
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Eco-friendly fabrication of organic solar cells: electrostatic stabilization of surfactant-free organic nanoparticle dispersions by illumination†
Philipp
Marlow
ab,
Felix
Manger
ab,
Karen
Fischer
ab,
Christian
Sprau
 a and
Alexander
Colsmann
*ab
a and
Alexander
Colsmann
*ab
aMaterial Research Center for Energy Systems, Karlsruhe Institute of Technology (KIT), Strasse am Forum 7, 76131 Karlsruhe, Germany. E-mail: alexander.colsmann@kit.edu
bLight Technology Institute, Karlsruhe Institute of Technology (KIT), Engesserstrasse 13, 76131 Karlsruhe, Germany
First published on 23rd March 2022
Abstract
Earlier reports have discussed the manifold opportunities that arise from the use of eco-friendly organic semiconductor dispersions as inks for printed electronics and, in particular, organic photovoltaics. To date, poly(3-hexylthiophene) (P3HT) plays an outstanding role since it has been the only organic semiconductor that formed nanoparticle dispersions with sufficient stability and concentration without the use of surfactants. This work elucidates the underlying mechanisms that lead to the formation of intrinsically stable P3HT dispersions and reveals prevailing electrostatic effects to rule the nanoparticle growth. The electrostatic dispersion stability can be enhanced by photo-generation of additional charges, depending on the light intensity and its wavelength. This facile, additive-free process provides a universal handle to also stabilize surfactant-free dispersions of other semiconducting polymers, which are frequently used to fabricate organic solar cells or other optoelectronic thin-film devices. The more generalized process understanding paves the way towards a universal synthesis route for organic nanoparticle dispersions.
1. Introduction
Among the emerging photovoltaic technologies, organic solar cells stand out with lowest environmental impact and an all-eco-friendly cradle-to-grave lifecycle.1 The light-harvesting bulk-heterojunctions (BHJs) comprise mostly abundant elements and their fabrication by printing enables an unsurpassed low energy payback time.2 Concepts are being discussed in the scientific literature on how to follow eco-friendly synthetic routes omitting toxic catalysts, e.g. by using direct arylation.3,4 Likewise, the deposition of the light-harvesting layers is increasingly accomplished by non-toxic solvents. Even though capture of toxic solvents evaporating from large areas is technically possible during printing or coating, this would require additional safety precautions and hence induce additional investments and operational costs, which conflict with the goal of low-cost manufacturing. Most developments on eco-friendly solution processing revolve around the use of aromatic and non-halogenated solvents. The solar cell performance is then controlled by the careful choice of solvents, solvent additives, thermal annealing and a set of other, often subtle, process parameters.5 A fundamentally different concept to the solution processing of BHJs utilizes nanoparticle dispersions, enabling the use of alcohols or water as processing agents. Some of the many advantages of this approach are: (i) The principal employment of polymers is not limited by the solubility of the polymer in a specific solvent anymore, providing access to a wider range of organic semiconductors. (ii) The decoupling of solubility from solution processing in nanoparticle dispersions readily enables the deposition of multi-layers, omitting the design of orthogonal solvent sequences.6 (iii) The formation of the BHJ depends less on the parameters of the solution deposition process but can be primed already during nanoparticle synthesis.7Probably the most important challenge for the use of nanoparticle dispersions is their stabilization against agglomeration. Agglomeration of dispersions is often mitigated by the use of surfactants. Following the miniemulsion method,8 a solution of an organic semiconductor is emulsified in an immiscible non-solvent in presence of a surfactant. After evaporation of the solvent, surfactant-stabilized organic nanoparticles remain in the dispersion. The miniemulsion route can also be used to synthesize blended-semiconductor inks for the production of organic solar cells with photoactive layers comprising a mixture of donor-type and acceptor-type semiconductors.9–11 However, the power conversion efficiencies (PCEs) of the solar cells most often did not live up to the performance of their solution processed counterparts, most likely due to prevailing surfactant in the photoactive layer.12,13 Better solar cell performance was only achieved after heavy dialysis of the nanoparticle dispersion in order to reduce the surfactant content.13 It was also reported that nanoparticles, which were prepared through miniemulsions, exhibit core–shell architectures which may hamper the charge carrier transport in the photoactive layers fabricated thereof.12
A different preparation technique was reported for semiconductor nanoparticle dispersions comprising poly(3-hexylthiophene) (P3HT) or blends of P3HT and indene-C60 bisadduct (ICBA). Stable dispersions of P3HT in water and in ethanol in absence of surfactants were synthesized via nanoprecipitation, a method which produces nanoparticles by mixing a polymer solution with a miscible non-solvent.14,15 In a nutshell, a chloroform solution (CHCl3, solvent) of a polymer or a polymer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :fullerene blend is injected into ethanol (EtOH, non-solvent). Due to the miscibility of chloroform and ethanol, the solubility of the semiconductors is reduced immediately and nanoparticles form within milliseconds. Having a lower boiling point, chloroform is then evaporated from the dispersion. Notably, the use of chloroform at this stage does not conflict with the goal of eco-compatible processing as this step can be carried out in a controlled and closed-cycle chemical environment, which is all different from the evaporation of solvents on square-meter-scale on an industrial coating setup. The method was successfully applied to produce composite nanoparticles of P3HT and ICBA in methanol or ethanol.16 The dispersions did not show notable aggregation for several weeks but were rather stable. The microstructure of the so-produced nanoparticles was close to the ideal BHJ morphology which helped to translate the nanoparticle dispersion into thin-films with well-crafted BHJ morphology.7 Remarkably, organic solar cells from these dispersions were close in efficiency to the respective solar cells processed from common halogenated solvents such as 1,2-dichlorobenzene.16
:fullerene blend is injected into ethanol (EtOH, non-solvent). Due to the miscibility of chloroform and ethanol, the solubility of the semiconductors is reduced immediately and nanoparticles form within milliseconds. Having a lower boiling point, chloroform is then evaporated from the dispersion. Notably, the use of chloroform at this stage does not conflict with the goal of eco-compatible processing as this step can be carried out in a controlled and closed-cycle chemical environment, which is all different from the evaporation of solvents on square-meter-scale on an industrial coating setup. The method was successfully applied to produce composite nanoparticles of P3HT and ICBA in methanol or ethanol.16 The dispersions did not show notable aggregation for several weeks but were rather stable. The microstructure of the so-produced nanoparticles was close to the ideal BHJ morphology which helped to translate the nanoparticle dispersion into thin-films with well-crafted BHJ morphology.7 Remarkably, organic solar cells from these dispersions were close in efficiency to the respective solar cells processed from common halogenated solvents such as 1,2-dichlorobenzene.16
So far, the surfactant-free nanoprecipitation faces a couple of challenges to overcome on the way to becoming a broadly deployable technology. First, the control over the nanoparticle size has been rather limited. The only way to achieve smaller nanoparticles has been a significant reduction of the initial polymer concentration in solution,15 after which the dispersion becomes useless for the deposition of thin-films with sufficient thickness. Second, to date, the synthesis of dispersions by nanoprecipitation only has produced satisfying results on P3HT or P3HT:ICBA blends, whereas the transfer to other semiconducting polymers only has been possible at very low concentrations. So far, it has been a mystery why P3HT plays this outstanding role. Most other polymers tend to form rather large particles or coagulate immediately when subjected to nanoprecipitation. Unveiling the underlying stabilization mechanisms against agglomeration would help to translate this fabrication concept to other organic semiconductors and allow a broader deployment of this eco-friendly process route.
In this work, we demystify the unique properties of P3HT nanoparticle formation in absence of surfactants and demonstrate that surfactant-free P3HT dispersions are stabilized electrostatically. We explore the generation of additional stabilizing charges by photoexcitation of P3HT and other semiconducting polymers during nanoprecipitation and find a handle to effectively gain control over the dispersion stability and the nanoparticle size.
