
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceParticle nanosizing and coating with an ionic liquid: two routes to improve the transport properties of Na3V2(PO4)2FO2†
Jacob
Olchowka
 *abc,
Runhe
Fang
bd,
Rafael
Bianchini Nuernberg
a,
Chloé
Pablos
abe,
Dany
Carlier
abc,
Sophie
Cassaignon
*bd and
Laurence
Croguennec
abc
*abc,
Runhe
Fang
bd,
Rafael
Bianchini Nuernberg
a,
Chloé
Pablos
abe,
Dany
Carlier
abc,
Sophie
Cassaignon
*bd and
Laurence
Croguennec
abc
aUniv. Bordeaux, CNRS, Bordeaux INP, ICMCB, UMR 5026, F-33600 Pessac, France. E-mail: jacob.olchowka@icmcb.cnrs.fr
bRS2E, Réseau Français sur le Stockage Electrochimique de l'Energie, CNRS 3459, 80039 Amiens Cedex 1, France
cALISTORE-ERI European Research Institute, CNRS 3104, 80039 Amiens Cedex 1, France
dSorbonne Université, CNRS, Laboratoire Chimie de la Matière Condensée de Paris, LCMCP, UMR 7574, 4 Place Jussieu, 75005 Paris, France
eLaboratoire de Réactivité et de Chimie des Solides, Université de Picardie Jules Verne, CNRS-UMR 7314, F-80039 Amiens Cedex 1, France
First published on 23rd May 2022
Abstract
Na3V2(PO4)2FO2 is a promising candidate for practical use as a positive electrode material in Na-ion batteries thanks to its high voltage and excellent structural stability upon cycling. However, its limited intrinsic transport properties limit its performance at fast charge/discharge rates. In this work, two efficient approaches are presented to optimize the electrical conductivity of the electrode material: particle nanosizing and particle coating with an ionic liquid (IL). The former reveals that particle downsizing from micrometer to nanometer range improves the electronic conductivity by more than two orders of magnitude, which greatly improves the rate capability without affecting the capacity retention. The second approch dealing with an original surface modification by applying an IL coating strongly enhances the ionic mobility and offers new perspectives to improve the energy storage performance by designing the electrode materials’ surface composition.
1. Introduction
Human-induced climate problem is already impacting the weather and producing extreme climate in every inhabited region across the world such as frequent heatwaves, heavy precipitation, droughts and tropical cyclones.1 To face this problem, the main solution is based on limiting the cumulative CO2 and other greenhouse gas emissions. To do so, one of the major methods is to convert the fossil fuel-based power station, led by coal-fired power plants, into alternative energy like solar and wind energy accompanied by performant energy storage systems, which can store the additional power during peak production periods and supply it to the grid during low hours to reduce burden on the electric grid. Complementary to the widely used lithium ion batteries, sodium ion batteries are environmentally friendly and a reliable choice for large-scale energy storage.2–5 Among the various families of positive electrode materials, despite weight penalty versus layered oxides, polyanionic ones attract much attention thanks to their high average operating potential and structural stability. The most studied and promising are NASICON Na3V2(PO4)3,6,7 Na4MnV(PO4)3,8,9 Na4FeV(PO4)3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10,11 and vanadium phosphate oxyfluoride series Na3V3+2−yV4+y(PO4)2F3−yOy (NVPFOy).12,13 Dealing with this latter, Na3V4+2(PO4)2FO2, the most oxidized member of NVPFOy family, exhibits a slightly lower average working potential than that of Na3V3+2(PO4)2F3 (∼3.8 V vs. Na/Na+ for NVPFO2versus ∼3.95 V vs. Na/Na+ for NVPF for a similar capacity).12,14 However, oxygen for fluorine substitution enhances Na+ mobility and stabilizes the structure at overcharged states, making NVPFO2 and mixed anionic NVPFOy still of interest for practical purposes.12,15
10,11 and vanadium phosphate oxyfluoride series Na3V3+2−yV4+y(PO4)2F3−yOy (NVPFOy).12,13 Dealing with this latter, Na3V4+2(PO4)2FO2, the most oxidized member of NVPFOy family, exhibits a slightly lower average working potential than that of Na3V3+2(PO4)2F3 (∼3.8 V vs. Na/Na+ for NVPFO2versus ∼3.95 V vs. Na/Na+ for NVPF for a similar capacity).12,14 However, oxygen for fluorine substitution enhances Na+ mobility and stabilizes the structure at overcharged states, making NVPFO2 and mixed anionic NVPFOy still of interest for practical purposes.12,15
Na3V3+2−yV4+y(PO4)2F3−yOy is composed of V2O8+yF3−y bi-octahedral units, which are connected by PO4 groups to generate a three-dimensional structure. Although the robust framework supported by PO43− groups leads to stable long cycling capability, the isolated transition metals in this structure result in rather low intrinsic electronic conductivity.16 Additionally, Notten et al. have recently demonstrated that the poor rate performance of NVPF is related to the low Na+ diffusion coefficient in the material.17 The authors have shown that hard carbon diffusion coefficient is one order of magnitude higher than that of the polyanionic material. Thus, the NVPFOy family materials suffer from significant capacity loss at high charging/discharging rates. In recent years, numerous methods such as the application of a carbon coating or the formation of composites with carbons (carbon nanotubes, reduced graphene oxides or amorphous carbons),18–21 partial transition metal substitution (Al, Cr, Mn, Y, Zr, Ga, etc.)22–28 and morphology design (flower, sand rose, nano/micro-sphere, nano/micro-cube, etc.)29–34 have been proposed to overcome this problem. Among the various synthesis methods, morphology control and hybridization with a carbon-based conductive phase are often carried out by hydrothermal/solvothermal synthesis.34,35 However, this synthesis process leads most often to partial oxidation of vanadium and to a V3+/V4+ ratio difficult to be controlled in the final composition, which drastically affects the energy storage performance and thus makes it difficult to evaluate the sole influence of morphology.34,36–39 Recently, Olchowka et al. have reported an original ionothermal synthesis method leading to NVPFO2 particles coated with an EMI TFSI ionic liquid (IL).40 Their energy storage performance appears promising, even if the effect of IL coating is not fully understood yet.40
In this work, Na3V2(PO4)2FO2 was synthesized by three different methods (solid-state reaction, ionothermal and solvothermal syntheses) to investigate the effects on the electrical properties of particle downsizing and of formation of an ionic liquid coating. For each compound, the final V4+ oxidation state, as well as the composition, was meticulously confirmed by combining X-ray diffraction and different spectroscopic characterization methods. The influence of the particle size and IL coating on electronic and ionic conductivities was investigated by electrochemical impedance spectroscopy, whereas electrochemical tests allowed us to evaluate the impact on the energy storage performance.
2. Experimental section
2.1 Material preparation
NVPFO2 materials were synthesized by three different methods. NVPFO2-Iono was obtained by ionothermal synthesis. α-VOPO4·2H2O (homemade) and NaF (molar ratio of 2/3) were placed in a Teflon Parr autoclave with 6 mL of EMI-TFSI (1-ethyl-3-methyl-imidazolium bis(trifluoromethylsulfonyl)imide) and heated at 220 °C for 10 hours, similar to the conditions described in our previous work.40 The polycrystalline powder was recovered by centrifugation and washed with ethanol. NVPFO2-Nano was obtained by solvothermal synthesis. A stoichiometric mixture of VO(acac)2 (Sigma-Aldrich), NaF (Sigma-Aldrich) and H3PO4 (molar ratio of 1/1.5/1) was placed in a 100 mL Teflon Parr autoclave with a mixture of 4.5 mL ethanol/4.5 mL distilled water and heated at 180 °C for 10 hours. NVPFO2-Bulk was prepared by a solid-state reaction of VPO4 (homemade), VOPO4 (homemade), NaF (Sigma-Aldrich) and Na2CO3 (Sigma-Aldrich) taken in a molar ratio of 1:1:1:1, as described earlier.41 Mechanical grinding was first applied in order to get a homogeneous mixture, and then the resulting powder was pelletized and heated under argon flow in a gold crucible at 550 °C for 2 hours. VPO4 and VOPO4 were obtained from an α-VOPO4·2H2O homemade precursor. First, α-VOPO4·2H2O was obtained by heating 5 g of V2O5 (Sigma-Aldrich) in a mixture composed of 40 mL of phosphoric acid, 110 mL of nitric acid (Sigma-Aldrich) and 130 mL of distilled water at 100 °C for 3 hours. The resulting yellowish powder was washed with acetone. Freshly obtained VOPO4·2H2O was dehydrated at 250 °C under O2 to produce VOPO4. The latter was then reduced under argon/hydrogen flow at 890 °C for 2 hours to synthesize VPO4.
2.2 Characterization techniques
X-ray diffraction (XRD) measurements were carried out using a BRUKER D8 ADVANCE diffractometer in θ–θ configuration, equipped with a Cu Kα1,2 X-ray source. The acquisition was performed in the 2θ angular range of 10°–140° with a step size of 0.0197°. The Rietveld refinements were performed using the FullProf Suite.42Scanning electron microscopic (SEM) images were acquired without any conductive deposition using a Hitachi Model S-3400N microscope. Gold deposition was made on the NVPFO2-Bulk sample to improve the quality of the images. SEM-FEG (field emission gun) images were obtained using a Hitachi SU-70 microscope. Transmission electron microscopy (TEM) was carried out using a Tecnai spirit G2 microscope. The powder was sonicated for 20 min in ethanol before the analysis.
