
Microstructural transition of poly(vinyl alcohol)-based aerogels in the presence of interpolymer complexes†
Chao
Wang
 ,
Guolong
Sang
,
Minghao
Yang
,
Ge
He
,
Yedong
Rong
and
Jinlong
Yang
*
,
Guolong
Sang
,
Minghao
Yang
,
Ge
He
,
Yedong
Rong
and
Jinlong
Yang
*
State Key Lab of New Ceramics and Fine Processing, School of Materials Science and Engineering, Tsinghua University, Beijing 100084, China. E-mail: jlyang@mail.tsinghua.edu.cn
First published on 18th November 2021
Abstract
Interpolymer interactions play a vital role of determining the microstructure and properties of polymer aerogels. In terms of poly(vinyl alcohol) (PVA) aerogels, the conformation and behaviour of polymer chains in semidilute aqueous solutions make considerable contribution to the evolution of polymer matrixes during preparation. In this work, we investigated the microstructure of a group of PVA-based aerogels from the view of interactions between polymer chains. In PVA/poly(ethylene oxide) (PEO) aerogels, disordered microstructures were formed with the increase in PEO amount, owing to the existence of weak hydrogen bonds and some existing crystalline domains of PVA and PEO. PVA/poly(acrylic acid) (PAA) samples, presented straight stripe-shaped structures, because of the strong interactions between PVA and PAA. In PVA/carboxyl methyl chitosan (CM-CTS) aerogels, high density of stripe microstructures were observed, due to the rigid conformation of CM-CTS in water and few hydrogen bonds existed between PVA and CM-CTS. PVA/PAA aerogels possess compressive strength as high as 0.94 MPa. The porosity of the PVA/PEO aerogel reached 98.6%. Factors such as chain length and pH value of the precursor solutions on the microstructure of aerogels were also investigated. A possible forming mechanism was put forward from the view of interpolymer interactions, accordingly. Such way of tuning the microstructure of PVA-based aerogels may provide a new understanding with respect to the microstructure of polymer aerogels and pave new ways for the practical use of this versatile light material.
Introduction
Structures at the molecular level dictate the properties of materials on a macro-scale. Desirable properties can be achieved via controllable preparation of particular materials. Aerogel, as a kind of light material, was first synthesized by sol–gel chemistry in the last century by Kistler.1 This unique material composed of either inorganic or polymeric networks exhibits high specific surface area, high porosity and extremely low density. The ease of fabricating aerogels with tuneable functionalities made this versatile material adopt a variety of applications such as thermal insulations,2 flame retardants,3,4 catalysts,5 energy storage,6 gas adsorption,7 and oil–water separation.8The microstructure of aerogels, however, determines the functionalities and mechanical properties of the final products. The fabricating process of aerogels generally contributes to their microstructures to a large extent. Freeze-drying is a widely used strategy for preparing polymer networks cross-linked by physical interactions between polymer chains. In the freezing process, the ice crystal grows in the amorphous regions of polymer chains, squeezing out polymer-rich phases to form physical cross-linking points, subsequently. A highly porous structure will thus form during the thawing process. This principle was adopted in preparing aerogels made up of a series of natural and synthetic polymers such as PVA, cellulose,9 xanthan,10 alginates,11 and amylopectin.12
Several decades ago, principles in the forming process of PVA gels by a freeze–thaw method were first studied by Ziabicki13 and Peppas.14 Since then, scientists in this field began to study the behaviour of PVA chains in both aqueous solutions and frozen states.15
PVA is a macromolecule that contains hydroxyl group as side groups. Multiple interactions including intra-molecular, inter-molecular and polymer-water interactions may occur in a PVA aqueous solution. In a dynamical view, chains will contact with each other when the concentration of PVA reaches about 3%. A peak of slow relaxation mode in dynamic light scattering (DLS) measurement will thus present.16 For the freeze–thaw method of making PVA aerogels, an enough concentrated precursor solution is usually used. When the first freeze step is applied, physical knots will form in the polymer-rich phase to support the whole network, which will then develop into isolated crystalline regions. The degree of crystalline will reach over 50% after three cycles of freeze–thaw. Further increasing the cycle times will not enhance the degree of crystallinity but only reinforce the crystal that already exists.17 The final PVA aerogel presents a parallel layered microstructure over the whole material. The microstructure rapidly changes with the concentration and molecular weight of PVA in solutions.18
Although homogenous PVA aerogels were invented and developed, aerogels composed of PVA and other functional polymers also play crucial roles in numerous functional materials. Non-covalent interactions between the polymer chains play essential roles dictating the properties of these PVA-based aerogels.
Plenty of work study the property of PVA-based aerogels made up of multiple components.8,19–21 Baglioni et al. studied the morphology of PVA/gelatin networks. The final aerogel showed irregular pores distributed inside the sample. They owed this phenomenon to the result of phase separation of the system and capillary force on a sub-micrometer scale.22 Jiang et al. reported a PVA/carbon nanotube aerogel membrane for the separation of oil-in-water emulsion. The membrane possesses a porosity up to 95%. The nano-sized pore inside made it exhibit high rejection for the oil-in-water emulsion with an ultrahigh permeation flux.20
Although the previous studies focused on the properties of PVA-based aerogel materials themselves, the intermolecular behaviours of polymer chains in precursor solutions are rare to be studied. It is indeed an important session prior to the freezing process of the PVA-based aerogel. The polymer chains will interact with each other in such semidilute solutions to form hierarchical morphologies and will accordingly have a significant impact on the properties of the resulting aerogels.
The term interpolymer complex (IPC) stands for a kind of aggregates formed by two types of polymers with non-covalent interactions.23–25 Hydrogen bonded IPC, for example, involved pairs of polymer chains with proton-donator/acceptor functional groups. Factors such as molecular weight, hydrophobic interaction and environmental conditions also play crucial roles in the formation of IPCs.26–28 The structure of IPC, either in dilute solutions or in bulk, was studied by means of numerous techniques, such as SLS,29–31 DLS,32–34 UV-Vis,35 SANS,36–38 and rheology.39
In this study, we explored the microstructures of aerogels made of PVA with three common polymers, PEO, PAA and CM-CTS from the view of both the solution state and the bulk material. The hydrogen-bonded interpolymer interactions of the aerogels were investigated by DLS and rheology from the perspective of polymer chain dynamic behaviours. ATR-FTIR spectroscopy was performed to study the hydrogen bonds among PVA and other polymers in the aerogels. Combined with necessary mechanisms provided, approaches for the formation of PVA-based aerogels with polymers in different properties were put forward.
