
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanics of migrating platelets investigated with scanning ion conductance microscopy†
Jan
Seifert‡
 ,
Johannes
Rheinlaender‡
*,
Hendrik
von Eysmondt
and
Tilman E.
Schäffer
*
,
Johannes
Rheinlaender‡
*,
Hendrik
von Eysmondt
and
Tilman E.
Schäffer
*
Institute of Applied Physics, University of Tübingen, Auf der Morgenstelle 10, 72076 Tübingen, Germany. E-mail: johannes.rheinlaender@uni-tuebingen.de; tilman.schaeffer@uni-tuebingen.de; Fax: +49 7071 29 5093; Tel: +49 7071 29 76030
First published on 27th May 2022
Abstract
Platelets are small blood cells involved in hemostasis, wound healing, and immune response. After adhesion and spreading, platelets can migrate at sites of injury inducing an early immune response to inflammation or infection. Platelet migration requires fibrinogen-integrin binding and fibrinogen depletion from the substrate inducing a self-generated ligand gradient guiding the direction of migration. This type of cellular motion is referred to as haptotactic migration. The underlying mechanisms of haptotactic platelet migration have just recently been discovered, but the connection to platelet mechanics has remained unknown yet. Using scanning ion conductance microscopy (SICM), we investigated the three-dimensional morphology and mechanics of platelets during haptotactic migration for the first time. Migrating platelets showed a polarized, anisotropic shape oriented in the direction of migration. This polarization goes hand in hand with a characteristic subcellular stiffness distribution showing a region of increased stiffness at the leading edge. Moreover, the mechanical properties of the leading edge revealed a highly dynamic stiffening and softening process with rapid changes of the elastic modulus by a factor of up to 5× per minute. Inhibition of actin polymerization stopped the dynamic stiffening and softening process and halted the migration. By combining SICM with confocal fluorescence microscopy, we found that the increased stiffness and mechanical dynamics at the leading edge coincided with an increased volumetric F-actin density. Our data provide a connection between platelet mechanics and the cytoskeletal contribution to the migration process of platelets.
Introduction
Platelets are small anucleate blood cells involved in hemostasis and wound healing.1,2 Platelets get activated and adhere at sites of injury,3 coagulate, and form a blood clot to close the damaged vessel wall.4–6 The role of platelets as immunological cells7 and the contribution of platelet migration8–11 has been a matter of debate and research for many years. Recently, it was shown that platelets are able to become motile and actively contribute to the innate immune response by migration.12 Essential for the migration of platelets are the adhesion to fibrinogen by integrin αIIbβ3 and the ability to break bonds between fibrinogen and the underlying surface by cellular force,13 leading to fibrinogen depletion and the generation of a ligand gradient on the surface.14 This type of cellular motion is driven by the actin cytoskeleton and is referred to as haptotactic migration.14The three-dimensional shape and the mechanical properties of migrating platelets have not been studied so far. We fill this gap by using scanning ion conductance microscopy (SICM)15–17 to investigate dynamics and mechanics of migrating platelets in vitro. SICM is a non-invasive scanning probe technique that is especially well suited for imaging the topography18–25 and the mechanics26–31 of living cells. We showed that migrating platelets exhibit a three-dimensional shape anisotropy, manifested by a shift of the center of volume (centroid) toward the trailing edge of the platelet. We found that migrating platelets have a characteristic subcellular stiffness distribution, including a region of high stiffness in the lamellipodium at the leading edge. Moreover, the expansion of the leading edge in the direction of migration was accompanied by a highly dynamic stiffening and softening process with an elastic modulus increase or decrease by a factor of up to 5× per minute. By combining SICM with confocal fluorescence microscopy we accurately determined the volumetric F-actin density of migrating platelets and associated the mechanical dynamics with an increased volumetric F-actin density at the leading edge. We thereby provide a connection between platelet mechanics and the cytoskeletal contribution to haptotactic platelet migration.
Experimental
Preparation of migrating platelets
All procedures were approved by the institutional ethics committee (273/2018BO2) and comply with the declaration of Helsinki. Informed consent was obtained from all subjects involved in the study. Washed human platelets were isolated from whole blood of healthy volunteers mixed with acid citrate dextrose at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7. Citrated blood was centrifuged at 200g for 20 min to gain platelet rich plasma (PRP). The PRP was then mixed with Tyrode-HEPES buffer (136.89 mM NaCl, 2.81 mM KCl, 11.9 mM NaHCO3, 1 g L−1D-glucose, 10 mM HEPES), pH 6.5, supplemented with 0.1 μg mL−1 prostacyclin (PGI2, ab120912, abcam, Cambridge, UK) and centrifuged again at 200g for 10 min to remove remaining red blood cells. The platelet-containing supernatant was transferred into a new tube and centrifuged at 880g for 10 min. Finally, washed platelets were gained by careful resuspension of the platelet pellet in Tyrode-HEPES buffer, pH 7.4.
:7. Citrated blood was centrifuged at 200g for 20 min to gain platelet rich plasma (PRP). The PRP was then mixed with Tyrode-HEPES buffer (136.89 mM NaCl, 2.81 mM KCl, 11.9 mM NaHCO3, 1 g L−1D-glucose, 10 mM HEPES), pH 6.5, supplemented with 0.1 μg mL−1 prostacyclin (PGI2, ab120912, abcam, Cambridge, UK) and centrifuged again at 200g for 10 min to remove remaining red blood cells. The platelet-containing supernatant was transferred into a new tube and centrifuged at 880g for 10 min. Finally, washed platelets were gained by careful resuspension of the platelet pellet in Tyrode-HEPES buffer, pH 7.4.
Human serum was gained from whole blood using serum monovettes (02.1063.001, Sarstedt, Nümbrecht, Germany). Blood was allowed to coagulate for 30 min at room temperature. Then, serum was gained in two centrifugation steps for 15 min at 2000g each.
For imaging, platelet suspension was added to a glass bottom dish (81218, ibidi, Gräfeling, Germany) containing Tyrode-HEPES migration buffer, consisting of Tyrode-HEPES buffer, pH 7.4, 3 μM U-46619 thromboxane analogon (TXa, Cay16450, biomol, Hamburg, Germany), 20 μM adenosine diphosphate (ADP, A2754, Sigma Aldrich, St. Louis, Missouri, USA), 0.1 mg mL−1 fibrinogen (F3879, Sigma Aldrich), and 5% human serum. Platelets were allowed to adhere and spread for 30 min. Non-adherent platelets were then carefully washed away by flushing with Tyrode-HEPES migration buffer. Afterwards, the sample was mounted in the SICM setup and migrating platelets were imaged at room temperature. For inhibition of actin polymerization, cytochalasin D was added at a concentration of 2 μM (C8273, Sigma Aldrich, stock solution 10 mM in DMSO). For control, DMSO was added to the imaging buffer at a dilution of 1:5000.
Scanning ion conductance microscopy (SICM)
We used custom-built SICM setups which are described elsewhere in detail.19,26,32 In brief, piezo scanners were used to move an electrolyte-filled nanopipette with respect to the sample in all three spatial dimensions (Fig. 1a). A voltage applied between two electrodes, one inside and one outside of the pipette, induced a distance-sensitive ion current through the tip of the pipette, which was used as a feedback signal for tracking the topography of the sample. Nanopipettes were fabricated from borosilicate glass capillaries using a CO2-laser based pipette puller (P2000, Sutter Instruments; see ESI Table 1† for parameters used). For topography imaging, nanopipettes had a typical inner opening diameter of approximately 80 nm and images were taken with a trigger setpoint of 99.5% of the saturation ion current and a typical image duration of 30 s. For mechanical measurements, nanopipettes had a typical diameter of 200 nm and measurements were performed with a trigger setpoint of 98% of the saturation current and a typical image duration of 3 min. Additionally, a constant pressure of 10 kPa was applied to the upper end of the nanopipette, which caused fluid to flow out of the tip to induce a local indentation of the sample surface (typically 50 nm, see ESI Fig. S1b, right panel†) when the nanopipette was close to the surface. Ion current–distance curves (I–z curves) were recorded for many positions on the sample and the local elastic modulus (ESI Fig. S1c†) was calculated from the slope in an I–z curve between 99% and 98% of the saturation current (ESI Fig. S1b, middle panel†).26,30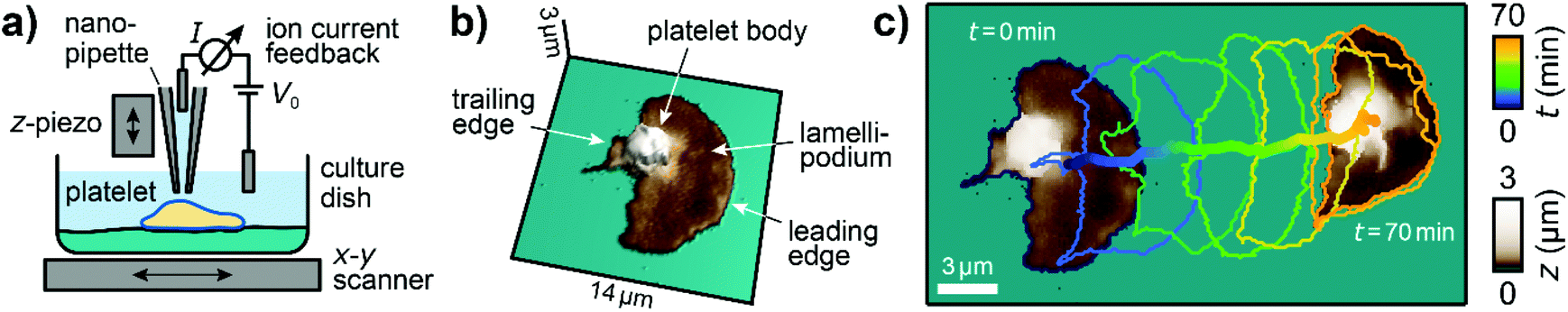 | ||
| Fig. 1 Imaging migrating platelets with SICM. (a) Schematic of the SICM setup. A nanopipette is positioned relative to the sample in the x-, y-, and z-directions with piezo scanners. A voltage V0 applied between two electrodes, inside and outside the pipette, induces an ion current I, which is used for topography imaging of the sample. (b) 3D relief of the first SICM topography image and (c) the first and the last SICM topography images (brown color) of an image sequence, time-coded cell contours of the intermediate images (every 12th contour is shown), and the trace of the center of area of a migrating platelet. Image resolution: 112 × 112 pixels (125 nm pixel size). Also see ESI Video 1† for the full image sequence. | ||
Confocal fluorescence imaging
For confocal fluorescence imaging, glass bottom dishes with an imprinted grid (81168, ibidi) were used to facilitate finding the location of interest after SICM imaging. Platelets were fixed immediately after SICM imaging in Tyrode-HEPES buffer containing 2% formaldehyde for 10 min. The samples were then carefully washed three times with PBS and permeabilized by incubation in PBS containing 0.1% Triton-X for 10 min. The samples were washed again three times and filamentous actin was stained by incubation in PBS with phalloidin i-fluor 488 (diluted by 1:1000, ab176753, abcam) for 30 min at room temperature. Confocal fluorescence images were recorded using an inverted microscope (Ti–E, Nikon, Tokyo, Japan) equipped with a confocal fluorescence setup (C2, Nikon) and a 100× oil immersion objective.Data analysis
Data analysis was performed in Igor Pro (Wavemetrics, Portland, Oregon, USA). Topography images were corrected for tilt and z-offset by a first-order plane fit.30,33 Platelets were identified as pixels with a height above h = 50 nm. The platelet area A was calculated as the sum of all single pixel areas ai of the platelet, . The platelet volume V was calculated as the sum of all single pixel volumes of the platelet,
. The platelet volume V was calculated as the sum of all single pixel volumes of the platelet, 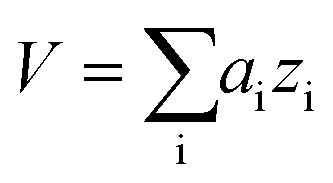 . The center of the platelet area CA was calculated as
. The center of the platelet area CA was calculated as 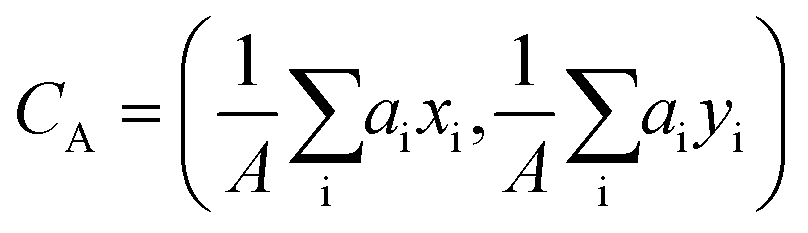 . The center of the platelet volume was calculated as
. The center of the platelet volume was calculated as 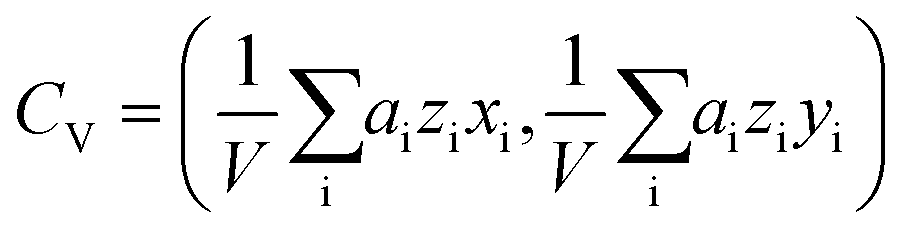 . With these two points, we determined the shape anisotropy vector s pointing from the center of volume to the center of area, s = CA − CV. The velocity vector v was calculated as the difference in the center of area of the platelet between two consecutively acquired images, divided by their time interval. To increase the robustness, the magnitudes of the shape anisotropy vector s and the migration velocity v, and the average angle Δφ were averaged over the full image sequence.
. With these two points, we determined the shape anisotropy vector s pointing from the center of volume to the center of area, s = CA − CV. The velocity vector v was calculated as the difference in the center of area of the platelet between two consecutively acquired images, divided by their time interval. To increase the robustness, the magnitudes of the shape anisotropy vector s and the migration velocity v, and the average angle Δφ were averaged over the full image sequence.
