
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSnapshot imprinting as a tool for surface mapping and identification of novel biomarkers of senescent cells†
Elena
Piletska
a,
Dana
Thompson
a,
Rebecca
Jones
a,
Alvaro Garcia
Cruz
 a,
Marta
Poblocka
b,
Francesco
Canfarotta
a,
Rachel
Norman
c,
Salvador
Macip
bc,
Donald J. L.
Jones
d and
Sergey
Piletsky
*a
a,
Marta
Poblocka
b,
Francesco
Canfarotta
a,
Rachel
Norman
c,
Salvador
Macip
bc,
Donald J. L.
Jones
d and
Sergey
Piletsky
*a
aChemistry Department, College of Science and Engineering, University of Leicester, Leicester, LE1 7RH, UK. E-mail: sp523@le.ac.uk
bMechanisms of Cancer and Aging Laboratory, Department of Molecular and Cell Biology, University of Leicester, Leicester, LE1 7RH, UK
cFoodLab, Faculty of Health Sciences, Universitat Oberta de Catalunya, 08018 Barcelona, Spain
dDepartment of Cancer Studies, RKCSB, University of Leicester, Leicester, LE2 7LX, UK
First published on 12th October 2022
Abstract
Cellular senescence has proved to be a strong contributor to ageing and age-related diseases, such as cancer and atherosclerosis. Therefore, the protein content of senescent cells is highly relevant to drug discovery, diagnostics and therapeutic applications. However, current technologies for the analysis of proteins are based on a combination of separation techniques and mass spectrometry, which require handling large sample sizes and a large volume of data and are time-consuming. This limits their application in personalised medicine. An easy, quick and inexpensive procedure is needed for qualitative and quantitative analysis of proteins expressed by a cell or tissue. Here, we describe the use of the “snapshot imprinting” approach for the identification of proteins differentially expressed by senescent cells. Molecularly imprinted polymer nanoparticles (MIPs) were formed in the presence of whole cells. Following trypsinolysis, protein epitopes protected by complex with MIPs were eluted from the nanoparticles and analysed by LC-MS/MS. In this work, “snapshot imprinting” was performed parallel to a standard proteomic “shaving approach”, showing similar results. The analysis by “snapshot imprinting” identified three senescent-specific proteins: cell division cycle 7-related protein kinase, partitioning defective three homolog B and putative ATP-dependent RNA helicase DHX57, the abundance of which could potentially make them specific markers of senescence. Identifying biomarkers for the future elimination of senescent cells grants the potential for developing therapeutics for age-related diseases.
Introduction
Replicative senescence was first proposed by Dr Leonard Hayflick in the early 1960s.1 The Hayflick limit states that a normal human diploid cell will divide for a maximum of 50 to 60 passages until it becomes senescent and can no longer divide. This limit occurs due to the irreversible shortening of the telomeres on the chromosomes.2 Unlike quiescence and exhaustion, senescent cells (SNCs) show a permanent growth arrest, proven so since usually no physiological stimuli can lead the SNCs to re-enter the cell cycle.3,4 Cellular senescence is thought to be more than just a regulator of proliferative lifespan, as it can also be triggered in response to stresses such as activated oncogenes, DNA replication stress, oxidative stress and cell–cell fusion.5,6 Cellular senescence is established and maintained by at least two known tumour suppressor pathways: p53/p21 and p16INK4a/retinoblastoma proteins (pRB).7,8 SNCs have particular features, which have already been used as biomarkers to detect them in vitro. However, they are neither sensitive nor specific enough.3,4 There are several widely-recognised characteristics of SNCs, including a senescence-associated secretory phenotype (SASP) formation, irreversible growth arrest, expression of anti-proliferative molecules, senescence-associated β-galactosidase (SAβ-gal) activity, senescence-associated heterochromatin foci (SAHF) formation and resistance to apoptosis.4,8,9Acute senescence can be considered as beneficial for the body, as it aids tissue regeneration. It is a product of specific stress and has a specific target to assist in the process of wound or injury healing and development, such as embryogenesis.4,10,11 Also, senescence can act as a cell-autonomous tumour suppressor and can therefore prevent the proliferation of cancerous cells due to the release of tumour suppressor genes, e.g. p53 and p16INK4a. Chronic senescence, on the other hand, can be detrimental to health, leading to disease and tumour formation.10 Previous reports have revealed that senescence has a clear link with ageing, and with this, age-related diseases and pathologies such asdiabetes, kidney disease, atherosclerotic plaques in arteries, skin ulcers, arthritic joints, inflammation, sarcopenia, adiposity, neurogenesis, fibrosis and glaucoma.4,8,10
Targeted elimination of SNCs can relieve symptoms of ageing and prolong lifespan.9 Several strategies have already been developed in order to interfere with the detrimental effects of cellular senescence, such as selectively inducing the death of SNCs (by the use of drugs classed as “senolytics” or the immune-mediated clearance of SNCs), preventing senescence (with “senoblockers”12) and SASP neutralisation (with drugs called “senostatics” or “senomorphics”).13,14 Current senolytics are not sufficiently specific, but have been proven to somewhat increase the health-span and alleviate diseases in mice, such as atherosclerosis, osteoarthritis, cataracts, sarcopenia and cancer,15 and some may be of natural origin.16 Efforts to optimise selectivity have been made by designing second generation senolytics that consist of a senolytic drug and a targeting factor such as a nanoparticle or an antibody against a specific exposed epitope.17,18 This involves identifying proteins that are overexpressed on the surface of SNCs.11,19 This approach, however, requires understanding of the difference in membrane proteome (or “surfaceome”) of the normal and senescent cells and identification of protein targets that can be used for specific delivery of cytotoxic agents.