Experimental
Materials
Poly(vinyl alcohol) (PVA) with a polymerization degree of 1700 and alcoholysis degree of 99% was purchased from Sinopharm Chemical Reagent Co. Ltd. Polyacrylic acid (PAA) with a molecular weight of 2.4 × 105 g mol−1 was obtained from J&K Chemical Ltd. Carboxymethyl chitosan (CM-CTS) with a molecular weight of 2 × 104 g mol−1 was purchased from Macklin reagent Ltd with an average degree of substitution (DS) of 92.4% (the specific calculation of the DS is shown in ESI† and Fig. S3). Poly(ethylene oxide) (PEO) with a molecular weight of 2 × 103 g mol−1, 6 × 103 g mol−1 and 2 × 104 g mol−1 was purchased from Macklin reagent Ltd. NaOH was purchased from Sinopharm Chemical Reagent Co. Ltd. Deionized water was generated using an Ulupure water purifier with an electrical resistivity of 18.2 MΩ. All reagents were used as received without further purification.Preparation of the polymer aerogels
The PVA-based aerogels were prepared by the classical freeze–thaw method. To ensure that the polymer chains were completely dissolved, different concentrations of polymer solutions were used, namely, 1% PEO-20k solution, 5% PAA-240k solution and 2.5% CM-CTS-20k solution. However, when higher concentrations of PEO and CM-CTS solutions were mixed with the PVA solution, precipitation will be formed due to the fast complexation of the polymer chains, which would strongly affect the formation of cross-linking points of PVA during the freeze–thawing process.40The preparation routine of the PVA/PEO aerogel is as follows: a stock aqueous solution of PVA with a concentration of 5% was made for use. Then, 1% aqueous solution of PEO with molecular weights of 2 kDa, 6 kDa and 20 kDa was made for further use. Following this, 0.2 ml, 0.4 ml, 0.6 ml and 0,8 ml of PEO solution was mixed with 0.8 ml, 0.6 ml, 0.4 ml and 0.2 ml of PVA solution, respectively. The mixed solutions were gently stirred using a glass stick to ensure the solutions were well mixed. The as-prepared solutions were poured into a 24-well plate. The samples were frozen at −24 °C in a fridge overnight. Then, the samples were unfrozen at room temperature for 6 h. After 2 cycles of freeze–thaw, the final products were obtained by freeze-drying.
The PVA/PAA and PVA/CM-CTS aerogel samples were prepared by a similar method.
The abbreviations of samples were noted according to the weight percentage of the second component polymer solution. For example, E20 stands for the sample in which there are 20% of PEO solution and 80% of PVA solution. A20 stands for the sample in which there are 20% of PAA solution and 80% of PVA solution. C20 stands for the sample in which there are 20% of CM-CTS solution and 80% of PVA solution.
Adjusting the pH value of PVA/PAA precursor solutions
The pH value of PVA/PAA precursor solutions was adjusted by adding 0.1 M NaOH aqueous solution dropwise, followed by stirring the solution gently. Considering the relaxation process of polymer chains, the pH of the samples was re-measured after being set aside for 2 h.Measuring the porosity of polymer aerogels
The porosity of the obtained polymer aerogels (P) was measured by calculating using the following equations:
In the equations, ρobj refers to the observed density, which was calculated by dividing the object mass with the object volume. ρmat refers to the density of the polymer itself, which is the weighted average density of the two components. vobj refers to the volume of the samples. m1 refers to the total mass of PVA and m2 to the total mass of the second type of polymer.
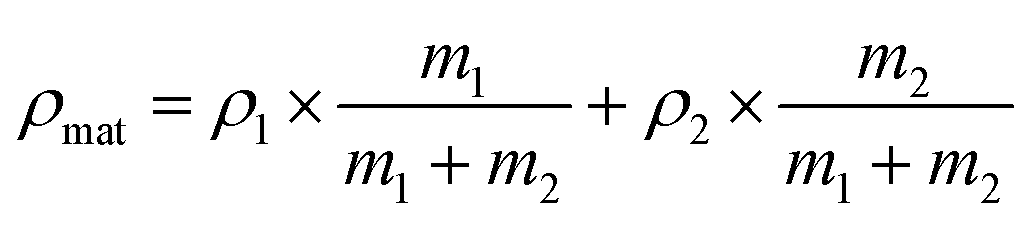
Characterization
Scanning electron microscopic (SEM) images with high magnification were acquired using a Merlin Compact instrument (Zeiss GmbH, Germany) at an accelerating voltage of 15 kV. The images with low magnification were acquired using a Hitachi SU8000 instrument working at an accelerating voltage of 30 kV. The aerogel samples were fixed on specimen mounts with adhesive tapes and then coated with platinum using a sputter coater (Leica, EM ACE600). Attenuated total reflection Fourier transform infrared (ATR-FTIR) spectra were obtained using a Tensor X70 Fourier spectrophotometer (Bruker, Germany) equipped with an ATR-FTIR accessary. The measured data ranged from 400 cm−1 to 4000 cm−1. The viscosity property of samples was measured using a Malvern-Kinexus instrument at 25 °C. Linear shearing mode was used at a shearing rate increased from 0 to 1000 rad s−1 within 2 min. The compressive strength was measured using a Shimadzu-AG2000 universal testing machine with a compressive speed of 2 mm min−1. The bottom diameter of aerogel samples was 9 mm. The height of samples was measured using a Vernier calliper and then input into the measuring software. The pH values were measured using a Mettler Toledo FE28 pH meter with a resolution value of 0.01 at room temperature. The X-ray diffraction (XRD) patterns were collected using a D8 Discover instrument with Cu Kα radiation produced in Bruker Company. The measured signals were collected from the 2 theta range of 10° to 80°.The dynamic light scattering measurement was carried out using a Malvern ZS Nanosizer. A 4 mW laser with a wavelength at 633 nm was used as an incident beam. The scattering angle was fixed at 173°. The refractive indices of PVA, PEO, PAA and CM-CTS were 1.4839, 1.458, 1.508 and 1.347. The scattering intensity and its fluctuations were recorded. The intensity autocorrelation function G(2)(τ) can be expressed as follows:

The normalized intensity autocorrelation function g(2)(τ) was connected to electric field time correlation function g(1)(τ) with the Seigert relation:
| g(2)(τ) = 1 + β|g(1)(τ)|2 |
Meanwhile, the average hydrodynamic radius (Rh) was obtained using the Einstein–Stokes equation:
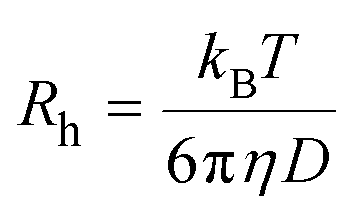
Results and discussion
Characterization of aerogel bulk materials
It is worth mentioning that the PVA-based aerogels in this work were prepared by a freeze–thaw method as pure PVA aerogels. Homogenous PEO, PAA and CT-CMS aerogels can hardly be made simply by this method due to the structure of their backbones. If homogenous aerogels of these materials were made with other strategies, it will be hard to contrast as parallels. Hence, we did not make those homogenous aerogels in this work.The profiles of the aerogel samples are listed in Table 1. To give better reference to understanding the interaction between polymer chains, the molar ratios of functional groups of each polymer are listed. However, the concentrations of polymer solutions we used were different. The densities of the obtained aerogel samples are listed as well. The difference in the concentration of polymer solutions made pronounced effects on the density of PVA/PEO samples. PVA/CT-CMS aerogels were also in different densities as the weight concentration of the CT-CMS solution was 2.5%. There was almost no impact on the density of PVA/PAA samples, as they were prepared in the same weight concentration.