The chemical analysis of the Na, P and V contents was performed by inductively coupled plasma-optical emission spectroscopy (ICP-OES) using a Varian Model 720-ES spectrometer, after a complete dissolution of the powders into a concentrated hydrochloric acid (HCl) solution.
X-ray photoelectron spectroscopy (XPS) spectra were recorded using an Omicron Argus X-ray photoelectron spectrometer for an applied power of 280 W and a monochromatic Al-Kα radiation source (1486.6 eV). Charge correction was done by calibrating the C 1s peak at a binding energy of 284.8 eV.
Raman spectra were recorded using a confocal LabRAM HR Evolution micro-spectrometer from Horiba, with a 633 nm laser source. They were collected in the range of 100–1300 cm−1, using a 10.6 mm (NA 0.5) focal length lens and with an acquisition time of 20 s and 40 accumulations.
Magic angle spinning nuclear magnetic resonance (MAS NMR) experiments were carried our using standard Bruker 2.5 mm MAS probes at a spinning frequency of 30 kHz. 31P MAS NMR spectra were recorded using a Bruker Avance III 100 MHz spectrometer at a resonance frequency of 40.6 MHz. A Hahn echo sequence was used synchronized with one rotor period rotation, with a 90° pulse of 1.1 μs and a recycle delay of 1 s. H3PO4 85% (Sigma-Aldrich) was used as an external reference for 0 ppm chemical shift. 23Na MAS-NMR spectra were recorded using a Bruker Avance 500 MHz spectrometer equipped with an 11.7 T widebore magnet (operating at a Larmor frequency of 132.3 MHz for 23Na). A short pulse length of 1.1 μs corresponding to a selective π/8 pulse determined using a 0.1 M NaCl aqueous solution was employed. Chemical shifts were referenced relative to an aqueous 0.1 M NaCl solution at 0 ppm. The spectral width was set to 1 MHz and the recycle delay of 0.5 s, which was long enough to avoid T1 saturation effects. The baseline distortions resulting from the spectrometer dead time (5–10 μs) were removed computationally by using a polynomial baseline correction routine.
The electrical properties of Na3V2(PO4)2FO2 compounds were investigated by electrochemical impedance spectroscopy (EIS). For this purpose, the powders were pressed into pellets of 13 mm diameter applying 200 MPa by means of a uniaxial press. Prior to pressing, NVPFO2-Nano and NVPFO2-Bulk powders were mixed with camphor (about 2 wt%) to provide enough mechanical strength allowing the extraction of the pellets from the die after pressing. Subsequently, the pellets were heat-treated at 250 °C for 1 h under air to evaporate the camphor. Contrary to the two other samples, NVPFO2-Iono could be well pelletized without camphor. Moreover, the evaporation of camphor at 250 °C would decompose the IL. Finally, gold electrodes were sputtered on the parallel and opposite faces of each pellet to ensure electrical contacts. EIS measurements were performed using a Solartron 1260 Impedance/Gain Phase Analyzer in the frequency range between 1 MHz and 1 Hz, with an applied root mean square voltage of 100 mV, 10 points per decade and 20 measures per point. Impedance of all the samples were measured under heating and cooling in air on a two-probe sample holder and over the temperature range of 100–300 °C. Among them, NVPFO2-Iono was left at 300 °C for 6 h to completely remove the IL and then taken for the cooling process.
The electrochemical performance of the materials were tested in CR2032-type coin cells. The cells were assembled in an argon-filled glovebox (H2O, O2 ≤ 0.1 ppm), with sodium metal as the counter electrode. The positive electrodes were prepared as a mixture containing an active material, carbon black, and polyvinylidene fluoride (PVDF) in NMP (N-methyl-2-pyrrolidone) in the ratio of 88/7/5 (by wt%). After two hours of thorough mixing, the black ink was casted as a flat film by a doctor blade on an aluminum foil and dried in an oven at 80 °C. Disks were cut, pressed under 5 tons and, finally, dried overnight at 80 °C under vacuum. The electrodes have a typical active mass loading around 4.5–5 mg cm−2. A homemade electrolyte containing a 1 mol L−1 solution of NaPF6 (Strem Chemical; 99%) in ethylene carbonate and dimethyl carbonate (EC/DMC = 1/1) with 2 wt% of fluoroethylene carbonate (FEC) was used for all the electrochemical tests. The assembled cells were cycled in a galvanostatic mode, from C/20 to 2C cycling rates between 2.5 and 4.3 V vs. Na+/Na. The theoretical capacity being 130 mA h g−1 (corresponding to 2 Na+ reversibly extracted from NVPFO2), the rate C/20 corresponds to the exchange of 2 Na+ in 20 hours. Galvanostatic intermittent titration technique (GITT) measurements were performed in half-cells versus Na after three cycles of formation at C/20. GITT measurements of NVPFO2 involved a series of 30 min galvanostatic charge/discharge at C/25 followed by relaxation up to 1 mV h−1 between 2.5 and 4.3 V to achieve a nearly steady state.
Full cells HC//NVPFO2-Nano were tested in CR2032-type coin cells with a mass ratio of m(NVPFO2)/m(HC) ≈ 2.35. The positive electrode possesses a diameter of 16 mm (∼9 mg), whereas the negative electrode is slightly smaller (15 mm). The assembled full cells were cycled in the galvanostatic mode, from C/20 to 2C cycling rates (based on NVPFO2 electrode) in the voltage range of 2.5 to 4.3 V and in the same electrolyte, as described previously.
3 Results and discussion
3.1 Structural and morphological characterization
The powder XRD patterns illustrated in Fig. 1 and the Rietveld refinements presented in Fig. S1† confirm that the three synthesis methods (solid-state reaction, solvothermal and ionothermal) lead to the synthesis of the pure Na3V4+2(PO4)2FO2 (NVPFO2) phase. For each compound, all the diffraction peaks could be indexed in the space group P42/mnm, and the lattice parameters, reported in Table 1, are in good agreement with those of Na3V4+2(PO4)2FO2, the end member composition of the Na3V3+2−yV4+y(PO4)2F3−yOy (0 ≤ y ≤ 2) solid solution.12,40,43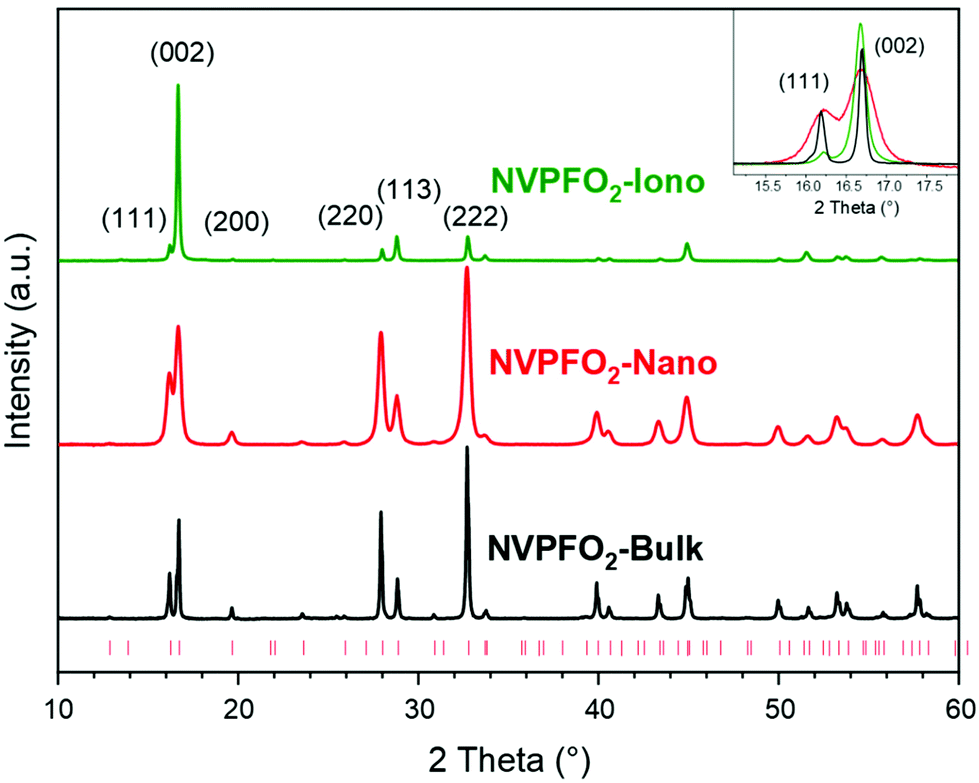 | ||
| Fig. 1 X-ray diffraction patterns of NVPFO2-Iono (green curve), NVPFO2-Nano (red curve) and NVPFO2-Bulk (black curve). The red ticks correspond to theoretical positions as reported in ICDD 01-083-7199. The inset shows the enlargement of the (111) and (002) reflections. | ||
| a (Å) | c (Å) | (220) (nm) | (002) (nm) | Space group | |
|---|---|---|---|---|---|
| NVPFO2-Iono | 9.0098(1) | 10.6278(1) | 52 | 52 | P42/mnm |
| NVPFO2-Nano | 9.0214(2) | 10.6216(3) | 23 | 21 | P42/mnm |
| NVPFO2-Bulk | 9.0330(1) | 10.6137(2) | 64 | 70 | P42/mnm |
Additionally, ICP analyses confirm the expected cationic ratio (Table S1†). A comparison of the X-ray diagrams (Fig. 1) clearly shows differences in the diffraction peak width and preferential orientation, suggesting huge variations in the crystallite size and morphology within the series of NVPFO2. Indeed, compared to observations made for NVPFO2-Nano and NVPFO2-Bulk, the very intense (002) reflection observed for NVPFO2-Iono suggests an oriented particle growth in the (ab) plane leading to platelet (2D) morphology. Furthermore, dissimilar diffraction peak broadening, highlighted in the inset of Fig. 1, unveils disparate crystallite sizes caused by the different synthesis conditions. The coherent domains calculated using the Scherrer equation are reported in Table 1, and they reveal that the high-temperature solid state reaction leads to the biggest crystallites (∼70 nm), whereas the solvothermal synthesis to the smallest ones (∼20 nm). Moreover, the similar coherent domains calculated with the (002) and (220) reflections suggest an isotropic crystal growth for all synthesis routes, despite an anisotropic particle growth by the ionothermal reaction. Finally, the internal strains generated by limited crystallite sizes distributed within the oriented primary particles result in difficulties to perfectly fit the peak profile during structural refinement, which could explain the slightly different lattice parameters within the NVPFO2 series (Table 1 and Fig. S1†). Consequently, the chemical composition has to be confirmed by local environmental characterization techniques such as ss-NMR, as it will be done later.