There are several ways to map the surface of SNCs for their epitopes. However, these are difficult and present drawbacks. An example of such a technique is X-ray diffraction analysis. Although it is the most precise method for epitope mapping, growing suitable crystals of membrane proteins is difficult and is marred by purity issues.16,17 Another example is the use of NMR, posing an advantage over X-ray diffraction by avoiding the need for crystals. However, this technique is of little use when it comes to mapping SNC surfaces, which can be explained by the fact that low metabolising SNCs will not possess a large enough excess of proteins on the cell surface that differentiate them from normal cells.18 For the same reason, generating antibodies against SNCs and using those for epitope mapping will be very difficult.
Cell surface mapping is a useful research technique in drug development and diagnostics.20,21 It consists of “shaving” a significant segment of a cell surface protein by digesting live, intact cells so the generated peptides can be analysed by LC-MS/MS. In the past, we have used the “shaving approach” for the identification of potential senescent biomarkers of the p16 pathway (manuscript in press). Although the results were successful, it is difficult to achieve a precise control of experimental conditions in the “shaving approach”, and for that reason, it cannot be readily applied in the clinical practice. Recently, we have described an experimental approach for using molecular imprinting to identify peptide sequences on the protein surface with potential antigenic properties.22 The method involves synthesis of MIP nanoparticles in the presence of whole protein, partial proteolysis of the protein bound to polymer, and subsequent sequencing of released peptides that were bound to the polymer. The important concept behind this principle relies on the assumption that MIPs synthesised in the presence of protein protect from proteolysis peptide sequences on the protein surface that are involved in MIP formation. This approach provides the possibility of identifying regions of the protein surface that have not yet been demonstrated to be antigenic in vivo, but which may offer improved affinity for natural and synthetic receptors such as antibodies, MIPs, aptamers, etc.
The same principle can be applied to take a “snapshot imprinting” of the cellular processes occurring in the cells, in order to map the topography of the cells and to identify peptide sequences and corresponding proteins that are expressed on the cell surface (Fig. 1).23 The obvious limitation to this procedure lies in the fact that cell populations might contain fractions representing different phases of the cell cycle. At the different phases, different proteins may be expressed to a higher degree. Hence, the ideal target for “snapshot imprinting” will be cells that are “frozen” in a perpetual phase of cell arrest, such as senescent cells. To prove this concept, MIPs were synthesised in the presence of cells undergoing p53-induced senescence. The proteins identified by snapshot imprinting for control and senescent cells were compared with the intention to identify abundant proteins characteristic to senescence that can be used as markers. The results of the “shaving approach” and “snapshot imprinting” were also compared, showing certain similarity of data obtained. We hope to use this knowledge in the future to identify and target senescent cells for diagnostic and therapeutic purposes, thus potentially delaying the onset of disease and aging.