PEO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PVA :PVA |
PAA:PVA |
CT-CMS:PVA |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mass ratio of polymers (%) | 20 | 40 | 60 | 80 | 20 | 40 | 60 | 80 | 20 | 40 | 60 | 80 |
| Molar ratio of monomer units | 1:20 |
2:15 |
3:10 |
4:5 |
11:72 |
11:27 |
11:12 |
22:9 |
1:50 |
4:75 |
3:25 |
8:25 |
| Density (10−3 g cm−3) | 71.6 | 65.1 | 50.6 | 31.9 | 91.2 | 93.9 | 96.0 | 97.9 | 90.8 | 83.8 | 76.9 | 69.4 |
 | ||
| Fig. 1 SEM images of the microstructure of PVA-based aerogels. (a–d) PVA/PEO aerogels with PEO20k, E20, E40, E60 and E80. Inset of (a) is the magnified image of the pore wall in the E20 sample. (e–h) PVA/PAA aerogels with PAA25k, A20, A40, A60 and A80. Inset of (g) is the cross section of the stripe structure in A60 (i–l) PVA/CMCTS aerogels. C20, C40, C60 and C80. | ||
In Fig. 1a, a pore structure with oriented edges was observed in the 20kE20 sample. The porous channel structure domains (marked with dashed line) showed some similarity to the microstructure of PVA aerogels.18 The diameter of the pores was about 50 μm and some fibres adhering on the pore walls can be found (inset of Fig. 1a, the fibre character was highlighted by dashed lines). This phenomenon was also reported by Baglioni et al.22 In the 20kE40 sample (Fig. 1b), regular porous structures containing larger pores with a diameter of 80–100 μm were found, with some fibrillar structures existing at the edge of the pores. This morphological transition revealed a distinct change at the scale of the polymer chains. When the ratio of the PEO solution reached 60%, the microstructure of the prepared aerogel became disordered. Curly patterns were randomly distributed inside the aerogel. The situation became more pronounced in the sample 20kE80 (the molar ratio of functional groups approaching 1:1). The scale of these patterns got smaller as well. In this work, although the weight concentration of PEO solutions was 1% (much lower than the concentration of PVA), the microstructural change brought about by the dilution of the PVA solution was shuttle if we compare the images of PVA/PEO aerogels with the images in pure PVA aerogel with different concentrations.18 Hence, we may deduce that the introduction of PEO destroyed the conformational arrangement of PVA chains at the molecular level. As we know that PVA is a semi-crystalline polymer. The intramolecular and intermolecular hydrogen bonds were distributed among the PVA matrix.41 A high ratio of PEO will hence interact with PVA easily and replace the forces between PVA themselves. In addition, PEO possesses a high ability to adsorb water molecules. On average, one repeat unit of PEO forms hydrogen bonds with 2–3 water molecules.42 Those bounded water will also modify the microstructure of aerogels as the free water sublimating from the polymer network.42–45
In terms of PVA/PAA aerogels, clear channel-like and pore structures are shown in Fig. 1e and f, which were similar to the case of PVA/PEO. Meanwhile, the fibrillar structure cannot be found neither at pore walls nor at pore edges. These morphological characters may be highly related with the interaction between PVA and PAA, which has been reported elsewhere.46,47 When forming IPC with PVA, PAA chains will destroy some hydrogen bonds between PVA chains and form new ones with them simultaneously. A synergism of those intra- and intermolecular hydrogen bonds will make the network strong and compact. When making the SEM sample, the cross-section will be neat and the pore walls will be smooth, accordingly. Fig. 1g shows the large pore structures formed. The inset of Fig. 1g shows hierarchical pores inside the stripe structure (the hierarchical character was highlighted by dashed lines). Baglioni et al. reported that when increasing the freeze–thaw cycles, pores tend to merge together, resulting in a disorder structure of the microstructure of aerogels.48 In the case of PVA/PAA aerogels, however, a more rigid polymer matrix with strong hydrogen bonds cannot be destroyed during repeated freeze–thaw cycles, leaving hierarchical structures inside the material. Interestingly, in the microstructure of A80, some straight stripe structures were formed inside the aerogel matrix. This revealed a fast and unidirectional growing of the ice crystals, which indicated a low viscosity of the solution and weak interactions between PAA and water molecules. Fu-sheng et al. reported that when the pH of the PVA/PAA interpolymer complex solution fall at 11.4, the obtained polymer film showed excellent ability of water absorbance owing to the deprotonation of –COOH groups. When the pH of the interpolymer complex solution fall at 6.5 or 2.7, the poor ability of water absorbance of the films presented, which was due to both the protonation of –COOH and a more dense complexation of PVA chains.40 In the case of A80 precursor solutions, the pH value was 3.16. The –COOH will hence be protonated and the interaction between –COOH of PAA and –OH of water will thus be weak.
In Fig. 1i–k, a high density of stripe microstructures is presented, corresponding to PVA/CM-CTS samples. The stripes were oriental distributed with a few of brunches on their edges. The length of these strips was in the scale of sub-micrometers. Space can be found between the stripes. A zoom-out SEM image is shown in Fig. S1 (ESI†). We know that the pKa value of CM-CTS is about 5.0, which depends on the deprotonation/protonation of the –NH2, –NH– and –COOH groups on their backbones. The pH of the precursor solution of these samples was above 9.5 (Table S1, ESI†). In this situation, the –COOH of CM-CTS will be deprotonated. The resulting –COO− group will be the H-acceptor rather than H-donor of the hydrogen bond between PVA and CM-CTS. The intensity of these hydrogen bonds will thus be weaker. In terms of the sample C80, curly strips presented connected with the surrounding structures. This non-oriented microstructure may result from the decreased amount and concentration of PVA (as the different concentrations between PVA and CM-CTS solutions). The physical cross-link points were thus decreased, leading to a lack of ordered structure of this sample.
Fig. 2a shows the photograph of the aerogel blocks of E40, A40 and C40, respectively. All of the aerogel samples were light in weight. It was quite stable to put them on a blade of grass.
