
A tumor microenvironment dual responsive contrast agent for contrary contrast-magnetic resonance imaging and specific chemotherapy of tumors†
Yudie
Lu‡
a,
Jie
Feng‡
b,
Zhiyu
Liang
b,
Xuanyi
Lu
a,
Shuai
Guo
a,
Lin
Huang
a,
Wei
Xiong
b,
Sijin
Chen
b,
Huimin
Zhou
c,
Xuehua
Ma
 d,
Yikai
Xu
b,
Xiaozhong
Qiu
c,
Aiguo
Wu
d,
Xiaoyuan
Chen
efg and
Zheyu
Shen
*abc
d,
Yikai
Xu
b,
Xiaozhong
Qiu
c,
Aiguo
Wu
d,
Xiaoyuan
Chen
efg and
Zheyu
Shen
*abc
aSchool of Biomedical Engineering, Southern Medical University, 1023 Shatai South Road, Guangzhou, Guangdong 510515, China. E-mail: sz@smu.edu.cn
bMedical Imaging Center, Nanfang Hospital, Southern Medical University, 1023 Shatai South Road, Guangzhou, Guangdong 510515, China
cGuangdong Provincial Key Laboratory of Construction and Detection in Tissue Engineering, School of Basic Medical Sciences, Southern Medical University, 1023 Shatai South Road, Guangzhou, Guangdong 510515, China
dCixi Institute of Biomedical Engineering, CAS Key Laboratory of Magnetic Materials and Devices, Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, 1219 Zhongguan West Road, Ningbo, Zhejiang 315201, China
eDepartments of Diagnostic Radiology, Surgery, Chemical and Biomolecular Engineering, and Biomedical Engineering, Yong Loo Lin School of Medicine and Faculty of Engineering, National University of Singapore, Singapore, 119074, Singapore
fClinical Imaging Research Centre, Centre for Translational Medicine, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 117599, Singapore
gNanomedicine Translational Research Program, NUS Center for Nanomedicine, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 117597, Singapore
First published on 9th February 2022
Abstract
Development of magnetic resonance imaging (MRI) contrast agents (CAs) is still one of the research hotspots due to the inherent limitations of T1- or T2-weighted CAs and T1/T2 dual-mode CAs. To dramatically enhance the MRI contrast between tumors and normal tissues, we propose a new concept of contrary contrast-MRI (CC-MRI), whose specific definition is that CC-MRI CAs present a positive or negative signal at normal tissues, but show contrary signals at diseased tissues. To realize CC-MRI of tumors, we designed and developed a tumor microenvironment (TME) dual responsive CA (i.e., SA-FeGdNP-DOX@mPEG), which is almost not responsive under normal physiological conditions, but highly responsive to the acidic and reductive TME. Our SA-FeGdNP-DOX@mPEG shows a negative MRI signal under normal physiological conditions due to the high r2 value (336.9 mM−1 s−1) and high r2/r1 ratio (18.4), but switches to a positive MRI signal in the TME because of the high r1 value (20.32 mM−1 s−1) and low r2/r1 ratio (7.2). Our TME dual responsive SA-FeGdNP-DOX@mPEG significantly enhanced the contrast of MR images between tumors and livers, and the ΔSNR difference reached 501%. In addition, our SA-FeGdNP-DOX2@mPEG2 with tumor targetability and controlled DOX release responding to the TME was also used for tumor-specific chemotherapy with reduced side effects.
New conceptsContrast agents (CAs) of magnetic resonance imaging (MRI) can potentially improve the accuracy of disease diagnosis to a higher level. In recent years, T1/T2 dual-mode MRI CAs have been developed to overcome the limitations of T1- and T2-weighted CAs. However, T1/T2 dual-mode CAs currently remain a challenge due to the quenching effects of the T2 contrast materials with strong magnetization on T1 relaxation. Herein, we propose a new concept of contrary contrast MRI (CC-MRI) to surmount the problems of T1- or T2-weighted CAs, and T1/T2 dual-mode CAs. Typically, in a single-mode of MRI sequence, the CAs of CC-MRI present a positive or negative signal at normal tissues, but show contrary signals at pathological tissues, which results in a significant enhancement of the MRI contrast between normal and pathological tissues. According to this concept, we designed and developed a tumor microenvironment dual responsive contrast agent (i.e., SA-FeGdNP-DOX@mPEG) that effectively realized CC-MRI acquiring a very high contrast of MR images between tumors and livers with 501% of ΔSNR. We believe that the new concept of CC-MRI can be regarded as an effective breakthrough in the field of MRI CAs. |
Introduction
Magnetic resonance imaging (MRI) has become a clinically highly dependent diagnostic apparatus with high spatial resolution, excellent soft-tissue contrast, non-invasiveness, safety and more.1 Meanwhile, the use of MRI contrast agents (CAs) has dramatically expanded the clinical scope of MRI technology.2,3At present, gadolinium (Gd) chelates are the most widely used MRI CAs in the clinic because the MRI images produced by T1-weighted CAs are brighter, are easier to be discerned by the naked eye and thus are preferred by physicians.4–7 The first Gd chelates approved by the US Food and Drug Administration (FDA) (i.e., Magnevist®) have been in clinical use for over 30 years. However, due to the potential risks in vivo (nephrogenic systemic fibrosis,8–10 deposition in the brain),11,12 non-specificity, and low longitudinal relaxivity (r1, ∼4 mM−1 s−1) of Gd chelates, the need to develop new alternatives to Gd chelates remains urgent.
Although T2-weighted CAs in the market (i.e., magnetic iron oxide nanoparticles, MIONs)13,14 have remarkable advantages (e.g., with tumor targetability15,16 and good biocompatibility),17,18 they were removed from the market due to the following problems: (1) the MIONs tend to cause susceptibility artifacts due to the high magnetic moment;19 (2) their inherent dark signals are not easy to be discerned by the naked eye and are easily confused with bleeding, calcification, metal deposition, etc.;20 (3) the long blood circulation and slow body clearance (60–180 nm in size) may lead to long-term toxicity;18 (4) the processing time of T2 imaging is long due to the long echo time (TE) and long repetition time (TR); (5) Eovist (Gd-EOB-DTPA), a liver-specific T1 contrast agent, approved in 2008, accelerated the elimination of T2 CAs that are mainly used for the diagnosis of liver disease from the market.21
Due to the inherent advantages and limitations of T1- and T2-weighted CAs, T1/T2 dual-mode MRI CAs have been proposed and developed in recent years.22–32 The T1/T2 dual-mode MRI continuously acquires T1- and T2-weighted images using a single MRI machine,24 providing two complementary images that cross-verify diagnostic information to reduce artifacts and misdiagnosis, while maintaining the complementary advantages of each single-mode CA.25,26 For example, Liu et al. synthesized novel intrapolymerization-doped manganese-eumelanin coordination nanocomposites that demonstrated satisfactory results for T1/T2 dual-mode MRI.26 Wang et al. developed an ultrahigh-field-tailored T1/T2 dual-mode MRI CA for high-performance vascular imaging.31
Although the T1/T2 dual-mode MRI CAs (with both high longitudinal relaxivity r1 and high transversal relaxivity r2) have certain advantages over single-mode MRI CAs, they still have some problems: (1) T1/T2 dual-mode MRI needs both T1- and T2-weighted sequences with different scanning parameters (low TE and TR for T1 imaging, but high TE and TR for T2 imaging) to be performed, which makes the busy clinical MRI machines even more engaged. (2) Diagnosis using both T1 and T2 images is more technically difficult for physicians than using single-mode images. (3) T1 and T2 signals from T1/T2 dual-mode CAs could be suppressed by each other to some extent because one is a positive signal and the other is a negative signal.
To overcome the inherent limitations of T1- or T2-weighted CAs, and T1/T2 dual-mode CAs, in this study, we propose a new concept of contrary contrast MRI (CC-MRI). In a single-mode MRI (single sequence), the CC-MRI CAs present a positive or negative signal at normal tissues, but show contrary signals at diseased tissues, which can significantly enhance the contrast between images of diseased tissues and normal tissues.
