
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNucleic acid-based supramolecular structures: vesicular spherical nucleic acids from a non-phospholipid nucleolipid†
Erik
Dimitrov
a,
Natalia
Toncheva-Moncheva
 *a,
Pavel
Bakardzhiev
a,
Aleksander
Forys
b,
Jordan
Doumanov
c,
Kirilka
Mladenova
c,
Svetla
Petrova
c,
Barbara
Trzebicka
b and
Stanislav
Rangelov
*a
*a,
Pavel
Bakardzhiev
a,
Aleksander
Forys
b,
Jordan
Doumanov
c,
Kirilka
Mladenova
c,
Svetla
Petrova
c,
Barbara
Trzebicka
b and
Stanislav
Rangelov
*a
aInstitute of Polymers, Bulgarian Academy of Sciences, Akad. G. Bonchev St. 103A, 1113 Sofia, Bulgaria. E-mail: ntoncheva@polymer.bas.bg; rangelov@polymer.bas.bg
bCentre of Polymer and Carbon Materials, Polish Academy of Sciences, M. Curie-Sklodowskiej 34, Zabrze, Poland
cDepartment of Biochemistry, Faculty of Biology, Sofia University St. Kliment Ohridski, Dragan Tsankov Blvd. 8, 1164 Sofia, Bulgaria
First published on 9th August 2022
Abstract
Vesicular spherical nucleic acids are dynamic nucleic acid-based supramolecular structures that are held together via non-covalent bonds. They have promising applications as drug and nucleic acid delivery materials, diagnostic and imaging tools and platforms for development of various therapeutic schemes. In this contribution, we report on vesicular spherical nucleic acids, constructed from a non-phospholipid nucleolipid – an original hybrid biomacromolecule, composed of a hydrophobic residue, resembling that of the naturally occurring phospholipids, and a DNA oligonucleotide strand. The nucleolipid was synthesized by coupling of dibenzocyclooctyne-functionalized oligonucleotide and azidated 1,3-dihexadecyloxy-propane-2-ol via an azide–alkyne click reaction. In aqueous solution it spontaneously self-associated into nanosized supramolecular structures, identified as unilamellar vesicles composed of a self-closed interdigitated bilayer. Vesicular structures were also formed upon intercalation of the nucleolipid via its lipid-mimetic residue in the phospholipid bilayer membrane of liposomes prepared from readily available and FDA-approved lipids (1,2-dipalmitoyl-rac-glycero-3-phosphocholine and cholesterol). The vesicular structures are thoroughly investigated by light scattering (dynamic, static, and electrophoretic) and cryogenic TEM and the physical characteristics, in particular, number of strands per particle, grafting density, and conformation of the strands, were compared to those of reference spherical nucleic acids. Finally, the vesicular structures were shown to exhibit cellular internalization with no need of transfection agents and enhanced colloidal and nuclease stability.
Introduction
Spherical nucleic acids (SNAs) are small 3D nanostructures originally composed of a gold core to the surface of which typically short (10–50 bases in length) oligonucleotide strands are covalently attached.1,2 The latter are closely arranged and highly oriented thus forming a dense monolayer shell around the core. The spherical three-dimensional architecture of the SNAs, their sub-100 nm size, the dense arrangement and high radial orientation of the oligonucleotide strands in the shell impart specific properties that are substantially different from those of their linear counterparts. They exhibit increased colloidal stability, ability to evade immune clearance from the bloodstream, prolonged circulation time, superior cellular uptake with no need of transfection agents, higher binding affinity to complementary sequences, minimal cytotoxicity and non-specific immunogenic responses.2–8 SNAs are a rapidly emerging class of nanoparticle-based therapeutics. They are attractive as nucleic acid delivery and gene regulation materials, intracellular diagnostics and imaging tools as well as platforms for development of various therapeutic schemes.8–13SNAs have been prepared with a variety of core compositions including gold, silver, iron oxide, silica, quantum dots2,14–22 and recent studies report on preparation of SNAs with organic cores8,23–28 as well as hollow SNA structures. The latter represent a novel class of SNAs based on liposomal cores that are stabilized with a shell of oligonucleotide strands, intercalated via a hydrophobic residue in the phospholipid bilayer of liposomes.23,29 In addition to the hallmark properties of the prototypical SNAs, the liposomal SNAs have been reported to exhibit superior stability compared to the native liposomes and to inhibit the degradation of the liposomal core in the presence of serum proteins.23 These structures are more dynamic than the prototypical SNAs, since they are held together via non-covalent bonds. The dynamic nature could lead to release of the hydrophobically modified oligonucleotides from the liposomal SNA structures and reach the nucleus to achieve gene regulation,29 which constitutes an important advantage over the SNAs made from the covalent chemistry.
The development of nucleolipids, that is, hybrid molecules bearing a lipophilic moiety covalently attached to a nucleobase, nucleoside, nucleotide or oligonucleotide, is of significant interest. Not only do these molecules combine the specific chemical and biophysical properties of lipids and nucleic acids/nucleosides, but they also can develop new characteristics. Some of them derive from the amphiphilic character and self-assembling properties of the nucleolipids: they have been reported to form a variety of supramolecular structures such as monolayer films, micelles, vesicles, fibers, hydrogels, multilamellar layers,30–36 in which the nucleobases are exposed to the aqueous phase. Via the lipophilic residue, they can anchor in lipidic bilayer membranes of vesicles and in the cores of micelles,8,23,29,37–39 thereby producing novel structures, altering their properties and introducing new ones. To date most of the nucleolipids are nucleoside- or nucleotide-based, that is, they contain a single nucleic acid base. Nucleolipids, carrying an oligonucleotide strand and particularly such with a lipophilic residue featuring the phospholipid double chain are rarely described.8,37,40,41
In this article we report vesicular spherical nucleic acids, prepared via self-assembly and co-assembly of an original nucleolipid. The nucleolipid was synthesized by covalently joining a phospholipid-mimetic double chain residue with a 21-bases long nucleic acid strand using an initiator-free click chemistry approach. By the original design and synthetic approach we expand both the library and methods of preparation of nucleolipids. The physicochemical characterization of the nucleolipid is addressed to the study of its self-assembling and co-assembling behavior and morphological investigation of the resulting vesicular structures by means of light scattering (dynamic, static, and electrophoretic) and cryogenic electron microscopy (cryo-TEM). Preliminary biological properties (stability to enzymatic degradation, cell internalization) are presented as well.