 | ||
| Fig. 2 (a) Photograph of PVA-based aerogels on a blade of grass. (b) Porosity of PVA/PEO, PVA/PAA and PVA/CM-CTS aerogels. (c) XRD curves of PVA/PEO, PVA/PAA and PVA/CM-CTS aerogels. (d) Mechanical properties of PVA-based aerogels. | ||
 | ||
| Fig. 3 ATR-FTIR curves of the PVA/PEO, PVA/PAA and PVA/CMCTS aerogel samples. (a) PVA/PEO aerogels, (b) PVA/PAA aerogels, (c)PVA/CMCTS aerogels and (d) powders of PVA, PEO, PAA and CM-CTS. | ||
In Fig. 3b, the curves of PVA/PAA aerogels are shown. In the PVA–PAA mix curve, two distinct peaks at 3250 cm−1 and 3393 cm−1 correspond to the free –OH of PAA. However, in ATR-FTIR data of aerogel samples, those two peaks developed into one broad peak, which is a sign of hydrogen bonds forming between PAA and PVA.53,57 Meanwhile, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration peak of PAA at 1699 cm−1 shifted to 1709 cm−1 in all the four PVA/PAA samples, indicating that strong hydrogen bonds formed between them.58 The peak at 1096 cm−1 in these curves corresponding to the stretching vibration of –OH of PVA molecules shifted to 1098 cm−1. This slight shifting may also be sign of IPC forming between PVA and PAA. In the broad band at 3000 to 3600 cm−1, the peak at 3306 cm−1 of A20 shift slightly to higher wavenumbers in A40 and A60 samples. Consulting the molar ratio of functional groups calculated in Table 1, we may find that this ratio in A60 approached the optimal functional group molar ratio of PAA to PVA (1:1). Intense hydrogen bonds will thus form, accordingly. A weak signal of this peak is shown in the A80 sample, owing to a high proportion of PAA in the mixture.58
O stretching vibration peak of PAA at 1699 cm−1 shifted to 1709 cm−1 in all the four PVA/PAA samples, indicating that strong hydrogen bonds formed between them.58 The peak at 1096 cm−1 in these curves corresponding to the stretching vibration of –OH of PVA molecules shifted to 1098 cm−1. This slight shifting may also be sign of IPC forming between PVA and PAA. In the broad band at 3000 to 3600 cm−1, the peak at 3306 cm−1 of A20 shift slightly to higher wavenumbers in A40 and A60 samples. Consulting the molar ratio of functional groups calculated in Table 1, we may find that this ratio in A60 approached the optimal functional group molar ratio of PAA to PVA (1:1). Intense hydrogen bonds will thus form, accordingly. A weak signal of this peak is shown in the A80 sample, owing to a high proportion of PAA in the mixture.58
Fig. 3c reveals the data of PVA/CM-CTS. The twin peak at 3400 cm−1 and 3500 cm−1 corresponds to the stretching vibration of primary amino group. The peak at 1590 cm−1 referred to the bending vibration of N–H.59 Almost no shifts were observed in the three amino peak at C20–C80 samples, indicating that few interactions exist between –NH– groups of CM-CTS and PVA. The peak at 1426 cm−1, corresponding to the symmetric stretching vibration of COO−, shifted to 1412 cm−1 in all of the aerogel samples. That should be a sign of the deprotonated carboxyl groups forming hydrogen bonds with –OH on PVA. Additionally, the broad peak at 3301 cm−1 became weaker as the amount of CT-CMS increased, which may be owing to the interaction between –OH on PVA and secondary –NH– group of CT-CMS.60
The ATR-FTIR curves of homogenous PVA, PEO, PAA and CM-CTS are showed in Fig. 3d. In the curve of PVA, there is no stretching signal of CO at 1735–1750 cm−1, indicating a fully alcoholise of PVA backbone.46 The broad peak at 3200–3570 cm−1 corresponds to the stretching mode of associated –OH. The double peak at 2840–3000 cm−1 corresponds to the stretching vibration of C–H bonds. The sharp peak at 998–1154 cm−1 represents the stretching vibration of C–O–C bonds. The peak at 1425 cm−1 corresponds to the bending mode of the CH2 group. In the curve of PEO, the peak at 2890 cm−1 referred to the methylene group on the PEO chains. The sharp peak at 1100 cm−1 corresponds to the stretching mode of C–O–C bonds. The surrounding peaks ranging from 839 cm−1 to 1464 cm−1 are assigned to the stretching of C–O–C as well. Because the infrared signals were sensitive to chain conformations of PEO and even the way of preparing the samples for measuring.61 In the curve of PAA, the sharp peak at 1709 cm−1 corresponds to the CO of the PAA. The broad band at 2750–3350 cm−1 represents the hydrogen bonds between the –COOH of two PAA chains. For the data of CM-CTS, the peak at 3490 cm−1 and 3415 cm−1 corresponds to the N–H extension vibration mode. The peaks at 1596 cm−1 and 1423 cm−1 correspond to the asymmetric and symmetric stretching vibration of CO bonds.
Interpolymer interactions in precursor solutions
The final morphology of aerogel microstructure was determined by the dynamic of polymer chains during the whole preparation process of aerogels. The IPC formed with the interaction between polymer chains dictates the morphology of polymer aggregates. We thus investigated the behaviours of polymer chains in the precursor solution of the aerogels. | ||
| Fig. 4 Viscosity and DLS measurement of the precursor solutions. (a–c) Viscosity profile of PVA/PEO, PVA/PAA and PVA/CM-CTS precursor solutions. (d–f) DLS data of PVA/PEO, PVA/PAA and PVA/CM-CTS precursor solutions. | ||
In the case of the PVA/PEO system (Fig. 4a), the estimated zero-shear viscosity decreased with the addition of PEO components. On the one hand, we can figure out that PEO chains weaken the intermolecular force between PVA themselves. The existed compact domains were loosened and the chains were oriented as the shearing rates increased. On the other hand, as the concentration of PEO was 1%, the addition of PEO solutions decreased the whole concentration of the mixed solution, making the viscosity become lower. We may also see a shear thinning phenomenon of E80 at low shearing rate. It is a sign of dissociation of the entangled polymer clusters.65 For the semidilute solution of PEO20k, the chain entanglements were often observed by DLS as a slow relaxation mode in numerous previous studies.66–69 Those chain entanglements of PEO will also retard the formation of inter- and intra-hydrogen bonds of PVA. In addition, although the pH values of PVA/PEO solutions were under acidic conditions (Table S1, ESI†), no significant influences were observed on the conformation of the PEO and PVA chains, as the alcoholysis degree of PVA was 99%.
In Fig. 4b, we found that the viscosities of A20, A40 and A60 were much higher than that of pure PVA. The highest viscosity value reached 0.08 Pa s, almost twice of the PVA solution itself. This was also noticeable when preparing the solutions. As we know that PVA and PAA formed strong IPCs. The optimal molar ratio of monomer units for IPC of PVA/PAA is 1:1. In general, the viscosity of the IPC system will become higher at optimal molar ratios owing to the synergetic effect of non-covalent bonds, according to the reports of other researchers.25,39,46 However, the PVA/PAA solution with a molar ratio approaching 1:1 (A60) did not present the highest viscosity. The A20 solution possesses the highest viscosity, which is an interesting result (the same trend can also be found in Fig. 1e–h). The reason may be supposed to be that PVA presents intermolecular hydrogen bonds, which is different from most of other polymers. The added PAA molecules formed interpolymer hydrogen bonds with PVA and destroyed the existing PVA intermolecular hydrogen bonds. Thus, the highest viscosity existed in the A20 solution instead of the A60 solution. Meanwhile, chain entanglement also exists as low PAA percentage as can be seen from the shearing thinning region of the A20 and A60 curves. However, the pH value of these PVA/PAA solutions were below the pKa value of the PVA solution (pKa = 4.570), and protonated –COOH groups presented at all the side groups. Hence, no significant changes were observed on the chain behaviours of PVA and PAA in this range of pH values.
In terms of PVA/CM-CTS polymer solutions, the viscosities of all the mixed component samples were not higher than that of the PVA solution. The shearing thinning behaviour was detected among all the mixed solutions. As we know that the pKa of CM-CTS is about 5.0, it depends on the degree of substitution. The pH value of the aqueous solution of PVA/CM-CTS was above 9.5 as mentioned before, higher than the pKa of CM-CTS. The carboxyl groups were deprotonated, accordingly. The chains will adopt a rigid conformation in the aqueous solution as the repulsive forces between the negative charged side groups. Therefore, at the onset of the shearing process, the CM-CTS chains will be oriented and may disturb the entanglements of PVA chains. The shearing thinning regions were thus formed. The viscosity of PVA/CM-CTS at medium and high shearing rates fell at 0.02 Pa s to 0.03 Pa s, indicating some non-covalent interactions with low intensities. These results were complementary to the previous findings.
