
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupport effects of metal–organic frameworks in heterogeneous catalysis
Masaaki
Sadakiyo

Department of Applied Chemistry, Faculty of Science Division I, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan
First published on 15th February 2022
Abstract
Catalytic support effects have been widely studied as a key factor for creating highly active heterogeneous catalysts with limited amounts of rare metal elements. Recently, support effects of metal–organic frameworks (MOFs) started to be investigated using their wide variety in pore size, electronic state, and selective adsorption property. Three types of support effects, namely molecular sieving, charge transfer, and substrate adsorption effects, have been reported on composite catalysts of metal nanoparticles supported on MOFs (M/MOFs). The current reports on heterogeneous catalysis in M/MOFs clearly demonstrated that both catalytic activity and product selectivity can be drastically enhanced and modulated by MOF supports through these support effects, and that application of MOFs as the supports is beneficial for creating novel high performance catalysts with metal nanoparticles. This minireview summarizes the catalytic properties and support effects observed on M/MOFs.
 Masaaki Sadakiyo | Dr Masaaki Sadakiyo is a Junior Associate Professor at Department of Applied Chemistry, Faculty of Science Division I, Tokyo University of Science. He received his Ph.D. from Kyoto University in 2012. He became an Assistant Professor at Kyushu University in 2012. He moved to Tokyo University of Science in 2019. His research interests include functionalities of metal–organic frameworks and related porous materials (e.g. ionic conduction, catalysis). |
1. Introduction
Metal–organic frameworks (MOFs) have emerged as a new class of crystalline porous materials; because of their unique features such as high materials variety and designable pores, they are considered for use in various applications such as separation,1,2 storage,3,4 delivery,5,6 and electrolytes.7–9 MOFs are organic–inorganic hybrid materials and thus have both organic and inorganic characters. For example, MOFs show a relatively high thermal stability (<500 °C)10,11 compared with pure crystalline organics that often decompose or sublimate below 200 °C,12,13 and this is one of the inorganic characteristics of MOFs due to their strongly bound organic components through metal–ligand bonds. By contrast, MOFs can be easily modified with some functional groups by using various functionalized ligands14 or through post-synthetic chemical reactions,15 allowing tuning of the electronic state or porous characters of the MOFs, which is one of the organic characteristics of the MOFs. Because of the relatively high thermal stability and designable architectures together with the high porosity, there has been a strong expectation for MOFs to be applied as new catalytic support materials for heterogeneous catalysis.16,17Catalytic support is one of the critical components of heterogeneous catalysts because of their important roles in improving the performance of catalysts. The main role of the catalytic support is to prevent aggregation of small-sized metal nanoparticles (NPs) including catalytically active sites on their surface, which allow the catalysts to show high activity for a prolonged time. Another important role is to improve the catalytic activity through the “support effects” of the support materials.18 Support effects are some of the most important ways to create high-performance catalysts using limited amounts of rare elements such as noble metals (e.g. Pt). In the traditional support materials such as metal oxides, the support effects have been widely studied using metal catalysts deposited on them.19–24 Recently, non-traditional support materials have been applied as catalytic supports to overcome the catalytic performance of traditional catalysts.25–27 Many researchers have tried to apply MOFs as catalytic support to enhance the catalytic performance using metal–MOF composites (M/MOFs). However, the catalytic support effects of MOFs have not been well clarified compared with traditional supports and this has become a current topic in recent years.
Recent progress in preparation methods for M/MOF catalysts having intended structures allows us to know how the MOF supports exactly affect to the catalytic property of M/MOFs. Recent reports on M/MOF catalysts clearly demonstrated that MOFs truly show three types of support effects, namely molecular sieving, charge transfer, and substrate adsorption effects, which have been observed in traditional oxide-based catalysts. There are non-negligible number of well-designed M/MOF catalysts that overcome the traditional catalysts by using these support effects especially in the reactions at relatively low temperature region (<300 °C).
In this review, we summarize recent studies on the support effects of MOFs in heterogeneous catalysis. First, we briefly describe recent advances in preparation methods for M/MOFs, which are strongly related to the clarification of the support effects of MOFs. Second, we present a classification of the support effects and give examples of each support effect observed in M/MOFs.
2. Preparation of M/MOFs
Considering that many important factors modulate the catalytic activity of metal-supported catalysts, such as particle size, loading amount, and the interaction between metal and support material, an ideal way to clarify the support effects is to compare catalytic activity among catalysts having different support materials with similar other factors (i.e. size, loading amount, direct contact with the support). However, for M/MOF catalysts, there are few rational methods of preparing such ideal samples because the general preparation method for oxide-based catalysts, such as impregnation with calcination at high temperature, cannot be widely applied for MOFs because of their moderate thermal stability (<500 °C). Therefore, to clarify the catalytic support effects of MOFs, the preparation method is of importance. This section summarizes recent advances and the most common preparation methods for M/MOFs.Previously, M/MOFs have been synthesized through various methods such as solution impregnation,28,29 chemical vapour deposition,30,31 and solid grinding,32,33 followed by a chemical reduction process. These methods are similar to some methods used for other metal-loaded catalysts. By contrast, some other processes have been developed as specific methods for M/MOFs. Lu et al. reported an encapsulation method that consists of the incorporation of polymer-coated NPs inside MOFs during the crystallization process of the MOFs (Fig. 1).34 Preliminarily-synthesized NPs, such as Ag, Au, Pt, Fe3O4, NaYF4, and CdTe, which are coated with poly(N-vinylpyrrolidone) (PVP) as a protecting reagent, were incorporated in ZIF-8. The framework of ZIF-8 is formed at room temperature (RT) and thus the NPs can be incorporated inside the ZIF-8 crystals at RT through the high affinity between ZIF-8 and PVP. Zhou et al. reported another encapsulation method using polydopamine (PDA).35 Crystals of ZIF-8 and UiO-66 were successfully grown around PDA-coated NPs. Similar to the case with PVP, the interaction between MOF and PDA is useful for preparing the core–shell-type structure of M/MOFs. Various NPs such as Au NPs and magnetic iron oxide were encapsulated in ZIF-8 and UiO-66. In the case of UiO-66, the reaction temperature for the encapsulation was 100 °C, which is different from ZIF-8 (RT).
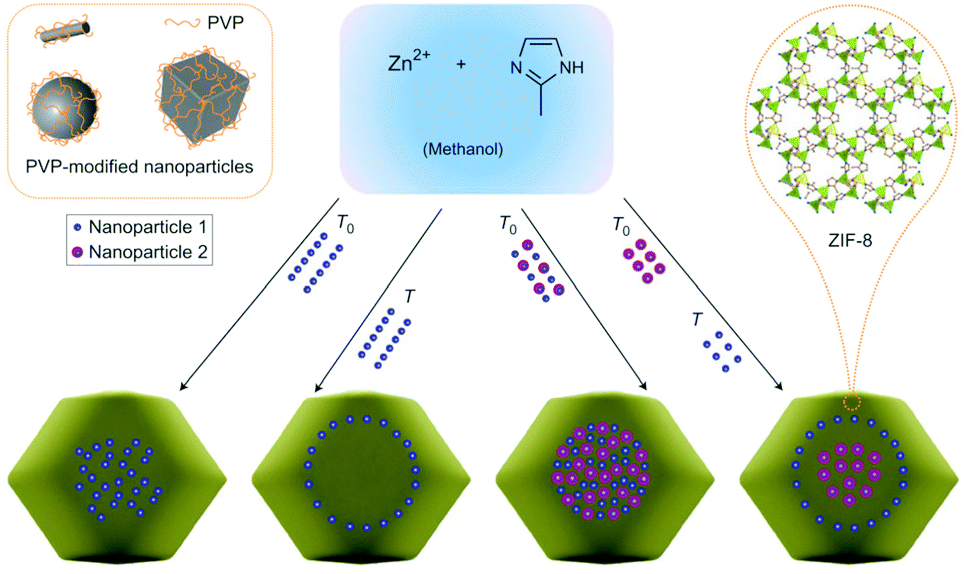 | ||
| Fig. 1 Schematic illustration of the encapsulation method for M/MOFs’ preparation. This figure has been reproduced from ref. 34 with permission from the Nature publishing group, copyright 2012. | ||
Mukoyoshi et al. reported another unique preparation method for M/MOFs, which is the partial decomposition of MOFs (Fig. 2).36 In this method, NPs are formed inside the MOF crystals during the thermal decomposition of the MOFs. The metal sources provided by the MOF are automatically reduced by decomposition of the ligands. Ni-MOF-74, which is composed of a central metal, Ni2+, and a reductive ligand, 2,5-dioxido-1,4-benzenedicarboxylate, was heated above 300 °C under vacuum to form Ni NPs in Ni-MOF-74 (Ni/Ni-MOF-74).
 | ||
| Fig. 2 Schematic illustration of the preparation of M/MOF composites through a partial decomposition method. | ||
For the use of M/MOFs for clarification of the support effects of MOFs in heterogeneous catalysis, these methods seem not to be ideal but have some problems. For instance, in wet chemistry such as solution impregnation with a chemical reduction process, only the MOFs that are highly stable against reagents can be applied. In addition, a synthetic recipe depends on the applied MOFs and thus similar-sized NPs have difficulty forming on different supports. In the encapsulation method, the size of the metal NPs can be easily changed because it is determined in the preliminary synthesis of the NPs. However, number of applicable MOF was limited (e.g. ZIF-8). In addition, this method requires protecting reagents that would prevent direct contact or strong interaction between the metallic NPs and the MOF support. In the partial decomposition method, the metal source is only from the MOF support and thus it is difficult to change the MOF support without changing the metal NPs.
Sadakiyo et al. recently reported the arc plasma deposition (APD) method that satisfies the conditions for the clarification of MOF support effects, i.e. similar-sized NPs, easy-to-change MOF supports, and direct contact between the NP and the MOF, at a higher level than in other methods (Fig. 3).37 In this method, metal atoms are deposited on support materials from a metal target in a vacuum chamber. Well-dispersed similar-sized metal NPs (approximately less than 2 nm in diameter at a low-loading amount such as below 1 wt%) are formed on the surface of any MOF crystals with direct contact between the NP and the MOF. This method does not require any chemicals, solvents, and protecting reagents, in contrast to other methods. Importantly, in this method, the well-dispersed metal NPs can be loaded on different MOFs using the same technique, which would be preferable for clarifying the support effects through a systematic comparison using different MOF supports.
 | ||
| Fig. 3 Schematic illustration of the preparation of M/MOFs through the arc plasma deposition method. | ||
3. Support effects of MOFs in heterogeneous catalysis
The support effects in heterogeneous catalysis can be roughly classified into three types (Fig. 4). First is the molecular sieving effect that prevents a non-target substrate from reaching an active site of the catalysts.19 Second is the charge-transfer effect that is caused by electronic interaction between the metal NP and the support material, resulting in modulation of the catalytic activity of the supported metal catalysts.20 Third is the substrate adsorption effect that is caused by the strong adsorption of the substrate by the support material. If the reaction includes multiple substrates, strong adsorption for one of the substrates could help to proceed the reaction.23 These support effects have been well investigated in traditional support materials such as oxide-based catalysts.19–24 Considering that MOFs have tuneable pore sizes, various electronic states, and various interactions with guest molecules, they should have a strong potential to show all types of these support effects (Fig. 4). Recent progress and examples of each support effect of MOFs are described here.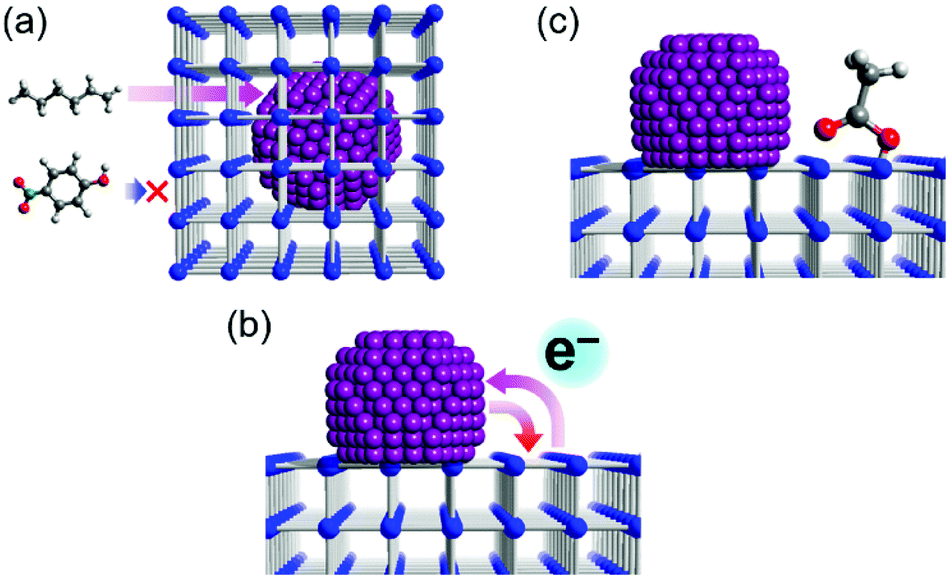 | ||
| Fig. 4 Schematic illustrations of support effects in heterogeneous catalysis, regarding (a) molecular sieving, (b) charge transfer, and (c) substrate adsorption, which are illustrated with porous MOFs as the support materials. | ||
3.1 Molecular sieving effect in M/MOFs
Because MOFs have a variety of porous structures, there is no doubt that M/MOF catalysts in which metal NPs are surrounded by MOFs show the molecular sieving effect. The encapsulation method described above is one of the most ideal methods to show the molecular sieving effect because the metal NPs are completely incorporated inside the MOF crystals. Lu et al. reported the molecular sieving effect in Pt NPs encapsulated by ZIF-8 (Pt/ZIF-8).34 They showed the difference in catalytic activity for hydrogenation reaction of n-hexene and cis-cyclooctene on Pt NPs among various Pt-loaded catalysts such as Pt/CNT (CNT: carbon nanotube), T-Pt@ZIF-8 (Pt-loaded catalysts prepared by the templating method, i.e. solution impregnation followed by chemical reduction), and Pt/ZIF-8. ZIF-8 itself did not show any catalytic activity because the active site is located only on the Pt NPs in this reaction (Fig. 5), Pt/CNT showed catalytic activity for both n-hexene and cis-cyclooctene. However, Pt/ZIF-8 showed almost no catalytic activity for cis-cyclooctene but showed activity for n-hexene, indicating that ZIF-8 acts as a molecular sieve selecting the substrates before they reach a catalytically active site on Pt NPs. Similarly, T-Pt@ZIF-8 showed a large difference in the catalytic activity for these substrates; however, the activity for cis-cyclooctene is not zero, which is indicative that T-Pt/ZIF-8 includes some of the Pt NPs completely exposed to the outside and that the encapsulation method is advantageous from the viewpoint of complete incorporation of the metallic NPs inside MOF crystals. Wang et al. also demonstrated the molecular sieving effect of ZIF-8 using the same reaction without protecting reagents for NP formation.38 These examples clearly demonstrate that the small window size of ZIF-8 is effective for preventing the migration of large molecules such as cyclic molecules. | ||
| Fig. 5 Catalytic activity for the hydrogenation of n-hexene and cis-cyclooctene. This figure has been reproduced from ref. 34 with permission from the Nature publishing group, copyright 2012. | ||
Guo et al. also reported the molecular sieving effect of Pt NPs confined in UiO-66-NH2 crystals (Pt@UiO-66-NH2).39 They showed the catalytic activity of Pt@UiO-66-NH2 for hydrogenation reactions with ethylene, 1-hexene, and cyclooctadiene (COD). The Pt@UiO-66-NH2 catalyst showed high catalytic activity for ethylene and 1-hexene, which is similar to Pt/SiO2 control catalyst. However, in the case of the bulky substrate of COD, the activity on Pt@UiO-66-NH2 was drastically lower than on Pt/SiO2 because of the limited pore size of UiO-66-NH2 (∼6 Å), which is smaller than the molecular size of the COD substrate (6.7 × 6.2 × 4.2 Å3).
Zhou et al. reported a clear molecular sieving effect using Au NPs encapsulated in ZIF-8 and UiO-66.40 The Au NPs were first embedded on PDA-coated magnetic iron oxide NPs (MagNP@PDA@AuNPs) and then the composite NPs, MagNP@PDA@AuNPs, were encapsulated in ZIF-8 (MagNP@PDA@AuNPs@ZIF-8) and UiO-66 (MagNP@PDA@AuNPs@UiO-66). These catalysts were applied to reduction reactions of 4-nitrophenol and methylene blue (Fig. 6). An apparent catalytic activity was observed for 4-nitrophenol on MagNP@PDA@AuNPs@UiO-66, whereas no such activity was observed on MagNP@PDA@AuNPs@ZIF-8. This indicates that the 4-nitrophenol cannot migrate in ZIF-8 but can freely pass through UiO-66 due to the larger pore size of UiO-66. By contrast, the catalytic activity for methylene blue, which is bulkier than 4-nitrophenol, was not observed even on MagNP@PDA@AuNPs@UiO-66, indicating the effective exclusion of the substrate by the surrounding MOFs around the Au NPs.
 | ||
| Fig. 6 Catalytic activity for (a) 4-nitrophenol and (b) methylene blue reduction reactions. This figure has been reproduced from ref 40 with permission from the American Chemical Society, copyright 2015. | ||
3.2 Charge transfer effect in M/MOFs
The various architectures of MOFs should provide a large variety of electronic structures. In particular, the organic components included as ligands allow tuning of the electronic states of MOFs, whereas the electronic states of oxide materials are normally difficult to widely tune. As described above, the electronic interaction between a metal NP and the support is one of the dominant factors in modulating catalytic activity on the metal NP, thus the support effect regarding the charge transfer in M/MOFs is also of interest in creating highly active catalysts.Li et al. reported on the difference in the catalytic activity of Pd NPs formed in UiO-66 frameworks having different functional groups (Pd@UiO-66-X; X = NH2, OMe, H).41 They applied Pd@UiO-66-X for the aerobic reaction between benzaldehyde and ethylene glycol. The possible products of this reaction are hemiacetal and ester (Fig. 7). The sample of X = NH2 showed low selectivity for ester, whereas both samples of X = OMe and X = H showed high selectivity for ester. The difference in oxidation capability between these catalysts was expected to be due to the difference in the electronic states of the included Pd NPs, which were modulated by the coordination by the functional groups on MOFs: Coordination of NH2 group to Pd NP seems to make an electron-rich surface of Pd NP. The difference in electronic states of the Pd NPs was investigated by diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and calculation with density functional theory (DFT). In the DRIFTS measurements under CO, the vibration peak of linearly adsorbed CO molecules at around 2020–2075 cm−1 in Pd@UiO-66-NH2 were observed at a lower wavenumber (2059 cm−1) than in Pd@UiO-66-OMe (2071 cm−1) and Pd@UiO-66 (2073 cm−1). This is indicative that Pd NPs in Pd@UiO-66-NH2 would show a stronger π back-donation to the CO due to the electron-rich surface. The DFT calculation revealed that d-band centres of Pd NPs were modulated by the coordination from the functional groups on the frameworks and that the Pd NP coordinated by the –NH2 group would give a higher level of d-band centres compared with that coordinated by the –OMe group, which led to a higher oxidation capability of the electron-poor surface of Pd@UiO-66-OMe to give a higher selectivity for ester.
 | ||
| Fig. 7 (a) Scheme of the aerobic reaction between benzaldehyde and ethylene glycol (EG). (b) Product selectivity on Pd@UiO-66-X (X = NH2, OMe, and H). This figure has been reproduced from ref. 41 with permission from the American Chemical Society, copyright 2016. | ||
Yoshimaru et al. performed a systematic study on the catalytic support effect regarding charge transfer.42 They showed a difference in the catalytic activity for the CO oxidation reaction on Pt NPs supported on different MOFs. As mentioned, there are various parameters for determining catalytic activity (e.g. size of the metal NPs). Therefore, to clarify the support effect, parameters other than the support material should preferably be almost the same. They prepared Pt/MOF catalysts including similar-sized Pt NPs, supported on different MOFs, with similar Pt content in the same preparation process by the APD method. Four MOFs, Zn-MOF-74, Mg-MOF-74, HKUST-1, and UiO-66-NH2, having different electronic states, were used as support materials. Ionization potentials that directly correspond to the top level of the valence band, i.e. electron-donating character, were evaluated using ultraviolet photoelectron spectroscopy measurements to identify the electronic character of these MOFs. The results showed that the order of electron-donating ability would be Zn-MOF-74 (ionization potential = 5.2 eV) > Mg-MOF-74 (5.7 eV) > HKUST-1 (6.0 eV) > UiO-66-NH2 (6.7 eV). The electronic states of the supported Pt NPs were also estimated using X-ray photoelectron spectroscopy (XPS) measurements. The binding energy (Pt0 4f7/2) of the Pt NPs completely depends on the MOFs and their order depends on the electron-donating character of the MOFs (Fig. 8), indicating the apparent charge-transfer interaction between the Pt NP and the MOF, i.e. electron donation from the MOF to the Pt NP or electron withdrawal from the Pt NP by the MOF.
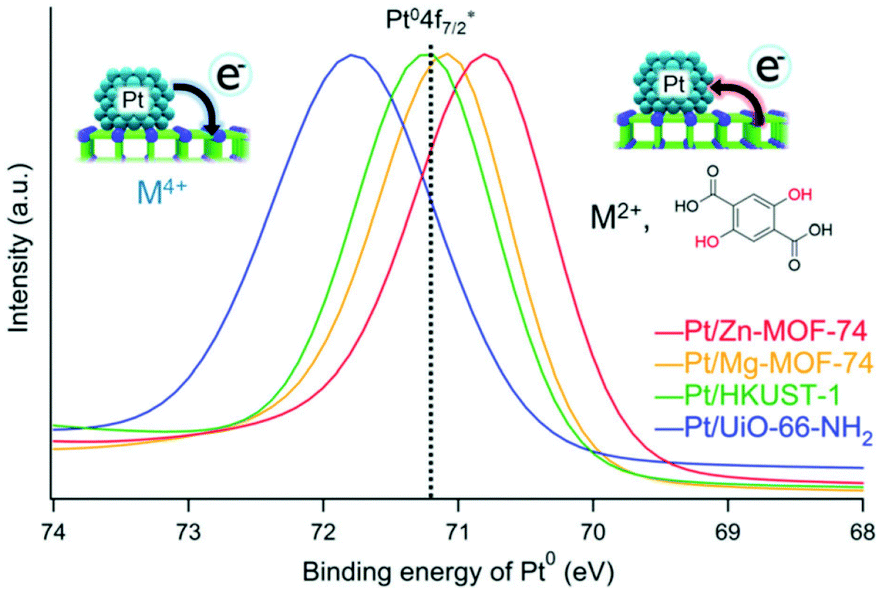 | ||
| Fig. 8 Fitted curves (Pt0 4f7/2) of XPS spectra of (red) Pt/Zn-MOF-74, (yellow) Pt/Mg-MOF-74, (green) Pt/HKUST-1, and (blue) Pt/UiO-66-NH2. The black dotted line indicates the binding energy of bulk Pt0 4f7/2. | ||
The catalytic support effects were demonstrated through the difference in catalytic activity for the CO oxidation reaction, which is known to be sensitive to the NP electronic state.43 The relationship between catalytic activity for CO oxidation (defined by T50, i.e. the temperature at 50% conversion of CO) and the electronic state of Pt NPs supported on different MOFs (i.e. the binding energy of Pt (Pt0 4f7/2)) is shown in Fig. 9. There is a clear relationship between catalytic activity and the electronic state of the Pt NPs and that the electron-rich surface of Pt NPs (i.e. Pt NPs on Zn-MOF-74) is preferable for the reaction. Considering that the order of electronic states and catalytic activity is the same as that of the electron-donating ability of the MOF supports, whereas the other factors (e.g. Pt NPs, synthetic procedure, Pt NPs amounts) are almost the same, the catalytic activity is truly controlled by the support materials through the charge-transfer interaction between Pt NP and MOF. They also compared the turnover frequency (TOF) of the catalysts, calculated by the number of active sites estimated through chemisorption of H2. The order of TOF at each temperature was also the same as that of electronic states of the Pt NPs, meaning that the catalytic activity of each active site on Pt NPs is modulated by the charge transfer with the MOFs.
 | ||
| Fig. 9 Relationship between catalytic activity (i.e. reaction temperature, T50) and electronic state of Pt NPs (i.e. binding energy of Pt0 4f7/2) of Pt/MOF catalysts. The black dotted line indicates the binding energy of bulk Pt0 4f7/2. | ||
Kobayashi et al. reported a charge transfer interaction between Cu and MOF, which relates to catalytic activity for methanol production through CO2 hydrogenation reaction.44 Cu NPs were prepared on various supports, γ-Al2O3, ZIF-8, MIL-100, and a series of UiO-66 based frameworks (UiO-66-X (X = H, COOH, NH2) and Hf-UiO-66). The charge transfer between Cu NP and support was evaluated through XPS measurements. Although the shift of XPS peaks of Cu NPs (Cu 2p) was not obvious, apparent shifts of the peaks of metal components included in the support materials (e.g. Zr 3d peak in UiO-66) were observed in Cu/UiO-66-X and Cu/Hf-UiO-66, while almost no shifts in the other catalysts. The Zr 3d peaks in Cu/UiO-66-X shifted to lower binding energy compared to blank MOFs, suggesting the existence of the charge transfer from Cu NPs to the MOFs. Fig. 10 shows the relationship between catalytic activity for methanol production at 220 °C on the Cu catalysts and the binding energy shifts of the metal components in the supports. Cu/UiO-66-COOH and Cu/Hf-UiO-66 showed high catalytic activity for the methanol production, and the activity seems to be correlated with the order of the shift of the XPS peaks (i.e., charge transfer), while the size of used Cu nanoparticles was not precisely the same (in range from 13 to 24 nm).
 | ||
| Fig. 10 Relationship between catalytic activity for methanol production and shift of binding energy of the metal component of support materials. | ||
3.3 Substrate adsorption effect in M/MOFs
Selective adsorption for various guest molecules is one of the most important and specific properties of MOFs because of their well-defined porous structure and interactive inner surface, where the interaction with guest molecules is easily tuned by changing the inner component such as the functional group. Therefore, the M/MOFs should have a strong potential to enhance catalytic activity around the direct contact between metal NP and the MOF due to the strong adsorption of the specific substrate by the MOFs.Yoshimaru et al. reported a systematic study of enhancement of the catalytic activity of metal NPs through the strong substrate adsorption by MOFs.45 They demonstrated the catalytic activity for ethanol (EtOH) production through the acetic acid (AcOH) hydrogenation (AAH) reaction on Pt NPs supported on seven different MOFs, MIL-125-NH2, UiO-66-NH2, HKUST-1, MIL-101, Zn-MOF-74, Mg-MOF-74, and MIL-121, having a high tolerance for AcOH. In the AAH reaction, it is known that the ability of substrate adsorption by support material is a critical factor for high activity and that TiO2 is the optimal support among traditional support materials.23 The difference in the adsorption ability of MOFs for the AcOH substrate was demonstrated using temperature-programmed desorption-mass spectrometry (TPD-MS). TPD-MS charts of MIL-125-NH2, UiO-66-NH2, HKUST-1, and MIL-101 at a mass number (m/z) 60, corresponding to AcOH molecule, are shown in Fig. 11. No desorption peak was observed in other MOFs, showing their very weak adsorption strength for AcOH. The order of adsorption strength (i.e. desorption temperature) for AcOH is MIL-125-NH2 > UiO-66-NH2 > HKUST-1 > MIL-101 > other MOFs, suggesting that MOFs having an amino group tend to show a high affinity for the AcOH molecule due to the acid–base interaction.
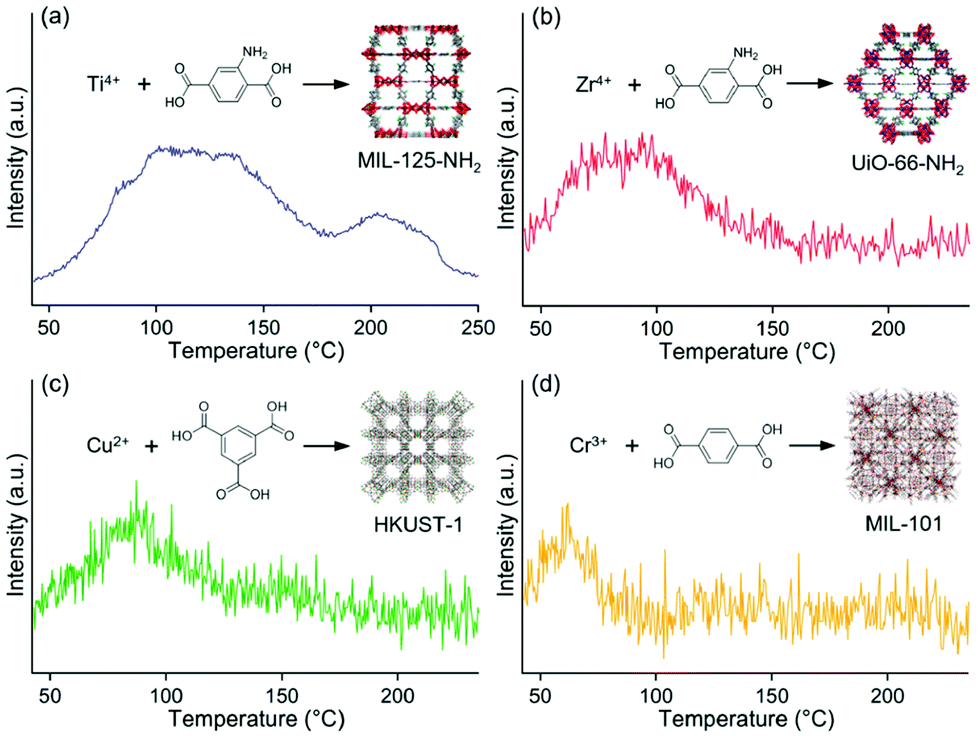 | ||
| Fig. 11 TPD-MS charts of (a) MIL-125-NH2, (b) UiO-66-NH2, (c) HKUST-1, and (d) MIL-101 at m/z = 60 (for AcOH molecule). This figure has been reproduced from ref. 45 with permission from the American Chemical Society, copyright 2021. | ||
The catalytic activity of Pt/MOF catalysts having a similar Pt diameter (∼2 nm) and Pt loading amount (∼0.5 wt%) is shown in Fig. 12. Pt/MIL-125-NH2 and Pt/UiO-66-NH2, in which the supports strongly adsorb AcOH, showed a high conversion of AcOH, reaching almost 100% above 260 °C, whereas Pt NPs on MOFs with a weak adsorption strength did not show an apparent catalytic activity. Considering the results of the adsorption strength for AcOH, this difference in catalytic activity clearly demonstrated a catalytic support effect regarding substrate adsorption of MOFs. Because the MOF supports (without Pt NPs) did not show catalytic activity, the active site was expected to be located around the interface between the Pt NP and the MOF or the area near the Pt NPs. The optimized traditional catalyst, Pt/TiO2, also showed a high activity similar to Pt/MIL-125-NH2 and Pt/UiO-66-NH2. Importantly, the product selectivities of these catalysts (Pt/MIL-125-NH2, Pt/UiO-66-NH2, and Pt/TiO2) were completely different (Fig. 13). The selectivity for the target product, EtOH, on Pt/UiO-66-NH2 is immediately decreased with increasing reaction temperature, instead of increasing the ethyl acetate (AcOEt) by-product. Pt/TiO2 showed a similar volcano-like curve of the selectivity for AcOEt with low selectivity for EtOH. By contrast, Pt/MIL-125-NH2 selectively produced EtOH as the main product at almost all temperature regions and showed suppressed selectivity for AcOEt.
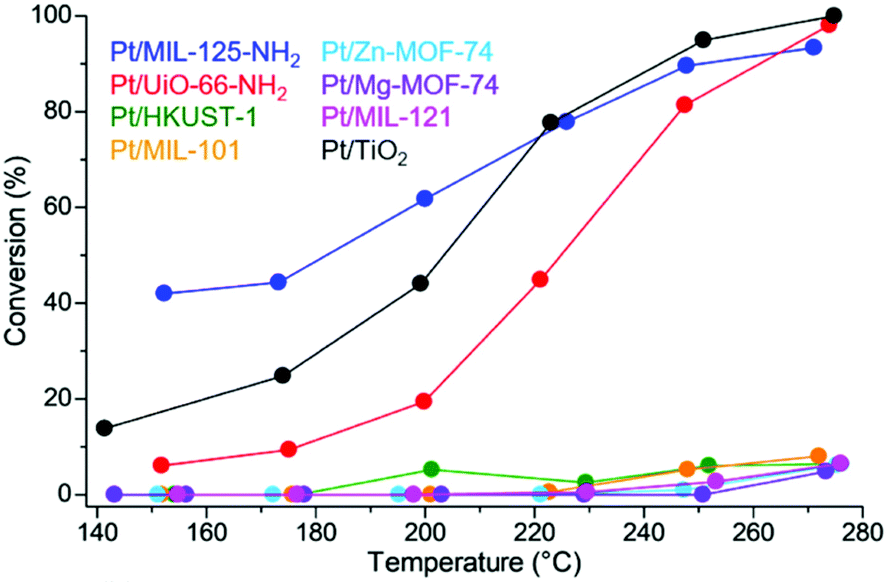 | ||
| Fig. 12 Conversion of AcOH on Pt/MOFs and Pt/TiO2. This figure has been reproduced from ref. 45 with permission from the American Chemical Society, copyright 2021. | ||
 | ||
| Fig. 13 Selectivity for each product on (a) Pt/UiO-66-NH2 and (b) Pt/MIL-125-NH2. This figure has been reproduced from ref. 45 with permission from the American Chemical Society, copyright 2021. | ||
Fig. 14 shows the yield of EtOH, i.e. total performance for EtOH production, on the catalysts. Because of both high EtOH selectivity and activity, Pt/MIL-125-NH2 showed the highest performance, which is apparently above the traditional optimal catalysts, Pt/TiO2, in almost all temperature regions. The reason for high selectivity for EtOH or low selectivity for AcOEt was studied using infrared spectroscopy and a theoretical study (DFT calculation). The results revealed that the adsorption strength of MIL-125-NH2 for EtOH is weaker than the other supports and thus the produced EtOH in Pt/MIL-125-NH2 is immediately released outside the catalysts, which would suppress the formation of AcOEt from the produced EtOH and AcOH substrate. These results clearly indicated that MOF-based catalysts have a great potential to overcome the traditional oxide-based catalysts by using the support effects of MOFs regarding substrate adsorption.
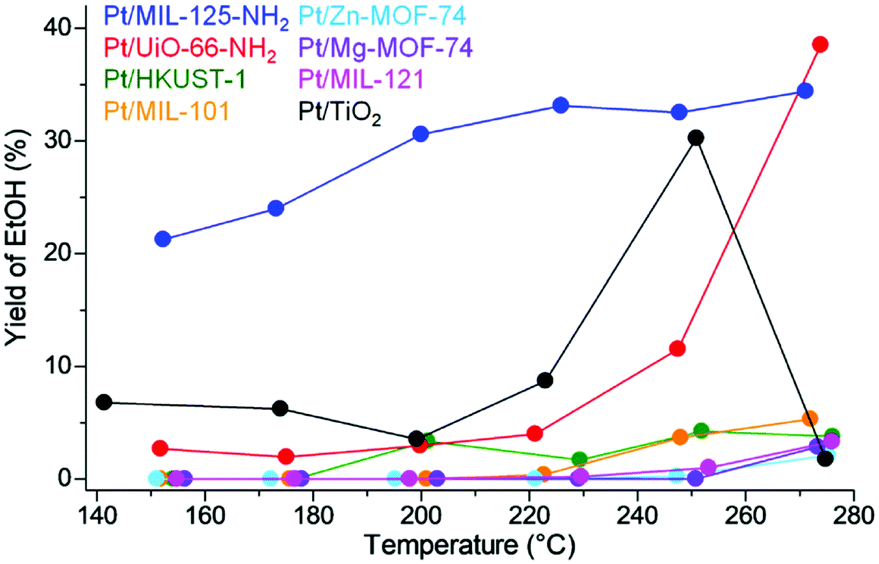 | ||
| Fig. 14 EtOH yield on Pt/MOFs and Pt/TiO2. This figure has been reproduced from ref. 45 with permission from the American Chemical Society, copyright 2021. | ||
Related to the guest adsorption, some researchers demonstrated that hydrophilicity or hydrophobicity of the MOF support is also an important factor for catalytic activity. Sun et al. reported a significant difference in catalytic activity for hydrodeoxygenation reaction of vanillin.46 They revealed that Pd NPs supported on hydrophilic MOF, MIL-101-SO3Na (Pd/MIL-101-SO3Na), showed higher catalytic performance for the reaction compared to that supported on hydrophobic supports of MIL-101 (Pd/MIL-101) and active carbon (Pd/C). The hydrophilic nature of the support allows the water soluble reactants to access the active sites easily and thus could enhance the catalytic activity. Another example was reported by Li et al.47 They showed higher catalytic activity of Pd@MIL-101-F5, having perfluoroalkyl groups, for dehydration coupling reaction of organosilane, compared to Pd@MIL-101-NH2 and other Pd catalysts. The hydrophobic character of the MIL-101-F5 contributes to construct well-accessible active sites. These examples clearly demonstrated that tuning of hydrophilicity or hydrophobicity of MOFs, i.e., tuning of guest–MOF interaction, is an important way for creating highly active catalysts.
4. Conclusions and outlook
Three types of catalytic support effects, which were previously observed in traditional support materials, have recently been reported on MOF supports. The number of reports on the support effects of MOFs is still limited; however, there should be a strong potential in expanding it as one of the important research areas of both MOFs and heterogeneous catalysts. MOF-based catalysts could show higher catalytic performance (i.e. reactivity, selectivity, and yield for target product) than traditional catalysts by choosing optimal MOF supports having a limited pore size, various electronic states, and selective adsorption properties. Considering that the thermal stability of MOFs (<500 °C) is lower than that of conventional oxide supports, the reaction temperature of target catalysis should be carefully considered; the main research area of the MOF-based catalysts should be catalysis proceeding below 300 °C. The stability of MOFs against substrates should also be carefully considered for their application as supports. Recently, many researchers have investigated the chemical stability (e.g. against solvents, acid, or base) of MOFs and thus we could roughly estimate the applicability of MOFs for various substrates before catalytic application. Optimization of MOF supports for various catalyses would create new high-performance catalysts and provide attractive insights on the interaction between metallic NPs and MOFs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is partly supported by JSPS Grant-in-Aid for Scientific Research No. 17H04890.References
- Q. Qian, P. A. Asinger, M. J. Lee, G. Han, K. M. Rodriguesz, S. Lin, F. M. Benedetti, A. X. Wu, W. S. Chi and Z. P. Smith, Chem. Rev., 2020, 120, 8161–8266 CrossRef CAS PubMed.
- Y. Belmabkhout, P. M. Bhatt, K. Adil, R. S. Pillai, A. Cadiau, A. Shkurenko, G. Maurin, G. Liu, W. J. Koros and M. Eddaoudi, Nat. Energy, 2018, 3, 1059–1066 CrossRef CAS.
- Y.-F. Zhang, Z.-H. Zhang, L. Ritter, H. Fang, Q. Wang, B. Space, Y.-B. Zhang, D.-X. Xue and J. Bai, J. Am. Chem. Soc., 2021, 143, 12202–12211 CrossRef CAS PubMed.
- D. E. Jaramillo, H. Z. H. Jiang, H. A. Evans, R. Chakraborty, H. Furukawa, C. M. Brown, M. H. Gordon and J. R. Long, J. Am. Chem. Soc., 2021, 143, 6248–6256 CrossRef CAS PubMed.
- H. D. Lawson, S. P. Walton and C. Chan, ACS Appl. Mater. Interfaces, 2021, 13, 7004–7020 CrossRef CAS PubMed.
- K. Suresh and A. J. Matzger, Angew. Chem., Int. Ed., 2019, 58, 16790–16794 CrossRef CAS PubMed.
- Y. Yoshida, K. Kato and M. Sadakiyo, J. Phys. Chem. C, 2021, 125, 21124–21130 CrossRef CAS.
- M. Sadakiyo and H. Kitagawa, Dalton Trans., 2021, 50, 5385–5397 RSC.
- M. Sadakiyo, H. Kasai, K. Kato, M. Takata and M. Yamauchi, J. Am. Chem. Soc., 2014, 136, 1702–1705 CrossRef CAS PubMed.
- H. G. T. Nguyen, N. M. Schweitzer, C.-Y. Chang, T. L. Drake, M. C. So, P. C. Stair, O. K. Farha, J. T. Hupp and S. T. Nguyen, ACS Catal., 2014, 4, 2496–2500 CrossRef CAS.
- R.-J. Li, M. Li, X.-P. Zhou, D. Li and M. O'Keeffe, Chem. Commun., 2014, 50, 4047–4049 RSC.
- W. Xie, Z. Gao, W.-P. Pan, D. Hunter, A. Singh and R. Vaia, Chem. Mater., 2001, 13, 2979–2990 CrossRef CAS.
- A. H. Jones, J. Chem. Eng. Data, 1960, 5, 196–200 CrossRef CAS.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- W. Morris, C. J. Doonan, H. Furukawa, R. Banerjee and O. M. Yaghi, J. Am. Chem. Soc., 2008, 130, 12626–12627 CrossRef CAS PubMed.
- Y. Zhao, J. Zhang, J. Song, J. Li, J. Liu, T. Wu, P. Zhang and B. Han, Green Chem., 2011, 13, 2078–2082 RSC.
- A. Aijaz, Q.-L. Zhu, N. Tsumori, T. Akita and Q. Xu, Chem. Commun., 2015, 51, 2577–2580 RSC.
- M. Comotti, W.-C. Li, B. Spliethoff and F. Schüth, J. Am. Chem. Soc., 2006, 128, 917–924 CrossRef CAS PubMed.
- J. C. S. Wu, J. G. Goodwin and M. Davis, J. Catal., 1990, 125, 488–500 CrossRef CAS.
- A. Bruix, J. A. Rodriguez, P. J. Ramirez, S. D. Senanayake, J. Evans, J. B. Park, D. Stacchiola, P. Liu, J. Hrbek and F. Illas, J. Am. Chem. Soc., 2012, 134, 8968–8974 CrossRef CAS PubMed.
- H.-J. Freund and G. Pacchioni, Chem. Soc. Rev., 2008, 37, 2224–2242 RSC.
- S. J. Tauster, S. C. Fung, R. T. K. Baker and J. A. Horsley, Science, 1981, 211, 1121–1125 CrossRef CAS PubMed.
- W. Rachmady and M. A. Vannice, J. Catal., 2000, 192, 322–334 CrossRef CAS.
- M. M. Schubert, S. Hackenberg, A. C. Veen, M. Muhler, V. Plzak and R. J. Behm, J. Catal., 2001, 197, 113–122 CrossRef CAS.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S. W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- T.-N. Ye, S.-W. Park, Y. Lu, J. Li, M. Sasase, M. Kitano and H. Hosono, J. Am. Chem. Soc., 2020, 142, 14374–14383 CrossRef CAS PubMed.
- D. Deng, K. S. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218–230 CrossRef CAS PubMed.
- Y. K. Hwang, D.-Y. Hong, J.-S. Chang, S. H. Jhung, Y.-K. Seo, J. Kim, A. Vimont, M. Daturi, C. Serre and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 4144–4148 CrossRef CAS PubMed.
- Q.-L. Xhu, J. Li and Q. Xu, J. Am. Chem. Soc., 2013, 135, 10210–10213 CrossRef PubMed.
- S. Hermes, M.-K. Schröter, R. Schmid, L. Khodeir, M. Muhler, A. Tissler, R. W. Fischer and R. A. Fischer, Angew. Chem., Int. Ed., 2005, 44, 6237–6241 CrossRef CAS PubMed.
- J. Hermannsdörfer, M. Friedrich, N. Miyajima, R. Q. Albuquerque, S. Kümmel and R. Kempe, Angew. Chem., Int. Ed., 2012, 51, 11473–11477 CrossRef PubMed.
- T. Ishida, M. Nagaoka, T. Akita and M. Haruta, Chem. – Eur. J., 2008, 14, 8456–8460 CrossRef CAS PubMed.
- H.-L. Jiang, B. Liu, T. Akita, M. Haruta, H. Sakurai and Q. Xu, J. Am. Chem. Soc., 2009, 131, 11302–11303 CrossRef CAS PubMed.
- G. Lu, S. Li, Z. Guo, O. K. Farha, B. G. Hauser, X. Qi, Y. Wang, X. Wang, S. Han, X. Liu, J. S. DuChene, H. Zhang, Q. Zhang, X. Chen, J. Ma, S. C. J. Loo, W. D. Wei, Y. Yang and J. T. Hupp, Nat. Chem., 2012, 4, 310–316 CrossRef CAS PubMed.
- J. Zhou, P. Wang, C. Wang, Y. T. Goh, Z. Fang, P. B. Messersmith and H. Duan, ACS Nano, 2015, 9, 6951–6960 CrossRef CAS PubMed.
- M. Mukoyoshi, H. Kobayashi, K. Kusada, M. Hayashi, T. Yamada, M. Maesato, J. M. Taylor, Y. Kubota, K. Kato, M. Takata, T. Yamamoto, S. Matsumura and H. Kitagawa, Chem. Commun., 2015, 51, 12463–12466 RSC.
- M. Sadakiyo, S. Yoshimaru, H. Kasai, K. Kato, M. Takata and M. Yamauchi, Chem. Commun., 2016, 52, 8385–8388 RSC.
- P. Wang, J. Zhao, X. Li, Y. Yang, Q. Yang and C. Li, Chem. Commun., 2013, 49, 3330–3332 RSC.
- Z. Guo, C. Xiao, R. V. Maligal-Ganesh, L. Zhou, T. W. Goh, X. Li, D. Tesfagaber, A. Thiel and W. Huang, ACS Catal., 2014, 4, 1340–1348 CrossRef CAS.
- J. Zhou, P. Wang, C. Wang, Y. T. Goh, Z. Fang, P. B. Messersmith and H. Duan, ACS Nano, 2015, 9, 6951–6960 CrossRef CAS PubMed.
- X. Li, T. W. Goh, L. Li, C. Xiao, Z. Guo, X. C. Zeng and W. Huang, ACS Catal., 2016, 6, 3461–3468 CrossRef CAS.
- S. Yoshimaru, M. Sadakiyo, A. Staykov, K. Kato and M. Yamauchi, Chem. Commun., 2017, 53, 6720–6723 RSC.
- Y. P. G. Chua, G. T. K. K. Gunasooriya, M. Saeys and E. G. Seebauer, J. Catal., 2014, 311, 306–313 CrossRef CAS.
- H. Kobayashi, J. M. Taylor, Y. Mitsuka, N. Ogiwara, T. Yamamoto, T. Toriyama, S. Matsumura and H. Kitagawa, Chem. Sci., 2019, 10, 3289–3294 RSC.
- S. Yoshimaru, M. Sadakiyo, N. Maeda, M. Yamauchi, K. Kato, J. Pirillo and Y. Hijikata, ACS Appl. Mater. Interfaces, 2021, 13, 19992–20001 CrossRef CAS PubMed.
- Q. Sun, M. Chen, B. Aguila, N. Nguyen and S. Ma, Faraday Discuss., 2017, 201, 317–326 RSC.
- L. Li, Z. Li, W. Yang, Y. Huang, G. Huang, Q. Guan, Y. Dong, J. Lu, S.-H. Yu and H.-L. Jiang, Chem, 2021, 7, 686–698 CAS.
| This journal is © The Royal Society of Chemistry 2022 |