
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePackaging of DNA origami in viral capsids: towards synthetic viruses†
Stanislav
Kler
*a,
Ran
Zalk
*b,
Alexander
Upcher
b and
Idit
Kopatz
 *a
*a
aGeza Ad Ltd, Givatayim 53251, Israel. E-mail: slavakler1@gmail.com; idit.kopatz@geza-ad.com
bIlse Katz Institute for Nanoscale Science & Technology, Ben-Gurion University of the Negev, Beer Sheva, 84105, Israel. E-mail: ranz@post.bgu.ac.il
First published on 21st July 2022
Abstract
We report a new type of nanoparticle, consisting of a nucleic acid core (>7500 nt) folded into a 35 nm DNA origami sphere, encapsulated by a capsid composed of all three SV40 virus capsid proteins. Compared to the prototype reported previously, whose capsid consists of VP1 only, the new nanoparticle closely adopts the unique intracellular pathway of the native SV40, suggesting that the proteins of the synthetic capsid retain their native viral functionality. Some of the challenges in the design of such near-future composite drugs destined for gene delivery are discussed.
The concept of an artificial virus – a synthetic entity that mimics key properties of a virus and which is destined to replace viral gene delivery vectors is not new – synthetic vectors have been under development since the early days of gene delivery.1,2
Synthetic approaches offer several important advantages over viral approaches including potentially improved safety profiles, lower cost and ease of production.3 Additionally, they provide significant flexibility with respect to the size, sequence and type of nucleic acid. Unlike viral vectors which are limited to only naturally occurring nucleic acids, non-viral systems can deliver a synthetic version of any nucleic acid, modified to exhibit superior activity.4,5 However, the scope of applications of these methods is typically restricted. While synthetic formulations of lipid and nucleic acid, have shown tremendous progress for delivery of mRNA and siRNA, most notably in vaccines, adapting these assemblies to a wider range of medical applications still requires significant improvements. Over the last three decades, extensive efforts, based mainly on macromolecular conjugate and lipid chemistry approaches, have been dedicated to improving the pharmacokinetics of these systems, as well as the efficiency of the cellular trafficking of the delivered nucleic acid. However, intracellular transport (1% escape from endosome), poor tissue/cell targeting and a marked tendency to be sequestered by blood-filtering organs like the liver and spleen, remain major challenges for systemic delivery by synthetic platforms.3,5–8
In contrast to synthetic systems, the viral capsid has evolved as a natural gene delivery agent, inherently capable of all the multiple functionalities that are required for efficient intracellular delivery of the viral nucleic acids.
In vitro encapsulation of nucleic acids by a functional viral capsid has been a long-time scientific ambition.9–18 The field has mostly been focused on encapsulation of flexible, unfolded nucleic acid cargos. However, packing of the relatively long, negatively charged nucleic acid chains into the constrained capsid-size cavity requires bending of the nucleic acid chain,19 which is energetically unfavorable.9,10 Consequently, double stranded DNA (dsDNA), a stiff polymer known for its long persistence length, is a poor substrate for packaging.9,10,12,14,15 In contrast, assembly of viral capsids around solid supports is typically simpler because these substrates are a priori compact. Previous studies on encapsulation of gold nanoparticles,20–22 have shown that a cargo has to withstand strict size and shape constraints to fit into the capsid cavity and allow for efficient encapsulation. The subject of our interest is the capsid of Simian Vacuolating virus 40 (SV40), whose native cargo, the SV40 genome, is to some extent characterized. According to early electron microscopy (EM) studies,23,24 SV40 genome, a nucleoprotein complex composed of a SV40 mini-chromosome and histones, can be isolated as a spherical compact structure with a diameter of ∼30–40 nm at the late stages of infection, suggesting that the genome is pre-folded prior to assembly of the capsid.24 Our approach to encapsulation of nucleic acids by a viral capsid using SV40 capsid as a model system is based on these considerations (Fig. 1).
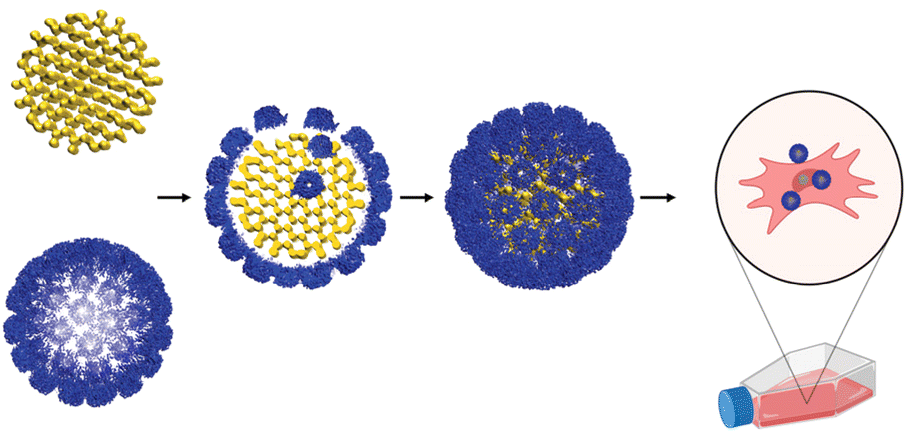 | ||
| Fig. 1 A schematic representation of a gene delivery system destined to combine the virtues of viral and non-viral vectors. Nucleic acid (shown in yellow), pre-folded by DNA origami technology to precisely fit into the capsid's cavity, is encapsulated by the viral capsid capsomers (blue) to form a virus-like nanoparticle. Individual capsomers produced by disassembly of recombinant empty capsids tend to spontaneously reassemble around negatively charged substrates.9–22 | ||
Previously, we have reported on the successful encapsulation of a large DNA molecule, packed as 35 nm diameter spherical DNA origami by VP1, the major SV40 capsid protein.25 The main assembly product was a 50 nm DNA origami-containing particle with an outer shell, composed of VP1 pentamers, that formed a regular SV40 lattice of T = 7 d icosahedral symmetry, as shown by cryo-electron microscopy (cryo-EM) analysis. Assembly of this new type of a nanoparticle was highly reproducible and efficient.