In the case of a semidilute solution of PVA/PAA, we found that the signal of isolate PAA chains existed at all sample solutions, despite the ratio of PVA and PAA, with an Rh around 4.9 nm to 7.8 nm (Fig. 4e). The molecular weight of PAA was much higher than PVA, and no isolated PVA chains existed after forming IPCs with PAA. Meanwhile, the rising trend of the second relaxation mode indicates the forming of IPCs between PVA and PAA chains. The optimal molar ratio of functional groups of PVA/PAA is 1:1, which corresponded to the sample A60. The rising size of IPCs in this group revealed this fact. The sizes of slow modes in A20 and A40 were much smaller than those of A60 and A80. This evidence corresponded to the viscosity results of A20 and A40. Aggregates with lower sizes make the viscosity of the liquid system higher. However, the slow mode of a pure PVA solution moved to much lower sizes (labelled by the arrow). We may thus suppose that PAA chains unfolded the PVA clusters by forming IPCs with them. This notable phenomenon indicated a strong hydrogen bonding between PVA and PAA owing to the H donor group of –COOH and the H-acceptor group of –OH.
The DLS results of PVA/CM-CTS showed two peaks in each sample. As mentioned before, the pH of the solutions were 9.5–9.9. Under this condition, the side chains of CM-CTS were negatively charged. The strong electrostatic interactions between the side chain groups make the backbone exhibit an elongated conformation. The Rh value of the single chain was hence 392 nm. Kalliola et al. reported that the diameter of CM-CTS in water will dramatically increase as the pH value goes higher than 6.5.71 It is worth noting that this size is not the real size of the chain. It is the radius of an equivalent sphere wrapped outside the chain. In terms of the PVA/CM-CTS sample solutions, the fast mode referred to the size of IPCs between them and the slow mode corresponds to the entanglement of polymer chains. We can see that the fast mode of C60 was 124.5 nm. The value was slightly decreased in the C80 sample. Meanwhile, as shown, the decreasing trend of slow mode in the PVA/CM-CTS solution was not as notable as that of PVA/PEO. Apart from the dilution effect of PVA solutions, the hydrogen bonding between –NH, –COO− of CM-CTS and –OH of PVA may make some contribution to making up for the size decrease of PVA chain clusters as the total concentration of PVA decreased.
Aerogel microstructures influenced by the pH of the system
The behaviour of interpolymer complexes in water was highly dependent on the pH value. H-donor groups will dissociate protons at a pH above pKa and will gain protons at a pH below pKa. This will have an impact on the formation of hydrogen bonds between polymer chains. Thus, we studied the pH effect on the microstructure of PVA/PAA aerogel samples.Fig. S2a and b (ESI†) show the chain dynamic profile of A40 and A60 at different pH values. It is obvious that all the peaks moved to higher values of sizes. The reason was not hard to speculate. PAA is an acid with partially dissociated –COOH groups. When evaluating the pH of the system, more negative charges will present at the side chains, leading to the stretched conformation of the chain resulting from the repelling forces of –COO− groups.
Fig. 5a–d show the morphology of A40 and A60 at pH = 4.2 and 6.0, respectively. Microstructures of the aerogels underwent considerable changes comparing Fig. 5a and b or Fig. 5c and d. Oriented structures were notable when the precursor solution was in a lower pH, while those structures were displaced by curling stripes when the pH of the precursor solution was higher than the pKa value of PAA. This phenomenon was in accordance with our previous analysis based on IPC. Meanwhile, when the intensity of hydrogen bonds decreased (in the case of sample with pH = 6.0), the ice crystal gained more spaces to expand their sizes, which will be another reason for the formation of the loosely packed microstructures.
 | ||
| Fig. 5 (a–d) SEM graphs of A40 and A60 aerogels with pH values of 4.2 (a and c) and 6.0 (b and d). (e) ATR-FTIR data of the A40 and A60 aerogels. (f) Porosity of A40 and A60 at pH values of 3.4, 4.2 and 6.0. | ||
Fig. 5e shows the ATR-FTIR curves of the A40 and A60 aerogels made in pH values higher than the original ones. It is obvious that the broad peaks from 3000 to 3600 cm−1 corresponding to the stretching vibration of –OH on PVA became stronger when the sample was made in a higher pH value. This may be due to the dissociation of –COOH and –OH, resulting in an exposure of –OH without hydrogen bonds. The peaks at 1705 cm−1 became weaker significantly and were shifted back to lower wavenumbers (refer to Fig. 3b), which was a sign of –COOH transferred to –COO− in a higher pH value. The newly developed peak at 1552 cm−1 corresponds to a stretching vibration of carboxyl salts, namely, deprotonated carboxyl groups. All of the evidence of ATR-FTIR indicated that the hydrogen bonds between PVA and PAA can be destroyed by tuning the pH value and thus dictate the microstructure of the obtained aerogels.
We measured the porosity data on samples prepared at pH values of 3.4, 4.2 and 6.0 (Fig. 5f). In terms of A40 and A60 formed at different pH values, the porosity went up with the increase in pH value. For A40, the average porosity increased from 94.4% to 96.5%, while the average porosity of A60 increased from 94.7% to 96.2%. The rising of porosity is mainly due to the decrease in hydrogen bonds and dissociation of IPCs. A microstructure with more flexible macro-phase domains will thus be formed as the corresponding SEM images revealed.
Aerogel microstructures influenced by molecular weight
Fig. 6 shows the microstructure of PVA/PEO aerogels dependent on the PEO molecular weight of 2 × 103 g mol−1 and 6 × 103 g mol−1 at different polymer solution ratios. Regular oriented stripe structures were presented in sample 2kE20, which possess some similarity to the microstructure of pure PVA aerogels.18 As the ratio of PEO increased, the stripes became thinner with more fibres on their edges (Fig. 6b). When the percentage of the PEO solution reached 60%, disordered structures became dominant. The stripes were not as straight as before (Fig. 6c). The edge of the stripes became thicker than 2kE20 and 2kE40. In aerogel sample 2kE80, curly stripes were in the whole vision of the SEM image, with some of the brunched structure presented. Fig. 6i shows the DLS data of the PVA/PEO2k mixed solution. The hydrodynamic radius of 2kPEO chain in water was 2.7 nm, much smaller than that of the PVA chain. In the solution of 2kE20, there were almost no changes in Rh of the PVA (or IPC of PVA/PEO2k). The largest size of IPC of PVA/PEO2k was only 37.3 nm, much smaller than the case of PVA/PEO20k.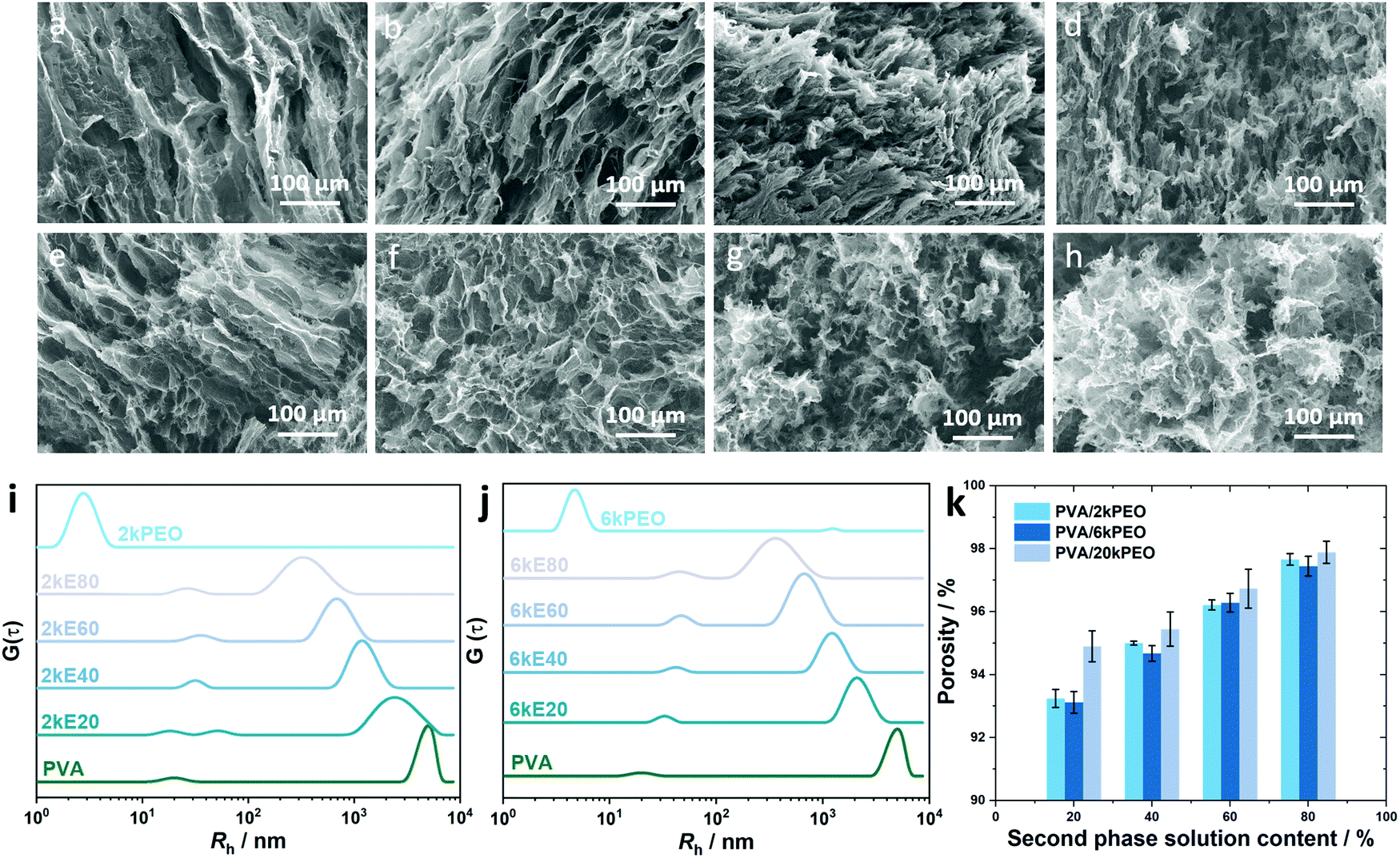 | ||
| Fig. 6 (a–h) SEM images of cross-section in PVA/PEO2k and PVA/PEO6k aerogels. (i and j) DLS data of PVA/PEO2k and PVA/PEO6k aerogel precursor solutions. (k) Porosity of the PVA/PEO aerogels with PEO molecular weights of 2 kDa, 6 kDa and 20 kDa. | ||
Fig. 6e–h show the microstructure of 6kE20–6kE80. As the microstructure of 6kE20, oriented porous stripes were shown. Interestingly, in Fig. 6f, porous structures presented in the whole vision with a pore diameter of 80 μm, which was similar to the case of 20kE40 (Fig. 1b). In Fig. 6g, curly structures began to show with a small portion of brunched structures. In the microstructure of 6kE80, highly brunched flower-like patterns presented. Meanwhile, we may see from the DLS data of PVA/PEO6k (Fig. 6j) that the Rh value of a single PEO6k chain was 4.8 nm. The largest size of IPC of PVA/PEO6k revealed from the first relaxation mode was 44.7 nm, corresponded to the sample 6kE60 and 6kE80.
Similarities can be easily found comparing the SEM images of PVA with PEO of different molecular weights. However, the chain length played an important role on influencing the resulting microstructure of those aerogels as well. PEO2k possesses the shortest chain. Hence, they exhibit poor ability to form IPC with PVA chains. That's the reason for the strand morphology of 2kE60, while E60 with other PEO molecular weights exhibiting disordered microstructures. PEO20k possesses a chain of 10 nm in Rh, half length of the PVA chains. The flexible PEO20k chain interspersed in PVA and made significant changes in the microstructure of PVA aerogels rather than PEO2k.
The porosity of these two groups of samples was also recorded. Compared with the data we measured using PVA/20kPEO samples, the porosities were much lower at E20 samples of PVA aerogels containing PEO2k (92.9%) and PEO6k (92.8%) molecules. This phenomenon may also result from the low capability of forming IPC between PEO2k and PVA. A large number of PEO chains were isolated in the solution, making few contributions to the microstructure of the aerogels. As the amount of PEO increased, the effect of molecular weight towards the IPC has weakened. The porosity went up to almost 98%.
Forming mechanism of aerogel microstructures from polymer solutions
To date, the forming mechanism of the PVA aerogel in the process of freeze–thaw has been illustrated extensively. The stranded microstructure was obtained attributed to many times of freeze–thaw cycles. In this work, freezing operation was done in the refrigerator with a 24-well board. The freezing process will begin from the top of the samples and will be much slower than those cases using liquid nitrogen as the cooling source. The growing direction and the size of ice crystals, however, are highly related to the viscosity of the polymer solutions, i.e. the interaction intensity among the polymer network.18,72 That might be a reason why PVA/PAA aerogels presented porous microstructures and PVA/CT-CMS aerogels present unidirectional microstructures. As for the PVA/PEO samples, low density of hydrogen bonds existed in the polymer network preventing the formation of aligned channels, although the viscosity of the mixed polymer solution is fairly low. However, when this density was decreased by adjusting the pH value, the size of the crystal increased, leading to loosely packed microstructure formed. We may thus suppose that the ice crystal will grow straight when the hydrogen bond density reaches a specific level, resulting in aligned channel microstructures of PVA-based aerogels. However, when the density of physical cross-linking is too high for the straight growth of the ice crystals, porous microstructures will be formed instead.Of course, the interactions between polymer chains in the precursor solution play crucial roles in different cases. When PEO, a flexible chain exhibiting weak hydrogen bonds with PVA, was introduced in the semi-dilute solution of PVA, a new pair of interpolymer interaction occurred (Fig. 7), which will decrease the amount of hydrogen bonds between PVA chains themselves. When the freeze was applied, less and weak interpolymer interactions presented, some crystalline domains of both PVA and PEO existed as cross-linking points of the network. After cycles of freeze–thaw, loose and irregular microstructure will be exhibited in the resulting aerogel samples. Changes in the chain length of PEO may alter the microstructure of aerogels by varying the intensity of hydrogen bonding between PEO and PVA as well.
 | ||
| Fig. 7 Mechanism scheme of PVA-based aerogels. The PVA polymer chains were first mixed with other polymers to form semi-dilute aqueous solutions. The IPCs were formed consequently in the present of hydrogen bonds between functional groups on polymer chains. After cycles of freeze–thaw, different microstructure presented with respect to the three types of polymers. The driving forces on the formation of morphologies within the polymer chains were mainly the IPCs and the crystalline domains. | ||
When a polymer chain adopting polar function groups was added into the PVA solution, such as PAA, strong hydrogen bonding interactions will occur between the polymer chains. When the temperature of the system becomes lower, the ice crystal will grow at those amorphous polymer domains, leaving abundant polymer-rich regions forming additional IPCs. Some stranded microstructures will thus form along the direction of growing ice after two cycles of freeze–thawing and a process of freeze-drying. Oriented morphology was thus formed in the PVA/PAA samples as multiple physical cross-linking points existed, ensuring the unidirectional aligned structure being kept after cycles of freeze–thaw. The mechanical property of these samples was enhanced to a large extent. However, when tuning the pH value of the precursor solution above the pKa of PAA, the resulting aerogel may possess different microstructures as the interpolymer hydrogen bonds became weaker. The mechanical properties will also decrease to some extent.
When semi-dilute solutions of PVA chains were mixed with CM-CTS chains, a small amount of hydrogen bonds were formed via –COO− groups and –NH– groups on CM-CTS and –OH on PVA. The IPCs formed between these two polymer chains were thus not strong enough. The added CM-CTS chains increased the concentration of polymer chains in the PVA matrix without bringing much intensity of interactions between polymer chains. Besides, the original crystalline domains of PVA were destroyed by introducing the rigid rings of CM-CTS. After the freeze–thaw process, unidirectional and high density of stripe microstructure was present. The mechanical strength of these samples was relatively weak due to the lack of interactions among polymer chains in micro-scale.
Conclusions
PVA-based aerogels were prepared by a freeze-drying strategy. The effect of interpolymer non-covalent bonds on the final morphology of PVA aerogels was investigated at a molecular level. Different types of microstructures in PVA-based aerogels were exhibited with respect to polymer chains from flexible to rigid. The formation of IPCs by means of non-covalent bonds between PVA and other polymer chains acted as physical cross-linking points in the final materials. Differences in microstructure between three groups of aerogels resulted from the interpolymer forces among the polymer network. Conformation of the polymers and IPCs in the solution state also has significant influences on the microstructure of aerogels before the freeze-drying process. Polymers with flexible chains tended to form a disordered morphology by being introduced into the PVA matrix. Polymers with polar groups interact with PVA to form strong hydrogen bonds, resulting in rigid structures similar to homogenous PVA aerogels. Polymers with rigid chains like derivatives of chitosan possessing 6 member-rings form fewer non-covalent bonds with PVA due to the low mass fraction of functional groups and adopt stretched chain conformation in solutions. A regular rod-like morphology presented in final aerogels, accordingly. A variation in the pH value of the PVA/PAA system may have an impact on the intensity of IPCs, which will consequently influence the microstructures of PVA-based aerogels. A change in chain length of PEO will alter the intensity of hydrogen bonds between PEO and PVA, which will result in different morphologies of the final aerogels.Studying the morphology of polymer aerogels altering with interactions between polymer chains may provide new understanding of the forming mechanism of PVA-based aerogels especially from the view of interpolymer behaviours. It will also be a convenient strategy to realize the versatility of aerogel materials as light materials in several practical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support by the National Natural Science Foundation of China (Grant No. 52072202).Notes and references
- S. S. Kistler, Nature, 1931, 127, 741 CrossRef CAS
.
- Y. Du, X. Zhang, J. Wang, Z. Liu, K. Zhang, X. Ji, Y. You and X. Zhang, ACS Nano, 2020, 14, 11919–11928 CrossRef CAS PubMed
- H. B. Chen, P. Shen, M. J. Chen, H. B. Zhao and D. A. Schiraldi, ACS Appl. Mater. Interfaces, 2016, 8, 32557–32564 CrossRef CAS
- H. B. Chen and D. A. Schiraldi, Polym. Rev., 2019, 59, 1–24 CrossRef CAS
- A. Lamy-Mendes, R. F. Silva and L. Durães, J. Mater. Chem. A, 2018, 6, 1340–1369 RSC
- S. Salimian, A. Zadhoush, M. Naeimirad, R. Kotek and S. Ramakrishna, Polym. Compos., 2018, 39, 3383–3408 CrossRef CAS
- B. Ren, J. Liu, Y. Rong, L. Wang, Y. Lu, X. Xi and J. Yang, ACS Nano, 2019, 13, 11603–11612 CrossRef CAS
- W. Luo, D. Sun, S. Chen, L. Shanmugam, Y. Xiang and J. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 57547–57559 CrossRef CAS
- H. Zhang, F. Zhang and J. Wu, React. Funct. Polym., 2013, 73, 923–928 CrossRef CAS
- P. Giannouli and E. R. Morris, Food Hydrocolloids, 2003, 17, 495–501 CrossRef CAS
- Y. Zhao, W. Shen, Z. Chen and T. Wu, Carbohydr. Polym., 2016, 148, 45–51 CrossRef CAS PubMed
- V. I. Lozinsky, L. G. Damshkaln, R. Brown and I. T. Norton, J. Appl. Polym. Sci., 2000, 75, 1740–1748 CrossRef CAS
- A. Łabudzińska and A. Ziabicki, Kolloid-Z. Z. Polym., 1971, 243, 21–27 CrossRef
- N. A. Peppas, Die Makromol. Chem., 1975, 176, 3433–3440 CrossRef CAS
- M. Bercea, S. Morariu and D. Rusu, Soft Matter, 2013, 9, 1244–1253 RSC
- A. Kjøniksen and B. Nystro, Macromolecules, 1996, 29, 7116–7123 CrossRef
- C. M. Hassan and N. A. Peppas, Macromolecules, 2000, 33, 2472–2479 CrossRef CAS
- M. C. Gutiérrez, Z. Y. García-Carvajal, M. Jobbágy, F. Rubio, L. Yuste, F. Rojo, M. L. Ferrer and F. Del Monte, Adv. Funct. Mater., 2007, 17, 3505–3513 CrossRef
- Z. Cheng, K. De Gracia and D. A. Schiraldi, Polymers, 2018, 10, 1–10 Search PubMed
- Y. Liu, Y. Su, J. Guan, J. Cao, R. Zhang, M. He and Z. Jiang, ACS Appl. Mater. Interfaces, 2018, 10, 26546–26554 CrossRef CAS
- A. Richter, S. Ludwig, J. Zimmermann, J. Kressler, D. Kuckling, T. Universita, F. Werkstoffwissenschaften and T. Universita, Acta Polym., 1999, 383–390 Search PubMed
- R. Gelli, S. Del Buffa, P. Tempesti, M. Bonini, F. Ridi and P. Baglioni, Colloids Surf., A, 2017, 532, 18–25 CrossRef CAS
