
Remarkably enhanced dynamic oxygen migration on graphene oxide supported by copper substrate†
Zihan
Yan
 ,
Wenjie
Yang
,
Hao
Yang
,
Chengao
Ji
,
Shuming
Zeng
,
Xiuyun
Zhang
,
Liang
Zhao
* and
Yusong
Tu
*
,
Wenjie
Yang
,
Hao
Yang
,
Chengao
Ji
,
Shuming
Zeng
,
Xiuyun
Zhang
,
Liang
Zhao
* and
Yusong
Tu
*
College of Physical Science and Technology, Yangzhou University, Jiangsu 225009, China. E-mail: zhaoliang@yzu.edu.cn; ystu@yzu.edu.cn
First published on 2nd July 2022
Abstract
The dynamic covalent properties of graphene oxide (GO) are of fundamental interest to a broad range of scientific areas and technological applications. It remains a challenge to access feasible dynamic reactions for reversibly breaking/reforming the covalent bonds of oxygen functional groups on GO, although these reactions can be induced by photonic or mechanical routes, or mediated by adsorbed water. Here, using density functional theory calculations, we demonstrate the remarkably enhanced dynamic oxygen migration along the basal plane of GO supported by copper substrate (GO@copper), with C–O bond breaking reactions and proton transfer between neighboring epoxy and hydroxyl groups. Compared to reactions on GO, the energy barriers of oxygen migrations on GO@copper are sharply reduced to be less than or comparable to thermal fluctuations, and meanwhile the crystallographic match between GO and copper substrate induces new oxygen migration paths on GO@copper. This work sheds light on understanding of the metal substrate-enhanced dynamic properties of GO, and evidences the strategy to tune the activity of two-dimensional-interfacial oxygen groups for various potential applications.
New conceptsThe emergence of dynamic covalent materials over the last few decades has gained tremendous attention and brought new ideas for the manufacturing of materials with novel mechanical and thermal features. However, the high reaction barriers of breaking/reforming the strong covalent bonds within dynamic covalent materials have hindered the widespread application. Searching for dynamic covalent materials with low reaction barriers has thus become an urgent need. In this work, novel aspects and new concepts have been proposed as follows: graphene oxide (GO) can be converted to a dynamic covalent material when supported by a copper substrate (GO@copper), and oxygen groups can migrate along the basal plane of GO via C–O bond breaking/reforming reactions and proton transfer between dangling C–O bonds and neighboring hydroxyl groups. Compared to that on free-standing GO, the energy barriers of dynamic oxygen migrations on GO@copper are significantly decreased to be less than or comparable to thermal fluctuations; and the crystallographic match between the substrate and GO induces new oxygen migration paths along the meta-positions of the aromatic rings. This work provides a simple and easy-to-implement method to access the dynamic properties of GO and evidences the strategy to tune the activity of two-dimensional-interfacial oxygen groups for various potential applications, which might provide possibilities for the realization of state-of-the-art high-performance (bio)sensors, biomedical devices, and electronic equipment in the future. |
The emergence of dynamic covalent materials over the last few decades has gained tremendous attention in a broad range of scientific areas such as chemical separation,1,2 biological imaging or sensing3 and drug delivery;4–6 they especially advance technological innovations in the manufacturing of materials with novel mechanical and thermal features including self-healing,4,7–11 shape-memory7,9,12,13 and thermoset reprocessing.4,7 These dynamic behaviors rely on reversible reactions of breaking/reforming strong covalent bonds within molecules, endowing materials with adaptivity in response to stimuli and variations in ambient conditions.14–17 However, the dynamic reactions usually require harsh conditions and take a long time to reach thermodynamic equilibrium,3,18 presumably attributed to the high reaction barriers. Thus, searching for feasible dynamic reactions within a reasonable system is the key to realizing dynamic covalent materials. Interestingly, we have found that when mediated by adsorbed water molecules, oxygen groups on graphene oxide (GO) can spontaneously migrate along the basal plane, and GO is converted to a dynamic covalent material with structural adaptivity to biomolecule adsorption.19 However, how to access this dynamic oxygen migration on GO without water remains unclear.