Here, we incorporated all three SV40 capsid proteins, VP1, VP2, and VP3, each carries out a distinct non-redundant function during cell entry (reviewed in ref. 26), towards the goal of assembling a synthetic nanoparticle with the functionality of an authentic virus.
SV40 is a small, nonenveloped virus belonging to the polyomavirus family. The capsid of SV40 is composed of 72 pentamers of VP1, the major capsid protein, that form a 50 nm diameter T = 7 d icosahedral outer shell around the 5200 bp dsDNA genome. Each pentamer harbors one of the hydrophobic minor capsid proteins, either VP2 or VP3, attached to its inward facing cavity.27–29 VP2 and VP3 differ only in their N-terminus where VP2 contains 118 additional amino acids.
To construct DNA origami-containing SV40-like nanoparticles with a shell composed of VP1, VP2, and VP3, we used our in vitro assembly system, previously defined for VP1.10,25 Empty viral-like particles (VLPs) produced from insect cells overexpressing either VP1 alone (VP1 VLPs) or VP1 with VP2 (VP1/2 VLPs) or VP1 with VP3 (VP1/3 VLPs) were purified and analyzed for their protein content by Coomassie blue staining (Fig. S1(a)†). Densitometric analysis of the Coomassie-stained gel showed that the molar ratios of VP1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :VP2 (in VP1/2 VLPs) and VP1:VP3 in (VP1/3 VLPs) are approximately 5:1, suggesting that most of the VP1 pentamers in the VP1/2 and VP1/3 VLPs are associated with one molecule of VP2 or VP3, respectively.
:VP2 (in VP1/2 VLPs) and VP1:VP3 in (VP1/3 VLPs) are approximately 5:1, suggesting that most of the VP1 pentamers in the VP1/2 and VP1/3 VLPs are associated with one molecule of VP2 or VP3, respectively.
VP1, VP1/2 and VP1/3 pentamers were produced, each from its respective empty VLPs, using our disassembly protocol.10 VP1 pentamers or a mixture of VP1/2 and VP1/3 pentamers at the ratio of 1:10 (representing VP1/2:VP1/3 ratio in the native SV40 virion and referenced below as VP1/2/3 pentamer mixture) were assembled with 35 nm spherical DNA origami at increasing pentamer/origami ratios. DNA origami core of 35 nm diameter was selected as it was found optimal for assembly of SV40-like particles in our previous study.25 Assembly products were analyzed by electrophoretic mobility shift assay (EMSA) on agarose gel to monitor migration (Fig. S1(b)†). In the presence of pentamers, both VP1 alone as well as VP1/2/3 pentamer mixture, the fast-migrating free DNA origami band essentially disappeared, already at the lower pentamer/origami ratio of 75:1, and instead appeared a distinct slower-migrating band, suggesting the formation of a stable, higher molecular weight SV40 pentamers/DNA origami complex. Further increase in pentamer/origami ratio did not result in further change in migration, suggesting that the final assembly product indeed comprises of a single capsid composed of 72 pentamers per one origami structure. In contrast, when using unfolded plasmid DNA as a substrate for assembly, multiplets of capsids are often formed on a single nucleic acid, resulting in electrophoretic mobility shift that does not approach saturation at increasing pentamer/DNA ratios (ref. 10 and Kler, unpublished data).
Analysis of VP1/2/3/DNA origami assembly products, formed at a molar ratio of 150:1 pentamers per DNA origami, by negative staining TEM (Fig. S1(c)†), revealed SV40-like particles. The large majority of the particles are in size and morphology similar to that of native SV40 virions and to the assembly product formed by VP1 alone.25
VP1/2/3/DNA origami assembly products were then fractionated by sedimentation velocity ultracentrifugation through 2.5 ml 20–60% sucrose gradient. 200 μl fractions were collected and analyzed for the presence of VP1, VP2 and VP3 by immunoblotting and for the presence of DNA by agarose gel (Fig. S1(d)†). All four components co-sedimented as a single particle, peaking at fractions 3 to 5.
