
Triggering the nanophase separation of albumin through multivalent binding to glycogen for drug delivery in 2D and 3D multicellular constructs†
Agata
Radziwon
 a,
Sukhvir K.
Bhangu
b,
Soraia
Fernandes
c,
Christina
Cortez-Jugo
a,
Robert
De Rose
a,
Brendan
Dyett
b,
Marcin
Wojnilowicz
a,
Petra
Laznickova
c,
Jan
Fric
cd,
Giancarlo
Forte
c,
Frank
Caruso
*a and
Francesca
Cavalieri
*be
a,
Sukhvir K.
Bhangu
b,
Soraia
Fernandes
c,
Christina
Cortez-Jugo
a,
Robert
De Rose
a,
Brendan
Dyett
b,
Marcin
Wojnilowicz
a,
Petra
Laznickova
c,
Jan
Fric
cd,
Giancarlo
Forte
c,
Frank
Caruso
*a and
Francesca
Cavalieri
*be
aDepartment of Chemical Engineering, The University of Melbourne, Parkville, Victoria 3010, Australia. E-mail: fcaruso@unimelb.edu.au
bSchool of Science, RMIT University, Melbourne, Victoria 3000, Australia. E-mail: francesca.cavalieri@rmit.edu.au
cInternational Clinical Research Center (ICRC), St Anne's University Hospital, CZ-65691 Brno, Czech Republic
dInstitute of Hematology and Blood Transfusion, Prague, Czech Republic
eDipartimento di Scienze e Tecnologie Chimiche, Università degli Studi di Roma Tor Vergata, Via della Ricerca Scientifica 1, 00133, Rome, Italy
First published on 24th January 2022
Abstract
Engineered nanoparticles for the encapsulation of bioactive agents hold promise to improve disease diagnosis, prevention and therapy. To advance this field and enable clinical translation, the rational design of nanoparticles with controlled functionalities and a robust understanding of nanoparticle–cell interactions in the complex biological milieu are of paramount importance. Herein, a simple platform obtained through the nanocomplexation of glycogen nanoparticles and albumin is introduced for the delivery of chemotherapeutics in complex multicellular 2D and 3D systems. We found that the dendrimer-like structure of aminated glycogen nanoparticles is key to controlling the multivalent coordination and phase separation of albumin molecules to form stable glycogen–albumin nanocomplexes. The pH-responsive glycogen scaffold conferred the nanocomplexes the ability to undergo partial endosomal escape in tumour, stromal and immune cells while albumin enabled nanocomplexes to cross endothelial cells and carry therapeutic agents. Limited interactions of nanocomplexes with T cells, B cells and natural killer cells derived from human blood were observed. The nanocomplexes can accommodate chemotherapeutic drugs and release them in multicellular 2D and 3D constructs. The drugs loaded on the nanocomplexes retained their cytotoxic activity, which is comparable with the activity of the free drugs. Cancer cells were found to be more sensitive to the drugs in the presence of stromal and immune cells. Penetration and cytotoxicity of the drug-loaded nanocomplexes in tumour mimicking tissues were validated using a 3D multicellular-collagen construct in a perfusion bioreactor. The results highlight a simple and potentially scalable strategy for engineering nanocomplexes made entirely of biological macromolecules with potential use for drug delivery.
 Francesca Cavalieri | Dr Francesca Cavalieri received her Laurea degree, ‘maxima cum laude’, in Industrial Chemistry from the University of Rome La Sapienza. She completed her PhD in Chemistry (2014) at the University of Melbourne. Since 2002, she has been a tenured Assistant Professor in Physical Chemistry of Macromolecules at the University of Rome ‘Tor Vergata’ Italy. In 2015, she was awarded the ARC Future Fellowship at the University of Melbourne and in 2019 she moved to RMIT University as Vice Chancellor Senior Research Fellow. Her research focuses on the engineering and characterization of nanostructured biopolymers for drug/gene delivery. |
Introduction
The delivery of conventional chemotherapeutics, biologics, and nucleic acids into solid tumours depends on their ability to cross numerous biological barriers including the ability to pass through vascular endothelium, penetrate the tumour microenvironment and enter the cancer cells.1 The direct administration of naked therapeutics to patients is often limited by their inherent instability in biological fluids and adverse side effects, highlighting the need for non-toxic and effective drug nanocarriers.2 Nanoparticles of various materials including lipids,3 polymers,4 metal,5 carbohydrates,6–9 proteins10–13 and DNA14 have been engineered to facilitate the transport of therapeutics across biological barriers and minimize nonspecific uptake by phagocytic cells and off-target in vivo biodistribution.15 However, it is widely reported that the nanoparticle delivery efficiency to solid tumours is very low and the majority of the nanotherapeutics generally accumulate in off-target tissues.16,17 The biologically informed design of highly efficient nanoparticles that are able to successfully overcome biological barriers still remains an elusive goal. After intravenous injection, nanoparticles access the solid tumour primarily by crossing the endothelial cells through an active process of transcytosis.18 In particular, a subset of tumour endothelial cells was found to act as gatekeepers to control the number of nanoparticles that enter the tumour tissue.18,19 Therefore, to enable entry and accumulation of nanoparticles in solid tumours, strategies to enhance the trans-endothelial transport of nanoparticles are required. Another biological barrier that hampers the efficient delivery of nanoparticles to cancer cells is the tumour microenvironment (TME).20,21 The heterogeneous TME is characterized by the presence of cells with distinct genetic, transcriptomic, epigenetic, and phenotypic properties and hence, different sensitivity towards drug treatment.22,23 In general, the TME consists of the tumour, endothelial, and immune cells, and tissue stroma.24 The stroma is composed of specialized connective tissue cells, including fibroblasts and the extracellular matrix (ECM). The ECM components can trap and sterically hinder nanoparticle diffusion to the cancer cells.24 Macrophages can not only phagocytose nanoparticles, thus preventing them from delivering the drugs to cancer cells but also lead to chemoresistance by secretion of interleukin-10 (IL-10) that causes suppression of tumour resident immune cells.25,26 Fibroblasts are known to remodel the ECM, by releasing the pro-angiogenic signals and immunosuppressive cytokines within the tumour matrix.25,26 Although anticancer therapies mediated by nanoparticles predominantly target cancer cells, recent preclinical studies27 suggest that chemotherapeutics should not only target cancer cells but all components of TME that support tumour growth. In addition, a rational approach for testing the drug delivery ability of nanoparticles requires the development of 3D-multicellular dynamic microenvironment to partially mimic the complex in vivo features of tumours, such as high cell density and cell-to-cell contacts, hypoxia, elevated interstitial pressure, resistance to drug treatment, and production of the extracellular matrix.Herein, we engineered a platform through the nano-complexation of two biomacromolecules, i.e., glycogen and serum albumin, for the delivery of chemotherapeutics in complex multicellular 2D and 3D systems, composed of cancer, endothelial, stromal, and immune cells. Glycogen is a dendrimer-like biological nanoparticle comprising of repeating units of glucose connected by linear a-D-(1,4) glycosidic linkages with a-D-(1,6) branching, which holds promise as a material for biomedical applications.28 Albumin, the most abundant protein in human plasma, is a biodegradable and non-immunogenic biopolymer, due to its intrinsic ability to carry hydrophobic drugs.29,30 In addition, albumin has high affinity to the gp60 receptor found on the surfaces of endothelial cells that line tumour vessels31 and the secreted protein acidic and rich in cysteine (SPARC), an extracellular protein overexpressed by many types of tumours.32 This potentially enables albumin-based nanoparticles to extravasate inside the tumour through the formation of transcytosis vesicles31 and accumulate in the intra-tumoral region.33 Different approaches for fabricating nanoparticles or submicron aggregates entirely made of albumin have been developed including desolvation,34–36 emulsification,37,38 thermal gelation,39,40 chemical conjugation41,42 or nanospray drying.43,44 These methods require the use of toxic crosslinkers (glutaraldehyde), denaturing agents (mercaptoethanol) and chlorinated solvents (chloroform), surfactants, thermal or high-pressure treatments that can partially denature the protein. Limited work has been undertaken on the nano-complexation of albumin with other macromolecules45,46 to obtain hybrid nanoparticles with controlled and predictable intracellular behaviour. In this work, we show that the unique dendrimer-like structure of cationic glycogen nanoparticles is key to controlling the multivalent coordination and phase separation of albumin molecules to form stable and multifunctional glycogen–albumin nanocomplexes (NCBGEDA/BSA). The nanocomplex was endowed with the ability to carry chemotherapeutics (i.e. paclitaxel and doxorubicin), cross endothelial cells by the albumin mediated transcytosis and overcome endo-lysosomal entrapment in cancer and stromal cells. When incubated with peripheral blood mononuclear cells (PBMCs) isolated from the blood of healthy donors, nanocomplexes were primarily phagocytosed by monocytes and showed limited interactions with T cells, B cells and natural killer cells. Chemotherapeutics were successfully delivered by the nanocomplexes in the co-culture of BT-474 breast cancer cells, NIH-3T3 stromal cells and RAW 264.7 macrophage cells. Penetration and cytotoxicity of the drug loaded nanocomplexes in tumour mimicking tissues was validated using a 3D multicellular-collagen construct in a perfusion bioreactor. The cytotoxicity of drug-loaded nanocomplexes in cells grown in a 3D perfusion bioreactor was significantly reduced compared to 2D cell co-cultures. Our results highlight the potential of glycogen–albumin nanocomplexes as a versatile drug delivery system and the utility of screening the nanomaterial bio-interactions and therapeutic activity in complex 3D multicellular systems that mimic the natural in vivo setting more closely than traditional monolayer (2D) cell cultures.
Experimental
Materials
Glycogen from bovine liver, sodium cyanoborohydride, ethylenediamine, α-amylase from Aspergillus oryzae, β-amylase from barley, Dulbecco's phosphate-buffered saline (DPBS), sodium potassium tartrate tetrahydrate, sodium carbonate, sodium bicarbonate, sodium sulfate, copper sulfate pentahydrate, ammonium molybdate, sulfuric acid, sodium arsenate dibasic pentahydrate, deuterated water, NaN3, filipin from S. filipinensis, EIPA, human serum and bovine serum albumin were purchased from Sigma-Aldrich (St Louis, MO, USA). Hydrochloric acid (HCl), sodium chloride (NaCl), Triton X-100 and Tween-20 were purchased from Chem-Supply (St Gillman, Australia). Pitstop 2 was purchased from Abcam (Cambridge, UK). Dialysis tubing (10 kDa molecular weight cutoff), LysoTracker™ Green DND-26, Hoechst 33342 solution, PrestoBlue™ cell viability reagent, AlamarBlue™ cell viability reagent, and Sytox Blue nucleic acid stain were obtained from Thermo Fisher Scientific (Scoresby, Australia). Alexa Fluor NHS dyes (AF488, AF555, and AF647), Alexa Fluor-phalloidin conjugates, AF647-wheat germ agglutinin conjugate and trypsin were purchased from Life Technologies (Scoresby, Australia). Dulbecco's modified Eagle medium (DMEM), EBM basal medium and EGM SingleQuot kit were purchased from Lonza (Allendale, USA). Fetal bovine serum (FBS) was purchased from Bovogen (Keilor East, Australia). Illustra NAP-10 columns were purchased from GE Healthcare and Life Sciences (Silverwater, Australia). G418 was obtained from InvivoGen (San Diego, USA). Raw 264.7 murine macrophage cell line (TIB-71™), BT-474 breast cancer cell line (HTB-20™) and NIH-3T3 cell line (CRL-1658™) were obtained from ATCC and cultured with the passage number between 10 and 40. The umbilical cord–vascular system HUVEC/TERT2 cell line was obtained from Evercyte (Vienna, Austria) and cultured with the passage number of less than 10. All cell lines were tested for mycoplasma on a regular basis. Rabbit anti-Rab7, rabbit anti-EEA1, rabbit anti-E-cadherin and rat CD11b/ITGAM monoclonal antibodies were purchased from Cell Signaling Technology (Danvers, United States). Mouse anti-LAMP-1 antibody and Alexa Fluor secondary conjugates were supplied by Invitrogen (Carlsbad, USA). CellTiter-Glo® 3D cell viability assay kit was purchased from Promega. Optimal cutting temperature (OCT) medium was purchased from ProSciTech (Kirwan, Australia). Alexa Fluor secondary conjugates were supplied by Invitrogen (Carlsbad, USA). All chemicals were used as received without further purification. High-purity water with a resistivity greater than 18.2 MU cm was obtained from a three-stage Millipore Milli-Q plus 185 purification system (Millipore Corporation, Billerica, MA).Synthesis of BGEDA nanoparticles
An aliquot of glycogen from bovine liver (100 mg, 0.6 mmol of glucose units) was dissolved in 0.5 M acetic buffer (pH = 5.5). Then, 26 mg (0.12 mmol) of sodium periodate was added and the reaction was performed for 4 h protected from the light. Afterwards, 36 mg (0.6 mmol) of ethylenediamine (EDA) and 10× excess of cyanoborohydride (76 mg; 1.2 mmol) was added. Following this, the reaction was degassed, the pH was adjusted to 5.5 and the mixture was stirred overnight. The product was purified by dialysis (14 kDa cut off) against Milli-Q water for 3 days (9 times water change) and then freeze dried. Yield: 85%. The degree of substitution of modified glycogen was determined by 1H NMR. 1H NMR spectra were recorded on a Varian INOVA 400 instrument, operating at 400 MHz after dissolution in deuterated water (D2O).Dynamic light scattering (DLS) and ζ-potential measurements
The hydrodynamic diameter and ζ-potential measurements were performed on a Zetasizer Nano-ZS (Malvern Instruments, Malvern, UK) equipped with He–Ne ion laser (λ = 633 nm). For the DLS measurements, solutions of 1 mg mL−1 of particles dissolved in Milli-Q water were measured in microcuvettes (ZEN0040, Malvern Instruments). ζ-Potential measurements were performed on 3 mg mL−1 solutions of particles in Milli-Q water in folded capillary cells (DTS1070, Malvern Instruments). All measurements were performed using standard procedure.NCBGEDA/BSA preparation
The complexation between BGEDA and BSA was monitored using DLS and ζ-potential. For the measurements, 1 mg mL−1 of BGEDA sample was titrated at BSA with increasing weight ratios (w/w 1–25) in Milli-Q H2O, followed by incubation for 20 min at 25 °C and subsequent DLS or ζ-potential measurement. The unbound protein was removed using Nanosep® Centrifugal Devices with Omega™ Membrane 300 kDa cut off, following the supplier's protocol.Atomic force microscopy (AFM)
AFM measurements (in air) were performed in AC mode with a Cypher ES atomic force microscope (Asylum Research, USA), using Tap300 cantilevers (Budget Sensors) with a resonance frequency of 300 kHz and a spring contrast of 40 Nm−1. NCBGEDA/BSA nanocomplexes were dispersed onto freshly cleaved mica and dried to a concentration 0.01 mg mL−1.Stochastic optical reconstruction microscopy (STORM)
For STORM analysis, BGEDA nanoparticles (10 mg) were dissolved in 2 mL of 0.1 M sodium bicarbonate buffer, pH = 8, mixed with 100 μL of 1 mg mL−1 Alexa Fluor 647 NHS dye and incubated in the dark with stirring overnight. Then, the product was purified on a NAP-10 filter column and the complexes with BSA were prepared using the protocol described in section NCBGEDA/BSA preparation. For imaging, AF647-BGEDA or NCAF647-BGEDA/BSA solutions were incubated on a glass slide or on a PEI-coated glass slide, respectively. After 30 min of incubation at 25 °C, unbound molecules were washed away with freshly prepared, standard imaging buffer with cysteamine (MEA).47STORM images were acquired using a Nikon N-STORM system equipped with a Nikon 100× 1.4 NA oil immersion objective. The focus and the total internal reflection fluorescence imaging angle were adjusted to obtain a high signal-to-noise ratio. A 647 laser was used for the excitation of the fluorophores. All time lapses were recorded within a 256 × 256 pixel region using an EMCCD camera. For each image, 5000 frames were acquired sequentially with full laser power. STORM images were first processed with the STORM module of the NIS Elements Nikon software, where drift correction was performed, and a list of particle localisations was prepared by Gaussian fitting of the fluorescence spots of blinking dyes. For all images, the identification threshold of 700 photons was kept constant. Blinking events that were detected in ≤5 consecutive frames were counted as single molecules, while events detected in more than 5 consecutive frames were discarded (trace length 1–5). The list of localisations was exported as a .txt file and analysed using a home-made clustering analysis script, where the BGEDA localizations were clustered using a kernel density estimation with a bandwidth of 50 nm. An ellipse was fitted to the obtained clusters with minimum 10 localizations and maximum elongation factor of 1.5 (ratio of long and short axes of the ellipse). Then, the circles containing 90% of detected spots in the cluster were fitted allowing for the determination of the imaged particles’ sizes.
Fluorescence correlation spectroscopy (FCS) measurements
FCS experiments were performed on a Nikon A1R confocal microscope combined with a MicroTime PicoQuant system with 40×/1.1 NA water immersion objective and 647 nm laser for illumination. The confocal volume (Veff) was calibrated by the measurements of AF 647 dye with the known DAF647 = 3.3 ± 0.1 × 10−6 cm2 s−1 at the beginning of each experiment. All measured solutions were diluted to 0.01 mg mL−1 of the labelled molecules before the measurement, following the same protocol as previously described for STORM imaging, with each measurement lasting 30 s and repeated at least 20 times. The generated autocorrelation function (ACF) curves were analysed using the SymPhoTime 64 software.Cell association studies
For cell association, BT-474, NIH-3T3 and Raw 264.7 cells were cultured at 37 °C in Dulbecco's modified Eagle medium (DMEM) with 10% FBS. The cells were seeded individually at a density of 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well on 24-well plates and incubated for 24 h prior to NCAF647-BGEDA/BSA addition at concentration of 0.01 mg mL−1 of BGEDA. The cell fluorescence intensity was acquired on a BD Accuri C6 flow cytometer and is expressed as the geometric mean of fluorescence intensity.
000 cells per well on 24-well plates and incubated for 24 h prior to NCAF647-BGEDA/BSA addition at concentration of 0.01 mg mL−1 of BGEDA. The cell fluorescence intensity was acquired on a BD Accuri C6 flow cytometer and is expressed as the geometric mean of fluorescence intensity.
Intracellular trafficking by confocal microscopy
BT-474, NIH-3T3 and Raw 264.7 cells were seeded at a density of 40000 cells per well in Labtek 8-well chamber slides and incubated overnight. The medium was replaced with fresh growth medium and the NCAF647-BGEDA/BSA were added to achieve a final concentration of 0.01 mg mL−1. After 4 h and 24 h of incubation, the medium was removed. The cells were gently washed twice with DPBS to remove excess NPs, and LysoTracker Green (100 nM) was added into the culture media and incubated for 5 min following the supplier's protocol to stain endo-lysosomes. Cells were gently washed three times with DPBS and live-imaged with a Nikon A1R confocal microscope equipped with a 60× 1.4 NA oil immersion objective.
Alternatively, for co-localization studies with early, late endosomes and lysosomes, cells were incubated with NCAF647-BGEDA/BSA for 3 h, 6 h and 24 h at 37 °C with 5% CO2. Cells were washed three times with DPBS and fixed with 4% paraformaldehyde (PFA) for 15 min, washed, permeabilized with 0.1% Triton X-100 solution in DPBS for 5 min, and washed again three times with DPBS. Samples were then incubated for 2 h with rabbit anti-EEA1 monoclonal antibody, rabbit anti-Rab7 monoclonal antibody or mouse anti-Lamp1 antibody at 1:200 dilution, followed by 1 h of incubation with goat anti-mouse or goat anti-rabbit AF647 conjugated antibody (2 μg mL−1). The cells were imaged with a Nikon A1R confocal microscope with a 60× 1.4 NA oil immersion objective. Pearson coefficient (R) and colour scatter plots were obtained using the WCIF ImageJ software. The experiments were repeated in triplicates and ten representative cell images were used to calculate the Pearson coefficient, displayed as mean ± SD.
Transcytosis in HUVEC endothelial cells
HUVEC cells were cultured on a culture dish coated with 0.1% gelatin diluted in PBS and incubated at 37 °C in EBM basal media supplemented with components of EGM SingleQuot kit (BBR, hEGF, hydrocortisone, ascorbic acid), G418 and 10% FBS. For transcytosis experiments, HUVEC and BT-474 cells were seeded at a density of 60000 cells per well on a 24-well plate and incubated for 24 h at 37 °C with 5% CO2. Then, NCAF647-BGEDA/BSA were added to the final concentration of 0.01 mg mL−1 of BGEDA and incubated for 2 h at 37 °C with 5% CO2. After incubation, the cells were gently washed three times with DPBS, placed in fresh growth media for additional 0 h, 2 h, 4 h and 6 h incubation at 37 °C with 5% CO2, prior to analysis by flow cytometry. Cell fluorescence was measured on a BD FACSCanto II flow cytometer and was expressed as the geometric mean of fluorescence intensity.
Mechanism of NCBGEDA/BSA internalization
BT-474 cells, NIH-3T3 cells and Raw 264.7 cells were seeded at a seeding density of 60000 cells per well in a 24-well plate and incubated at 37 °C overnight. Then, endocytosis inhibitors pitstop2 (12 μg mL−1), filipin from S. filipinensis (5 μg mL−1), EIPA (15 μg mL−1) and sodium azide (120 mM) were added to the cells and incubated for 15 min, followed by addition of NCAF647-BGEDA/BSA (0.01 mg mL−1) and another 2 h of incubation. Afterwards, the culture medium was removed, cells were washed twice with DPBS, trypsinized and centrifuged at 400g for 5 min, and washed twice with fresh DPBS. Then, cell fluorescence intensity was measured on a BD Accuri C6 flow cytometer and is expressed as the geometric mean of fluorescence intensity.
Cell association of NCBGEDA/BSA with BT-474, NIH-3T3 and Raw 264.7 cells in 2D co-culture
For cell association in 2D co-culture, BT-474 cells were labelled with CellTracker™ Blue CMAC Dye, 3T3 cells were labelled using the PKH67 green fluorescent cell linker kit and Raw 264.7 cells were labelled using the PKH26 red fluorescent cell linker kit according to the manufacturers’ protocols. The cells were then seeded on 24-well plates and incubated for 24 h prior to NCBGEDA/BSA addition at a concentration of 0.01 mg mL−1 of BGEDA. The association was measured on a BD FACSCanto™ flow cytometer and expressed as the geometric mean fluorescence intensity. Data were analyzed using FlowJo.Paclitaxel loading on the NCBGEDA/BSA
NCBGEDA/BSA obtained at the 1:20 w/w ratio were incubated with 250 μg mL−1 PTX dissolved in ethanol. After 30 min, the unbound drug was removed by centrifugation using Nanosep® centrifugal devices with Omega™ Membrane 30 kDa (9000g, 5 min). The purifed nanoparticles were dissolved in MilliQ/acetonitrile 50/50 and were analysed by an Agilent 1260 Infinity II high-performance liquid chromatography with a UV detector using a Jupiter 300 5 μm (250 × 4.6 mm) C18 column. The injection volume was 20 μL and the detector wavelength was set at 254 nm. The concentration of PTX in nanoparticles was calculated from the standard curve obtained by HPLC measurements of PTX at various concentrations.
Doxorubicin loading on the NCBGEDA/BSA
NCBGEDA/BSA at 1:20 w/w ratio were incubated with 156.6 μg mL−1 of DOX dissolved in ethanol. After 30 min, the unbound drug was removed by centrifugation using Nanosep® centrifugal devices with Omega™ Membrane 30 kDa (9000g, 5 min). The purifed nanoparticles were analyzed by UV-Vis spectrophotometry (SPECORD 250 PLUS) and the absorbance value at 593 nm was used to estimate the loading of DOX on the NCBGEDA/BSA.
Cell viability in 2D co-culture
For cell viability studies in 2D co-cultures, BT-474 cells were labelled using a PKH67 green fluorescent cell linker kit, NIH-3T3 cells were labelled with AF647-WGA conjugate, while Raw 264.7 cells were unstained. The cells were then seeded on 24-well plates and incubated for 24 h prior to NCBGEDA/BSA-PTX, NCBGEDA/BSA-DOX, free PTX or free DOX addition at final concentrations of 200 nM of PTX or 10 μg mL−1 of DOX. After 24 h, 48 h and 72 h of incubation, cells were washed three times with DPBS and detached from the culture dish, followed by centrifugation (400g, 5 min). The SytoxBlue dye was then added according to the supplier's protocol. Cell viability was measured on a BD FACSCanto™ flow cytometer by measuring the percent of cells with Sytox-Blue fluorescence. Data were analyzed using FlowJo.3D co-culture in perfusion bioreactor
A commercially available perfused bioreactor system (CELLEC Biotek AG, Basel, Switzerland) was used to form 3D co-cultures. The collagen scaffold (Ultrafoam, Avitene; Davol Inc., Warwick, RI, USA) of 8 mm diameter and 2 mm thickness was installed in a bioreactor, and 1 × 106 NIH-3T3, 1 × 106 BT-474 cells and 0.5 × 106 Raw 264.7 cells were then seeded and perfused overnight at 400 μm s−1 superficial velocity, 37 °C. After a 24 h cell seeding phase, superficial velocity was reduced to 100 μm s−1 and perfused for another 2 weeks, with twice a week medium change. The bioreactor was then disassembled and the collagen scaffold was retrieved for further processing. The cell viability was measured using CellTiter-Glo® 3D cell viability assay from Promega as per the manufacturer's protocol. For hematoxylin and eosin (H&E) staining and immunostaining, the scaffold was fixed with 4% PFA for 1 h at room temperature, embedded in OCT and sliced on cryostat. Then, the slides were washed with PBS and the Raw 264.7 cells were stained with rat anti-CD11b-AF488 antibody, NIH-3T3 cells were stained with anti-vimentin-AF555 antibody and BT-474 cells were stained with rabbit anti-E-cadherin-AF647 antibody. For the association studies, the nuclei were stained with Hoechst 33342 and F-actin was stained with phalloidin-AF488 conjugate, according to the supplier's protocol. Cells were imaged with a Nikon A1R confocal microscope.Isolation and analysis of peripheral blood mononuclear cells (PBMCs)
PBMCs from healthy donors were isolated from fresh buffy-coats by density gradient centrifugation on Lymphoprep medium according to the supplier's protocol. Isolated PBMCs were plated in 24-well plates with a seeding density of 1 × 106 cells per well in 1 mL of X-Vivo media (Lonza) and incubated for 30 min at 37 °C to allow cell recovery after isolation. Then, PBMCs were treated with NCAF488-BGEDA/BSA at a final concentration of 10 μg mL−1 for 2 h. Cells were then transferred to a V-bottom 96-well plate, centrifuged (350g, 5 min) and washed twice with FACS buffer (1% FBS in PBS). Cells were stained on ice for 30 min, using titrated concentrations of antibodies CD19, CD20 (biotin, streptavidin BV421), CD14 (PE), CD57 (PE_Cy7) and CD 3 (APC_Cy7). Cells were washed twice with cold PBS and the association data were acquired with a FACSCanto flow cytometer and analysed using FlowJo.For confocal microscopy imaging, cells were incubated with NCAF488-BGEDA/BSA and stained in Ibidi channel slides (6-well μ-Slide VI 0.5) according to the same protocol. After incubation, cells were fixed with 4% paraformaldehyde for 15 min and the nuclei were stained with DAPI according to the supplier's protocol. Cells were imaged with a LSM870 confocal microscope.
Results and discussion
Nano-complexation of cationic glycogen nanoparticles and serum albumin
Glycogen from bovine liver (BG), approximately 20 ± 4 nm in size (Fig. 1A, Mw = 3.6 × 105 Da)48 was chemically modified to obtain amine functionalized cationic glycogen nanoparticles, hereafter referred to as BGEDA. Briefly, BG nanoparticles in aqueous suspension were modified by reductive amination (Fig. S1A†)48–50 to incorporate ethylene diamine (EDA) residues. The degree of substitution (DS = 27%) of BGEDA after purification was evaluated by 1H NMR (Fig. S1B†) from the integration of EDA proton signals in the range 3.2–2.4 ppm. The synthesized BGEDA nanoparticles maintained an average size of 26 ± 4 nm, as determined by dynamic light scattering (Fig. 1A) and exhibited a ζ-potential of approximately 38 ± 6 mV. The biodegradability of BGEDA nanoparticles (Fig. 1B) was evaluated by treating them with both exo- and endo-enzymes for 3 h and measuring the concentration of reducing sugars via the Somogyi–Nelson method. While native BG nanoparticles were degraded up to approximately 40% by α-amylase, BGEDA nanoparticles had increased resistance to both α- and β-amylase digestion, undergoing only 15 ± 2% and 3 ± 1% degradation, respectively (Fig. 1B). This suggests that the chemical modification of polymer chains in BGEDA dramatically reduced its affinity for hydrolytic enzymes. However, we cannot rule out that the resistance of BGEDA against amylases might be correlated to the electrostatic interactions between the positively charged BGEDA nanoparticles and the positively charged enzymes (isoelectric point, pI = 6.5–7), which might interfere with the activity of the enzymes. To give a deeper insight into the mechanism which makes BGEDA resistant to degradation, we modified BG nanoparticles using periodate oxidation and NaBH4 mediated reduction at different substitution degrees from 5 to 20% (Fig. S1A†). The size, surface charge and degradability of the resulting nanoparticles, BGoxred, were analysed. Data reported in Table S1† show that BGoxred are slightly negatively charged nanoparticles, and that the higher the DS, the greater the degradability of BG. This suggests that the oxidation of the glucose rings itself made the glycogen dendritic structure more accessible to the enzymes, whereas the positively charged moieties on BGEDA seem to repel the enzymes and prevent nanoparticle degradation under the experimental conditions used.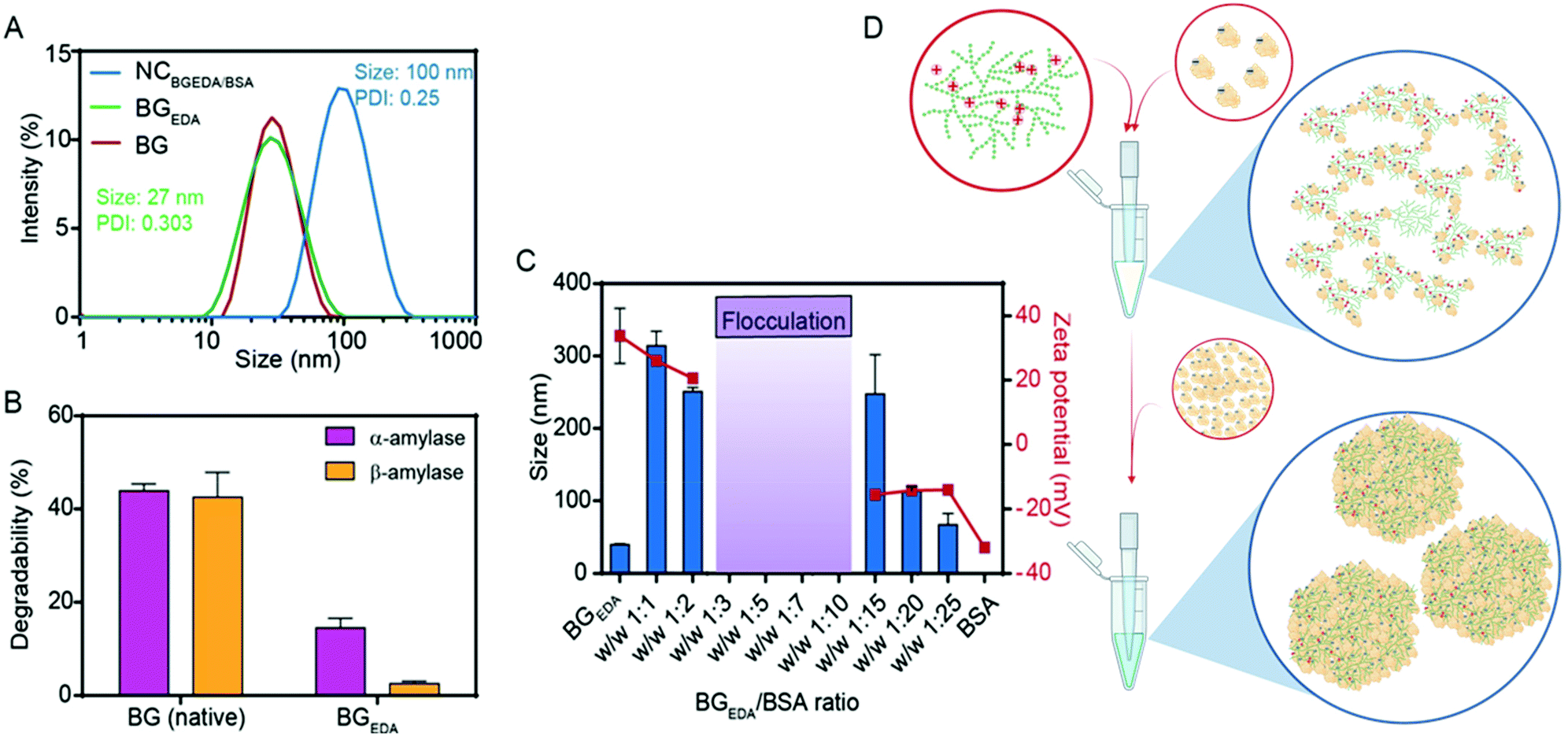 | ||
| Fig. 1 The nanocomplexation of BGEDA and BSA to induce BSA phase separation. (A) Intensity size distributions of BG, BGEDA and NCBGEDA/BSA as determined by DLS. (B) Comparison of the degradability of unmodified BG and BGEDA by treatment with α- and β-amylase. (C) Monitoring hydrodynamic diameter and ζ-potential of NCBGEDA/BSA at increasing weight ratio by DLS and electrophoretic mobility measurements, respectively. (D) Schematics of nanocomplexation of BGEDA and BSA at ratio 1:1 (top) and 1:20 (bottom). Created with BioRender.com. | ||
To exploit the ability of bovine serum albumin (BSA) to bind hydrophobic drugs, and to investigate the capacity of BGEDA to carry BSA, the nano-complexation of BGEDA and BSA was assessed by mixing the two biomacromolecules at different w/w ratios (Fig. 1C). As the complexation is driven by electrostatic interactions between BGEDA amine groups and the negatively charged subdomains of the protein (BSA pI = 4.7), it is crucial to optimize the biomacromolecular weight ratio to avoid their uncontrolled flocculation and formation of large aggregates. An aqueous solution of BGEDA nanoparticles (1 mg mL−1) was mixed with solutions of BSA at various concentrations and allowed to equilibrate for 30 min 25 °C. Fig. 1C shows the size and surface charge of the obtained nanocomplexes as a function of the BGEDA/BSA weight ratio. At the weight ratios 1:1 (1:5 mol ratio) and 1:2 (1:10 mol ratio), the nano-complexation of BGEDA and BSA resulted in the formation of 200–300 nm well-dispersed nanoparticles stabilized by their positively charged surface (20 mV) (Fig. 1C). This indicates that the albumin molecules could adsorb onto the BGEDA surface and bridge the nanoparticles to form nanoclusters. When BGEDA and BSA were mixed at ratios from 1:3 to 1:10, flocculation of large micrometre sized aggregates was observed (Fig. 1C). Therefore, we can infer that at ratios higher than 1:2 the positive charges exposed by BGEDA were neutralized by the BSA adsorption and the van der Waals short-range attraction prevailed over Coulomb electrostatic repulsion, leading to the uncontrolled and rapid aggregation process. When the nano-complexation process was performed at the ratio 1:15, charge reversal occurred. The larger aggregates disassembled to form polydisperse nanoparticles with a diameter of approximately 250 nm and a ζ-potential of −14 mV (Fig. 1C and D). At the highest ratios (1:20 and 1:25) uniform and stable nanocomplexes were obtained with diameters in the range of 50–100 nm and a ζ-potential of −14 mV (Fig. 1C). We selected the BGEDA/BSA nanocomplex, hereafter referred to as NCBGEDA/BSA, obtained at 1:20 weight ratio (1:100 molar ratio) for further studies and characterization as they have an optimal size and charge to penetrate the extracellular matrix in tumours.16Fig. 1A shows that NCBGEDA/BSA displayed a hydrodynamic diameter of size ∼100 nm (PDI = 0.25 ± 0.04) from DLS measurement. AFM images (Fig. 2A) of air-dried nanocomplexes indicate that the drying process induced a significant size shrinkage of the NCBGEDA/BSA to approximately 70 nm. The stability and integrity of NCBGEDA/BSA incubated with PBS containing 10% foetal bovine serum for 5 h was verified by super-resolution stochastic optical reconstruction microscopy (STORM) (Fig. 2B) with nanoscale resolution (30–50 nm). The accurate and comparative analysis of single molecule localizations by cluster analysis (Fig. S2†) indicates that NCBGEDA/BSA maintained their integrity and size distribution upon incubation with serum proteins. NCBGEDA/BSA exhibited an average size of 90 ± 30 nm in PBS (Fig. 2C and S2B†) and 100 ± 40 nm after 5 h of incubation in 10% FBS (Fig. 2D and S2C†). No evidence of aggregation or disassembly induced by either the screening effect of salt or competitive binding of other serum proteins was observed. Overall, these results suggest that NCBGEDA/BSA are stable when exposed to cell culture media.
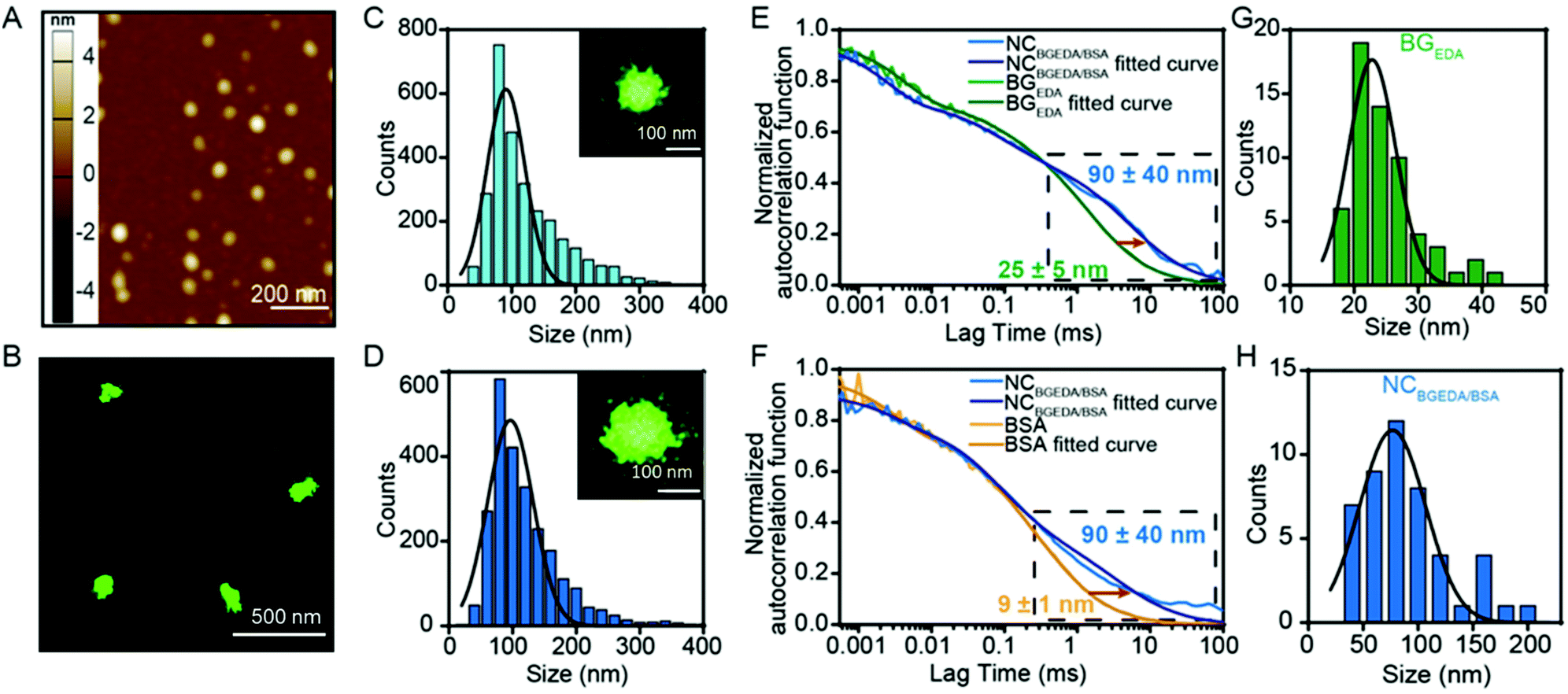 | ||
| Fig. 2 AFM and single molecule characterisation of the nanocomplexation of BGEDA and BSA. (A) Representative AFM image of dried NCBGEDA/BSA obtained at 1:20 w/w ratio. (B) Representative STORM image of NCAF647BGEDA/BSA incubated in human serum for 5 h with (C) Size distributions (n = 2400) of NCAF647BGEDA/BSA complex in PBS and (D) serum determined by cluster analysis of single molecules localizations. Insets show individual NCBGEDA/BSA. (E) Probing the nanocomplexation of BGEDA and BSA by fluorescence correlation spectroscopy; experimental autocorrelation curves and fitting for AF647-BGEDA and NCAF647-BGEDA/BSA, (F) experimental autocorrelation curves and fitting for AF647-BSA and NCBGEDA/AF647-BSA. The decreased diffusion is observed within the long lag region of the NCBGEDA/BSA curve (black-dashed box), demonstrating formation of complexes. (G) Size distribution of BGEDA and (H) NCBGEDA/BSA measured by FCS. | ||
To provide insight into the mechanism of complexation between BGEDA and BSA at the weight ratio 1:20, fluorescence correlation spectroscopy (FCS), a spectroscopic technique with single-molecule sensitivity was employed. FCS enabled quantitative monitoring of the formation of single NCBGEDA/BSA, in a microscopic detection volume of approximately 1 femtoL and could discriminate between free and complexed BGEDA and BSA based on their different diffusional properties. The diffusion coefficients (D) of AF647-BGEDA (0.01 mg mL−1) and AF647-BSA (0.2 mg mL−1) were first determined by the autocorrelation analysis of the fluorescence emission, using the AF647 dye as reference (DAF647 = 3.3 × 10−6 cm2 s−1 at 25 °C) for FCS calibration. The autocorrelation curves (Fig. 2E and F) were best fitted with a Brownian diffusion model including triplet dynamic and two components. The recorded autocorrelation functions of both BGEDA and BSA exhibited a two-component profile (Fig. S3†), with the fast component corresponding to the free dye and a slower component corresponding to BGEDA (DBGEDA = 20 × 10−8 cm2 s−1) and BSA (DBSA = 50 × 10−8 cm2 s−1), respectively. The corresponding size of BGEDA was approximately 25 ± 5 nm (Fig. 2G and H) by FCS, which is in fair agreement with the DLS measurement shown in Fig. 1A. The size of BSA was measured at 9 ± 1 nm, in agreement with the literature data.51 Next NCBGEDA/BSA were prepared at the 1:20 weight ratio and diluted 100 times to acquire the FCS autocorrelation function. To separately probe the free and bound species in equilibrium with the nanocomplex, we prepared and analysed NCAF647-BGEDA/BSA and NCBGEDA/AF647-BSA by labelling either BGEDA or BSA, respectively. In both cases, the autocorrelation functions (Fig. 2E and F) shifted towards the longer lag time region and the fitting of the curve yielded a slow diffusing species with a DBGEDA/BSA = 6 × 10−8 cm2 s−1 corresponding to the size of 90 nm (Fig. 2H), indicating the formation of the NCBGEDA/BSA. Neither free BGEDA nor free BSA were detected in the analysed nanocomplex suspension. This indicates the complexation at 1:20 weight ratio had a high association constant and the nanocomplexes were thermodynamically stable.
Based on these findings, a mechanism of complexation between BGEDA and BSA can be hypothesized. At first, the nano-complexation is driven by electrostatic interactions until the saturation of the binding sites on BGEDA is reached and flocculation occurs. Then, BSA molecules act as molecular glue between the protein-coated BGEDA nanoparticles and protein–protein phase separation occurs within the nanocomplexes. The nanophase separation is likely mediated by either intermolecular hydrophobic interactions or hydrogen bonds. The unique hyperbranched and dendrimer like flexible structure of glycogen is key to controlling the multivalent coordination and the phase separation of albumin molecules, hence resulting in the formation of uniform and stable glycogen–protein nanocomplexes. By considering the volume of BGEDA, BSA and NCBGEDA/BSA, we estimated that a single nanocomplex consisted of approximately 12 BGEDA nanoparticles and approximately 1200 BSA molecules. A schematic of the proposed mechanism is depicted in Fig. 1D. This simple approach offers a strategy to prepare hybrid multifunctional nanoparticles where BGEDA can potentially provide a scaffold for triggering intracellular endosomal escape via pH responsive amine moieties48 and albumin can provide the ability to extravasate into solid tumours by active transcytosis through endothelial cells.
NCBGEDA/BSA are endocytosed by epithelial, stromal, immune cells, and cross the endothelial barrier
The potential of NCBGEDA/BSA as a drug delivery system was investigated in a complex tumour microenvironment (TME) in vitro. We first studied the interactions of NCBGEDA/BSA with different cell lines that represent different components of the TME, including BT-474 breast cancer cells (epithelial), NIH-3T3 fibroblast cells (stromal) and Raw 264.7 macrophage cells (immune). Of note, BT-474 is a breast cancer cell line, derived from an invasive ductal carcinoma52 and is widely used as a breast cancer model, because it forms dense 3D spheroids, thus exhibiting relative resistance to chemotherapeutics in 3D-culture.53 Flow cytometry analysis showed a rapid association of nanocomplexes with Raw 264.7 and NIH-3T3 cells which reached a plateau (saturation) in the first 15 h of incubation (Fig. 3A). The slow uptake kinetics in BT-474 cells observed in the first 6 h of incubation (Fig. 3A) was likely due to their cluster-like morphology, which might slow down the permeation of NCBGEDA/BSA.54 Live-cell confocal microscopy images of BT-474 cells acquired after 4 h (Fig. 3B and S4A†) and 24 h (Fig. 3C and S4B†) of incubation with NCBGEDA/BSA confirmed that in the first 4 h of incubation the nanocomplexes remained associated on the outer layer of the cellular cluster (Fig. 3B and S4A†), whereas at 24 h of incubation the diffusion and uptake of the NCBGEDA/BSA across the entire cellular cluster occurred (Fig. 3C and S4B†). Next, we tested the ability of NCBGEDA/BSA to enter all components of TME in 2D co-cultures of BT-474 breast cancer cells, NIH-3T3 fibroblasts and Raw 264.7 cells mixed at the ratio 4:2:1. The cell ratio was optimized considering the proliferation rates of the different cell lines and the typical tumour:stroma cell ratio.55 The presence of the three cellular components was first verified by flow cytometry and confocal microscopy after labelling them with different fluorescent dyes: BT-474 breast cancer cells (green), NIH-3T3 fibroblasts (white) and Raw 264.7 (red, Fig. S5†). The association of NCAF647-BGEDA/BSA with cells was investigated by flow cytometry. Fig. 3D shows the rapid association of NCAF647-BGEDA/BSA with Raw 264.7 and NIH-3T3 cells in the first 10 h of incubation as previously observed in monoculture studies. After 10 h of incubation, the rate of association significantly decreased for NIH-3T3 cells and the association levelled off, indicating that fibroblasts were saturated with NCAF647-BGEDA/BSA. Raw 264.7 cells associated with the nanoparticles over time although at a slower rate post 10 h (Fig. 3D). Compared to NIH-3T3 and Raw 264.7, the association of NCAF647-BGEDA/BSA with BT-474 cells was significantly lower (one order of magnitude). This indicates that the three cell lines compete for the uptake of nanocomplexes and the cluster-like morphology of BT-474 cancer cells hinders the access to nanocomplexes that are instead rapidly internalized by macrophages and fibroblasts.
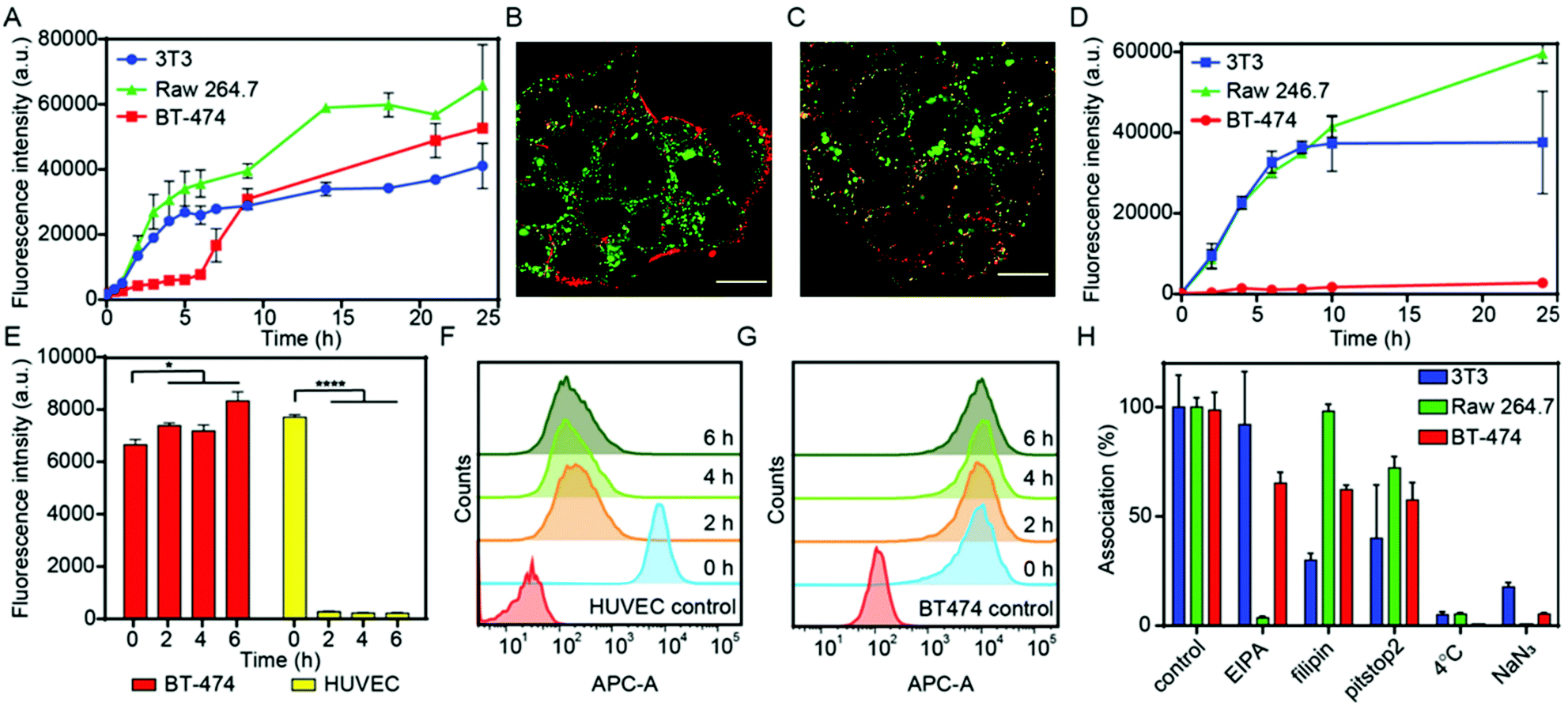 | ||
| Fig. 3 The interaction of NCBGEDA/BSA with cancer cells, fibroblasts, macrophages, and endothelial cells. (A) Cell association of NCBGEDA/BSA performed in monocultures of NIH-3T3 fibroblast cell line (blue line), BT-474 breast cancer cell line (red line) and Raw 264.7 macrophage cell line (green line). The data are presented as the geometric mean of AF647 fluorescence (n = 3 independent experiments). (B) Representative CLSM images of BT-474 cells after 4 h and (C) 24 h incubation with NCBGEDA/BSA. LysoTracker Green for staining endo/lysosomal vesicles and NCAF647-BGEDA/BSA (red) were used. Scale bars 20 μm. (D) Cell association of NCBGEDA/BSA performed in 2D co-cultures of NIH-3T3 (blue line), BT-474 (red line) and Raw 264.7 macrophages (green line). Before seeding, each cell line were individually stained with spectrally distinct dye and the association with NCAF647-BGEDA/BSA was measured by flow cytometry. Data are plotted as mean fluorescence intensity. (E) Comparison of the geometric mean fluorescence intensity of the untreated HUVEC and BT-474 cells with cells incubated with NCAF647-BGEDA/BSA for 2 h, followed by 0 h, 2 h, 4 h and 6 h culture in fresh media. Statistical analyses were performed using two-way ANOVA with Tukey's multiple comparison test. ****p < 0.0001 (n = 3 independent experiments). (F) Corresponding histograms of untreated HUVEC and (G) BT-474 cells and cells incubated with NCAF647-BGEDA/BSA for 2 h, followed by 0 h, 2 h, 4 h and 6 h cultivation in fresh media. (H) Effect of endocytic inhibitors on the uptake of NCBGEDA/BSA by NIH-3T3, BT-474 and Raw 264.7 cells after 2 h incubation with the nanocomplex. | ||
To evaluate the capability of NCBGEDA/BSA to cross the endothelial cell layer, human umbilical vein endothelial cells (HUVEC) were pre-incubated with NCAF647-BGEDA/BSA for 2 h, followed by incubation in fresh growth medium for different times and analysed by flow cytometry. The nanocomplexes were readily internalized by HUVEC cells during the first 2 h of incubation (equivalent to t = 0 h in fresh growth medium post particle incubation) as indicated by the increase in the AF647 fluorescence of the cells (Fig. 3E and F). However, the fluorescence of the HUVEC cells dramatically decreased with time (Fig. 3E and F), suggesting excretion/transcytosis of the NCBGEDA/BSA out of the cells. In contrast, BT-474 breast cancer cells did not show evidence of nanoparticles transcytosis (Fig. 3E and G), indicating that different pathways were involved in the intracellular trafficking of NCBGEDA/BSA. This finding suggests that NCBGEDA/BSA can potentially pass-through blood vessels and be internalized in the different cellular components of the TME.
Mechanisms of internalization and intracellular trafficking of NCBGEDA/BSA
Since the cellular pathways and mechanisms involved in the uptake and trafficking of NCBGEDA/BSA strongly influence their ability to deliver therapeutic agents, we investigated the pathways involved in the internalization and endo-lysosomal trafficking of the nanocomplex. Of note, recent studies have shown that BSA-based nanoparticles loaded with docetaxel enter breast cancer cell lines through a clathrin-mediated process and accumulate in the endo-/lysosomal vesicles.56 We first assessed the effect of sodium azide (NaN3) and low temperature incubation, which are both inhibitors of endocytosis, on the internalization efficiency of NCBGEDA/BSA. Both the presence of NaN3 and low temperature incubation significantly inhibited the entry of NCBGEDA/BSA in cells (Fig. 3H). This signifies that NCBGEDA/BSA were internalized by endocytic pathways rather than via membrane fusion. Next, we investigated the influence of metabolic inhibitors like 5-(N-ethyl-N-isopropyl) amiloride (EIPA), filipin from Streptomyces filipinensis and pitstop2, which are inhibitors of macropinocytosis, caveolae- and clathrin-dependent endocytosis, respectively. Fig. 3H shows that in Raw 264.7 cells, the most significant effect was observed upon treatment of cells with EIPA, indicating that macropinocytosis is involved in the internalization process.57 No inhibition in the uptake of NCBGEDA/BSA in NIH-3T3 cells was detected upon incubation with EIPA. In contrast, treatment of NIH-3T3 cells with filipin and pitstop2 caused a significant reduction in NCBGEDA/BSA association from 100% to approximately 30% and 40% cell internalization. This suggests that the internalization of NCBGEDA/BSA in NIH-3T3 cells was mediated by a combination of clathrin- and caveolae-dependent processes. In BT-474 cells, all three inhibitors led to a decrease in NCBGEDA/BSA association to approximately 60%, indicating the interplay of different internalization mechanisms.To investigate the intracellular trafficking of NCBGEDA/BSA, specific antibodies which can distinguish the different stages of endosome maturation in Raw 264.7, NIH-3T3 and BT-474 cell lines were used. Staining of early endosomes was performed with an antibody directed against early endosome antigen 1 (EEA1), a protein exclusively present on those vesicles. Late endosomes were identified using an antibody against Rab7, a terminal marker of the late endosome to lysosome maturation.58 For lysosome identification, an antibody raised against lysosomal-associated membrane protein 1 (LAMP-1), residing across lysosomal membranes was used. Additionally, Lysotracker™ was used to stain all acidic endo-lysosomal compartments of Raw 264.7 cells. The representative confocal microscopy images of Raw 264.7 incubated with NCAF647-BGEDA/BSA and Lysotracker (Fig. S6†) show that NCBGEDA/BSA were rapidly internalized and the signal was primarily colocalized with endo-lysosomal vesicles. This was also indicated by the intensity correlation analysis of confocal images (Fig. S6†), which showed high Pearson's correlation coefficients (PCC) of 0.50 ± 0.05, 0.63 ± 0.06 and 0.64 ± 0.04 after 1 h, 5 h, and 24 h, respectively. Note that a PCC of 1 indicates 100% co-localization of the particles and vesicles. Taken together, these data suggest that after internalization by macropinocytosis, NCBGEDA/BSA were partially directed to lysosomes in Raw 264.7 cells.
Next, to investigate the intracellular trafficking of NCBGEDA/BSA in NIH-3T3 and BT-474 cells, we performed co-localization studies with early endosomes (Fig. 4A, B and S7, S10†), late endosomes (Fig. 4C, D and S8, S11†) and lysosomes (Fig. 4E, F and S9, S12†) by confocal microscopy at different incubation time points. Representative confocal microscopy images shown in Fig. 4 and S7–S12† clearly indicated the efficient uptake of nanocomplexes in both cancer cells and fibroblasts. Only partial colocalization of the NCBGEDA/BSA with the endocytic vesicles up to 24 h was observed. Pearson's correlation coefficients (PCC, Fig. S13A and B†) showed that while the co-localization with early endosome decreased, the co-localization with both late endosome and lysosome increased with the incubation time. This suggests that after internalization, the NCBGEDA/BSA underwent intracellular trafficking from early endosomes to lysosomes. However, the PPC values for all endocytic vesicles were relatively low (below 0.5) indicating that while a fraction of internalized NCBGEDA/BSA remained entrapped inside endosomes or lysosomes, a significant fraction of NCBGEDA/BSA escaped from the endo-lysosomal vesicles. We thus postulate, that the endosomal escape of NCBGEDA/BSA was likely mediated by the buffering capacity of BGEDA, which has one of the pKa values (pKa1 6 and pKa2 10 as measured by potentiometric titration on Fig. S14†) in the range of endosomal pH. We have shown by direct STORM imaging that the buffering properties of BGEDA can induce the disruption of endosomal membranes by the proton sponge mechanism.50 In addition, the internalization mechanism studies revealed that NCBGEDA/BSA can undergo caveolae-mediated endocytosis avoiding lysosomal trafficking.59
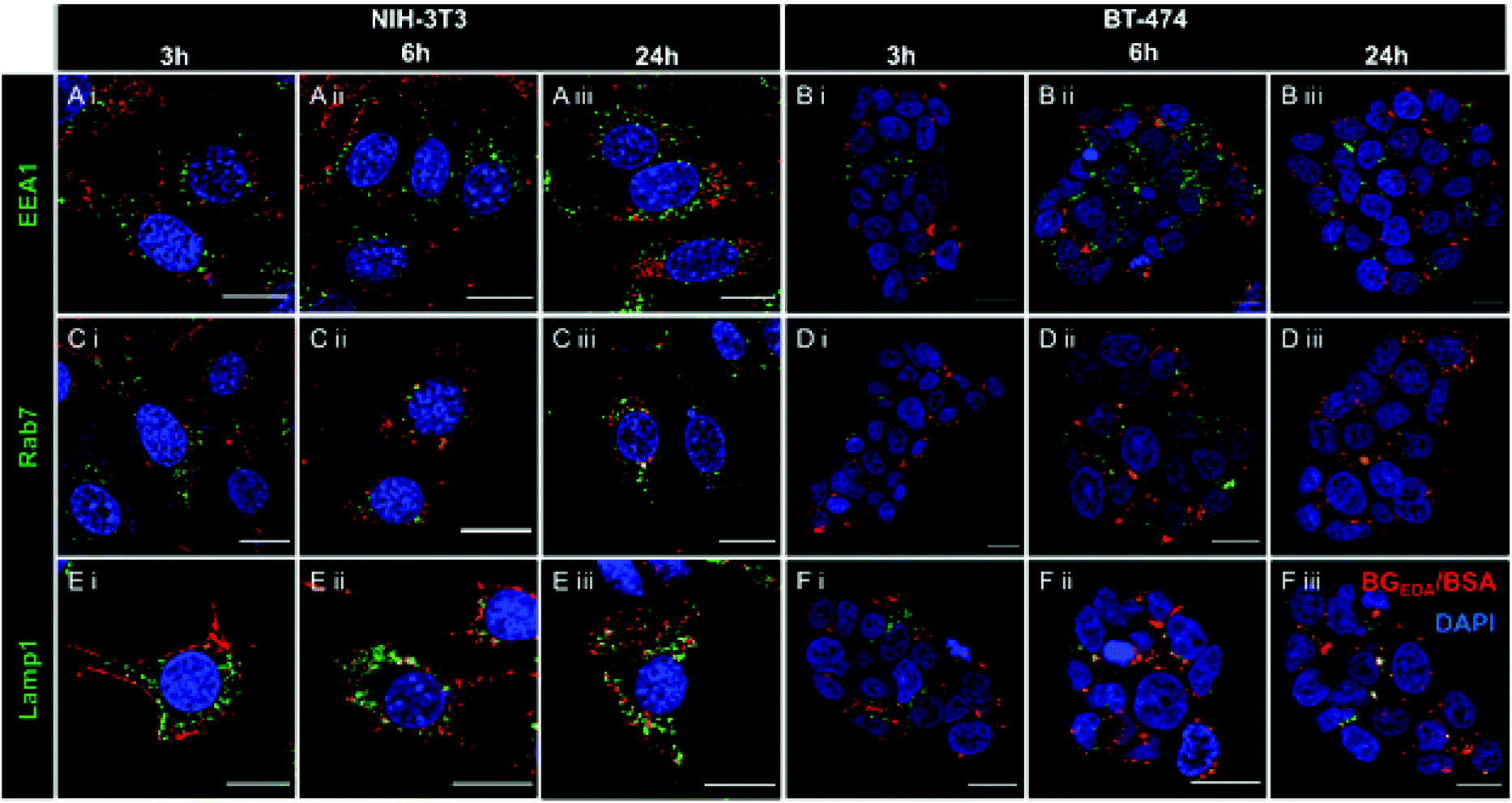 | ||
| Fig. 4 (A) Representative CLSM images of colocalization studies of NCAF647BGEDA/BSA (red) and early endosomes (labelled with anti-EEA1 antibody) in NIH-3T3 and (B) BT-474 cells taken (i) 3 h, (ii) 6 h, and (iii) 24 h post nanocomplexes transfection. (C) Representative CLSM images of colocalization studies of NCAF647BGEDA/BSA (red) and late endosomes (labelled with anti-Rab7 antibody) in NIH-3T3 and (D) BT-474 cells taken after i, 3 h, ii, 6 h, and iii, 24 h transfection. (E) Representative CLSM images of colocalization studies of NCAF647BGEDA/BSA (red) and lysosomes (labelled with anti-Lamp1 antibody) in NIH-3T3 and (F) BT-474 cells taken after i, 3 h, ii, 6 h, and iii, 24 h transfection. Nuclei are stained with DAPI (blue). Scale bar = 20 μm. | ||
We also investigated the interactions of NCBGEDA/BSA with immune cells present in human blood. We isolated peripheral blood mononuclear cells (PBMCs) from buffy coats from five blood donors and then incubated the PBMCs with NCAF488-BGEDA/BSA. The PBMCs were then stained with cell subtype-specific markers (to identify monocytes, T-cells, B-cells and natural killer (NK)) to determine cell association with NCAF488-BGEDA/BSA by flow cytometry (Fig. 5A and S15†). We observed a significant association of NCAF488-BGEDA/BSA with monocytes and limited interaction was detected with other lymphocytes (Fig. 5A and S15†). The internalization of NCAF488-BGEDA/BSA in monocytes was confirmed by CLSM imaging (Fig. 5B). These data demonstrate that cells with active phagocytosis and endocytosis machinery, like monocytes, efficiently take up NCBGEDA/BSA, while lymphocytes (T cells and B cells) only slightly associated with NCBGEDA/BSA.
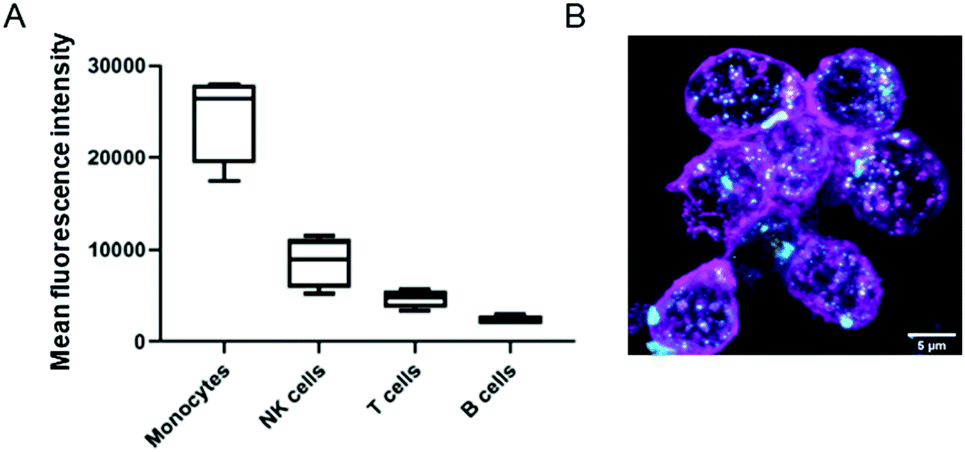 | ||
| Fig. 5 (A) Mean fluorescence intensity of different PBMC subsets: CD14+ monocytes, CD3+ T cells, CD19+CD20+ B cells and CD57+ NK cells after 2 h incubation with NCAF488-BGEDA/BSA. (B) Maximum intensity projections showing the internalization of NCAF488-BGEDA/BSA (cyan) by monocytes isolated from human blood. The cell membrane was stained with wheat germ agglutinin (magenta). Scale bar is 5 μm. | ||
NCBGEDA/BSA enable release of chemotherapeutics in 2D mono – and co-cultures
Next, NCBGEDA/BSA were loaded with either paclitaxel (PTX) or doxorubicin (DOX), two conventional chemotherapeutic agents used for the treatment of breast cancer. The drugs were readily adsorbed by the nanocomplexes after 30 min of incubation. The nanocomplexes maximum loading capacities for paclitaxel (7.27 μg mg−1 of BSA) and doxorubicin (35 μg mg−1 of BSA) were estimated by UV spectroscopy and HPLC analysis. The kinetics of release of the two drugs from the NCBGEDA/BSA was assessed in DPBS at pH 7 and 5 (Fig. S16†). A limited release of DOX and PTX from the nanocomplexes (Fig. S16B†) was observed in the first 7 h. As the NCBGEDA/BSA have been already internalised after 7 h, this indicates that the drug is primarily released inside the cells rather than in the extracellular space. The cytotoxic activity of NCBGEDA/BSA loaded with PTX and DOX was first evaluated on individual NIH-3T3, BT-474 and Raw 264.7 cells (Fig. S17†). For all cell lines, the cytotoxicity of the PTX either as free or encapsulated drug was similar. Up to 48 h of incubation with the PTX-loaded NCBGEDA/BSA, BT-474 cancer cells showed resistance to the drug treatment. However, after 72 h of incubation with the PTX-loaded NCBGEDA/BSA, BT-474 cancer cells became more sensitive to the drug with an IC50 of 50 nM. Raw 264.7 cell viability decreased to approximately 70% irrespective of the dose and incubation time, whereas the cell viability of NIH-3T3 cells was reduced to approximately 60–70% at 72 h incubation at 400 nM drug concentration. Overall, these results indicate that at long exposure time both free PTX and PTX-loaded NCBGEDA/BSA are more effective against BT-474 cancer cells than fibroblasts and macrophages. The antiproliferative activity of DOX-loaded NCBGEDA/BSA and free DOX towards NIH-3T3 and Raw 264.7 cell lines was also determined after 24 h incubation (Fig. S18A†) and towards BT-474 cells after 24 h, 48 h and 72 h of incubation (Fig. S18B†). BT-474 cancer cells also showed resistance to DOX and DOX loaded NCBGEDA/BSA treatment with an IC50 of approximately 0.93 μg mL−1 after 72 h treatment, whereas DOX loaded NCBGEDA/BSA exhibited an IC50 of 5 and 3 μg mL−1 against NIH-3T3 and Raw 264.7 cells, respectively.To study the influence of cell–cell interactions on the toxicity of NCBGEDA/BSA loaded with PTX and DOX, we measured the viability of BT-474, NIH-3T3 and Raw 264.7 cells grown in 2D co-culture after treatment with 200 nM PTX and 10 μg mL−1 DOX. Fig. 6A shows that after 48 h of treatment with PTX NIH-3T3 cells and Raw 264.7 cells in co-culture exhibited similar viability of approximately 60–70% to that obtained in monoculture. Fig. 6B shows the flow cytometry histograms of treated NIH-3T3, BT-474 and Raw 263.7 cells where the extent of the fluorescence shift is used to estimate the number of dead cells in the population.
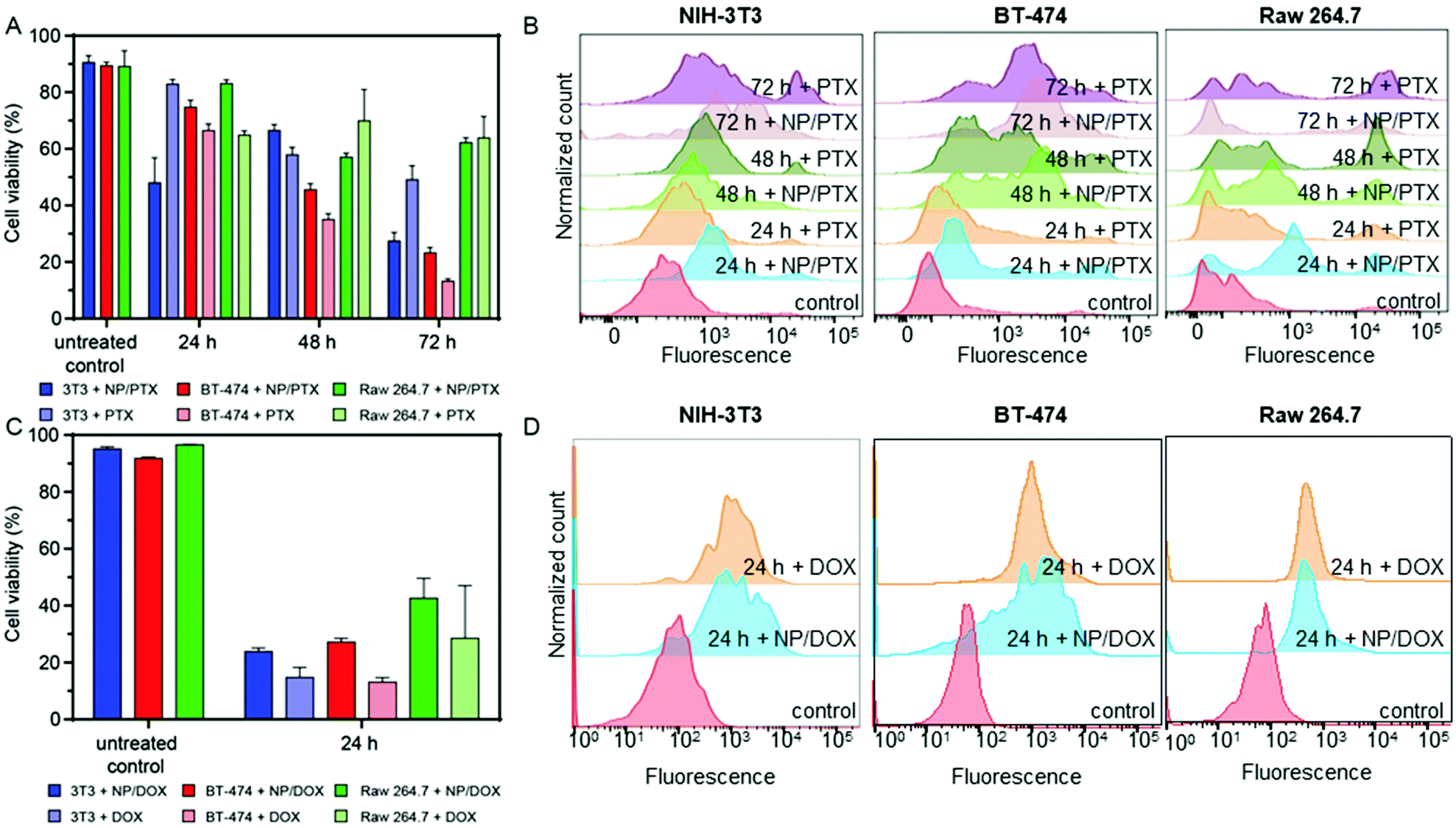 | ||
| Fig. 6 (A) Cytotoxicity of NCBGEDA/BSA-PTX nanocomplexes in NIH-3T3 cells, BT-474 cells and Raw 264.7 cells grown in 2D co-culture after 24, 48 and 72 h incubation compared with the cytotoxicity of free PTX at the final PTX concentration of 200 nM. Cytotoxicity was determined using the Alamar Blue viability assay. Data are shown as the mean ± standard deviation (SD) (n = 3). (B) Corresponding histograms of NIH-3T3, BT-474 and Raw 263.7 cells incubated with a dead cell stain (Sytox Blue). The fluorescence shift is representative of the number of dead cells in the population. (C) Cytotoxicity of NCBGEDA/BSA-DOX nanocomplexes in NIH-3T3 cells, BT-474 cells and Raw 264.7 cells grown in co-culture after 24 h incubation compared with the cytotoxicity of free DOX at the final DOX concentration of 10 μg mL−1. The data are shown as the mean ± standard deviation (SD) (n = 3). (D) Corresponding histograms of NIH-3T3, BT-474 and Raw 264.7 cells incubated with dead cell stain. The fluorescence shift is representative of the number of dead cells in the population. | ||
Interestingly the viability of BT-474 cells treated with the free and loaded PTX in co-culture was 41% and 37% lower at 48 h and 72 h, respectively when compared with the monoculture. A similar effect was observed when co-cultured BT-474 cells were treated with both free DOX and DOX loaded NCBGEDA/BSA. The viability of co-cultured BT-474 and NIH-3T3 cells after 24 h treatment was approximately 80% and 55% lower, respectively, than the one observed in monoculture (Fig. 6C). On the other hand, the toxicity towards Raw 264.7 cells was similar in both monoculture and co-culture. This reduction in viability observed in cancer cells may be attributed to the soluble factors secreted by macrophages. Fig. 6D shows the flow cytometry histograms of NIH-3T3, BT-474 and Raw 263.7 cells after 24 h treatment with both free DOX and DOX loaded NCBGEDA/BSA. It has been reported, that DOX increases the production of tumour necrosis factor (TNF), interleukin-1 (IL-1), interferon γ (IFNγ) and NO, therefore, increasing the tumoricidal potential of macrophage cells.60,61 Another study62 reported that the co-culture with Raw 264.7 cells reduced the viability and susceptibility to DOX of the three different cancer cell lines, which is consistent with our earlier findings.
Overall, these results indicate that the drugs loaded on the NCBGEDA/BSA retained their cytotoxic activity, which is comparable with the activity of the free drugs. In addition, BT-474 cells were more sensitive to the free and loaded chemotherapeutic drugs when cultured with the other cells.
NCBGEDA/BSA can permeate a 3D dynamic cell culture system mimicking tumour microenvironment
The 2D co-culture model still lacks many important tumour microenvironment (TME) characteristics, like heterogeneity, the oxygen and nutrient gradient, cell–matrix interaction, cell–cell signalling and fluid shear stress found in vivo.63–65 Those dynamic conditions can be mimicked by the use of a commercially available U-CUP perfusion bioreactor device (Fig. 7A). In fact, the U-Cup bioreactor was used to study the toxicity of PTX-loaded keratin nanoparticles towards a 3D monoculture of breast cancer cell lines, proving that the cells were more resistant to drug toxicity than in 2D culture.66 We used this model to establish 3D, dynamic co-cultures, where the BT-474, NIH-3T3 and Raw 264.7 cells at 4:2:1 ratio were seeded onto the porous collagen scaffold and subsequently grown for 2 weeks under continuous flow in cell culture media (10% FBS/DMEM) at 37 °C, 5% CO2. The morphology of the 3D multicellular constructs was investigated by hematoxylin and eosin (H&E) staining (Fig. 7B) and confocal microscopy (Fig. 7C), which showed tissue-like morphology. Next, the multicellular 3D construct was treated with NCAF647-BGEDA/BSA for 4 h and 24 h, fixed and cryo-sectioned before imaging. Fig. 7D, E and S19† show representative confocal microscopy images of samples after 4 h and 24 h of incubation with NCBGEDA/BSA. It can be observed that NCAF647-BGEDA/BSA penetrated and accumulated in the tissue-like structures after 4 h and 24 h circulation. Furthermore, NCBGEDA/BSA loaded with DOX and PTX were injected and remained in circulation up to 24 h in the 3D multicellular construct. Then the cell viability was assessed with a 3D viability assay (Fig. 7F). We found that the cells incubated with NCBGEDA/BSA loaded with DOX and PTX exhibited 30% and 20% reduction in viability, respectively. These results are comparable with the data obtained when the multicellular construct was treated with free DOX and PTX at a similar concentration (Fig. 7F). Of note the toxic effect exerted by free and encapsulated drugs is significantly lower than the one in 2D co-culture. In fact, many studies have shown big discrepancies between the 2D and 3D cell culture response to treatment and have indicated that the 3D model provides a more reliable prediction of treatment efficacy in vivo.67–69
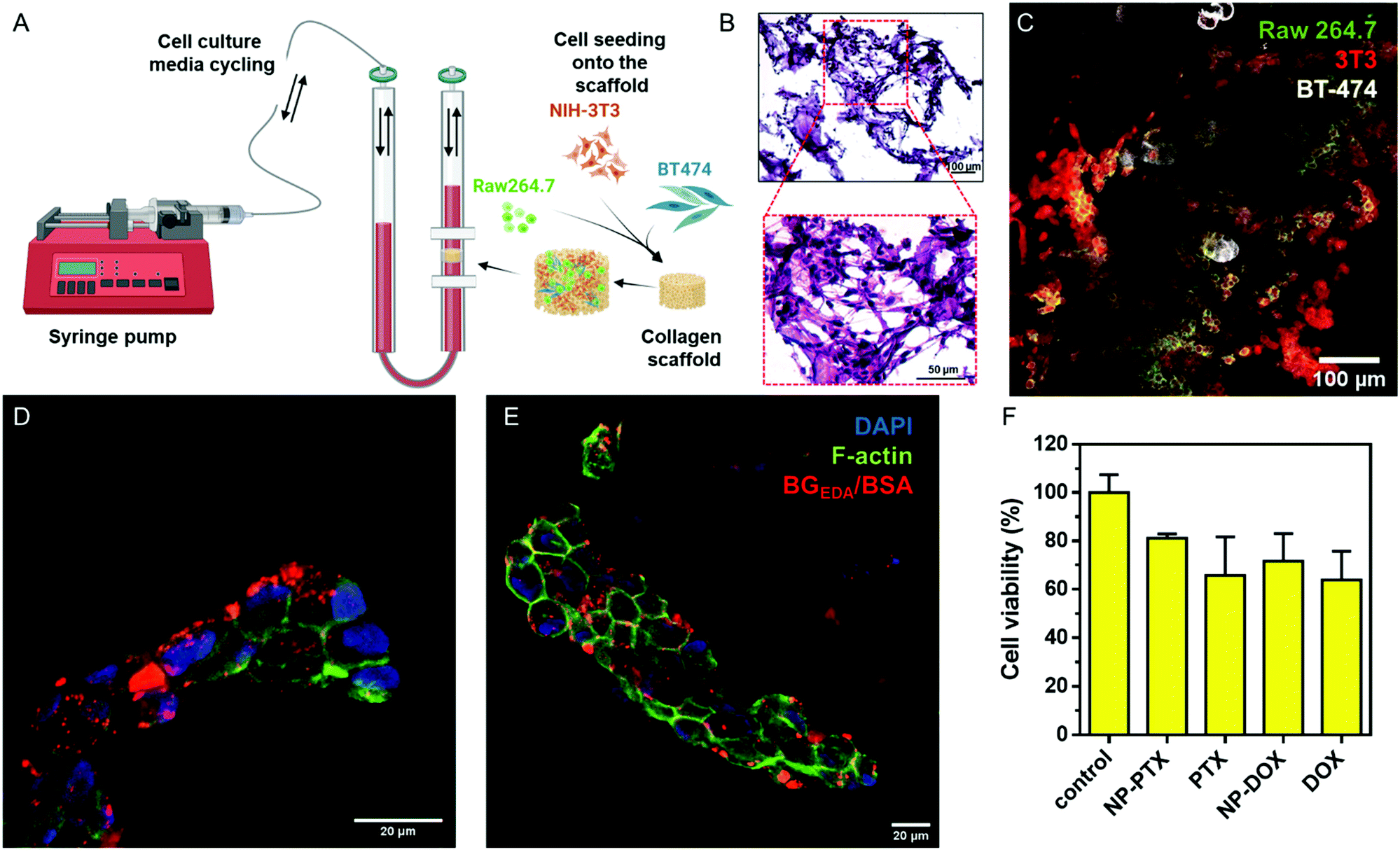 | ||
| Fig. 7 (A) Schematic showing the U-Cup bioreactor, where cells seeded onto collagen scaffold are grown under a continuous flow of culture media. (B) Hematoxylin and eosin staining of the sectioned collagen scaffold after 2 weeks of cell growth. (C) CLSM image of BT-474, NIH-3T3 and Raw 264.7 cells seeded onto the collagen scaffold. Raw 264.7 cells are stained with rat anti CD11b-AF488 antibody (green), NIH-3T3 cells are stained with mouse anti-vimentin-AF555 antibody (red) and BT-474 cells are stained with rabbit anti-E-cadherin-AF647 antibody (white). (D) Representative CLSM images of collagen scaffold co-cultured with NIH-3T3, BT-474 and Raw 264.7 cells and stained for F-actin (phalloidin; green) and nucleus (DAPI; blue) after incubation with NCAF647-BGEDA/BSA (red) for 4 and (E) 24 h. (F) Cytotoxicity of nanocomplexes loaded with PTX (NCBGEDA/BSA-PTX) and DOX (NCBGEDA/BSA-DOX) in NIH-3T3 cells, BT-474 cells and Raw 264.7 cells grown in a 3D perfusion bioreactor after 24 h incubation compared with the cytotoxicity of free PTX at the final PTX concentration of 200 nM and free DOX with the final concentration of 10 μg mL−1. Data are shown as the mean ± standard deviation (SD) (n = 3). Created with BioRender.com. | ||
Overall, these results show that, NCBGEDA/BSA were able to cross the extracellular matrix, penetrate the 3D tumour-like structure and effectively deliver the cytotoxic drugs. The 3D multicellular construct developed in perfusion bioreactor provided the important characteristics to mimic in vivo tumour microenvironment, like the presence of different cell lines, cell–cell and cell–ECM interactions in 3D, shear stress due to the continuous flow of growth medium as well as the collagen scaffold mimicking the extracellular matrix. The comparison of drug toxicity in 2D and 3D cell culture models unveil the increased resistance of cancer cells towards the drugs when grown in 3D.
Conclusions
Stable nanocomplexes of aminated glycogen nanoparticles with serum albumin were formed for the delivery of hydrophobic drugs. The nanocomplexes are internalized by a wide range of cells present in the TME and can effectively penetrate 3D tumour-like structure. Transcytosis was observed in an endothelial cell line, which could potentially provide a mechanism for the complexes to access the TME. In human blood, the nanocomplexes interact mainly with monocytes. The ability of the BSA component to complex and deliver cytotoxic, including DOX and PXT was demonstrated in 2D monolayers, 2D co-cultures and 3D co-cultures. The highest toxicity in breast cancer cells was observed in 2D co-cultures compared with 2D monolayers and 3D co-cultures. While this work shows that the engineered nanocomplex is a promising vehicle for the delivery of hydrophobic drugs in complex multicellular environments, a dedicated in vivo pre-clinical study is required to show the efficacy, safety, and clinical potential of this system.This work also highlights the importance of performing studies on the interaction of nanoparticles in 3D multicellular cultures, which more accurately mimic the in vivo conditions than conventional 2D monolayer cultures.
Author contributions
AG carried out the overall design and structural and biological characterization of the nanocomplexes and wrote a draft of manuscript. SKB carried out the drug release kinetics, serum stability and cytotoxicity assays in bioreactor. SF carried out the optimization of 3D cell culture systems and immunostaining of multicellular co-culture. CCJ and RDR performed studies on the cytotoxicity and cellular distribution of nanocomplexes. BD developed a clustering analysis script. MW contributed to the development of the bioreactor system. PL and JF contributed to the analysis of nanocomplex isolation and analysis of peripheral blood mononuclear cells, GF contributed to the optimization of 3D cell culture systems. F. Cavalieri contributed to the experimental design and wrote the manuscript. F. Caruso contributed to experimental design and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Australian Research Council (ARC) under a Future Fellowship (F. Cavalieri, FT140100873). F. Caruso acknowledges the award of a National Health and Medical Research Council Senior Principal Research Fellowship (GNT1135806). F. Cavalieri acknowledges the award of an RMIT Vice Chancellor Senior Research Fellowship. This project received funding from the European Union Horizon 2020 Research and Innovation Program under the H2020 Marie Skłodowska-Curie Actions grant agreement no. 872233 (“PEPSAMATE”). This project received funding by the European Regional Development Fund – project ENOCH (no. CZ.02.1.01/0.0/0.0/16_019/0000868), by the European Regional Development Fund Project MAGNET (CZ.02.1.01/0.0/841 0.0/15_003/0000492) and the Ministry of Health of the Czech Republic – DRO (Institute of Hematology and Blood Transfusion, IHBT, 00023736). This work was performed in part at the Materials Characterisation and Fabrication Platform (MCFP) and Melbourne Histology Platform (MHP) at The University of Melbourne. We acknowledge Haitao Yu for providing help with the STORM analysis.Notes and references
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed
.
- W. He, X. Xing, X. Wang, D. Wu, W. Wu, J. Guo and S. Mitragotri, Adv. Funct. Mater., 2020, 30, 1910566 CrossRef CAS
- R. Tenchov, R. Bird, A. E. Curtze and Q. Zhou, ACS Nano, 2021, 15, 16982–17015 CrossRef CAS PubMed
- A. Zielinska, F. Carreiro, A. M. Oliveira, A. Neves, B. Pires, D. N. Venkatesh, A. Durazzo, M. Lucarini, P. Eder, A. M. Silva, A. Santini and E. B. Souto, Molecules, 2020, 25, 3731 CrossRef CAS PubMed
- A. A. Yaqoob, H. Ahmad, T. Parveen, A. Ahmad, M. Oves, I. M. I. Ismail, H. A. Qari, K. Umar and M. N. M. Ibrahim, Front. Chem., 2020, 8, 341 CrossRef CAS PubMed
- Y. Zeng, Y. Xiang, R. Sheng, H. Tomas, J. Rodrigues, Z. Gu, H. Zhang, Q. Gong and K. Luo, Bioact. Mater., 2021, 6, 3358–3382 CrossRef CAS PubMed
- J. Pushpamalar, A. K. Veeramachineni, C. Owh and X. J. Loh, ChemPlusChem, 2016, 81, 504–514 CrossRef CAS PubMed
- Z. Liu, Y. Jiao, Y. Wang, C. Zhou and Z. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1650–1662 CrossRef CAS PubMed
- A. Sood, A. Gupta and G. Agrawal, Carbohydr. Polym. Technol. Appl., 2021, 2, 100067 Search PubMed
- Q. Zhang, J. Zhang, J. Song, Y. Liu, X. Ren and Y. Zhao, ACS Nano, 2021, 15, 8001–8038 CrossRef CAS PubMed
- M. L. Verma, B. S. Dhanya, Sukriti, V. Rani, M. Thakur, J. Jeslin and R. Kushwaha, Int. J. Biol. Macromol., 2020, 154, 390–412 CrossRef CAS PubMed
- A. Jain, S. K. Singh, S. K. Arya, S. C. Kundu and S. Kapoor, ACS Biomater. Sci. Eng., 2018, 4, 3939–3961 CrossRef CAS PubMed
- D. Verma, N. Gulati, S. Kaul, S. Mukherjee and U. Nagaich, J. Pharm., 2018, 2018, 9285854 Search PubMed
- Q. Hu, H. Li, L. Wang, H. Gu and C. Fan, Chem. Rev., 2019, 119, 6459–6506 CrossRef CAS PubMed
- J. Lazarovits, Y. Y. Chen, E. A. Sykes and W. C. Chan, Chem. Commun., 2015, 51, 2756–2767 RSC
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS
- Y. H. Cheng, C. He, J. E. Riviere, N. A. Monteiro-Riviere and Z. Lin, ACS Nano, 2020, 14, 3075–3095 CrossRef CAS PubMed
- S. Sindhwani, A. M. Syed, J. Ngai, B. R. Kingston, L. Maiorino, J. Rothschild, P. MacMillan, Y. Zhang, N. U. Rajesh, T. Hoang, J. L. Y. Wu, S. Wilhelm, A. Zilman, S. Gadde, A. Sulaiman, B. Ouyang, Z. Lin, L. Wang, M. Egeblad and W. C. W. Chan, Nat. Mater., 2020, 19, 566–575 CrossRef CAS PubMed
- B. R. Kingston, Z. P. Lin, B. Ouyang, P. MacMillan, J. Ngai, A. M. Syed, S. Sindhwani and W. C. W. Chan, ACS Nano, 2021, 15, 14080–14094 CrossRef CAS PubMed
- Y. R. Zhang, R. Lin, H. J. Li, W. L. He, J. Z. Du and J. Wang, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2019, 11, e1519 Search PubMed
- Y. L. Su and S. H. Hu, Pharmaceutics, 2018, 10, 193 CrossRef CAS PubMed
- A. Palumbo Jr., O. Da Costa Nde, M. H. Bonamino, L. F. Pinto and L. E. Nasciutti, Mol. Cancer, 2015, 14, 145 CrossRef PubMed
- A. Dongre and R. A. Weinberg, Nat. Rev. Mol. Cell Biol., 2019, 20, 69–84 CrossRef CAS PubMed
- M. R. Junttila and F. J. de Sauvage, Nature, 2013, 501, 346–354 CrossRef CAS PubMed
- B. Son, S. Lee, H. Youn, E. Kim, W. Kim and B. Youn, Oncotarget, 2017, 8, 3933–3945 CrossRef PubMed
- M. Shen and Y. Kang, Front. Med., 2018, 12, 426–439 CrossRef PubMed
- K. C. Valkenburg, A. E. de Groot and K. J. Pienta, Nat. Rev. Clin. Oncol., 2018, 15, 366–381 CrossRef PubMed
- Q. A. Besford, F. Cavalieri and F. Caruso, Adv. Mater., 2020, 32, e1904625 CrossRef PubMed
- A. Spada, J. Emami, J. A. Tuszynski and A. Lavasanifar, Mol. Pharm., 2021, 18, 1862–1894 CrossRef CAS PubMed
- E. N. Hoogenboezem and C. L. Duvall, Adv. Drug Delivery Rev., 2018, 130, 73–89 CrossRef CAS PubMed
-
N. Desai, in Albumin in Medicine, Springer, 2016, pp. 101–119 Search PubMed
- I. T. Tai and M. J. Tang, Drug Resist. Updates, 2008, 11, 231–246 CrossRef CAS PubMed
- N. P. Desai, V. Trieu, L. Y. Hwang, R. Wu, P. Soon-Shiong and W. J. Gradishar, Anticancer Drugs, 2008, 19, 899–909 CrossRef CAS PubMed
- H. Wartlick, B. Spankuch-Schmitt, K. Strebhardt, J. Kreuter and K. Langer, J. Controlled Release, 2004, 96, 483–495 CrossRef CAS PubMed
- A. Jahanban-Esfahlan, S. Dastmalchi and S. Davaran, Int. J. Biol. Macromol., 2016, 91, 703–709 CrossRef CAS PubMed
- B. Storp, A. Engel, A. Boeker, M. Ploeger and K. Langer, J. Microencapsulation, 2012, 29, 138–146 CrossRef PubMed
- B. Demirkurt, G. Cakan-Akdogan and Y. Akdogan, J. Mol. Liq., 2019, 295, 111713 CrossRef CAS
- A. N. Kovács, N. Varga, G. Gombár, V. Hornok and E. Csapó, J. Flow Chem., 2020, 10, 497–505 CrossRef
- J. I. Boye, I. Alli and A. A. Ismail, J. Agric. Food Chem., 1996, 44, 996–1004 CrossRef CAS
- J. Qi, P. Yao, F. He, C. Yu and C. Huang, Int. J. Pharm., 2010, 393, 176–184 CrossRef CAS PubMed
- P. J. Patel, N. S. Acharya and S. R. Acharya, Drug Delivery, 2013, 20, 143–155 CrossRef CAS PubMed
- V. Kushwah, A. K. Agrawal, C. P. Dora, D. Mallinson, D. A. Lamprou, R. C. Gupta and S. Jain, Pharm. Res., 2017, 34, 2295–2311 CrossRef CAS PubMed
- R. C. Pedrozo, E. Antônio, N. M. Khalil and R. M. Mainardes, Braz. J. Pharm. Sci., 2020, 56, 17692 CrossRef
- S. H. Lee, D. Heng, W. K. Ng, H. K. Chan and R. B. Tan, Int. J. Pharm., 2011, 403, 192–200 CrossRef CAS PubMed
- W. Du and Y. Wang, J. Appl. Polym. Sci., 2013, 127, 4256–4261 CrossRef CAS
- A. Papagiannopoulos and A. Sklapani, Carbohydr. Polym. Technol. Appl., 2021, 2, 100075 Search PubMed
- G. T. Dempsey, J. C. Vaughan, K. H. Chen, M. Bates and X. Zhuang, Nat. Methods, 2011, 8, 1027–1036 CrossRef CAS PubMed
- M. Wojnilowicz, Q. A. Besford, Y. L. Wu, X. J. Loh, J. A. Braunger, A. Glab, C. Cortez-Jugo, F. Caruso and F. Cavalieri, Biomaterials, 2018, 176, 34–49 CrossRef CAS PubMed
- E. Czuba-Wojnilowicz, S. Miellet, A. Glab, S. Viventi, F. Cavalieri, C. Cortez-Jugo, M. Dottori and F. Caruso, Biomacromolecules, 2020, 21, 3186–3196 CrossRef CAS PubMed
- M. Wojnilowicz, A. Glab, A. Bertucci, F. Caruso and F. Cavalieri, ACS Nano, 2019, 13, 187–202 CrossRef CAS PubMed
- E. S. Bronze-Uhle, B. C. Costa, V. F. Ximenes and P. N. Lisboa-Filho, Nanotechnol., Sci. Appl., 2017, 10, 11–21 CrossRef CAS PubMed
- E. Y. Lasfargues, W. G. Coutinho and A. S. Dion, In Vitro, 1979, 15, 723–729 CrossRef CAS PubMed
- Y. Imamura, T. Mukohara, Y. Shimono, Y. Funakoshi, N. Chayahara, M. Toyoda, N. Kiyota, S. Takao, S. Kono, T. Nakatsura and H. Minami, Oncol. Rep., 2015, 33, 1837–1843 CrossRef CAS PubMed
- K. Liang, S. Mei, X. Gao, S. Peng and J. Zhan, Drug Des., Dev. Ther., 2021, 15, 5135–5150 CrossRef PubMed
- K. Stock, M. F. Estrada, S. Vidic, K. Gjerde, A. Rudisch, V. E. Santo, M. Barbier, S. Blom, S. C. Arundkar, I. Selvam, A. Osswald, Y. Stein, S. Gruenewald, C. Brito, W. van Weerden, V. Rotter, E. Boghaert, M. Oren, W. Sommergruber, Y. Chong, R. de Hoogt and R. Graeser, Sci. Rep., 2016, 6, 28951 CrossRef CAS PubMed
- V. Kushwah, S. S. Katiyar, C. P. Dora, A. K. Agrawal, D. A. Lamprou, R. C. Gupta and S. Jain, Acta Biomater., 2018, 73, 424–436 CrossRef CAS PubMed
- A. Fernandez and M. Vendrell, Chem. Soc. Rev., 2016, 45, 1182–1196 RSC
- Y. Feng, B. Press and A. Wandinger-Ness, J. Cell Biol., 1995, 131, 1435–1452 CrossRef CAS PubMed
- J. P. X. Cheng and B. J. Nichols, Trends Cell Biol., 2016, 26, 177–189 CrossRef CAS PubMed
- P. Ujhazy, G. Zaleskis, E. Mihich, M. J. Ehrke and E. S. Berleth, Cancer Immunol. Immunother., 2003, 52, 463–472 CrossRef CAS PubMed
- S. V. Kalivendi, S. Kotamraju, H. Zhao, J. Joseph and B. Kalyanaraman, J. Biol. Chem., 2001, 276, 47266–47276 CrossRef CAS PubMed
- H. Feng, A. Kishimura, T. Mori and Y. Katayama, Anal. Sci., 2020, 36, 1279–1283 CrossRef CAS PubMed
- Y. Wang, F. Cuzzucoli, A. Escobar, S. Lu, L. Liang and S. Wang, Nanotechnology, 2018, 29, 332001 CrossRef PubMed
- G. Rijal and W. Li, Sci. Adv., 2017, 3, e1700764 CrossRef PubMed
- C. Hirt, A. Papadimitropoulos, M. G. Muraro, V. Mele, E. Panopoulos, E. Cremonesi, R. Ivanek, E. Schultz-Thater, R. A. Droeser, C. Mengus, M. Heberer, D. Oertli, G. Iezzi, P. Zajac, S. Eppenberger-Castori, L. Tornillo, L. Terracciano, I. Martin and G. C. Spagnoli, Biomaterials, 2015, 62, 138–146 CrossRef CAS PubMed
- F. Foglietta, G. C. Spagnoli, M. G. Muraro, M. Ballestri, A. Guerrini, C. Ferroni, A. Aluigi, G. Sotgiu and G. Varchi, Int. J. Nanomed., 2018, 13, 4847–4867 CrossRef CAS PubMed
- R. Edmondson, J. J. Broglie, A. F. Adcock and L. Yang, Assay Drug Dev. Technol., 2014, 12, 207–218 CrossRef CAS PubMed
- J. C. Fontoura, C. Viezzer, F. G. Dos Santos, R. A. Ligabue, R. Weinlich, R. D. Puga, D. Antonow, P. Severino and C. Bonorino, Mater. Sci. Eng., C, 2020, 107, 110264 CrossRef CAS PubMed
- D. Yip and C. H. Cho, Biochem. Biophys. Res. Commun., 2013, 433, 327–332 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr08429a |
| This journal is © The Royal Society of Chemistry 2022 |