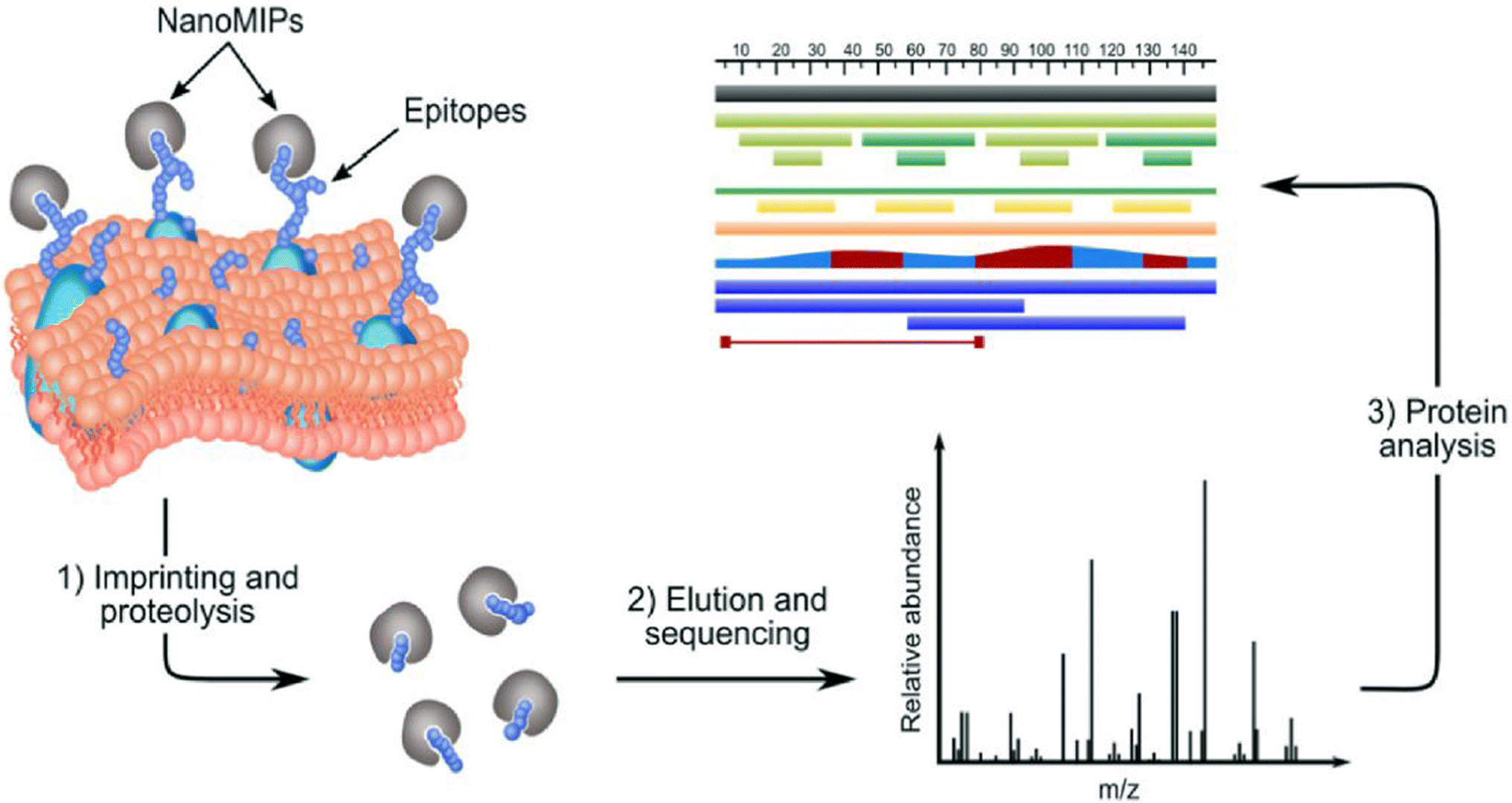 | ||
| Fig. 1 .Identification of membrane proteins in a surfaceome analysis by “snapshot imprinting”.23 | ||
Results and discussion
In order to characterize the senescent surfaceome using novel techniques, we took advantage of the well characterized cellular model of senescence, EJp53.24 These are EJ bladder cancer cells with a tetracyline(tet)-regulatable p53 expression system that undergo senescence 2 to 5 days after the removal of tet, as shown in Fig. 2. | ||
| Fig. 2 .Induction of senescence in EJp53. (A) Representative images of a SA-βgal staining of EJp53 cultured in the presence of tet (control, proliferating) or five days after the removal of tet (senescent). (B) Quantitation of the staining shown in A. Bars represent mean values of three independent experiments and error bars represent standard deviation. *** = p ≤ 0.001. (C) Representative western blot (and its quantitation, normalised to actin and the control cells) showing the induction of p53 in the cells shown in A. Actin is shown as a loading control. Bars represent mean values of two independent experiments and error bars represent standard deviation. | ||
The MIP synthesis, enzyme proteolysis and peptide sequencing were performed as described earlier.22 The monomeric mixture, comprised of NIPAm, TBAm, MBAA, AA and 3-aminopropyl methacrylamide, was optimised for protein imprinting.25,26 The synthesised nanoparticles are nontoxic to cultured cells, although the monomer mixture itself might induce stress.11,27,28 In our experiments, we did not notice cell lysis triggered by imprinting, which is essential for enriching fraction of membrane proteins in collected samples (data not shown). After approximately 1 hour of polymerisation, the non-polymerised monomers were washed away and cells/MIPs complexes treated with trypsin. After tryptic digestion, the MIP/epitope complexes were separated from unwanted cellular components and trypsin by centrifugal filtration and washing. Separated MIPs have average diameters of 170–180 nm (Table 1), which are significantly smaller than cells (5–20 μm). It is expected that each particle can be templated with only one epitope for a single protein. It is possible to describe cell imprinting as “freezing” exposed fragments of cell proteins in their complex with polymeric networks. This allows performing subsequent cell lysis without worrying whether cells remain intact during trypsinolysis. The epitope templates were removed from MIPs by heating. Peptide epitopes were eluted, concentrated and sequenced by LC/MS-MS. The epitopes sequences were analysed, providing information about types of the proteins exposed on cell membranes, and which contributed to the formation of MIPs, and their abundance. The LC-MS/MS results collected from three experiments with EJp53 cells are summarised in ESI Tables 1 and 2.†
| Control cells | Z-Ave | PdI | Senescent cells | Z-Ave | PdI |
|---|---|---|---|---|---|
| Mean (nm) | 178.8 | 0.4 | Mean | 169.8 | 0.5 |
| Std Dev (nm) | 6.0 | 0.1 | Std Dev | 11.2 | 0.1 |
The size of MIPs collected after elution of peptides was characterised using dynamic light scattering (DLS) and transmission electron microscopy (TEM) techniques. It was found that the hydrodynamic diameter of the MIPs in water was on average 128 nm ± 39 nm (polydispersity index—0.02). This size corresponds to the value reported for MIPs imprinted with proteins.29 The typical appearance of the MIPs under the TEM was spherical with their diameter approximately two times smaller than measured by DLS (Fig. 3).