The 3D map of the VP1/2/3/DNA origami particle was reconstructed by cryo-EM single particle analysis. Particles, formed at a molar ratio of 150:1 pentamers per DNA origami, were auto-picked (RELION3.1) from 2446 micrographs (Fig. 2). Particles were subjected to 2D classification that demonstrated the presence of both empty and DNA origami-filled capsid classes, similarly to the assembly products of DNA origami and VP1 alone.25 The empty capsid class was refined to a resolution of ∼12 Å (consisting of 9682 particles, with imposed icosahedral symmetry, Fig. 2(B)). The lower resolution of the structure, compared to that of the empty particle produced by VP1 alone,25 despite the larger number of the particles in the dataset and the smaller pixel size, is likely due to structural heterogenicity introduced by the presence of VP2 and VP3. The internal capsid proteins are incorporated asymmetrically into the capsid in a relative arrangement and molar ratio that may vary from particle to particle, resulting in deviation from the idealized icosahedral symmetry.30,31 Additionally, VP2 and VP3 are believed to be intrinsically highly flexible,29 further affecting resolution.30,31 The T = 7 icosahedral organization of SV40 lattice was readily discernible in the VP1/2/3 empty capsid, however, with some kind of distortion (Fig. 2(B), please see below).
 | ||
| Fig. 2 VP1, VP2 and VP3 containing SV40-like particles assembled on 35 nm DNA origami. (A) Representative micrograp of SV40-like particles composed of VP1, VP2, and VP3. Inset is showing origami-filled (left) and empty (right) particles. (B) 3D reconstruction of the empty particles with imposed icosahedral symmetry at ∼12 Å, showing a cut-through the capsid surface. (C) 3D reconstruction of the origami-filled particles at 18 Å with no imposed symmetry, showing a cut through the capsid surface (DNA origami in yellow). (D) 2D class averages showing the presence of both empty and DNA origami-filled particles (scale bar 100 nm). | ||
The 2D classes of the SV40-like capsid containing the origami in its cavity were further reconstructed into a 3D map from 11781 particles, with no imposed symmetry due to the asymmetric nature of the DNA origami, (Fig. 2(C)). The structure, resolved at 18 Å, revealed the DNA origami in its details as originally designed and produced,25 including the parallel helices and distinct honeycomb lattice organization, the structural features previously observed in our earlier study.25 The DNA origami appeared encapsulated by a spherical shell, lacking icosahedral features (Fig. 2(C) and ESI Movie,† please see discussion below).
The ratio between empty and origami-filled particles (observed in 2D classification), 17770 and 27020 respectively, suggests ∼60% encapsulation efficiency.
The unique intracellular trafficking pathway of SV40, part of which takes place in the ER, an unusual destination in the entry pathway of animal viruses, has been extensively studied (reviewed in ref. 26). To enter cells, SV40 binds to its ganglioside GM1 host cell receptor,32–34 internalized mainly via caveolae-dependent endocytosis and possibly through other entry mechanisms,35–38 transported to endosomal compartments including the early and late endosomes39,40 from where it is sorted to the ER.39–41
In the ER SV40 undergoes initial capsid disassembly.42,43 As a consequence, the internal hydrophobic proteins VP2 and VP3 become exposed44 and are thought to promote insertion of the partially disassembled virion into the ER membrane.45,46
Subsequent steps lead to translocation across ER membrane in order to deliver the genome into the nucleus to cause infection. SV40 induces the formation of membranal foci, a structure where several selected ER membrane proteins including BAP3145,46 and other critical host ER membrane proteins,47 but not ER membrane proteins dispensable for SV40 infection,45 are reorganized. A large body of evidence suggests that that these virus-induced foci serve as SV40's cytosolic gateway, from where it escapes into the cytosol,48–52 to subsequently reach the nucleus by classical nucleocytoplasmic transport.53
A critical question with respect to our nanoparticle is how closely it mimics the interaction of SV40 with host cells. To function as a gene delivery agent, it should retain the efficient trafficking pathway of the native virus. To examine how closely our nanoparticle mimics SV40 cellular interactions, we constructed fluorescent SV40-encapsulated DNA origami particles by assembling VP/1/2/3 capsids around Cy5-labeled 35 nm DNA origami. CV-1 cells were transfected with the fluorescent particles, at a ratio of 15000 particles per cell, and observed by confocal microscopy. Several observations suggest that the VP1/2/3/DNA origami particle uses the same cellular pathway as the native SV40. When viewed in live cells, 6 hours after transfection, VP1/2/3/DNA origami could be seen as numerous, intracellular, bright foci, of variable sizes, dispersed throughout the cytosol, suggesting that VP1/2/3/DNA origami particles effectively enter CV1-cells, the natural host cells of SV40 (Fig. 3(a)). Larger foci may represent organelles that contain multiple particles, as often observed for SV40 infection when a large number of virions is added to cells.38,41