To prove the feasibility of the CC-MRI strategy, we designed and developed a CC-MRI CA, which is almost not responsive under acidic or reductive in vivo conditions, but highly responsive to the tumor microenvironment (TME) with unique acidity and high GSH content. Typically, we first synthesized core–shell Fe/Gd hybrid nanoparticles (FeGdNPs), and then developed a smart CC-MRI CA (i.e., SA-FeGdNP-DOX@mPEG) based on the FeGdNPs (Scheme 1). In detail, 4-formylbenzoic acid (FBA) was reacted with methoxy poly(ethylene glycol) (mPEG) in the presence of dicyclohexylcarbodiimide (DCC) and 4-(dimethylamino)pyridine (DMAP) to result in mPEG-FBA. Cystamine dihydrochloride (CA) was then connected to mPEG-FBA (Scheme 1a), and the obtained mPEG-FBA-CA was coupled onto the surface of core–shell nanoparticle FeGdNP to form FeGdNP@mPEG. After that, doxorubicin (DOX) was loaded onto FeGdNP@mPEG, generating well-dispersed FeGdNP-DOX@mPEG in DMF. After being transferred to pure water, FeGdNP-DOX@mPEG particles were aggregated to form SA-FeGdNP-DOX@mPEG (Scheme 1b). As shown in Scheme 1c, under normal physiological conditions, the SA-FeGdNP-DOX@mPEG has the following characteristics: (1) the circulation in the blood is long without renal excretion and with reduced liver and spleen uptake due to the ideal particle size (∼100 nm); (2) DOX release is slow due to the self-assembled structure; (3) a negative MRI signal is on due to the high r2 and high r2/r1. However, in the both acidic and reductive TME (pH ∼6.8, 2–20 mM of GSH), both the acid-labile linker benzoic imine and the GSH-labile linker disulfide bond would be broken to shed mPEG, and thus disassemble the SA-FeGdNP-DOX@mPEG nanoparticles. Therefore, (1) the tumor penetration of the disassembled nanoparticles (i.e., FeGdNP-DOX) is deep due to the small size (∼5 nm); (2) the DOX release is fast due to the disassembled structures; (3) a positive MRI signal is on due to the high r1 and low r2/r1 of FeGdNP. As a consequence, in a single-mode of MRI sequence (TE = 30 ms, TR = 1000 ms), our SA-FeGdNP-DOX@mPEG realized CC-MRI, which significantly enhanced the contrast of MR images between tumors and livers, and the ΔSNR difference reached 501%.
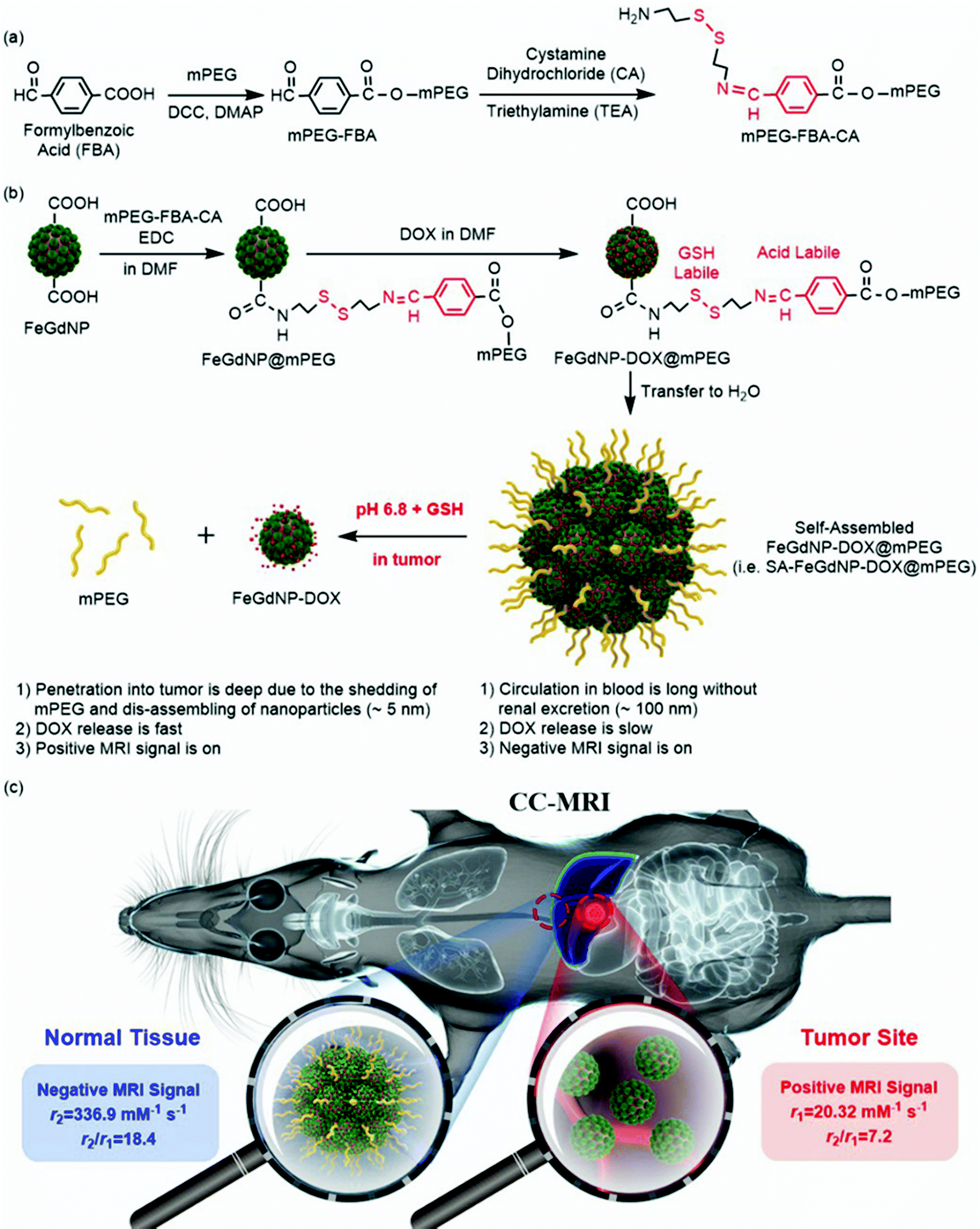 | ||
| Scheme 1 (a and b) Schematic illustration of the synthesis of mPEG-FBA-CA (a) and the CC-MRI CA (b). The FeGdNP-DOX@mPEG is well dispersed in DMF, and self-assembled after being transferred to pure water forming self-assembled FeGdNP-DOX@mPEG (i.e., SA-FeGdNP-DOX@mPEG). (c) Schematic illustration of the CC-MRI of tumors using our SA-FeGdNP-DOX@mPEG. | ||
Results and discussion
Synthesis and characterization of functional polymers
The carboxyl group (–COOH) of FBA was reacted with the hydroxyl group (–OH) of mPEG in the presence of DCC and DMAP to generate mPEG-FBA. The DCC activates the –COOH and the DMAP catalyzes the reaction, whose mechanism is shown in Fig. S1 (ESI†). Before the reaction of mPEG-FBA and CA, we studied the influence of solvents on the reaction of FBA and CA in the presence of triethylamine (TEA). Fig. S2a (ESI†) shows photos of the resulting FBA-CA1-4 solutions reacted in DMF, DMSO, THF, or ethanol. The yellow color of the FBA-CA3 solution indicates much higher recovery in THF than in the other solvents. Fig. S2b (ESI†) shows emission curves at the excitation of 405 nm, and Fig. S2c (ESI†) shows excitation curves at the emission of 510 nm. The maximum excitation and emission of FBA-CA3 are measured to be 412 and 492 nm, respectively. These fluorescence spectra demonstrate that FBA-CA1–4 are auto-fluorescent. The strongest fluorescence of FBA-CA3 indicates that THF is the best solvent for the reaction between –CHO of FBA and –NH2 of CA. Therefore, the reaction of mPEG-FBA and CA was conducted in THF to form mPEG-FBA-CA. In THF, the reaction of the aldehyde group (–CHO) on the benzene ring of mPEG-FBA and primary amino group (–NH2) at the end of the CA produced a benzoic imine through the Schiff base reaction in the presence of TEA. Thus, the obtained polymer mPEG-FBA-CA possesses both the acid-labile linker benzoic imine33 and GSH-labile linker disulfide bond.34 The chemical shifts of hydrogen atoms in the 1H NMR spectrum match the polymer formula of mPEG-FBA-CA (Fig. S3, ESI†), indicating the successful synthesis of mPEG-FBA-CA. Fig. S4 (ESI†) shows the Fourier transform infrared spectroscopy (FT-IR) spectrum of mPEG-FBA-CA in comparison with those of FBA, mPEG, CA, and mPEG-FBA. FBA has a characteristic peak at 1690 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration in carboxyl group). mPEG has a characteristic peak at 1470 cm−1 (C–H stretching vibration in methylene). Both the characteristic peaks at 1700 cm−1 (CO stretching vibration in ester) and 1470 cm−1 demonstrate the successful synthesis of mPEG-FBA. CA has a characteristic peak at 3430 cm−1 (N–H stretching vibration in primary amine). Both the characteristic peaks at 3430 cm−1 and 1650 cm−1 (CN stretching vibration) demonstrate the successful formation of mPEG-FBA-CA.