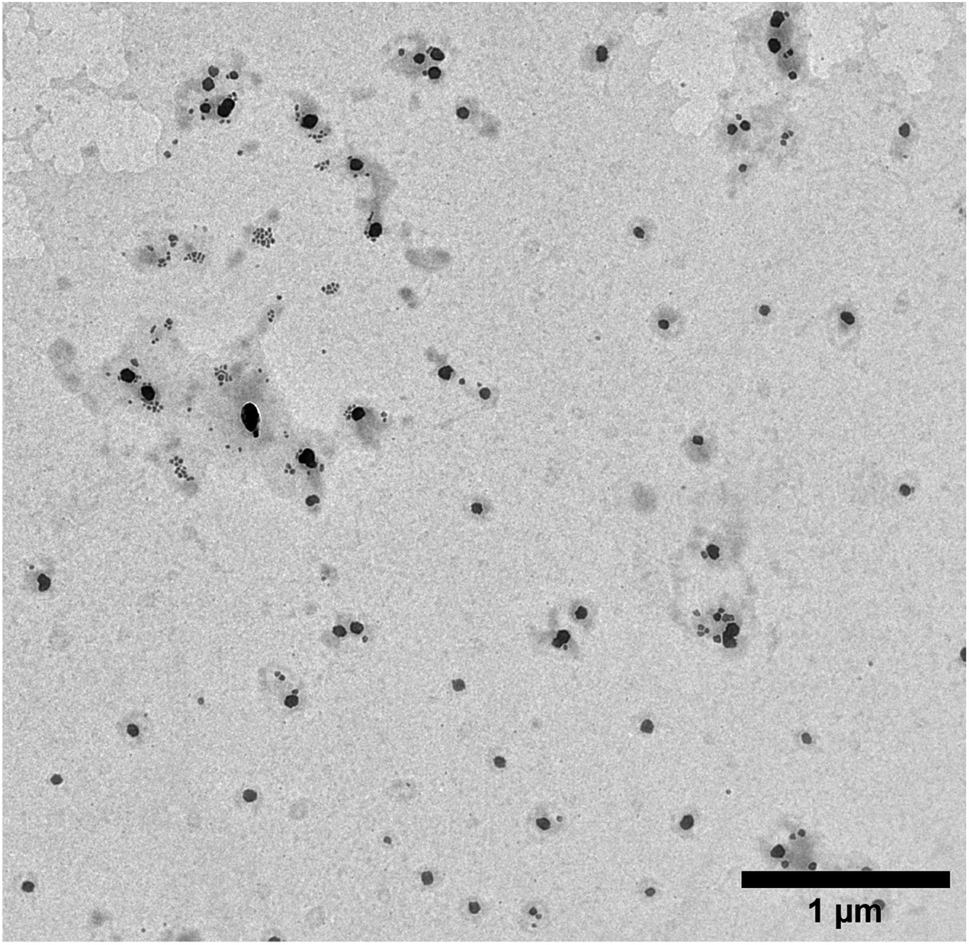 | ||
| Fig. 3 TEM image of the MIP nanoparticles obtained after surface mapping (magnification: 5000×). | ||
The analysis of surface proteome by “snapshot imprinting” and by the “shaving approach” showed certain similarities. “Snapshot imprinting” allowed the identification of 112 proteins, versus 93 in the “shaving approach” (ESI Tables 1 and 2†). Approximately 33% of these proteins were identical for both approaches tested here. It is useful to notice that the concentration of peptide epitopes discovered in “snapshot imprinting” is substantially higher, by almost two orders of magnitude. This is probably a result of pre-concentration of peptides on the surface of MIP nanoparticles. The additional benefits of “snapshot imprinting” lies in its ability to link sequences of identified epitopes with their ability to generate MIP nanoparticles. This is in contrast to established protocols, e.g. the “shaving approach”, where information obtained about peptides/protein structure cannot be used for generating antibodies due to lack of correlation between abundance of proteins and their immunogenicity. These binders can be used for future elimination of senescent cells, granting the potential for the development of therapeutics for age-related diseases. In “snapshot imprinting” and also in the proteolytic “shaving approach” less than half of the identified proteins are actually membrane proteins. This phenomenon, observed in classical surfaceome research, is not completely understood, and could be due to the following possibilities: (i) cytoplasmic proteins come from cell lysis, thus contaminating the surfaceome fraction; (ii) cytoplasmic proteins have reached the surface by unspecified exporting/secretory machinery.30 Whether cytoplasmic proteins without any canonical secretion/exporting or retention signal are really translocated across the membrane is still unresolved, although there are many evidences both in prokaryotes and eukaryotes.31–34
As expected, uninduced (control) cells showed a large excess of identified proteins that were imprinted successfully. The proteins prevalent in the senescent cells are presented in Table 2. It is useful to notice that no reliable senescent markers were discovered by the “shaving approach”. The peptides belonging to two histone proteins, which are nuclear proteins but show a relative abundance in senescent cells, were measured with too large error margins (Table 2). This does not allow us to stipulate with confidence that these proteins are indeed present predominantly in senescent cells. The reliability of peptide measurement by “snapshot imprinting” is substantially higher. Three proteins were identified that show higher abundance in senescent cells by a factor of more than 5: cell division cycle 7-related protein kinase, partitioning defective 3 homolog B and putative ATP-dependent RNA helicase DHX57. The brief discussion of the role of these proteins in senescence is provided below.
| Accession | Description | Senescent cells, fmol | Control cells, fmol |
|---|---|---|---|
| Snapshot imprinting | |||
| B2R6V2; B7Z5H7; O00311; Q6JSD6 | Cell division cycle 7-related protein kinase | 5721 ± 234 | 0.00 |
| A0A087WYL6; A0A087WZG6 | Partitioning defective 3 homolog B | 6748 ± 735 | 27 ± 16 |
| A0A087X1S9; Q587I4; Q8TEW8 | |||
| Q6P158; A0A087WZ11; B4DKW2 | Putative ATP-dependent RNA helicase DHX57 | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 675 ± 660 675 ± 660 |
2150 ± 238 |
|
|||
| Shaving approach | |||
| P04908; P0C0S8; P0C0S9; P20671; Q16777; Q3ZBX9; Q6FI13; Q7L7L0; Q93077; Q96KK5; Q99878; Q9BTM1; P0C0S4; P0C0S5; P16104; Q32LA7; Q71UI9; Q8IUE6; Q96QV6 | Histone H2A type 2-C | 17.2 ± 14 | 0.1 ± 0.1 |
| O60814; P06899; P23527; P33778; P58876; P62807; P62808; Q16778; Q2M2T1; Q32L48; Q5QNW6; Q93079; Q99877; Q99879; P57053; Q96A08; Q99880; Q8N257 | Histone H2B type 1-B | 19 ± 15.2 | 1.7 ± 1.1 |
The cell division cycle 7-related protein kinase (CDC7) is a serine/threonine kinase that phosphorylates minichromosome maintenance protein 2 (MCM2) of the eukaryotic pre-replication complex.35 This protein is found in the nucleus of the cell and is implicated in cell division, cell-cycle checkpoint mechanisms, and cancer progression and its overexpression is present in different cancers.36 The activity of the catalytic subunit CDC7 is positively regulated by a complex formation with its activation subunit, the Dbf4 protein, also known as ASK (activator of S-phase kinase in human).37–39 The CDC7-ASK/Dbf4 complex also commonly referred to as the Dbf4 dependent kinase (or DDK) regulates the timing of DNA replication origin firing throughout S phase mainly by phosphorylation of MCM proteins, the major components of replicative helicase.40,41 The overexpression of CDC7 noted in our experiments could be directly caused by p53 expression,42 and this would have to be investigated using other cellular models of senescence. Of note, increased levels of CDC7 may not be per se a specific marker of senescence, since it is detected in different cancers.36
The partitioning defective 3 homolog B (PARD3B) is a membrane protein found to be much more abundant in SNCs. Atypical protein kinase C zeta (PKC zeta) forms a complex with PARD3 and PARD6 to regulate normal epithelial cell apico-basolateral polarization.43 The dissociation of the PKC zeta/PPARD3/PARD6 complex is essential for the disassembly of the tight/adherent junction and epithelial–mesenchymal transition (EMT) that is critical for tumour spreading.44 Suppression of PARD3 is associated with altered expression of genes regulating wound healing, cell apoptosis/death and cell motility, and particularly upregulation of MAP3K1 and fibronectin, which are known to contribute to cancer progression.43 Experimental evidence suggest that reduced expression of PKC zeta/Pard3/Pard6 contributes to EMT, invasion, and chemoresistance.45 It is possible that PARD3 protein can function as a senescence marker, although more experiments are needed to compare expression of PARD3 in senescent cells versus proliferating non-cancer cells, which also have increased level of PARD3 expression, as compared to cancer cells.46
The Putative ATP-dependent RNA helicase (DHX57) is part of the ATP-dependent RNA helicases, which are involved in diverse cellular functions such as RNA splicing, unwinding of a DNA or RNA helix, ribosome assembly, initiation of translation, spermatogenesis, embryogenesis, and cell growth and division. The precise mechanism and the substrates of these enzymes have not been defined. It is known that they play important roles in all types of processes in RNA metabolism and its expression might be related to biotic and abiotic stresses.47 DHX57 expression levels are also positively correlated with chronological age in the cerebellum.48 This supports our finding that this protein could be a marker of the presence of senescent cells.
Conclusions
Overall, our results demonstrate the utility of a new protocol for identifying potential candidates for biomarkers-related research. Before the identified proteins can be considered as senescence biomarkers, more research and validation experiments are required. Three proteins were identified in “snapshot imprinting” that show abundance in SNCs with a factor more than 5: CDC7, PARD3B and DHX57. This would suggest that these proteins could be useful as biomarkers for in vivo detection and targeting of senescent cells. The “snapshot imprinting” has shown similar results to the “shaving approach”, allowing identification of a broader range of proteins with a better confidence level. Compared to the classical proteomics methods, the proposed novel approach will enable us to shorten the timetable to discover new candidates. The additional benefits of “snapshot imprinting” lies in its ability to link sequences of identified epitopes with their ability to generate synthetic binders—MIP nanoparticles. In contrast to established protocols, e.g. the “shaving approach”, the information obtained related to peptides/protein structure cannot be used for generating antibodies due to a lack of correlation between the abundance of proteins and their immunogenicity. These binders can be used for the future elimination of senescent cells, granting the potential to develop therapeutics for age-related diseases.Material and methods
Cell culture
Cells were grown at 37 °C in Dulbecco's Modified Eagles Medium (DMEM) supplemented with 10% foetal bovine serum, penicillin-streptomycin [50 unit per mL], antibiotics (hygromycin [100 μg mL−1] and genticin [750 μg mL−1]) and tetracycline every 3 days [1 μg mL−1]. Senescence was induced by removing tetracycline by washing the cells 3 times with PBS and adding new growth medium with the same composition apart from tetracycline. The medium was aspirated from the cells. 2 mL of PBS (PBS with calcium and magnesium) was added to the plate, ensuring cells were covered. This was then aspirated. 1 mL of 0.25% trypsin–EDTA solution was added. The plate was incubated at 37 °C for 5 minutes. 8 mL of DMEM were added, using the pipette to break up the cells.Western blots
For lysate extraction, the medium was removed from cells and plates were washed once with 1× PBS. Cells were trypsinised, collected, centrifuged and the pellets kept on ice. The cell pellet was re-suspended in 100 μL of ice cold RIPA lysis buffer containing a 1 μg mL−1 Protease and Phosphatase Inhibitor Cocktail Set III (Calbiochem) and incubated for 20 min on ice. Cells were ruptured by passing through a syringe 10 times or with sonication, and centrifuged at 14000g for 15 min at 4 °C. The supernatant was transferred into Eppendorf tubes and protein concentrations were determined using the Bradford protein assay (Fermentas). 4× Laemmli buffer was added in 1:4 ratio and samples were heated at 95 °C for 5 min. 20 μg of total protein per sample were subjected to 10% or 15% SDS-PAGE and transferred to Immobilon-P membranes (Millipore). An Odyssey CL× Infrared Imaging System (Li-COR, Lincoln, NE, USA) was used to visualise the results. Primary antibodies used were: β-actin (Abcam, #ab8227), and p53 (Santa Cruz Biotechnology, #sc-126).