 | ||
| Fig. 3 Fluorescence analysis of CV-1 cells transfected with Cy5 labeled VP1/2/3/DNA origami particle. (a) Live CV-1 cells transfected for 6 h with VP1/2/3/DNA origami particles (red), at a ratio of 15000 particles per cell, and counterstained with Hoechst nuclear dye (blue), showing effective internalization. A merge of 4 internal cellular sections (scale bar 14 μm). (b) BAP31 staining (green) in untreated CV-1 cells (scale bar 14 μm). (c) Top: CV-1 cells transfected for 18 h with VP1/2/3/DNA origami particle (15000 particles per cell) and stained with BAP31 antibodies (green) showing induction of BAP31 foci. A merge of 4 internal cellular sections (scale bar 14 μm). Bottom: an enlargement of the square indicated in the merged fluorescence image in the upper row. BAP31 is seen at few spots, mostly perinuclear, in which VP1/2/3/DNA origami particle is strongly colocalized. | ||
To visualize the effects of VP1/2/3/DNA origami particle on BAP31 distribution, CV-1 cells were either left untreated or transfected with the fluorescent particle for 18 hours, and then fixed and stained with BAP31 antibodies. After exposure of cells to VP1/2/3/DNA origami particles, the BAP31 staining dramatically changed with a fraction of the protein now reorganized in discrete foci (compare Fig. 3(b) and (c)). BAP31 foci were seen in relatively few perinuclear spots (Fig. 3(c) bottom, and Fig. 4(b) below), as often observed for BAP31 foci induced by SV40 infection.45,47,48,54 Notably, nearly every detectable BAP31 foci strongly co-localized with the VP1/2/3/DNA origami particle (Fig. 3(c)). In contrast, VP1/2/3/DNA origami foci only partially colocalized with BAP31 foci (Fig. 3(c) bottom, arrowheads in the merged image), consistent with the previous observations that during SV40 infection, not all the bulk of SV40 that enters cells is contained in the SV40-induced BAP31 foci but only the viral particles in which disassembly has initiated and the internal proteins are exposed.47,51
 | ||
| Fig. 4 Fluorescence analysis of CV-1 cells transfected with Cy5 labeled VP1/DNA origami particle. (a) Live CV-1 cells transfected for 6 h with VP1/DNA origami particles (red), at a ratio of 15000 particles per cell, and counterstained with Hoechst nuclear dye (blue), showing effective internalization, suggesting that VP2/3 is dispensable for the initial stages of cell entry. A merge of 4 internal cellular sections (scale bar 14 μm). (b) CV-1 cells transfected for 18 h with VP1/2/3/DNA origami particle (15000 particles per cell) and stained with BAP31 antibodies (green), showing Bap31 as few perinuclear spots, each strongly colocalizes with VP1/2/3/DNA origami. Images are a merge of 3 internal cellular sections (scale bar 14 μm). (c) CV-1 cells transfected for 18 h with VP1/DNA origami particles (15000 particles per cell) and stained with BAP31 antibodies showing small and disperse BAP31 foci which are not associated with VP1/DNA origami, suggesting that VP2/3 mediates the interaction with BAP31 foci and may also control their size and location. Images are a merge of 2 internal sections (scale bar 14 μm). | ||
In previous studies, the abundant ER membrane protein, calnexin, was found to be dispensable for SV40 infection and is not recruited to BAP31 foci.45 In agreement, we observed no major reorganization in calnexin staining patten following transfection with VP1/2/3/DNA origami (compare Fig. S2(a) and S2(b)),† suggesting indeed that the dramatic effects of VP1/2/3/DNA origami on ER membrane reorganization are specific to BAP31. Some VP1/2/3/DNA origami foci stained faintly for calnexin (Fig. S2(b)† bottom, arrowheads in the merged image).
The interactions of the SV40 with BAP31 foci are thought to be mediated by VP2/3.45,47 To examine the effects of depletion of VP2 and VP3 from the capsid on formation of BAP31 foci, we assembled VP1 only capsids around Cy5-labeled 35 nm DNA origami. CV-1 cells were transfected with the fluorescent VP1/DNA origami particles, at a ratio of 15000 particles per cell, and observed in live cells by confocal microscopy. When cells were viewed 6 hours after transfection, we observed no major differences in intracellular amount, location or appearance of VP1/DNA origami particle compared to the particle that harbors all three proteins (Fig. 4(a)). VP1/DNA origami particle enters CV-1 cells as effectively as VP1/2/3 DNA origami particle, suggesting that VP2/3 is dispensable for the early stages of cell entry. In agreement, SV40 mutant lacking VP3 enters cells and is also properly transported to the ER,47,52 suggesting no involvement of VP3 up to ER arrival.
However, when CV-1 cells, transfected with a fluorescent particle devoid of VP2 and VP3, were fixed and immunostained for BAP31 18 hours after transfection, we observed dramatically different effects on BAP31 distribution than observed for the VP1/2/3 particle. Instead of the large perinuclear foci that strongly colocalizes with VP1/2/3/DNA origami particle (Fig. 4(b)), BAP31 foci now lost their perinuclear location and appeared more scattered and smaller (Fig. 4(c)). Strikingly, in the absence of VP2 and VP3, VP1/DNA origami particles were absolutely absent from these foci.