SEM, SEM-FEG (Fig. 2) and TEM images (Fig. S2†) further confirm the various morphologies of NVPFO2 materials obtained by the different synthesis methods. NVPFO2-Iono, which was obtained by a topochemical reaction from layered α-VOPO4·2H2O,40 was characterized by aggregated flakes with an average length of around 5–10 μm and a thickness of 100–200 nm (Fig. 2a, b and S2†). The small fragments that can be distinguished probably come from pieces of big flakes. HRTEM analyses further revealed that the platelets exhibit the (220) crystallographic planes through lattice fringes observation (Fig. S3a and b†), which perfectly fit to the exalted intensity of the (002) diffraction peak observed by XRD. Thus, it can be concluded that the Na+ diffusion channels along the [110] and [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] directions are situated in the plane of the flakes (Fig. S4†). NVPFO2-Nano synthesized by a solvothermal process presents a quite different morphology (Fig. 2c and d). It was characterized by aggregates made of randomly agglomerated spherical secondary particles of diameter ∼ 500 nm. These latter were constructed with smaller primary particles of size around 30 nm, which is in good agreement with the crystallite size determined by XRD (Table 1). The different lattice fringes observed by HRTEM (Fig. S3†) for NVPFO2-Nano (0.32 nm (220), 0.53 nm (002) and 0.55 nm (111)) support the spherical morphology of the small primary particles and the absence of preferential orientation. Finally, the shape of NVPFO2-Bulk obtained by the high-temperature solid-state reaction approximates to cubes varying from hundreds of nanometers to several micrometers (Fig. 2e and f). Moreover, the thickness of NVPFO2-Bulk cubes makes the lattice fringe measurement impossible.
0] directions are situated in the plane of the flakes (Fig. S4†). NVPFO2-Nano synthesized by a solvothermal process presents a quite different morphology (Fig. 2c and d). It was characterized by aggregates made of randomly agglomerated spherical secondary particles of diameter ∼ 500 nm. These latter were constructed with smaller primary particles of size around 30 nm, which is in good agreement with the crystallite size determined by XRD (Table 1). The different lattice fringes observed by HRTEM (Fig. S3†) for NVPFO2-Nano (0.32 nm (220), 0.53 nm (002) and 0.55 nm (111)) support the spherical morphology of the small primary particles and the absence of preferential orientation. Finally, the shape of NVPFO2-Bulk obtained by the high-temperature solid-state reaction approximates to cubes varying from hundreds of nanometers to several micrometers (Fig. 2e and f). Moreover, the thickness of NVPFO2-Bulk cubes makes the lattice fringe measurement impossible.
 | ||
| Fig. 2 (a and b) SEM images of NVPFO2-Iono. (c and d) SEM-FEG images of NVPFO2-Nano. (e and f) SEM-FEG images of NVPFO2-Bulk. | ||
The series of NVPFO2 was further characterized by various spectroscopic techniques to probe the Na, P and V local environments and confirm the composition deduced from Rietveld refinements. As illustrated in the Raman spectra in Fig. 3, all materials possess the typical signature of Na3V2(PO4)FO2 with peaks at 539 cm−1, 943 cm−1, 1010 cm−1 and 1052 cm−1 attributed to the vibrations of V–O, V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and phosphate groups respectively.39,44,45 As reported in our previous work on the topochemical reaction mechanism happening during the ionothermal synthesis of NVPFO2, the ionic liquid functionalizes the particle surface to create an IL coating, which can be seen as an “artificial solid electrolyte interface”.40 This latter, which in some cases stabilizes the electrode material upon cycling and favors ionic adsorption on the surface and/or alkaline diffusion through the material,46–48 is observed on the NVPFO2-Iono spectrum (Fig. 3) and on HRTEM images (Fig. S5†). Indeed, the band marked with a star on the Raman spectrum is in rather good agreement with those expected for the EMI TFSI ionic liquid.49–52 IL can also be detected on the surface of NVPFO2-Iono by XPS analysis that reveals the presence of N 1s and S 2p peaks originating from the imidazolium ring and TFSI respectively (as shown by the surveys in the energy range of 0–1100 eV in Fig. S6† and by the quantification given in Table S2†). Meanwhile, the F 1s spectra (Fig. 4a) clearly show a signal at 684.1 eV for all the compounds, corresponding to fluorine in the polyanionic materials, whereas NVPFO2-Iono presents a second peak at 688.6 eV, which is in agreement with the presence of CF3 from EMI TFSI.47,53,54 However, this IL coating does not seem to affect the inorganic core phase. The V 2p XPS spectra of the NVPFO2 series are presented in Fig. 4b, and the spectrum of each compound exhibits two broad peaks at 517.3 eV and 524.8 eV corresponding to the V 2p3/2 and V 2p1/2 transitions. These binding energies are consistent with the expected ones for this polyanionic material with the V4+ environment and demonstrate for NVPFO2-Iono that the IL coating does not affect the electronic structure of vanadium present near the surface.55
O and phosphate groups respectively.39,44,45 As reported in our previous work on the topochemical reaction mechanism happening during the ionothermal synthesis of NVPFO2, the ionic liquid functionalizes the particle surface to create an IL coating, which can be seen as an “artificial solid electrolyte interface”.40 This latter, which in some cases stabilizes the electrode material upon cycling and favors ionic adsorption on the surface and/or alkaline diffusion through the material,46–48 is observed on the NVPFO2-Iono spectrum (Fig. 3) and on HRTEM images (Fig. S5†). Indeed, the band marked with a star on the Raman spectrum is in rather good agreement with those expected for the EMI TFSI ionic liquid.49–52 IL can also be detected on the surface of NVPFO2-Iono by XPS analysis that reveals the presence of N 1s and S 2p peaks originating from the imidazolium ring and TFSI respectively (as shown by the surveys in the energy range of 0–1100 eV in Fig. S6† and by the quantification given in Table S2†). Meanwhile, the F 1s spectra (Fig. 4a) clearly show a signal at 684.1 eV for all the compounds, corresponding to fluorine in the polyanionic materials, whereas NVPFO2-Iono presents a second peak at 688.6 eV, which is in agreement with the presence of CF3 from EMI TFSI.47,53,54 However, this IL coating does not seem to affect the inorganic core phase. The V 2p XPS spectra of the NVPFO2 series are presented in Fig. 4b, and the spectrum of each compound exhibits two broad peaks at 517.3 eV and 524.8 eV corresponding to the V 2p3/2 and V 2p1/2 transitions. These binding energies are consistent with the expected ones for this polyanionic material with the V4+ environment and demonstrate for NVPFO2-Iono that the IL coating does not affect the electronic structure of vanadium present near the surface.55