Elastic modulus values were averaged by using the geometric average to account for the underlying log–normal distribution of the mechanical properties of cells.34,35 Images of the stiffening rate SR were calculated as the gradient of the log-transformed elastic modulus images, multiplied (scalar product) with the current migration velocity of the platelet and a reference time of 1 minute, and re-transformed by exponential transformation: 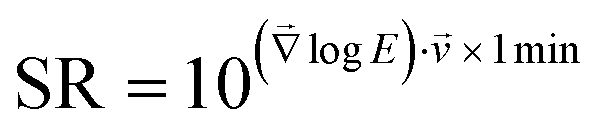 . Thereby, the stiffening rate images indicate an x-fold change per minute of the platelet's elastic modulus in the direction of migration in the reference frame of the underlying substrate. Here we assume that the subcellular stiffness distribution is time-constant in the reference frame of the platelet, as observed in ESI Fig. S2 and ESI Video 4.† Quickly migrating platelets with a high local stiffness variation therefore show a high stiffening rate. For calculation of the volumetric F-actin density, the pixel resolution of the SICM images was adjusted to the pixel resolution of the confocal fluorescence images by linear interpolation. The SICM and confocal images were then manually aligned and images of the volumetric F-actin density were calculated by dividing the F-actin fluorescence intensity by the corresponding local height of the platelet.
. Thereby, the stiffening rate images indicate an x-fold change per minute of the platelet's elastic modulus in the direction of migration in the reference frame of the underlying substrate. Here we assume that the subcellular stiffness distribution is time-constant in the reference frame of the platelet, as observed in ESI Fig. S2 and ESI Video 4.† Quickly migrating platelets with a high local stiffness variation therefore show a high stiffening rate. For calculation of the volumetric F-actin density, the pixel resolution of the SICM images was adjusted to the pixel resolution of the confocal fluorescence images by linear interpolation. The SICM and confocal images were then manually aligned and images of the volumetric F-actin density were calculated by dividing the F-actin fluorescence intensity by the corresponding local height of the platelet.
Statistical analysis
Data are presented as average ± standard deviation, unless stated otherwise. For elastic modulus and stiffening rate data, averaging and statistics were performed with the log-transformed data. Data were tested for statistical significance using un-paired, two-sided Student's t-test or Tukey's range test, unless stated otherwise. For circular statistics, data were tested for uniformity using Rayleigh's test. Data were considered significantly different for P < 0.05.Results
Scanning ion conductance microscopy (SICM) of migrating platelets
The three-dimensional topography of platelets undergoing haptotactic migration was imaged with SICM (Fig. 1a). Platelets in Tyrode-HEPES migration buffer started to migrate after adhering and spreading, as described by Gaertner et al.12 Migrating platelets had a polarized shape with a flat lamellipodium (Fig. 1b, dark brownish color) at the leading edge and a high, sphere-like body (light brown-white color) near the trailing edge. During migration, platelets constantly changed their shape and position (Fig. 1c, contour plot; ESI Video 1†). The shown platelet migrated a Euclidean distance of about 17 μm during a time of 70 min.Migrating platelets exhibit a three-dimensional shape anisotropy
The lamellipodium of migrating platelets was shifted toward the direction of migration, forming a polarized topography with a leading edge (Fig. 2a). As a quantitative parameter that characterizes this polarized topography, we define the shape anisotropy vector s as the vector from the center of volume (centroid, Fig. 2b, marked with □) to the center of area (marked with ○) of the platelet (Fig. 2b, red arrow). The length of this vector is therefore a measure of the three-dimensional asymmetry of the cell and typically ranged from 0 to 1 μm for the platelets in this study. To quantify the migration movement, we define the velocity vector v as the velocity of the center of area of the platelet relative to the substrate (Fig. 2b, blue arrow).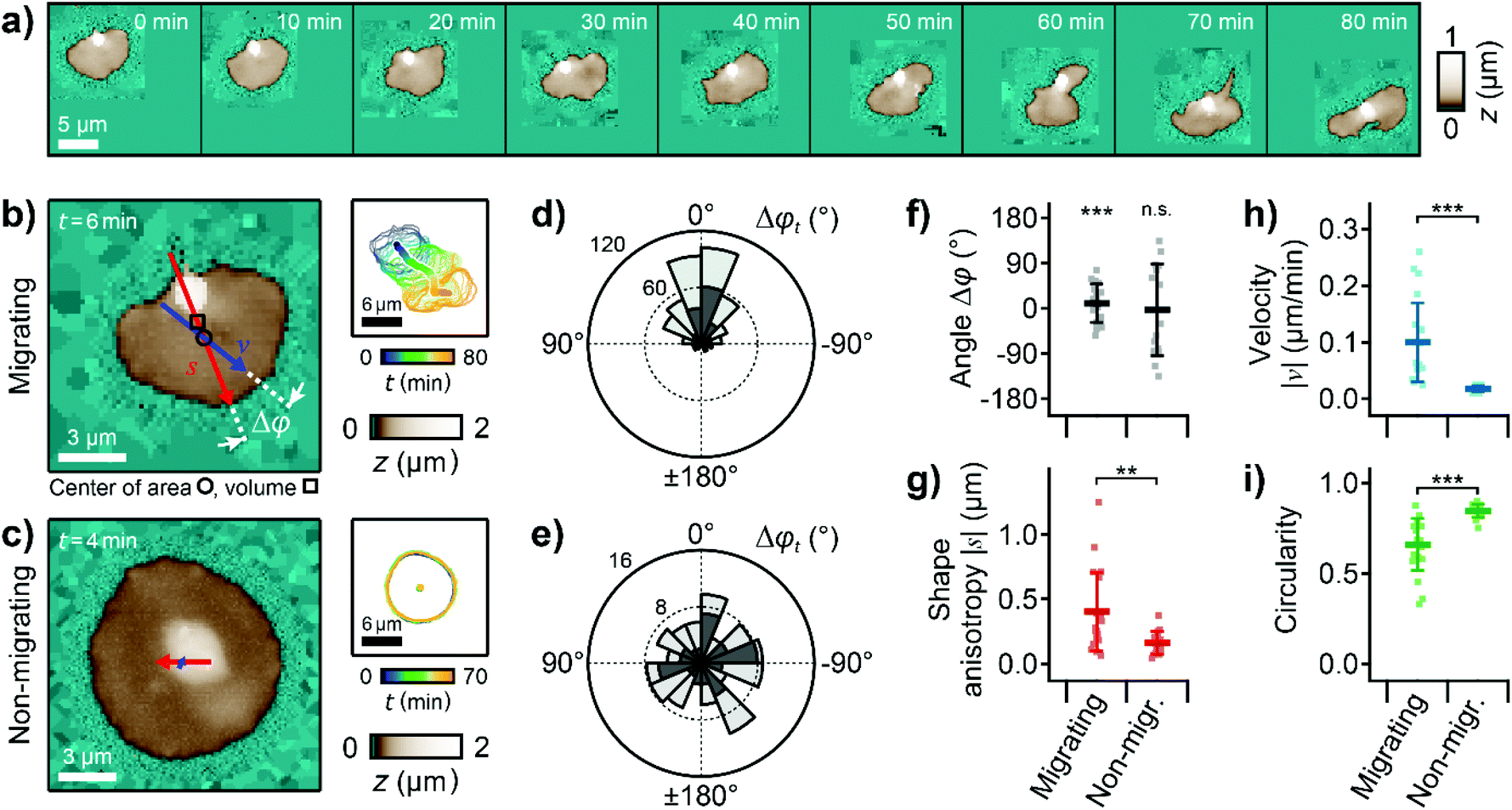 | ||