Small and scattered BAP31 foci can be observed in SV40-infected CV-1 cells, in which the expression of the motor protein kinesin-1 has been downregulated,54 implicating kinesin-1 in maturation of small BAP31 foci into a large perinuclear one.54 Our studies with the synthetic particle may have therefore enlightened an additional and unexpected role for VP2/3 in mediating the interactions of SV40 with kinesin-1, in addition to affirming their role in engaging BAP31.45,47
Conclusions
Based on the present work, together with our previous work,25 we propose an entirely new type of synthetic gene delivery vectors, enabled by our newly developed technology for in vitro packaging of nucleic acid in an authentic viral capsid. The purpose of the capsid is to confer onto the synthetic vector extracellular and intracellular delivery mechanisms, that approach the efficiency of viral delivery, thus overcome many of the limitations of the current synthetic vectors, essentially assemblies of nucleic acid/lipids/polymers that are used for gene delivery today.3,5,6,8 We reported previously on the assembly of a prototype, the core of which consists of a large nucleic acid (>7500 nt) folded into a 35 nm DNA origami sphere, encapsulated by a capsid consisting of VP1, the major capsid protein of SV40.25 Here, we made a step further in this direction by incorporating all three SV40 capsid proteins into the particle's capsid and assessing whether the particle preserves some of the functional properties of native SV40.Our biochemical evidence shows that all three capsid components, are successfully incorporated into the particle's capsid (Fig. S1†). However, the structural analysis of VP1/2/3 capsids by cryo-EM/single particle analysis was not as straightforward as that of a capsid composed of VP1 alone.25 Empty VP1/2/3 capsids are resolved at significantly lower resolution than that of empty VP1 capsids (Fig. 2(B) and ref. 25), which to some extent is expected due to the heterogeneity introduced by VP2 and VP3, as discussed above. Adding to this heterogeneity is the rather unexpected lateral distortion in the pentamers’ position, which may be indicative of undesirable interactions within the capsid. Such interactions may stem, for example, from the excess of post-translation modification of VP2 and VP3 produced in insect cells, compared to VP2 and VP3 from mammalian cells (Stanislav Kler, PhD thesis) or from sub-optimal ratio of VP2/VP3 in the capsid.
The lateral distortion in pentamers’ coordinates is even more excessive in origami-filled capsids (Fig. 2(C)), an effect that may result from a combination of several factors: first, the two lattices, the DNA origami and the capsid may not be oriented in a fixed position with respect to each other. Our previous study25 of the same DNA origami, encapsulated in a capsid composed of VP1 only, showed that the DNA origami had only limited amount of freedom in its position with respect to the capsid. It is possible that the addition of VP2 and VP3 to the capsid allowed multiple orientations of the DNA origami with respect to the capsid symmetry which may result in the “blurry” capsid observed in the 2D class averages. In addition, interactions within the capsid, including interactions of the capsid proteins with each other and/or their interactions with the DNA origami, a non-native substrate, may further accentuate these blurring effects. Importantly, as VP2/3 directly interact with the viral genome in the native capsid,55,56 they are likely to be also involved in the interactions with the DNA origami in the VP1/2/3/DNA origami particle. In this regard, we would like to acknowledge the cryo-EM study of the native BK polyomavirus, a SV40 family member, in which the icosahedral structure of a native capsid incorporating all three capsid proteins was fully resolved.56
Nonetheless, we have made several critical observations that support the notion that VP1/2/3/DNA origami particle closely adopts SV40 intracellular pathway: 1. VP1/2/3/DNA origami particle induces reorganization of the ER membrane protein BAP31 into discrete foci (Fig. 3(c)). 2. VP1/2/3/DNA origami particle strongly localizes in the foci, similar to the native virus (Fig. 3(c)). 3. Induction of foci seems to be selective for ER membrane proteins that promote SV40 infection. The ER membrane protein calnexin which is dispensable for SV40 infection45 is not reorganized in these foci (Fig. S2(b)†). 4. VP1/DNA origami particle, devoid of VP2 and VP3, effectively enters CV1 cells (Fig. 4(a)), but is not recruited into the membranal BAP31 foci (Fig. 4(c)), consistent with the role proposed for VP2/3 in mediating virus insertion into the ER membrane44,46 and the interactions with BAP31.45
This correlation between the entry pathway of our nanoparticle and the native SV40 virus positions our particle as a valuable tool for studying SV40 and may also provide a first proof of concept for feasibility of using synthetic viruses as a general approach for gene delivery. Further studies towards this goal will aim at optimizing the structure of the capsid and modulating the properties of the DNA origami core of our nanoparticle to unfold inside cells to allow access to cell machineries.
Conflicts of interest
IK and SK are employees of Geza Ad Ltd, a biotech company developing gene delivery systems.Acknowledgements
The authors thank the teams of of the Protein Expression and the Bio-imaging Facilities of the Hebrew University of Jerusalem, in particular Yael Keren, Dr. Tsafi Danieli and Dr. Naomi Melamed-Book. The authors thank Dr. Jean-Philippe Sobczak and Dr. Tamara Aigner of tilibit nanosystems GmbH for their support. The authors would like to express their special appreciation for Prof. Ariella Oppenheim and the late prof. Amos Oppenheim for inspiring the idea of synthetic viruses.Notes and references
- G. Zuber, E. Dauty, M. Nothisen, P. Belguise and J. P. Behr, Towards synthetic viruses, Adv. Drug Delivery Rev., 2001, 52(3), 245–253 CrossRef CAS PubMed.