Preparation of monomer mixture
Phosphate Buffer Solution (PBS) was produced using one PBS tablet per 100 mL of HPLC (High Performance Liquid Chromatography) grade water. This was swirled and sonicated for approximately 10 minutes. The monomeric solution was then prepared. Into 1.5 mL Eppendorf tubes, the following compounds were weighed: 19.5 mg of N-isopropylacrylamide (NIPAm), 3 mg of N,N′-methylene-bisacrylamide (MBAA), 15 mg of n-tert-butylacrylamide (TBAm) with 200 μL ethanol, 3 mg of 3-aminopropyl methacrylate. 1 mL of PBS was added to each of these tubes and the mixed using the Vortexer. Then, in turn, each were added to 50 mL of PBS using a pipette. 50 μL of acrylic acid in PBS (22 μL mL−1) was also added to the solution. The monomeric solution was purged with nitrogen for 20 minutes. An initiation mixture was produced in an Eppendorf tube, using 12 mg ammonium persulfate (APS), 6 μLl tetramethylethylenediamine (TEMED), 400 μL PBS. This was mixed using the Vortexer. 200 μL of the initiation solution was then added to each flask containing the monomeric mixture, swirled and left to stand for 1 hour.Preparation of the cells
The liquid from each of the flasks (NuncЙ EasYFlask™ Cell Culture Flask T175 or T225 Format) was decanted. The cells (which line one side of the flask) were washed with 20 mL of PBS. This was again decanted. Washing of the cells was completed twice in total. 20 mL of monomeric mixture was then added to each flask and purged under N2 for 5 minutes.Washing and digestion of cell cultures
Each flask was washed three times with the same procedure as step 4. A digestion solution was produced using (for each flask): 1 mg Trypsin dissolved in 0.5 mL PBS. This was added to the flasks containing 15 mL of PBS solution. The solution was swirled and left to stand for 3 days at room temperature.Separation of the epitopes of the cells and MIPs
The solution in the flasks were poured into 100 kDa centrifuge tubes (Millipore Amicon Ultra-15 Centrifugal Filter) and balanced with HPLC grade water before being put onto the centrifuge (Centurion Scientific 1010 Centrifuge). This was turned on until the solution had been filtered into the bottom of the cartridge, leaving the MIPs in the filter. The centrifuge ran at 2236 g for 20 minutes. Wash one involved pipetting 10 mL of water into the flask and decanting into the centrifuge cartridge, then centrifuging again until the liquid had been passed through the filter, accumulating in the bottom of the cartridge. Emptying the cartridge after each centrifuge cycle. Washes 2, 3 and 4 involved adding 10 mL of water into the filter of the cartridge and then centrifuged. HPLC water had been heated to around 95 °C. For each flask, using a Pasteur pipette, 1 mL of the heated water was added to the filter, mixed with the MIPs and then transferred to an Eppendorf tube. This was placed into an Eppendorf Thermomixer® (C109073 Eppendorf Thermomixer) for 10 minutes. The solution was transferred back to the filter of the corresponding centrifuge cartridge and centrifuged for 5 minutes. Hot elution step was repeated two more times. All eluted fractions (3 × 1 mL) were combined and kept frozen.LC-MS/MS procedure
Empore™ Solid Phase Extraction (SPE) desalting. The column was primed with ethanol and washed four times with 0.1% formic acid. Samples were acidified with 1% formic acid and mixed using a Vortexer. Samples were spun for 10 minutes at 12550 rpm (14087 G). 95% of the sample was then added to the column and washed 4 times with 0.1% formic acid. The elutions were collected in one tube. 750 μL 60% acetonitrile and 0.1% formic acid was eluted. 750 μL 80% acetonitrile and 0.1% formic acid was eluted. Samples were then placed on the SpeedVac for 2 hours. The samples were then lyophilised overnight. Samples were reconstituted in 20 μL 0.1% formic acid/3% acetonitrile and 20 μL 100 fmol μL−1 alcohol dehydrogenase (ADH) and transferred into a HPLC vial. In a Waters nanoACQUITY UPLC (Ultra Performance Liquid Chromatography), the samples were injected onto a Waters 2G-V/M Symmetry C18 trap column (180 μm × 20 mm, 5 μm) to ensure accurate focus of the peptides before elution onto the UPLC column. They were then eluted onto a Waters Acquity HSS T3 analytical UPLC column (75 μm × 250 mm, 1.8 μm). Trapping method: single pump trapping with 99.9% solvent A and 0.1% solvent B at a flow rate of 5 μL min−1 for 3 min. Analytical column gradient: 0 min—3% B, 30 min—40% B, 32 min—85% B, 40 min—85% B and 41 min—3% B. The flow rate was 0.3 μL min−1 and the temperature of the column was 40 °C. Solvent A consisted of LC-MS grade water with 0.1% formic acid and solvent B—acetonitrile with 0.1% formic acid. The NanoAcquity UPLC was coupled to a Waters Synapt G2 HDMS mass spectrometer, which was in positive electrospray ionisation mode; capillary voltage—2.4 kV; cone voltage—30 V. A helium gas flow of 180 mL min−1 and ion mobility separator nitrogen gas flow of 90 mL min−1 with a pressure of 2.5 mbar were used. The IMS wave velocity was set at 650 m s−1 and the IMS wave height at 40 V. [Glu1]-Fibrinopeptide (GFP) with m/z 785.8427 was used as a lockspray to maintain mass accuracy. Argon was used as t collision gas. For low energy acquisition, the collision energy was set at 4 V and for high energy acquisition, a voltage gradient from 20 to 40 V was used.