Experimental
Materials
1-Hexadecanol (HAD, ReagentPlus®, 99%, Sigma-Aldrich), glycidyl hexadecyl ether (GHE, technical grade, Sigma-Aldrich), SnCl4 (Sigma-Aldrich, 99.995% trace metals basis), potassium (cubes in mineral oil, 99.5% trace metals basis, Sigma-Aldrich), ethylene oxide (EO, ≥99.5%, Sigma-Aldrich), MgSO4 (anhydrous, ReagentPlus®, ≥99.5%, Sigma-Aldrich), triethylamine (TEA, ≥99.5%, Aldrich), 4-dimethylaminopyridine (DMAP, ReagentPlus®, ≥99%, Sigma-Aldrich), methanesulfonyl chloride (MsCl, ≥99.7%, Aldrich), sodium azide (ReagentPlus®, ≥99.5%, Sigma-Aldrich), 1,2-dipalmitoyl-rac-glycero-3-phosphocholine (DPPC, ∼99%, Sigma-Aldrich), cholesterol (Chol, Sigma Grade, ≥99% Sigma-Aldrich), 1,6-diphenyl-1,3,5-hexatriene (DPH, 98%, Sigma-Aldrich) were used as received. Hexane (anhydrous, 95% Sigma-Aldrich), methylene chloride (>99.98%, Fisher Scientific), toluene (>99.8%, Fisher Scientific), and tetrahydrofuran (THF, >99.5%, Fisher Scientific) were dried with calcium hydride and freshly distilled before use. N,N-Dimethylformamide, (DMF, ACS reagent, ≥99.8%) and dimethyl sulfoxide (DMSO, ACS reagent, ≥99.9%) were dried by molecular sieves. A dibenzocyclooctyne-functionalized oligonucleotide (DBCO-oligonucleotide) was purchased from (https://Biomers.net) GmbH. The sequences and composition of DBCO-oligonucleotide as well as a MALDI spectrum and a HPLC chromatogram are presented in the ESI (Table SI1 and Fig. SI1).† Deionized water was obtained by a Millipore Milli-Q system and was additionally filtered through a 220 nm PTFE filter and a 20 nm cellulose filter. Chemicals for biological evaluation are analytical grade purchased from Merck (Kenilworth, USA): Dulbecco's modification of Eagle's medium/nutrient mixture F-12 Ham (DMEM F-12 Ham), Fetal Calf Serum (FCS), 2-(4,5-dimethyl-2-thiazolyl)-3,5-diphenyl-2H-tetrazolium bromide (MTT), Penicillin, Streptomycin, Crystal violet dye and DNases.Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DPPC:Chol molar ratio, 2 mol% of NucL2 as follows: 0.04 μmol (304.88 μg) of NucL2, 1.30 μmol (954.2 μg) of DPPC, and 0.65 μmol (251.55 μg) of Chol. The solvent ratio was kept in the 1:9–1:10 range with respect to methanol in order to avoid precipitation. The solvent was removed under a gentle stream of argon leaving a thin film. All traces of solvent were removed under vacuum overnight at room temperature. The dry thin lipid film was hydrated with 1 mL of Mili-Q water and the resulting dispersion was subjected to ten freeze–thaw cycles and then extruded 15 times through polycarbonate filters (100 nm pore size) using a handle type extruder (Avanti Polar Lipids, USA).
:1 DPPC:Chol molar ratio, 2 mol% of NucL2 as follows: 0.04 μmol (304.88 μg) of NucL2, 1.30 μmol (954.2 μg) of DPPC, and 0.65 μmol (251.55 μg) of Chol. The solvent ratio was kept in the 1:9–1:10 range with respect to methanol in order to avoid precipitation. The solvent was removed under a gentle stream of argon leaving a thin film. All traces of solvent were removed under vacuum overnight at room temperature. The dry thin lipid film was hydrated with 1 mL of Mili-Q water and the resulting dispersion was subjected to ten freeze–thaw cycles and then extruded 15 times through polycarbonate filters (100 nm pore size) using a handle type extruder (Avanti Polar Lipids, USA).
Methods
| Rh = kT/(6πηD0) | (1) |
Static light scattering (SLS) measurements were carried out in the interval of angles from 40 to 140° at 25 °C using the same light scattering set. The SLS data were analyzed using the Berry plot software provided by Brookhaven Instruments. Information on the weight-average molar mass, Mw, the radius of gyration, Rg, and the second virial coefficient, A2, was obtained from the dependence of the quantity (Kc/Rθ)1/2 on the concentration (c) and scattering angle (θ). Here, K is the optical constant given by K = 4π2n02(dn/dc)2/NAλ4, where n0 is the refractive index of the solvent, NA is Avogadro's constant, λ is the laser wavelength, and Rθ is the Rayleigh ratio at θ. dn/dc is the refractive index increment measured in water in separate experiments on an Orange GPC19 DNDC refractometer. The dn/dc value of the investigated system was 0.142 mL g−1.
Static and dynamic light scattering parameters of DPPC/Chol/NucL2 SNAs were determined at a single concentration.
| ζ = 4πηυ/ε | (2) |
Results and discussion
Nucleolipid synthesis and characterization
To develop vesicular structures, stabilized by a shell of oligonucleotides, we designed a novel nucleolipid (hereinafter, NucL2), composed of a hydrophobic lipid-mimetic double chain moiety and a hydrophilic DNA oligonucleotide strand. The novel nucleolipid is amphiphilic: it is expected to self-associate into nanosized aggregates and to intercalate via its lipid-mimetic residue (anchor) between the phospholipids. The synthesis of NucL2 was realized by an azide–alkyne click reaction, which is a straightforward and high yielding approach.In a typical synthesis, the lipid-mimetic anchor, 1,3-dihexadecyloxy-propane-2-ol (DHP), prepared as described elsewhere42,43 (see also ESI, Fig. SI2 and SI3),† was modified by ethoxylation of the hydroxyl group, thus introducing a short spacer of an average of 6 EO units – DHP-(EO)6-CH2CH2OH (Scheme 1a). This intermediate was obtained by anionic polymerization of EO and the average number of EO units of the spacer was determined from the 1H NMR spectrum of the purified product (Fig. 1). DHP-(EO)6-CH2CH2OH was mesylated (Scheme 1b) by a reaction with methanesulfonyl chloride and, in the final step, the mesylate group was converted into azide to obtain DHP-(EO)6-CH2CH2N3 (Scheme 1b). The successful transformations of the end hydroxyl group into mesylate and azide groups were proved by 1H NMR (Fig. 1). Together with all characteristic signals assigned for the structure of DHP-(EO)6-CH2CH2OH (Fig. 1a), a new signal at 3.09 ppm corresponding to methylene protons adjacent to the mesylate end group appeared (Fig. 1b). Upon azidation, a new signal at 3.39 ppm, assigned to the methylene protons adjacent to the azide end group, was detected, while the signal at 3.09 ppm completely disappeared (Fig. 1c). In addition, the integral ratio of the proton signals from CH2N3 (2H at 3.39 ppm) and the end methyl end groups of DHP (6H at 0.88 ppm) was almost equal to the theoretical value 2:6.