Metal substrate-supported GO, is a promising building block of engineering materials and electronic devices, which shows superior mechanical strength,20–23 high electronic and thermal conductivity,24,25 and sensitive reaction activity.26 The presence of metal substrates creates metal–GO interactions and lattice matching. In particular, the (111) surface of the copper substrate has been found to assist the epitaxial growth of graphene and shows a better lattice match with the honeycomb plane of GO in experiments.27,28 Here, we perform density functional theory (DFT) calculations to investigate the dynamic oxygen migrations on GO supported by a copper substrate (GO@copper).
Unexpectedly, we find that the copper substrate remarkably enhances the dynamic migration of oxygen groups on the basal plane of GO. The energy barriers of oxygen migrations are significantly decreased to be less than or comparable to thermal fluctuations. Meanwhile, the crystallographic match between GO and copper substrate induces new oxygen migration paths on GO@copper. To the best of our knowledge, this is the first report of copper substrate-enhanced dynamic oxygen migration on GO, which is important for tuning the activity of two-dimensional-interfacial oxygen groups for various potential applications.
Results and discussion
The dominant oxygen groups on the basal plane of GO, such as hydroxyl and epoxy groups, are distributed with high correlation according to our previous DFT calculations.19,29 As shown in Fig. 1(a), we choose a pair of hydroxyl and epoxy groups to represent the correlated distribution of the oxygen groups. Considering the lattice match between GO and the copper substrate, we denote the hydroxyl-occupied carbon atom on the top of a copper atom and at the face-centered position, by top site and fcc site, respectively (see Fig. 1(b)). This configuration has been shown to be the most stable adsorption configuration of graphene on the copper substrate,30,31 and results for the other two configurations with top and hcp sites, as well as the bridge site can be found in PS. 1 of ESI.†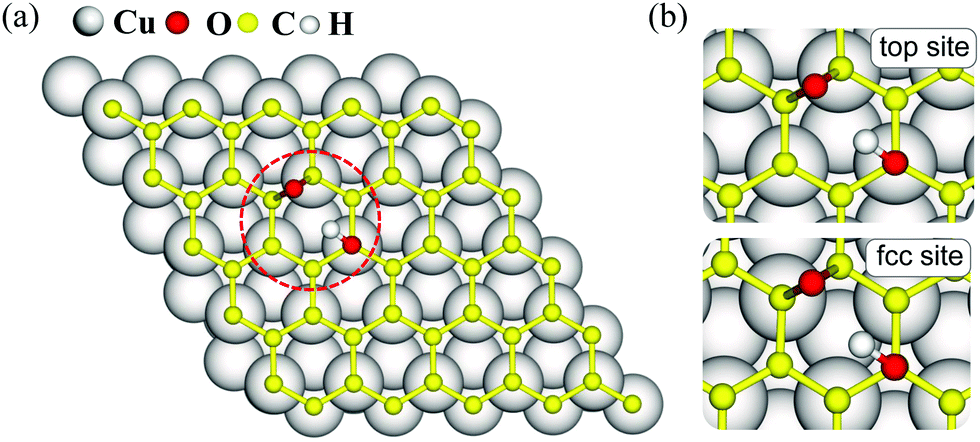 | ||
| Fig. 1 (a) Top view of GO@copper with a pair of hydroxyl and epoxy groups. (b) Zoomed-in snapshots of two different reactant configurations depending on the lattice matching between GO and copper substrate. Top site: hydroxyl-occupied carbon atom is on the top of copper atoms; fcc site: hydroxyl-occupied carbon atom is on the face-centered position of copper atoms. | ||
The presence of the copper substrate significantly decreases the energy barriers of oxygen migrations. Fig. 2 presents the oxygen migration pathways and state configurations on the free-standing GO (fr-GO) and GO@copper. The oxygen migration assisted by the hydroxyl includes two successive reactions: the C–O bond breaking reaction and the proton transfer between the dangling oxygen and neighboring hydroxyl for the exchange. Without the copper substrate, the C–O bond breaking reaction has an energy barrier of 3.9 kcal mol−1 whereas there is a relatively high barrier of 14.7 kcal mol−1 for the proton transfer,19 indicating that it is difficult for oxygen groups to migrate spontaneously at ambient conditions.32,33 However, for the top site of GO@copper, the energy barrier for the C–O bond breaking reaction reduces to 1.3 kcal mol−1; and for the proton transfer, we can see a significant decrease of the barrier from 14.7 kcal mol−1 to 0.6 kcal mol−1. These energy barrier values are lower than or comparable to thermal fluctuations, and for the case for the fcc site (3.1 kcal mol−1 and 1.7 kcal mol−1), suggest that the migration of oxygen groups can be accessed through the C–O bond breaking reaction and proton transfer on GO@copper. In addition, we have also tested the oxygen migration pathway on fr-GO using other functionals (see PS. 3, ESI†). The oxygen migration pathway on fr-GO obtained with the B3LYP functional is used since this method has been considered to be more reliable in the description of transition states.34 It can also be seen that PBE-based methods (PBE, PBE-D3 and PBE + optB86b-vdW) give better predictions of energy for fr-GO than LDA.