2. Stabilization mechanisms in surfactant-free P3HT dispersions
To date, P3HT is the only reported organic semiconductor for solar cell applications to form nanoparticle dispersions of sufficient concentration (≥ 10 g L−1) and shelf stability (>weeks) in the absence of stabilization agents. Other polymers only form stable, surfactant-free nanoparticle dispersions with either much lower concentrations or much lower shelf stability. It would be of great practical use to understand the stabilization mechanisms in P3HT nanoparticles, to enhance control over the nanoparticle size and to reliably translate the process to other polymers.2.1. Electrostatic stabilization
A fundamental requirement for obtaining nanoparticle dispersions is sufficient stabilization against agglomeration.17 In general, stability of colloidal dispersions against agglomeration can be achieved by steric or electrostatic repulsion between nanoparticles.18,19 Steric stabilization is achieved by adsorption of soluble macromolecules onto the nanoparticle surface. Electrostatic repulsion occurs between nanoparticles of the same charge. Since the reported synthesis of P3HT (and P3HT:ICBA) nanoparticle dispersions by nanoprecipitation omitted any surfactants and since the dispersion medium was a poor solvent for P3HT, steric stabilization was unlikely to occur.
In order to probe the dispersion for any electrostatic stabilization effects, i.e. electrical charging of nanoparticles, we performed an electrophoretic separation experiment on a P3HT dispersion. The P3HT dispersion was synthesized by nanoprecipitation following established experimental protocols.16 Therefore, we rapidly injected a chloroform solution of P3HT (5 g L−1) into the non-solvent ethanol (1:4 v/v). After the chloroform was removed by thermal annealing, the dispersion was diluted to finally yield a P3HT concentration of 0.1 g L−1 in ethanol and filled into a capillary cell. Fig. 1 depicts a series of photos of the capillary cell inside the electrophoretic measurement setup at t = 0, 15, 30, 45 and 90 min. Under a constant DC driving voltage of 50 V, we observed migration of the P3HT nanoparticles towards the (negatively charged) cathode and concomitant depletion of the volume near the anode. From this migration of the nanoparticles in an electric field, we conclude that the P3HT nanoparticles in ethanol are positively charged. This positive charge is well in agreement with the general deployment of P3HT as an electron donor. Thus we conclude that the stabilization of the surfactant-free P3HT dispersions is likely of electrostatic nature, as proposed in earlier reports.20,21
 | ||
| Fig. 1 Photo series of the electrophoresis setup showing a P3HT dispersion (0.1 g L−1 in EtOH) in a plastic capillary cell (length 5 cm) under a constant DC voltage of 50 V after t = 0, 15, 30, 45 and 90 min. The nanoparticles migrate from the anode (right side, red line) to the cathode (left side, black line), indicating a positive nanoparticle charge. | ||
The nature of the chemical species carrying the countercharge in the electrophoresis experiments is less obvious. Besides unknown impurities in the ethanol non-solvent (typically 0.5–1.0 vol%), redox-active impurities such as dissolved oxygen, can act as electron acceptors for P3HT.22 Yet, a control nanoprecipitation experiment with degassed solvents in the inert nitrogen atmosphere of a glovebox and comparison with the nanoparticle size of an otherwise equal process in air showed no significant differences (Table S1†), which led us to conclude that adventitious oxygen has no significant influence on the process. Other known processes to charge nanoparticles in non-aqueous dispersions include specific adsorption of ions from the medium or from surfactants, proton transfer reactions (Brønsted acid–base-reactions) or charge transfer reactions (redox reactions or extreme cases of Lewis acid–base-reactions).23 Also protonation appears feasible: polythiophenes can be protonated by acids24 and conjugated π-electron systems can stabilize their protonated form by delocalization of the positive charge.25 Ethanol itself is a weak acid and may therefore act as a proton donor for P3HT. However, identification of the electron acceptor or proton donor in the dispersion medium is not needed for the discussion in this work and thus this challenging task remains for further investigations.
2.2. The effect of positive charges on the P3HT nanoparticle formation and stabilization
The important role of the positive charges for the stabilization of the P3HT nanoparticle dispersions can be studied by deliberately introducing ions to the final dispersion, which foster nanoparticle aggregation due to screening of the nanoparticle charges and eventually complete coagulation of the dispersion.19 Such ionic charges can be introduced to the system by some soluble salt (an electrolyte), which prompted us to add sodium bromide (NaBr, 10−3 mol L−1) to a stable P3HT nanoparticle dispersion with a concentration of 0.1 g L−1. As depicted in Fig. 2a, the addition of NaBr led to coagulation of the dispersion within minutes. While electrolytes at low concentrations are unlikely to affect any steric stabilization, they represent an efficient way to screen electric charges and reduce the mutual repulsion of charged nanoparticles. Thus, the immediate coagulation of the P3HT dispersions upon addition of NaBr confirmed the electrostatic stabilization which we previously concluded from the electrophoretic experiments.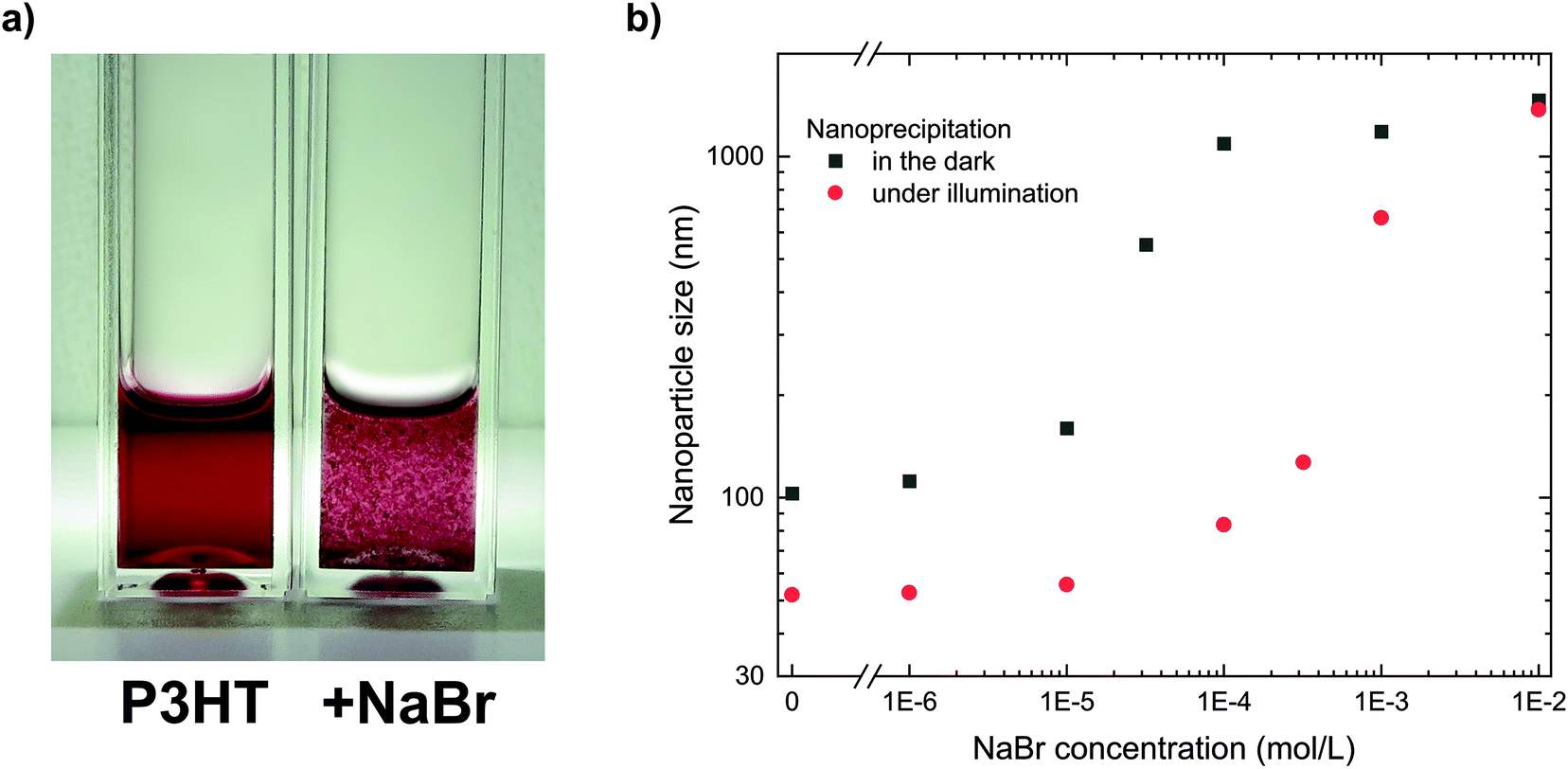 | ||