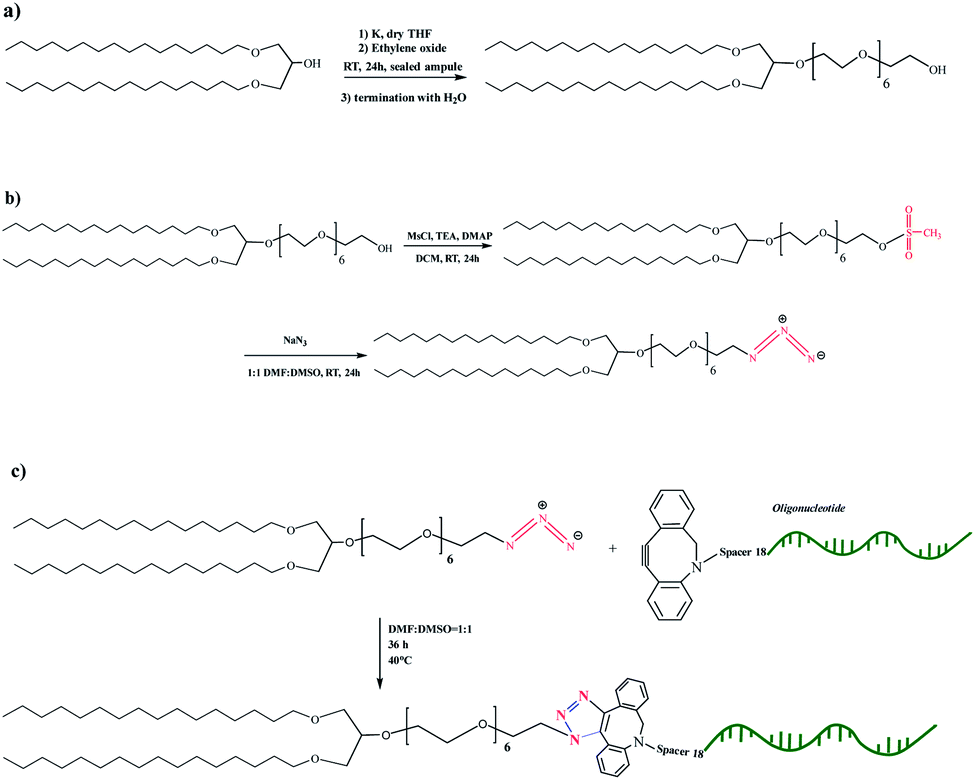 | ||
| Scheme 1 Synthetic scheme for the preparation of the intermediates DHP-(EO)6-CH2CH2OH (a), DHP-(EO)6-CH2CH2OSO2CH3 and DHP-(EO)6-CH2CH2N3 (b), and the nucleolipid NucL2 (c). | ||
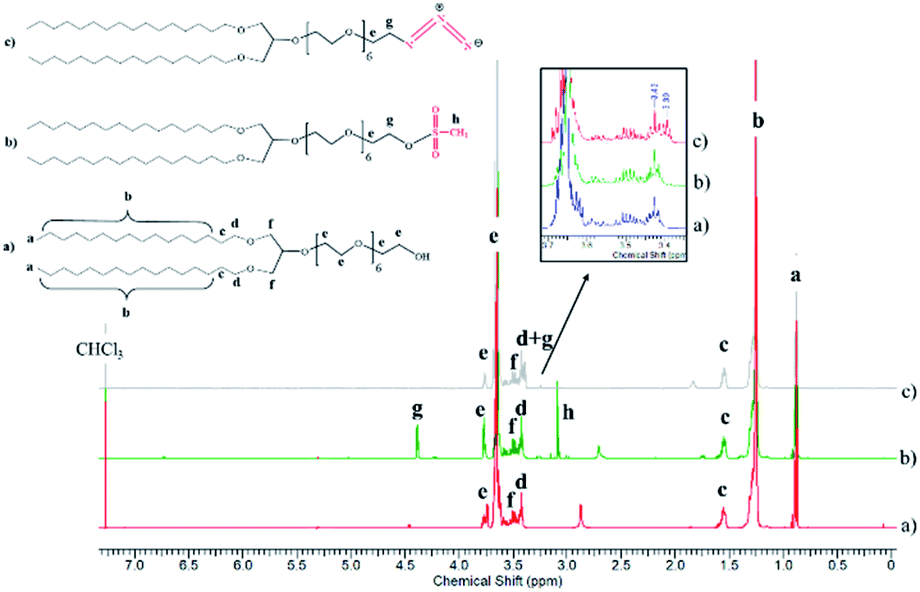 | ||
| Fig. 1 1HNMR spectra of the intermediates DHP-(EO)6-CH2CH2OH (a), DHP-(EO)6-CH2CH2OSO2CH3 (b) and DHP-(EO)6-CH2CH2N3 (c) in CDCl3 (600 MHz). | ||
The introduction of a spacer was of key importance because the transformation of the secondary hydroxyl group of DHP into mesylate and azide groups has proven unsuccessful (see ESI, Fig. SI4–SI6).† To obtain the nucleolipid, a proper excess of the intermediate DHP-(EO)6-CH2CH2N3 and dibenzocyclooctyne-terminated DNA oligonucleotide strands, both in a solvent mixture DMSO/DMF (v/v 1/1), were added to initiate the copper-free azide–alkyne click reaction (Scheme 1c). The resulting conjugate (nucleolipid NucL2) was purified by dialysis and ultrafiltration by which the excess of the azidated compound was removed.
Agarose gel electrophoresis was used to analyze the resulting nucleolipid (Fig. 2). Before that, the mixed DMSO/DMF solvent was displaced by dialysis against ultrapure water, which yielded a slightly opalescent aqueous dispersion of NucL2. As evident from Fig. 2, the electrophoretic mobility of NucL2 was retarded compared to that of DBCO-oligonucleotide, used as control, which was consistent with the enlargement of the molecular weight of NucL2. Notably, a second band was clearly observed, indicating formation of another population of particles with mobility that was further reduced compared to that of the major band.
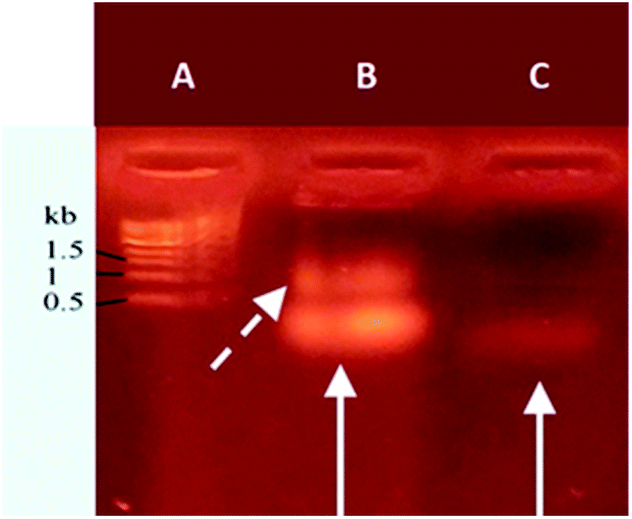 | ||
| Fig. 2 Agarose gel retardation analysis of DBCO-functionalized oligonucleotide (lane C) and NucL2 (lane B). The solid arrows indicate the different retardations of the functionalized oligonucleotide and nucleolipid. The dashed arrow indicates appearance of a fraction of supramolecular particles. Nucleic acids were stained with ethidium bromide. Overall duration of the experiment: 20 min. Lane A: molecular weight marker. Total amounts of 6 μg and 0.6 μg were loaded in B and C, respectively. | ||
Aqueous solution properties
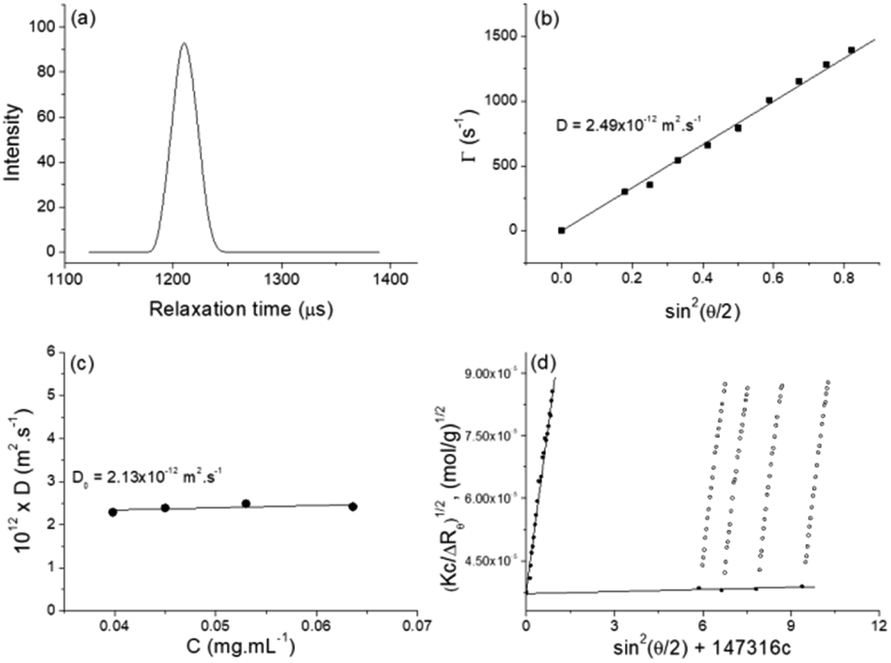 | ||
| Fig. 3 (a) Representative relaxation time distribution (τ), measured at an angle of 90° for aqueous solution of NucL2 at concentration of 0.0398 mg mL−1. (b) Relaxation rate (Γ) as a function of sin2(θ/2) for NucL2 at concentration of 0.053 mg mL−1. (c) Concentration dependence of diffusion coefficients for NucL2. The lines through the data points in (b) and (c) represent the linear fit to the data. (d) Berry plot of NucL2 in aqueous solution. Open symbols and closed symbols represent experimental points and extrapolated points to zero concentration and zero angle, respectively. Measurements were performed at 25 °C. | ||
| Parameters | R h (nm) | R g (nm) | R g/Rh | 107 × A2 (mL mol g−2) | 10−6 × Mw g mol−1 | 10−4 × Nagg | ζ (mV) |
|---|---|---|---|---|---|---|---|
| Value | 115.1 ± 3.4 | 112.0 ± 2.8 | 0.97 | 7.20 ± 0.39 | 709.0 ± 33.0 | 8.8 ± 0.4 | −31.9 |
SLS was performed in the same concentration range as DLS. The static parameters – weight-average molar mass (Mw), radius of gyration (Rg), and second virial coefficient (A2) – were evaluated by the Berry plot method. The Berry diagram for NucL2 is presented in Fig. 3d, whereas the derived static light scattering parameters are collected in Table 1. Evident from the results in Table 1 is that NucL2 in aqueous solution formed relatively large particles (radii of above 110 nm) with a weight-average molar mass reaching 709 × 106 g mol−1, corresponding to aggregation number, Nagg, of about 88000 molecules per particle. In accordance with the high Mw, A2 is very small (of the order of 10−7) but positive, indicating favorable particle–solvent interactions. The particles exhibited strongly negative ζ potential (−31.9 mV, Table 1), which was consistent with the anionic nature of the hydrophilic portion of the nucleolipid – the oligonucleotide strands.