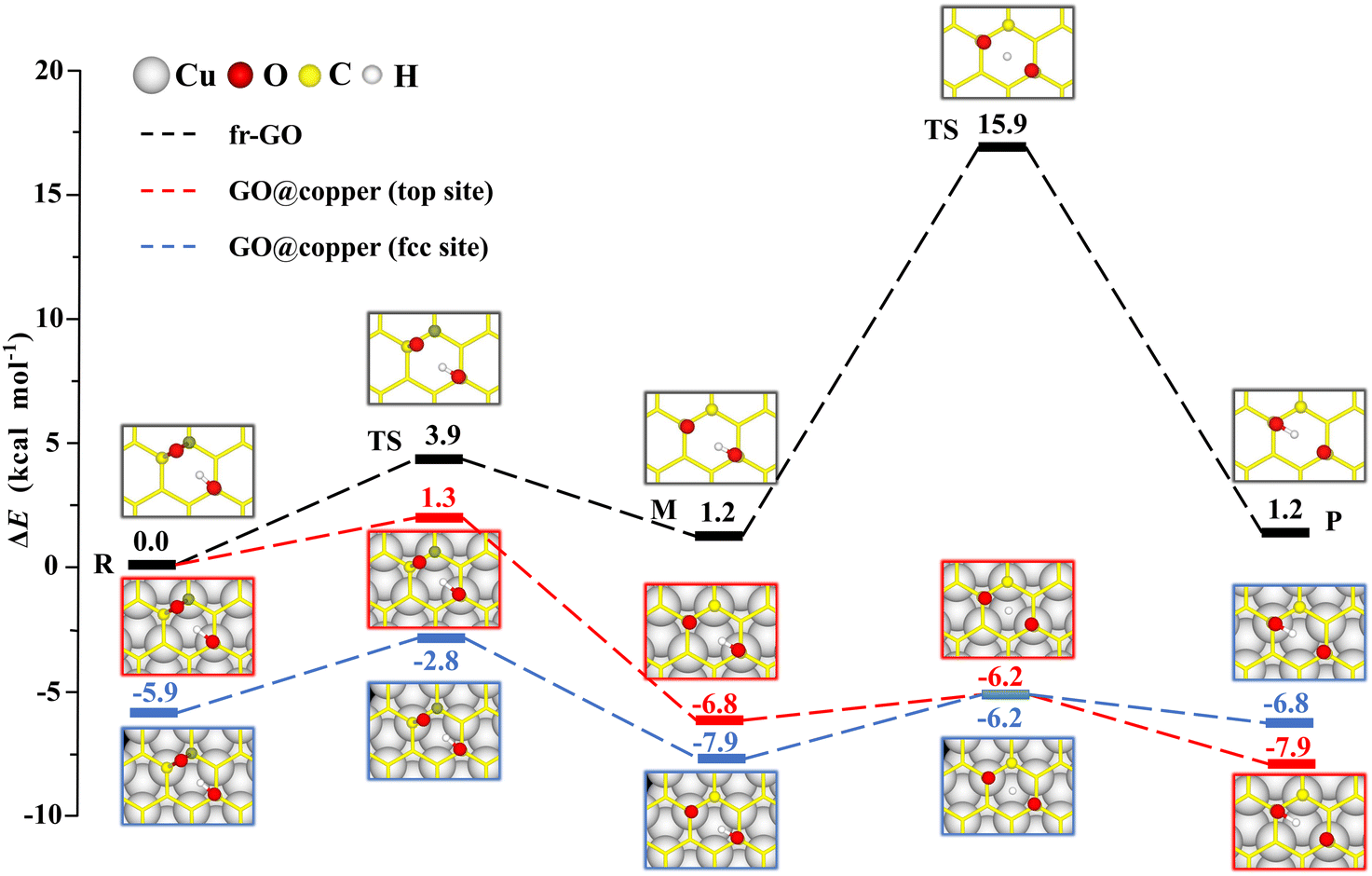 | ||
| Fig. 2 Reaction pathways and state configurations for oxygen migration on fr-GO and GO@copper. The hydroxyl-assisted oxygen migration includes the C–O bond breaking reaction and the proton transfer between the dangling oxygen and neighboring hydroxyl for the exchange on fr-GO without the substrate (black lines), on the top site of GO@copper (red lines) and fcc site of GO@copper (blue lines). Notations: reactants (R), intermediates (M), transition states (TS), and products (P). The energy levels of R on fr-GO and GO@copper (top site) are shifted for a better comparison. The reaction pathway on fr-GO is cited from our previous work.19 Side views of all state configurations can be found in PS. 2 of ESI.† | ||
The intermediate (M) is more stable than the reactant (R) due to the presence of the copper substrate. On fr-GO, M is 1.2 kcal mol−1 higher than R, indicating that the epoxy configuration with two C–O bonds is more stable than the dangling C–O bond. Whereas on GO@copper, the energy of M is 6.8 kcal mol−1 (top site) and 2.0 kcal mol−1 (fcc site) lower than that of R. We further analyze the charge density difference and Bader charge of R and M for the top site: as shown in Fig. 3, the oxygen groups gain charge while the three carbon atoms bonded to oxygen groups and the copper atoms near the GO, lose the charge. The Bader charge further shows that the charge distribution is almost unchanged on the hydroxyl group between R and M on fr-GO or GO@copper. However, the dangling oxygen gains more charge, +1.03e in the presence of the copper substrate, compared to +0.80e on fr-GO. This enhanced electronegativity of the dangling oxygen atom induces a stronger electrostatic interaction between the dangling oxygen atom and the hydroxyl group, and further makes M on GO@copper more stable. This can also be seen from the shortened dangling C–O bond, the elongated O–H bond, and closer distance