O stretching vibration in carboxyl group). mPEG has a characteristic peak at 1470 cm−1 (C–H stretching vibration in methylene). Both the characteristic peaks at 1700 cm−1 (CO stretching vibration in ester) and 1470 cm−1 demonstrate the successful synthesis of mPEG-FBA. CA has a characteristic peak at 3430 cm−1 (N–H stretching vibration in primary amine). Both the characteristic peaks at 3430 cm−1 and 1650 cm−1 (CN stretching vibration) demonstrate the successful formation of mPEG-FBA-CA.
Preparation and characterization of smart theranostic nanoplatforms
The obtained polymer mPEG-FBA-CA was then coupled on the surface of core–shell FeGdNP nanoparticles by stable amide bonds to construct FeGdNP@mPEG nanoparticles (Scheme 1b). FeGdNP@mPEG1–5 nanoparticles were prepared with different conjugation densities of polymer mPEG-FBA-CA, whose mPEG-FBA-CA/Gd mass ratio ranges from 8.0 to 0.5 (Table S1, ESI†). The FT-IR spectrum of FeGdNP@mPEG2 at the peaks of 1650 cm−1 (CN stretching vibration) and 3400 cm−1 (O–H stretching vibration in carboxyl group) demonstrates the successful conjugation of mPEG-FBA-CA on the surface of FeGdNP (Fig. S4c, ESI†).
Subsequently, MilliQ-water was added dropwise into the above FeGdNP@mPEG1–5 nanoparticle dispersions at 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio to trigger self-assembly. After dialysis purification, the self-assembled FeGdNP@mPEG1–5 (i.e., SA-FeGdNP@mPEG1–5) were obtained. Table S1 (ESI†) shows the synthesis conditions and characterization results of SA-FeGdNP@mPEG1–5. Due to the different feeding ratios of mPEG-FBA-CA/Gd, the Gd recovery calculated from the molar ratio of Gd in SA-FeGdNP@mPEG to that in the feeding FeGdNP ranged from 76% to 84%. Among them, SA-FeGdNP@mPEG2 has the highest Gd recovery of 84%.
:1 volume ratio to trigger self-assembly. After dialysis purification, the self-assembled FeGdNP@mPEG1–5 (i.e., SA-FeGdNP@mPEG1–5) were obtained. Table S1 (ESI†) shows the synthesis conditions and characterization results of SA-FeGdNP@mPEG1–5. Due to the different feeding ratios of mPEG-FBA-CA/Gd, the Gd recovery calculated from the molar ratio of Gd in SA-FeGdNP@mPEG to that in the feeding FeGdNP ranged from 76% to 84%. Among them, SA-FeGdNP@mPEG2 has the highest Gd recovery of 84%.
It is possible that one CA reacts with two mPEG-FBA generating (mPEG-FBA)2-CA, which has no –NH2 and cannot be conjugated to FeGdNP. Therefore, the (mPEG-FBA)2-CA can be removed from the nanoparticles via dialysis (Mw cut-off 12–14 kDa).
The fluorescence spectra of our autofluorescent SA-FeGdNP@mPEG2 nanoplatforms were then determined by fluorescence spectrophotometry (Fig. S5, ESI†). At 650 nm of emission, the maximum excitation of the nanoparticles was recorded at 425 nm (Fig. S5a, ESI†). The fluorescence spectra of the SA-FeGdNP@mPEG2 with various Gd concentrations (CGd) from 3.63 to 116 μM were then measured at 425 nm of excitation (Fig. S5b, ESI†). The relationship between the fluorescence intensities (Ex 425 nm, Em 646 nm) of our SA-FeGdNP@mPEG2 nanoplatforms and CGd (μM) or CmPEG-FBA-CA (μg mL−1) is linear (Fig. S5c, ESI†). The SA-FeGdNP@mPEG2 nanoplatform is autofluorescent without loading any fluorescent agents due to the CN bonds in the Schiff base, which had been demonstrated to be autofluorescent by Wei et al.35 The autofluorescence property of our SA-FeGdNP@mPEG2 nanoplatforms is beneficial for in vitro and in vivo trace studies.
SA-FeGdNP-DOX1–5@mPEG2 nanoplatforms were finally prepared utilizing a procedure similar to that of the above SA-FeGdNP@mPEG at different mass ratios of feeding DOX to Gd in FeGdNP. The synthesis conditions and characterization results of SA-FeGdNP-DOX1–5@mPEG2 are summarized in Table S2 (ESI†). TEM images of FeGdNP and SA-FeGdNP-DOX1–5@mPEG2 (with 1.6–0.1 of DOX/Gd mass ratio) are shown in Fig. 1. Clearly, FeGdNP is well-dispersed without any aggregates at all. SA-FeGdNP-DOX1@mPEG2 nanoplatforms aggregate seriously, indicating that the DOX/Gd mass ratio of 1.6 is too high. SA-FeGdNP-DOX5@mPEG2 nanoplatforms are not uniform due to the low DOX/Gd mass ratio of 0.1. SA-FeGdNP-DOX2–4@mPEG2 are well-dispersed assemblies of FeGdNP because of the appropriate DOX/Gd mass ratio of 0.8–0.2. The average particle sizes of SA-FeGdNP-DOX2–4@mPEG2 were measured to be 77.8, 71.3, and 66.6 nm, respectively (Fig. S6, ESI†). The DOX loading content was determined to be 20.6, 13.2, and 7.1% for SA-FeGdNP-DOX2–4@mPEG2 (Table S2, ESI†), which indicates that the DOX/Gd mass ratio of 0.8 should be the optimal condition.
 | ||
| Fig. 1 TEM images of FeGdNP (a), and SA-FeGdNP-DOX1-5@mPEG2 (b–f). FeGdNP is well-dispersed without any aggregates at all. SA-FeGdNP-DOX1@mPEG2 aggregates seriously, indicating that 1.6 DOX/Gd mass ratio is too high. SA-FeGdNP-DOX2-5@mPEG2 (with 0.8–0.1 of DOX/Gd mass ratio) shows well-dispersed assemblies of FeGdNP. | ||
The T1 and T2 relaxation rates were measured as a function of CGd for SA-FeGdNP-DOX1–5@mPEG2 (Fig. S7, ESI†). There is no linear relationship between 1/T2 and CGd for SA-FeGdNP-DOX1@mPEG2 (R2 = 0.932) because a 1.6 DOX/Gd mass ratio is too high resulting in unstable aggregates. The r1, r2, and r2/r1 of SA-FeGdNP-DOX2–5@mPEG2 and FeGdNP are summarized in Table S2 (ESI†). In general, a low r2/r1 ratio brings T1-dominant contrast, and the higher the r1 value, the more conducive to showing positive MRI signals. Conversely, a high r2/r1 ratio results in T2-dominant contrast, and a higher r2 value is preferable to give negative MRI signals.36,37 The high r1 value (20.32 mM−1 s−1) and low r2/r1 ratio (7.2) indicate that our FeGdNP is an excellent positive MRI CA. In addition, the high r2 value and high r2/r1 ratio demonstrate that the negative MRI signal of SA-FeGdNP-DOX2–5@mPEG2 is on. Due to the highest r2 value and r2/r1 ratio, SA-FeGdNP-DOX2@mPEG2 should be the optimal choice, which is consistent with the above conclusion drawn from the DOX loading content.
In vitro disassembly and release behavior of SA-FeGdNP-DOX2@mPEG2
Our smart theranostic nanoplatform SA-FeGdNP-DOX2@mPEG2 contains both benzoic imine and a disulfide bond, whose labile properties under the acidic and reductive tumor microenvironment (TME, pH ∼6.8, CGSH = 2–20 mM) were subsequently investigated.Fig. 2a–d show the TEM images of SA-FeGdNP-DOX2@mPEG2 after 24 h of incubation at in vitro simulated TME (pH = 6.8, CGSH = 10 mM) or other conditions. Compared to the group of pH 7.4 without GSH (normal physiological condition, Fig. 2a), both pH 6.8 without GSH (Fig. 2b) and pH 7.4 with 10 mM GSH (Fig. 2c) almost did not lead to the disassembling of SA-FeGdNP-DOX2@mPEG2. However, after incubation at pH 6.8 with 10 mM GSH (TME mimic), SA-FeGdNP-DOX2@mPEG2 was almost completely disassembled (Fig. 2d). Therefore, although the acidic condition can trigger the breakage of acid-labile linker benzoic imine and the reductive condition can induce the breakage of the GSH-labile linker disulfide bond (Scheme 1), acidic or reductive conditions cannot easily disconnect the SA-FeGdNP-DOX2@mPEG2 nanoparticles due to the tight assembly. However, both acidic and reductive conditions (dual responsive) can result in rapid shedding of mPEG and quick disassembly of SA-FeGdNP-DOX2@mPEG2 nanoparticles. From this perspective, the real TME with unique acidity and high GSH content (pH ∼6.8, CGSH = 2–20 mM) in vivo should be able to induce the whole disassembly of SA-FeGdNP-DOX2@mPEG2 to realize CC-MRI of tumors, but other acidic or reductive in vivo conditions cannot (e.g., intercellular reductive conditions of the liver, or acidic environment of Kupffer cell endosomes).
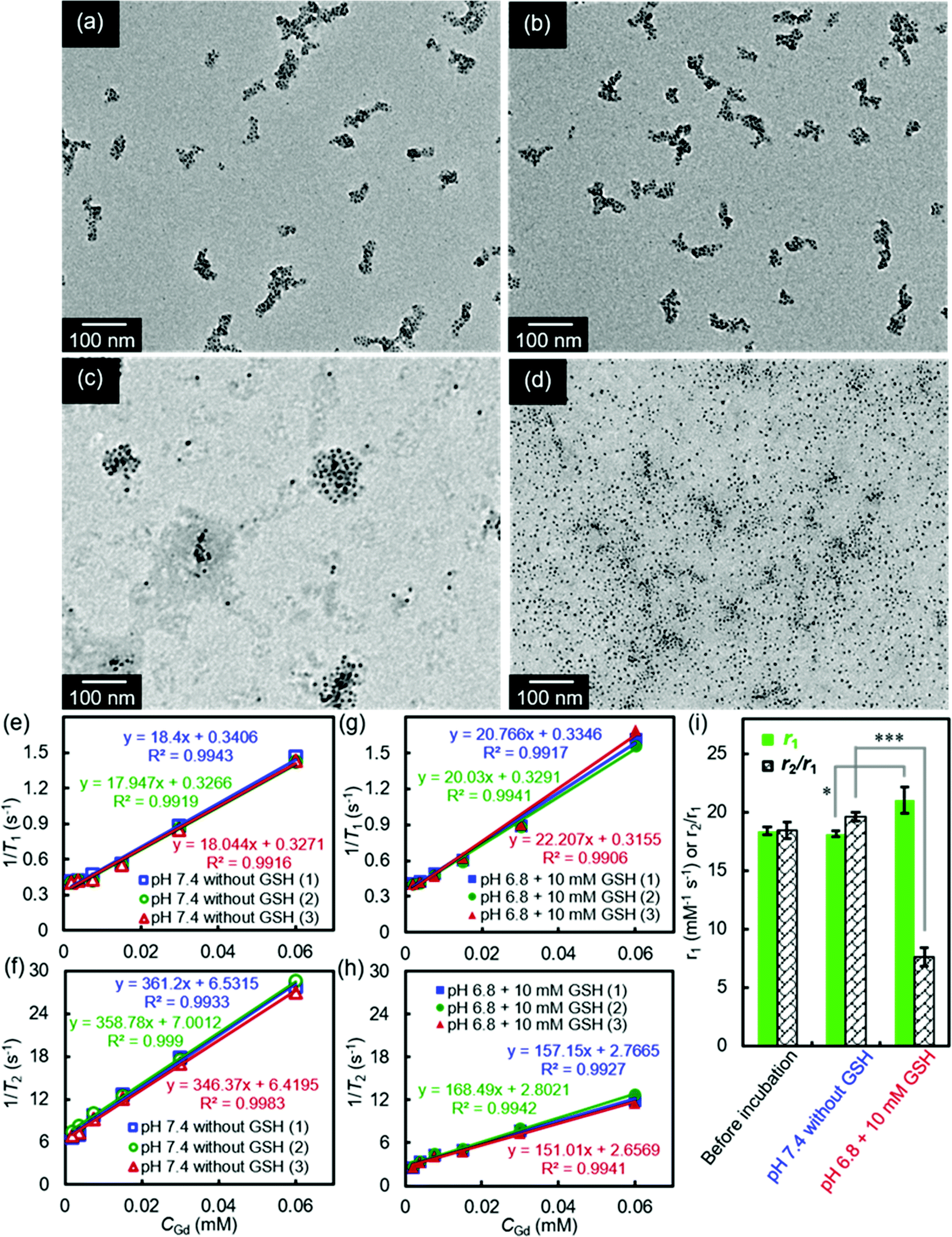 | ||
| Fig. 2 (a–d): TEM images of SA-FeGdNP-DOX2@mPEG2 after 24 h of incubation at pH 7.4 without GSH (a), at pH 6.8 without GSH (b), at pH 7.4 with 10 mM of GSH (c), or at pH 6.8 with 10 mM of GSH (d), showing the disassembling of the SA-FeGdNP-DOX2@mPEG2 in a mimetic tumor microenvironment (pH = 6.8, CGSH = 10 mM). (e–h) T1 relaxation rates (1/T1) (e, g) and T2 relaxation rates (1/T2) (f, h) plotted as a function of CGd for SA-FeGdNP-DOX2@mPEG2 after 24 h of incubation at pH 7.4 without GSH, or at pH 6.8 with 10 mM of GSH. (i) Comparison of r1 or r2/r1 of SA-FeGdNP-DOX2@mPEG2 nanoparticles before incubation, after 24 h of incubation at pH 7.4 without GSH, and at pH 6.8 with 10 mM of GSH. (1), (2) and (3) indicate the same samples from 3 different batches. *P < 0.05; ***P < 0.001. | ||
The MRI relaxation properties of SA-FeGdNP-DOX2@mPEG2 nanoplatforms after 24 h of incubation at mimetic TME or normal physiological conditions were then evaluated. Fig. 2e–h show the T1/T2 relaxation rates plotted as a function of CGd of SA-FeGdNP-DOX2@mPEG2 nanoparticles. The r2 value of SA-FeGdNP-DOX2@mPEG2 nanoparticles after 24 h of incubation at pH 7.4 without GSH is very different from that after 24 h of incubation at pH 6.8 with 10 mM of GSH. The r2 value of the disassembled nanoparticles is so much smaller than that before disassembly because the r2 value is proportional to saturation magnetization and particle size.38 The r1 and r2/r1 of SA-FeGdNP-DOX2@mPEG2 nanoparticles before incubation, after 24 h of incubation at pH 7.4 without GSH (normal physiological conditions), or at pH 6.8 with 10 mM GSH (mimetic TME) are compared in Fig. 2i. It was found that incubation at pH 7.4 without GSH almost cannot induce any changes of r1 and r2/r1. However, incubation at pH 6.8 with 10 mM GSH results in significant enhancement of r1 (*P < 0.05) and huge decrease of r2/r1 (***P < 0.001). This result reinforces that the acidic and GSH-abundant TME can trigger complete disassembly of the SA-FeGdNP-DOX2@mPEG2 nanoplatform to release FeGdNP. Since the MRI performance of magnetic nanoparticles is dependent on their aggregation state,39,40 the assembly and disassembly of our smart SA-FeGdNP-DOX@mPEG nanoplatform shows a negative MRI signal under normal physiological conditions, but switches to a positive MRI signal under TME, whose smart signal switch can be used for high contrast MRI of tumors.
The DOX release behavior of SA-FeGdNP-DOX2@mPEG2 nanoplatforms at pH 7.4 without GSH or pH 6.8 with 10 mM GSH is shown in Fig. S8 (ESI†). The DOX release under a neutral environment without GSH was less than 20% even after 72 h. However, in an acidic and reductive environment, SA-FeGdNP-DOX2@mPEG2 released over 80% of the loaded DOX at 72 h (***P < 0.001), suggesting that SA-FeGdNP-DOX2@mPEG2 allows sustainable DOX release under the TME. This result further demonstrates that our SA-FeGdNP-DOX2@mPEG2 is stable under normal physiological conditions, but can be disassembled to release the loaded DOX under TME facilitating tumor treatment.
Cellular uptake and chemotherapeutic efficacy of SA-FeGdNP-DOX2@mPEG2
We observed the cellular uptake behavior of the SA-FeGdNP-DOX2@mPEG2 nanoplatform under simulated TME or normal physiological conditions. Fig. S9 and S10 (ESI†) show the laser scanning confocal microscopy (LSCM) images of U-87 MG and HepG2 cells incubated with SA-FeGdNP-DOX2@mPEG2 post-incubation at pH 6.8 with 10 mM of GSH, or pH 7.4 without GSH for 24 h. The merged LSCM images of the U-87 MG cells and HepG2 cells are summarized in Fig. 3. It was found that for both U-87 MG and HepG2 cells, the uptake amount of the nanoplatforms post-incubation at pH 6.8 with 10 mM GSH is much higher than that at pH 7.4 without GSH. Most likely the simulated TME (pH 6.8 with 10 mM GSH) can result in mPEG shedding and disassembly of SA-FeGdNP-DOX2@mPEG2 to release DOX-loaded FeGdNP and free DOX, which become much easier to be taken up by cells than SA-FeGdNP-DOX2@mPEG2 due to the smaller sizes.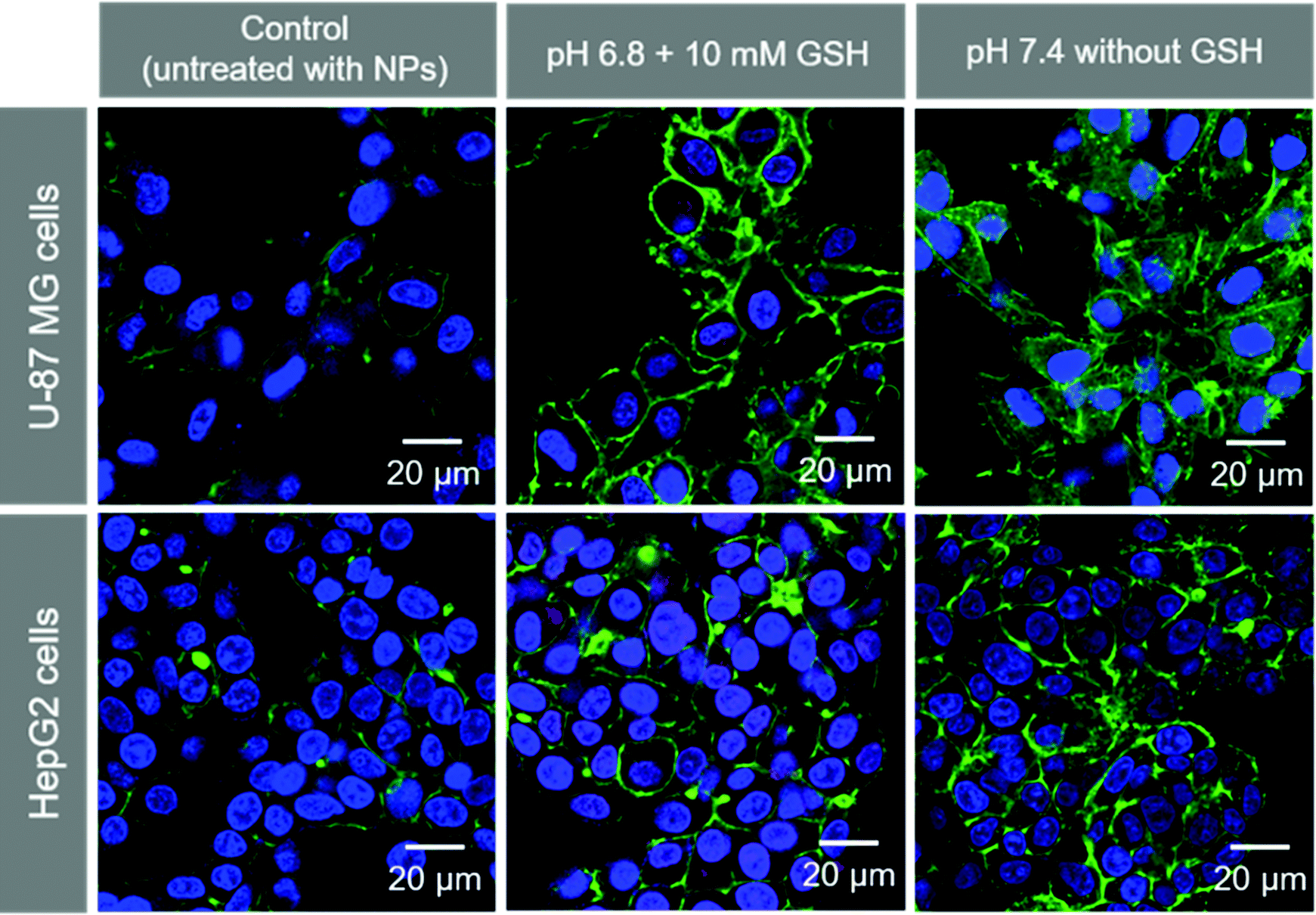 | ||
| Fig. 3 LSCM images of U-87 MG and HepG2 cells incubated with SA-FeGdNP-DOX2@mPEG2 nanoparticles. Before incubation with cells, the nanoparticles were incubated at 37 °C for 24 h at pH 6.8 with 10 mM of GSH, or at pH 7.4 without GSH. The cells untreated with nanoparticles are used as the control. DOX is red. The cytoskeleton stained with phalloidin-FITC is green. The nucleus stained with Hoechst 33258 is blue. | ||
Fig. S11 (ESI†) shows the LSCM images of U-87 MG cells incubated with SA-FeGdNP-DOX2@mPEG2 nanoparticles. Before incubation with cells, the nanoparticles were incubated at 37 °C for 24 h at pH 6.8 with 10 mM of GSH. The red signal (DOX) overlaps with the green signal (endosome and lysosome stained with LysoTrackerTM) and blue signal (nucleus stained with Hoechst 33258), which demonstrates that the released DOX-loaded FeGdNP and free DOX were internalized into endosomes via an endocytosis mechanism, and some of them escaped from the endosome and lysosome and entered the nucleus.
The cytotoxicity of SA-FeGdNP@mPEG2 and SA-FeGdNP@DOX2@mPEG2 at pH 7.4 without GSH or pH 6.8 with 10 mM of GSH was further directly demonstrated by MTT assay on U-87 MG cells (Fig. S12a, ESI†) and HepG2 cells (Fig. S12b, ESI†). Cells incubated with SA-FeGdNP@mPEG2 at both pH 7.4 without GSH and pH 6.8 with 10 mM of GSH showed almost no cytotoxicity, indicating good biocompatibility of SA-FeGdNP@mPEG2. However, cells incubated with SA-FeGdNP@DOX2@mPEG2 at pH 6.8 in the presence of 10 mM GSH show much higher cytotoxicity than that at pH 7.4 without GSH (***P < 0.001). The cell viability of SA-FeGdNP@DOX2@mPEG2 treatment at pH 6.8 with 10 mM GSH was similar to that of free DOX at any DOX concentration in the range from 0.6 to 10 μg mL−1 on both U-87 MG and HepG2 cells. The high-performance chemotherapy of SA-FeGdNP-DOX2@mPEG2 can be ascribed to the high DOX loading content and efficient DOX release in the TME.
These results reinforce that our SA-FeGdNP-DOX2@mPEG2 nanoplatforms should be able to darken normal tissues (high r2 value and high r2/r1 ratio under normal physiological conditions) in MR images with low DOX side effects (low cellular uptake and low DOX release), but brighten tumors (high r1 value and low r2/r1 ratio under TME) with high-performance chemotherapy (high cellular uptake and high DOX release).