An obvious question that arose from the results was what the structure of the particles that NucL2 formed is. The nucleolipids are known to form a variety of supramolecular structures, including particulate structures such as micelles, vesicles, and fibers.30–36 The NucL2 aggregates cannot be core–corona micelles because of their large size: the radius of the particles is much larger than the length of the fully stretched molecule of NucL2. Furthermore, huge molar masses and aggregation numbers have been observed for vesicles, liposomes, and bilayer structures.43,47,48 In addition Rg/Rh attained a value close to unity (Table 1), which was consistent with vesicular structures.49,50 In order to resolve the morphology of the particles formed by NucL2, we performed cryo-TEM. Fig. 4 shows single objects with clearly distinguishable rims at the outermost periphery, evidencing the vesicular structure of the particles. They are of spherical (though not perfect) morphology with dimensions in a reasonable agreement with those obtained by DLS. The vesicles are single-walled with no internal structure. The bilayer membrane looked intact, that is, openings and perforations were not observed.
 | ||
| Fig. 4 cryo-TEM micrographs of vesicles taken from aqueous dispersions of NucL2. | ||
After having established that the aggregates were vesicles, from DLS and SLS results, we may calculate the mean area occupied by NucL2 molecule in the bilayer. From the total area of the vesicular bilayer and aggregation number, a value of 3.8 nm2 per molecule was obtained, which might look higher than what is realistic even for nucleolipids with a relatively long oligonucleotide strand. To rationalize the results, we may speculate on interdigitation of the bilayer. A schematic representation of an interdigitated bilayer is given in Scheme 2. A prerequisite for formation of this phase is strong repulsion between the lipid heads, which seemed to be present in the system: the oligonucleotide strands (as lipid heads) are negatively charged and may experience repulsive electrostatic interactions. Furthermore, 100% of the lipid heads are oligonucleotide strands (no other lipid molecules in the bilayer). Hristova and Needham in an earlier paper predicted that interdigitation was likely to occur for PEG (poly(ethylene glycol) as a hydrophilic head of PEG-lipids) of molar mass of 350 at contents of about 30 mol% and at lower molar percent for higher molar mass PEG.51 With molar mass of oligonucleotide strands of more than 7000 (see Table SI1†) and 100% grafting, it is very likely that the oligonucleotide strands are in the brush regime. Thus, interdigitation of the bilayer would increase the area per NucL2 molecule and would relax the lateral pressure between the strands.
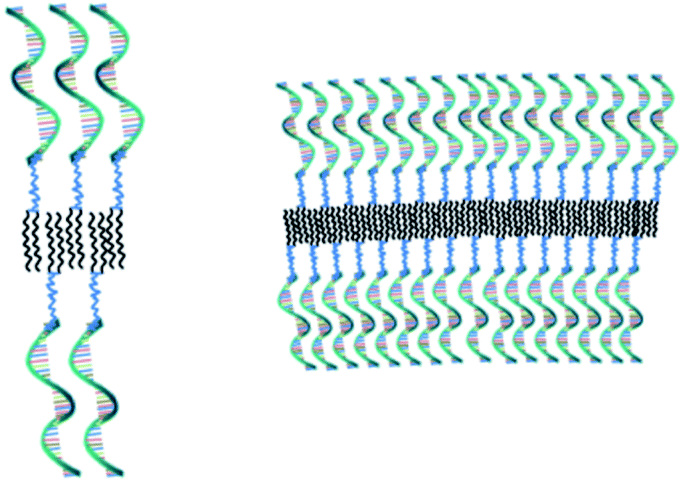 | ||
| Scheme 2 Schematic representation of small (left) and large (right) sections of an interdigitated bilayer of vesicular SNAs, prepared from the nucleolipid NucL2. | ||
:1 DPPC:Chol molar ratio, whereas the NucL2 content was fixed to 2 mol%. The dispersions were prepared by hydration of a dry thin film. Afterwards, they were subjected to 10 freeze–thaw cycles and extruded through polycarbonate filters of pore size 100 nm. The dispersions appeared slightly/moderately opalescent and remained optically clear for weeks. Defining factors to avoid individual self-assembly of the components and/or their separation within the bilayer membrane were the Chol content along with the method of preparation of the structures. It has been shown that at concentration of 33.3 mol% Chol is completely miscible with DPPC and induce formation of a liquid-ordered phase.52–54 Furthermore, the preparation procedure ensures effective mixing of all constituents thereby maximizing the equal distributions of various polymer-derivatized lipids in the outer and inner leaflets of the bilayer and homogeneity of the liquid-ordered phase.55–58
DLS study showed monomodal size distributions with very narrow polydispersity indices typically below 0.1. A representative size distribution is presented in Fig. 5a. More particle size distributions are shown in Fig. SI8.† The diffusion coefficient was determined from the angular dependence of the relaxation rate as shown above (see also Fig. SI9 in the ESI†) and then converted into apparent hydrodynamic radius. The radius of gyration was determined from the partial Berry plot (Fig. 5b). The two parameters, along with the Rg/Rh ratio and ζ potential, are presented in Table 2.
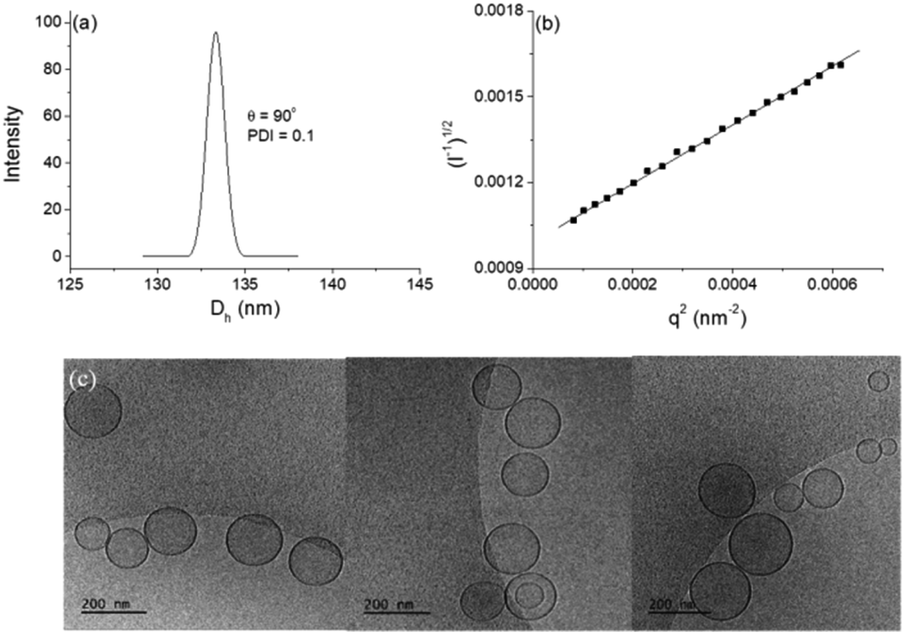 | ||
| Fig. 5 Particle size distribution from DLS, measured at an angle θ = 90° (a) and partial Berry plot (b) of DPPC/Chol/NucL2 vesicles at c = 2 mM total lipid. Measurements were performed at 25 °C. (c) cryo-TEM images of DPPC/Chol/NucL2. | ||
| Parameter | R h (nm) | R g (nm) | R g/Rh | ζ (mV) | Total bilayer area (nm2) | N agg | Number of oligo-nucleotides per vesicle | σ (nm−2) |
|---|---|---|---|---|---|---|---|---|
| a Determined from the angular dependence of the relaxation rate at a single concentration. b Determined from the partial Berry plot at a single concentration. c Aggregation number. d Grafting density. | ||||||||
| Value | 66.6 ± 0.9 | 78.0 ± 0.3 | 1.17 | −17.8 ± 0.5 | 111480 ± 3000 |
171500 ± 4600 |
3430 ± 92 | 0.031 |
The particles were visualized by cryo-TEM (Fig. 5c). More images as well as particle size distributions are presented in the ESI (Fig. SI10).† The dominating structures are clearly unilamellar vesicles of spherical morphology. Bilamellar vesicles and vesicles of irregular (non-spherical, e.g., ellipsoidal) morphology were only occasionally observed. The average size (average diameter of about 140 nm), measured from cryo-TEM, was in very good agreement with that obtained from DLS (cf.Fig. 5a, SI11, and Table 2 in ESI†). The bilayer membrane looked intact, i.e., openings or any kind of damages were not detected. It is of constant thickness of about 5 nm (see the ESI†), which is compatible with DPPC systems.59,60 The background of the images was clear and small material, e.g., bilayer fragments or other, non-bilayer structures, was not observed.
The method for preparation of the dispersions, in particular, the step of multiple extrusion through filters of pore size 100 nm, is known to yield unilamellar liposomes with a mean hydrodynamic radius of about 50–60 nm.55,56 In that aspect, the incorporation of NucL2 in the bilayer membrane was marginal, since it resulted only in a slight increase of Rh (Table 2). However, the other two parameters – the quantity Rg/Rh and ζ potential – were more strongly affected. The former increased from a unity, which is typically obtained for large unilamellar vesicles,49,50 to a value of 1.17, whereas the latter shifted to a more negative value (Table 2), compared to DPPC/Chol liposomes.61,62 The changes in these parameters can apparently be associated with successful incorporation of the nucleolipid in the bilayer membrane and formation of an outer shell of oligonucleotide strands. The bilayer membrane, however, must be grafted on both sides as the vesicles were obtained by simultaneous association of the lipids and NucL2 via rehydration of a mixed DPPC/Chol/NucL2 thin film. In other words, there must be a portion of oligonucleotide strands in the interior of the vesicles forming an inner shell. A schematic illustration of a vesicle is presented in Scheme 3.
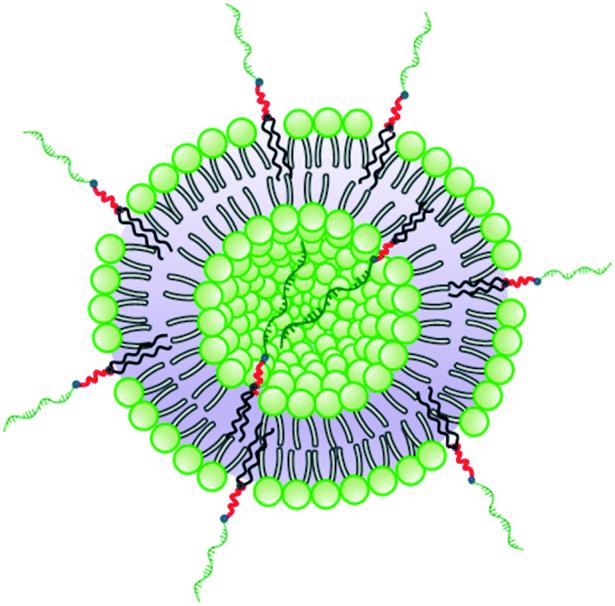 | ||
| Scheme 3 Schematic illustration of a vesicular SNA formed by co-assembly of DPPC, Chol, and NucL2. | ||
Since the unique properties of the prototypical SNAs stem from the density and orientation of the oligonucleotides in the outer region of the nanostructures, questions arise what is the grafting density of the oligonucleotide strands in the present vesicular SNAs, how dense is the shell, and what is the conformation/orientation of the strands in the shell. The grafting density, σ, was determined from the number of oligonucleotide strands and total bilayer area. The total bilayer area was calculated from the hydrodynamic radius from DLS. Then, assuming an average area of 0.65 nm2 per both DPPC and nucleolipid, which is a reasonable value,60 we calculated the aggregation number, Nagg, from which the number of nucleolipid molecules (hence, oligonucleotide strands) was determined knowing the nucleolipid content in the formulation (2 mol%) upon the assumption that the nucleolipid is equally distributed in all vesicles. The results are collected in Table 2. As seen, the average number of oligonucleotide strands per vesicle is about 3430, which is relatively higher than that of reference SNAs, including micellar and liposomal SNAs as well as gold nanoparticle SNAs26,63–65 and can be attributed to the larger size of the present vesicular SNAs. This value was converted into grafting density, expressed as a number of strands per nm2. The resulting value of σ = 0.031 nm−2 was slightly lower than those of reference samples,26,63–65 implying somewhat lower density of the shell. It is noteworthy that both the number of oligonucleotide strands per vesicle and the grafting density refer to this particular formulation (DPPC/Chol/Nucl2 65.3/32.7/2.0 mol ratio) and would increase upon increasing nucleolipid content.
An interesting although somewhat expected finding was that the grafting density was apparently lower than the critical grafting density, σtr, implying that the oligonucleotide strands are isolated and non-interacting each other in the shell. σtr is the grafting density at which surface-anchored polymer coils (or the oligonucleotide strands in the present system) start to interact laterally and a transition from mushroom to brush conformation takes place. σtr (= 0.117 nm−2) for the nucleolipid was estimated from the Flory radius of the oligonucleotide strand as shown in the ESI.† It must be noted here that the critical values of σtr have never been reached even for reference SNAs as discussed elsewhere.66 Nevertheless, these structures have been shown to manifest the unique properties of SNAs.
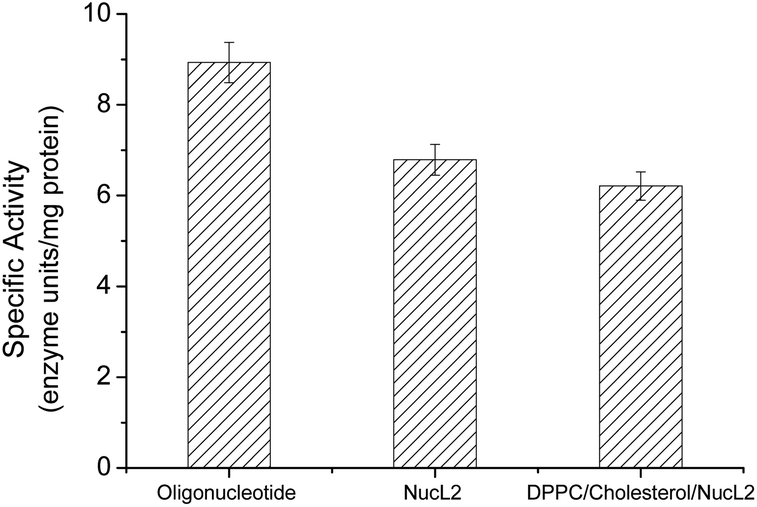 | ||
| Fig. 6 DNase enzyme specific activity against oligonucleotide, nucleolipid NucL2, and DPPC/cholesterol/NucL2 (65.3/32.7/2.0 mol ratio) vesicles. | ||
The rapid cellular uptake without the need of transfection agents is another, probably the most important, property of the SNAs.2 To examine this property, A549 cells were incubated with DPPC/Chol/NucL2 vesicles. The vesicles were firstly treated with 1,6-diphenyl-1,3,5-hexatriene (DPH) – a fluorescent compound widely utilized in studies of membrane interiors and fluidity.70,71 It is practically non-fluorescent in water, but when intercalated into membranes, exhibits a strong increase in fluorescence. Thus, by labeling the bilayer membranes of the DPPC/Chol/NucL2 vesicles it was possible to follow the in vitro cell uptake. Remarkably, the vesicles entered A549 cells readily even after a very short (30 min) incubation time (Fig. 7). After internalization, the negatively charged DPPC/Chol/NucL2 vesicles were observed as small granules (blue dots), denoted by arrows in Fig. 7. Scavenger receptor-mediated and caveolin-mediated endocytotic pathways are the two processes by which most SNAs enter cells.29 A549 cells normally take up nanoparticles via the caveolin-mediated pathway and the internalized nanoparticles follow the common route: they mature in the late endosomes and are degraded in the lysosomes.72 Therefore, it is essential to avoid enzymatic degradation in lysosomal compartments. There are evidences that the nanoparticles could bypass lysosomal degradation and vesicles' cargo can be delivered to various cellular destinations (endoplasmic reticulum, Golgi apparatus, etc.).72 However, larger particles (e.g., structures greater than 25 nm in diameter and over 500 kDa in molar mass), even if able to escape the lysosomes, typically do not enter the nucleus. Despite of their relatively large size (see Fig. 5 and Table 2), the DPPC/Chol/NucL2 vesicles have the advantage of their non-covalent architecture and dynamic structure, which could lead to release of intact nucleolipid molecules that could reach and enter the nucleus. Incorporating of hydrophobically modified antisense oligonucleotides into liposomal SNAs have recently been hypothesized and evaluated in the context of antisense regulation of long-noncoding RNAs.29
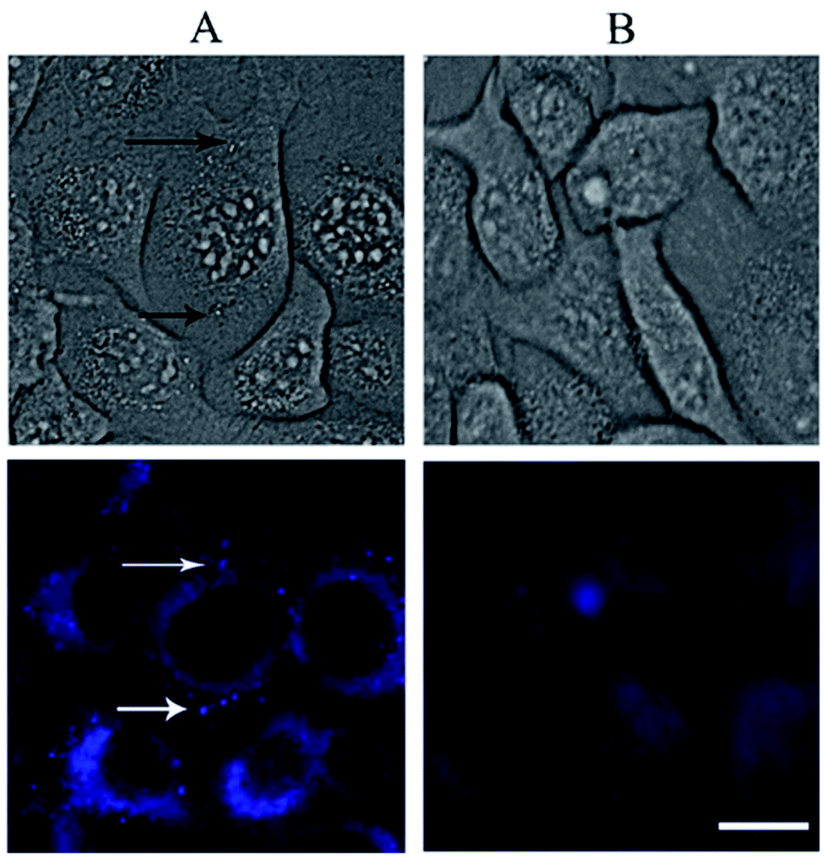 | ||
| Fig. 7 White light (up) and fluorescent (low) micrographs of A549 cells following 30 min exposure to DPPC/Chol/NucL2 vesicles (panel A). The vesicles taken up by the cells are indicated by arrows. Imaging results with A549 cells that have not been treated with the vesicles (control) are shown in panel B. Scale bar 15 μm applies for all images. | ||
Conclusions
We have designed and synthesized an original nucleolipid, composed of a lipid-mimetic residue covalently attached to a DNA oligonucleotide strand. The novel nucleolipid was synthesized via an initiator-free azide–alkyne click coupling reaction of appropriately functionalized species – a lipid-mimetic anchor DHP (1,3-dihexadecyloxy-propane-2-ol) and a short (21 bases) single stranded oligonucleotide with non-specific sequence. The functionalization of DHP required introduction of a short spacer of six EO units, because the direct conversion of the secondary hydroxyl group in DHP to mesylate and, then, azide groups was found to be ineffective. Importantly, the strain of the alkyne ring of dibenzocyclooctyne-terminated oligonucleotide was employed to conjugate the latter to the azido-finctionalized DHP thus avoiding copper catalysis. Upon conjugation of species with opposite water solubility, amphiphilic properties of the resulting nucleolipid were conferred. In aqueous solution, the nucleolipid was found to self-associate into supramolecular structures identified as self-closed interdigitated bilayers densely (100%) grafted with oligonucleotide strands and characterized by a relatively large size (Rh = 115.1 nm), molar mass (Mw = 709.0 × 106 g mol−1) and aggregation number (Nagg = 8.8 × 104), and strongly negative (−31.9 mV) ζ potential. Another type of vesicular structures were successfully prepared by co-assembly of the novel nucleolipid with readily available lipids 1,2-dipalmitoyl-sn-glycero-phosphocholine and cholesterol. They were obtained by the common route for preparation of small unilamellar liposomes (thin film hydration, freeze-thawing, and multiple extrusion through small pore size filters) and their vesicular structure was revealed by cryo-TEM. The vesicles were found to carry thousands (an average of 3430 ± 92) of oligonucleotide strands per particle anchored in the bilayer membrane at a grafting density that was slightly lower than the grafting density at the transition to the brush regime implying unextended conformation of the strands. The grafting density, however, can be increased and the strands can adopt a more extended conformation upon increasing nucleolipid content in the formulations. The shell of oligonucleotide strands endowed the two vesicular structures with properties similar to those of the spherical nucleic acids. They exhibited enhanced nuclease stability and ability to cross the cell membranes without the need of transfection agents. Considering the resistance of the structures to enzymatic degradation, one may anticipate enhanced stability in biological media at physiological conditions that would increase their lifetime.Author contributions
Conceptualization, Stanislav Rangelov and Natalia Toncheva-Moncheva; data curation, Erik Dimitrov, Pavel Bakardzhiev, Aleksander Forys, Kirilka Mladenova; formal analysis, Erik Dimitrov, Pavel Bakardzhiev, Aleksander Forys, Kirilka Mladenova, and Jordan Doumanov; funding acquisition, Stanislav Rangelov, Jordan Doumanov, and Natalia Toncheva-Moncheva; investigation, Erik Dimitrov, Natalia Toncheva-Moncheva, Pavel Bakardzhiev, Aleksander Forys, Kirilka Mladenova, Svetla Petrova, Jordan Doumanov, Barbara Trzebicka, and Stanislav Rangelov; methodology, Erik Dimitrov, Aleksander Forys, Svetla Petrova, Kirilka Mladenova, Pavel Bakardzhiev; project administration, Stanislav Rangelov, Natalia Toncheva-Moncheva, and Jordan Doumanov; resources, Stanislav Rangelov and Jordan Doumanov; supervision, Stanislav Rangelov, Barabara Trzebicka, Svetla Petrova, and Jordan Doumanov; validation, Erik Dimitrov, Pavel Bakardzhiev, Aleksander Forys, Kirilka Mladenova and Natalia Toncheva-Moncheva; writing – original draft, Natalia Toncheva-Moncheva and Stanislav Rangelov; writing – review and editing, Erik Dimitrov, Natalia Toncheva-Moncheva, Pavel Bakardzhiev, Aleksander Forys, Jordan Doumanov, Kirilka Mladenova, Svetla Petrova, Barbara Trzebicka, and Stanislav Rangelov.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Fund (Bulgaria) project no DN19/8-2017. Research equipment of Distributed Research Infrastructure INFRAMAT, part of the Bulgarian National Roadmap for Research Infrastructures, supported by the Bulgarian Ministry of Education and Science, was used in this investigation.References
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607 CrossRef CAS PubMed.
- J. I. Cutler, E. Auyeung and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 1376 CrossRef CAS PubMed.
- C. H. Chung, J. H. Kim, J. Jung and B. H. Chung, Biosens. Bioelectron., 2013, 41, 827 CrossRef CAS PubMed.
- C. H. Choi, L. Hao, S. P. Narayan, E. Auyeung and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7625 CrossRef CAS PubMed.
- M. D. Massich, D. A. Giljohann, A. L. Schmucker, P. C. Patel and C. A. Mirkin, ACS Nano, 2010, 4, 5641 CrossRef CAS PubMed.
- C. Guan, N. Chernyak, D. Dominguez, L. Cole, B. Zhang and C. A. Mirkin, Small, 2018, 14, e1803284 CrossRef PubMed.
- W. Ruan, M. Zheng, Y. An, Y. Liu, D. B. Lovejoy, M. Hao, Y. Zou, A. Lee, S. Yang, Y. Lu, M. Morsch, R. Chung and B. Shi, Chem. Commun., 2018, 54, 3609 RSC.
- R. J. Banga, B. Meckes, S. P. Narayan, A. J. Sprangers, S. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 2017, 139, 4278 CrossRef CAS PubMed.
- J. R. Melamed, N. L. Kreuzberger, R. Goyal and E. S. Day, Mol. Ther.--Nucleic Acids, 2018, 12, 207–219 CrossRef CAS PubMed.
- S. A. Jensen, E. S. Day, C. H. Ko, L. A. Hurley, J. P. Luciano, F. M. Kouri, T. J. Merkel, A. J. Luthi, P. C. Patel, J. I. Cutler, W. L. Daniel, A. W. Scott, M. W. Rotz, T. J. Meade, D. A. Giljohann, C. A. Mirkin and A. H. Stegh, Sci. Transl. Med., 2013, 5, 1 Search PubMed.
- C. A. Mirkin and S. H. Petrosko, Clin. Chem., 2018, 64, 971–972 CrossRef CAS PubMed.
- G. Yamankurt, E. J. Berns, A. Xue, A. Lee, N. Bagheri, M. Mrksich and C. A. Mirkin, Nat. Biomed. Eng., 2019, 3, 318 CrossRef CAS PubMed.
- D. Bousmail, L. Amrein, J. J. Fakhoury, H. H. Fakih, J. C. C. Hsu, L. Panasci and H. F. Sleiman, Chem. Sci., 2017, 8, 6218 RSC.
- L. Hao, P. C. Patel, A. H. Alhasan, D. A. Giljohann and C. A. Mirkin, Small, 2011, 7, 3158 CrossRef CAS PubMed.
- J. S. Lee, A. K. R. Lytton-Jean, S. J. Hurst and C. A. Mirkin, Nano Lett., 2007, 7, 2112 CrossRef CAS PubMed.
- J. L. Cutler, D. Zheng, X. Y. Xu, D. A. Giljohann and C. A. Mirkin, Nano Lett., 2010, 10, 1477 CrossRef CAS PubMed.
- G. P. Mitchell, C. A. Mirkin and R. L. Letsinger, J. Am. Chem. Soc., 1999, 121, 8122 CrossRef CAS.
- C. Zhang, R. J. Macfarlane, K. L. Young, C. H. J. Choi, L. Hao, E. Auyeung, G. Liu, X. Zhou and C. A. Mirkin, Nat. Mater., 2013, 12, 741 CrossRef CAS PubMed.
- K. L. Young, A. W. Scott, L. Hao, S. E. Mirkin, G. Liu and C. A. Mirkin, Nano Lett., 2012, 12, 3867 CrossRef CAS PubMed.
- C. Xue, X. Chen, S. J. Hurst and C. A. Mirkin, Adv. Mater., 2007, 19, 4071 CrossRef CAS.
- C. H. Rische, A. Goel, A. F. Radovic-Moreno and S. M. Gryaznov, Mater. Today Commun., 2016, 9, 30 CrossRef CAS.
- D. Yao, T. Song, X. Sun, S. Xiao, F. Huang and H. Liang, J. Am. Chem. Soc., 2015, 137, 14107 CrossRef CAS PubMed.
- R. J. Banga, N. Chernyak, S. P. Narayan, S. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 9866 CrossRef CAS PubMed.
- J. D. Brodin, E. Auyeung and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4564 CrossRef CAS PubMed.
- K. J. Watson, S. J. Park, J. H. Im, S. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 2001, 123, 5592 CrossRef CAS PubMed.
- Z. Li, Y. Zhang, P. Fullhart and C. A. Mirkin, Nano Lett., 2004, 4, 1055 CrossRef CAS.
- X. Tan, X. Lu, F. Jia, X. Liu, Y. Sun, J. K. Logan and K. Zhang, J. Am. Chem. Soc., 2016, 138, 10834 CrossRef CAS PubMed.
- R. J. Banga, S. A. Krovi, S. P. Narayan, A. J. Sprangers, G. Liu, C. A. Mirkin and S. T. Nguyen, Biomacromolecules, 2017, 18, 483 CrossRef CAS PubMed.
- A. J. Sprangers, L. Hao, R. J. Banga and C. A. Mirkin, Small, 2017, 13, 1602753 CrossRef PubMed.
- H. Rosemeyer, Chem. Biodiversity, 2005, 2, 977 CrossRef CAS PubMed.
- L. Simeone, G. Mangiapia, C. Irace, A. Di Pascale, A. Colonna, O. Ortona, L. De Napoli, D. Montesarchio and L. Paduano, Mol. BioSyst., 2011, 7, 3075 RSC.
- A. Gissot, M. Camplo, W. G. Mark and P. Barthelemy, Org. Biomol. Chem., 2008, 6, 1324 RSC.
- S. Milani, D. Berti, S. Dante, T. Hauss and P. Baglioni, Langmuir, 2009, 25, 4084 CrossRef CAS PubMed.
- N. Taib, A. Aimé, S. Houmadi, S. Castano, Ph. Barthélémy, M. Laguerre and I. Bestel, Langmuir, 2012, 28, 7452 CrossRef CAS PubMed.
- E. Lepeltier, C. Bourgaux, V. Rosilio, J. H. Poupaert, F. Meneau, F. Zouhiri, S. Lepetre-Mouelhi, D. Desmaele and P. Couvreur, Langmuir, 2013, 29, 14795 CrossRef CAS PubMed.
- V. Allain, C. Bourgaux and P. Couvreur, Nucleic Acids Res., 2012, 40, 1891 CrossRef CAS PubMed.
- C. Gosse, A. Boutorine, I. Aujard, M. Chami, A. Kononov, E. Cogné-Laage, J.-F. Allemand, J. Li and L. Jullien, J. Phys. Chem. B, 2004, 108, 6485 CrossRef CAS PubMed.
- J. S. Nowick, J. S. Chen and G. Noronha, J. Am. Chem. Soc., 1993, 115, 7636 CrossRef CAS.
- J. S. Nowick, T. Cao and G. Noronha, J. Am. Chem. Soc., 1994, 116, 3285 CrossRef CAS PubMed.
- R. G. Shea, J. C. Marsters and N. Bischofberger, Nucleic Acids Res., 1990, 18, 3777 CrossRef CAS PubMed.
- E. T. Rump, R. L. A. de Vrueh, L. A. J. M. Sliedregt, E. A. L. Biessen, T. J. C. van Berkel and M. K. Bijsterbosch, Bioconjugate Chem., 1998, 9, 341 CrossRef CAS PubMed.
- S. Rangelov, E. Petrova, I. Berlinova and C. Tsvetanov, Polymer, 2001, 42, 4483 CrossRef CAS.
- P. Bakardzhiev, S. Rangelov, B. Trzebicka, D. Momekova and G. Lalev, RSC Adv., 2014, 4, 37208 RSC.
- S. W. Provencher, Macromol. Chem., 1979, 180, 201 CrossRef CAS.
- Q. He, J. Liu, X. Sun and Z. R. Zhang, World J. Gastroenterol., 2004, 10, 660 CrossRef CAS PubMed.
- H. Li, B. Zhang, X. Lu, X. Tan, F. Jia, Y. Xiao, Z. Cheng, Y. Li, D. O. Silva, H. S. Schrekker, K. Zhang and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4340 CrossRef CAS PubMed.
- T. Enoki, V. M. Henriques and T. V. M. Lamy, Chem. Phys. Lipids, 2012, 165, 826 CrossRef CAS PubMed.
- L. N. d. M. Ribeiro, V. M. Couto, L. F. Fraceto and d. E. Paula, Sci. Rep., 2018, 8, 982 CrossRef PubMed.
- W. Burchard, Adv. Polym. Sci., 1983, 48, 1 CrossRef CAS.
- A. Thurn, W. Burchard and R. Niki, Colloid Polym. Sci., 1987, 265, 653 CrossRef CAS.
- K. Hristova and D. Needham, Macromolecules, 1995, 28, 991 CrossRef CAS.
- J. Y. Huang and G. W. Feigenson, Biophys. J., 1999, 76, 2142 CrossRef CAS PubMed.
- J. A. Clarke, A. J. Heron, J. M. Seddon and R. V. Law, Biophys. J., 2006, 90, 2383 CrossRef CAS PubMed.
- P. Bakardzhiev, A. Forys, B. Trzebicka, T. Andreeva and S. Rangelov, Nanoscale, 2021, 13, 15210 RSC.
- D. D. Lasic, Biochem. J., 1988, 256, 1 CrossRef CAS PubMed.
- Y. P. Patil and S. Jadhav, Chem. Phys. Lipids, 2014, 177, 8 CrossRef CAS PubMed.
- Y. Barenholz, Curr. Opin. Colloid Interface Sci., 2001, 6, 66 CrossRef CAS.
- W. M. Li, L. Xue, L. D. Mayer and M. B. Bally, Biochim. Biophys. Acta, 2001, 1513, 193 CrossRef CAS.
- A. Kenworthy, K. Hristova, D. Needham and T. J. McIntosh, Biophys. J., 1995, 68, 1921 CrossRef CAS PubMed.
- J. F. Nagle and S. Tristram-Nagle, Biochim. Biophys. Acta, 2000, 1469, 159 CrossRef CAS.
- A. Magarkar, V. Dhawan, P. Kallinteri, T. Viitala, M. Elmowafy, T. Róg and A. Bunker, Sci. Rep., 2014, 4, 5005 CrossRef CAS PubMed.
- P. Bakardzhiev, D. Momekova, K. Edwards, S. Konstantinov and S. Rangelov, J. Drug Delivery Sci. Technol., 2015, 29, 90 CrossRef CAS.
- T. A. Taton, C. A. Mirkin and R. L. Letsinger, Science, 2000, 289, 1757 CrossRef CAS PubMed.
- C. M. Calabrese, T. J. Merkel, W. E. Briley, P. S. Randeria, S. P. Narayan, J. L. Rouge, D. A. Walker, A. W. Scott and C. A. Mirkin, Angew. Chem., Int. Ed., 2015, 54, 476 CAS.
- S. J. Hurst, A. K. R. Lytton-Jean and C. A. Mirkin, Anal. Chem., 2006, 78, 8313 CrossRef CAS PubMed.
- E. Haladjova, I. Ugrinova and S. Rangelov, Soft Matter, 2020, 16, 191 RSC.
- N. L. Rosi, D. A. Giljohann, C. S. Thaxton, A. K. Lytton-Jean, M. S. Han and C. A. Mirkin, Science, 2006, 312, 1027 CrossRef CAS PubMed.
- W. Briley, T. L. Halo, P. S. Randeria, A. H. Alhasan, E. Auyeung, S. J. Hurst, and C. A. Mirkin, in Nanomaterials for Biomedicine, ACS Symposium Series, ed. R. Nagarajan, American Chemical Society, Washington, DC, 2012, pp. 1–20.
- X. A. Wu, C. H. J. Choi, C. Zhang, L. Hao and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 7726 CrossRef CAS PubMed.
- F. Sakurai, T. Nishioka, H. Saito, T. Baba, A. Okuda, O. Matsumoto, T. Taga, F. Yamashita, Y. Takakura and M. Hashida, Gene Ther., 2001, 8, 677 CrossRef CAS PubMed.
- C. Poojari, N. Wilkosz, R. B. Lira, R. Dimova, P. Jurkiewicz, R. Petka, M. Kepczynski and T. Róg, Chem. Phys. Lipids, 2019, 223, 104784 CrossRef CAS PubMed.
- Sh. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreadene, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Chem. Soc. Rev., 2017, 46, 4218 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: MALDI spectrum, HPLC chromatogram, and other characterization data of DBCO-oligonucleotide; procedures for synthesis and functionalization of 1,3-dihexadecyloxy-propane-2-ol, 1H NMR and FTIR spectra; DLS data for vesicular SNAs, prepared by self-assembly of NucL2 in aqueous solution, particle size distributions by intensity, volume, and number from DLS; additional and supporting data for vesicular SNAs, prepared by co-assembly of NucL2 with DPPC and Chol, particle size distributions by intensity, volume, and number from DLS, angular dependence of relaxation rate, cryo-TEM images, particle size distribution from cryo-TEM; calculation of the Flory radius, RF, and determination of the critical grafting density (σtr) at the transition from mushroom to brush conformation. See https://doi.org/10.1039/d2na00527a |
| This journal is © The Royal Society of Chemistry 2022 |