between the two oxygen atoms in M on GO@copper (see PS. 4, ESI†). Besides, we find that the p-band centers of carbon atoms or oxygen in hydroxyl are far from the Fermi level in R and M states on GO@copper, indicating the decreased energy barriers of C–O bond breaking and proton transfer reactions and the enhanced oxygen migration on GO@copper (see PS. 5, ESI†).
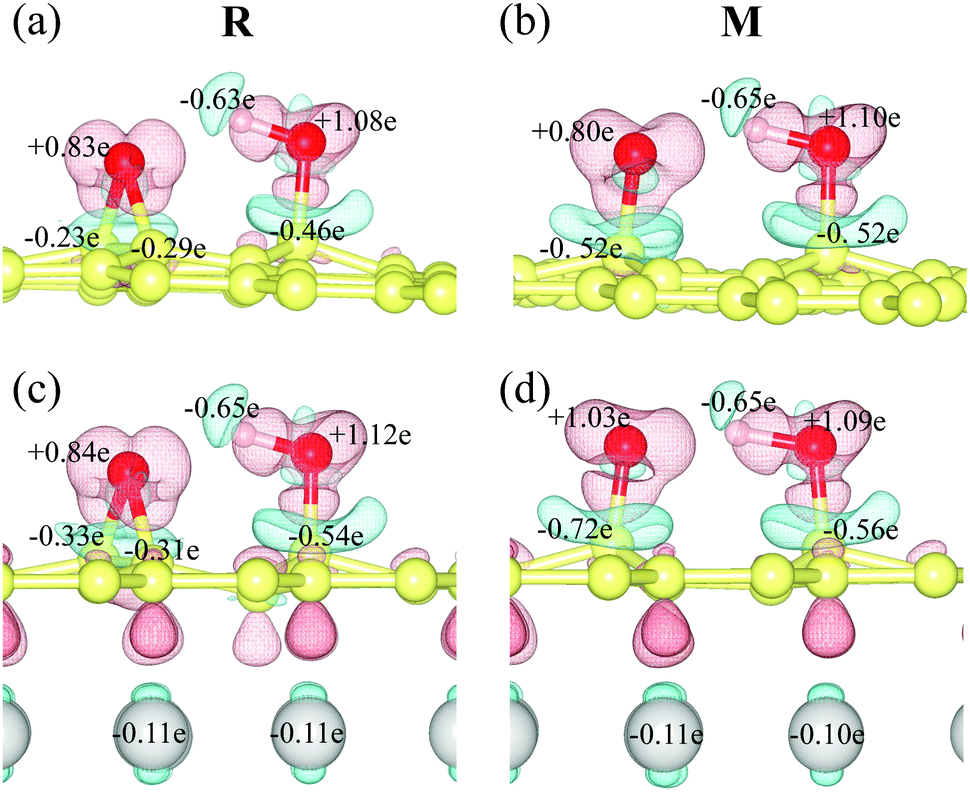 | ||
| Fig. 3 The charge density difference and Bader charge of reactants (R) and intermediates (M) on (a and b) fr-GO and on (c and d) GO@copper (top site). Blue and red colors represent the charge depletion and accumulation regions with an isovalue of 0.01 e bohr−3, respectively. The number indicates the Bader charge value, with positive and negative values denoting the gain and loss of charge. | ||
The presence of the copper substrate induces new oxygen migration pathways on GO. Generally, the dangling C–O bond and the hydroxyl cannot coexist at the meta-position of an aromatic ring of GO due to steric effects.19,29 However, when GO is supported by the copper substrate, a dangling C–O bond is formed at the meta-position of the hydroxyl (see M in Fig. 4). In this new oxygen migration pathway, a C–O bond in epoxy breaks, followed by proton transfer. The energy barriers for the C–O breaking reaction and proton transfer are 3.3 kcal mol−1 and 0.1 kcal mol−1 for the top site, 2.4 kcal mol−1 and 0.6 kcal mol−1 for the fcc site. These values are all less than or comparable to thermal fluctuations, an indication of feasible migrations of oxygen groups on GO@copper. In particular, the barrier of the proton transfer is only 0.1 kcal mol−1, indicating that the proton can transfer rapidly between the dangling oxygen and hydroxyl groups, while the position exchange of these two groups is enabled. We also note that the hydroxyl can migrate within an aromatic ring of GO@copper, via the combination of oxygen migration paths in Fig. 2 and 4. The new oxygen migration path enriches the understanding of the dynamic behavior of oxygen groups on GO@copper.
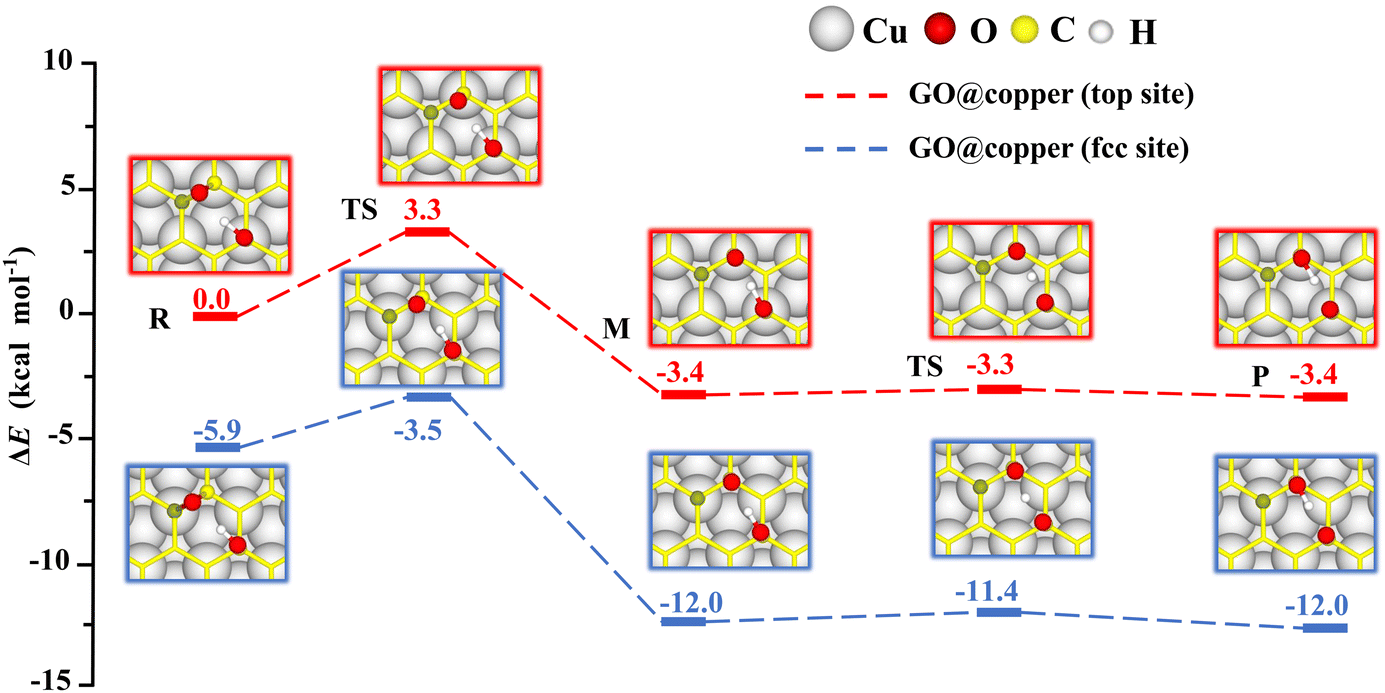 | ||
| Fig. 4 Copper-substrate-induced new reaction pathways of oxygen migration, which occur at the meta-positions of an aromatic ring, on the top site (red lines) and fcc site (blue lines) of GO@copper. Notations: reactants (R), intermediates (M), transition states (TS) and products (P). | ||
Conclusions
In summary, we have demonstrated that the copper substrate can remarkably enhance dynamic oxygen migration on GO@copper based on DFT calculations. The oxygen migration paths are all accessed by C–O bond breaking reaction and proton transfer between neighboring epoxy and hydroxyl groups. Compared to that on GO, the energy barriers of oxygen migrations on GO@copper are sharply reduced to be less than or comparable to thermal fluctuations. Besides, the crystallographic match between the GO and copper substrate induces new oxygen migration paths on GO@copper, where oxygen groups can migrate along the meta-positions of an aromatic ring.Our work demonstrates the significant dynamic behavior of oxygen groups on GO@copper. The significant dynamic behavior and the enhanced interfacial oxygen activity result from the significantly decreased energy barriers and crystallographic match-induced new oxygen migration paths, which may allow structural adaptation of GO@copper to interfacial oxygen-related reactions. Moreover, we also tested the dynamic oxygen migration on GO@copper with the distribution of the coexistence of the oxidized and unoxidized regions (see PS. 6, ESI†), the energy barriers of dynamic oxygen migration are still comparable to thermal fluctuations. It should be noted that the high-quality and stable growth of GO, and other similar two-dimensional (2D) materials have been fabricated on metal substrates in experiments.27,28,35 Therefore, this work evidences the strategy to tune the activity of 2D-interfacial oxygen groups for various potential applications in electrolysis,36 capacitors,37 fuel cells,38–41 sensors,40 catalysis,42,43 and might provide possibilities for the realization of state-of-the-art high-performance (bio)sensors, biomedical devices, and electronic equipment in the future.
Methods
The DFT calculations are carried out using the Vienna Ab initio Simulation Package (VASP).44,45 The exchange–correlation energy is described by the local density approximation (LDA),46 which has been proved to be more reasonable for the system of graphene supported by metal substrates.30,31,47 The Perdew–Burke–Ernzerhof (PBE) functional48 with van der Waals corrections, PBE + optB86b-vdW and PBE-D3 methods,49,50 are also employed to test the dynamic oxygen migration on fr-GO and GO@copper, and the results are consistent with those using LDA (see PS.7, ESI†). The convergence accuracy for energy and force in the geometry optimization and transition state (TS) search are 10−5 eV and 0.01 eV Å−1, respectively. The projector augmented wave (PAW) is used with the plane wave cutoff of 500 eV. The Monkhorst–Pack51 mesh in the k-space is 1 × 1 × 1. The system consists of a GO sheet supported on four layers of the (111) surface of the copper substrate, including 100 copper atoms and 50 carbon atoms, in a periodic 5 × 5 supercell. The GO is stretched about 3.7% to match the lattice of the copper substrate. The vacuum layer in the z-axis direction is set to 15 Å to avoid interactions between periodic images. The copper atoms in the bottom-most layer of the substrate are fixed in all the calculations (see PS. 8 for the effects of vacuum and substrate layers on the oxygen migration pathways, ESI†). The TS is searched via the climbing image-nudged elastic-band (CI-NEB) method.52 The VASPKIT code53 and VESTA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 54 are used for data processing and graphics production.
54 are used for data processing and graphics production.
Author contributions
Yusong Tu conceived, designed, and guided the research; Zihan Yan, Wenjie Yang, Hao Yang, and Chengao Ji performed the simulations; Zihan Yan, Shuming Zeng, Liang Zhao, and Yusong Tu analyzed the data; Zihan Yan and Liang Zhao wrote the paper; all the authors participated in discussions of the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for the helpful suggestions given by PhD candidates Zhijing Huang (Yangzhou University), Guangyu Du (The Hong Kong Polytechnic University), and Xiaoxue Liu (Jilin University). This work was funded by the National Natural Science Foundation of China (No. 12075201, 11675138), the Natural Science Foundation of Jiangsu Province (No. BK20201428), the Special Program for Applied Research on Supercomputation of the NSFC-Guangdong Joint Fund (the second phase) and Jiangsu Students’ Innovation and Entrepreneurship Training Program (No. 202111117008Z).References
- N. Hafezi and J.-M. Lehn, J. Am. Chem. Soc., 2012, 134, 12861–12868 CrossRef CAS PubMed.
- Y. Jin, Q. Wang, P. Taynton and W. Zhang, Acc. Chem. Res., 2014, 47, 1575–1586 CrossRef CAS PubMed.
- Y. Zhang, Y. Qi, S. Ulrich, M. Barboiu and O. Ramström, Mater. Chem. Front., 2020, 4, 489–506 RSC.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Adv. Mater., 2017, 29, 1606100 CrossRef PubMed.
- S. J. Rowan, S. J. Cantrill, G. R. Cousins, J. K. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- H. Otsuka, Polym. J., 2013, 45, 879–891 CrossRef CAS.
- N. Zheng, Y. Xu, Q. Zhao and T. Xie, Chem. Rev., 2021, 121, 1716–1745 CrossRef CAS PubMed.
- J.-M. Lehn, Angew. Chem., Int. Ed., 2015, 54, 3276–3289 CrossRef CAS PubMed.
- M. M. Perera and N. Ayres, Polym. Chem., 2020, 11, 1410–1423 RSC.
- N. Roy, B. Bruchmann and J.-M. Lehn, Chem. Soc. Rev., 2015, 44, 3786–3807 RSC.
- B. J. Blaiszik, S. L. Kramer, S. C. Olugebefola, J. S. Moore, N. R. Sottos and S. R. White, Annu. Rev. Mater. Res., 2010, 40, 179–211 CrossRef CAS.
- G. Zhang, W. Peng, J. Wu, Q. Zhao and T. Xie, Nat. Commun., 2018, 9, 1–7 CrossRef PubMed.
- W. Miao, W. Zou, B. Jin, C. Ni, N. Zheng, Q. Zhao and T. Xie, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- K. Imato, M. Nishihara, T. Kanehara, Y. Amamoto, A. Takahara and H. Otsuka, Angew. Chem., 2012, 124, 1164–1168 CrossRef.
- P. Chakma and D. Konkolewicz, Angew. Chem., 2019, 131, 9784–9797 CrossRef.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- G. Zhu, Z. Huang, L. Zhao and Y. Tu, Nanoscale, 2021, 13, 15231–15237 RSC.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- Y. Tu, L. Zhao, J. Sun, Y. Wu, X. Zhou, L. Chen, X. Lei, H. Fang and G. Shi, Chin. Phys. Lett., 2020, 37, 066803 CrossRef CAS.
- I. Ovid’Ko, Rev. Adv. Mater. Sci., 2014, 38, 190–200 Search PubMed.
- D.-B. Xiong, M. Cao, Q. Guo, Z. Tan, G. Fan, Z. Li and D. Zhang, ACS Nano, 2015, 9, 6934–6943 CrossRef CAS PubMed.
- J. Hwang, T. Yoon, S. H. Jin, J. Lee, T.-S. Kim, S. H. Hong and S. Jeon, Adv. Mater., 2013, 25, 6724–6729 CrossRef CAS PubMed.
- J. Wang, Z. Li, G. Fan, H. Pan, Z. Chen and D. Zhang, Scr. Mater., 2012, 66, 594–597 CrossRef CAS.
- H. Xu, X. Wu, X. Li, C. Luo, F. Liang, E. Orignac, J. Zhang and J. Chu, Carbon, 2018, 127, 491–497 CrossRef CAS.
- V. Goyal and A. A. Balandin, Appl. Phys. Lett., 2012, 100, 073113 CrossRef.
- I. Khalil, S. Rahmati, N. M. Julkapli and W. A. Yehye, J. Ind. Eng. Chem., 2018, 59, 425–439 CrossRef CAS.
- O. Frank, J. Vejpravova, V. Holy, L. Kavan and M. Kalbac, Carbon, 2014, 68, 440–451 CrossRef CAS.
- L. Gao, J. R. Guest and N. P. Guisinger, Nano Lett., 2010, 10, 3512–3516 CrossRef CAS PubMed.
- J. Yang, G. Shi, Y. Tu and H. Fang, Angew. Chem., Int. Ed., 2014, 126, 10354–10358 CrossRef.
- P. Khomyakov, G. Giovannetti, P. Rusu, G. V. Brocks, J. Van den Brink and P. J. Kelly, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 195425 CrossRef.
- Z. Xu and M. J. Buehler, J. Phys. Condens. Matter, 2010, 22, 485301 CrossRef PubMed.
- P. V. Kumar, N. M. Bardhan, S. Tongay, J. Wu, A. M. Belcher and J. C. Grossman, Nat. Chem., 2014, 6, 151–158 CrossRef CAS PubMed.
- S. Kim, S. Zhou, Y. Hu, M. Acik, Y. J. Chabal, C. Berger, W. De Heer, A. Bongiorno and E. Riedo, Nat. Mater., 2012, 11, 544–549 CrossRef CAS PubMed.
- L. Simón and J. M. Goodman, Org. Biomol. Chem., 2011, 9, 689–700 RSC.
- P. Hidalgo-Manrique, X. Lei, R. Xu, M. Zhou, I. A. Kinloch and R. J. Young, J. Mater. Sci., 2019, 54, 12236–12289 CrossRef CAS.
- T. Kida, Y. Kuwaki, A. Miyamoto, N. L. Hamidah, K. Hatakeyama, A. T. Quitain, M. Sasaki and A. Urakawa, ACS Sustainable Chem. Eng., 2018, 6, 11753–11758 CrossRef CAS.
- W. Gao, N. Singh, L. Song, Z. Liu, A. L. M. Reddy, L. Ci, R. Vajtai, Q. Zhang, B. Wei and P. M. Ajayan, Nat. Nanotechnol., 2011, 6, 496–500 CrossRef CAS PubMed.
- R. P. Pandey, G. Shukla, M. Manohar and V. K. Shahi, Adv. Colloid Interface Sci., 2017, 240, 15–30 CrossRef CAS PubMed.
- W. Gao, G. Wu, M. T. Janicke, D. A. Cullen, R. Mukundan, J. K. Baldwin, E. L. Brosha, C. Galande, P. M. Ajayan and K. L. More, et al. , Angew. Chem., Int. Ed., 2014, 53, 3588–3593 CrossRef CAS PubMed.
- M. R. Karim, K. Hatakeyama, T. Matsui, H. Takehira, T. Taniguchi, M. Koinuma, Y. Matsumoto, T. Akutagawa, T. Nakamura and S.-I. Noro, et al. , J. Am. Chem. Soc., 2013, 135, 8097–8100 CrossRef CAS PubMed.
- F. Li, X. Jiang, J. Zhao and S. Zhang, Nano Energy, 2015, 16, 488–515 CrossRef CAS.
- W. Yu, L. Sisi, Y. Haiyan and L. Jie, RSC Adv., 2020, 10, 15328–15345 RSC.
- X. Jiang, J. Nisar, B. Pathak, J. Zhao and R. Ahuja, J. Catal., 2013, 299, 204–209 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- J. P. Perdew and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23, 5048 CrossRef CAS.
- Z. Zhang, J. Yin, X. Liu, J. Li, J. Zhang and W. Guo, J. Phys. Chem. Lett., 2016, 7, 867–873 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. Klimeš, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188 CrossRef.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nh00041e |
| This journal is © The Royal Society of Chemistry 2022 |