- P. Belguise-Valladier and J. P. Behr, Nonviral gene delivery: Towards artificial viruses, Cytotechnology, 2001, 35(3), 197–201 CrossRef CAS PubMed.
- M. L. Guevara, F. Persano and S. Persano, Advances in Lipid Nanoparticles for mRNA-Based Cancer Immunotherapy, Front. Chem., 2020, 8, 589959 CrossRef CAS PubMed.
- J.-P. Behr, Synthetic Gene Transfer Vectors II: Back to the Future, Acc. Chem. Res., 2012, 45(7), 980–984 CrossRef CAS PubMed.
- M. Ericsona, K. Ricea and G. Zuberb, Macromolecular Conjugates for Non-Viral Nucleic Acid Delivery, in Advanced Textbook On Gene Transfer, Gene Therapy And Genetic Pharmacology: Principles, Delivery And Pharmacological And Biomedical Applications Of Nucleotide-based Therapies, 2019, p. 223 Search PubMed.
- V. Escriou, N. Mignet and A. Miller, Auto-Associative Lipid-Based Systems for Non-Viral Nucleic Acid Delivery, in Advanced Textbook on Gene Transfer, Gene Therapy and Genetic Pharmacology, WORLD SCIENTIFIC, EUROPE, 2018, pp. 237–270 Search PubMed.
- I. Kopatz, J.-S. Remy and J.-P. Behr, A model for non-viral gene delivery: through syndecan adhesion molecules and powered by actin, J. Gene Med., 2004, 6(7), 769–776 CrossRef CAS PubMed.
- X. Hou, T. Zaks, R. Langer and Y. Dong, Lipid nanoparticles for mRNA delivery, Nat. Rev. Mater., 2021, 6(12), 1078–1094 CrossRef CAS PubMed.
- A. Zlotnick, J. Z. Porterfield and J. C. Wang, To build a virus on a nucleic acid substrate, Biophys. J., 2013, 104(7), 1595–1604 CrossRef CAS PubMed.
- S. Kler, J. C. Wang, M. Dhason, A. Oppenheim and A. Zlotnick, Scaffold properties are a key determinant of the size and shape of self-assembled virus-derived particles, ACS Chem. Biol., 2013, 8(12), 2753–2761 CrossRef CAS PubMed.
- S. Mukherjee, S. Kler, A. Oppenheim and A. Zlotnick, Uncatalyzed assembly of spherical particles from SV40 VP1 pentamers and linear dsDNA incorporates both low and high cooperativity elements, Virology, 2010, 397(1), 199–204 CrossRef CAS PubMed.
- M. S. Dhason, J. C. Wang, M. F. Hagan and A. Zlotnick, Differential assembly of Hepatitis B Virus core protein on single- and double-stranded nucleic acid suggest the dsDNA-filled core is spring-loaded, Virology, 2012, 430(1), 20–29 CrossRef CAS PubMed.
- C. Li, A. R. Kneller, S. C. Jacobson and A. Zlotnick, Single Particle Observation of SV40 VP1 Polyanion-Induced Assembly Shows That Substrate Size and Structure Modulate Capsid Geometry, ACS Chem. Biol., 2017, 12(5), 1327–1334 CrossRef CAS PubMed.
- G. Saper, S. Kler, R. Asor, A. Oppenheim, U. Raviv and D. Harries, Effect of capsid confinement on the chromatin organization of the SV40 minichromosome, Nucleic Acids Res., 2013, 41(3), 1569–1580 CrossRef CAS PubMed.
- S. Mukherjee, C. M. Pfeifer, J. M. Johnson, J. Liu and A. Zlotnick, Redirecting the coat protein of a spherical virus to assemble into tubular nanostructures, J. Am. Chem. Soc., 2006, 128(8), 2538–2539 CrossRef CAS PubMed.
- S. Mukherjee, M. Abd-El-Latif, M. Bronstein, O. Ben-nun-Shaul, S. Kler and A. Oppenheim, High cooperativity of the SV40 major capsid protein VP1 in virus assembly, PLoS One, 2007, 2(8), e765 CrossRef PubMed.
- T. Enomoto, I. Kukimoto, M. A. Kawano, Y. Yamaguchi, A. J. Berk and H. Handa, In vitro reconstitution of SV40 particles that are composed of VP1/2/3 capsid proteins and nucleosomal DNA and direct efficient gene transfer, Virology, 2011, 420(1), 1–9 CrossRef CAS PubMed.
- S. Kler, R. Asor, C. Li, A. Ginsburg, D. Harries, A. Oppenheim, A. Zlotnick and U. Raviv, RNA encapsidation by SV40-derived nanoparticles follows a rapid two-state mechanism, J. Am. Chem. Soc., 2012, 134(21), 8823–8830 CrossRef CAS PubMed.
- M. G. M. v. Rosmalen, D. Kamsma, A. S. Biebricher, C. Li, A. Zlotnick, W. H. Roos and G. J. L. Wuite, Revealing in real-time a multistep assembly mechanism for SV40 virus-like particles, Sci. Adv., 2020, 6(16), eaaz1639 CrossRef PubMed.
