell, Jr., J. Comput. Chem., 2010, 31, 671–690 CAS.
- K. Vanommeslaeghe and A. D. MacKerell, J. Chem. Inf. Model., 2012, 52, 3144–3154 CrossRef CAS PubMed.
- K. Vanommeslaeghe, E. P. Raman and A. D. MacKerell, J. Chem. Inf. Model., 2012, 52, 3155–3168 CrossRef CAS PubMed.
- A. Kumar, O. Yoluk and A. D. MacKerell, Jr., J. Comput. Chem., 2020, 41, 958–970 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- A. D. MacKerell Jr., D. Bashford, M. Bellott, R. L. Dunbrack Jr., J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo and S. Ha, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef PubMed.
- R. M. Venable, Y. Luo, K. Gawrisch, B. Roux and R. W. Pastor, J. Phys. Chem. B, 2013, 117, 10183–10192 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1, 19–25 CrossRef.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- B. Hess, J. Chem. Theory Comput., 2008, 4, 116–122 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- G. M. Torrie and J. P. Valleau, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS.
- J. S. Hub, B. L. De Groot and D. Van Der Spoel, J. Chem. Theory Comput., 2010, 6, 3713–3720 CrossRef CAS.
- D. Orthaber, A. Bergmann and O. Glatter, J. Appl. Crystallogr., 2000, 33, 218–225 CrossRef CAS.
- I. Bressler, J. Kohlbrecher and A. F. Thünemann, J. Appl. Crystallogr., 2015, 48, 1587–1598 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr01648f |
| This journal is © The Royal Society of Chemistry 2022 |