| Fig. 2 (a) The addition of NaBr to the P3HT dispersion (0.1 g L−1 in EtOH) promotes coagulation of the nanoparticle dispersions. The cuvette on the left side contains neat P3HT dispersion while the cuvette on the right side shows the same P3HT dispersion 30 min after addition of NaBr (10−3 mol L−1). (b) Influence of NaBr dissolved in EtOH prior to nanoprecipitation on the nanoparticle size (P3HT in CHCl3, 0.5 g L−1, nanoprecipitated in EtOH plus NaBr, 1:4 v/v). Higher NaBr concentrations produced larger nanoparticles, both in the dark (black squares) and under illumination (100 W m−2, white-light LED, red circles). Under illumination, smaller nanoparticles are obtained and, up to 10−4 mol L−1, their size is less sensitive to the NaBr concentration. | ||
The addition of NaBr to the non-solvent ethanol also offers a facile tool to study the role of electric charges during the nanoprecipitation process. By systematically adding minute amounts of NaBr to the non-solvent ethanol before nanoprecipitation, we gradually reduced the electrostatic stabilization in a series of nanoprecipitations and monitored the size of the nanoparticles. The nanoparticle size distributions of the dispersions were obtained by dynamic light scattering (DLS). As depicted in Fig. 2b (black symbols, “in the dark”), the nanoparticle size increased towards higher NaBr concentrations. At a low NaBr concentration of 10−6 mol L−1, the increase in nanoparticle size was negligible, which we attribute to some intrinsic (counter)ions in ethanol with a concentration of similar order of magnitude (both, pure ethanol and ethanol with 10−6 mol L−1 of NaBr, exhibited a conductivity of less than 1 μS cm−1). At NaBr concentrations of 10−5 mol L−1 or beyond, gradually larger nanoparticles formed. NaBr concentrations of 10−4 mol L−1 or more did not allow the preparation of stable dispersions. We note that the time-critical coagulation process within minutes in presence of NaBr prompted us to skip the chloroform-removal process in these experiments. Instead we investigated the dispersions by DLS right after injection of P3HT/chloroform into ethanol. At the investigated semiconductor concentrations, this process modification does not affect the validity of the results since preliminary experiments have shown that the solvent-removal process did not change the nanoparticle size distribution of stable dispersions (Table S2†). Only less stable polymer dispersions or dispersions with significantly higher semiconductor concentrations exhibited some minor increase in nanoparticle size during solvent removal, but this does not affect the conclusions from this experiment.
To explain the correlation between electrostatic stabilization and nanoparticle size in dispersions synthesized by nanoprecipitation, we use a model of nanoparticle growth by agglomeration and coalescence. Such models were suggested, experimentally investigated and theoretically described in several earlier publications.17,26–28 Upon mixing of the polymer solution with the non-solvent, the polymer chains collapse into globules.26 Due to attractive van der Waals forces, these initial globules agglomerate to form dimers, trimers and larger clusters, eventually leading to complete coagulation.17 The synthesis of stable nanoparticle dispersions requires the defined termination of the agglomeration process by a repulsive force which counteracts the van der Waals attraction. Here, this repulsive force is of electrostatic nature and stems from charges on the nanoparticles. In aqueous dispersions with moderate or large electrolyte concentration, the repulsive forces are commonly attributed to an overlap of the electrical double layers of each nanoparticle. In media with low ion concentrations, such as ethanol as used in our experiments, the origin of the repulsion may be a superposition of coulombic and other effects.29 In both cases, the repulsive forces between nanoparticles increase with an increasing number of surface charges (or an increasing surface potential). Likewise, at a constant surface potential, the repulsive forces increase with increasing nanoparticle sizes.17 Altogether, the interaction of van der Waals forces and electrostatic forces determines the sizes of the nanoparticles: under sufficient electrostatic stabilization, the particle growth slows down towards increasing nanoparticle sizes until a (meta-)stable size distribution is obtained. Hence, the nanoparticle size is a measure of the colloidal stability of the dispersion. Smaller nanoparticles (with a larger total surface) indicate better stabilization by larger amounts of charges.
Notably, the remarkable destabilization of the dispersions even by minute amounts of soluble ions demonstrates the importance of using ultra-clean glassware and dispersion media (here: ethanol) for a reproducible nanoparticle synthesis. Smallest (ionic) impurities stemming from P3HT, the non-solvent or glassware may well explain the inconsistent experimental results on P3HT (and P3HT:fullerene) nanoparticle syntheses in the scientific literature. Table S3 of the ESI† exemplifies the divergence of nanoparticle sizes in dependence of the ethanol batch used for nanoprecipitation.