- S. E. Aniagyei, C. Dufort, C. C. Kao and B. Dragnea, Self-assembly approaches to nanomaterial encapsulation in viral protein cages, J. Mater. Chem., 2008, 18(32), 3763–3774 RSC.
- J. Sun, C. DuFort, M. C. Daniel, A. Murali, C. Chen, K. Gopinath, B. Stein, M. De, V. M. Rotello, A. Holzenburg, C. C. Kao and B. Dragnea, Core-controlled polymorphism in virus-like particles, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(4), 1354–1359 CrossRef CAS PubMed.
- M. C. Daniel, I. B. Tsvetkova, Z. T. Quinkert, A. Murali, M. De, V. M. Rotello, C. C. Kao and B. Dragnea, Role of surface charge density in nanoparticle-templated assembly of bromovirus protein cages, ACS Nano, 2010, 4(7), 3853–3860 CrossRef CAS PubMed.
- J. D. Griffith, Chromatin structure: deduced from a minichromosome, Science, 1975, 187(4182), 1202–1203 CrossRef CAS PubMed.
- A. J. Varshavsky, S. A. Nedospasov, V. V. Schmatchenko, V. V. Bakayev, P. M. Chumackov and G. P. Georgiev, Compact form of SV40 viral minichromosome is resistant to nuclease: possible implications for chromatin structure, Nucleic Acids Res., 1977, 4(10), 3303–3325 CrossRef CAS PubMed.
- I. Kopatz, R. Zalk, Y. Levi-Kalisman, E. Zlotkin-Rivkin, G. A. Frank and S. Kler, Packaging of DNA origami in viral capsids, Nanoscale, 2019, 11(21), 10160–10166 RSC.
- Y. J. Chen, X. Liu and B. Tsai, SV40 Hijacks Cellular Transport, Membrane Penetration, and Disassembly Machineries to Promote Infection, Viruses, 2019, 11(10), 917 CrossRef CAS PubMed.
- R. C. Liddington, Y. Yan, J. Moulai, R. Sahli, T. L. Benjamin and S. C. Harrison, Structure of simian virus 40 at 3.8-A resolution, Nature, 1991, 354(6351), 278–284 CrossRef CAS PubMed.
- T. Stehle, S. J. Gamblin, Y. Yan and S. C. Harrison, The structure of simian virus 40 refined at 3.1 A resolution, Structure, 1996, 4(2), 165–182 CrossRef CAS.
- X. S. Chen, T. Stehle and S. C. Harrison, Interaction of polyomavirus internal protein VP2 with the major capsid protein VP1 and implications for participation of VP2 in viral entry, EMBO J., 1998, 17(12), 3233–3240 CrossRef CAS PubMed.
- D. J. Goetschius, H. Lee and S. Hafenstein, Chapter Three - CryoEM reconstruction approaches to resolve asymmetric features, in Advances in Virus Research, ed. F. A. Rey, Academic Press, 2019, vol. 105, pp. 73–91 Search PubMed.
- S. H. Scheres, Processing of Structurally Heterogeneous Cryo-EM Data in RELION, Methods Enzymol., 2016, 579, 125–157 CAS.
- B. Tsai, J. M. Gilbert, T. Stehle, W. Lencer, T. L. Benjamin and T. A. Rapoport, Gangliosides are receptors for murine polyoma virus and SV40, EMBO J., 2003, 22(17), 4346–4355 CrossRef CAS PubMed.
- A. E. Smith, H. Lilie and A. Helenius, Ganglioside-dependent cell attachment and endocytosis of murine polyomavirus-like particles, FEBS Lett., 2003, 555(2), 199–203 CrossRef CAS.
- M. A. Campanero-Rhodes, A. Smith, W. Chai, S. Sonnino, L. Mauri, R. A. Childs, Y. Zhang, H. Ewers, A. Helenius, A. Imberty and T. Feizi, N-glycolyl GM1 ganglioside as a receptor for simian virus 40, J. Virol., 2007, 81(23), 12846–12858 CrossRef CAS.
- E. M. Damm, L. Pelkmans, J. Kartenbeck, A. Mezzacasa, T. Kurzchalia and A. Helenius, Clathrin- and caveolin-1-independent endocytosis: entry of simian virus 40 into cells devoid of caveolae, J. Cell Biol., 2005, 168(3), 477–488 CrossRef CAS PubMed.
- L. C. Norkin, H. A. Anderson, S. A. Wolfrom and A. Oppenheim, Caveolar endocytosis of simian virus 40 is followed by brefeldin A-sensitive transport to the endoplasmic reticulum, where the virus disassembles, J. Virol., 2002, 76(10), 5156–5166 CrossRef CAS PubMed.
- H. A. Anderson, Y. Chen and L. C. Norkin, Bound simian virus 40 translocates to caveolin-enriched membrane domains, and its entry is inhibited by drugs that selectively disrupt caveolae, Mol. Biol. Cell, 1996, 7(11), 1825–1834 CrossRef CAS PubMed.
- L. Pelkmans, J. Kartenbeck and A. Helenius, Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER, Nat. Cell Biol., 2001, 3(5), 473–483 CrossRef CAS PubMed.
- M. Qian, D. Cai, K. J. Verhey and B. Tsai, A lipid receptor sorts polyomavirus from the endolysosome to the endoplasmic reticulum to cause infection, PLoS Pathog., 2009, 5(6), e1000465 CrossRef PubMed.
- S. Engel, T. Heger, R. Mancini, F. Herzog, J. Kartenbeck, A. Hayer and A. Helenius, Role of endosomes in simian virus 40 entry and infection, J. Virol., 2011, 85(9), 4198–4211 CrossRef CAS PubMed.
- J. Kartenbeck, H. Stukenbrok and A. Helenius, Endocytosis of simian virus 40 into the endoplasmic reticulum, J. Cell Biol., 1989, 109(6 Pt 1), 2721–2729 CrossRef CAS PubMed.
- M. Schelhaas, J. Malmström, L. Pelkmans, J. Haugstetter, L. Ellgaard, K. Grünewald and A. Helenius, Simian Virus 40 depends on ER protein folding and quality control factors for entry into host cells, Cell, 2007, 131(3), 516–529 CrossRef CAS PubMed.
- C. P. Walczak and B. Tsai, A PDI family network acts distinctly and coordinately with ERp29 to facilitate polyomavirus infection, J. Virol., 2011, 85(5), 2386–2396 CrossRef CAS PubMed.
- R. Daniels, N. M. Rusan, P. Wadsworth and D. N. Hebert, SV40 VP2 and VP3 insertion into ER membranes is controlled by the capsid protein VP1: implications for DNA translocation out of the ER, Mol. Cell, 2006, 24(6), 955–966 CrossRef CAS PubMed.
- R. Geiger, D. Andritschke, S. Friebe, F. Herzog, S. Luisoni, T. Heger and A. Helenius, BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol, Nat. Cell Biol., 2011, 13(11), 1305–1314 CrossRef CAS PubMed.
- K. M. Giorda, S. Raghava, M. W. Zhang and D. N. Hebert, The viroporin activity of the minor structural proteins VP2 and VP3 is required for SV40 propagation, J. Biol. Chem., 2013, 288(4), 2510–2520 CrossRef CAS PubMed.
- P. Bagchi, C. P. Walczak and B. Tsai, The endoplasmic reticulum membrane J protein C18 executes a distinct role in promoting simian virus 40 membrane penetration, J. Virol., 2015, 89(8), 4058–4068 CrossRef CAS PubMed.
- C. P. Walczak, M. S. Ravindran, T. Inoue and B. Tsai, A cytosolic chaperone complexes with dynamic membrane J-proteins and mobilizes a nonenveloped virus out of the endoplasmic reticulum, PLoS Pathog., 2014, 10(3), e1004007 CrossRef PubMed.
- A. Dupzyk and B. Tsai, Bag2 Is a Component of a Cytosolic Extraction Machinery That Promotes Membrane Penetration of a Nonenveloped Virus, J. Virol., 2018, 92(15), e00607-18 CrossRef PubMed.
- A. Dupzyk, J. M. Williams, P. Bagchi, T. Inoue and B. Tsai, SGTA-Dependent Regulation of Hsc70 Promotes Cytosol Entry of Simian Virus 40 from the Endoplasmic Reticulum, J. Virol., 2017, 91(12), e00232-17 CrossRef.
- E. C. Goodwin, A. Lipovsky, T. Inoue, T. G. Magaldi, A. P. Edwards, K. E. Van Goor, A. W. Paton, J. C. Paton, W. J. Atwood, B. Tsai and D. DiMaio, BiP and multiple DNAJ molecular chaperones in the endoplasmic reticulum are required for efficient simian virus 40 infection, mBio, 2011, 2(3), e00101–e00111 CrossRef PubMed.
- T. Inoue and B. Tsai, A large and intact viral particle penetrates the endoplasmic reticulum membrane to reach the cytosol, PLoS Pathog., 2011, 7(5), e1002037 CrossRef CAS PubMed.
- A. Nakanishi, D. Shum, H. Morioka, E. Otsuka and H. Kasamatsu, Interaction of the Vp3 nuclear localization signal with the importin alpha 2/beta heterodimer directs nuclear entry of infecting simian virus 40, J. Virol., 2002, 76(18), 9368–9377 CrossRef CAS PubMed.
- M. S. Ravindran, M. F. Engelke, K. J. Verhey and B. Tsai, Exploiting the kinesin-1 molecular motor to generate a virus membrane penetration site, Nat. Commun., 2017, 8, 15496 CrossRef CAS PubMed.
- D. A. Dean, P. P. Li, L. M. Lee and H. Kasamatsu, Essential role of the Vp2 and Vp3 DNA-binding domain in simian virus 40 morphogenesis, J. Virol., 1995, 69(2), 1115–1121 CrossRef CAS PubMed.
- D. L. Hurdiss, E. L. Morgan, R. F. Thompson, E. L. Prescott, M. M. Panou, A. Macdonald and N. A. Ranson, New Structural Insights into the Genome and Minor Capsid Proteins of BK Polyomavirus using Cryo-Electron Microscopy, Structure, 2016, 24(4), 528–536 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr01316a |
| This journal is © The Royal Society of Chemistry 2022 |