In vivo MR imaging and tumor chemotherapy
To evaluate the MRI efficiency of CC-MRI CA SA-FeGdNP-DOX2@mPEG2 in vivo, a U-87 MG tumor model was established in nude mice. SA-FeGdNP-DOX2@mPEG2 and commercial Dotarem® were injected into the tumor-bearing mice via the tail vein, and MR images were obtained at different time points post-injection (Fig. 4a–h). As shown in Fig. 4c, the positive MRI signal of the tumors reached the maximum at around 20 min post-injection of Dotarem®. The tumors showed the highest positive signals at around 24 h post-injection of SA-FeGdNP-DOX2@mPEG2 (Fig. 4g), which is much higher than that at 20 min post-injection of Dotarem®. After that, we used ΔSNR to quantify the MR signals of the tumors at different time points (Fig. 4i and j). The highest tumor ΔSNR of SA-FeGdNP-DOX2@mPEG2 was 357.3 ± 34.2% (at 24 h post-injection), which is significantly higher than that of Dotarem® (87.3 ± 12.0%, at 20 min post-injection, ***P < 0.001). The much stronger MRI efficiency of our SA-FeGdNP-DOX2@mPEG2 compared with commercial Dotarem® can be ascribed to the following two aspects: (1) the nanoparticles can passively target tumors via extravasation from the high-density and leaky tumor blood vessels, and retention in solid tumors due to the ineffective lymphatic drainage (known as the enhanced permeability and retention (EPR) effect), but commercial Gd-chelates do not;41,42 (2) the SA-FeGdNP-DOX2@mPEG2 can be disassembled in the TME to release FeGdNP, whose r1 (20.32 mM−1 s−1) is much higher than that of Dotarem® (3.68 mM−1 s−1).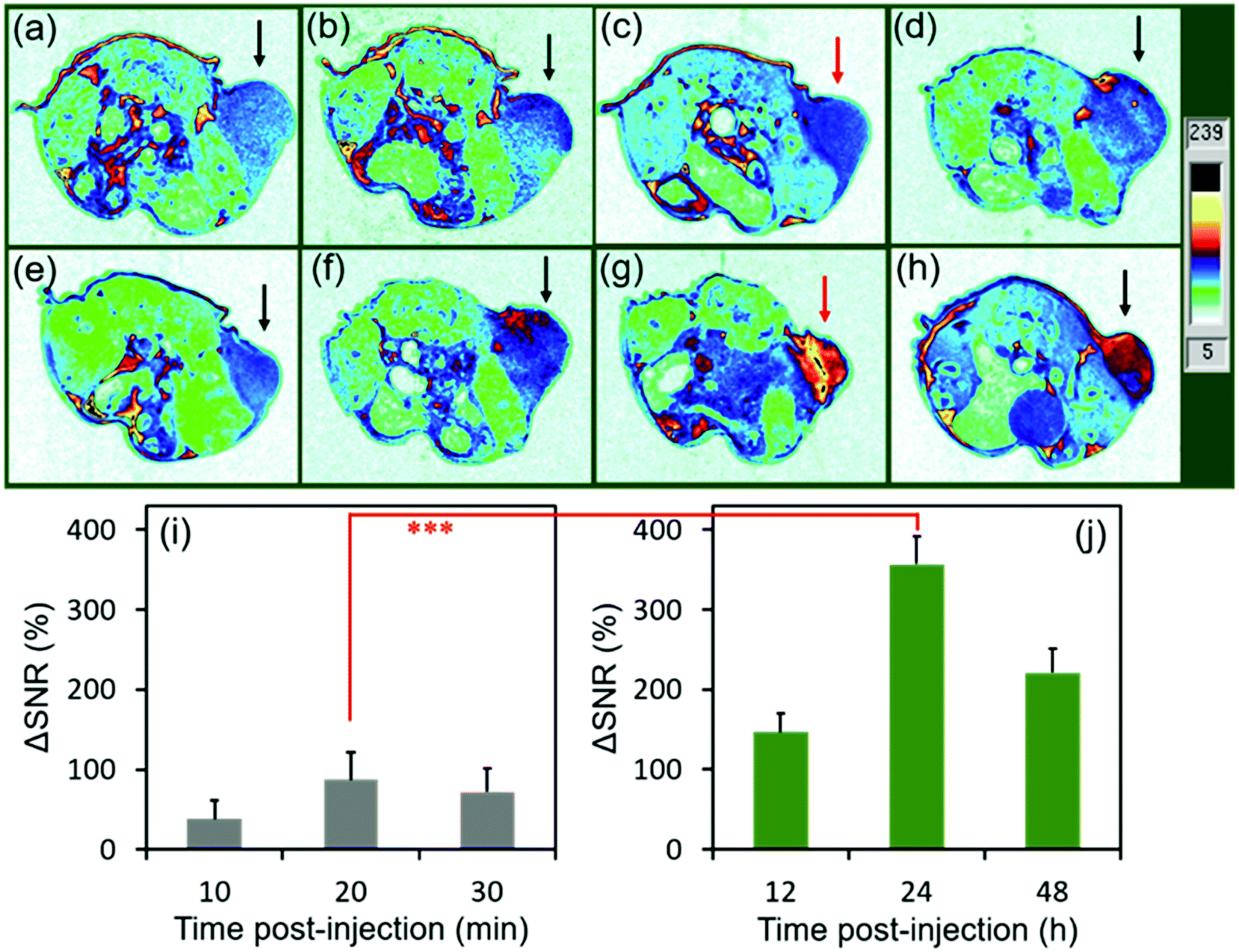 | ||
| Fig. 4 MR imaging and quantitative analysis of mice bearing U-87 MG tumors pre- or post-injection (i.v.). The slice orientation is axial. (a–d) The mouse showing tumors pre- (a), or at 10 min (b), 20 min (c), and 30 min (d) post-injection of commercial Dotarem® (r1 = 3.68 mM−1 s−1, r2 = 5.05 mM−1 s−1,and r2/r1 = 1.37). (e–h) The mouse showing tumors pre- (e), or at 12 h (f), 24 h (g), or 48 h (h) post-injection of SA-FeGdNP-DOX2@mPEG2 nanoparticles. (i, j) Quantitative analysis of the tumors after injection of Dotarem® (i) or SA-FeGdNP-DOX2@mPEG2 nanoparticles (j) using ΔSNR. The Gd injection dosage is 5.0 mg kg−1. The magnetic field is 7.0 T. TE = 30 ms, TR = 1000 ms. ***P < 0.001. SNR = SImean/SDnoise. ΔSNR = (SNRpost − SNRpre)/SNRpre × 100%. | ||
To evaluate the efficiency of CC-MRI, SA-FeGdNP-DOX2@mPEG2 nanoparticles were intravenously injected into U-87 MG tumor mice and the tumor signal was compared with that of the liver. For fair comparison, Fig. 5a–h show the slices with the most strong MRI signal, and not slices with the same depth. At 24 h post-injection, the MRI signal of the tumors reached a maximum, but that of the livers was minimum (Fig. 5a–h). That is because both an acidic and reductive condition (i.e., TME) can trigger the disassembly of SA-FeGdNP-DOX2@mPEG2, but other acidic or reductive in vivo conditions (e.g., intercellular reductive conditions of livers, or acidic environment of Kupffer cell endosomes) cannot (Fig. 2a–d). The corresponding ΔSNR values are shown in Fig. 5i and j. The maximum ΔSNR of tumors is 302 ± 32%, and the minimum ΔSNR of livers is −199 ± 30%. The smart MRI signal switch of our SA-FeGdNP-DOX2@mPEG2 nanoplatforms significantly enhanced the contrast of MR images between a tumor and the liver, whose ΔSNR difference was as high as 501%. These results show that our SA-FeGdNP-DOX2@mPEG2 nanoparticles with smart signal switch can be used for CC-MRI of tumors.
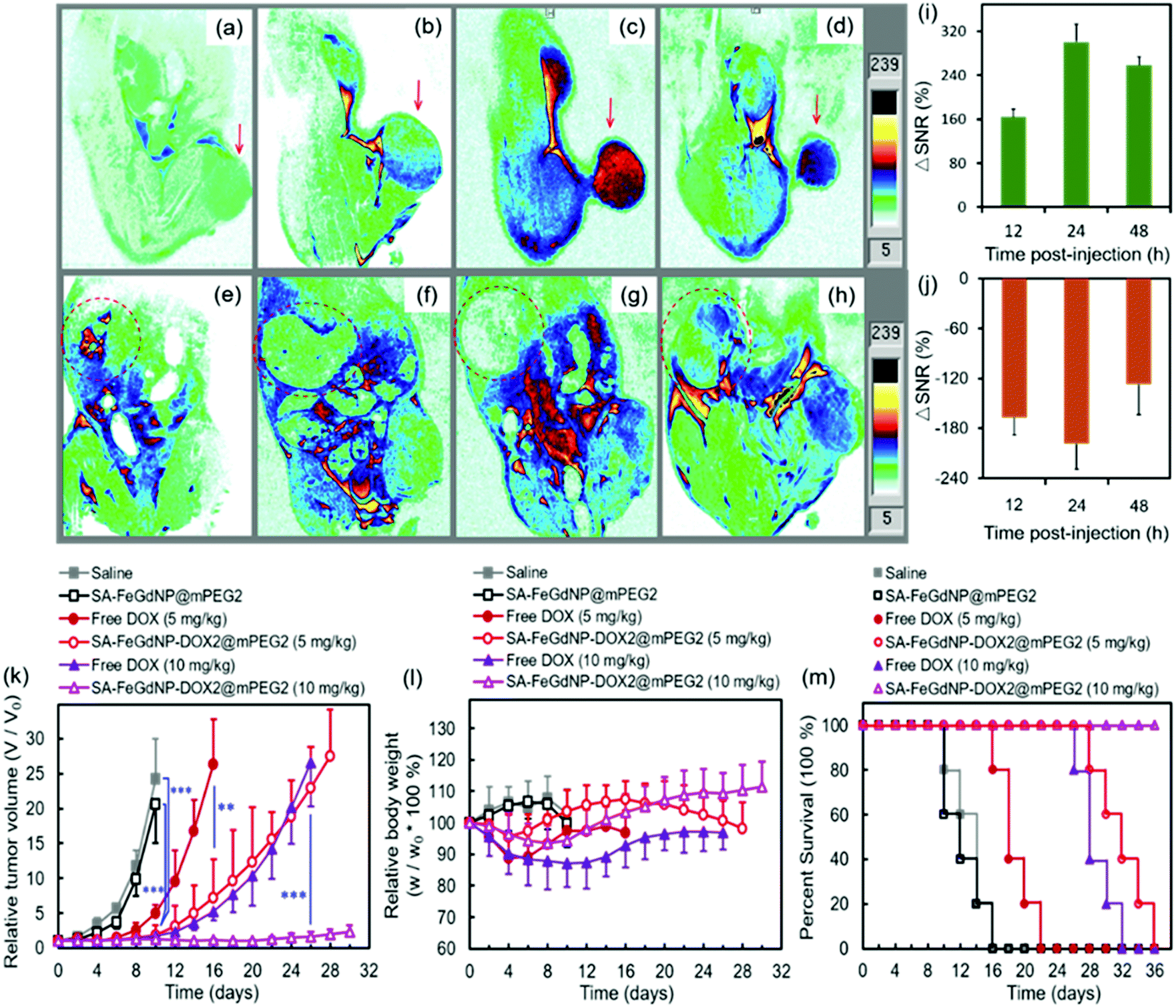 | ||
| Fig. 5 (a–j) MR imaging and quantitative analysis of mice bearing U-87 MG tumors pre- and post-injection (i.v.) of SA-FeGdNP-DOX2@mPEG2. The slice orientation is coronal. The mouse showing tumors (a–d) or livers (e–h) before injection (a and e), and at 12 h (b and f), 24 h (c and g), or 48 h (d and h) post-injection. Quantitative analysis of the tumors (i) or livers (j) using ΔSNR. The Gd injection dosage is 5.0 mg kg−1. The magnetic field is 7.0 T. TE = 30 ms, TR = 1000 ms. (k–m) Anti-tumor therapy of saline, SA-FeGdNP@mPEG2, and free DOX and SA-FeGdNP-DOX2@mPEG2 with 5 mg kg−1 or 10 mg kg−1 of DOX dosage on nude mice bearing U-87 MG tumors (mean ± SD, n = 5). (k) Tumor growth curves of the mice after treatment. (l) Changes of the mouse body weights after treatment. (m) The mouse percent survival rates after treatment. **P < 0.01, ***P < 0.001. | ||
The therapeutic efficacies of our SA-FeGdNP-DOX2@mPEG2 nanoplatforms in vivo with 5 mg kg−1 or 10 mg kg−1 of DOX dosage were compared with free DOX, SA-FeGdNP@mPEG2, and PBS in U-87 MG tumor-bearing nude mice. The therapeutic agents were administered via intravenous injection. Quantitative analysis of tumor growth (Fig. 5k) shows that the tumors in both the PBS group and SA-FeGdNP@mPEG2 group grew exponentially, whose relative tumor volumes on day 10 were both significantly larger than those of the SA-FeGdNP-DOX2@mPEG2 group (5 mg kg−1, ***P < 0.001). Tumor growth in the free DOX group was initially delayed, but relapsed over time, whose relative tumor volume on day 16 was also much larger than that of the SA-FeGdNP-DOX2@mPEG2 group (5 mg kg−1, **P < 0.01). With 10 mg kg−1 of DOX dosage, the tumor growth in the SA-FeGdNP-DOX2@mPEG2 group was almost completely suppressed. On day 26 post-treatment, the average relative tumor volume of the SA-FeGdNP-DOX2@mPEG2 (10 mg kg−1) group was significantly different from that of the free DOX (10 mg kg−1) group (***P < 0.001). Therefore, the tumor treatment of our SA-FeGdNP-DOX2@mPEG2 nanoplatforms is highly efficient.
Fig. 5l shows the changes of mouse body weight after treatment. The initial body weight loss and overall lower body weight of the free DOX group compared with the other 3 groups indicate the serious side effects of free DOX. However, the obviously higher body weight of our SA-FeGdNP-DOX2@mPEG2 group demonstrates the much-reduced side effects. In addition, on day 20 post-injection, the survival rate of mice treated with SA-FeGdNP-DOX2@mPEG2 (5 mg kg−1) was still 100%, while those treated with free DOX (5 mg kg−1), SA-FeGdNP@mPEG2, and saline were 20%, 0%, and 0%, respectively (Fig. 5m). On day 36 post-injection, the tumor-bearing mice treated with SA-FeGdNP-DOX2@mPEG2 (10 mg kg−1) were all alive, but the mice of other groups all died. The excellent survival rate benefits from the high-performance tumor therapy.
Fig. S13 (ESI†) shows histological analysis of the major organs and tumors (H&E staining) of subcutaneous tumor-bearing nude mice on day 2 after intravenous administration of saline, SA-FeGdNP@mPEG2, and SA-FeGdNP-DOX2@mPEG2. The main organs (heart, liver, spleen, lungs, and kidneys) and tumors had no apparent toxicity after the injection of saline and SA-FeGdNP@mPEG2. Besides, no toxicity was found for the principal organs, including the heart, liver, spleen, lungs, and kidneys, in tumor-bearing nude mice treated with SA-FeGdNP-DOX2@mPEG2. However, significant toxicity was found for the tumors, indicating efficient tumor treatment and potential biocompatibility of our SA-FeGdNP-DOX2@mPEG2. These results show that our SA-FeGdNP-DOX2@mPEG2 nanoplatforms can be used for high performance tumor chemotherapy with reduced side effects due to the tumor targetability and controlled DOX release responding to TME.
Conclusions
In summary, we proposed a new concept of single-mode CC-MRI to solve the problems of T1/T2 dual-mode MRI. We first synthesized polymer mPEG-FBA-CA containing both the acid-labile linker benzoic imine and a GSH-labile linker disulfide bond, whose successful synthesis was proved by the 1H NMR spectrum and FT-IR spectrum. The mPEG-FBA-CA was then conjugated onto core–shell nanoparticles FeGdNPs followed by DOX loading and self-assembly to construct the CC-MRI CA (i.e., SA-FeGdNP-DOX@mPEG). TEM images show that the acidic and reductive TME triggered the breakage of the acid-labile linker benzoic imine and GSH-labile linker disulfide bond, resulting in rapid shedding of mPEG and quick disassembly of SA-FeGdNP-DOX2@mPEG2 nanoparticles to release FeGdNP. The assembly of our smart SA-FeGdNP-DOX@mPEG can switch on a negative MRI signal under normal physiological conditions due to the high r2 value (336.9 mM−1 s−1) and high r2/r1 ratio (18.4), but the disassembly switches on a positive MRI signal in the TME because of the high r1 value (20.32 mM−1 s−1) and low r2/r1 ratio (7.2), whose smart signal switch can be used for CC-MRI of tumors. The DOX release behavior reinforces that our SA-FeGdNP-DOX2@mPEG2 is stable under normal physiological conditions, but can be disassembled to release the loaded DOX under TME facilitating tumor treatment. In a single-mode of MRI sequence (TE = 30 ms, TR = 1000 ms), the MRI studies on tumor-bearing mice show that the maximum ΔSNR of tumors is 302 ± 32%, and the minimum ΔSNR of livers is −199 ± 30%. As a consequence, our SA-FeGdNP-DOX@mPEG with smart MRI signal switch realized CC-MRI, which significantly enhanced the contrast of MR images between tumors and livers, and the ΔSNR difference reached 501%. The treatment studies on tumor-bearing mice demonstrate that our CC-MRI CA SA-FeGdNP-DOX2@mPEG2 can also be used for high-performance tumor chemotherapy with reduced side effects due to the tumor targetability and controlled DOX release responding to the TME.Author contributions
Yudie Lu: methodology, investigation, validation, data curation, writing – original draft, and writing –review and editing. Jie Feng: methodology, investigation, resources, software, and writing – review and editing. Zhiyu Liang: methodology, investigation, and validation. Xuanyi Lu: investigation, methodology, and validation. Shuai Guo: investigation, methodology. Lin Huang: investigation, methodology. Wei Xiong: investigation. Sijin Chen: investigation. Huimin Zhou: investigation. Xuehua Ma: investigation. Yikai Xu: resources, supervision, and writing – review and editing. Xiaozhong Qiu: resources, supervision, and writing – review and editing. Aiguo Wu: resources, supervision, and writing – review and editing. Xiaoyuan Chen: resources, supervision, and writing – review and editing. Zheyu Shen: conceptualization, formal analysis, resources, funding acquisition, supervision, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the Guangzhou Key Research and Development Program of China (202103000094), Guangdong Provincial Natural Science Foundation of China (2021A1515010605), Zhejiang Provincial Natural Science Foundation of China (LR19E030001), and National Natural Science Foundation of China (51761145021).Notes and references
- X. Hu, Y. Tang, Y. Hu, F. Lu, X. Lu, Y. Wang, J. Li, Y. Li, Y. Ji, W. Wang, D. Ye, Q. Fan and W. Huang, Theranostics, 2019, 9, 4168–4181 CrossRef CAS PubMed.
- X. Sun, Y. Cai, Z. Xu and D. Zhu, Molecules, 2019, 24, 1477 CrossRef CAS PubMed.
- G. Angelovski, Angew. Chem., Int. Ed., 2016, 55, 7038–7046 CrossRef CAS PubMed.
- F. De Sarno, A. M. Ponsiglione, M. Russo, A. M. Grimaldi, E. Forte, P. A. Netti and E. Torino, Theranostics, 2019, 9, 1809–1824 CrossRef CAS PubMed.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS PubMed.
- T. Anani, S. Rahmati, N. Sultana and A. E. David, Theranostics, 2011, 11, 579–601 CrossRef PubMed.
- Y. Miao, Q. Xie, H. Zhang, J. Cai, X. Liu, J. Jiao, S. Hu, A. Ghosal, Y. Yang and H. Fan, Theranostics, 2019, 9, 1764–1776 CrossRef CAS PubMed.
- Z. Zou, H. L. Zhang, G. H. Roditi, T. Leiner, W. Kucharczyk and M. R. Prince, JACC Cardiovasc. Imaging, 2011, 4, 1206–1216 CrossRef PubMed.
- A. S. Peak and A. Sheller, Ann. Pharmacother., 2007, 41, 1481–1485 CrossRef PubMed.
- I. A. Mendichovszky, S. D. Marks, C. M. Simcock and Ø. E. Olsen, Pediatr. Radiol., 2008, 38, 489–496 CrossRef PubMed.
- T. Kanda, T. Fukusato, M. Matsuda, K. Toyoda, H. Oba, J. I. Kotoku, T. Haruyama, K. Kitajima and S. Furui, Radiology, 2015, 276, 228–232 CrossRef PubMed.
- V. Gulani, F. Calamante, F. G. Shellock, E. Kanal and S. B. Reeder, Lancet Neurol., 2017, 16, 564–570 CrossRef PubMed.
- D. Ni, W. Bu, E. B. Ehlerding, W. Cai and J. Shi, Chem. Soc. Rev., 2017, 46, 7438–7468 RSC.
- Z. Gao, Y. Hou, J. Zeng, L. Chen, C. Liu, W. Yang and M. Gao, Adv. Mater., 2017, 29, 1701095 CrossRef PubMed.
- F. Rety, O. Clement, N. Siauve, C. A. Cuenod, F. Carnot, M. Sich, A. Buisine and G. Frija, J. Magn. Reson. Imaging, 2000, 12, 734–739 CrossRef CAS PubMed.
- T. Allkemper, C. Bremer, L. Matuszewski, W. Ebert and P. Reimer, Radiology, 2002, 223, 432 CrossRef PubMed.
- K. Briley-Saebo, A. Bj Rnerud, D. Grant, H. K. Ahlstrom, T. Berg and G. M. R. Kindberg, Cell Tissue Res., 2004, 316, 315–323 CrossRef CAS PubMed.
- L. Gu, R. H. Fang, M. J. Sailor and J. Park, ACS Nano, 2012, 6, 4947–4954 CrossRef CAS PubMed.
- J. W. M. Bulte and D. L. Kraitchman, NMR Biomed., 2004, 17, 484–499 CrossRef CAS PubMed.
- H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133–2148 CrossRef CAS.
- Y. X. Wang, World J. Gastroenterol., 2015, 21, 13400–13402 CrossRef CAS PubMed.
- J. Choi, J. Lee, T. Shin, H. Song, E. Y. Kim and J. Cheon, J. Am. Chem. Soc., 2010, 132, 11015–11017 CrossRef CAS PubMed.
- T. Shin, Y. Choi, S. Kim and J. Cheon, Chem. Soc. Rev., 2015, 44, 4501–4516 RSC.
- Z. Zhou, R. Bai, J. Munasinghe, Z. Shen, L. Nie and X. Chen, ACS Nano, 2017, 11, 5227–5232 CrossRef CAS PubMed.
- N. Lee, D. Yoo, D. Ling, M. H. Cho, T. Hyeon and J. Cheon, Chem. Rev., 2015, 115, 10637–10689 CrossRef CAS PubMed.
- H. Liu, C. Chu, Y. Liu, X. Pang, Y. Wu, Z. Zhou, P. Zhang, W. Zhang, G. Liu and X. Chen, Adv. Sci., 2018, 5, 1800032 CrossRef PubMed.
- T. Shin, J. Choi, S. Yun, I. Kim, H. Song, Y. Kim, K. I. Park and J. Cheon, ACS Nano, 2014, 8, 3393–3401 CrossRef CAS PubMed.
- X. Sun, G. Zhang, R. Du, R. Xu, D. Zhu, J. Qian, G. Bai, C. Yang, Z. Zhang, X. Zhang, D. Zou and Z. Wu, Biomaterials, 2019, 194, 151–160 CrossRef CAS PubMed.
- Z. Zhou, D. Huang, J. Bao, Q. Chen, G. Liu, Z. Chen, X. Chen and J. Gao, Adv. Mater., 2014, 24, 6223–6228 CrossRef PubMed.
- L. Gao, J. Yu, Y. Liu, J. Zhou, L. Sun, J. Wang, J. Zhu, H. Peng, W. Lu, L. Yu, Z. Yan and Y. Wang, Theranostics, 2018, 8, 92–108 CrossRef CAS PubMed.
- J. Wang, Y. Jia, Q. Wang, Z. Liang, G. Han, Z. Wang, J. Lee, M. Zhao, F. Li, R. Bai and D. Ling, Adv. Mater., 2021, 33, 2004917 CrossRef CAS PubMed.
- X. Li, S. Lu, Z. Xiong, Y. Hu, D. Ma, W. Lou, C. Peng, M. Shen and X. Shi, Adv. Sci., 2019, 6, 1901800 CrossRef CAS PubMed.
- Y. Gao, R. Ma, Y. An and L. Shi, J. Controlled Release, 2011, 152, e81–e82 CrossRef CAS PubMed.
- L. Huang, J. Feng, W. Fan, W. Tang, X. Rong, W. Liao, Z. Wei, Y. Xu, A. Wu, X. Chen and Z. Shen, Nano Lett., 2021, 21, 9551–9559 CrossRef CAS PubMed.
- W. Wei, L. Wang, L. Yuan, Q. Wei, X. Yang, Z. Su and G. Ma, Adv. Funct. Mater., 2007, 17, 3153–3158 CrossRef CAS.
- Z. Zhou, Z. Zhao, H. Zhang, Z. Wang, X. Chen, R. Wang, Z. Chen and J. Gao, ACS Nano, 2014, 8, 7976–7985 CrossRef CAS PubMed.
- Z. Hai, Y. Ni, D. Saimi, H. Yang, H. Tong, K. Zhong and G. Liang, Nano Lett., 2019, 19, 2428–2433 CrossRef CAS PubMed.
- Z. Shen, W. Fan, Z. Yang, Y. Liu, V. I. Bregadze, S. K. Mandal, B. C. Yung, L. Lin, T. Liu, W. Tang, L. Shan, Y. Liu, S. Zhu, S. Wang, W. Yang, L. H. Bryant, D. T. Nguyen, A. Wu and X. Chen, Small, 2019, 15, 1903422 CrossRef CAS PubMed.
- Z. Zhou, R. Tian, Z. Wang, Z. Yang, Y. Liu, G. Liu, R. Wang, J. Gao, J. Song, L. Nie and X. Chen, Nat. Commun., 2017, 8, 15468 CrossRef CAS PubMed.
- F. Li, J. Lu, X. Kong, T. Hyeon and D. Ling, Adv. Mater., 2017, 29, 1605897 CrossRef PubMed.
- U. Prabhakar, H. Maeda, R. K. Jain, E. M. Sevick-Muraca, W. Zamboni, O. C. Farokhzad, S. T. Barry, A. Gabizon, P. Grodzinski and D. C. Blakey, Cancer Res., 2013, 73, 2412–2417 CrossRef CAS PubMed.
- J. L. Perry, K. G. Reuter, J. C. Luft, C. V. Pecot, W. Zamboni and J. M. DeSimone, Nano Lett., 2017, 17, 2879–2886 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nh00632k |
| ‡ These authors contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2022 |