- M. L. Huggins, J. Chem. Educ., 1957, 34, 480–488 CrossRef CAS
- K. L. Smith, A. E. Winslow and D. E. Petersen, Ind. Eng. Chem., 1959, 51, 1361–1364 CrossRef CAS
- F. E. Bailey, R. D. Lundberg and R. W. Callard, J. Polym. Sci., Part A: Gen. Pap., 1964, 2, 2975 CrossRef
- H. T. Oyama, W. T. Tang and C. W. Frank, Macromolecules, 1987, 20, 474–480 CrossRef CAS
- T. Song, S. H. Goh and S. Y. Lee, Macromolecules, 2002, 35, 4133–4137 CrossRef CAS
- T. Ikawa, K. Abe, K. Honda and E. Tsuchida, J. Polym. Sci., Polym. Chem. Ed., 1975, 13, 1505–1514 CrossRef CAS
- C. Wu and S. Zhou, Macromolecules, 1995, 28, 5388–5390 CrossRef CAS
- C. Wu and S. Zhou, Macromolecules, 1995, 28, 8381–8387 CrossRef CAS
- X. Liu, S. Luo, J. Ye and C. Wu, Macromolecules, 2012, 45, 4830–4838 CrossRef CAS
- J. Hao, G. Yuan, W. He, H. Cheng, C. C. Han and C. Wu, Macromolecules, 2010, 43, 2002–2008 CrossRef CAS
- R. Deng, S. Diao, Y. Yue, T. Ngai, C. Wu and F. Jin, Biopolymers, 2010, 93, 571–577 CAS
- Y. Lu, K. Zhou, Y. Ding and C. Wu, Phys. Chem. Chem. Phys., 2010, 12, 3188–3194 RSC
- S. Ma, X. Qi, Y. Cao, S. Yang and J. Xu, Polymer, 2013, 54, 5382–5390 CrossRef CAS
- M. Shibayama, Polym. J., 2010, 43, 18–34 CrossRef
- Y. Katsumoto and M. Shibayama, Macromolecules, 2013, 46, 6225–6232 CrossRef
-
K. Mortensen, Advanced functional molecules and polymers, 2001, ch. 8, pp. 223–269 Search PubMed
- M. Bercea, S. Morariu and M. Teodorescu, J. Polym. Res., 2006, 13, 387–395 Search PubMed
- F. S. Wang, T. F. Wang, H. H. Lu, W. S. Ao-Ieong, J. Wang, H. L. Chen and C. H. Peng, Macromolecules, 2017, 50, 6054–6063 CrossRef CAS
- M. A. Darabi, A. Khosrozadeh, Y. Wang, N. Ashammakhi, H. Alem, A. Erdem, Q. Chang, K. Xu, Y. Liu, G. Luo, A. Khademhosseini and M. Xing, Adv. Sci., 2020, 7, 1902740 CrossRef CAS
- X. Zhang, X. Guo, S. Yang, S. Tan, X. Li, H. Dai, X. Yu, X. Zhang, N. Weng, B. Jian and J. Xu, J. Appl. Polym. Sci., 2009, 112, 3063–3070 CrossRef CAS
- R. Cheng, Macromol. Symp., 1997, 34, 27–34 CrossRef
- A. Higuchi and T. Iijima, Polymer, 1985, 26, 1207–1211 CrossRef CAS
- Z. Lian and L. Ye, J. Thermoplast. Compos. Mater., 2013, 26, 912–922 CrossRef
- N. X. Chen and J. H. Zhang, Chin. J. Polym. Sci., 2010, 28, 903–911 CrossRef CAS
- M. Lim, D. Kim and J. Seo, Polym. Int., 2016, 65, 400–406 CrossRef CAS
- R. Mastrangelo, D. Chelazzi, G. Poggi, E. Fratini, L. P. Buemi, M. L. Petruzzellis, P. Baglioni, R. Mastrangelo, D. Chelazzi, G. Poggi, E. Fratini and L. Pensabene, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 7011–7020 CrossRef CAS PubMed
- R. Ricciardi, F. Auriemma, C. De Rosa and F. Lauprêtre, Macromolecules, 2004, 37, 1921–1927 CrossRef CAS
- R. T. Abdulwahid, O. G. Abdullah, S. B. Aziz, S. A. Hussein, F. F. Muhammad and M. Y. Yahya, J. Mater. Sci.: Mater. Electron., 2016, 27, 12112–12118 CrossRef CAS
- N. Zhang, J. He, W. Han and Y. Wang, J. Mater. Sci., 2019, 54, 9603–9612 CrossRef CAS
- F. Saeedi, A. Montazeri, Y. Bahari, M. Pishvaee and M. Ranjbar, Int. J. Chemoinf. Chem. Eng., 2018, 7, 1–12 CAS
- S. Zou, R. Lv, Z. Tong, B. Na, K. Fu and H. Liu, Polymer, 2019, 183, 121878 CrossRef CAS
- H. S. Mansur, C. M. Sadahira, A. N. Souza and A. A. P. Mansur, Mater. Sci. Eng., C, 2008, 28, 539–548 CrossRef CAS
- B. Gupta, R. Agarwal and M. Sarwar Alam, J. Appl. Polym. Sci., 2013, 127, 1301–1308 CrossRef CAS
- E. F. dos Reis, F. S. Campos, A. P. Lage, R. C. Leite, L. G. Heneine, W. L. Vasconcelos, Z. I. P. Lobato and H. S. Mansur, Mater. Res., 2006, 9, 185–191 CrossRef
-
E. Tsuchida and K. Abe, Interactions between macromolecules in solution and intermacromolecular complexes, 1982 Search PubMed
- T. Liu, C. Jiao, X. Peng, Y. N. Chen, Y. Chen, C. He, R. Liu and H. Wang, J. Mater. Chem. B, 2018, 6, 8105–8114 RSC
- J. M. Joshi and V. K. Sinha, J. Polym. Res., 2006, 13, 387–395 CrossRef CAS
- K. Choo, Y. C. Ching, C. H. Chuah, S. Julai and N. S. Liou, Materials, 2016, 9, 644 CrossRef
- I. Pucić and T. Jurkin, Radiat. Phys. Chem., 2012, 81, 1426–1429 CrossRef
- S. I. Song and B. C. Kim, Polymer, 2004, 45, 2381–2386 CrossRef CAS
- S. Li and X. H. Yang, Adv. Mater. Sci. Eng., 2014, 2014, 163678 Search PubMed
- H. Wang, H. Wen, B. Hu, G. Fei, Y. Shen, L. Sun and D. Yang, Sci. Rep., 2017, 7, 1–12 CrossRef
- A. Giuntoli, F. Puosi, D. Leporini, F. W. Starr and J. F. Douglas, Sci. Adv., 2020, 1–10 Search PubMed
- L. H. Derek, H. Boualem and R. K. Steven, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 135–138 Search PubMed
- M. Duval and D. Sarazin, Polymer, 2000, 41, 2711–2716 CrossRef CAS
- W. F. Polik and W. Burchard, Macromolecules, 1983, 16, 978–982 CrossRef CAS
- W. Brown, Macromolecules, 1984, 17, 66–72 CrossRef CAS
- K. F. Tjipangandjara, Adv. Powder Technol., 1992, 3, 119–127 CrossRef CAS
- S. Kalliola, E. Repo, V. Srivastava, F. Zhao, J. P. Heiskanen, J. A. Sirviö, H. Liimatainen and M. Sillanpää, Langmuir, 2018, 34, 2800–2806 CrossRef CAS
- A. H. Kang, K. Shang, D. D. Ye, Y. T. Wang, H. Wang, Z. M. Zhu, W. Liao, S. M. Xu, Y. Z. Wang and D. A. Schiraldi, Chem. Eng. J., 2017, 327, 992–999 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nj04646b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |