
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural and ethylene oligomerization studies of chelating (imino)phenol Fe(II), Co(II) and Ni(II) complexes: an experimental and theoretical approach†
Makhosonke
Ngcobo
a,
Holliness
Nose
b,
Arumugam
Jayamani
a and
Stephen O.
Ojwach
 *a
*a
aSchool of Chemistry and Physics, University of KwaZulu-Natal, Private Bag X01, Scottsville, Pietermaritzburg 3209, South Africa. E-mail: ojwach@ukzn.ac.za
bSchool of Chemistry and Material Science, Technical University of Kenya, Haile Selasie Avenue, P.O. Box 52428-00200, Nairobi, Kenya
First published on 11th March 2022
Abstract
The metal complexes [Fe(L1)2] (Fe1); [Fe(L2)2] (Fe2); [Fe(L3)2] (Fe3); [Co(L1)2] (Co1); [Co(L2)2] (Co2); [Co(L3)3] (Co3); [Ni(L1)2] (Ni1); [Ni(L2)2] (Ni2) and [Ni(L3)3] (Ni3); where L = 2,4-dibromo-6-((pyridin-2-ylimino)methyl)phenol (L1H), 2,4-dibromo-6-(((4-methylpyridin-2-yl)imino)methyl)phenol (L2H) and 2,4-dibromo-6-((quinolin-8-ylimino)methyl)phenol (L3H), were synthesized in good yields. The complexes were characterized using IR spectroscopy, UV-visible spectroscopy, mass spectrometry, magnetic moment measurements, elemental analysis, and X-ray crystallography. The molecular structures of complexes Fe3a (oxidised form of Fe3) and Ni3 confirmed the isolation of bis(chelated) tridentate bound octahedral compounds. Activation of the complexes with the EtAlCl2 co-catalyst produced active catalysts in the ethylene oligomerization reactions to afford mainly C4 and C6 oligomers. The catalytic activities and product distribution were largely controlled by the nature of the ligand and the metal atom. Density functional theory calculations were used to investigate the influence of complex properties and global descriptors in the ethylene oligomerization reactions. The stability and magnitude of the charge of the metal atom appear to drive the overall catalytic activities of the complexes.
1. Introduction
Ethylene oligomerization reactions are major industrial processes, producing tonnes of olefins which are raw materials extensively used in the production of detergents, lubricants, plasticizers and polyolefins among others.1–4 In the matrix of olefin production, late transition metal catalysts have shown high catalytic activities and selectivities, as well as tolerance to polar monomers, compared to early transition metal counterparts.5 As a result, the past decades have witnessed significant progress in the use of late transition metal complexes as ethylene oligomerization catalysts.6,7 For example, Ni(II) complexes are currently applied industrially in the Shell Higher Olefin Process (SHOP).8 The pioneering work of Brookhart et al.9 on the application of α-diimine nickel(II) complexes in olefin oligomerization and polymerization reactions has stimulated the development of new modified Ni(II) complexes based on nitrogen-donor ligands with varied outcomes.10–15Other late transition metal catalysts, most notably, Fe(II) and Co(II) systems ligated on nitrogen-donor ligands, independently discovered by Brookhart and Gibson are also known for their high catalytic activities and selectivity towards the formation of linear and branched polyethylenes.16,17 Subsequent research studies have thus focused on modifying these catalyst models to achieve a balance between the catalytic activity, selectivity and stability.18–22 Schiff base ligands display interesting and varied coordination chemistries and, consequently, have attracted much attention in the design of ethylene polymerization and oligomerization catalysts.23–25 For example, the use of bidentate salicylaldehyde ligands and their derivatives with late transition metal complexes has led to the development of active and selective ethylene oligomerization catalysts.26,27
We recently reported the use of Fe(II) and Co(II) complexes bearing 2-[(ethylimino)methyl]phenol ligands as catalysts in ethylene oligomerization reactions.28,29 Driven by their promising catalytic performances, we hereby report on the syntheses and structural elucidation of Fe(II), Co(II) and Ni(II) complexes of bromo-substituted salicylaldimine ligands and investigate their potential as catalysts in ethylene oligomerization reactions. Density Functional theory studies were performed to gain insight into the effect of the complex structure and other global descriptors on the catalytic behaviour of these complexes and are herein discussed.
2. Experimental section
2.1. Materials and instrumentation
All solvents were of analytical grade and were dried and distilled prior to use. The reagents 3,5-dibromosalicylaldehyde (98%), 2-aminopyridine (99%), 2-amino-4-methylpyridine (99%) and 8-aminoquinoline (98%) were purchased from Sigma-Aldrich; anhydrous CoCl2 (98%), FeCl2·4H2O (98%) and [NiBr2(DME)] (DME = ethylene glycol dimethyl ether) (97%) were purchased from Merck and used as received without further purification. The synthetic protocols and spectral data of the (imino)phenol ligands (L1H-L3H) are given in the ESI.† The infrared spectra were recorded on a PerkinElmer Spectrum 100 in the 4000–400 cm−1 range. ESI-mass spectra were recorded on an LC premier micro mass spectrometer. The magnetic moments were determined using an Evans balance (Sherwood MK-1). The ultraviolet absorption spectra of the metal complexes were recorded using a PerkinElmer LAMBDA 35 spectrometer (2002). The scanning region is from 150–700 nm and the spectral bandwidth is 1 nm. GC analyses were performed on a Varian CP-3800 gas chromatograph equipped with a flame ionization detector and a 30 m (0.2 mm i.d., 0.25 μm film thickness) CP-Sil5 CB capillary column.2.2. Synthesis of Fe(II), Co(II) and Ni(II) complexes
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3063 (νOH), 1613 (νC=N); μobs = 5.60 BM. Anal. calcd for C24H14Br4FeN4O2: C, 37.49; H, 1.84; N, 7.29. Found (%): C, 37.85; H, 1.76, N, 7.19.
:3063 (νOH), 1613 (νC=N); μobs = 5.60 BM. Anal. calcd for C24H14Br4FeN4O2: C, 37.49; H, 1.84; N, 7.29. Found (%): C, 37.85; H, 1.76, N, 7.19.
Complexes Fe2, Fe3, Co1, Co2, and Co3 and Ni1, Ni2 and Ni3 were synthesized following the procedure described for complex Fe1.
:3063 (νOH), 1623 (νC=N); μobs = 3.26 BM. Anal. calcd for C24H14Br4CoN4O2: C, 37.64; H, 1.84; N, 7.32. Found (%): C, 37.32, H, 1.56; N, 7.58.
:3288 (νOH), 1634 (νC=N); μobs = 2.82 BM. Anal. calcd for C24H14Br4NiN4O2: C 37.50; H, 1.84; N, 7.64. Found (%): C 37.49, H 1.54, N 7.94.
:3063 (νOH), 1609 (νC=N); μobs = 5.15 BM. Anal. calcd for C26H18Br4FeN4O2: C, 39.33; H, 2.29; N, 7.06. Found (%): C 39.22, H, 2.36, N 7.09.
:1613 (νC=N); μobs = 3.99 BM. Anal. calcd for C26H18Br4CoN4O2: C, 39.18; H, 2.28; N, 7.03. Found (%): C, 38.98, H, 2.60; N, 7.00.
:3063 (νOH), 1623 (νC=N); μobs = 3.05 BM. Anal. calcd for C26H18Br4NiN4O2: C, 39.13; H, 2.28; N, 7.03. Found (%): C, 38.92; H, 2.66; N, 6.92.
:1614 (νC=N); μobs = 5.43 BM. Anal. calcd for C32H18Br4FeN4O2: C, 44.38; H, 2.10; N, 6.47. Found (%): C, 44.52; H, 2.13; N, 6.59. Recrystallization of the complex in methanol solvent by a slow evaporation afforded green single crystals suitable for X-ray analyses.
:1619 (νC=N); μobs = 3.59 BM. Anal. calcd for C16H10Br2Cl2CoN2O: C, 35.86; H, 1.88; N, 5.23. Found (%): C, 35.82; H, 1.66; N, 5.29.
:1635 (νC=N), 572 (νNi-O); μobs = 2.78 BM. Anal. calcd for C32H18Br4NiN4O2: C, 44.24; H, 2.09; N, 6.45. Found (%): C, 43.82; H, 2. 66; N, 6.29. Recrystallization of the complex in methanol solvent by slow evaporation afforded green single crystals suitable for X-ray analyses.
2.3. X-ray crystallography data collection
Single orange block-shaped crystals of Fe3a and green rod-shaped crystals of Ni3 were recrystallized from methanol solvent by slow evaporation. A suitable crystal of each complex was selected and mounted on a MITIGEN holder in Paratone oil on a Bruker APEX-II CCD diffractometer. The crystal was kept at T = 100(2) K during data collection. Using Olex2,30 the structure was solved using ShelXS-201331 structure solution program, using the direct solution method. The model was refined using version 2016/6 of ShelXL,32 using least squares minimisation.2.4. Density functional theory calculations
All computational calculations were carried out using the Gaussian 16 suite of programs.33 Density functional theory with Becke's three-parameter hybrid functional (B3),34,35 for the exchange part and the Lee–Yang–Parr (LYP) correlation function,36 accepted as a cost-effective approach has been used to compute optimized structural geometrical parameters, energies, natural bond orbitals (NBOs) and UV-vis spectra of the complexes. The LanL2DZ (Los Alamos International 2 Double ζ) basis set was used for the metals throughout the study, and 6-311G(d,p) basis set was used for all other atoms in geometry optimizations, NBO analysis, and excited state calculations. To model the UV-vis spectrum, electronic transitions, vertical excitation energies, absorbance and oscillator strengths of metal complexes, time-dependent density functional theory (TD–DFT) method was performed. The frontier molecular orbital (such as the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO)) energies were determined by the TD−DFT approach.2.5. Ethylene oligomerization experiments
The ethylene oligomerization reactions were carried out utilizing a 100 mL stainless steel reactor equipped with a stirrer bar. A pre-weighed amount of the synthesized pre-catalyst (10.0 μmol) in chlorobenzene (10 mL) was transferred into a dry Schlenk tube via a cannula under a nitrogen atmosphere. This was followed by the addition of the respective co-catalyst (EtAlCl2 or MMAO) using a syringe. The resultant solution in the Schlenk tube was then transferred using a cannula into the pre-evacuated reactor, followed by the addition of 20 mL of chlorobenzene or toluene solvent to give a total volume of 30 mL. The reactor was then purged with ethylene gas and pressure and temperature were set at 10 bar and 30 °C, respectively, and the reaction was initiated by switching on the magnetic stirrer for 1 h. Upon completion of the oligomerization process, the reactor was cooled to about 10 °C using liquid nitrogen and the unreacted ethylene was drained. This was followed by quenching the mixture through the addition of 10% HCl (2 mL). Aliquots of the reaction sample were placed in GC-vials and analyzed to determine both the catalytic activity and oligomer compositions. Heptane was used as the internal standard37 while GC–MS data were used to establish the molecular weights and identities of the oligomer products.3. Results and discussion
3.1. Synthesis and spectroscopic characterization of pyridine/quinoline (imino)phenol Fe(II), Co(II) and Ni(II) metal complexes
Schiff's base ligands (L1H–L3H) were synthesized in high yields (81–89%) by condensation reactions of stoichiometric amounts of 3,5-dibromo salicylaldehyde with 2-aminopyridine, 4-methyl-2-aminopyridine and 8-aminoquinoline, respectively, as depicted in Scheme 1. The reactions of ligands L1H–L3H with the respective metal salts, FeCl2, CoCl2 or [Ni(DME)Br2] resulted in the formation of the respective Fe(II), Co(II) and Ni(II) complexes in good to high yields (65%–84%) as shown in Scheme 2. | ||
| Scheme 1 Syntheses of bromo substituted pyridine and quinolone Schiff base ligands. | ||
 | ||
| Scheme 2 Syntheses of pyridine and quinoline (imino)phenol Fe(II), Co(II) and Ni(II) complexes. | ||
The compounds were characterized using 1H NMR (Fig. S1–S3, ESI†) 13C NMR (Fig. S4–S6, ESI†) for the ligands, IR spectroscopy (Fig. S7–S18, ESI†), mass spectrometry (Fig. S19–S30, ESI†), elemental analyses, magnetic moment measurements and single crystal X-ray crystallography. IR spectra of complexes Fe(II), Co(II) and Ni(II) showed sharp absorption band characteristic of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching frequency in the 1609–1617 cm−1 region (Fig. S13–S21, ESI†). Notably, the CN diagnostic absorption band was recorded at lower frequencies for the corresponding ligands, which pointed to the successful complexation of the ligands. Deprotonation of ligands L1H–L3H to form anionic ligands (L1−–L3−) upon coordination was derived from the absence of the OH stretching vibrations in the coordinated ligands. This data is also consistent with the absence of the halides in the metal coordination sphere.38 For example, the quinoline ligand L3H showed the OH vibration signal at 3062 cm−1 (Fig. S9, ESI†), while this signal is absent in the corresponding complexes Fe3, Co3 and Ni3 (Fig. S16–S18, ESI†).
N stretching frequency in the 1609–1617 cm−1 region (Fig. S13–S21, ESI†). Notably, the CN diagnostic absorption band was recorded at lower frequencies for the corresponding ligands, which pointed to the successful complexation of the ligands. Deprotonation of ligands L1H–L3H to form anionic ligands (L1−–L3−) upon coordination was derived from the absence of the OH stretching vibrations in the coordinated ligands. This data is also consistent with the absence of the halides in the metal coordination sphere.38 For example, the quinoline ligand L3H showed the OH vibration signal at 3062 cm−1 (Fig. S9, ESI†), while this signal is absent in the corresponding complexes Fe3, Co3 and Ni3 (Fig. S16–S18, ESI†).
Mass spectrometry was also used in confirmation of the molecular masses and identities of the Fe(II), Co(II) and Ni(II) complexes (Fig. S22–S30, ESI†). From the mass spectral data, the formation of the bis(ligated) complexes could be deduced as depicted in Scheme 2. For instance, ESI mass spectrum of complex Fe1 showed a molecular ion peak at m/z = 764 amu (10%) and agrees with the proposed formula of [Fe(L1)2] and molar mass of 765.85 g mol−1 (Fig. S22, ESI†). In addition, there was good agreement between the theoretical and experimental isotopic mass distributions of the complexes (Fig. S22–30, ESI†).
The magnetic moments of complexes Fe1–Fe3 were recorded as 5.60 BM, 5.15 BM and 5.43 BM respectively. These values are notably higher than the spin-only value of 4.90 BM for d6 Fe(II) complexes at 300 K. Nevertheless, the values fall within the expected range of high spin Fe(II) complexes of 5.10–5.70 BM at 300 K.39 Similarly, values of 3.26 BM, 3.99 BM and 3.59 BM for the Co(II) complexes Co1–Co3 were recorded respectively, and are comparable to the spin-only value of 3.87 BM for high spin d7 Co(II) complexes at 300 K. Finally, the magnetic moment values of 2.82 BM, 3.05 BM and 2.78 BM recorded for the Ni(II) complexes Ni1–Ni3 respectively, were consistent with spin-only value of 2.83 BM and fall within the expected range of 2.90 BM–4.20 BM at 300 K for high-spin d8 Ni(II) complexes.40 One interesting import from the magnetic moments data is that the Fe(II) complexes showed relatively high observed magnetic moments, pointing to larger crystal field effects (orbital contribution) for the Fe(II) complexes in comparison to the Co(II) and Ni(II) complexes.41 The micro-analyses data of the complexes were consistent with two ligand motifs per metal atom, as illustrated in Scheme 2 and confirmed the purity of the bulk materials.
3.2. Molecular structures of complexes Fe3a and Ni3
Single crystals suitable for X-ray analyses of complexes Fe3 and Ni3 were obtained by slow evaporation of their methanol solutions at room temperature. The resolved molecular structure of complex Fe3, however, contains an Fe(III) metal center with a Br− counter anion, and two anionic ligand L3− units (Fe3a), pointing to in situ oxidation during crystallization (Fig. 1). Oxidation of Fe(II) to Fe(III) complexes is common, and has been associated with the presence of trace amounts of air or moisture.42–44 Table S1 (ESI†) gives a summary of the crystallographic data and structure refinement parameters, while Fig. 1 and 2 represent molecular structures and selected bond parameters for complexes Fe3a and Ni3 respectively. The molecular structures of complexes Fe3a and Ni3 confirm the presence of bis(chelated) mononuclear complexes in which the two tridentate N^N^O anionic ligand (L3−) lie perpendicular to each other around the metal centre. The crystal structures of the complexes Fe3a and Ni3 exhibit two five- and two six-membered chelate rings occupying meridional positions of the octahedral structures.45 The four coordinating atoms that make up the basal plane are the two phenoxy–O atoms and two quinoline-N atoms, while the axial sites are occupied by the two imine N-donor atoms.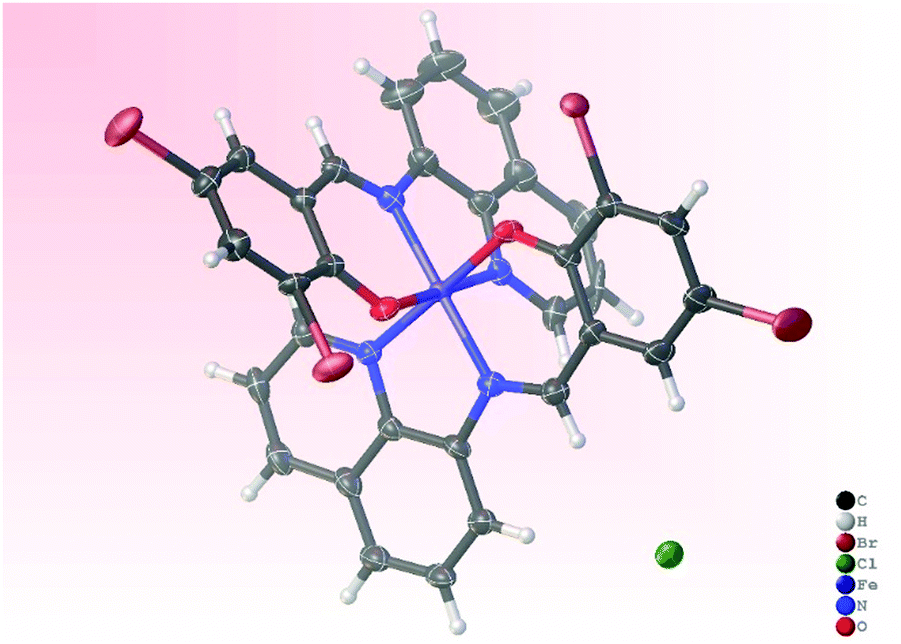 | ||
| Fig. 1 Molecular structure of complex Fe3a. Ellipsoids are illustrated at 50% probability. Selected bond lengths (Å) and angles (°): Fe(1)-N(5), 1.897(5); Fe(1)-N(4), 1.931(5); Fe(1)-N(3), 1.955(5); Fe(1)-N(1), 1.912(5); Fe(1)-O(2), 1.885(4); Fe(1)-O(3), 1.881(4); O(3)-Fe(1)-N(4), 178.2(2); O(2)-Fe(1)-N(3), 178.6(2); O(2)-Fe(1)-O(3), 88.97(18); O(2)-Fe(1)-N(5), 88.27(18); O(3)-Fe(1)-N(5), 95.64(18); N(3)-Fe(1)-N(5), 91.9(2); N(1)-Fe(1)-N(5), 175.3(2); N(4)-Fe(1)-N(5), 84.6(2). | ||
 | ||
| Fig. 2 Molecular structure of complex Ni3 with atoms shown as 30% thermal ellipsoids, hydrogen atoms are removed for more clarity. Selected bond lengths (Å) and angles (°): Ni(1)-O(2), 2.039(7); Ni(1)-N2(2), 2.069(8); Ni(1)-N(1), 2.088(8); Ni(1)-N(3), 2.112(8); Ni(1)-N(4), 2.047(8); Ni(1)-O(1), 2.088(7); O(1)-Ni(1)-N(1), 171.2(3); O(2)-Ni(1)-N(3), 169.7(3); N(4)-Ni(1)-N(2), 174.0(3); N(2)-Ni(1)-O(1), 91.0(3); N(2)-Ni(1)-N(1), 80.2(3); N(4)-Ni(1)-O(1), 92.9(3); and N(4)-Ni(1)-N(1), 95.8(3). | ||
The average Ni–Nquinoline bond distance of 2.0795 Å of complex Ni3 is longer than the mean Ni–Nimine bond distance of 2.0785 Å. In a related study, Insiti et al.46 reported a Ni–Nquinoline bond length of 2.097 Å for the bis(4-bromo-2-(((quinolin-8-)imino)methyl)phenolato)-Ni(II) complex, which is 0.0175 Å longer than the mean Ni–Nquinoline bond length of 2.0795 Å for complex Ni3. In addition, the mean Ni–Nquinoline (2.0795 Å) and Ni–Nimine (2.0785 Å) bond distances obtained for compound Ni3 are longer than the mean bond lengths of 2.104 ± 0.027 Å and 2.055 ± 0.060 Å reported in 47 and 51 similar structures respectively.47 The average Ni–Ophenolic bond length of 2.0635 Å is shorter than the average bond length of 2.024 ± 0.026 Å calculated for 64 similar structures.47 It is also noteworthy to mention that the reported Ni–Nquinoline (2.0795 Å), Ni–Nimine (2.0785 Å) and the Ni–Ophenolic (2.0635 Å) mean bond lengths all lie within the reported minimum and maximum bond distances of 2.044 Å–2.182 Å, 1.977 Å–2.239 Å and 1.956 Å–2.092 Å, reported in 47, 51 and 64 similar structures, respectively.48 For complex Fe3a, the average Fe–Nimine bond distance of 1.926 Å, is slightly longer than the average Fe–Nquinoline bond length of 1.9216 Å. In addition, the average Fe–Nquinoline, Fe–Nimine and Fe–Ophenolic bond lengths of 1.9216 Å, 1.926 Å and 1.883 Å are all shorter than the mean Fe–Nquinoline, Fe–Nimine and Fe–Ophenolic bond lengths of 2.036 ± 0.085 Å, 2.066 ± 0.087 Å and 1.905 ± 0.037 Å respectively, reported for 396, 323 and 453 similar structures respectively.48 More significantly, it appears that since the reported mean Fe–Nimine bond distance of 1.926 Å has been the shortest reported to date, the minimum Fe–Nimine bond length calculated from 323 similar structures is 1.933 Å.48
The bond angles for O(3)-Fe(1)-N(4), O(2)-Fe(1)-N(3), and N(1)-Fe(1)-N(5) of 175.3(2)°, 178.6(2)° and 178.2(2)°, respectively, in complex Fe3a and the bond angles for O(1)-Ni(1)-N(1), O(2)-Ni(1)-N(3), and N(4)-Ni(1)-N(2) of 171.2(3)°, 169.7(3)° and 174.0(3)°, respectively in complex Ni3 deviate from the expected 180° linear geometry by 2–11,° revealing distorted octahedral geometries. For example, the average N^N five-membered bite angles in complexes Fe3a and Ni3 were calculated to be 88.25° and 88.0° respectively, while the mean N^O six-membered bite angle was found to be 91.55° in both cases. In addition, the packing of Ni3 was observed to be stabilized by edge-to-edge π⋯π stacking, Br⋯H, C–H⋯π and nonconventional C–H⋯O hydrogen bonding interactions. In contrast to the crystal structure of the Ni3 complex, no hydrogen bonding interactions and structure stabilizations could be quantified for the Fe(II) complex Fe3a.
3.3. Experimental and theoretical UV-visible spectral analyses
The electron transfer from bonding or nonbonding orbitals to antibonding orbitals induces electronic absorption behaviour in the Fe(II), Co(II), and Ni(II) metal complexes. In general, π → π*, n → π*, n → σ*, d → d, and L ↔ M transitions are possible in metal–organic materials.49–51 In this study, the absorption UV spectrum of the metal compounds was calculated by the TD–DFT method using the LanL2DZ basis set for metal ions and 6-311G (d,p) basis set for all other atoms. The computed wavelength of absorption (λ), the value of oscillator strength (f), excitation energy (E), transition assignments and the predominant transitions along with the experimental UV data are listed in Table 1 and Fig. S2, S3 (ESI†). The calculated wavelengths are also plotted against the oscillator strengths using a Gaussian line shape as shown for complex Co1 in Fig. 3 and Fig. S31 (ESI†) for the remaining complexes. Table S3 (ESI†) shows the HOMO/LUMO diagrams, corresponding to the peaks displayed in the spectra of the metal complexes calculated using the B3LYP density functional theory (DFT) as shown in Table S2 (ESI†). The calculated UV spectrum of the Co1 complex shows four electronic transitions at 404, 310, 244, and 201 nm, corresponding to experimental values at 402, 298, 271, and 232 nm as shown in Table 1. The electronic transitions observed are assigned as follows: 402 nm is due to d → d transitions, 298 nm is due to n → π* transitions, 271 nm is due to π → π* transitions and 232 nm and are due to n → σ* transitions. These bands were also consistent with high-spin distorted tetrahedral complexes of ligands L1 and L2.52| B3LYP/6-311G(d,p) | Experimental λ (nm) | Assignments | |||
|---|---|---|---|---|---|
| Metal compound | λ (nm) | (f) | E (eV) | ||
| Fe1 | 392 | 0.0843 | 3.1632 | — | d → d |
| 305 | 0.3037 | 4.0695 | 302 | n → π* | |
| 234 | 0.1967 | 5.2927 | 259 | π → π* | |
| 197 | 0.1948 | 6.2847 | 232 | n → σ* | |
| Co1 | 404 | 0.1174 | 3.0677 | 402 | d → d |
| 310 | 0.2124 | 3.9940 | 298 | n → π* | |
| 244 | 0.2457 | 5.0901 | 271 | π → π* | |
| 201 | 0.1716 | 6.1662 | 232 | n → σ* | |
| Ni1 | 409 | 0.0964 | 3.0293 | 414 | d → d |
| 294 | 0.2178 | 4.2121 | 297 | n → π* | |
| 237 | 0.0882 | 5.2234 | 269 | π → π* | |
| 198 | 0.1338 | 6.2652 | 234 | n → σ* | |
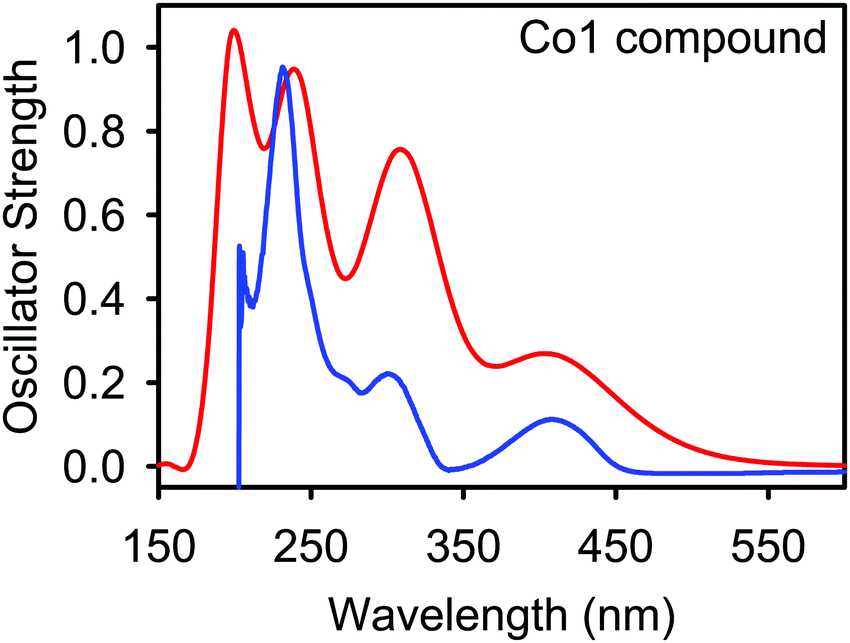 | ||
| Fig. 3 Absorption spectra of Co1 compound. The red curve shows the absorption spectra generated theoretically by using B3LYP functional whereas the blue curve shows the absorption spectra generated experimentally. | ||
The low intensity band at 402 nm is consistent with the d → d transitions of the metal cation. However, the major contributor to this metal compound involved pure π → π* excitations within the aromatic rings. This is followed by intra-ligand n → π* transitions that may occur by nitrogen, oxygen and Br atom lone pair electrons. Small transitions involving n → σ* transitions are also observed during excitation. A comparison between B3LYP theoretically determined wavelength values and experimentally determined wavelength values showed good agreements. A similar trend was observed for the remaining metal complexes, as shown in the supporting information, Table S2 and Fig. S31 (ESI†). In all the cases, the experimental and theoretical UV-vis spectra of the M2+ complexes agree, confirming that the complexes exist as M2+ in the bulk state. It is therefore reasonable to assert that the crystal structure of Fe3a (obtained from a solution of Fe3), which has a Fe3+ metal atom (Fig. 1), was formed via an in situ oxidation process during crystallization.
3.4. Ethylene oligomerization reactions catalyzed by the Fe(II), Co(II) and Ni(II) complexes
| Entry | Catalyst | Yieldb (g) | Activity (kg mol−1 h−1) | Product distribution (%)c | |
|---|---|---|---|---|---|
| C4 (α-C4) | C6 (α-C6) | ||||
| a Reaction conditions: complex, 10 μmol; solvent, chlorobenzene/toluene, 30 mL; temperature, 30 °C; time, 1 h; pressure, 10 bar; EtAlCl2: Al/M = 250. b Determined using GC and n-heptane served as an internal standard. c Determined by GC and GC-MS. d Using the MMAO-12 co-catalyst. nd denotes not detected. e In toluene solvent, remaining products were alklytoluenes. | |||||
| 1 | Fe1 | 9.18 | 918 | 39 (92) | 61 (14) |
| 2 | Co1 | 10.36 | 1036 | 48 (95) | 52 (18) |
| 3 | Ni1 | 16.08 | 1608 | 60 (95) | 40 (17) |
| 4 | Fe2 | 8.65 | 865 | 41 (90) | 59 (7) |
| 5 | Co2 | 9.14 | 915 | 53 (98) | 47 (26) |
| 6 | Ni2 | 12.82 | 1282 | 54 (96) | 46 (10) |
| 7 | Fe3 | 6.84 | 684 | 65 (93) | 35 (19) |
| 8 | Co3 | 8.82 | 882 | 62 (93) | 38 (15) |
| 9 | Ni3 | 9.95 | 995 | 61 (88) | 39 (15) |
| 10d | Ni1 | nd | 0 | nd | nd |
| 11e | Ni1 | 12.86 | 1286 | 32 (>99) | <1 |
| 12e | Ni2 | 10.48 | 1048 | 37 (76) | <1 |
| 13e | Ni3 | 7.21 | 721 | 5 (96) | <1 |
The steric and electronic properties of the coordinated ligands also played a crucial role in controlling the catalytic activity of the complexes. For example, comparisons of complexes supported on the aldimine ligand L1H (Fe1, Co1 and Ni1) with their counterparts (Fe2, Co2 and Ni2) supported on the ketimine ligand L2−, revealed that introduction of the methyl substituent in the pyridyl ring reduced the catalytic activities of the complexes. For example, complex Fe1 displayed catalytic activity of 918 kg mol−1 h−1 whilst its analogous complex Fe2, showed lower catalytic activities of 865 kg mol−1 h−1 (Table 2, entries 1 and 4). This trend may be attributed to electronic factors, where the unsubstituted complex Fe1 results in a more electropositive Fe(II) metal centre, than its methyl substituted counterpart Fe2.58
Furthermore, the catalytic activities of the complexes were compromised when the pyridyl ring in complexes Fe1, Co1 and Ni1 was changed to the quinoline group as in complexes Fe3, Co3 and Ni3. For example, complex Fe3 displayed a lower catalytic activity of 684 kg mol−1 h−1 compared to the catalytic activity of complex Fe1 of 918 kg mol−1 h−1. This may be linked to the degree of π-back-bonding abilities of the pyridyl group (L1) in comparison to the quinoline group (L3).59 This trend agrees with the electronic effects observed in complexes Fe1 and Fe2. In addition, the lower catalytic activities of the complexes Fe3, Co3 and Ni3 (anchored on the tridentate ligand L3−) could be due to steric crowding. As an illustration, complexes Fe3, Co3 and Ni3 are six-coordinate, while those of anionic ligands L1− and L2− are four-coordinate. This has the overall effect of hindering ethylene coordination to the active metal site in the tridentate bound complexes, consistent with their diminished catalytic activities.60
The product distribution of the Fe(II), Ni(II) and Co(II) complexes was also driven by the structural variations in the complexes. Generally, Ni(II) complexes produced more C4 oligomers than their Co(II) and Fe(II) counterparts. For example, the percentage compositions of C4 oligomers of 60%, 48% and 39% were produced by Ni1, Co1 and Fe1 catalysts respectively (Table 2, entries 1–3). The nature of the coordinated ligand was also witnessed to control the product distribution. In general, the more sterically hindered complexes Fe3, Co3 and Ni3 (ligated by tridentate L3−), favoured the formation of C4 oligomers, when compared to for instance complexes Fe1, Co1 and Ni1 supported on the less hindered L1− ligand (Table 2, entries 1–3 vs. 7–9). This trend agrees with the literature findings and fundamental concepts, where sterically hindered catalysts favour chain propagation over chain termination.61,62 While there was no significant isomerism of the preformed C4 fractions greater isomerism of the C6 oligomers was observed. This is consistent with our previous reports and the proposed mechanism in Scheme S1 (ESI†).63
3.5. Density functional theory (DFT) studies
In attempts to gain in-depth understanding of the effects of the complex structure on the catalytic performance of these complexes in ethylene oligomerization reactions, we carried out density functional theory (DFT) computational studies on the structure–activity relationships of the complexes. The computational calculations were performed using the split basis set LANL2DZ (Los Alamos International 2 Double ζ) for Fe(II), Co(II) and Ni(II) complexes, and 6-311G(dp) for the remaining atoms.33,35,36 The ground state electronic structures of the complexes (Fig. S36, ESI†) were employed to probe the effect of various global descriptors of the metal complexes on their catalytic performance in ethylene oligomerization reactions. First, the energy gaps were analysed to study their influence on the catalytic activities of the respective complexes (Table 3 and Fig. S37, ESI†). Two opposing trends were observed, controlled by the nature of the coordinated ligands (L1–L3−). Comparison between the HOMO–LUMO energy gap and catalytic activities of the complexes (within a metal series) bearing the bidentate ligands (L1− and L2−) reveal that the smaller the HOMO–LUMO energy gap, the higher the catalytic activity (Fig. S37, ESI†). For example, complexes Fe1 and Fe2 displayed HOMO–LUMO gaps of 0.1280 eV and 0.1289 eV, and catalytic activities of 918 kg mol−1 h−1 and 865 kg mol−1 h−1 respectively. This is consistent with the ethylene oligomerization reaction involving a nucleophilic attack to the metal centre by the ethylene monomer.64 Thus smaller energy difference between the HOMO and LUMO should promote the coordination of the ethylene substrate to the metal centre. A similar observation was observed for Co1, Co2, Ni1 and Ni2 complexes (Fig. S37, ESI†). However, for complexes (Fe3, Ni3 and Co3) anchored on the tridentate ligand L3, lower catalytic activities than, for example, Fe1, Ni1 and Co1 were reported despite lower HOMO–LUMO gaps. This points to structural effects being the major contributor here. For instance, complexes Fe3, Co3 and Ni3 of L3 are six-coordinate and more sterically hindered, and thus limit facile coordination of the ethylene monomer. This argument is well supported by the results obtained for the global softness and hardness (Fig. S38, ESI†), where complexes Fe3, Co3 and Ni3 showed an opposite trend to the four-coordinate complexes.| Parameter | Fe1 | Co1 | Ni1 | Fe2 | Co2 | Ni2 | Fe3 | Co3 | Ni3 |
|---|---|---|---|---|---|---|---|---|---|
| a E LM and EHM are the LUMO and HOMO orbital energy values of the metal complexes and ethylene molecule, respectively. b Reaction conditions: complex, 10 μmol; solvent, chlorobenzene (30 mL), temperature, 30 °C; time, 1 h; pressure, 10 bar; EtAlCl2, Al/M = 250. | |||||||||
| HOMO-LUMO Energy Gap | 0.1280 | 0.1259 | 0.1262 | 0.1289 | 0.1267 | 0.1281 | 0.1062 | 0.1036 | 0.1013 |
| Global Softness | 7.8113 | 7.9441 | 7.9221 | 7.7568 | 7.8959 | 7.8082 | 9.4127 | 9.6571 | 9.8707 |
| Chemical Hardness | 0.0640 | 0.0629 | 0.0631 | 0.0645 | 0.0633 | 0.0640 | 0.0531 | 0.0517 | 0.0507 |
| NBO Charge | 1.1730 | 1.0956 | 1.0438 | 1.1733 | 1.0960 | 1.0421 | 1.0846 | 1.0015 | 0.9293 |
| Ionization Potential | 0.2229 | 0.2222 | 0.2236 | 0.2217 | 0.2209 | 0.2230 | 0.2004 | 0.1979 | 0.1965 |
| Chemical Potential | −0.1589 | −0.1593 | −0.1605 | −0.1572 | −0.1575 | −0.1589 | −0.1473 | −0.1460 | −0.1458 |
| Activity/Kg mol−1 h−1 | 918 | 1036 | 1608 | 865 | 915 | 1282 | 684 | 882 | 995 |
In the ethylene oligomerization mechanism, coordination of the ethylene monomer to the metal active site is considered as the rate determining step and is largely controlled by the metal charge.65 We thus used DFT studies to probe the possible role of the NBO charges of Fe, Ni and Co atoms in controlling the catalytic activities of these complexes (Table 3 and Fig. 4). From Fig. 4, the role of the coordinated ligand was evident within a given metal series, where the higher the NBO charge, the higher the catalytic activity. For example, complexes Ni1, Ni2 and Ni3 carrying NBO charges of 1.044, 1.042 and 0.929, exhibited catalytic activities of 1608 kg mol−1 h−1, 1282 kg mol−1 h−1 and 995 kg mol−1 h−1 respectively. A similar trend was reported for the Fe and Co catalysts (Fig. 4). Comparable NBO charges of the complexes anchored on ligands L1 and L2 is chemically sound since both the aldimine and ketimine units are remotely located from the metal atoms. On the other hand, the higher metal charges, on complexes supported on ligands L1− and L2−, in comparison to those of ligand L3−, may be attributed to the pyridine ring being a better π-acceptor than the quinoline ring.59,66 An opposite trend was interestingly observed on examinations of the different metal atoms, containing the same ligand. For example, Fe1, Ni1 and Co1, exhibited NBO charges of 1.173, 1.096 and 1.044 and catalytic activities of 918 kg mol−1 h−1, 1036 kg mol−1 h−1 and 1608 kg mol−1 h−1, respectively (Fig. 4). This could be assigned to the different charge/ionic size ratios, where the smaller Ni2+ has the highest charge/ionic size ratio, and hence was more susceptible to nucleophilic attack by the ethylene monomer.
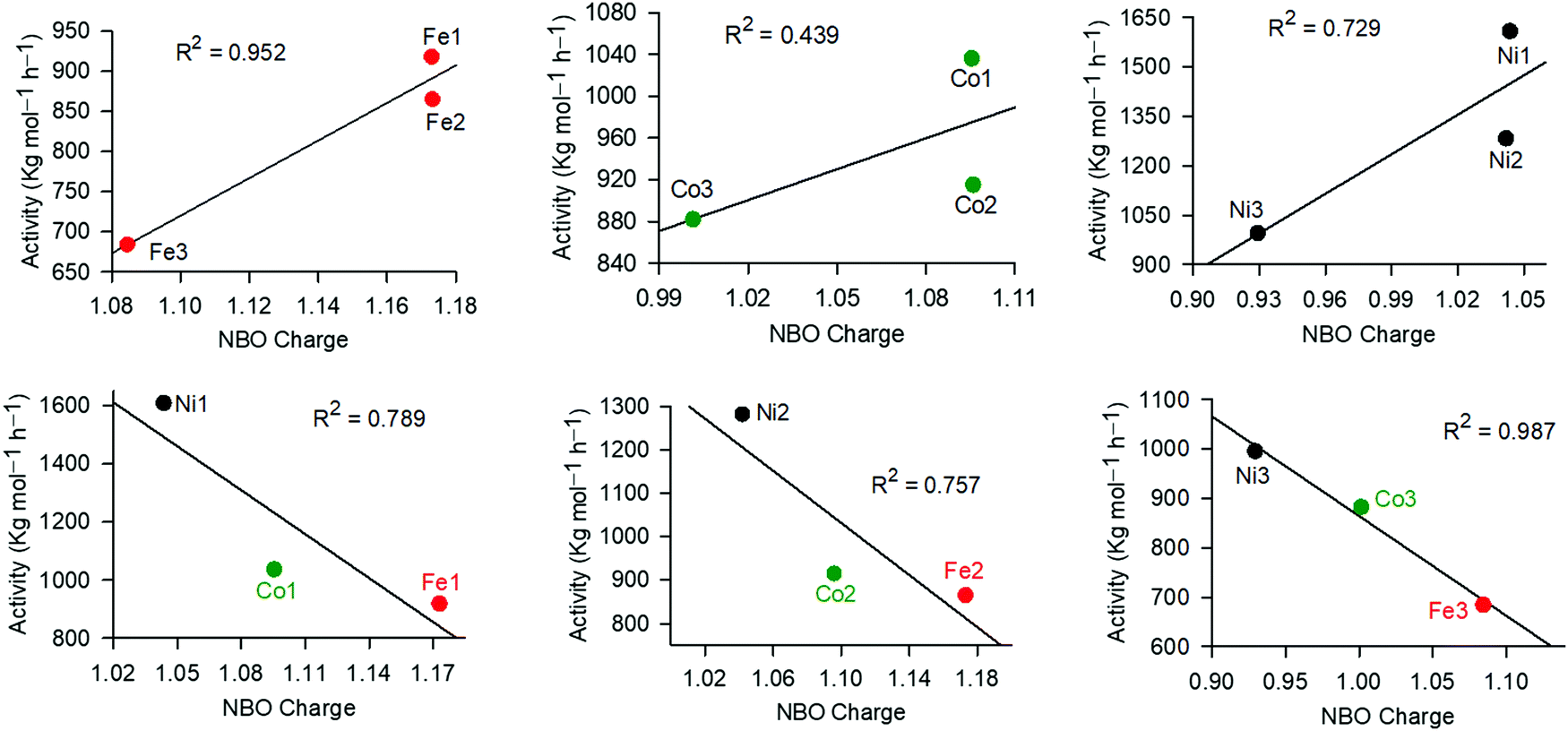 | ||
| Fig. 4 Influence of the metal NBO charge on the catalytic activities of the complexes in ethylene oligomerization reactions. | ||
Next, we investigated the possible role of ionization and chemical potentials of the complexes in ethylene oligomerization reactions. From the data presented in Fig. S39, (ESI†) all the complexes followed a similar trend, where the most negative complexes within the metal series (high tendency to lose an electron) were the most active. For example, the chemical potentials of Ni1, Ni2 and Ni3 complexes were −0.1605, −0.1589 and −0.1458, respectively (Table 3). This followed the order of their catalytic activities of 1608 kg mol−1 h−1, 1282 kg mol−1 h−1 and 995 kg mol−1 h−1 respectively. A similar trend was observed for the other complexes and the ionization potential data (Fig. S39, ESI†). The negative chemical potential of the complexes is indicative of their relative stability. This is fundamentally reasonable since the less sterically crowded four coordinate complexes of ligands L1− and L2− are expected to be more stable than the six-coordinate complexes Fe3, Ni3 and Co3.
We finally sort to understand the relationship between catalytic stability and activity, by analysing the dependence of catalytic activities on the M–N/O bond lengths (Fig. 5). A shorter bond length is associated with enhanced stability of the compounds, and vice versa. From Fig. 5, it was apparent that in all cases, longer bond lengths (M–N/O) were associated with decreased catalytic activities. This asserts the earlier data (chemical potential, hardness and softness), and points to the stability of the complexes as the driving force in controlling catalytic activities. Thus, longer bond distances led to the decomposition of the active species, consistent with the lower catalytic activities observed with the increase in M–N/O bond distances (Fig. 5).
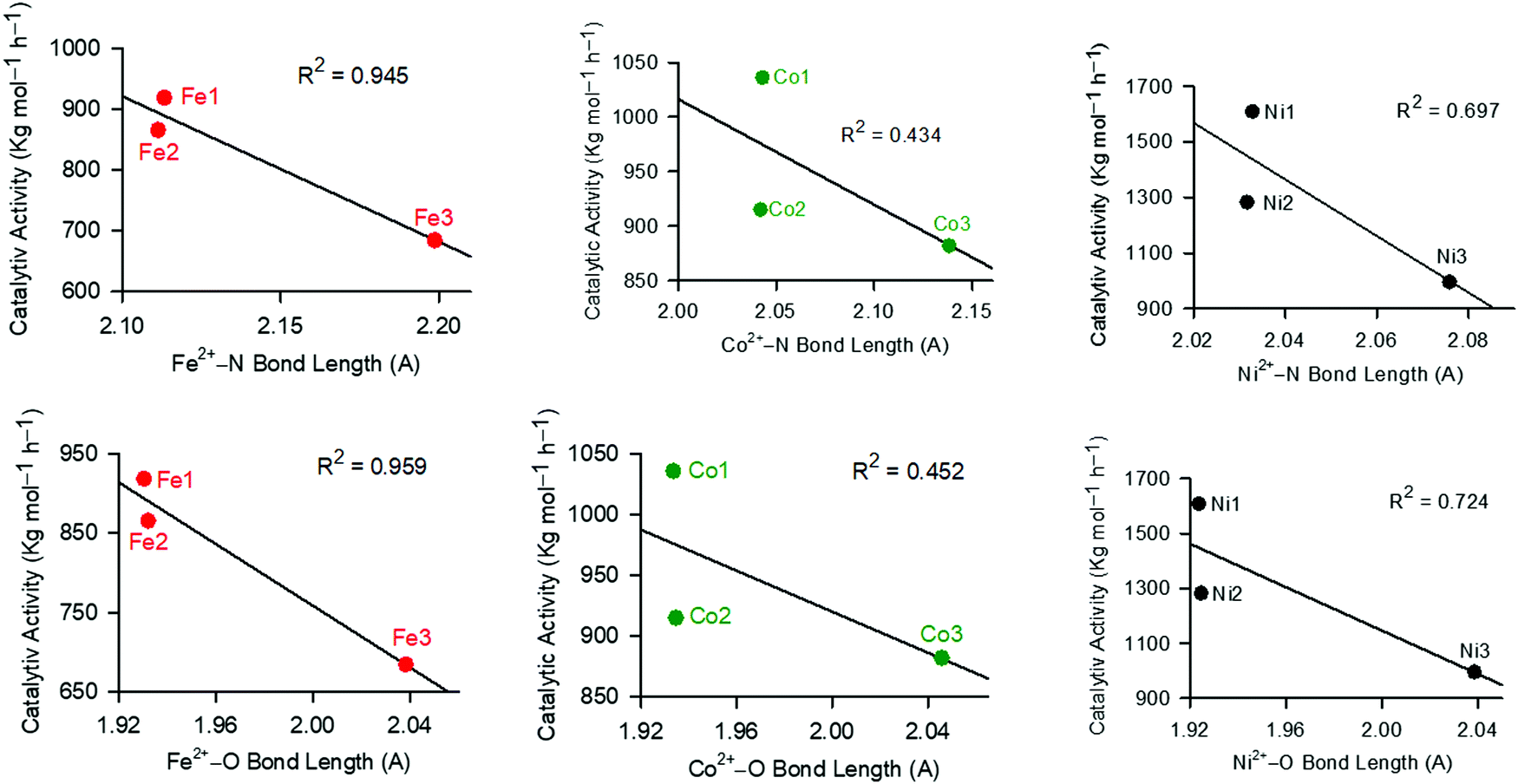 | ||
| Fig. 5 The influence of the M–N/O bond length on the resultant catalytic activities of the complexes showing that higher bond lengths result in lower catalytic activities. | ||
4. Conclusions
Fe(II), Co(II) and Ni(II) complexes of bidentate and tridentate (imino)phenol ligands bearing pyridine and quinoline motifs were successfully synthesized and characterized using IR and UV-visible spectroscopies, mass spectrometry, magnetic moment measurements, and micro-, and X-ray crystallography analyses. The solid-state structures of complexes Fe3a and Ni3 confirmed the formation of six-coordinate bis(chelated) tridentate bound complexes. Experimental and theoretical UV-visible spectra were consistent with the presence of M(II) oxidation state complexes in the bulk state. Activation of the Fe(II), Co(II) and Ni(II) complexes with the EtAlCl2 co-catalyst afforded active species in ethylene oligomerization reactions to give predominantly C4 and C6 oligomers. The catalytic activities and product distribution of the catalyst relied significantly on the complex structure. Theoretical DFT calculations point to the stability and metal charge of the active species as the main factors controlling the resultant catalytic activities of the complexes.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the University of KwaZulu-Natal, National Research Foundation (NRF-South Africa) for providing doctorate bursary to M. Ngcobo, Kenya Education Network for funding for providing financial support to H. Nose and Mr Sizwe Zamisa for solving the structures of the complexes.References
- S. Damavandi, N. Samadieh, S. Ahmadjo, Z. Etemadinia and G. H. Zohuri, J. Eur. Polym., 2015, 64, 118–125 CrossRef CAS.
- A. P. R. Ehlert, E. M. Carvalho, D. Thiele, C. Favero, I. Vicente, K. B. Gusmao, R. Stieler, R. F. de Souza and M. O. de Souza, Catal. Today, 2017, 296, 272–276 CrossRef CAS.
- A. Ghisolfi, C. Fliedel, V. Rosa, K. Y. Manakhov and P. Braunstein, Organometallics, 2014, 33, 2523–2534 CrossRef CAS.
- W.-H. Sun, S. Jie, W. Zhang, Y. Song, H. Ma and J. Chen, Organometallics, 2006, 25, 666–677 CrossRef CAS.
- W. Hang, Y. Weidong, J. Tao, L. Binbin, X. Wenqing, M. Jianjiang and H. Youliang, J. Chin. Sci. Bull., 2002, 47, 616 Search PubMed.
- K. P. Bryliakov and A. A. Antonov, J. Organomet. Chem., 2018, 867, 55–61 CrossRef CAS.
- L. Xiao, R. Gao, M. Zhang, Y. Li, X. Cao and W.-H. Sun, Organometallics, 2009, 28, 2225–2233 CrossRef CAS.
- K. Kumar, T. Godeto and J. Darkwa, J. Organomet. Chem., 2016, 818, 137–144 CrossRef CAS.
- M. Brookhart, B. L. Small and A. M. A. Bennet, J. Am. Chem. Soc., 1998, 120, 4049–4054 CrossRef.
- S. O. Ojwach, I. A. Guzei, L. L. Benade, S. F. Mapolie and J. Darkwa, Organometallics, 2009, 28, 2127–2133 CrossRef CAS.
- G. M. Adams and A. S. Weller, Coord. Chem. Rev., 2018, 355, 150–172 CrossRef CAS.
- J. M. Benito, E. de Jesus, F. J. de La Mata, J. C. Flores, R. Gomez and P. Gomez-Sal, Organometallics, 2006, 25, 3876–3887 CrossRef CAS.
- C. Bianchini, G. Giambastiani, I. G. Rios, G. Mantovani, A. Meli and M. A. Segarra, Coord. Chem. Rev., 2006, 250, 1391–1418 CrossRef CAS.
- M. Zada, L. Guo, R. Zhang, W. Zhang, Y. Ma, G. A. Solan, Y. Sun and W.-H. Sun, Appl. Organomet. Chem., 2019, e4749 CrossRef.
- R. Gao, W.-H. Sun and C. Redshaw, Catal. Sci. Technol., 2013, 3, 1172–1179 RSC.
- Z. Wang, Q. Liu, G. A. Solan and W.-H. Sun, Coord. Chem. Rev., 2017, 350, 68–83 CrossRef CAS.
- Z. Wang, G. A. Solan, W. Zhang and W.-H. Sun, Coord. Chem. Rev., 2018, 363, 92–108 CrossRef CAS.
- P. Boulens, E. Pellier, E. Jeanneau, J. N. H. Reek, H. Olivier-Bourbigou and P.-A. R. Breuil, Organometallics, 2015, 34, 1139–1142 CrossRef CAS.
- G. J. P. Britovsek, W. Keim, S. Mecking, D. Sainz and T. Wagner, J. Am. Chem. Soc., 1993, 23, 1632–1634 Search PubMed.
- M. O. de Souza, L. R. Rodrigues, R. M. Gauvin, F. R. de Souza, H. O. Pastore, L. Gengembre, J. A. C. Ruiz, J. M. R. Gallo, T. S. Milanes and M. A. Milani, Catal. Commun., 2010, 11, 597–600 CrossRef CAS.
- A. Tavman, I. Boz and A. Seher Birteksoz, Spectrochim. Acta, Part A, 2010, 77, 199–206 CrossRef PubMed.
- W. Yang, J. Yi, Z. Ma and W.-H. Sun, Catal. Commun., 2017, 101, 40–43 CrossRef CAS.
- M. K. Ainooson, S. O. Ojwach, I. A. Guzei, L. C. Spencer and J. Darkwa, J. Organomet. Chem., 2011, 696, 1528–1535 CrossRef CAS.
- O. S. Trofymchuk, D. V. Gutsulyak, C. Quintero, M. Parvez, C. G. Daniliuc, W. E. Piers and R. S. Rojas, Organometallics, 2013, 32, 7323–7333 CrossRef CAS.
- H. Makio, H. Terao, A. Iwashita and T. Fujita, Chem. Rev., 2011, 111, 2363–2449 CrossRef CAS PubMed.
- Z. Sun, F. Huang, M. Qu, E. Yue, I. V. Oleynik, I. I. Oleynik, Y. Zeng, T. Liang, K. Li, W. Zhang and W.-H. Sun, Rsc. Adv., 2015, 5, 77913–77921 Search PubMed.
- S. Wang, W. Zhang, S. Du, S. Asuha, Z. Flisak and W.-H. Sun, J. Organomet. Chem., 2015, 798, 408–413 CrossRef CAS.
- M. Ngcobo and S. O. Ojwach, J. Organomet. Chem., 2017, 846, 33–39 CrossRef CAS.
- M. Ngcobo and S. O. Ojwach, Inorg. Chim. Acta, 2017, 467, 1–5 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 339–341 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 12, 3–8 Search PubMed.
- M. J. T. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT., 2016 Search PubMed.
- A. D. Becke, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, A38, 3098–3100 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- K. Rome and A. Mclntyre, Chromatogr. Today, 2012, 52, 52–55 Search PubMed.
- J. Wang, N. Zhang, C. Li, W. Shi and Z. Lin, Adv. Polym. Technol., 2016, 35, 21704–21712 Search PubMed.
- G. S. Nyamato, M. G. Alam, S. O. Ojwach and M. P. Akerman, J. Organomet. Chem., 2015, 783, 64–72 CrossRef CAS.
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemisty, Wiley, New York, 6th edn, 1999, p. 835 Search PubMed.
- P. W. Atkins, T. L. Overton, J. P. Rourke, M. T. Weller and F. A. Armstrong, Inorganic Chemistry, W. H. Freeman and Co., New York, 5th edn, 2010 Search PubMed.
- R. K. Chaggar, J. Fawcett and G. A. Solan, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, 462–463 CrossRef.
- L. Guo, X. Jing, S. Xiong, W. Liu, Y. Liu, Z. Liu and C. Chen, Polymers, 2016, 8, 389–401 CrossRef PubMed.
- K. Nienkemper, V. V. Kotov, G. Kehr, G. Erker and R. Fröhlich, Eur. J. Inorg. Chem., 2006, 366–379 CrossRef CAS.
- P. Ghorai, A. Chakraborty, A. Panja, T. K. Mondal and A. Saha, Rsc. Adv., 2016, 6, 36020–36030 Search PubMed.
- P. Insiti, P. Jitthiang, P. Harding, K. Chainok, R. Chotima, J. Sirirak, S. Blackwood, A. Alkas, S. G. Telfer and D. J. Harding, Polyhedron, 2016, 114, 242 CrossRef CAS.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson, R. Taylor, I. R. Cooper, S. E. Harris and A. G. Orpen, J. Chem. Inf. Comput. Sci., 2004, 44, 2133–2144 CrossRef CAS PubMed.
- M. J. M. Chambell, Coord. Chem. Rev., 1975, 15, 279 CrossRef.
- Diwaker, Spectrochim. Acta, Part A, 2014, 128, 819–829 CrossRef CAS PubMed.
- M. M. Mostafa, A. E. Hammid, M. Shallaby and A. A. El-Asmy, Trans. Mat. Chem., 1981, 6, 303 CrossRef CAS.
- J. Ghannam, T. A. Assil, T. C. Pankratz, R. L. Lord, M. Zeller and W.-T. Lee, Inorg. Chem., 2018, 57, 8307–8316 CrossRef CAS PubMed.
- A. R. Karam, E. L. Catari, F. Lopez-Linares, F. Agrifoglio, G. Agrifoglio, C. L. Albano, A. Diaz-Barrios, T. E. Lehmann, S. V. Pekerar, L. A. Albornoz, R. Atencio, T. Gonzalez, H. B. Ortega and P. Joskowics, Appl. Catal., A, 2005, 280, 165–173 CrossRef CAS.
- S. M. Nelana, K. Kumar, A. L. Guzei, T. Mahamo and J. Darkwa, J. Organomet. Chem., 2017, 323, 45–54 Search PubMed.
- J. Wang, S. Hou, L. Wang, H. Hou, N. Zhang and W. Shi, Appl. Catal., A, 2019, 55–61 CAS.
- G. S. Nyamato, S. O. Ojwach and M. P. Akerman, J. Mol. Catal. A: Chem., 2014, 394, 274–282 CrossRef CAS.
- M. Ngcobo, G. S. Nyamato and S. O. Ojwach, Mol. Catal., 2019, 478, 110590 CrossRef.
- J. Yu, X. Hu, Y. Zeng, L. Zhang, C. Ni, X. Hao and W.-H. Sun, New J. Chem., 2011, 35, 178–183 RSC.
- G. A. Kinunda, Tanz. J. Sci., 2018, 44, 45–63 Search PubMed.
- G. E. Bekmukhamedov, A. V. Sukhov, A. M. Kuchaev and D. G. Yakhvarov, Catalysts, 2020, 10, 498–518 CrossRef CAS.
- L. K. Johnson, S. Mecking and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 267 CrossRef CAS.
- I. Pierro, G. Zanchin, E. Parisini, J. Marti-Rujas, M. Canetti, G. Ricci, F. Bertini and G. Leone, Macromelecules, 2018, 51, 801–814 CrossRef CAS.
- M. Ngcobo and S. O. Ojwach, Mol. Catal., 2021, 508, 111583 CrossRef CAS.
- G. S. Nyamato, S. O. Ojwach and M. P. Akerman, Dalton Trans., 2016, 45, 3407–3416 RSC.
- L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414 CrossRef CAS.
- P. O. Ongoma and D. Jaganyi, Dalton Trans., 2012, 44, 10724–10730 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional spectroscopy and mass spectral data (NMR, FT-IR and LC–MS spectroscopy, GC and GC–MS) in addition to these catalysis data. Crystallographic data of compounds Fe3a and Ni3 in this article can be accessed via Cambridge Crystallographic Data Centre respectively. CCDC 2126949 (Fe3a) and 2126951 (Ni3). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1nj06065a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |