
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ liquid transmission electron microscopy reveals self-assembly-driven nucleation in radiolytic synthesis of iron oxide nanoparticles in organic media†
Nathaly
Ortiz Peña
 ab,
Dris
Ihiawakrim
a,
Sorina
Creţu
a,
Geoffrey
Cotin
a,
Céline
Kiefer
a,
Sylvie
Begin-Colin
a,
Clément
Sanchez
cd,
David
Portehault
c and
Ovidiu
Ersen
*a
ab,
Dris
Ihiawakrim
a,
Sorina
Creţu
a,
Geoffrey
Cotin
a,
Céline
Kiefer
a,
Sylvie
Begin-Colin
a,
Clément
Sanchez
cd,
David
Portehault
c and
Ovidiu
Ersen
*a
aInstitut de Physique et Chimie des Matériaux de Strasbourg (IPCMS), UMR 7504 CNRS Université de Strasbourg, BP 43 Strasbourg Cedex 2, France. E-mail: ovidiu.ersen@ipcms.unistra.fr
bUniversité Paris Cité, CNRS, Laboratoire Matériaux et Phénomènes Quantiques, 75013 Paris, France
cSorbonne Université, CNRS, Collège de France, Laboratoire de Chimie de la Matière Condensée de Paris (CMCP), 4 place Jussieu, F-75005, Paris, France
dUniversity of Strasbourg Institute for Advanced Studies (USIAS), 67083 Strasbourg, France
First published on 4th July 2022
Abstract
We have investigated the early stages of the formation of iron oxide nanoparticles from iron stearate precursors in the presence of sodium stearate in an organic solvent by in situ liquid phase transmission electron microscopy (IL-TEM). Before nucleation, we have evidenced the spontaneous formation of vesicular assemblies made of iron polycation-based precursors sandwiched between stearate layers. Nucleation of iron oxide nanoparticles occurs within the walls of the vesicles, which subsequently collapse upon the consumption of the iron precursors and the growth of the nanoparticles. We then evidenced that fine control of the electron dose, and therefore of the local concentration of reactive iron species in the vicinity of the nuclei, enables controlling crystal growth and selecting the morphology of the resulting iron oxide nanoparticles. Such a direct observation of the nucleation process templated by vesicular assemblies in a hydrophobic organic solvent sheds new light on the formation process of metal oxide nanoparticles and therefore opens ways for the synthesis of inorganic colloidal systems with tunable shape and size.
Introduction
Understanding crystallization pathways is crucial to controlling the size and shape of nanoparticles. This is particularly important in the case of the thermal decomposition method developed by Hyeon et al.1 and Sun et al.2 to design iron oxide nanoparticles with hand-picked sizes and morphologies. The synthesis consists of the thermal decomposition of a metal precursor (e.g., iron stearate, iron oleate or iron pentacarbonyl) in the presence of a surfactant (often oleic acid) in a high boiling point organic solvent.3 The heating rates, reagents and surfactants molar ratio, water content, and the nature of surfactants and reagents are efficient experimental parameters to achieve nano-objects with narrow size distribution and controlled morphology that are promising for the biomedical field,4–10 especially as magnetic resonance imaging contrast agents11–13 or for hyperthermia cancer treatment.14–17 Although there is a good understanding of how a high supersaturation regime drives monodispersity in the heating-up thermal decomposition method,3 the atom-scale and nanoscale mechanisms responsible for the particle size and shape control are mostly a black box. This is especially the case for events occurring before nucleation, the so-called pre-nucleation stage that impacts deeply the kinetics of precursor production, hence the nucleation and growth rates.In situ liquid TEM complements other in situ techniques18–22 for probing the nucleation and growth stages by providing direct visualization of solid formation in real space, with nanoscale or even atomic-scale resolution, and has been already instrumental in deciphering the processes of nucleation and growth of solids in liquids.23–35 Indeed, beyond the classical nucleation theory that relies on monomer-by-monomer aggregation in a supersaturated solution,36–38 a myriad of other mechanisms are now recognized to occur during the formation of various solids in liquids.29,39,40 These processes are coined as non-classical crystallization pathways.41,42 They also encompass events occurring during the pre-nucleation stage.43,44
The mechanisms of particle formation by the thermal decomposition method have been mostly inferred from ex situ analyses, which are not adapted to assess the pre-nucleation processes.45 More recently, in situ small angle X-ray scattering (SAXS) studies5,46 have demonstrated the importance of the pre-nucleation stage by unveiling the assembly of inorganic clusters preceding particle nucleation. The size and concentration of these clusters depend on the precursor-to-surfactant ratio and heating rate, which in turn leads to differences in the onset and rate of nucleation. However, the local structure of the pre-nucleation assemblies and their dynamics are yet to be determined. In an effort to get insights into these assemblies, we have adapted the conditions of synthesis to trigger the radiolytic decomposition of an iron precursor in a transmission electron microscope. The present work focuses on unveiling by in situ liquid TEM the self-assembly processes that occur during pre-nucleation in radiolysis-induced colloidal synthesis, under conditions close to the thermal decomposition method. We probed the reaction in situ by using the electron beam as a trigger, thus allowing the processes to be observed before and after nucleation.
Results and discussion
We focus here on the decomposition of iron(II) stearate FeSt2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 47,48 in the presence of 80% of sodium oleate and 20% of oleic acid surfactants in octadecene, conditions that lead ex situ to spinel iron oxide nanoplates at ca. 315 °C.9,47,49,50 The mixture was first stirred and heated at 120 °C for 1 h without a reflux condenser in order to homogenize the solution. Thereafter, one drop was added on the observation window of the in situ liquid TEM cell. In this setup (ESI Figure†), a thin layer of liquid is enclosed between two electron transparent observation windows. In our case, the enclosed liquid was not heated up within the holder. Therefore, the reaction was triggered by the electron beam. The temperature rise of a liquid irradiated by an electron beam depends on the stopping power of the medium and the thickness of the liquid. We calculated the increase in temperature due to the electron beam to be negligible (see the ESI†).51 Therefore, the in situ decomposition does not arise from a temperature increase, as in the usual thermal decomposition method,52 but relates to radiolytic processes. Although the in situ nucleation and growth are beam induced, computational studies49,53,54 have pointed out that the decomposition of the precursor into reactive intermediates starts at temperatures close to the homogenization conditions used in our protocol, before in situ observations. Thus, upon homogenization and beam irradiation, the reaction medium should contain complexed single iron cations, free iron cations and reduced iron species (Fig. 1). Two processes might be at the origin of the decomposition of the precursors: electron beam-induced reduction and direct cleavage of the iron stearate precursor. This protocol yields crystalline spinel iron oxide and metallic iron nanoparticles by the end of the reaction, as demonstrated by high resolution TEM (HRTEM) and selected area electron diffraction (SAED) analyses of the sample recovered after drying (Fig. S1†).
47,48 in the presence of 80% of sodium oleate and 20% of oleic acid surfactants in octadecene, conditions that lead ex situ to spinel iron oxide nanoplates at ca. 315 °C.9,47,49,50 The mixture was first stirred and heated at 120 °C for 1 h without a reflux condenser in order to homogenize the solution. Thereafter, one drop was added on the observation window of the in situ liquid TEM cell. In this setup (ESI Figure†), a thin layer of liquid is enclosed between two electron transparent observation windows. In our case, the enclosed liquid was not heated up within the holder. Therefore, the reaction was triggered by the electron beam. The temperature rise of a liquid irradiated by an electron beam depends on the stopping power of the medium and the thickness of the liquid. We calculated the increase in temperature due to the electron beam to be negligible (see the ESI†).51 Therefore, the in situ decomposition does not arise from a temperature increase, as in the usual thermal decomposition method,52 but relates to radiolytic processes. Although the in situ nucleation and growth are beam induced, computational studies49,53,54 have pointed out that the decomposition of the precursor into reactive intermediates starts at temperatures close to the homogenization conditions used in our protocol, before in situ observations. Thus, upon homogenization and beam irradiation, the reaction medium should contain complexed single iron cations, free iron cations and reduced iron species (Fig. 1). Two processes might be at the origin of the decomposition of the precursors: electron beam-induced reduction and direct cleavage of the iron stearate precursor. This protocol yields crystalline spinel iron oxide and metallic iron nanoparticles by the end of the reaction, as demonstrated by high resolution TEM (HRTEM) and selected area electron diffraction (SAED) analyses of the sample recovered after drying (Fig. S1†).
 | ||
| Fig. 1 Schematic representation of the reaction mixture upon the homogenization step and initial irradiation. The process is expected to go through the formation of complexed single iron ions, free iron ions and reduced species.44–46 | ||
Fig. 2 shows an image sequence acquired at the beginning of the in situ reaction using a liquid reaction mixture immediately after its homogenization by a heat treatment at 120 °C for 1 h. No nucleus could be detected just after starting the electron irradiation. Nucleation occurs after several minutes (ESI Movie S1†). Before nucleation started, we observed globule-like structures with diameters ranging from ca. 20 to 80 nm (Fig. 2). The presence of iron in these globules was confirmed by energy-dispersive spectroscopy (Fig. S2†). These globules do not contain yet nanoparticles. They exhibit a very dynamic behavior (circles in Fig. 2a–g) as they move around in the liquid reaction mixture, deforming and undergoing coalescence as well as Ostwald-like ripening. Their walls are darker than their cores, suggesting that the iron precursor molecules are rather localized in the walls. Different from typical beam-induced bubbles,55 these globules finally burst to form nuclei and then nanoparticles, observed as dark spots in Fig. 2 and 3. Furthermore, when octadecene without precursors was observed under the same irradiation conditions, any bubbles formed in the media did not present darker, more contrasted walls like in the case of the reaction mixture (ESI Movie S2†).
 | ||
| Fig. 2 (a–g) In situ liquid-phase TEM image sequence from a reaction mixture in octadecene. (h) Evolution of the globule diameter within the yellow circle over time. The electron dose was 90 e A−2 s−1. | ||
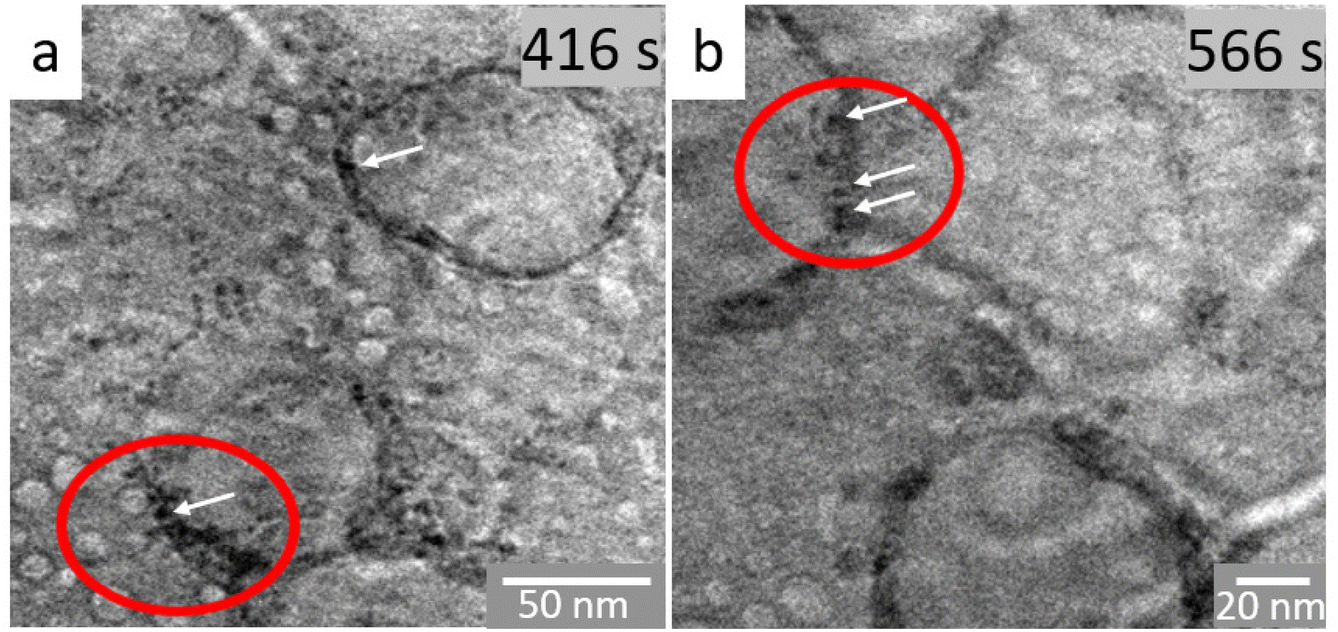 | ||
| Fig. 3 In situ TEM image sequence of the reaction mixture in octadecene (a) after 416 and (b) after 566 s of irradiation. The ovals and arrows highlight the walls of the vesicles where the nuclei form. The electron dose was 90 e A−2 s−1. | ||
In order to probe the local structure of the globule-like structures and their evolution, we have focused on their dynamic behavior over several minutes before and after nanoparticles appear, hence in the time scale of nucleation (ESI Movie S3† and corresponding snapshots in Fig. 3). As already observed in the previous image sequence, objects ranging from 10 to 20 nm in diameter coalesce or collapse and new ones are formed continuously. This behavior is typical of vesicle-like assemblies. The diameters of these globules increase up to several tens of nanometers until they collapse (Fig. 2h). A close look at the walls of the vesicles highlights the presence of some more contrasted, darker areas of ca. 1–2 nm, which underline that nucleation occurs in the walls of the vesicles. Therefore, these vesicles trap in their corona some iron oxo oligomers. Such self-assembled structures are related to lamellar iron soaps observed in aqueous media.56 Thus, we hypothesize that such globules could be attributed to self-assembled iron precursors.57 This assumption is supported by previous works on simulations,10,58 which showed that bilayers form by self-assembly of the iron precursors used herein. Iron cations are expected to be located in the center part of these bilayers.10,58 Because these hybrid self-assemblies form here in an organic medium, they yield closed spherical entities with vesicular shapes and with iron cations segregated into the walls.59 The increase in the number of nuclei in the walls over time destabilizes the vesicles, which then dissociate, releasing nuclei in the continuous medium. These nuclei further aggregate and form nanoparticles. Ex situ and cryo-electron microscopy experiments have recently confirmed the presence of iron in similar structures.60
These observations point to a two-step pre-nucleation/nucleation process61 taking place for the radiolytically induced formation of iron oxide in organic media. Mechanisms involving self-assembly prior to nucleation have been enlisted to explain the crystallization of proteins, some organic molecules62,63 and a few minerals and metallic nanoparticles.64 Although we could not solve the local structure of the inverse vesicle walls, it is instructive to note that the decomposition process involves iron complexes with long chain oleate ligands, which are prone to self-assembly into layered mesophases.65–67 Such species could then spontaneously evolve into a vesicular mesophase, thus locating iron cations within the vesicle walls. This hypothesis is in line with the observation that the iron oxide nuclei are forming only in the walls, and not in the core of the reverse vesicles. Our observations clearly indicate the presence of globule-like structures prior to the nucleation burst. Thus we provide direct evidence for the self-organization of the iron precursors in the pre-nucleation step. Furthermore, we reveal the formation and accumulation of nuclei within the restrained volume of the globule's walls. Such inverse vesicle-like structures have been intuited from in situ X-ray absorption and mass spectrometry studies that have shown evidence of poly-oxo-iron bridges in the pre-nucleation stage of a heating-up synthesis.60 The fact that we directly observe this phenomenon from radiolytic synthesis shows that at least the pre-nucleation and nucleation stages are similar, whatever the stimulus used for the decomposition of iron precursors.
To further understand the subsequent course of nanoparticle formation, we have evaluated the impact of the electron dose on the radiolytic decomposition synthesis. In fact, it has already been observed that the electron dose may provide a direct control of the formation kinetics and the morphology of Au, Ag, Pt and mixed oxides from aqueous solutions.68–74 First, we used the conventional TEM mode to provide a high electron dose, since the observation area is continuously irradiated. With a beam current of 150 pA, the electron dose is about 9400 e− nm−2 s−1. Under these conditions, the formation of nanoparticles is very fast: nucleation takes place in less than 10 s as evidenced in Fig. 4 (ESI Movie S4†). The nuclei grow rapidly until reaching a critical radius. The particle size stabilizes at this critical radius (Fig. 4b), then growth continues before the particle reaches a second plateau corresponding to the final particle size. It should be noted that with this amount of electron dose and energy input, neither the morphology nor the size distribution can be controlled.
 | ||
| Fig. 4 (a–f) In situ image sequence of the evolution of iron oxide nanoparticles in octadecene recorded under an electron dose of 9400 e− nm−2 s−1. (g) Evolution of the size of one nanoparticle (white arrow) over time. It is possible to identify the different formation regimes, from the nucleation burst until the critical size around 25 nm, after which growth continues until reaching the final size of around 60 nm. | ||
Under the second set of conditions, the electron dose was decreased to a value of 6900 e− nm−2 s−1 in TEM mode, almost a third less than under the previous conditions, in order to delay the nucleation step (Fig. 5 and ESI Movie S5†). Contrary to the observations made with the highest electron dose (Fig. 4), pre-nucleation vesicles are detected (Fig. 5a–e). The nanoparticles released by the collapsing vesicles tend to further aggregate in order to form large particles (Fig. 5f–h), possibly by oriented attachment growth already evidenced for iron oxides.29
 | ||
| Fig. 5 (a–h) In situ TEM sequence using an electron dose of 6900 e− nm−2 s−1. (i) Schematic representation of the nucleation within a vesicle. The nucleation takes place in the walls of the vesicle, then the particles released upon vesicle collapse aggregate and form larger nanoparticles. | ||
Under the third set of exposure conditions, the electron dose used was 2500 e− nm−2 s−1 in the TEM mode. We were then able to monitor the size evolution of two nanoparticles over time (Fig. 6 and ESI Movie S6†). Three regimes are observed (Fig. 6g): (i) in the first one, the size of the nanoparticles increases; (ii) the second region shows the relative stability of the particle sizes; and (iii) in the last stage, the size of small nanoparticles decreases for the benefit of larger particles. This observation could account for the Ostwald ripening mechanism caused by the change in the solubility of nanoparticles depending on their size. Due to the high surface energy of the smaller particles, they have high solubility and re-dissolve within the solution, which in turn allows the larger particles to grow further.39
 | ||
| Fig. 6 (a–f) Image sequence from a video acquired by in situ liquid-phase TEM under an electron dose of 2500 e− nm−2 s−1 for monitoring the size evolution of two neighbouring nanoparticles NP1 and NP2. (g) Evolution of NP1 (in red in the first image) particle size over time. Nuclei under the critical size re-dissolve in the solution, probably in favour of the growth of other surrounding particles. | ||
Under the last set of illumination conditions, we used the scanning TEM (STEM) mode for which the beam irradiation conditions are very different in terms of the total electron dose and the image acquisition protocol. In this case, even after two hours of electron beam irradiation, the reaction did not take place. This can be explained by the fact that the energy provided by the electron probe during the rastering of the area of interest is rapidly dissipated and therefore insufficient to exceed the threshold required for the decomposition reaction of iron stearate. Thus, to trigger the reaction, we used the spot mode, in which the beam is focused and blocked for a while (generally a few seconds) at a unique position. Hence, this mode allows concentrating the energy input at a chosen location, in the center of the observation area. Thanks to this highly concentrated beam on a small area (tens of pm),75 it was possible to trigger the reaction. In addition, this mode provides a fine control of the beam current and then the energy input and its impact on the formation of the nanoparticles and their final morphologies. When the electron dose was 900 e− nm−2 s−1 (Fig. 7), the nanoparticles grew into an isotropic, faceted polyhedral morphology (ESI Movie S7†). In contrast, at a lower dose of 230 e− nm−2 s−1, anisotropic growth was observed (ESI Movie S8†).
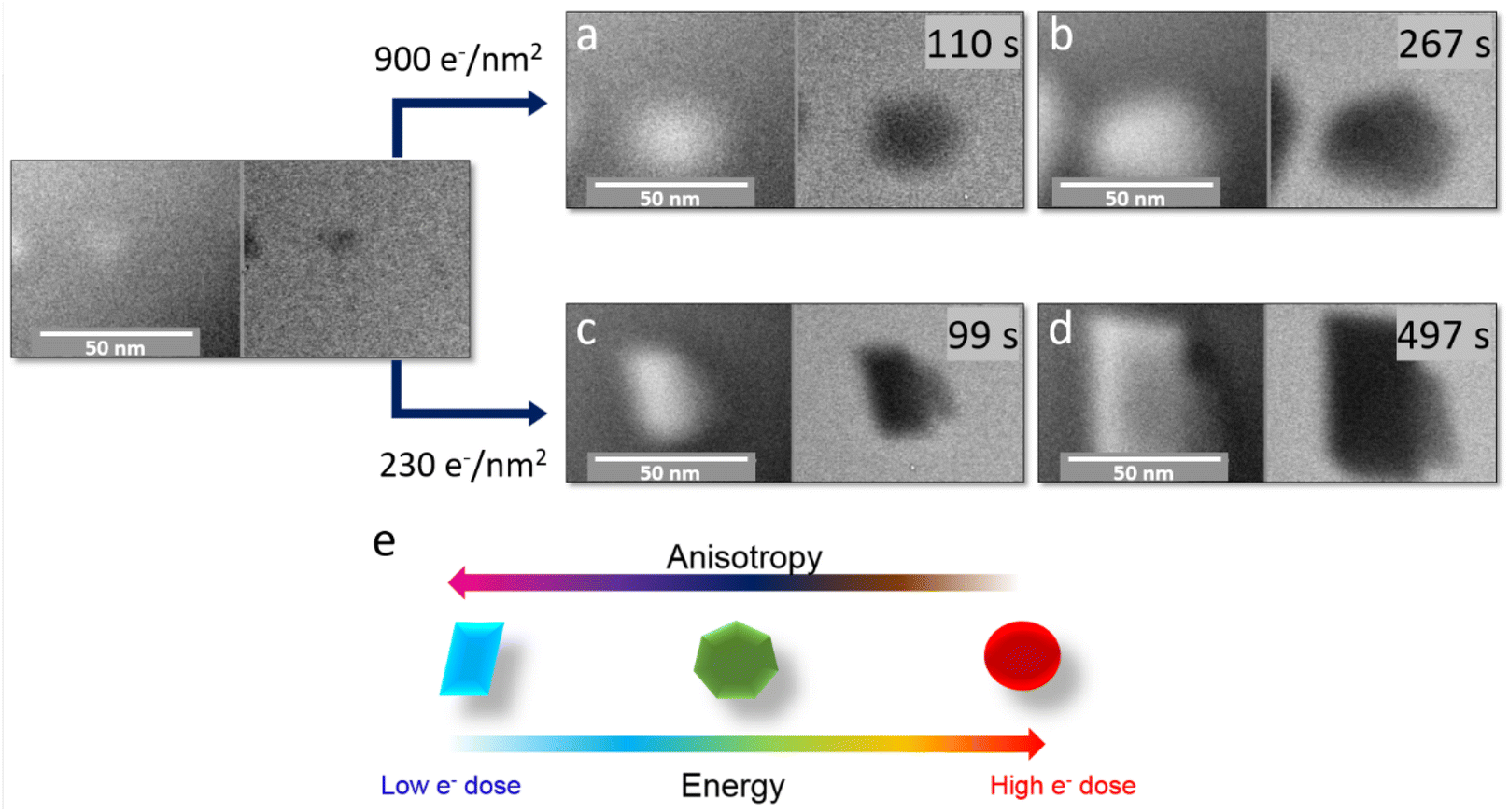 | ||
| Fig. 7 In situ scanning TEM analysis of the radiolytic growth of iron oxide particles induced by electron irradiation using an electron dose of 900 e− nm−2 s−1 (a and b) and of 230 e− nm−2 s−1 (c and d). The influence of the amount of energy provided by the electron beam on the growth of nanoparticles is schematized in (e). At a higher electron dose, the nanoparticles tend to be polyhedral and spherical; at lower electron doses, a faceting effect is unambiguously observed. | ||
Contrary to the observations in the TEM mode, the STEM mode enables triggering nanoparticle nucleation and growth only in the focused electron beam area, so that the observed nanoparticle evolution can be ascribed only to the objects within the observation area. Therefore, we can hypothesize that under the lowest dose conditions, the amount of reactive species produced is low, so that the nanoparticle growth rate is limited by the flux of reagents and the depletion of monomers near the growing nanoparticles.76 Under these conditions, the difference in the reactivity of the various crystal facets is exacerbated and yields anisotropic nano-objects. On the other hand, a large energy input (provided either by the electron beam or by the thermal energy) generates larger amounts of monomers, so that the growth rates of the different facets are levered, yielding isotropic particles. The fact that the nanoparticle morphology can be tailored as a function of energy input, without varying the nature of the capping agents, opens exciting paths in the development of nanoparticles with very specific and well identified morphologies, possibly by using radiolytic synthesis.73,74 Complementary insight has been achieved as well by correlating different characterization methods, both in situ and post mortem in similar systems, in a recent publication.60
Experimental
Materials
All chemicals were purchased from Sigma-Aldrich and used as received.Iron oleate reaction mixture preparation
The reaction mixtures used in these experiments were the ones usually employed for thermal decomposition synthesis of iron oxide Fe3−xO4 nanoparticles in organic media. First, 2.32 mmol of iron stearate was mixed with 3 mmol of the ligands, oleic acid and sodium oleate, in 15 mL of octadecene used as a solvent. The mixture was stirred and heated at 120 °C for 60 min without a reflux condenser in order to dissolve the reactants and remove the water residues.In situ liquid TEM and STEM observations and post-mortem analysis
For in situ liquid TEM analysis, a Protochips liquid cell holder (Poseidon Select 550) was used. It consists of disposable silicon chips onto which the sample is deposited. The experiments were performed in a corrected JEOL 2100F/Cs (S)TEM at 200 kV. Selected area electron diffraction (SAED) analysis was performed post in situ over the dry e-chips.Conclusions
In situ liquid-phase transmission electron microscopy allowed triggering by radiolysis and tracking in real space and in real time with nanometre resolution the formation of iron oxide nanoparticles from a thermal decomposition reaction mixture. We provided evidence that the reaction, especially the pre-nucleation step, follows a non-classical crystallization route, through the formation of elusive and dynamic inverse vesicular-like assemblies with the iron precursor confined within the walls. Nucleation occurs in the walls of the vesicles where the subsequent accumulation of the nuclei agrees with a soft-templating nucleation and growth process. After a significant increase in the size of the inorganic nuclei, the vesicles collapse and release nuclei into the continuous medium. By scrutinizing the effect of the energy input through the electron dose, we observed a strong impact of the electron dose on the morphology of the nanoparticles, which we attributed to different regimes of growth depending on the local concentration of reactive iron species near the growing particles and the stability of vesicles. This work contributes not only to the comprehension of the formation mechanism of iron oxide nanoparticles by an electron beam, but we hope it will also help in the overall understanding of the nanometer-scale processes taking place probably in thermal decomposition synthesis as well.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the French National Agency for Research (ANR) under the project InSiChem ANR-16-CE05-0011 and by the USIAS Chair of Prof. Clément Sanchez.Notes and references
- J. Park, K. An, Y. Hwang, J.-G. Park, H.-J. Noh, J.-Y. Kim, J.-H. Park, N.-M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed.
- S. Sun and H. Zeng, J. Am. Chem. Soc., 2002, 124, 8204–8205 CrossRef CAS PubMed.
- S. G. Kwon and T. Hyeon, Small, 2011, 7, 2685–2702 CrossRef CAS PubMed.
- M. V. Kovalenko, M. I. Bodnarchuk, R. T. Lechner, G. Hesser, F. Schäffler and W. Heiss, J. Am. Chem. Soc., 2007, 129, 6352–6353 CrossRef CAS PubMed.
- A. Lassenberger, T. A. Grünewald, P. D. J. Van Oostrum, H. Rennhofer, H. Amenitsch, R. Zirbs, H. C. Lichtenegger and E. Reimhult, Chem. Mater., 2017, 29, 4511–4522 CrossRef CAS PubMed.
- J. Park, K. An, Y. Hwang, J. E. G. Park, H. J. Noh, J. Y. Kim, J. H. Park, N. M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed.
- R. Hufschmid, H. Arami, R. M. Ferguson, M. Gonzales, E. Teeman, L. N. Brush, N. D. Browning and K. M. Krishnan, Nanoscale, 2015, 7, 11142–11154 RSC.
- W. Baaziz, B. P. Pichon, S. Fleutot, Y. Liu, C. Lefevre, J. M. Greneche, M. Toumi, T. Mhiri and S. Begin-Colin, J. Phys. Chem. C, 2014, 118, 3795–3810 CrossRef CAS.
- A. Walter, C. Billotey, A. Garofalo, C. Ulhaq-Bouillet, C. Lefèvre, J. Taleb, S. Laurent, L. Vander Elst, R. N. Muller, L. Lartigue, F. Gazeau, D. Felder-Flesch and S. Begin-Colin, Chem. Mater., 2014, 26, 5252–5264 CrossRef CAS.
- G. Cotin, C. Kiefer, F. Perton, M. Boero, B. Özdamar, A. Bouzid, G. Ori, C. Massobrio, D. Begin, B. Pichon, D. Mertz and S. Begin-Colin, ACS Appl. Nano Mater., 2018, 1, 4306–4316 CrossRef CAS.
- L. Lartigue, P. Hugounenq, D. Alloyeau, S. P. Clarke, M. Lévy, J. C. Bacri, R. Bazzi, D. F. Brougham, C. Wilhelm and F. Gazeau, ACS Nano, 2012, 6, 10935–10949 CrossRef CAS PubMed.
- A. Boni, A. M. Basini, L. Capolupo, C. Innocenti, M. Corti, M. Cobianchi, F. Orsini, A. Guerrini, C. Sangregorio and A. Lascialfari, RSC Adv., 2017, 7, 44104–44111 RSC.
- L. Li, W. Jiang, K. Luo, H. Song, F. Lan, Y. Wu and Z. Gu, Theranostics, 2013, 3, 595–615 CrossRef PubMed.
- A. Espinosa, R. Di Corato, J. Kolosnjaj-Tabi, P. Flaud, T. Pellegrino and C. Wilhelm, ACS Nano, 2016, 10, 2436–2446 CrossRef CAS PubMed.
- M. Bañobre-López, A. Teijeiro and J. Rivas, Rep. Pract. Oncol. Radiother., 2013, 18, 397–400 CrossRef PubMed.
- C. Martinez-Boubeta, K. Simeonidis, A. Makridis, M. Angelakeris, O. Iglesias, P. Guardia, A. Cabot, L. Yedra, S. Estradé, F. Peiró, Z. Saghi, P. A. Midgley, I. Conde-Leborán, D. Serantes and D. Baldomir, Sci. Rep., 2013, 3, 1–8 Search PubMed.
- C. Blanco-Andujar, A. Walter, G. Cotin, C. Bordeianu, D. Mertz, D. Felder-Flesch and S. Begin-Colin, Nanomedicine, 2016, 11, 1889–1910 CrossRef CAS PubMed.
- P. J. M. Smeets, A. R. Finney, W. J. E. M. Habraken, F. Nudelman, H. Friedrich, J. Laven, J. J. De Yoreo, P. M. Rodger and N. A. J. M. Sommerdijk, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E7882–E7890 CrossRef CAS PubMed.
- B. Fleury, M. A. Neouze, J. M. Guigner, N. Menguy, O. Spalla, T. Gacoin and D. Carriere, ACS Nano, 2014, 8, 2602–2608 CrossRef CAS PubMed.
- S. L. P. Wolf, L. Caballero, F. Melo and H. Cölfen, Langmuir, 2017, 33, 158–163 CrossRef CAS PubMed.
- J. Baumgartner, R. K. Ramamoorthy, A. P. Freitas, M.-A. Neouze, M. Bennet, D. Faivre and D. Carriere, Nano Lett., 2020, 20, 5001–5007 CrossRef CAS PubMed.
- J. Baumgartner, A. Dey, P. H. H. Bomans, C. Le Coadou, P. Fratzl, N. A. J. M. Sommerdijk and D. Faivre, Nat. Mater., 2013, 12, 310–314 CrossRef CAS PubMed.
- J. J. De Yoreo, Prog. Cryst. Growth Charact. Mater., 2016, 62, 69–88 CrossRef CAS.
- M. H. Nielsen, D. Li, H. Zhang, S. Aloni, T. Y.-J. Han, C. Frandsen, J. Seto, J. F. Banfield, H. Cölfen and J. J. De Yoreo, Microsc. Microanal., 2014, 20, 425–436 CrossRef CAS PubMed.
- M. M. Longuinho, V. Ramnarain, N. Ortiz Peña, D. Ihiawakrim, R. Soria-Martínez, M. Farina, O. Ersen and A. L. Rossi, CrystEngComm, 2022, 24, 2602–2614 RSC.
- W. Dachraoui, D. Keller, T. R. Henninen, O. J. Ashton and R. Erni, Nano Lett., 2021, 21, 2861–2869 CrossRef CAS PubMed.
- T. R. Henninen, D. Keller and R. Erni, ChemNanoMat, 2021, 7, 110–116 CrossRef CAS.
- G. Zhu, H. Reiner, H. Cölfen and J. J. De Yoreo, Micron, 2019, 118, 35–42 CrossRef CAS PubMed.
- J. J. De Yoreo, P. U. P. A. Gilbert, N. A. J. M. Sommerdijk, R. L. Penn, S. Whitelam, D. Joester, H. Zhang, J. D. Rimer, A. Navrotsky, J. F. Banfield, A. F. Wallace, F. M. Michel, F. C. Meldrum, H. Cölfen and P. M. Dove, Science, 2015, 349, aaa6760 CrossRef PubMed.
- H. Zheng, R. K. Smith, Y. W. Jun, C. Kisielowski, U. Dahmen and A. P. Alivisatos, Science, 2009, 324, 1309–1312 CrossRef CAS PubMed.
- M. R. Hauwiller, X. Zhang, W. I. Liang, C. H. Chiu, Q. Zhang, W. Zheng, C. Ophus, E. M. Chan, C. Czarnik, M. Pan, F. M. Ross, W. W. Wu, Y. H. Chu, M. Asta, P. W. Voorhees, A. P. Alivisatos and H. Zheng, Nano Lett., 2018, 18, 6427–6433 CrossRef CAS PubMed.
- H. G. Liao, L. Cui, S. Whitelam and H. Zheng, Science, 2012, 336, 1011–1014 CrossRef CAS PubMed.
- A. S. Kashin and V. P. Ananikov, Nat. Rev. Chem., 2019, 3, 624–637 CrossRef CAS.
- N. Bhattarai, D. L. Woodall, J. E. Boercker, J. G. Tischler and T. H. Brintlinger, Nanoscale, 2019, 11, 14573–14580 RSC.
- W. Dachraoui, T. R. Henninen, D. Keller and R. Erni, Sci. Rep., 2021, 11, 1–10 CrossRef PubMed.
- D. B. K. Chu, J. S. Owen and B. Peters, J. Phys. Chem. A, 2017, 121, 7511–7517 CrossRef CAS PubMed.
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847 CrossRef CAS.
- H. Mehranpour, M. Askari and M. Sasani Ghamsari, LaMer theory approach to study the nucleation and growth of sol-gel derived TiO2 nanoparticles, I.R. Iran, 2012, pp. 1710–1712 Search PubMed.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS PubMed.
- J. Lee, J. Yang, S. G. Kwon and T. Hyeon, Nat. Rev. Mater., 2016, 1, 16034 CrossRef CAS.
- H. Cölfen and M. Antonietti, Mesocrystals and Nonclassical Crystallization, John Wiley & Sons, Ltd, Chichester, UK, 1st edn, 2008 Search PubMed.
- Z. Ou, Z. Wang, B. Luo, E. Luijten and Q. Chen, Nat. Mater., 2020, 19, 450–455 CrossRef CAS PubMed.
- D. Gebauer, M. Kellermeier, J. D. Gale, L. Bergström and H. Cölfen, Chem. Soc. Rev., 2014, 43, 2348–2371 RSC.
- W. J. E. M. Habraken, J. Tao, L. J. Brylka, H. Friedrich, L. Bertinetti, A. S. Schenk, A. Verch, V. Dmitrovic, P. H. H. Bomans, P. M. Frederik, J. Laven, P. van der Schoot, B. Aichmayer, G. de With, J. J. DeYoreo and N. A. J. M. Sommerdijk, Nat. Commun., 2013, 4, 1507 CrossRef PubMed.
- W. Wu, Z. Wu, T. Yu, C. Jiang and W.-S. Kim, Sci. Technol. Adv. Mater., 2015, 16, 023501 CrossRef PubMed.
- Y. Tan, D. Sun, H. Yu, S. Jiao, Y. Gong, S. Yan, Z. Chen, X. Xing, G. Mo, Q. Cai and Z. Wu, J. Phys. Chem. C, 2018, 122, 16397–16405 CrossRef CAS.
- G. Cotin, F. Perton, C. Petit, S. Sall, C. Kiefer, V. Begin, B. Pichon, C. Lefevre, D. Mertz, J. M. Greneche and S. Begin-Colin, Chem. Mater., 2020, 32, 9245–9259 CrossRef CAS.
- F. Perton, Architecturation de nanoparticules hybrides pour une imagerie et/ou thérapie multimodale, Université de Strasbourg, 2019 Search PubMed.
- G. Cotin, C. Kiefer, F. Perton, M. Boero, B. Özdamar, A. Bouzid, G. Ori, C. Massobrio, D. Begin, B. Pichon, D. Mertz and S. Begin-Colin, ACS Appl. Nano Mater., 2018, 1, 4306–4316 CrossRef CAS.
- G. Cotin, C. Kiefer, F. Perton, D. Ihiawakrim, C. Blanco-Andujar, S. Moldovan, C. Lefevre, O. Ersen, B. Pichon, D. Mertz and S. Bégin-Colin, Nanomaterials, 2018, 8, 881 CrossRef PubMed.
- N. M. Schneider, in Liquid Cell Electron Microscopy, Cambridge University Press, 2017, pp. 140–163 Search PubMed.
- G. K. Soon, Y. Piao, J. Park, S. Angappane, Y. Jo, N. M. Hwang, J. G. Park and T. Hyeon, J. Am. Chem. Soc., 2007, 129, 12571–12584 CrossRef PubMed.
- S. Palchoudhury, W. An, Y. Xu, Y. Qin, Z. Zhang, N. Chopra, R. A. Holler, C. H. Turner and Y. Bao, Nano Lett., 2011, 11, 1141–1146 CrossRef CAS PubMed.
- A. López-Cruz and G. E. López, Mol. Phys., 2009, 107, 1799–1804 CrossRef.
- J. M. Grogan, N. M. Schneider, F. M. Ross and H. H. Bau, Nano Lett., 2014, 14, 359–364 CrossRef CAS PubMed.
- R. Corkery, Artificial biomineralisation and metallic soaps, Australian National University, 1998 Search PubMed.
- H. Kunieda, K. Nakamura, M. R. Infante and C. Solans, Adv. Mater., 1992, 4, 291–293 CrossRef CAS.
- B. Özdamar, A. Bouzid, G. Ori, C. Massobrio and M. Boero, J. Chem. Theory Comput., 2018, 14, 225–235 CrossRef PubMed.
- F. Evans and H. Wennerström, The colloidal domain where physics, chemistry, biology and technology meet, Wiley-VCH, USA, 2nd edn, 1999 Search PubMed.
- G. Cotin, B. Heinrich, F. Perton, C. Kiefer, G. Francius, D. Mertz, B. Freis, B. Pichon, J. Strub, S. Cianférani, N. Ortiz Peña, D. Ihiawakrim, D. Portehault, O. Ersen, A. Khammari, M. Picher, F. Banhart, C. Sanchez and S. Begin-Colin, Small, 2022, 18, 2200414 CrossRef CAS PubMed.
- P. G. Vekilov, Cryst. Growth Des., 2010, 10, 5007–5019 CrossRef CAS PubMed.
- P. G. Vekilov, Cryst. Growth Des., 2004, 4, 671–685 CrossRef CAS.
- A. Sauter, F. Roosen-Runge, F. Zhang, G. Lotze, R. M. J. Jacobs and F. Schreiber, J. Am. Chem. Soc., 2015, 137, 1485–1491 CrossRef CAS PubMed.
- Y. Mikhlin, A. Karacharov, M. Likhatski, T. Podlipskaya, Y. Zubavichus, A. Veligzhanin and V. Zaikovski, J. Colloid Interface Sci., 2011, 362, 330–336 CrossRef CAS PubMed.
- C. V. Nikiforidis, E. P. Gilbert and E. Scholten, RSC Adv., 2015, 5, 47466–47475 RSC.
- M. Delample, F. Jérôme, J. Barrault and J. P. Douliez, Green Chem., 2011, 13, 64–68 RSC.
- C. Larquet, D. Carriere, A. M. Nguyen, T. K. C. Le, X. Frogneux-Plé, I. Génois, P. Le Griel, A. Gauzzi, C. Sanchez and S. Carenco, Chem. Mater., 2020, 32, 1131–1139 CrossRef CAS.
- Y. Wu, X. Chen, C. Li, J. Fang and H. Liu, New J. Chem., 2019, 43, 12548–12554 RSC.
- M. Ge, M. Lu, Y. Chu and H. Xin, Sci. Rep., 2017, 7, 16420 CrossRef PubMed.
- D. Alloyeau, W. Dachraoui, Y. Javed, H. Belkahla, G. Wang, H. Lecoq, S. Ammar, O. Ersen, A. Wisnet, F. Gazeau and C. Ricolleau, Nano Lett., 2015, 15, 2574–2581 CrossRef CAS PubMed.
- V. Beermann, M. E. Holtz, E. Padgett, J. F. de Araujo, D. A. Muller and P. Strasser, Energy Environ. Sci., 2019, 12, 2476–2485 RSC.
- T. J. Woehl, J. E. Evans, I. Arslan, W. D. Ristenpart and N. D. Browning, ACS Nano, 2012, 6, 8599–8610 CrossRef CAS PubMed.
- M. Mirdamadi-Esfahani, M. Mostafavi, B. Keita, L. Nadjo, P. Kooyman and H. Remita, Gold Bull., 2010, 43, 49–56 CrossRef CAS.
- W. Abidi, P. R. Selvakannan, Y. Guillet, I. Lampre, P. Beaunier, B. Pansu, B. Palpant and H. Remita, J. Phys. Chem. C, 2010, 114, 14794–14803 CrossRef CAS.
- H. Sawada, N. Shimura, F. Hosokawa, N. Shibata and Y. Ikuhara, Microscopy, 2015, 64, 213–217 CrossRef CAS PubMed.
- R. Kröger and A. Verch, Minerals, 2018, 8, 21 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr01511k |
| This journal is © The Royal Society of Chemistry 2022 |