Data analysis
Data was acquired using the MassLynx 4.1 software. The raw data was analysed using Progenesis QI for proteomics (Non-Linear Dynamics (Waters), UK). This aligns the chromatography for each run to one selected run, performs peak picking and identifies peptides and proteins from the peptide and fragment m/z values and searching against a human database from Uniprot (https://www.uniprot.org—downloaded in 2017). A between-subject experiment design was used to compare senescent and normal cells.Dynamic light scattering (DLS) procedure
After elution of the peptides-specific MIPs were characterised using DLS. Samples were re-suspended in HPLC grade water in a glass vial and sonicated for 5 minutes (0.95 mL HPLC grade water, 0.05 mL sample). The solution was then transferred to a disposable cuvette which was placed into the DLS machine for analysis. Preparation of the software—Zetasizer, ran on Microsoft Windows XP combined with a using a Zetasizer Nano Range DLS machine. Size graphs can be extracted from the computer software, and these show each of the six runs that were produced in each measurement. An average can be found for each of the peak sizes for the MIP data.Transmission electron microscopy (TEM) analysis
TEM images were obtained in a JEOL JEM-1400 TEM with an accelerating voltage of 120 kV, equipped with EMSIS Xarosa digital camera with Radius software. Samples were sonicated for 2 min immediately prior to adsorption to the grid. Then, 10 μL of the sample was applied to a freshly glow discharged carbon film grid (400 mesh, AGS160-Agar Scientific Ltd). Grids were glow discharged in a Quorum GloQube System for 15 s at 20 mA. Then, the sample was left to adsorb and dry for 25 min at a room temperature. The statistical analysis of the images was made using open source software ImageJ.Conflicts of interest
There are no conflicts to declare.References
- L. Hayflick, Exp. Cell Res., 1965, 37, 614–636 CrossRef CAS PubMed.
- G. P. Dimri, X. Lee, G. Basile, M. Acosta, G. Scott, C. Roskelley, E. E. Medrano, M. Linskens, I. Rubelj and O. Pereira-Smith, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 9363–9367 CrossRef CAS PubMed.
- J. Mendelsohn, P. M. Howley, M. A. Israel, J. W. Gray and C. B. Thompson, The Molecular Basis of Cancer E-Book, Elsevier Health Sciences, 2014 Search PubMed.
- S. He and N. E. Sharpless, Cell, 2017, 169, 1000–1011 CrossRef CAS PubMed.
- D. G. Burton and V. Krizhanovsky, Cell. Mol. Life Sci., 2014, 71, 4373–4386 CrossRef CAS PubMed.
- D. Muñoz-Espín and M. Serrano, Nat. Rev. Mol. Cell Biol., 2014, 15, 482–496 CrossRef PubMed.
- B. G. Childs, M. Gluscevic, D. J. Baker, R.-M. Laberge, D. Marquess, J. Dananberg and J. M. Van Deursen, Nat. Rev. Drug Discovery, 2017, 16, 718–735 CrossRef CAS PubMed.
- J. Gil and D. J. Withers, Nature, 2016, 530, 164–165 CrossRef CAS PubMed.
- J. Campisi, Annu. Rev. Physiol., 2013, 75, 685–705 CrossRef CAS PubMed.
- J. A. Sorrentino, H. K. Sanoff and N. E. Sharpless, Trends Mol. Med., 2014, 20, 375–384 CrossRef CAS PubMed.
- M. Althubiti, L. Lezina, S. Carrera, R. Jukes-Jones, S. Giblett, A. Antonov, N. Barlev, G. Saldanha, C. Pritchard and K. Cain, Cell Death Dis., 2014, 5, e1528 CrossRef CAS PubMed.
- A. E. Ekpenyong-Akiba, M. Poblocka, M. Althubiti, M. Rada, D. Jurk, S. Germano, G. Kocsis-Fodor, Y. Shi, J. J. Canales and S. Macip, Aging Cell, 2020, 19, e13079 CrossRef CAS PubMed.
- O. H. Jeon, C. Kim, R.-M. Laberge, M. Demaria, S. Rathod, A. P. Vasserot, J. W. Chung, D. H. Kim, Y. Poon and N. David, Nat. Med., 2017, 23, 775–781 CrossRef CAS PubMed.
- E.-C. Kim and J.-R. Kim, BMB Rep., 2019, 52, 47 CrossRef CAS PubMed.
- A. F. Tabasso, D. J. Jones, G. D. Jones and S. Macip, Clin. Oncol., 2019, 31, 283–289 CrossRef CAS PubMed.
- A. Kaur, S. Macip and C. M. Stover, Front. Cell Dev. Biol., 2020, 8, 218 CrossRef PubMed.
- M. Poblocka, A. L. Bassey, V. M. Smith, M. Falcicchio, A. S. Manso, M. Althubiti, X. Sheng, A. Kyle, R. Barber and M. Frigerio, Sci. Rep., 2021, 11, 1–10 CrossRef PubMed.
- A. E. Ekpenyong-Akiba, F. Canfarotta, B. Abd H, M. Poblocka, M. Casulleras, L. Castilla-Vallmanya, G. Kocsis-Fodor, M. E. Kelly, J. Janus and M. Althubiti, Nanoscale Horiz., 2019, 4, 757–768 RSC.
- A. E. Ekpenyong-Akiba, M. Poblocka and S. Macip, in Senolytics in Disease, Ageing and Longevity, Springer, 2020, pp. 103–130 Search PubMed.