| Fig. 2 Migrating platelets exhibit a three-dimensional shape anisotropy. (a) Sequence of topography images of a migrating platelet. Pixels outside the scan range were set to a height of z = 0. (b) Topography image (left) and time-coded plot of the cell contours (right, every 5th contour shown) of a migrating and (c) a non-migrating platelet. The red and blue arrows represent the shape anisotropy vector s and the velocity vector v, respectively. The shape anisotropy vector s points from the center of volume (centroid, marked with □) to the center of area (marked with ○). The length of the arrows indicates the vector magnitude multiplied with a factor of 20× for s and 20 × 1 min for v (|s| = 0.36 μm, |v| = 0.24 μm min−1 in (b) and |s| = 0.12 μm, |v| = 0.03 μm min−1 in (c)). (d) Circular histogram of the angle, Δφt, between the shape anisotropy and velocity vectors for the migrating and (e) for the non-migrating platelet. Dark grey histograms represent data of platelets shown in b and c, light grey histograms represent data of all platelets pooled (number of images: n = 481 and n = 111, respectively). (f) Temporal averages per platelet within a whole image sequence: angle Δφ, (g) magnitude of the shape anisotropy vector s, (h) magnitude of the velocity vector v, and (i) circularity for migrating and non-migrating platelets. Plots show average (marker), standard deviation (error bar), data points for individual platelets (dots); number of platelets N = 22 for migrating and 15 for non-migrating; ** P < 0.01, *** P < 0.001 from Rayleigh test for uniformity (f) or Student's t-test (g–i). Also see ESI Videos 2 and 3† for the whole image sequences of the platelets shown in (a)–(c). Image resolution: (a) and (b) 60 × 60 pixels (200 nm pixel size), (c) 112 × 112 pixels (125 nm pixel size). | ||
We investigated the correlation between the shape anisotropy vector s and the velocity vector v for migrating and for non-migrating platelets (Fig. 2b and c, ESI Videos 2 and 3, exemplary platelets shown†). For migrating platelets, the flat regions of the lamellipodium were asymmetrically distributed around the platelet body, which was located near the trailing edge (Fig. 2b). For non-migrating platelets, the platelet body was located near the platelet center, isotropically surrounded by the lamellipodium (Fig. 2c). The angle Δφt(s,v,t) for each time step between the shape anisotropy vector s and the velocity vector v of the migrating platelet was distributed in a narrow range around 0°, typically between ±60° (Fig. 2d, dark grey histogram, P < 10−10, Rayleigh test for uniformity), which means that both vectors roughly pointed in the same direction. For the non-migrating platelet, the angle Δφt was randomly distributed from 0° to ±180° (Fig. 2e, dark grey histogram, P = 0.78, Rayleigh test for uniformity). Pooling all platelets in this study gave similar distributions of Δφt (Fig. 2d and e, light grey histograms). Similarly, the temporal average Δφt of all angles Δφt within a full image sequence followed a non-uniform distribution around 0° for migrating platelets (Fig. 2f, P = 2.0 × 10−7, Rayleigh test for uniformity), whereas Δφ was uniformly distributed for non-migrating platelets (P = 0.84, Rayleigh test for uniformity). The magnitude of the shape anisotropy vector s was significantly (P = 0.0019) larger for migrating platelets compared to non-migrating platelets (Fig. 2g). Consistent with previous studies, migrating platelets moved with a typical velocity of |v| = 0.1 μm min−1 (Fig. 2h, P = 4.1 × 10−5 compared to the non-migrating platelets), and had a less roundish shape as indicated by the significantly (P = 4.1 × 10−5) smaller circularity of the platelet outline (Fig. 2i).12,36
Migrating platelets have a characteristic subcellular stiffness distribution
Next, we investigated the mechanical properties of migrating platelets with SICM. A constant pressure of 10 kPa was applied to the upper end of the nanopipette and I–z curves (ion current as a function of the z-position of the nanopipette) were recorded at many lateral positions on a platelet. The platelet's local elastic modulus was extracted from the respective I–z curve.26 We thereby obtained a map of the local elastic modulus of the platelet simultaneously with its topography (Fig. 3a and b, ESI Video 4†). The calcium signal of platelets was unaffected by SICM (ESI Fig. S3†), which indicates that the platelet behavior was not altered by the imaging process. The shown platelet was continuously imaged for 3 h, migrating a Euclidean distance of 30 μm and exhibiting the typical shape anisotropy. The platelet had a non-homogeneous subcellular stiffness distribution with soft