 | ||
| Fig. 3 Raman spectra of NVPFO2-Iono (green), NVPFO2-Nano (red) and NVPFO2-Bulk (black). The band marked with a star on the Raman spectrum corresponds to the EMI TFSI ionic liquid. | ||
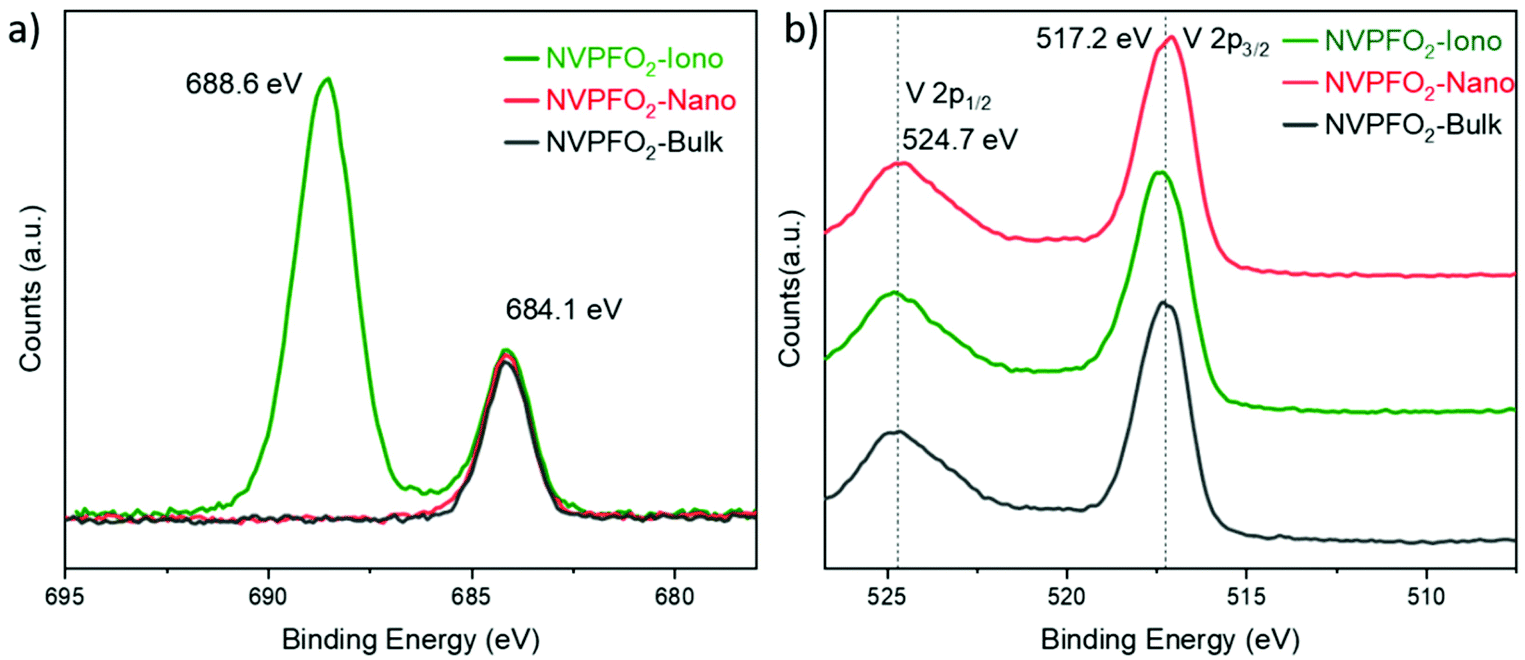 | ||
| Fig. 4 (a) F 1s XPS spectra and (b) V 2p XPS spectra for all the NVPFO2 materials. | ||
For all compounds, the 23Na ss-NMR spectrum (Fig. 5a) reveals a main peak at ∼77 ppm, whose position is mainly due to the hyperfine interaction between the Na+ nucleus and the unpaired electron of paramagnetic V4+ ions and is characteristic of the Na(OV4+)2 environment.12,56 The absence of signals at a higher isotropic shift confirms the absence of V3+ in these phases and validates the Na3V4+2(PO4)FO2 composition.14,36,41,56 Meanwhile, the broader signal for NVPFO2-Nano could be the consequence of disorder due to particle downsizing.3431P ss-NMR spectra obtained for all NVPFO2 show a very intense signal at ∼0 ppm corresponding to the P(OV4+)4 local environment (Fig. 5b).56 Once again the absence of signals induced by the Fermi contact shift at ∼1500, 3000 ppm, 4500 ppm or 6000 ppm, as previously observed for the Na3V2(PO4)2F3–yOy series, excludes the existence of V3+ in the compounds.22,56 The enlarged area in Fig. 5c unfolds more details on the signal located around 0 ppm. Three distinct signals are observed for NVPFO2-Bulk that most probably correspond to the inequivalent local environments for the phosphorus. However, the signals of NVPFO2-Nano and NVPFO2-Iono are broader, asymmetric and less resolved due to the small changes on the local environment brought by the nanosizing/specific morphology.57 Therefore, the use of different synthesis methods allows to obtain Na3V4+2(PO4)FO2 materials with extremely different particle size, with and without coating, thus offering an ideal series to study the particle downsizing and IL coating effects on the transport properties and energy storage performance.
 | ||
| Fig. 5 (a) 23Na solid-state MAS NMR spectra of NVPFO2-Iono (green), NVPFO2-Nano (red) and NVPFO2-Bulk (black). (b) Corresponding 31P solid-state MAS NMR spectra. (c) Enlargement of the signals around 0 ppm. | ||
3.2 Transport kinetics and energy storage performance
The energy storage performance of the three Na3V2(PO4)2FO2 samples was first evaluated by cyclic voltammetry and galvanostatic measurements in the half-cell configuration. The electrode formulation (88 wt% active material/7 wt% carbon black/5 wt% PVDF) and the mass loading of ∼5 mg cm−2 were expected to emphasize on the influence of different NVPFO2 electrode materials. Fig. 6a–c shows the galvanostatic charge–discharge curves obtained within the potential window 2.5–4.3 V vs. Na+/Na at different current intensities, whereas the reversible capacity evolution depending on the C-rate is represented in Fig. 6d. All electrode materials exhibit two reversible plateaus or pseudo-plateaus at around 3.6 and 4 V vs. Na+/Na, attributed to the V5+/V4+ redox couple that is activated by reversible Na+ extraction and in perfect agreement with the values reported in the literature.12,58 Sharma et al. reported that the reaction mechanisms upon charge and discharge are composed of first-order transitions (two-phase transition) separated by second-order ones (solid solution).59 At C/20, NVPFO2-Nano and NVPFO2-Iono exhibit similar reversible high capacities (∼115 mA h g−1 and ∼110 mA h g−1 respectively), close to the theoretical one (130 mA h g−1). However, NVPFO2-Nano possesses a much better capacity retention when the charge rate increases up to 2C. Meanwhile, the discharge capacity of NVPFO2-Bulk already merely reaches 67 mA h g−1 at C/20 and quickly drops when the current becomes higher. The full initial capacity recovering after several cycles of fast charge/discharge clearly shows that the difference in performance mainly originates from the kinetics limitations specific to the electrode material. | ||
| Fig. 6 (a–c) Charge and discharge curves of all the NVPFO2 materials at different cycling rates. (d) Rate capabilities obtained for all these materials, from C/20 to 2C. | ||
This is supported by the evolution of polarization when the charge rate increases, as illustrated in Fig. 7, which shows the first derivative curves of the 2nd galvanostatic cycle (Fig. 6) performed at C/20 and C/5.
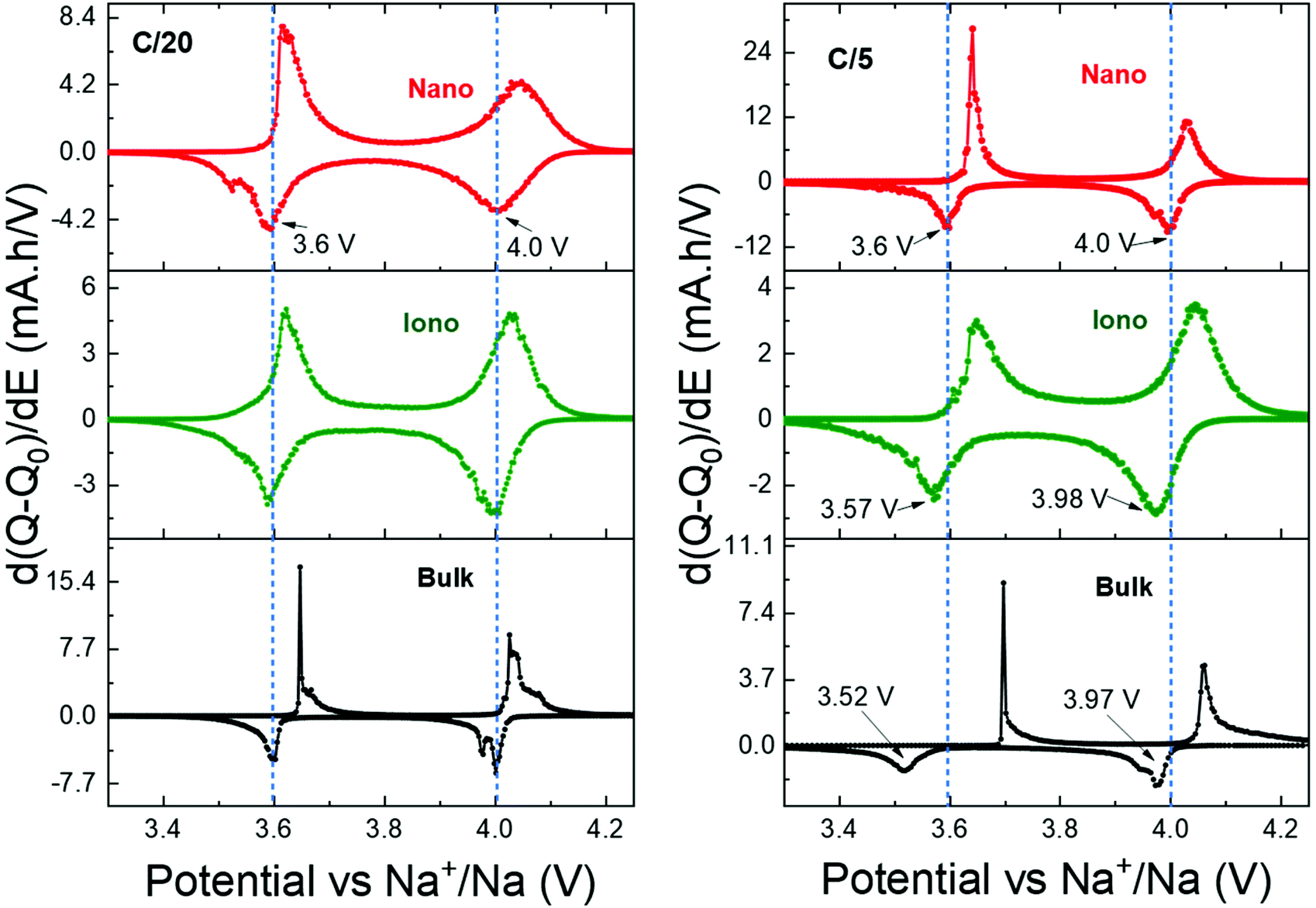 | ||
| Fig. 7 First derivative curves of the second cycle of galvanostatic charge discharge process of all the NVPFO2 materials at C-rates of C/20 and C/5. | ||
At C/20, the average discharge redox potentials for all electrode materials are observed at 4 V and 3.6 V vs. Na+/Na. These potentials remain unchanged for NVPFO2-Nano when the charge rate increases to C/5, whereas the values drop to 3.98 V and 3.57 V for NVPFO2-Iono and 3.97 V and 3.52 V for NVPFO2-Bulk. Similarly, the ΔEp (potential difference between charge/discharge) increases for NVPFO2-Iono and more significantly for NVPFO2-Bulk, which leads to a system with slower kinetics, lower average discharge potential and, thus, lower energy density. This clearly shows that the resistance R (sum of electrical and ionic resistances) follows the trend RBulk > RIono > RNano and, thus, strongly depends on the active material, as it is the only variable parameter in this electrode series. Therefore, the transport properties were further investigated to get a deeper insight into the influence of the particle size and IL coating.
The complex impedance data of all the NVPFO2 compounds reveal quite distinct electrical behaviors. Fig. 8 depicts the complex impedance plots of each NVPFO2 sample recorded at 200 °C under heating, and also upon cooling for NVPFO2-Iono. Here, the complex impedance data (Z*) have already been normalized regarding the shape factor (l/A, thickness over area) of each sample, according to the relation Zs* = Z*A/l giving rise to the specific impedance (Zs*). This approach allows a straight comparison between the impedance responses of samples. For samples NVPFO2-Nano (Fig. 8a) and NVPFO2-Bulk (Fig. 8b), the overall electrical resistivity can be promptly read at the low-frequency intercept of the impedance data with the real part of the specific impedance (Zs′). Thus, sample NVPFO2-Bulk is about one order of magnitude more resistive than sample NVPFO2-Nano. Indeed, sample NVPFO2-Bulk is so resistive that its overall impedance is out of the equipment impedance range at this temperature, thus explaining the dispersion of impedance data in the low-frequency range. A common feature between the electrical behaviors of these samples is the distorted semi-circles probably caused by a joint contribution of grains and grain boundaries in the high- and middle-frequency ranges, respectively.39 In contrast, NVPFO2-Iono (Fig. 8c) presents an impedance response completely different from that of the other samples and even less resistive than that of the sample NVPFO2-Nano. Its impedance response combines a high-frequency semicircle probably linked to the ionic transport enhanced by the IL coating and a low-frequency highly depressed semicircle due to the electrode polarization phenomenon. In theory, if the ionic liquid is rigidly attached onto the surface of NVPFO2 particles and only Na+ ions are mobile, a steep increase of the imaginary part of the impedance should take place in low-frequency range.47 Since this is not the case, we hypothesized that the ionic molecular species that constitute the ionic liquid could also be relatively mobile either because they are not grafted or because of the high temperature employed for the impedance spectroscopy measurements. In any case, the dependence of the real part of permittivity on frequency reveals a logarithmic increase in the relative permittivity in the low-frequency range typical of an ion blocking effect, precluding the hypotheses of a majority electronic contribution to the overall charge transport (Fig. S7†). Therefore, the electrical resistivity related to the Na+ transport can be roughly read at the middle-frequency real-part projection of the specific impedance (Zs′) between the two semicircles. Furthermore, once this sample is heated up to 300 °C and then cooled down, its electrical behavior changes entirely (Fig. 8d) due to the degradation of the IL, whereas the electrical conductivity is nearly the same upon heating and cooling for the two other electrode materials (Fig. 9). In this case, the final electrical behavior of NVPFO2-Iono is much like that of NVPFO2-Bulk, highly resistive and with two impedance contributions likely coming from grain and grain boundary contributions. The polarization phenomenon caused by the grain boundaries and the ion blocking effect can be visualized respectively as a middle-frequency pseudo plateau and low-frequency logarithmic increase of the real part of permittivity (Fig. S7†).
 | ||
| Fig. 8 EIS analyses of Na3V2(PO4)2FO2 samples recorded at 200 °C under heating for NVPFO2-Nano (a) and NVPFO2-Bulk (b) as well as for NVPFO2-Iono under heating (c) and under cooling (d). The complex impedance data shown here have already been normalized regarding the shape factor of each sample for comparison purposes. | ||
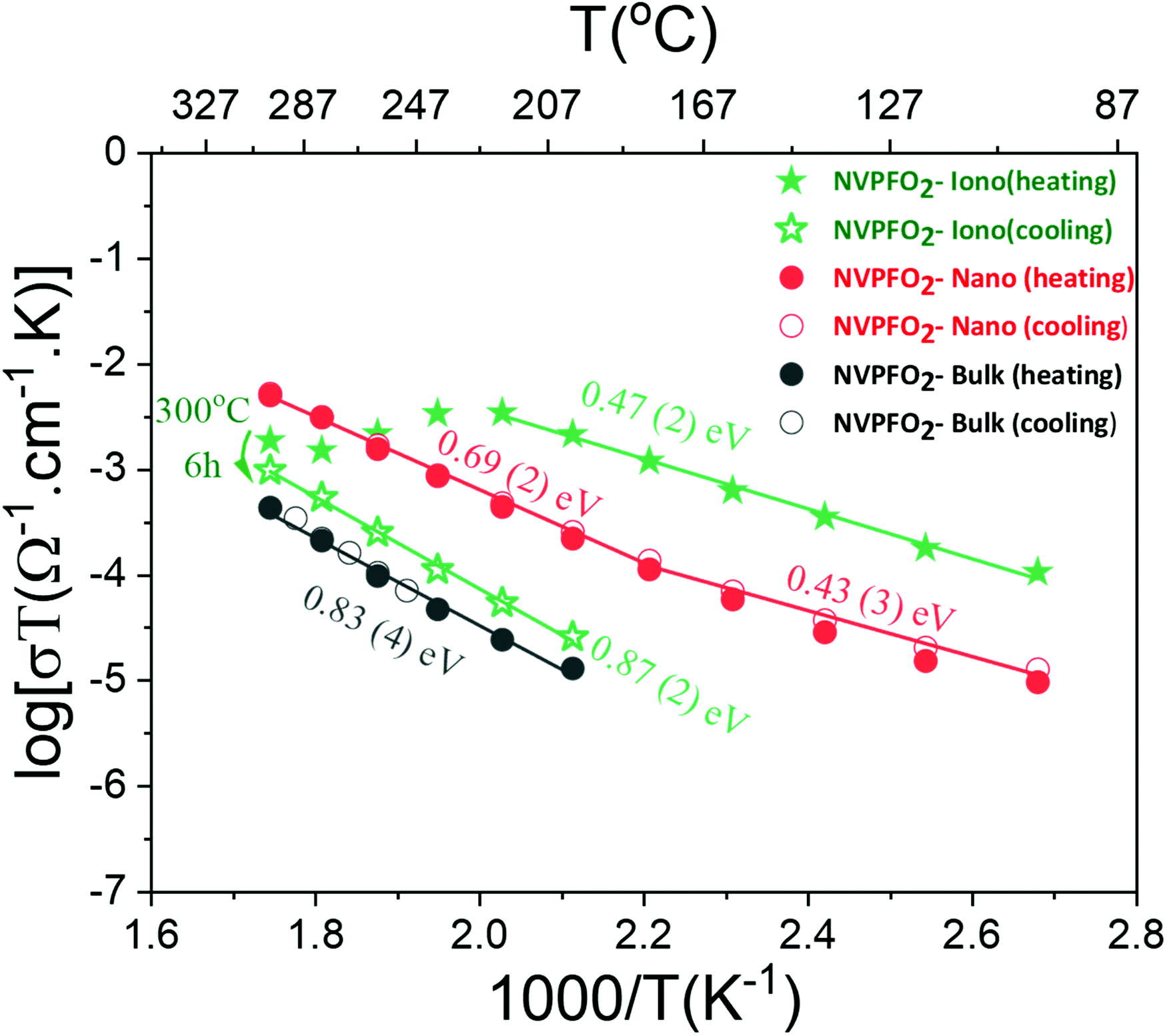 | ||
| Fig. 9 Arrhenius-like plots of the dependence of the overall electric conductivity on the inverse temperature for NVPFO2 samples synthesized under different conditions. | ||
The dependence of the overall electrical conductivity on the inverse of the temperature was plotted by applying the linearized form of the Arrhenius-like relation (eqn (1)), which is mostly used for ionic conductors but sometimes also applied for electronic conductors:60–62
 | (1) |
The sample coated with the IL shows at low temperatures an overall conductivity even higher than that of nanometric NVPFO2 (2.7 × 10−7 S cm−1vs. 3 × 10−8 S cm−1 at 100 °C) and a similar activation energy within errors of ∼0.45 eV. However, above 220 °C, the conductivity drops considerably probably due to the IL decomposition. After six hours at 300 °C, the conductivity of the sample gradually decreases upon cooling to values two orders of magnitude smaller than those observed upon heating (Fig. 9). The activation energy is almost doubled compared with that observed under heating, suggesting a totally different conduction mechanism. Meanwhile, the electrical conductivity of NVPFO2-Iono under cooling is in the same order of magnitude of that observed for NVPFO2-Bulk with similar activation energies of ∼0.85 eV, which indicates the strong ionic conductivity improvement brought by IL coating. Barpanda et al. have demonstrated a lithium conductivity enhancement by IL grafting on lithium fluoride sulfate compounds,47 thus in our case Na+ conductivity in NVPFO2-Iono would be strongly improved.
The Na+ diffusion during redox reactions was investigated through the CV analysis at different scan rates from 0.025 mV s−1 to 0.8 mV s−1 in the potential range between 2.5 V and 4.3 V vs. Na+/Na to avoid possible electrolyte degradation (Fig. 10a–c). Two oxidation and two reduction peaks, representing respectively the two plateaus in galvanostatic charge and discharge curves, are observed for all the NVPFO2 materials. Interestingly, at high scan rates, the two oxidation and reduction peaks strongly overlap for NVPFO2-Iono, which suggests the absence of Na+ ordering and a single-phase reaction mechanism. This could be explained by the enhanced Na+ conductivity determined by impedance spectroscopy. Furthermore, the overlap is globally less pronounced for NVPFO2-Nano and even less for NVPFO2-Bulk, which, for instance, exhibits two well-defined reduction peaks for all scan rate ranges. Fig. 10d and Fig. S8† depict the evolution of the peak current intensity (ip) versus the square root of the scan rate (v1/2) that allows to estimate Na+ diffusion coefficients using the Randles–Sevcik equation (ESI Part S8† and Table 2).
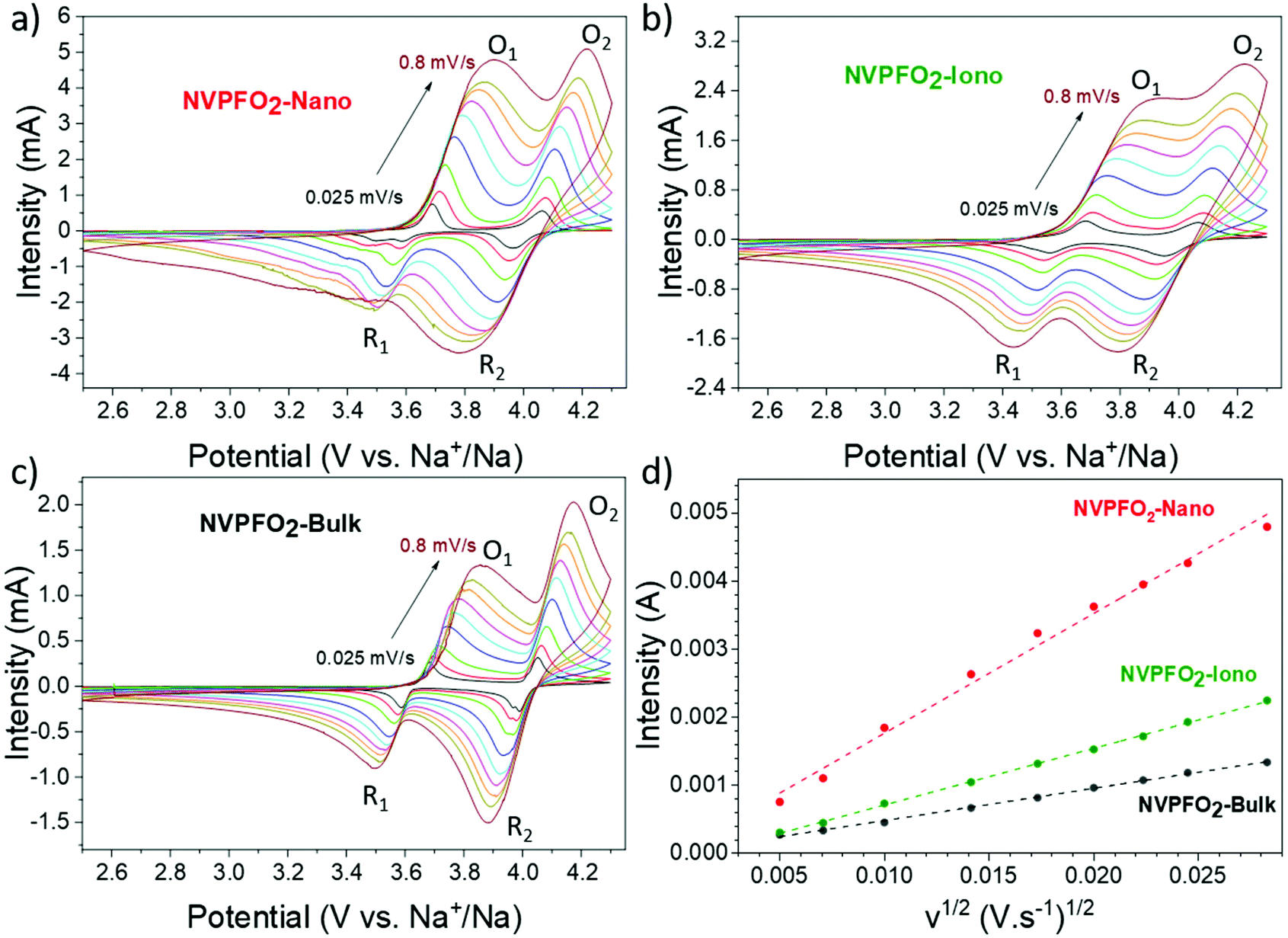 | ||
| Fig. 10 (a–c) CV curves at different scan rates and (d) the corresponding linear fitting curves between the maximum current (ip) and the square root of the scan rate (v1/2) for the O1 peak of each compound. | ||
| D (cm2 s−1) | NVPFO2-Nano | NVPFO2-Iono | NVPFO2-Bulk |
|---|---|---|---|
| O2 | 4.06 × 10−11 | 1.32 × 10−11 | 5.73 × 10−12 |
| O1 | 3.28 × 10−11 | 7.67 × 10−12 | 2.43 × 10−12 |
| R2 | 1.78 × 10−11 | 5.16 × 10−12 | 3.10 × 10−12 |
| R1 | 1.73 × 10−11 | 4.66 × 10−12 | 1.08 × 10−12 |
Nanospherical NVPFO2-Nano reveals the best diffusion capability of 4.06 × 10−11 cm2 s−1 at the O2 oxidation peak, nearly one order of magnitude higher than the cubic NVPFO2-Bulk with a calculated diffusion coefficient of 5.73 × 10−12 cm2 s−1. Quite surprisingly, IL-coated NVPFO2 exhibits only an intermediate diffusion coefficient during redox reactions (e.g. 1.32 × 10−11 cm2 s−1 for higher potential Na+ extraction), whereas a high ionic mobility is measured by impedance spectroscopy. It is thus possible that the ionic liquid coating only enhances the Na+ mobility on the surface of the electrode material and at grain boundaries. During electrochemical impedance spectroscopy measurements, NaTFSI can be formed at the surface of NVPFO2-Iono, while the cationic part of the ionic liquid attached on the NVPFO2 surface transfers its charge to an inorganic material.64 Thus, the measured conductivity would be that of Na+ in the IL coating. However, this coating does not affect the Na+ diffusion in the bulk of the NVPFO2 material. Thus, Na+ diffusion during oxidation/reduction (CV or galvanostatic charge/discharge) is still limited by solid-state diffusion within the bulk of NVPFO2. Na+ extraction or insertion necessarily involves an electron transfer to compensate the charge (vanadium oxidation or reduction), and as the electronic conductivity of micrometric NVPFO2 is very low, this limits the intercalation/extraction processes and thus Na+ diffusion. Besides, GITT measurements confirm this trend, as detailed in ESI Parts S8 and S9.†
According to Fig. 11a, all the positive electrode materials reveal good reversible capacity retentions after 80 cycles at C/5. Overall, all NVPFO2 possess stable capacity retention showing that particle downsizing and IL coating does not lead to electrode degradation or irreversible reaction that can be detrimental to practical application. However, the coulombic efficiency of NVPFO2-Bulk is not stable (Fig. 11b). Starting from the 20th cycle, the positive electrode suffers frequent coulombic efficiency loss that might be due to the electrolyte degradation, electric noise or the contact problem in the coin cell. However, its worse electrochemical performance than NVPFO2-Iono and NVPFO2-Nano at this charge and discharge rate emphasizes the benefits of nanosizing and surface coating.
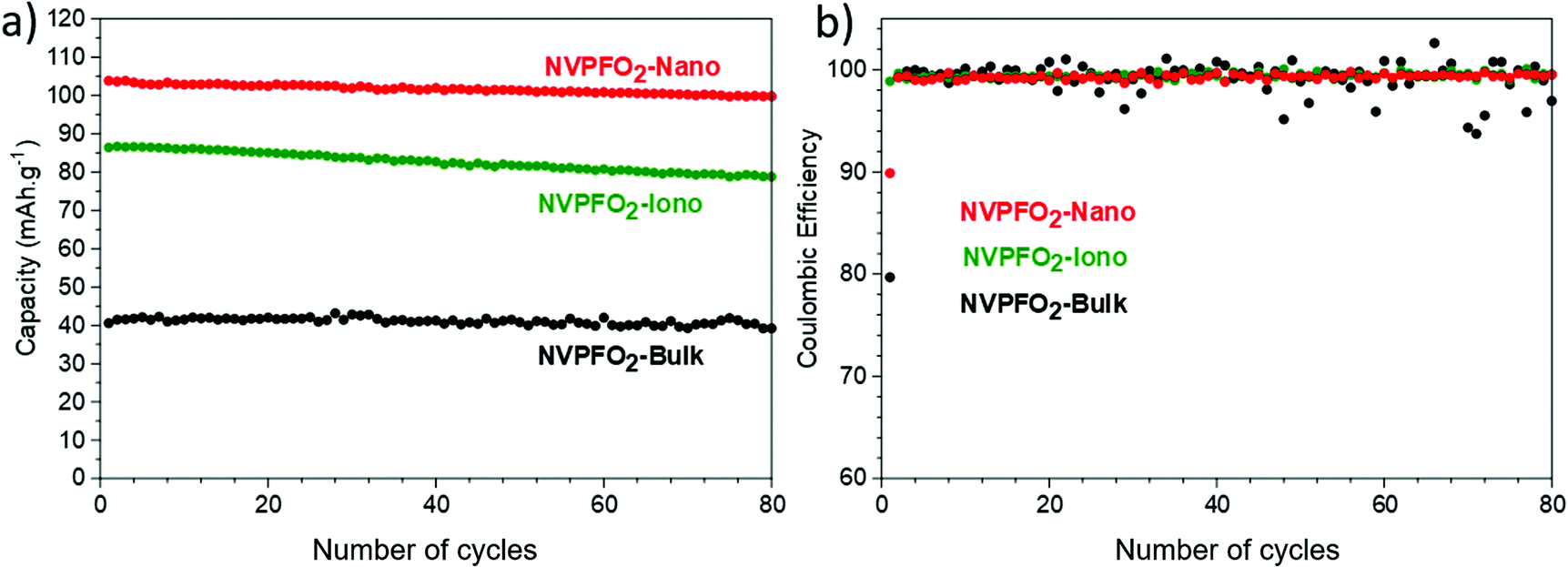 | ||
| Fig. 11 (a) Evolution of reversible capacity upon long-term cycling at C/5 and (b) related coulombic efficiency of all the NVPFO2 materials. | ||
Bearing in mind the very promising performance obtained for nanospherical NVPFO2, a full cell NVPFO2-Nano versus hard carbon was assembled and tested. Considering the electrode composition (88% active material/7% C/5% PVDF), the full cell shows excellent performance with an energy density of 320 W h kg−1 at C/20 and 310 W h kg−1 at C/10 (Fig. 12a). For comparison, Iarchuk et al. reported the same energy density for NVPFO2 but with electrode loadings containing only 70% of active material.65 Moreover, the full cell still exhibits 70% of its initial capacity at 1C (Fig. 12b). The Ragone plot given in Fig. 12c demonstrates the variation in energy density along with the increase in power density. Compared to a recent study reported by Notten et al., NVPFO2-Nano displays a similar high-rate performance in full cells to an optimized carbon-coated NVPF,17 which validates the extremely promising performance of our electrode nano-material. Meanwhile, the long-term cycling stability of full cells needs further optimisation as the capacity retention remains limited around 70% after 56 cycles (Fig. S11†). A better balancing between positive and negative electrode masses and other optimisations such as electrolyte formulation would allow us to improve the long-term capacity retention.
 | ||
| Fig. 12 (a) Energy density calculated for NVPFO2-Nano at C/20 and C/10. (b) Specific capacity retention in % upon increasing the C-rate. (c) Ragone plots of the NVPFO2-Nano full cell; the blue points correspond to the capacity calculated based on the NVPFO2 mass and the red points on both electrode masses. | ||
4. Conclusion
In summary, NVPFO2 electrode materials have been synthesized by three different synthesis methods, leading to a series of NVPFO2 with different characteristics, which allows the investigation of particle size impact and the effect of an IL coating on the electronic conductivity, diffusion capacity of Na+ and the energy storage performance separately. A set of spectroscopic characterizations (NMR, XPS and Raman) confirm the Na3V2(PO4)2FO2 composition for each compound and reveal that the ionic liquid coating on NVPFO2-Iono does not affect the vanadium nor the sodium local environment. Electrochemical impedance spectroscopy measurements demonstrate that particle downsizing improves the electronic conductivity by more than two orders of magnitude in the low temperature range, whereas the IL coating strongly enhances the ionic mobility. For instance, NVPFO2-Nano and NVPFO2-Iono present electrical conductivities around 3 × 10−8 S cm−1 and 2.5 × 10−7 S cm−1 at 100 °C, whereas the one of NVPFO2-Bulk is estimated below 10−10 S cm−1. The broad and overlapping redox peaks on CV curves for NVPFO2-Iono, in addition to the slopping profile on GCD curves, suggest the absence of Na+ ordering during cycling which may be due to the enhanced ionic mobility caused by the IL coating. Moreover, when NVPFO2-Iono loses its IL coating upon thermal degradation, the conductivity and activation energy become similar to those of NVPFO2-Bulk, which confirms the beneficial role of IL coating. Concerning energy storage performance, NVPFO2-Nano demonstrates the best rate capacity retention, confirming that a good electronic conductivity of positive electrode materials is primordial for high rate cycling, and its absence cannot be fully compensated by improving the ionic diffusion. Besides, NVPFO2-Nano performance in full cells is very promising considering the absence of carbon-coating or formulation optimization. Finally, it was shown that particle downsizing strongly increases the electronic conductivity, whereas an ionic liquid coating enhances the ionic mobility, thus the next step would be to combine both approaches to develop a more performant electrode material.Author contributions
The manuscript was written by J. O., R. F., R. B. N., S. C. and L. C. through support of all the co-authors. The study was conceptualized by J. O., S. C. and L. C. The synthesis of the different materials were done by R. F. J. O. and C. P.; R. B. N. performed all the electrochemical impedance spectroscopy measurements and analyzed the data; C. P. prepared all the electrode as well as the half and full cells; D. C. performed ss-NMR acquisitions and did data interpretation. Raman measurements and interpretation were done by J. O.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors thank the Materials Physics and Chemistry Doctoral School (ED397) of Sorbonne University for the funding of Runhe Fang's PhD thesis, as well as the financial support of Région Nouvelle Aquitaine, of the French National Research Agency (STORE-EX Labex Project ANR-10-LABX-76-01 and the Project Nano-INSPIRE ANR-21-CE50-0006) and of the European Union's Horizon 2020 research and innovation program under grant agreement no. 875629 (NAIMA project). The authors also thank Emmanuel PETIT, Cathy DENAGE, Eric LEBRAUD (ICMCB) for their technical support.References
- IPCC Working Group I, Climate Change 2021: The Physical Science Basis, 2021 Search PubMed.
- Y. Hu, S. Komaba, M. Forsyth, C. Johnson and T. Rojo, Small Methods, 2019, 3, 1900184 CrossRef.
- I. Hasa, S. Mariyappan, D. Saurel, P. Adelhelm, A. Y. Koposov, C. Masquelier, L. Croguennec and M. Casas-Cabanas, J. Power Sources, 2021, 482, 228872 CrossRef CAS.
- J.-M. Tarascon, Joule, 2020, 4, 1616–1620 CrossRef.
- K. M. Abraham, ACS Energy Lett., 2020, 3544–3547 CrossRef CAS.
- J.-N. Chotard, G. Rousse, R. David, O. Mentré, M. Courty and C. Masquelier, Chem. Mater., 2015, 27, 5982–5987 CrossRef CAS.
- C. V. Manohar, A. Raj K, M. Kar, M. Forsyth, D. R. MacFarlane and S. Mitra, Sustainable Energy Fuels, 2018, 2, 566–576 RSC.
- W. Zhou, L. Xue, X. Lü, H. Gao, Y. Li, S. Xin, G. Fu, Z. Cui, Y. Zhu and J. B. Goodenough, Nano Lett., 2016, 16, 7836–7841 CrossRef CAS PubMed.
- F. Chen, V. M. Kovrugin, R. David, O. Mentré, F. Fauth, J.-N. Chotard and C. Masquelier, Small Methods, 2019, 3, 1800218 CrossRef CAS.
- S. Park, J. N. Chotard, D. Carlier, I. Moog, M. Courty, M. Duttine, F. Fauth, A. Iadecola, L. Croguennec and C. Masquelier, Chem. Mater., 2021, 33, 5355–5367 CrossRef CAS.
- C. Xu, J. Zhao, E. Wang, X. Liu, X. Shen, X. Rong, Q. Zheng, G. Ren, N. Zhang, X. Liu, X. Guo, C. Yang, H. Liu, B. Zhong and Y. Hu, Adv. Energy Mater., 2021, 11, 2100729 CrossRef CAS.
- L. H. B. Nguyen, T. Broux, P. S. Camacho, D. Denux, L. Bourgeois, S. Belin, A. Iadecola, F. Fauth, D. Carlier, J. Olchowka, C. Masquelier and L. Croguennec, Energy Storage Mater., 2019, 20, 324–334 CrossRef.
- M. Bianchini, F. Fauth, N. Brisset, F. Weill, E. Suard, C. Masquelier and L. Croguennec, Chem. Mater., 2015, 27, 3009–3020 CrossRef CAS.
- Y.-U. Park, D.-H. Seo, H. Kim, J. Kim, S. Lee, B. Kim and K. Kang, Adv. Funct. Mater., 2014, 24, 4603–4614 CrossRef CAS.
- Y. Qi, L. Mu, J. Zhao, Y. Hu, H. Liu and S. Dai, Angew. Chem., Int. Ed., 2015, 54, 9911–9916 CrossRef CAS.
- L. Zhu, H. Wang, D. Sun, Y. Tang and H. Wang, J. Mater. Chem. A, 2020, 8, 21387–21407 RSC.
- K. Chayambuka, M. Jiang, G. Mulder, D. L. Danilov and P. H. L. Notten, Electrochim. Acta, 2022, 404, 139726 CrossRef CAS.
- N. Eshraghi, S. Caes, A. Mahmoud, R. Cloots, B. Vertruyen and F. Boschini, Electrochim. Acta, 2017, 228, 319–324 CrossRef CAS.
- F. Li, Y. Zhao, L. Xia, Z. Yang, J. Wei and Z. Zhou, J. Mater. Chem. A, 2020, 8, 12391–12397 RSC.
- L. Zhu, Q. Zhang, D. Sun, Q. Wang, N. Weng, Y. Tang and H. Wang, Mater. Chem. Front., 2020, 4, 2932–2942 RSC.
- T. Broux, F. Fauth, N. Hall, Y. Chatillon, M. Bianchini, T. Bamine, J. Leriche, E. Suard, D. Carlier, Y. Reynier, L. Simonin, C. Masquelier and L. Croguennec, Small Methods, 2019, 3, 1800215 CrossRef CAS.
- J. Olchowka, L. H. B. Nguyen, T. Broux, P. Sanz Camacho, E. Petit, F. Fauth, D. Carlier, C. Masquelier and L. Croguennec, Chem. Commun., 2019, 55, 11719–11722 RSC.
- A. Criado, P. Lavela, C. Pérez-Vicente, G. F. Ortiz and J. L. Tirado, J. Electroanal. Chem., 2020, 856, 113694 CrossRef CAS.
- M. Wang, K. Wang, X. Huang, T. Zhou, H. Xie and Y. Ren, Ceram. Int., 2020, 2, 1–9 Search PubMed.
- C. Guo, J. Yang, Z. Cui, S. Qi, Q. Peng, W. Sun, L.-P. Lv, Y. Xu, Y. Wang and S. Chen, J. Energy Chem., 2022, 65, 514–523 CrossRef.
- I. L. Matts, S. Dacek, T. K. Pietrzak, R. Malik and G. Ceder, Chem. Mater., 2015, 27, 6008–6015 CrossRef CAS.
- Y. Zhang, S. Guo and H. Xu, J. Mater. Chem. A, 2018, 6, 4525–4534 RSC.
- L. H. B. Nguyen, J. Olchowka, S. Belin, P. Sanz Camacho, M. Duttine, A. Iadecola, F. Fauth, D. Carlier, C. Masquelier and L. Croguennec, ACS Appl. Mater. Interfaces, 2019, 11, 38808–38818 CrossRef CAS PubMed.
- C. Shen, H. Long, G. Wang, W. Lu, L. Shao and K. Xie, J. Mater. Chem. A, 2018, 6, 6007–6014 RSC.
- J. Xun, Y. Zhang and H. Xu, Inorg. Chem. Commun., 2020, 115, 107884 CrossRef CAS.
- J. Zhao, L. Mu, Y. Qi, Y. S. Hu, H. Liu and S. Dai, Chem. Commun., 2015, 51, 7160–7163 RSC.
- A. Mukherjee, T. Sharabani, R. Sharma, S. Okashy and M. Noked, Batteries Supercaps, 2020, 3, 510–518 CrossRef CAS.
- Y. Qi, L. Mu, J. Zhao, Y.-S. Hu, H. Liu and S. Dai, J. Mater. Chem. A, 2016, 4, 7178–7184 RSC.
- R. Fang, J. Olchowka, C. Pablos, P. S. Camacho, D. Carlier, L. Croguennec and S. Cassaignon, Batteries Supercaps, 2022, 5, e202100179 CAS.
- Y. Qi, J. Zhao, C. Yang, H. Liu and Y.-S. Hu, Small Methods, 2019, 3, 1800111 CrossRef CAS.
- P. Serras, V. Palomares, J. Alonso, N. Sharma, J. M. López del Amo, P. Kubiak, M. L. Fdez-Gubieda and T. Rojo, Chem. Mater., 2013, 25, 4917–4925 CrossRef CAS.
- P. R. Kumar, K. Kubota, Y. Miura, M. Ohara, K. Gotoh and S. Komaba, J. Power Sources, 2021, 493, 229676 CrossRef CAS.
- D. Semykina, O. Podgornova and N. Kosova, Mater. Today: Proc., 2020, 25, 497–500 CAS.
- R. Fang, J. Olchowka, C. Pablos, R. Bianchini Nuernberg, L. Croguennec and S. Cassaignon, ACS Appl. Energy Mater., 2022, 5, 1065–1075 CrossRef CAS.
- J. Olchowka, L. H. B. Nguyen, E. Petit, P. S. Camacho, C. Masquelier, D. Carlier and L. Croguennec, Inorg. Chem., 2020, 59, 17282–17290 CrossRef CAS PubMed.
- T. Broux, T. Bamine, F. Fauth, L. Simonelli, W. Olszewski, C. Marini, M. Ménétrier, D. Carlier, C. Masquelier and L. Croguennec, Chem. Mater., 2016, 28, 7683–7692 CrossRef CAS.
- J. Rodríguez-Carvajal, Physica B: Condens. Matter, 1993, 192, 55–69 CrossRef.
- N. Sharma, P. Serras, V. Palomares, H. E. A. Brand, J. Alonso, P. Kubiak, M. L. Fdez-Gubieda and T. Rojo, Chem. Mater., 2014, 26, 3391–3402 CrossRef CAS.
- F. D. Hardcastle and I. E. Wachs, J. Phys. Chem., 1991, 95, 5031–5041 CrossRef CAS.
- C. J. Antony, A. Aatiq, C. Y. Panicker, M. J. Bushiri, H. T. Varghese and T. K. Manojkumar, Spectrochim. Acta, Part A, 2011, 78, 415–419 CrossRef CAS.
- J. Olchowka, R. Invernizzi, A. Lemoine, J. Allouche, I. Baraille, D. Flahaut and L. Guerlou-Demourgues, J. Electrochem. Soc., 2020, 167, 100527 CrossRef CAS.
- P. Barpanda, R. Dedryvère, M. Deschamps, C. Delacourt, M. Reynaud, A. Yamada and J.-M. Tarascon, J. Solid State Electrochem., 2012, 16, 1743–1751 CrossRef CAS.
- J. Olchowka, T. Tailliez, L. Bourgeois, M. A. Dourges and L. Guerlou-Demourgues, Nanoscale Adv., 2019, 1, 2240–2249 RSC.
- R. Baddour-Hadjean and J. P. Pereira-Ramos, Chem. Rev., 2010, 110, 1278–1319 CrossRef CAS.
- A. Criado, P. Lavela, G. Ortiz, J. L. Tirado, C. Pérez-Vicente, N. Bahrou and Z. Edfouf, Electrochim. Acta, 2020, 332, 135502 CrossRef CAS.
- Y. Cai, X. Cao, Z. Luo, G. Fang, F. Liu, J. Zhou, A. Pan and S. Liang, Adv. Sci., 2018, 2, 1800680 CrossRef PubMed.
- K. Fujii, T. Fujimori, T. Takamuku, R. Kanzaki, Y. Umebayashi and S. I. Ishiguro, J. Phys. Chem. B, 2006, 110, 8179–8183 CrossRef CAS PubMed.
- C. J. Jafta, X. G. Sun, H. Lyu, H. Chen, B. P. Thapaliya, W. T. Heller, M. J. Cuneo, R. T. Mayes, M. P. Paranthaman, S. Dai and C. A. Bridges, Adv. Funct. Mater., 2021, 31, 2008708 CrossRef CAS.
- T. Sugimoto, M. Kikuta, E. Ishiko, M. Kono and M. Ishikawa, J. Power Sources, 2008, 183, 436–440 CrossRef CAS.
- Z. Tong, Y. Qi, J. Zhao, L. Liu, X. Shen and H. Liu, Waste Biomass Valorization, 2020, 11, 2201–2209 CrossRef CAS.
- L. H. B. Nguyen, P. Sanz Camacho, T. Broux, J. Olchowka, C. Masquelier, L. Croguennec and D. Carlier, Chem. Mater., 2019, 31, 9759–9768 CrossRef CAS.
- W. Papawassiliou, J. P. Carvalho, N. Panopoulos, Y. Al Wahedi, V. K. S. Wadi, X. Lu, K. Polychronopoulou, J. B. Lee, S. Lee, C. Y. Kim, H. J. Kim, M. Katsiotis, V. Tzitzios, M. Karagianni, M. Fardis, G. Papavassiliou and A. J. Pell, Nat. Commun., 2021, 12, 1–11 CrossRef.
- L. H. B. Nguyen, A. Iadecola, S. Belin, J. Olchowka, C. Masquelier, D. Carlier and L. Croguennec, J. Phys. Chem. C, 2020, 124, 23511–23522 CrossRef CAS.
- N. Sharma, P. Serras, V. Palomares, H. E. A. Brand, J. Alonso, P. Kubiak, M. L. Fdez-Gubieda and T. Rojo, Chem. Mater., 2014, 26, 3391–3402 CrossRef CAS.
- J.-L. Souquet, Encyclopedia of Glass Science, Technology, History, and Culture, Wiley, 2021, pp. 453–463 Search PubMed.
- M. C. Ungureanu, M. Lévy and J. L. Souquet, Ionics, 1998, 4, 200–206 CrossRef.
- R. B. Nuernberg, Ionics, 2020, 26, 2405–2412 CrossRef CAS.
- T. Broux, B. Fleutot, R. David, A. Brüll, P. Veber, F. Fauth, M. Courty, L. Croguennec and C. Masquelier, Chem. Mater., 2018, 30, 358–365 CrossRef CAS.
- A. Paolella, G. Bertoni, W. Zhu, D. Campanella, A. La Monaca, G. Girard, H. Demers, A. C. Gheorghe Nita, Z. Feng, A. Vijh, A. Guerfi, M. Trudeau, M. Armand and S. A. Krachkovskiy, J. Am. Chem. Soc., 2022, 144, 3442–3448 CrossRef CAS PubMed.
- A. R. Iarchuk, D. V. Sheptyakov and A. M. Abakumov, ACS Appl. Energy Mater., 2021, 4, 5007–5014 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr01080a |
| This journal is © The Royal Society of Chemistry 2022 |