- K.-H. Kim, G.-T. Park, Y.-B. Lim, S.-W. Rue, J.-C. Jung, J.-K. Sonn, Y.-S. Bae, J.-W. Park and Y.-S. Lee, Biochem. Biophys. Res. Commun., 2004, 318, 819–825 CrossRef CAS PubMed.
- U. Reineke and M. Schutkowski, Epitope Mapping Protocols, Springer, 2009 Search PubMed.
- C. Popescu, A. D. Zamfir and N. Dinca, Applications of Mass Spectrometry in Life Safety, Springer Science & Business Media, 2008 Search PubMed.
- S. S. Piletsky, E. Piletska, M. Poblocka, S. Macip, D. J. Jones, M. Braga, T. H. Cao, R. Singh, A. C. Spivey and E. O. Aboagye, Nano Today, 2021, 41, 101304 CrossRef CAS.
- S. Macip, M. Igarashi, P. Berggren, J. Yu, S. W. Lee and S. A. Aaronson, Mol. Cell. Biol., 2003, 23, 8576–8585 CrossRef CAS PubMed.
- S. Özcan, N. Alessio, M. B. Acar, E. Mert, F. Omerli, G. Peluso and U. Galderisi, Aging, 2016, 8, 1316 CrossRef PubMed.
- R. L. Juliano and V. Ling, Biochim. Biophys. Acta, Bioenerg., 1976, 455, 152–162 CrossRef CAS.
- S. Piletsky, E. Piletska, F. Canfarotta and D. Jones, Methods and kits for determining binding sites, 2017, GB1704823.2 Search PubMed.
- K. Yoshimatsu, H. Koide, Y. Hoshino and K. J. Shea, Nat. Protoc., 2015, 10, 595–604 CrossRef CAS PubMed.
- E. Moczko, A. Guerreiro, C. Cáceres, E. Piletska, B. Sellergren and S. A. Piletsky, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2019, 1124, 1–6 CrossRef CAS PubMed.
- A. Poma, A. Guerreiro, S. Caygill, E. Moczko and S. Piletsky, RSC Adv., 2014, 4, 4203–4206 RSC.
- Y. Hoshino, H. Koide, T. Urakami, H. Kanazawa, T. Kodama, N. Oku and K. J. Shea, J. Am. Chem. Soc., 2010, 132, 6644–6645 CrossRef CAS PubMed.
- L. Wang, L. Xiao, R.-Z. Zhang, L.-Z. Qiu, R. Zhang and H.-X. Shi, Biomed. Mater., 2018, 14, 015003 CrossRef PubMed.
- M. Gallorini, A. Cataldi and V. Di Giacomo, Int. Endod. J., 2014, 47, 813–818 CrossRef CAS PubMed.
- L. Gómez-Gascón, I. Luque, A. Olaya-Abril, I. Jiménez-Munguía, R. A. Orbegozo-Medina, E. Peralbo, C. Tarradas and M. J. Rodríguez-Ortega, J. Proteomics, 2012, 75, 5654–5666 CrossRef PubMed.
- L. Aguilera, E. Ferreira, R. Giménez, F. J. Fernández, M. Taulés, J. Aguilar, M. C. Vega, J. Badia and L. Baldomà, Int. J. Biochem. Cell Biol., 2012, 44, 955–962 CrossRef CAS PubMed.
- J. D. Bendtsen, L. Kiemer, A. Fausbøll and S. Brunak, BMC Microbiol., 2005, 5, 1–13 CrossRef PubMed.
- C. Nombela, C. Gil and W. L. Chaffin, Trends Microbiol., 2006, 14, 15–21 CrossRef CAS PubMed.
- E. Zografos, A. K. Anagnostopoulos, A. Papadopoulou, E. Legaki, F. Zagouri, E. Marinos, G. T. Tsangaris and M. Gazouli, Cancer Genomics Proteomics, 2019, 16, 129–137 CrossRef CAS PubMed.
- M. Lei, Y. Kawasaki, M. R. Young, M. Kihara, A. Sugino and B. K. Tye, Genes Dev., 1997, 11, 3365–3374 CrossRef CAS PubMed.
- Z. Erbayraktar, B. Alural, R. S. Erbayraktar and E. P. Erkan, Cancer Cell Int., 2016, 16, 1–10 CrossRef PubMed.
- W. Jiang, D. McDonald, T. J. Hope and T. Hunter, EMBO J., 1999, 18, 5703–5713 CrossRef CAS PubMed.
- A. Jackson, P. Pahl, K. Harrison, J. Rosamond and R. Sclafani, Mol. Cell. Biol., 1993, 13, 2899–2908 CAS.
- L. Matthews and A. Guarné, Cell Cycle, 2013, 12, 1180–1188 CrossRef CAS PubMed.
- S. Matsumoto and H. Masai, Biochem. Soc. Trans., 2013, 41, 1712–1719 CrossRef CAS PubMed.
- K. Labib, Genes Dev., 2010, 24, 1208–1219 CrossRef CAS PubMed.
- M. Yamada, H. Masai and J. Bartek, Cell Cycle, 2014, 13, 1859–1866 CrossRef CAS PubMed.
- Q. Zhou, J. Dai, T. Chen, L. A. Dada, X. Zhang, W. Zhang, M. M. DeCamp, R. A. Winn, J. I. Sznajder and G. Zhou, Cell. Signalling, 2017, 38, 49–59 CrossRef CAS PubMed.
- B. Ozdamar, R. Bose, M. Barrios-Rodiles, H.-R. Wang, Y. Zhang and J. L. Wrana, Science, 2005, 307, 1603–1609 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2na00424k |
| This journal is © The Royal Society of Chemistry 2022 |