regions of approximately 5 kPa for the platelet body and for the inner lamellipodium (Fig. 3b, purple color and purple arrow in the profile) and a 10-fold stiffer region at the leading edge with a maximum elastic modulus of approximately 50 kPa (yellow color and yellow arrow in the profile), located at a distance of about 0.5 μm from the softer outer edge. In comparison, non-migrating platelets usually showed a more homogenous subcellular stiffness distribution without a stiff region near the edge (ESI Fig. S4†). To assess the change of the mechanical properties during migration, we calculated the local stiffening rate (SR) during the movement. The platelet showed regions of dynamic stiffening (SR > 1) at the outer leading edge (Fig. 3c, red color and red arrow in the profile) followed by regions of dynamic softening (SR < 1) toward the platelet body (Fig. 3c, blue color and blue arrow in the profile). The stiffening rates reached values of up to 5× per minute, showing that the mechanical properties of the leading edge were subject to highly dynamic changes.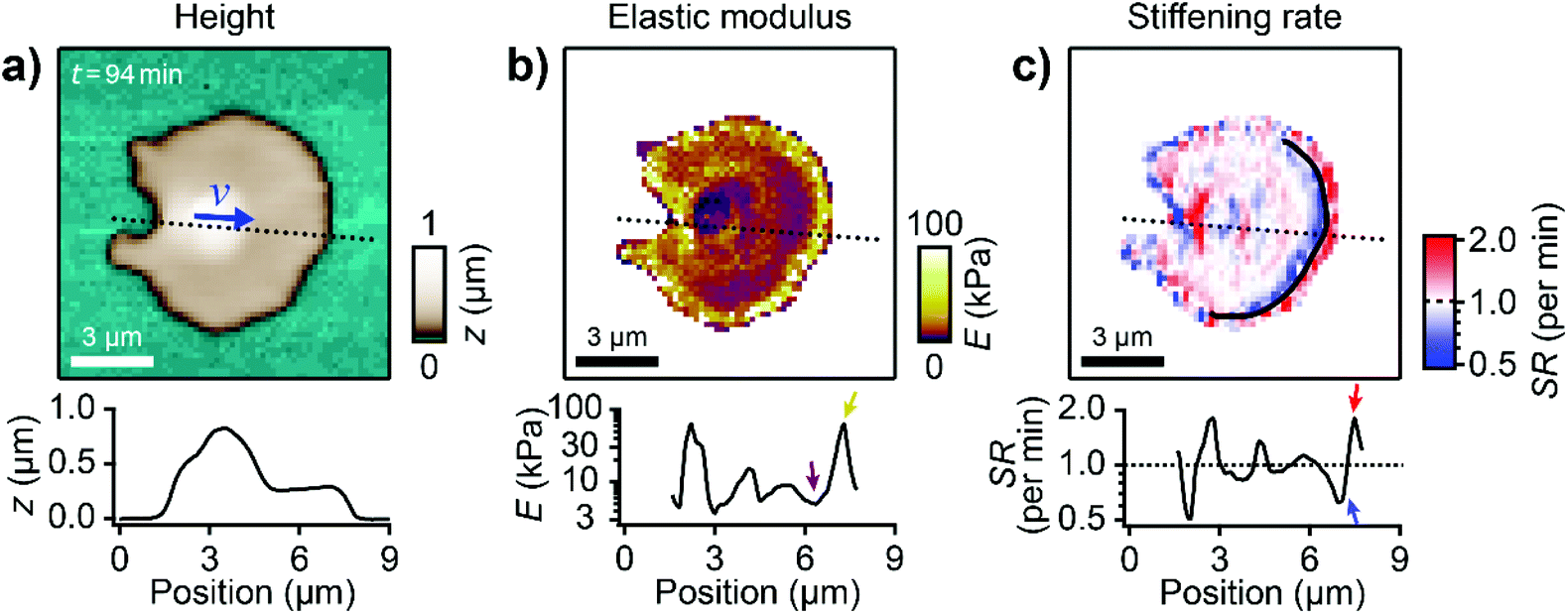 | ||
| Fig. 3 Migrating platelets show a characteristic subcellular stiffness distribution. (a) Topography image and (b) elastic modulus map of a migrating platelet. The blue arrow in a) indicates the velocity vector v. The length of the arrow indicates the vector magnitude (|v| = 0.11 μm min−1) multiplied with a factor of 20 × 1 min. (c) Stiffening rate (SR) in the direction of migration (indicated by the blue arrow in a)). The black curve indicates the region of high stiffness (≈50 kPa) in the elastic modulus map in (b). The plots show profiles of height, elastic modulus, and stiffening rate, respectively (location indicated in the images by dotted lines). Also see ESI Video 4 for the whole image sequence.† Image resolution: 64 × 64 pixels (187.5 nm pixel size). | ||
Actin depolymerization leads to a loss of the subcellular stiffness distribution
To investigate the role of the actin cytoskeleton, we treated migrating platelets with the actin polymerization inhibitor cytochalasin D.37 Before the treatment, a migrating platelet exhibited the characteristic shape anisotropy, subcellular stiffness distribution, and stiffening regions at the leading edge (Fig. 4a, ESI Video 5). Following treatment with 2 μM cytochalasin D, the platelet retained its polarized shape and shape anisotropy (Fig. 4b, left). However, the platelet had a more homogenous stiffness distribution with softer regions throughout the platelet. The stiff regions and the high stiffening rate at the leading edge could not be observed anymore (Fig. 4b, center and right). The migration velocity decreased from |v| ≈ 0.3 μm min−1 before to |v| ≈ 0.0 μm min−1 after the addition of cytochalasin D, showing that migration had stopped after the treatment (Fig. 4c). The average elastic modulus (Fig. 4d) and the stiffening rate variation (geometric standard deviation of all pixel values in the stiffening rate map, Fig. 4e) decreased, indicating an overall softening of the platelet and the loss of the characteristic subcellular stiffness distribution, respectively. Note that the decrease in the stiffening rate variation is mainly caused by the decrease in migration velocity. On average, the elastic modulus of the platelets was significantly (P = 2.2 × 10−6) decreased after the treatment with cytochalasin D (Fig. 4f). Compared to non-migrating platelets, however, the elastic modulus of migrating platelets was not significantly different (P = 0.77). The stiffening rate variation of non-migrating and cytochalasin D-treated migrating platelets was similar (P = 0.99) and significantly lower (P = 1.2 × 10−5 and 1.7 × 10−5, respectively) compared to untreated migrating platelets (Fig. 4g).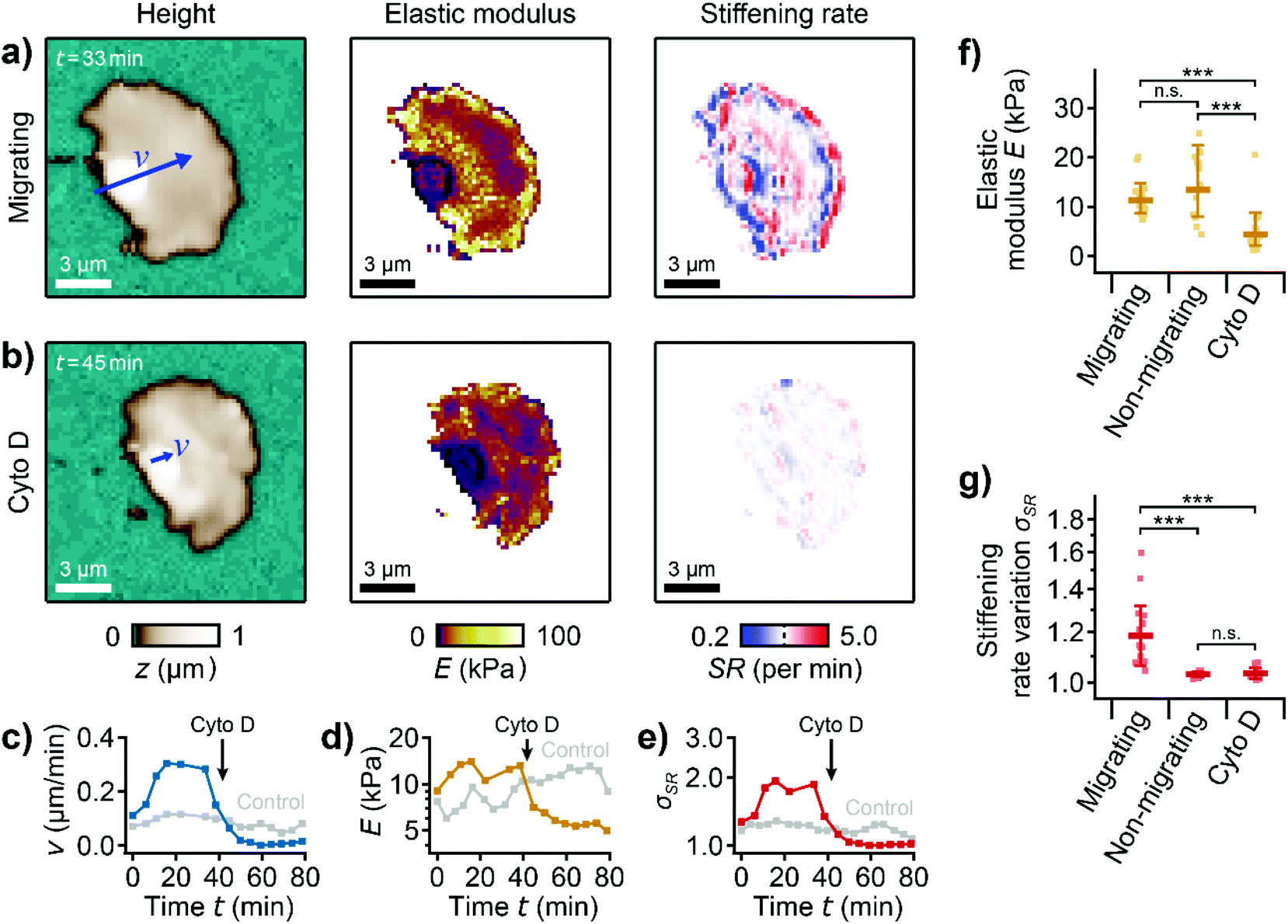 | ||
| Fig. 4 Platelet migration is driven by active polymerization of the actin cytoskeleton. (a) Topography image (left), elastic modulus map (center), and stiffening rate map (right) of a migrating platelet before and (b) after the addition of 2 μM cytochalasin D. The blue arrows indicate the velocity vectors v. The length of the arrows indicates the vector magnitude multiplied with a factor of 20 × 1 min (|v| = 0.29 μm min−1 in (a) and |v| = 0.07 μm min−1 in (b)). (c) Time traces of migration velocity, (d) mean elastic modulus, and (e) stiffening rate variation, σSR, before and after the addition of cytochalasin D. (f) Elastic modulus and (g) stiffening rate variation for migrating, non-migrating, and cytochalasin D-treated platelets. Plots show geometric average (marker), geometric standard deviation (error bar), data points (dots); number of cells N = 20 for migrating, 14 for non-migrating, and 14 for cytochalasin D; * P < 0.05, ** P < 0.01, *** P < 0.001 from Tukey's range test. Also see ESI Video 5 for the whole image sequence.† Image resolution: 64 × 64 pixels (218.75 nm pixel size). | ||