3. Controlling the nanoparticle size by illumination
3.1. Photoexcitation of nanoparticles
We have seen that P3HT nanoparticle dispersions can be stabilized against agglomeration by nanoparticle charging and that the amount of (positive) charges provides a handle to control their size. Generating charges beyond the intrinsic charge density on the nanoparticles would therefore allow the further reduction of the nanoparticle size. One way to produce additional charges on photoactive semiconductors is their photoexcitation. Hence, we synthesized dispersions of the semiconducting polymer P3HT and gradually increased the irradiance during nanoprecipitation from batch to batch, i.e. from the dark (<0.01 W m−2) to bright white light (1000 W m−2), using a tunable cold-white high-power LED. As depicted in Fig. 3a, we observed a gradual reduction of the nanoparticle size from 77 ± 2 nm in the dark to 37 ± 1 nm under illumination (1000 W m−2). Thus, adjusting the irradiance provides a handle to precisely control the nanoparticle size via photogenerated charges. While the positive charges remain on the nanoparticles, the negative charges are likely detached as discussed in section 2.1 with the soluble anion yet unknown. The enhanced dispersion stability by larger numbers of stabilizing charges also shows upon addition of NaBr, following the experimental procedure described in section 2.2. As depicted in Fig. 2b (red data points), under illumination, a larger amount of NaBr is required to destabilize the nanoparticle dispersion and to foster coagulation than in the dark, which is indicative for a larger number of (photogenerated) stabilizing charges.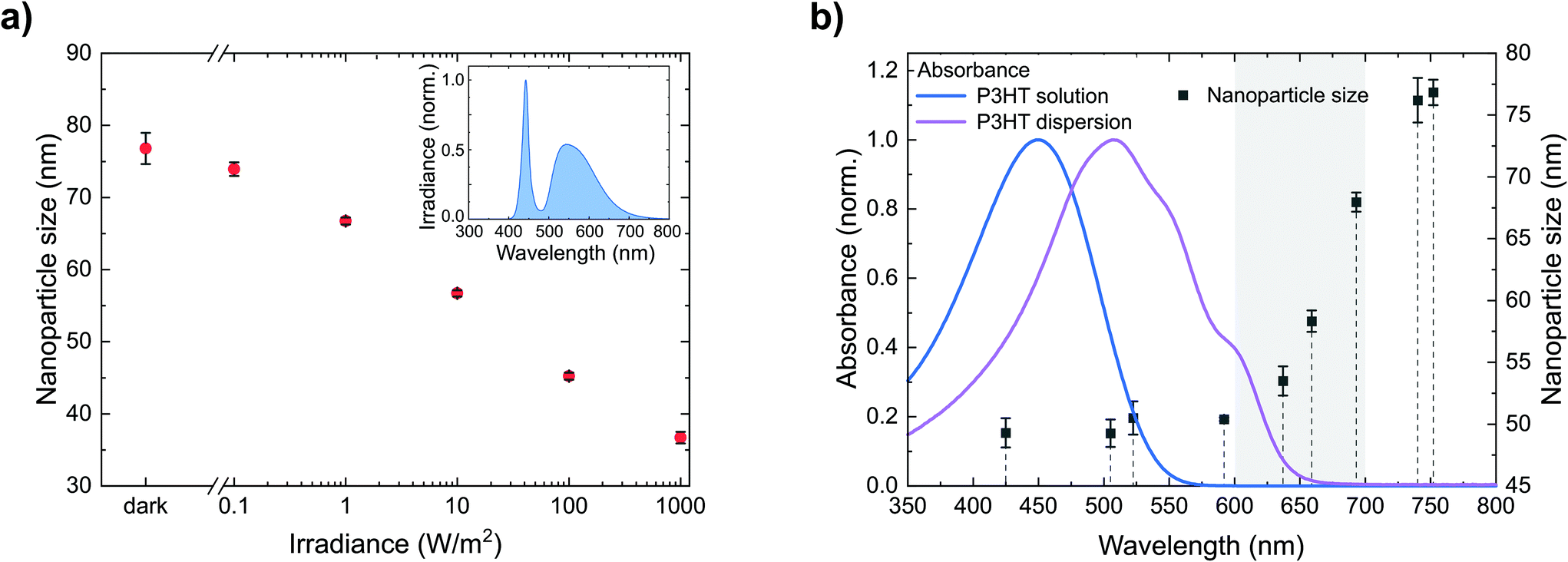 | ||
| Fig. 3 (a) Dependence of the nanoparticle size on the irradiance during nanoprecipitation (P3HT in CHCl3, 0.5 g L−1, precipitated in EtOH, 1:4 v/v). Inset: spectrum of the light source, a white LED. (b) Absorbance of a typical P3HT solution and dispersion as well as dependence of the nanoparticle size on the illumination wavelength during precipitation (P3HT in CHCl3, 0.5 g L−1, precipitated in EtOH, 1:4 v/v). The different wavelengths were realized by different LEDs – indicated by their peak wavelength (dashed lines). All LEDs were powered to emit the same photon flux of 1020 m−2 s−1. The error bars represent the standard deviation of the nanoparticle size of three nanoprecipitation experiments. | ||
More detailed insights into the fundamental origin of this effect can be gained from separating the broad white-light spectrum into wavelength regimes. Therefore, we investigated the nanoparticle formation under illumination from LEDs of different wavelengths with a constant photon flux of 1020 m−2 s−1. Fig. 3b depicts the resulting nanoparticle sizes versus the peak wavelengths of the respective LEDs, indicating three wavelength regimes. Upon illumination from LEDs with peak wavelengths shorter than 600 nm, the reduction of the nanoparticle size to below 50 nm is most pronounced. In contrast, the effect is negligible upon illumination from LEDs with a peak wavelength of more than 700 nm. Using LEDs with intermediate wavelengths of 600–700 nm, the nanoparticle size increases with increasing wavelength. Fig. 3b also depicts the absorbance of the P3HT/chloroform solution that is injected into the ethanol, as well as the absorbance of the P3HT dispersion that forms upon injection, the latter of which is red-shifted due to P3HT aggregation inside the nanoparticles. The nanoparticle size correlates very well with the absorption of the P3HT dispersion, which indicates that the stabilizing effect occurs after the initial aggregation of P3HT, i.e. the formation of polymer globules.
Fig. 4a depicts the calculated absorption rates r of photons in the P3HT dispersions (photons absorbed per second) during nanoprecipitation. First, we calculated the offset-corrected absorption (1-transmittance) occurring within the first 1 cm of the P3HT dispersion (5 mL of a 0.1 g L−1 P3HT dispersion). Multiplying the absorption of the dispersion and the emission spectrum of each LED yields the absorption rate r. LEDs with short peak wavelengths and hence large spectral overlap with the P3HT absorption yield a large absorption rate while the absorption rate of LEDs with peak wavelengths above 700 nm is (almost) zero. In Fig. 4b, we translated the plot of nanoparticle size over irradiance (Fig. 3a) into a plot of nanoparticle size over absorption rate. In both experiments, the nanoparticle sizes follow the same trend – no matter whether the absorption rate is tuned by the irradiance of the white LED or by the spectral overlap of monochromatic LEDs and nanoparticle absorption. This observation underlines that the absorption rate, and hence the number of photogenerated charges on the nanoparticles, is indeed the relevant quantity to influence the nanoparticle size during precipitation. In contrast, it is rather unlikely that illumination reduces the particle size by affecting the nucleation of the P3HT molecules or by conformational changes of the P3HT chains in solution as we did not see any correlation with the absorption of P3HT solutions (Fig. S1†).
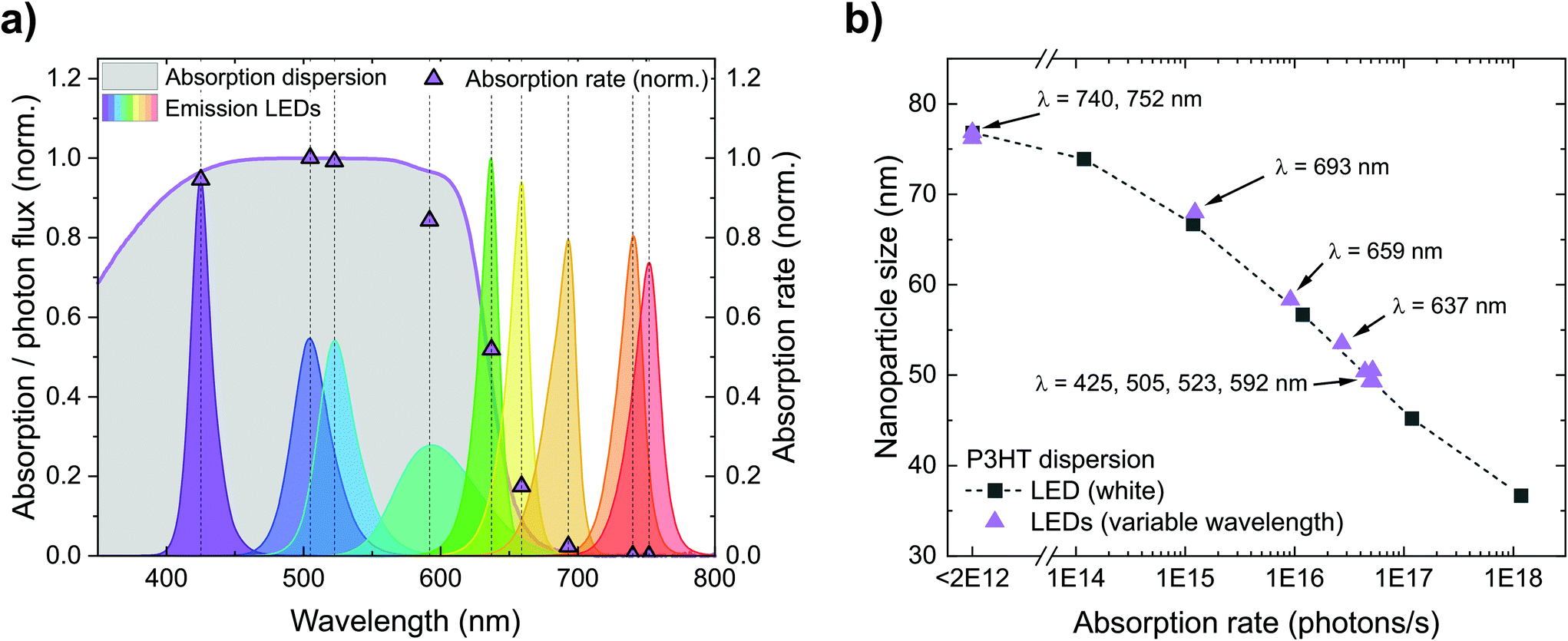 | ||
| Fig. 4 (a) Normalized absorption rates r during nanoprecipitation, depending on the LED used for illumination of the setup. The rates were calculated from the overlap between the absorption of the P3HT dispersion in the beaker and the spectrum of the respective LED. For LEDs with peak wavelengths below 600 nm, the normalized absorption rate r is close to 1, for wavelengths above 700 nm, the absorption rate is close to 0. (b) Both experiments from Fig. 3 (variable irradiance with white light or variable emission spectra) yield the same trend in nanoparticle size versus absorption rate, confirming that the absorption rate of the nanoparticles is indeed the relevant quantity to influence the nanoparticle size during nanoprecipitation. | ||
3.2. Transfer to other organic semiconductors
The photoexcitation of P3HT nanoparticle dispersions provides a handle to their stabilization. The question that remains to be answered is whether this stabilization of nanoparticle dispersions against agglomeration by photoexcitation is a singular observation on P3HT or if it can be generalized to be used with other organic semiconductor nanoparticle dispersions. Therefore, we repeated the nanoprecipitation experiment using a set of broadly commercially available semiconducting polymers, i.e. poly[2-methoxy-5-(3′,7′-dimethyloctyloxy)-1,4-phenylene vinylene] (MDMO-PPV), poly [[4,8-bis[(2-ethylhexyl)oxy]benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl][3-fluoro-2-[(2-ethylhexyl)carbonyl]thieno[3,4-b]thiophenediyl]] (PTB7), poly[(2,6-(4,8-bis(5-(2-ethylhexyl)thiophene-2-yl)-benzo[1,2-b:4,5-b′]dithiophene))-alt-(5,5-(1′,3′-di-2-thienyl-5′,7′-bis(2-ethylhexyl)benzo[1′,2′-c:4′,5′-c′]dithiophene-4,8-dion)] (PBDB-T, “PCE12”) and poly[2,2′′′′-bis[[(2-butyloctyl)oxy]carbonyl][2,2′:5′,2′′:5′′,2′′′-quaterthiophene]-5,5′′′-diyl] (PDCBT). Fig. 5 depicts the sizes of the nanoparticles that formed under illumination (white light, 750 W m−2) and in the dark (polymer in chloroform, 0.1 g L−1, nanoprecipitated in EtOH, 1:5 v/v). P3HT is shown for reference. The time elapsed between nanoprecipitation and the first DLS measurement was one minute in all experiments. All four polymer dispersions that were synthesized under illumination, exhibited remarkably reduced nanoparticle sizes of around 100 nm. In contrast, upon nanoprecipitation in the dark, all dispersions exhibited “nanoparticle” sizes of 300–1000 nm. Altogether, this experiment demonstrates that the concept of electrostatic stabilization by photoexcitation during nanoprecipitation is broadly applicable to other organic semiconductors, too, which is an important leap forward in the synthesis of surfactant-free organic semiconductor dispersions. Yet, for the fabrication of some optoelectronic thin-film devices, which require even smaller nanoparticles and higher dispersion concentrations, the stabilization by photoexcitation must be paired with other concepts to further reduce the nanoparticle sizes in the future.
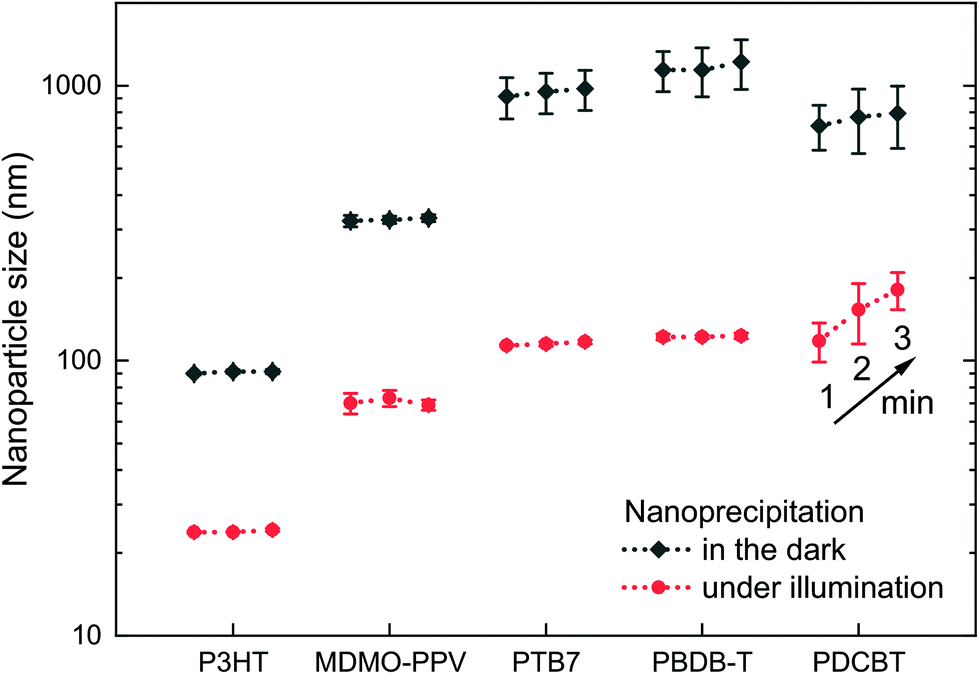 | ||
| Fig. 5 Sizes of nanoparticles synthesized from a range of different polymers under illumination (white light, 750 W m−2) and in the dark (polymer in CHCl3, 0.1 g L−1, nanoprecipitated in EtOH, 1:5 v/v, 50 °C). Consecutive measurements of the nanoparticle sizes after 1, 2 and 3 minutes indicate the initial dispersion stability. The error bars represent the standard deviation of the nanoparticle size of three nanoprecipitation experiments. | ||
Among the tested polymers, PDCBT dispersions showed the strongest tendency to coagulate: the nanoparticles already grew during the DLS measurements, showing varying size distributions throughout three consecutive measurements after 1, 2 and 3 minutes. The other polymer dispersions remained stable for a couple of days, even though no stabilizing agent was used. Continuous illumination helped to extend the time to coagulation of the dispersion. Yet, further volume reduction of the dispersions by evaporation of the solvent after nanoparticle synthesis, as it is required for later device fabrication, was not possible for either of the other polymers than P3HT. Dispersions and concentrated dispersions of P3HT exhibited shelf-lifetimes of several weeks or even months, no matter whether they were synthesized under residual light or under illumination. Obviously, the long-term stability of the dispersion depends on the ability of the polymer to maintain its (positive) charge created by photoexcitation, i.e. on the ionization potential of the semiconductor. We determined the ionization potentials of the polymers by photoelectron spectroscopy in air (PESA) (Fig. S2†). Among the investigated polymers, P3HT has a particularly low ionization potential, which might explain its outstanding stability in comparison to other surfactant-free organic semiconductor dispersions. Yet, other parameters than the ionization potential may also have influence on the intrinsic stability of polymer nanoparticles.
3.3. On the measurement of the zeta potential
The observations above pose some severe implications for state-of-the-art nanoparticle characterization routines, in first place for measurements of the zeta potential. The zeta potential is often used to describe the electrostatic stabilization of nanoparticle dispersions. A common technique for the determination of the zeta potential is electrophoretic light scattering. In a nutshell, this measurement principle uses an electrophoresis setup where the nanoparticle velocities are determined by analyzing the Doppler shift of scattered light.30 Hence, the measured quantity represents the mean nanoparticle velocity or, with respect to the applied electric field, the mean electrophoretic mobility of the nanoparticles. Using standard zeta potential measurement techniques, we confirmed the positive nanoparticle charge via electrophoretic light scattering and obtained electrophoretic mobilities for P3HT dispersions on the nominal order of 10−8 m2 V−1 s−1, which corresponds to a nominal zeta-potential of +80 mV (linear Hückel approximation). While the zeta potential reliably exhibited a positive sign, indicating a positive nanoparticle charge, its magnitude showed significant variations throughout the measurements, indicating a fundamental measurement limitation: as (to some extent persistent) charges are generated on the nanoparticles under illumination, the measurement is severely affected by the probe laser of the measurement setup (here: wavelength 633 nm, i.e. within the absorption range of P3HT nanoparticles). Hence, in our experiments on P3HT (and other) nanoparticle dispersions, electrophoretic light scattering appeared not to be suitable for the quantitative determination of the zeta potential. A better indication for the degree of stabilization of the dispersions is the nanoparticle size as discussed above. Likewise, the zeta potential measurements of other future light-harvesting organic nanoparticle dispersions must be interpreted carefully.4. The role of the fullerene
4.1. Nanoparticle formation in presence of ICBA
So far, we have observed that P3HT forms stable nanoparticle dispersions. Under illumination, additional photogenerated charges enhance the electrostatic stabilization and enable the formation of smaller nanoparticles. The latter effect also allows the synthesis of nanoparticles of other polymers which are not accessible without illumination. Common BHJ solar cells, however, contain a blend of polymers and fullerenes. So far, BHJ nanoparticle dispersions with best colloidal stability were realized employing the fullerene ICBA.To investigate the role of ICBA in the precipitation process, we performed nanoprecipitations of mixtures of P3HT and ICBA under illumination as well as in the dark (Fig. 6a). The initial P3HT concentration (1 g L−1) was kept constant and different amounts of ICBA were added to the P3HT/chloroform solution, up to a P3HT:ICBA ratio of 1:1 (w/w). Similar to the previous experiments with neat P3HT, illumination decreased the nanoparticle size significantly. Interestingly, already the addition of a small amount of ICBA (0.05 g L−1) to the initial P3HT solution yields a reduced nanoparticle size, indicating an enhanced charging of the nanoparticles. We observed this effect under illumination and in the dark but it is more pronounced under illumination. Towards higher ICBA content (P3HT:ICBA 1:1 w/w), the nanoparticle sizes increased somewhat again.
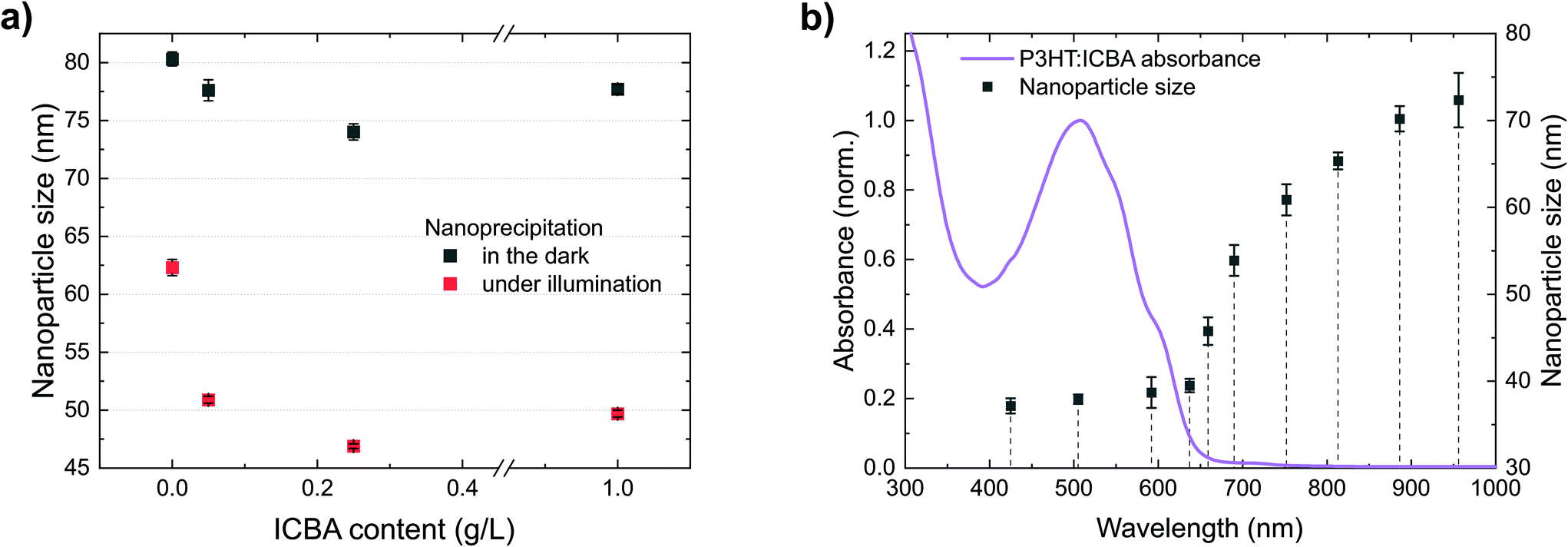 | ||
| Fig. 6 (a) Precipitation of P3HT:ICBA solutions with different amounts of ICBA (P3HT/CHCl3, 1.0 g L−1, plus ICBA, 0–1 g L−1, nanoprecipitated in EtOH, 1:4 v/v). (b) Dependence of the nanoparticle size on the illumination wavelength during nanoprecipitation (P3HT/CHCl3, 0.5 g L−1, plus ICBA, 0.5 g L−1, nanoprecipitated in EtOH, 1:4 v/v). The dependence of the nanoparticle size on the wavelength resembles the observations in Fig. 3b, but the addition of ICBA leads to smaller nanoparticles and the effect extends further into the infrared. The error bars represent the standard deviation of the nanoparticle size of three nanoprecipitation experiments. | ||
We attribute reduction of the nanoparticle size upon addition of ICBA to a fundamental property of BHJs: immediately after photoexcitation, electrons transfer from the polymers to ICBA.31,32 We found that ICBA exhibits a notable solubility of about 0.1 g L−1 in the ethanol:chloroform mixture (4:1 v/v). Hence, ICBA can carry the negative countercharge away from the nanoparticle into solution, providing additional stabilization support for the nanoparticle dispersion. This effect further enhances up to an ICBA content of 0.25 g L−1 in the initial solution (corresponding to 0.05 g L−1 in the dispersion). Beyond an ICBA content of 0.5 g L−1 in the initial solution (i.e. 0.1 g L−1 in the dispersion), the ICBA solubility limit in the ethanol:chloroform mixture is exceeded which may account for the nanoparticles to grow. The insoluble fraction of ICBA is then likely to be incorporated into the nanoparticles instead of providing additional floating and stabilizing countercharges (anions). The decreased nanoparticle size upon addition of ICBA in the dark could either be caused by residual light (<0.01 W m−2) or rely on intrinsic ground-state charge transfer. This interpretation is in accordance with an earlier report on the stabilization of P3HT nanoparticles by dissolved PCBM in dimethylsulfoxide (DMSO), yet illumination conditions were not considered in that study.33 The theory of floating countercharges to stabilize P3HT:ICBA dispersions is also supported by a recent report on P3HT:ICBA nanoparticle dispersions where P3HT was chemically modified for pyridine end-capping. Acetic acid, which was added to the non-solvent ethanol, protonated the pyridine group and then formed a floating (negatively charged) anion.34 After nanoparticle formation and layer deposition, the process reverted back to the uncharged polymer under evaporation of acetic acid.
The P3HT:ICBA nanoparticle size in Fig. 6b shows a wavelength-dependence similar to neat P3HT but the stabilizing effect extends further into the infrared. We expect the excitation of charge-transfer states by absorption, that is an electron transfer from ground state P3HT to the electronically excited fullerene,35 to account for the generation of stabilizing charges at wavelengths beyond 750 nm.36
4.2. Increasing the concentration of the dispersion
For the fabrication of inks for organic solar cells or other optoelectronic applications, typically, concentrations higher than 1 or 2 g L−1 are needed, otherwise imposing too many consecutive process steps during thin-film deposition. In earlier reports, concentrations around 10 g L−1 (5 g L−1 P3HT and 5 g L−1 ICBA) were used for this purpose.6,16,21 When increasing the concentration of the initial P3HT solution, we observed only little gain in nanoparticle stability under illumination (white light, 10 W m−2) versus nanoprecipitation in the dark (Fig. S3†). From an initial polymer concentration of 5 g L−1 and beyond, illumination did no longer affect the nanoparticle size, which we attribute to the strong light attenuation by the nanoparticles close to the surface of the vial and hence lower illumination of buried nanoparticles. For mixed solutions of P3HT and ICBA (each 5 g L−1, total 10 g L−1), the nanoparticle size was reduced from 84 nm upon nanoprecipitation in dark to 71 nm upon nanoprecipitation under white-light illumination (1000 W m−2). However, using smaller vials for the nanoprecipitation and even stronger illumination, we achieved significantly smaller nanoparticles compared to the nanoprecipitation in the dark. This demonstrates the general feasibility to affect the nanoparticle size even for the preparation of concentrated dispersions, but proper light penetration into the dispersion is essential.5. Conclusions
If handles to the control of the nanoparticle sizes and the dispersion stability can be identified, surfactant-free organic nanoparticle dispersions will become a viable route to the eco-friendly processing of organic solar cells and organic printed electronics in general. In this work, we have shown that the surfactant-free P3HT nanoparticle dispersions, which have been used for the fabrication of organic solar cells so far, are stabilized electrostatically. The electrostatic stabilization of the dispersions can be enhanced by illumination during the nanoprecipitation and the formation of nanoparticles. The nanoparticle size was used as an indicator for the dispersion stability, since smaller nanoparticles require stabilization of an overall larger surface. The dispersion stability depends on the illumination intensity as well as its wavelength. The countercharges float away from the P3HT nanoparticles, with the effect being more pronounced by intermixing the electron acceptor ICBA which exhibits residual solubility in the dispersion medium ethanol or ethanol:chloroform. Intriguingly, the stabilization of dispersions by light lets us omit any process additives, which may later hamper the semiconductor performance in devices such as solar cells. The use of electron acceptors such as ICBA is particularly appealing for the synthesis of solar inks and the fabrication of organic solar cells, where acceptors are incorporated anyway. Not least, ambient illumination of the nanoparticle synthesis platform is an important hidden process parameter which must be controlled for reproducible results.
We conclude that the stability of surfactant-free (organic) semiconductor dispersions can be enhanced by the generation of charges on the nanoparticles. While the powerful concept presented in this work is readily applicable to a wide range of polymers, further stabilization of the dispersions may be needed to also achieve long-term stability of the surfactant-free dispersions. Future efforts to further enhance the dispersion stability should therefore encompass the identification of processes that generate even more charges on the nanoparticle surface.
6. Methods
All experiments were carried out in a cleanroom (class 10000) to reduce the risk of contamination with dust particles.
6.1. Materials
Regioregular poly(3-hexylthiophene-2,5-diyl) (P3HT, “4002-EE”, Mw = 50–70 kg mol−1, regioregularity ≥90%) was purchased from Rieke Metals. Indene-C60 bisadduct (ICBA) was purchased from Solenne, poly [[4,8-bis[(2-ethylhexyl)oxy]benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl][3-fluoro-2-[(2-ethylhexyl)carbonyl]thieno[3,4-b]thiophenediyl]] (PTB7) and poly[2,2′′′′-bis[[(2-butyloctyl)oxy]carbonyl][2,2′:5′,2′′:5′′,2′′′-quaterthiophene]-5,5′′′-diyl] (PDCBT) from 1-material, poly[2-methoxy-5-(3′,7′-dimethyloctyloxy)-1,4-phenylene vinylene] (MDMO-PPV) from Sigma Aldrich and poly[(2,6-(4,8-bis(5-(2-ethylhexyl)thiophene-2-yl)-benzo[1,2-b:4,5-b′]dithiophene))-alt-(5,5-(1′,3′-di-2-thienyl-5′,7′-bis(2-ethylhexyl)benzo[1′,2′-c:4′,5′-c′]dithiophene-4,8-dion)] (PBDB-T, “PCE-12”) from Brilliant Matters. Chloroform (CHCl3, analytical grade) and ethanol (EtOH, analytical grade) were purchased from Merck and were used without further purification unless indicated otherwise.6.2. Nanoparticle Synthesis
Organic semiconductor nanoparticles were prepared via surfactant-free nanoprecipitation. Initial solutions were prepared by dissolving the respective polymers under magnetic stirring in CHCl3 at 60 °C. The nanoprecipitation was conducted at room-temperature in small glass beakers following established protocols.16 Therefore, the polymer/CHCl3 solution was injected with a pipette into the non-solvent EtOH under vigorous stirring (1:4 or 1:5 v/v, total volumes of 2–5 mL, injection time of 1 s per mL of polymer solution). Nanoparticle sizes were measured right after the precipitation. Where indicated, the remaining CHCl3 was removed by heating in a water bath (75 °C). While removing the CHCl3, the concentration of the dispersions was increased to 5 g L−1. Nanoprecipitation under illumination was carried out using a white-light LED (Cree XM-L2 LED, Fig. 3a). Illumination with different wavelength regimes was realized with an LED-solar simulator (VeraSol-2, Oriel, Fig. 4a). Either light source was mounted above the beaker during nanoprecipitation. The irradiance was monitored using a spectrometer (CAS 140CT-156, Instrument Systems).
6.3. Electrophoresis measurements
The electrophoretic measurements were conducted in a folded plastic capillary cell (Malvern Panalytical) at a DC voltage of 50 V. The respective P3HT dispersion was prepared by surfactant-free nanoprecipitation of a CHCl3 solution (5 g L−1) in the non-solvent EtOH (1:4 v/v). Subsequently CHCl3 was removed by heating in a water bath. To enhance the measurement quality, the dispersion was then diluted to a concentration of 0.1 g L−1 with pure EtOH.
6.4. Solubility of ICBA in EtOH
To determine the solubility of ICBA in ethanol or in the ethanol:chloroform mixture (4:1 v/v), we added ICBA (2 mg) to the solvent (2 mL) and stirred the mixture at room temperature for three days. Then we separated the undissolved fullerene from the solution by filtering through a syringe filter (0.2 μm). The solubility was then determined by comparing the absorption spectrum of the filtrate with the absorption of a reference solution (UV-Vis-NIR spectrophotometer Cary 5000, Agilent Technologies).
6.5. Probing the electrostatic stability
To probe the dispersion stability after nanoprecipitation, we added NaBr (10−3 mol L−1, i.e. 10 vol% of a 10−2 mol L−1 NaBr solution in EtOH) to a 0.1 g L−1 P3HT dispersion. To probe the influence of electrostatic forces during nanoparticle growth, we dissolved small amounts of NaBr (10−6–10−2 mol L−1) in EtOH prior to nanoprecipitation.6.6. Dynamic light scattering
The size distribution of the nanoparticles in dispersion was determined by dynamic light scattering (DLS, Zetasizer Nano ZS, Malvern Panalytical). The reported nanoparticle sizes in this work are the “z-average” diameters that were calculated by the measurement software. We diluted all samples prior to the measurement, so that dispersions contained only minor amounts of CHCl3 (2 vol%). For the calculation of the nanoparticle size, we assumed pure EtOH, a temperature of 20 °C, with a dynamic viscosity of 1.14 mPa s and a refractive index of 1.361.6.7. Zeta-potential
The electrophoretic mobility or zeta-potential of the nanoparticle dispersions was determined by electrophoretic light scattering (ELS, Zetasizer Nano ZS, Malvern Panalytical) at a semiconductor concentration of 0.05 g L−1 in pure EtOH after removal of CHCl3 and dilution with EtOH, using an electrophoretic “dip” cell (Malvern Panalytical) at a voltage of 10 V.6.8. UV-Vis absorption spectrometry
UV-Vis absorption spectra of semiconductor dispersions or solutions in quartz cuvettes were recorded with a UV-Vis-NIR spectrophotometer (Cary 5000, Agilent Technologies). The P3HT solution was measured at a concentration of 0.05 g L−1 in CHCl3, the ICBA solution at a concentration of 0.02 g L−1 in CHCl3. The P3HT dispersions were nanoprecipitated from a solution with a concentration of 5 g L−1 in EtOH (1:4 v/v). After removal of CHCl3, we diluted the dispersions with EtOH (0.05 g L−1).
6.9. Photoelectron spectroscopy in air (PESA)
Ionization potentials of polymers were obtained on thin films by photoelectron spectroscopy in air using a photoelectron spectrometer (AC-2, Riken Keiki Co., Ltd). The thin-film samples were prepared by spincoating of a 5–10 g L−1 solution of the respective polymer in o-dichlorobenzene onto a glass substrate.Author contributions
P. M., F. M. and A. C. conceived the experiments. P. M., F. M. and K. F. carried out the experiments. C. S. and A. C. supervised the work. P. M., C. S. and A. C. jointly wrote the manuscript. All authors discussed the data and approved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Federal Ministry of Education and Research (BMBF), Germany, under contract no 03EK3571 (project TAURUS II). The authors acknowledge fruitful discussions with David Jones (University of Melbourne) and Holger Röhm (Karlsruhe Institute of Technology).References
- M. P. Tsang, G. W. Sonnemann and D. M. Bassani, Sol. Energy Mater. Sol. Cells, 2016, 156, 37–48 CrossRef CAS.
- N. Espinosa, M. Hösel, D. Angmo and F. C. Krebs, Energy Environ. Sci., 2012, 5, 5117–5132 RSC.
- D. Zimmermann, C. Sprau, J. Schröder, V. G. Gregoriou, A. Avgeropoulos, C. L. Chochos, A. Colsmann, S. Janietz and H. Krüger, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1457–1467 CrossRef CAS.
- Y. Huang and C. K. Luscombe, Chem. Rec., 2019, 19, 1039–1049 CrossRef CAS PubMed.
- C. Sprau, J. Kattenbusch, Y. Li, E. Müller, D. Gerthsen, R. Berger, J. J. Michels and A. Colsmann, Sol. RRL, 2021, 5, 2100238 CrossRef CAS.
- S. Gärtner, S. Reich, M. Bruns, J. Czolk and A. Colsmann, Nanoscale, 2016, 8, 6721–6727 RSC.
- S. Gärtner, A. J. Clulow, I. A. Howard, E. P. Gilbert, P. L. Burn, I. R. Gentle and A. Colsmann, ACS Appl. Mater. Interfaces, 2017, 9, 42986–42995 CrossRef PubMed.
- K. Landfester, R. Montenegro, U. Scherf, R. Güntner, U. Asawapirom, S. Patil, D. Neher and T. Kietzke, Adv. Mater., 2002, 14, 651–655 CrossRef CAS.
- T. Kietzke, D. Neher, K. Landfester, R. Montenegro, R. Güntner and U. Scherf, Nat. Mater., 2003, 2, 408–412 CrossRef CAS PubMed.
- S. Ulum, N. Holmes, M. Barr, A. L. D. Kilcoyne, B. B. Gong, X. Zhou, W. Belcher and P. Dastoor, Nano Energy, 2013, 2, 897–905 CrossRef CAS.
- N. P. Holmes, M. Marks, P. Kumar, R. Kroon, M. G. Barr, N. Nicolaidis, K. Feron, A. Pivrikas, A. Fahy, A. D. de Z. Mendaza, A. L. D. Kilcoyne, C. Müller, X. Zhou, M. R. Andersson, P. C. Dastoor and W. J. Belcher, Nano Energy, 2016, 19, 495–510 CrossRef CAS.
- D. Darwis, N. Holmes, D. Elkington, A. L. D. Kilcoyne, G. Bryant, X. Zhou, P. Dastoor and W. Belcher, Sol. Energy Mater. Sol. Cells, 2014, 121, 99–107 CrossRef CAS.
- C. Xie, T. Heumüller, W. Gruber, X. Tang, A. Classen, I. Schuldes, M. Bidwell, A. Späth, R. H. Fink, T. Unruh, I. McCulloch, N. Li and C. J. Brabec, Nat. Commun., 2018, 9, 1–11 CrossRef.
- H. Shimizu, M. Yamada, R. Wada and M. Okabe, Polym. J., 2008, 40, 33–36 CrossRef CAS.
- J. E. Millstone, D. F. J. Kavulak, C. H. Woo, T. W. Holcombe, E. J. Westling, A. L. Briseno, M. F. Toney and J. M. J. Fréchet, Langmuir, 2010, 26, 13056–13061 CrossRef CAS PubMed.
- S. Gärtner, M. Christmann, S. Sankaran, H. Röhm, E. M. Prinz, F. Penth, A. Pütz, A. E. Türeli, B. Penth, B. Baumstümmler and A. Colsmann, Adv. Mater., 2014, 6653–6657 CrossRef PubMed.
- J. Polte, CrystEngComm, 2015, 17, 6809–6830 RSC.
- R. J. Hunter, Introduction to Modern Colloid Science, Oxford University Press, Oxford, 1993 Search PubMed.
- J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press, Waltham, 3rd edn, 2011 Search PubMed.
- H. Shimizu, M. Yamada, R. Wada and M. Okabe, Polym. J., 2008, 40, 33–36 CrossRef CAS.
- C. Xie, X. Tang, M. Berlinghof, S. Langner, S. Chen, A. Späth, N. Li, R. H. Fink, T. Unruh and C. J. Brabec, ACS Appl. Mater. Interfaces, 2018, 10, 23225–23234 CrossRef CAS PubMed.
- S. Bellani, D. Fazzi, P. Bruno, E. Giussani, E. V. Canesi, G. Lanzani and M. R. Antognazza, J. Phys. Chem. C, 2014, 118, 6291–6299 CrossRef CAS.
- J. B. Rosenholm, Adv. Colloid Interface Sci., 2018, 259, 21–43 CrossRef CAS PubMed.
- D. Valencia, G. T. Whiting, R. E. Bulo and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2016, 18, 2080–2086 RSC.
- A. Knežević and Z. B. Maksić, New J. Chem., 2006, 30, 215–222 RSC.
- C. Zhang, V. J. Pansare, R. K. Prud'homme and R. D. Priestley, Soft Matter, 2012, 8, 86–93 RSC.
- A. Nikoubashman, V. E. Lee, C. Sosa, R. K. Prud'homme, R. D. Priestley and A. Z. Panagiotopoulos, ACS Nano, 2016, 10, 1425–1433 CrossRef CAS PubMed.
- N. Li, A. Nikoubashman and A. Z. Panagiotopoulos, J. Chem. Phys., 2018, 149, 084904 CrossRef PubMed.
- I. D. Morrison, Colloids Surf., A, 1993, 71, 1–37 CrossRef CAS.
- S. Bhattacharjee, J. Controlled Release, 2016, 235, 337–351 CrossRef CAS PubMed.
- N. S. Sariciftci, L. Smilowitz, A. J. Heeger and F. Wudl, Science, 1992, 258, 1474–1476 CrossRef CAS PubMed.
- P. Heinrichova, M. Vala and M. Weiter, Chem. Phys. Lett., 2014, 592, 314–319 CrossRef CAS.
- A. Palacio Valera, C. Schatz, E. Ibarboure, T. Kubo, H. Segawa and S. Chambon, Front. Energy Res., 2019, 6, 1–11 Search PubMed.
- S. Saxena, P. Marlow, J. Subbiah, A. Colsmann, W. W. H. Wong and D. J. Jones, ACS Appl. Mater. Interfaces, 2021, 13, 36044–36052 CrossRef CAS PubMed.
- J. W. Arbogast, C. S. Foote and M. Kao, J. Am. Chem. Soc., 1992, 114, 2277–2279 CrossRef CAS.
- R. A. Street, D. Davies, P. P. Khlyabich, B. Burkhart and B. C. Thompson, J. Am. Chem. Soc., 2013, 135, 986–989 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr00095d |
| This journal is © The Royal Society of Chemistry 2022 |