Volumetric actin density is increased in the leading edge
To verify that this subcellular distribution is indeed caused by the actin cytoskeleton, we combined SICM with confocal fluorescence imaging to directly investigate the spatial correlation between cell stiffness and the actin cytoskeleton. A migrating platelet was fixed immediately after SICM imaging (Fig. 5a and b), F-actin was stained using fluorescently labelled phalloidin, and fluorescence images were recorded (Fig. 5c). Please note that the imaging duration (120 s for the SICM image) before the platelet fixation imposed a short effective time delay (on average 60 s) between the SICM image and the confocal image. We then calculated an image of the volumetric F-actin density (Fig. 5d) by normalizing the F-actin fluorescence intensity by the local height of the platelet, which allows the accurate determination of the F-actin content per volume in the platelet. The platelet exhibited the typical regions with high stiffness at the leading edge (Fig. 5b, green arrows). These regions of high stiffness correlated with an increased F-actin fluorescence intensity (Fig. 5c, green arrows) and an increased volumetric F-actin density (Fig. 5d, green arrows). Note that the high F-actin fluorescence intensity in the platelet body is not present in the height-normalized volumetric F-actin density map. The volumetric F-actin density in the platelet lamellipodium and body is low and is increased only at the platelet edges.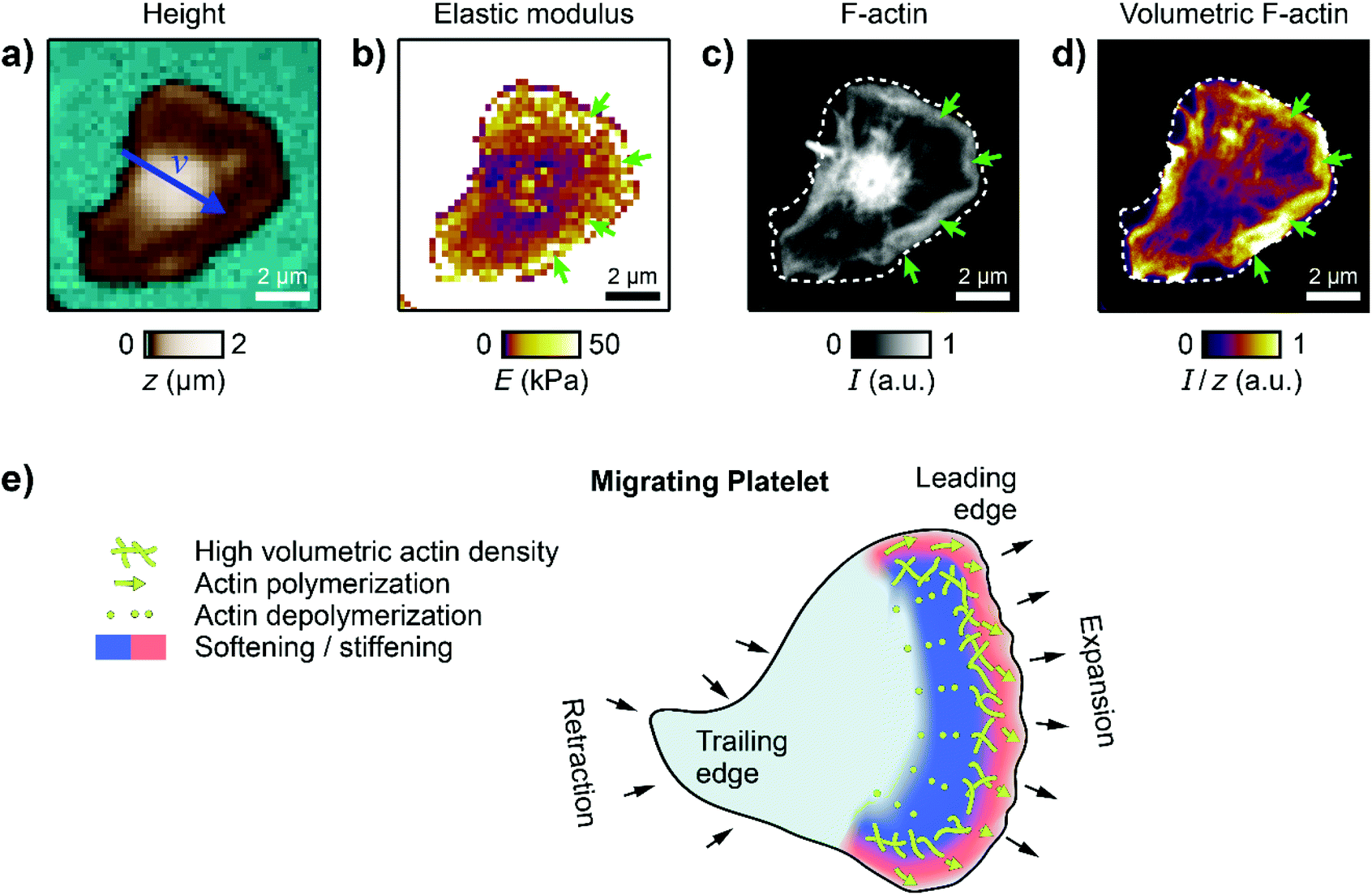 | ||
| Fig. 5 Cytoskeletal organization in migrating platelets. (a) SICM topography and (b) elastic modulus image of a migrating platelet. The blue arrow in (a) indicates the velocity vector v. The length of the arrow indicates the vector magnitude (|v| = 0.22 μm min−1) multiplied with a factor of 20 × 1 min. (c) Confocal fluorescence image of the F-actin cytoskeleton of the platelet in (a) and (b). (d) Volumetric F-actin density calculated as the F-actin fluorescence intensity divided by the local height of the platelet. Dashed white curves in (c) and (d) represent the platelet contour from SICM. Green arrows indicate the leading edge. F-actin was stained using fluorescently labelled phalloidin. (e) Schematic of the platelet mechanics during haptotactic migration. Actin polymerization drives the expansion of the leading edge in the direction of migration, which leads to an increase of the elastic modulus at the edge. Actin depolymerization leads to a decrease of the elastic modulus towards the lamellipodium. Image resolution: (a) and (b) 42 × 42 pixels (238 nm pixel size), (c) and (d) 880 × 880 pixels (11.4 nm pixel size). | ||
Discussion and conclusion
We investigated the topographical and mechanical behavior of human platelets during migration using SICM. This imaging technique has proven to be a versatile tool for the morphological and mechanical investigation of living platelets.20–22,30,38,39 Here, we provided the first direct mechanical measurements and the first scanning probe-based images of migrating platelets in vitro. Migrating platelets exhibit a characteristic shape anisotropy (Fig. 2), showing a polarization of their shape in the direction of migration. Shape polarization is generally essential for cell migration.40 The mechanical investigation showed a similar average elastic modulus for migrating and for non-migrating platelets. However, migrating platelets had a characteristic subcellular stiffness distribution with an increased stiffness at the leading edge (Fig. 3). An increased membrane tension in the leading edge41 may contribute to this increased stiffness. A dynamic analysis of the mechanics at the leading edge revealed a highly dynamic stiffening and softening process with changes of the elastic modulus by a factor of up to 5× per minute. These results demonstrate a huge mechanical influence of the dynamic actin polymerization and depolymerization process induced by the Arp2/3 complex in the leading edge in actin-driven cell migration (Fig. 5e).14,42–44 Accordingly, inhibition of actin polymerization by cytochalasin D decreased the overall stiffness, eliminated the stiffening region at the leading edge, and stopped platelet migration (Fig. 4). Using combined SICM and confocal fluorescence microscopy, we were able to accurately determine the volumetric F-actin density in the platelet, which is usually not possible by the sole use of optical methods. We identified regions of increased volumetric F-actin density at the leading edge (Fig. 5). The peripheral edge of platelets can generate high forces,45,46 which may be crucial for mechanical platelet function including haptotactic migration. Actin-driven migration is found in many mammalian cells.44,47,48 However, different types of migration like membrane-driven migration49 have been observed for immune cells,50 which do not require a strong integrin-ligand connection. Haptotactic platelet migration goes hand in hand with fibrinogen depletion and incorporation from the surface, which requires an active contribution of the actin cytoskeleton.13 We identified this contribution as a highly dynamic stiffening and softening processes in the leading edge (Fig. 5e). Our study thereby provides a direct connection between haptotactic migration and platelet mechanics.Author contributions
J. S, J. R., and H. v. E. performed the experiments. J. S., J. R., and T. E. S. designed the study and analyzed data. J. S. and J. R. drafted the manuscript. J. S., J. R., H. v. E., and T. E. S. interpreted data, discussed results, and revised the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This research was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) - Projektnummer 374031971 - TRR 240 and project number 335549539/GRK2381.References
- A. T. Nurden, Thromb. Haemostasis, 2011, 105, S13–S33 CrossRef CAS PubMed.
- M. R. Thomas and R. F. Storey, Thromb. Haemostasis, 2015, 114, 449–458 CrossRef PubMed.
- J. E. Aslan, A. Itakura, J. M. Gertz and O. J. T. McCarty, in Platelets and Megakaryocytes: Volume 3, Additional Protocols and Perspectives, ed. M. J. Gibbins and P. M. Mahaut-Smith, Springer, New York, 2012, Platelet Shape Change and Spreading, pp. 91–100, DOI:10.1007/978-1-61779-307-3_7.
- J. C. Loftus, J. Choate and R. M. Albrecht, J. Cell Biol., 1984, 98, 2019–2025 CrossRef CAS PubMed.
- J. G. White, E. L. Leistikow and G. Escolar, Blood Cells, 1990, 16, 43–70 CAS.
- S. P. Watson, Curr. Pharm. Des., 2009, 15, 1358–1372 CrossRef CAS PubMed.
- O. Garraud and F. Cognasse, Front. Immunol., 2015, 6, 70 Search PubMed.
- R. W. Lowenhaupt, M. A. Miller and H. I. Glueck, Thromb. Res., 1973, 3, 477–487 CrossRef.
- D. Feng, J. A. Nagy, K. Pyne, H. F. Dvorak and A. M. Dvorak, Int. Arch. Allergy Immunol., 1998, 116, 188–195 CrossRef CAS PubMed.
- S. C. Pitchford, S. Momi, S. Baglioni, L. Casali, S. Giannini, R. Rossi, C. P. Page and P. Gresele, Am. J. Respir. Crit. Care Med., 2008, 177, 604–612 CrossRef CAS PubMed.
- A. Witte, A. K. Rohlfing, B. Dannenmann, V. Dicenta, M. Nasri, K. Kolb, J. Sudmann, T. Castor, D. Rath, O. Borst, J. Skokowa and M. Gawaz, Cardiovasc. Res., 2021, 117, 903–917 CrossRef CAS PubMed.
- F. Gaertner, Z. Ahmad, G. Rosenberger, S. Fan, L. Nicolai, B. Busch, G. Yavuz, M. Luckner, H. Ishikawa-Ankerhold, R. Hennel, A. Benechet, M. Lorenz, S. Chandraratne, I. Schubert, S. Helmer, B. Striednig, K. Stark, M. Janko, R. T. Böttcher, A. Verschoor, C. Leon, C. Gachet, T. Gudermann, Y. S. M. Mederos, Z. Pincus, M. Iannacone, R. Haas, G. Wanner, K. Lauber, M. Sixt and S. Massberg, Cell, 2017, 171, 1368–1382.e1323 CrossRef CAS PubMed.
- A. Sarkar, D. N. LeVine, N. Kuzmina, Y. Zhao and X. Wang, Curr. Biol., 2020, 30, 4022–4032.e4025 CrossRef CAS PubMed.
- L. Nicolai, K. Schiefelbein, S. Lipsky, A. Leunig, M. Hoffknecht, K. Pekayvaz, B. Raude, C. Marx, A. Ehrlich, J. Pircher, Z. Zhang, I. Saleh, A.-K. Marel, A. Löf, T. Petzold, M. Lorenz, K. Stark, R. Pick, G. Rosenberger, L. Weckbach, B. Uhl, S. Xia, C. A. Reichel, B. Walzog, C. Schulz, V. Zheden, M. Bender, R. Li, S. Massberg and F. Gaertner, Nat. Commun., 2020, 11, 5778 CrossRef CAS PubMed.
- P. K. Hansma, B. Drake, O. Marti, S. A. Gould and C. B. Prater, Science, 1989, 243, 641–643 CrossRef CAS PubMed.
- Y. E. Korchev, C. L. Bashford, M. Milovanovic, I. Vodyanoy and M. J. Lab, Biophys. J., 1997, 73, 653–658 CrossRef CAS PubMed.
- C. Zhu, K. Huang, N. P. Siepser and L. A. Baker, Chem. Rev., 2021, 121, 11726–11768 CrossRef CAS PubMed.
- P. Novak, C. Li, A. I. Shevchuk, R. Stepanyan, M. Caldwell, S. Hughes, T. G. Smart, J. Gorelik, V. P. Ostanin, M. J. Lab, G. W. J. Moss, G. I. Frolenkov, D. Klenerman and Y. E. Korchev, Nat. Methods, 2009, 6, 279–281 CrossRef CAS PubMed.
- J. Seifert, J. Rheinlaender, P. Novak, Y. E. Korchev and T. E. Schäffer, Langmuir, 2015, 31, 6807–6813 CrossRef CAS PubMed.
- X. Liu, Y. Li, H. Zhu, Z. Zhao, Y. Zhou, A.-M. Zaske, L. Liu, M. Li, H. Lu, W. Liu, J.-F. Dong, J. Zhang and Y. Zhang, Platelets, 2015, 26, 480–485 CrossRef CAS PubMed.
- Y. Zhang, X. Liu, L. Liu, A. M. Zaske, Z. Zhou, Y. Fu, X. Yang, J. L. Conyers, M. Li, J. F. Dong and J. Zhang, Thromb. Haemostasis, 2013, 110, 331–339 CrossRef CAS PubMed.
- J. Seifert, J. Rheinlaender, F. Lang, M. Gawaz and T. E. Schäffer, Sci. Rep., 2017, 7, 4810 CrossRef PubMed.
- M.-J. Kraus, J. Seifert, E. F. Strasser, M. Gawaz, T. E. Schäffer and J. Rheinlaender, Platelets, 2016, 27, 541–546 CrossRef CAS PubMed.
- Y. Takahashi, Y. Zhou, T. Miyamoto, H. Higashi, N. Nakamichi, Y. Takeda, Y. Kato, Y. Korchev and T. Fukuma, Anal. Chem., 2020, 92, 2159–2167 CrossRef CAS PubMed.
- S. M. Leitao, B. Drake, K. Pinjusic, X. Pierrat, V. Navikas, A. P. Nievergelt, C. Brillard, D. Djekic, A. Radenovic, A. Persat, D. B. Constam, J. Anders and G. E. Fantner, ACS Nano, 2021, 15, 17613–17622 CrossRef CAS PubMed.
- J. Rheinlaender and T. E. Schäffer, Soft Matter, 2013, 9, 3230–3236 RSC.
- L. Sachs, C. Denker, A. Greinacher and R. Palankar, Res. Pract. Thromb. Haemostasis, 2020, 4, 386–401 CrossRef PubMed.
- R. W. Clarke, P. Novak, A. Zhukov, E. J. Tyler, M. Cano-Jaimez, A. Drews, O. Richards, K. Volynski, C. Bishop and D. Klenerman, Soft Matter, 2016, 12, 7953–7958 RSC.
- Y. Chen, G. B. Sukhorukov and P. Novak, Nanoscale, 2018, 10, 16902–16910 RSC.
- J. Rheinlaender, S. Vogel, J. Seifert, M. Schächtele, O. Borst, F. Lang, M. Gawaz and T. E. Schäffer, Thromb. Haemostasis, 2015, 113, 305–311 CrossRef PubMed.
- V. S. Kolmogorov, A. S. Erofeev, E. Woodcock, Y. M. Efremov, A. P. Iakovlev, N. A. Savin, A. V. Alova, S. V. Lavrushkina, I. I. Kireev, A. O. Prelovskaya, E. V. Sviderskaya, D. Scaini, N. L. Klyachko, P. S. Timashev, Y. Takahashi, S. V. Salikhov, Y. N. Parkhomenko, A. G. Majouga, C. R. W. Edwards, P. Novak, Y. E. Korchev and P. V. Gorelkin, Nanoscale, 2021, 13, 6558–6568 RSC.
- S. Simeonov and T. E. Schäffer, Anal. Chem., 2019, 91, 9648–9655 CrossRef CAS PubMed.
- J. Seifert, J. Rheinlaender and T. E. Schäffer, Jpn. J. Appl. Phys., 2018, 57, 08NB02 CrossRef.
- C. Rotsch, F. Braet, E. Wisse and M. Radmacher, Cell Biol. Int., 1997, 21, 685–696 CrossRef CAS PubMed.
- Y. Shimizu, T. Kihara, S. M. A. Haghparast, S. Yuba and J. Miyake, PLoS One, 2012, 7, e34305 CrossRef CAS PubMed.
- J. Seifert, H. von Eysmondt, M. Chatterjee, M. Gawaz and T. E. Schäffer, Cells, 2021, 10, 2932 CrossRef CAS PubMed.
- J. F. Casella, M. D. Flanagan and S. Lin, Nature, 1981, 293, 302–305 CrossRef CAS PubMed.
- S. Vogel, R. Bodenstein, Q. Chen, S. Feil, R. Feil, J. Rheinlaender, T. E. Schäffer, E. Bohn, J.-S. Frick, O. Borst, P. Münzer, B. Walker, J. Markel, G. Csanyi, P. J. Pagano, P. Loughran, M. E. Jessup, S. C. Watkins, G. C. Bullock, J. L. Sperry, B. S. Zuckerbraun, T. R. Billiar, M. T. Lotze, M. Gawaz and M. D. Neal, J. Clin. Invest., 2015, 125, 4638–4654 CrossRef PubMed.
- S. Simeonov and T. E. Schäffer, Nanoscale, 2019, 11, 8579–8587 RSC.
- W.-J. Rappel and L. Edelstein-Keshet, Curr. Opin. Syst. Biol., 2017, 3, 43–53 CrossRef PubMed.
- A. D. Lieber, Y. Schweitzer, M. M. Kozlov and K. Keren, Biophys. J., 2015, 108, 1599–1603 CrossRef CAS PubMed.
- A. Mogilner and L. Edelstein-Keshet, Biophys. J., 2002, 83, 1237–1258 CrossRef CAS PubMed.
- N. Watanabe and T. J. Mitchison, Science, 2002, 295, 1083–1086 CrossRef CAS PubMed.
- T. D. Pollard and G. G. Borisy, Cell, 2003, 112, 453–465 CrossRef CAS PubMed.
- S. Schwarz Henriques, R. Sandmann, A. Strate and S. Köster, J. Cell Sci., 2012, 125, 3914–3920 Search PubMed.
- J. M. Brockman, H. Su, A. T. Blanchard, Y. Duan, T. Meyer, M. E. Quach, R. Glazier, A. Bazrafshan, R. L. Bender, A. V. Kellner, H. Ogasawara, R. Ma, F. Schueder, B. G. Petrich, R. Jungmann, R. Li, A. L. Mattheyses, Y. Ke and K. Salaita, Nat. Methods, 2020, 17, 1018–1024 CrossRef CAS PubMed.
- A. Mogilner and G. Oster, Biophys. J., 1996, 71, 3030–3045 CrossRef CAS PubMed.
- T. J. Mitchison and L. P. Cramer, Cell, 1996, 84, 371–379 CrossRef CAS PubMed.
- P. R. O'Neill, J. A. Castillo-Badillo, X. Meshik, V. Kalyanaraman, K. Melgarejo and N. Gautam, Dev. Cell, 2018, 46, 9–22.e24 CrossRef PubMed.
- N. Garcia-Seyda, V. Seveau, F. Manca, M. Biarnes-Pelicot, M.-P. Valignat, M. Bajénoff and O. Theodoly, Biol. Cell, 2021, 113, 28–38 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr01187e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |