
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioengineering a glucose oxidase nanosensor for near-infrared continuous glucose monitoring†
Vitalijs
Zubkovs
 ab,
Hanxuan
Wang
a,
Nils
Schuergers
ac,
Astrid
Weninger
d,
Anton
Glieder
de,
Stefano
Cattaneo
b and
Ardemis A.
Boghossian
*a
ab,
Hanxuan
Wang
a,
Nils
Schuergers
ac,
Astrid
Weninger
d,
Anton
Glieder
de,
Stefano
Cattaneo
b and
Ardemis A.
Boghossian
*a
aEcole Polytechnique Fédérale de Lausanne (EPFL), Institute of Chemical Sciences and Engineering, CH-1015 Lausanne, Switzerland. E-mail: ardemis.boghossian@epfl.ch
bSwiss Center for Electronics and Microtechnology (CSEM), Landquart, Switzerland
cInstitute of Biology III, University of Freiburg, Freiburg, Germany
dInstitute of Molecular Biotechnology, Graz University of Technology, Graz, Austria
ebisy GmbH, Hofstaetten, Austria
First published on 4th May 2022
Abstract
Single-walled carbon nanotubes (SWCNTs) emit photostable near-infrared (NIR) fluorescence that is conducive for optical glucose monitoring. Such SWCNT-based optical sensors often require the immobilization of proteins that can confer glucose selectivity and reactivity. In this work, we immobilize a glucose-reactive enzyme, glucose oxidase (GOx), onto SWCNTs using a N-(1-pyrenyl)maleimide (PM) crosslinker via thiol bioconjugation of engineered cysteine residues. We compare the conjugation of several glucose oxidase variants containing rationally-engineered cysteines and identify a D70C variant that shows effective bioconjugation. The bioconjugation was characterized through both absorption and fluorescence spectroscopy. Furthermore, we demonstrate an application for continuous glucose monitoring in the NIR-II optical region using the bioconjugated reaction solution, which shows a reversible response to physiological concentrations of glucose. Finally, we develop a miniaturized NIR-II reader to be used for cell cultures that require continuous glucose monitoring.
Introduction
Optical technologies represent a leading and growing field for biomedical diagnosis, analysis, and even treatment.1 Many of the in vivo technologies rely on fluorescent dyes and biomolecules that can receive or transmit signals deep within biological tissue. Although conventional fluorescent dyes often undergo photodegradation under prolonged illumination, certain fluorophores such as quantum dots (QDs) and single-walled carbon nanotubes (SWCNTs) have photostabilities that can extend the lifespan of optical devices for continuous measurements.2 Through proper functionalization and encapsulation, SWCNTs have shown long-term biocompatability in vivo.3 While the near-infrared (NIR) fluorescence of SWCNTs allows deep optical penetration, the sensitivity of this fluorescence to biomolecules, even down to the single molecule, allows SWCNTs to be used as in vivo optical sensors for continuous biomedical monitoring.4The selectivity of these SWCNT-based sensors can be modulated by functionalizing the surface of the nanotubes with proteins, such as glucose-binding protein, glucose dehydrogenase, concanavalin A, or glucose oxidase (GOx).5–8 Among these proteins, GOx represents the gold standard for glucose sensing because of its stability and high specificity to glucose.9 The oxidation of glucose and release of free electrons occur at the reaction center of the protein in the presence of a flavin adenine dinucleotide (FAD) cofactor. The key challenge for effective glucose sensing lies with the bioconjugation strategy of the protein.10 While non-specific protein adsorption provides a facile, scalable approach towards achieving adequate immobilization, the adsorption efficiency is protein-specific and depends on the surface chemistry of the proteins.11,12 The disruption in adsorbed protein conformation may also result in the loss of activity. Moreover, the sensor response is limited to only correctly oriented proteins. In the case of GOx, proteins oriented with active sites in proximity to the SWCNT, for example, could be involved in optical modulation through enzymatic pocket doping.5 Compared to conjugative methods that rely on naturally available residues that are often abundant on protein surfaces,6 such controlled orientation often requires the introduction of rare or mutated sites that are uniquely susceptible to the reaction of interest.13,14 Addressing these challenges through improved and novel bioconjugation strategies is the focus of recent studies.7,13,15,16
In this work, we immobilize GOx using a hydrophobic crosslinker that is covalently conjugated to engineered cysteine residues. While wild type GOx has three native cysteines, these residues are not accessible for conjugation; two cysteines form a disulfide bond (Cys164–Cys206), and the third (Cys521) is buried within the protein.17,18 We engineered four variants with single-cysteine mutations (Fig. 1a). The selected ampholytic crosslinker, PM, contains a maleimide group that can covalently conjugate to the cysteine residues, while the pyrene moiety at the other end can stack onto a SWCNT surface with a binding energy of 32.34 kcal mol−1 (Fig. 1b).19 As this binding energy exceeds that of aromatic or aliphatic amino acids (which are in order of 10 kcal mol−1 and 5 kcal mol−1, respectively), this linker can provide a favorable approach for oriented protein binding.19–21 In addition, linker-based protein immobilization has been shown to improve the retention of protein structure and function in the vicinity of the SWCNT.14 Using the resulting GOx-PM-SWCNT complex shown in Fig. 1b, we are able to demonstrate a continuous, reversible glucose sensor in the NIR-II optical region.
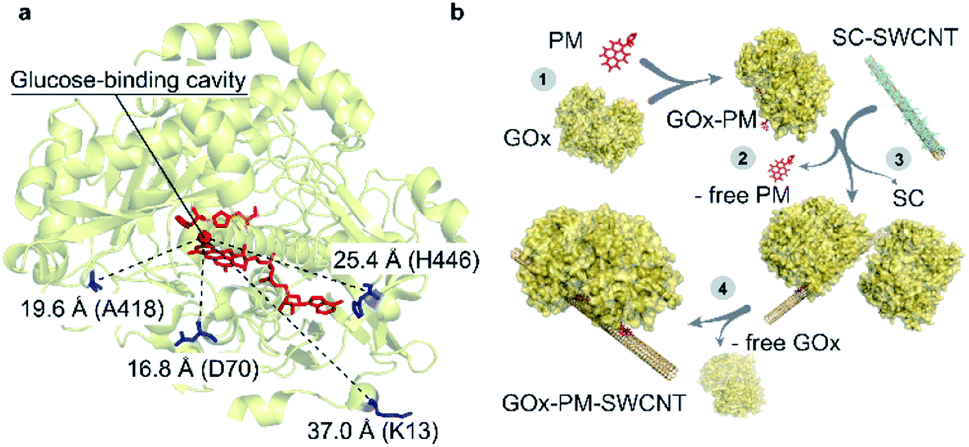 | ||
| Fig. 1 Selected residues for site-directed GOx mutagenesis. (a) Indicated distances are shown between the glucose-binding cavity above the si-face of the flavin ring (tricyclic isoalloxazine),22 which is shown as a red sphere, and the mutated cysteine residues shown in blue. The FAD and glucose-capturing amino acids above the glucose-binding cavity (His559 and His516) are shown in red (PDB: 3QVP). (b) Illustration of GOx-PM-SWCNT synthesis procedure. (1) Reduction of a GOx mutant and its conjugation to PM. (2) Purification of GOx-PM from the unreacted PM. (3) Wrapping of sodium cholate (SC)-suspended SWCNTs with GOx-PM through removal of SC. (4) Removal of free GOx in the final dialysis step. | ||
Results and discussion
Mutagenesis and expression of GOx
The mutation sites were selected based on non-conserved surface-exposed residues that were identified using sequence alignments of GOx homologues and the GOx crystal structure (PDB: 3QVP).22 We selected four positions (K13, D70, A418, and H446) with variable distances from the glucose-binding cavity of the enzyme (Fig. 1a).Wild type GOx and the K13C, D70C, A418C, and H446C variants were expressed in an optimized P. pastoris (Komagataella phaffii) production yeast strain.23,24 The SDS-PAGE analysis results shown in Fig. 2a confirm an expected protein size of ∼85 kDa. The recombinant GOx from P. pastoris shows a slight increase in the apparent molecular weight compared to the commercial GOx from A. niger (∼80 kDa). This difference is attributed to the greater degree of glycosylation.25 The secondary structures were characterised with the circular dichroism (CD) measurements shown in Fig. 2b. All structures, including the expressed wild type, commercial wild type, and mutants, share similar CD spectra. The expressed proteins contain 17–28% α-helices and 16–22% β-strands (Fig. S1†), in accordance with values reported for the commercial GOx (24% and 17% for α-helixes and β-strands, respectively) and with the protein crystal structure (PDB: 1CF3, 27% and 19% α-helixes and β-strands, respectively).17 These results confirm that the mutations did not significantly disrupt protein folding. The preservation of enzymatic activity was also confirmed by comparing activities measured with the colorimetric 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) assay (Fig. 2c). As shown in the figure, the enzymatic activity towards glucose does not significantly differ between wild type GOx and the mutants, indicating that the mutations had no significant effect on activity.
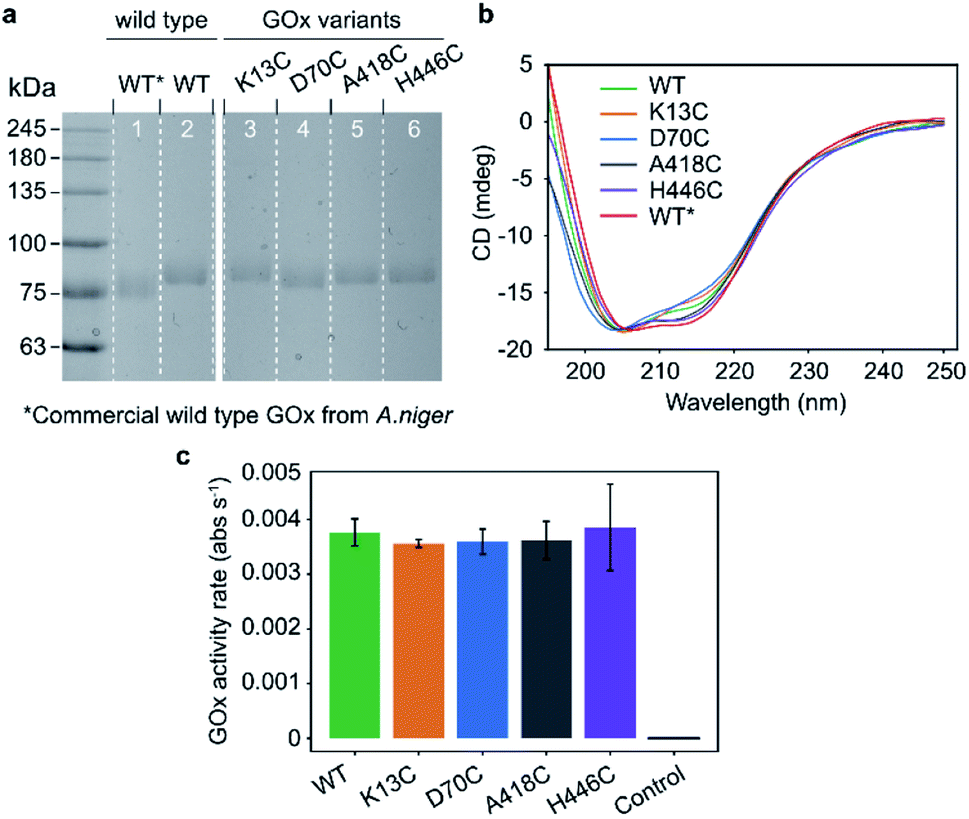 | ||
| Fig. 2 Characterization of expressed and purified GOx variants. (a) Coomassie-stained SDS-PAGE of commercial wild type GOx from A. niger (lane 1, denoted as WT*) compared to purified wild type (lane 2, denoted as WT) and mutated GOx (lanes 3–6) from P. pastoris. (b) CD spectra of GOx variants from P. pastoris and a commercially available wild type GOx from A. niger. (c) Relative activities of GOx towards glucose as measured by the ABTS colorimetric assay. The control sample contains the reaction mixture without GOx. The error bars indicate standard deviations among technical triplicates. | ||
Bioconjugation of GOx with thiol-reactive crosslinkers
The accessibility of the targeted residues was compared based on their reactivity towards 3-(2-pyridyldithio)propionic acid N-hydroxysuccinimide ester (SPDP), which releases a conjugation reaction product, pyridine-2-thione, that absorbs at 343 nm.26 Absorption spectra were measured over 120 minutes (Fig. S2†), and the reactivity of free thiols was monitored through the increase of absorption at 343 nm (Fig. 3a). A418C was identified as the most reactive cysteine mutant, with 0.4 moles of SPDP reacted per mole of GOx, followed by the D70C (∼0.3 moles of SPDP reacted per mole of GOx), and the K13C and H446C mutants (∼0.2 moles of SPDP reacted per mole of GOx). As expected, the reduced wild type GOx did not react with SPDP.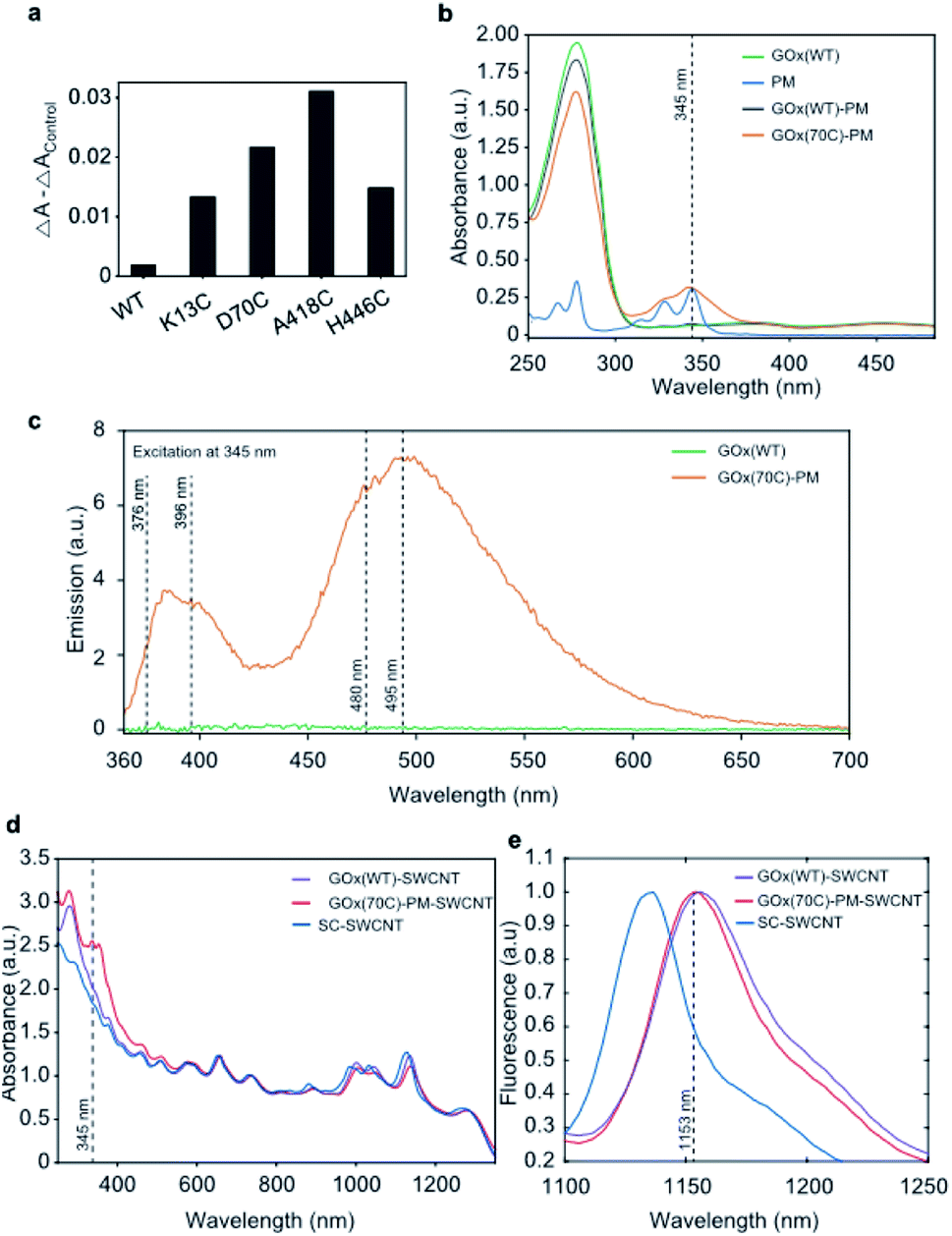 | ||
| Fig. 3 Preparation and characterization of GOx-PM-SWCNTs. (a) Changes in absorbance at 343 nm for GOx variants measured before and after the addition of SPDP. WT denotes purified wild type GOx. The control sample contained the reaction mixture without GOx, and its absorbance value was subtracted from the measured absorbance of the variants. (b) UV-Vis absorbance spectra of wild type GOx (GOx(WT)), PM, and purified samples following reaction of PM with wild type GOx (GOx(WT)-PM) and the D70C GOx mutant (GOx(70C)-PM). The absorption of the aromatic protein residues is seen at 280 nm. The broad peak at 450 nm corresponds to the absorption of FAD. The dotted line shows the PM absorbance peak maximum at 345 nm. The GOx concentration in the samples was 0.5 mg mL−1. (c) Emission spectra of GOx(WT) and GOx(70C)-PM upon excitation at 345 nm. The dotted lines show the pyrene fluorescence peak maxima at 376 and 396 nm and the pyrene excimer fluorescence at 480 and 495 nm. The GOx concentration in the samples was 0.5 mg mL−1. (d) UV-Vis-NIR absorbance spectra of GOx(WT)-SWCNTs, GOx(70C)-PM-SWCNTs, and SC-SWCNTs. The spectra were normalized to the SWCNT absorbance at 739 nm. The dotted line at 345 nm shows the PM absorbance peak. (e) Normalized fluorescence emission spectra of GOx(WT)-SWCNTs, GOx(70C)-PM-SWCNTs, and SC-SWCNTs. The dotted line shows GOx(70C)-PM-SWCNT fluorescence maxima of the E11 (7,6) SWCNT peak at 1153 nm. Excitation at 660 ± 5 nm. | ||
To create an enzymatic sensor, we applied a previously reported protocol,14 and we selected one of the maleimide-reactive variants (D70C) for immobilization onto the SWCNT using the conjugation strategy shown in Fig. 1b. We note that despite its greater reactivity, the A418 was not selected for further experiments due to its low expression yield. The absorbance measurements shown in Fig. 3b confirm the presence of PM in the purified, conjugated D70C mutant. In contrast, no PM absorption peak was observed from the wild type protein following PM conjugation. The conjugation of the PM linker to the GOx mutant is also confirmed by the PM fluorescence measurements shown in Fig. 3c. In agreement with previous observations, the PM linker emits fluorescence at 376 and 396 nm upon 345 nm excitation when a covalent bond is formed between the PM maleimide and the protein thiol.27 Broad emission peaks at 480 and 495 nm also appear due to the proximity of the two pyrenes to one another (10 Å),28 indicating that the PM-conjugated D70C GOx (GOx(70C)-PM) monomers are folded into dimers. In contrast, the wild type GOx showed no fluorescence following the PM conjugation reaction and purification. These results confirm PM conjugation of the D70C mutant through the engineered cysteine residue.
Protein conjugation to the SWCNT surface was confirmed through both absorbance and fluorescence spectroscopy. Compared to sodium cholate suspended SWCNTs (SC-SWCNTs), SWCNTs suspended with non-specifically adsorbed wild type GOx (GOx(WT)-SWCNTs) and GOx(70C)-PM (GOx(70C)-PM-SWCNTs) show red-shifted absorption peaks for the (7,5) and (7,6) chiralities (Fig. 3d). The convoluted E22 (7,5) and E22 (7,6) SWCNT absorption peak shifted from 657 to 660 nm. The E11 (7,5) and E11 (7,6) SWCNT peaks shifted from 1045 to 1047 nm and from 1135 to 1138 nm, respectively. This shifting is attributed to the change in solvation due to the protein corona. Compared to the GOx(WT)-SWCNTs, the GOx(70C)-PM-SWCNT sample shows additional absorption peaks at 345 nm that correspond to the PM linker. Fig. 3e similarly shows distinct SWCNT fluorescence spectra for the three samples on 660 nm excitation. Most notably, the protein-suspended samples show a significant red-shift in fluorescence compared to the SC-SWCNTs. This red-shifting is in agreement with previous studies that have reported red-shifting due to the decreased nanotube surface coverage by proteins compared to SC.29,30 Furthermore, the GOx(WT)-SWCNTs show an even greater 4 ± 1 nm red-shift of the E11 (7,6) chirality emission peak relative to the corresponding peak observed at 1153 ± 1 nm for the GOx(70C)-PM-SWCNTs. This further red-shifting suggests greater interaction or shielding of the nanotube surface with the conjugated GOx(70C)-PM compared to the non-specifically adsorbed GOx.
Glucose detection using the GOx(70C)-PM-SWCNT sensor
The performance of the GOx(70C)-PM-SWCNT sensor was evaluated by monitoring the (7,6) fluorescence intensity peak at 1153 ± 1 nm upon glucose addition (Fig. 4a). In the presence of glucose, we observe an increase in the fluorescence intensity. On the other hand, no fluorescence response was observed for the SC-SWCNTs. These measurements confirm an enzymatic dependence on the glucose response, in agreement with previous measurements performed using non-specifically adsorbed GOx.5 However, as the concentration of the immobilized GOx remains unknown in these samples, the relative contribution of the linker to the glucose response cannot be quantified. Nonetheless, the performance of the bioconjugated construct was further demonstrated in a cell culture incubator at 37 °C using the setup shown in Fig. 4b and c. The GOx(70C)-PM-SWCNT sensor was deposited inside a well and covered with a semipermeable membrane. The top of the membrane was attached to a fluidic cross-flow channel that allowed convective fluid exchange through the membrane. The fluorescence response of the sensor was monitored from the glass substrate using a focusing lens and fiber optics probe. The sensor demonstrated an operating range of 1 to 10 mM (Fig. 4d). This range falls within the typical concentration limit of glucose levels in blood or mammalian cell cultures.31 The sensor response was further measured continuously for 12 h over six rinsing cycles consisting of 1 h flow of 10 mM glucose solution in PBS and 1 h flow of PBS in the absence of glucose (Fig. 4e). The fluorescence intensity of the GOx(70C)-PM-SWCNT sensor increased during the flow of glucose and recovered when the device was rinsed with PBS. The increase and recovery of intensity over multiple cycles confirms the activity and reversibility of the sensor at physiological temperatures.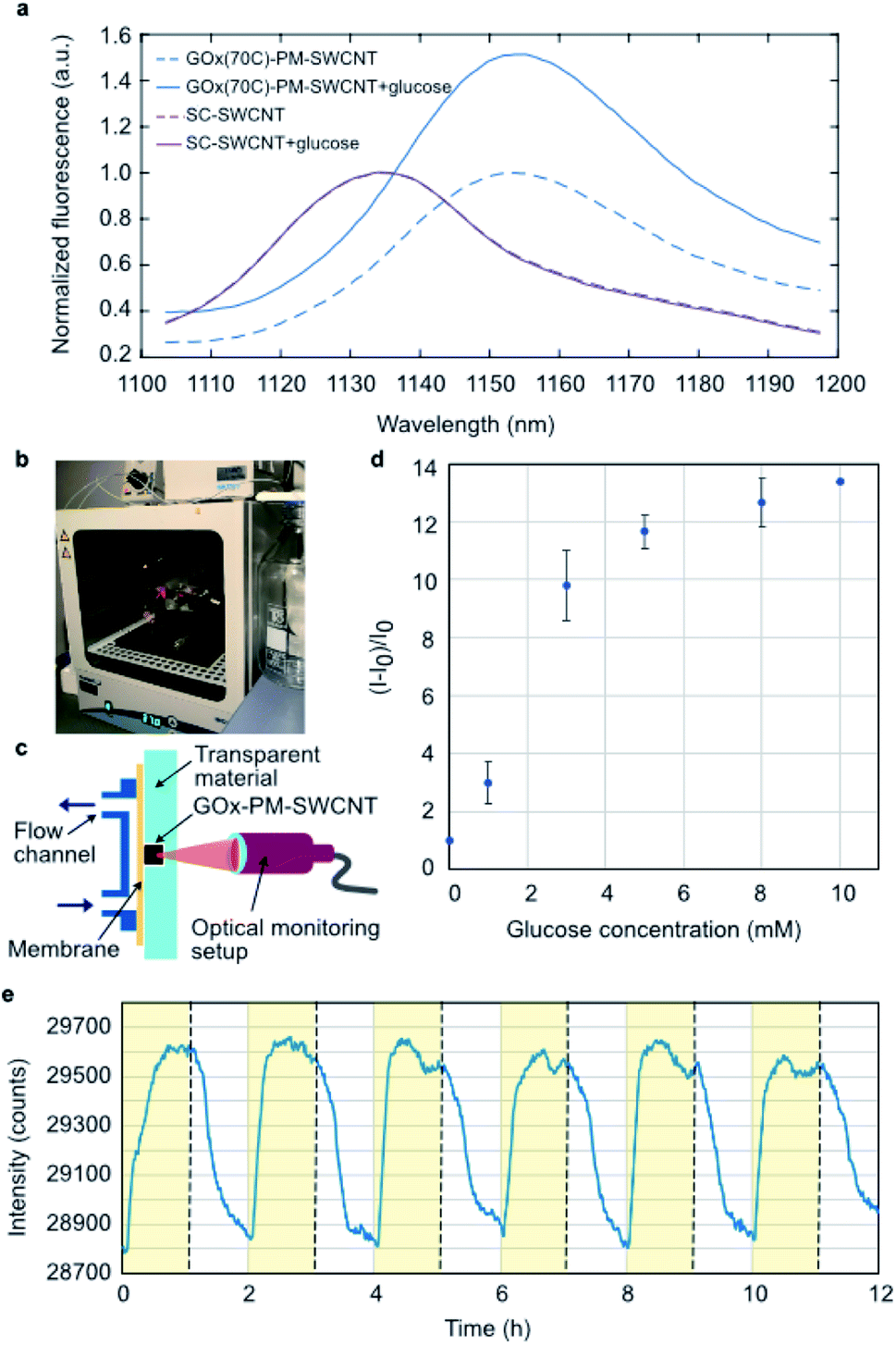 | ||
| Fig. 4 NIR fluorescence response of the GOx-PM-SWCNT sensor towards glucose. (a) NIR fluorescence spectra of the GOx(70C)-PM-SWCNTs and SC-SWCNTs before (dashed lines) and 2 min after (solid line) the addition of 20 mM glucose. The (7,6) peaks were normalized to their intensity maxima before the addition of glucose. Excitation at 660 ± 5 nm. (b) The setup used for monitoring sensor activity at 37 °C. (c) A schematic of the fluid setup. (d) Calibration curve showing the intensity change of the GOx-PM-SWCNT sensor to varying glucose concentrations ranging from 1 to 10 mM. Standard glucose solutions were prepared in PBS. (e) The (7,6) peak intensity of GOx(70C)-PM-SWCNTs sensors monitored in the fluidic setup with a semipermeable membrane at 37 °C. | ||
The reversibility demonstrated herein provides a promising basis for long-term glucose monitoring. GOx-based SWCNTs have been shown to be stable for up to several months.32–34 Similarly, the constructs developed in this study can be stored in PBS and re-used for over 6 months at 4 °C. However, the sensor irreversibly diminishes following dehydration, in agreement with previous observations that have reported irreversible agglomeration of solution-phase SWCNT samples.35 These limitations have been circumvented using alternative encapsulation methods based on hydrogels.2,36–39 These hydrogel-based constructs were shown to be stable for 300 days after subcutaneous implantation,2 providing an avenue for long-term measurements in vivo.
Conclusions and outlook
Effective bioconjugation is key to engineering optical sensors based on SWCNT fluorescence. We herein demonstrate the bioconjugation of GOx to SWCNTs using a PM linker. The stacking of the linker's pyrene group on the nanotube surface reflects a non-covalent immobilization approach that can retain the nanotube's fluorescence. Further, the thiol-specific reactivity of the linker allows one to conjugate the protein in a site-specific, oriented manner. This control over protein orientation is crucial for sensing mechanisms that rely on protein conformational changes or active site distance from the nanotube surface. For the GOx-based sensor developed in this work, the primary sensing mechanism is proposed to be based on enzymatic pocket doping.5,40,41 Such mechanisms often rely on proximity of the enzyme's active site to the nanotube surface for effective charge transfer. Though this study focuses on bioengineering attachment sites that show sufficient bioconjugation to the PM linker, the platform can be extended to focus on engineering attachment sites that optimize the optical response to glucose. This optimization can be achieved by comparing the NIR response of the purified engineered bioconjugates. In addition to optimizing the attachment site, this bioconjugation approach can also be used to immobilize proteins that have been otherwise engineered for improved sensor responsivity through, for example, chimera design or non-cysteine-specific mutations. This conjugation method therefore allows the controlled immobilization of different bioengineered enzymes, an aspect that is important for developing SWCNT-based optical sensors.In addition to its advantages in controlling immobilization, this method is also compatible for use as a commercial NIR glucose sensor. Glucose sensing is necessary for applications such as in vivo monitoring of diabetics as well as cell culture monitoring. Such sensors are especially useful for monitoring mammalian cultures like Chinese hamster ovary (CHO) cells, which are used for commercial therapeutic protein production. Since these cells consume glucose during proliferation, the glucose concentration must be continuously monitored to maintain sufficient levels for growth and viability. Compared to conventional electrochemical technologies, the NIR sensors demonstrated herein benefit from cost-effective fabrication and non-invasive measurements. Although NIR sensors currently lack a commercially available, portable device for detecting the NIR emissions, recent advancements in camera technologies have opened the doors to miniaturized devices. Most recently, we designed a miniaturized cell culture monitoring sensor shown in Fig. 5a. The setup consists of a 660 nm LED excitation source for illuminating the sensor, visible light photodiode (PD1) for monitoring the excitation light intensity, and an InGaAs photodiode (PD2) for monitoring the NIR fluorescence of GOx(70C)-PM-SWCNTs (Fig. 5b and c). The LED excitation wavelength has been selected to overlap with the excitation wavelengths used in this study. A NIR-transmitting filter is placed between the sensing material and PD2. All optoelectronic components are imbedded in an epoxy resin that provides hermetic packaging. A semi-permeable membrane is glued above the compartment with GOx(70C)-PM-SWCNTs to prevent leakage of the sensing material. The construct can sample cell culture media from a flask using a fluidics system. The sensor readout is then collected using a custom microcontroller unit, as demonstrated in Fig. S5.† Although the demonstration of this portable device for glucose monitoring of a cell culture is the focus of ongoing efforts, its construction presents a significant advancement in overcoming a major bottleneck in the commercialization of NIR sensors. The construction of this portable device therefore provides a powerful avenue for the commercial realization of NIR sensors, starting with the glucose sensor demonstrated herein.
 | ||
| Fig. 5 Miniaturized device for continuous glucose monitoring. (a) Demonstration of the sensor application for continuous glucose monitoring in a fluidics device. The sensor printed circuit board (PCB) is connected with a cable to the microcontroller PCB. (b) Illustration and (c) photograph of the sensor setup. | ||
Methods
Construction of recombinant plasmids for GOx expression
Positions for non-conserved amino acids were identified using ConSurf software.42 GOx expression plasmids were constructed using a pUC57 vector harboring a synthetic gene of A. niger GOx (GeneBank X16061, Table S1†) that lacks the signal peptide. The pUC57 vector harboring the synthetic gene was provided by bisy GmbH. Site-directed mutagenesis for the amino-acid exchanges was performed by two single-primer reactions according to a protocol by Oded Edelheit et al.,43 with the exception of the Pwo polymerase, which was substituted by Q5 polymerase. The wild type and mutant variants of the synthetic GOx gene were amplified using primer pairs from Table S2† that add a SapI restriction site, and the gene was ligated into the SapI digested expression vector, pBSYA3S1Z, which was provided by bisy GmbH. The ligation fused the coding sequence to an α-factor secretion signal. The resulting vectors were subcloned into E. coli and verified by sequencing (GATC, Germany). The polymerase chain reaction (PCR) was performed in 0.2 mL MicroAmp reaction tubes (Applied Biosystems, Life Technologies) in a SimpliAmp thermal cycler (Thermo Fisher Scientific). Briefly, the 50 μL reaction mix included 5 μL of a 40 pM primer, 4 μL (or 500 ng) of the plasmid deoxyribonucleic acid (DNA), 2 μL of 0.2 mM dNTPs, 1 μL (or 1.5 U) of Q5 polymerase, 10 μL of 5 × Q5 buffer from a cloning kit (M0491S, New England Biolabs), and 28 μL water. The PCR protocol included denaturation for 15 s at 98 °C, annealing for 15 s at 60 °C, and elongation for 2 h 30 min at 72 °C. This procedure was repeated over 33 cycles. Next, the 40 μL of two single-primer PCR products with forward and reverse primers were combined in a PCR tube, and they were denatured for 5 min at 98 °C to separate the synthesized DNA from the plasmid template DNA. The tubes were gradually cooled from 98 °C to 16 °C. The non-mutated DNAs were digested by 1 μL of 20 U DpnI in 2.5 μL of FD buffer for 3 h at 37 °C and incubated overnight at 10 °C. The digested PCR products were analyzed by agarose gel electrophoresis and purified from the gel using a QIAEX II gel extraction kit. The purified sample contained 140 ng μL−1 of the expression plasmid, as measured with a NanoDrop 2000 spectrometer (Thermo Fisher Scientific). The plasmids were stored at 20 °C.P. pastoris transformation
The glycoengineered P. pastoris (Komagataella phaffii) BSY11-M81 strain with full deletions of the AOX1 and OCH1 genes was obtained from bisy GmbH. Prior to transformation, P. pastoris BSY11-M81 competent cells were prepared according to a protocol by Joan Lin-Cereghino et al.44 100 μL of competent P. pastoris cells were mixed with 3 μg of pBSYA3-GOx-K13C, pBSYA3-GOx-D70C, pBSYA3-GOx-A418C, pBSYA3-GOx-H446C plasmids and wild type pBSYA3-GOx plasmid. Competent P. pastoris cells were transformed with 3 μg episomal plasmid DNA by electroporation at 1.25 kV (Eppendorf Eporator), and transformed colonies were selected on YPD agar plates with 100 μg mL−1 Zeocin. In accordance with Weis et al.,12 randomly selected clones from each strain were inoculated in 250 μL of BMD 1% growth medium in a sterile 96-deep well plate covered with a gas-permeable 114 μm rayon film (VWR International) at 30 °C while shaking at 300 rpm.45 After glucose depletion, 250 μL of BMM2 medium were added to each of the wells. After 12 additional hours, 50 μL of BMM10 medium were added, and this step was repeated two times after an additional 12 and 24 h of incubation. The media preparation protocols are shown in Table S3.† After four days, the optical densities of the cultures were measured at 600 nm, and the cultures were centrifuged at 3220×g for 10 min. The supernatants were collected to test for GOx activity, and the cell pellets were stored at 4 °C.Colorimetric GOx enzymatic activity assay
Microtiter plates were used to measure enzymatic activity based on the colorimetric change of ABTS (BioChemica, ITW Reagents)46 in the presence of horseradish peroxidase (HRP) and the generated hydrogen peroxide. Briefly, 50 mM sodium citrate buffer pH 5.75 (119.8 μL per well) was mixed with 1 M glucose (β-D-glucose, AB 136302, ABCR GmbH & Co. KG) (80 μL per well), and 20 mM ABTS dissolved in sodium citrate buffer (40 μL per well). 0.2 μL HRP type VI (P6782, Sigma, 2 mg mL−1) per well were added to the reaction mixture. 15 μL of the cell culture supernatant or dilutions thereof were transferred to a clear microtiter plate. 240 μL of the reaction mixture were added to the microtiter plate directly before measuring the absorbance at 414 nm continuously in a plate reader (Varioskan LUX). Changes in absorbance were normalized to the optical density (OD600) of the original expression cultures in order to compensate for differences in growth.GOx expression in P. pastoris
Recombinant GOx was produced in P. pastoris, which can achieve yields greater than those using A. niger strains.47 Selected GOx clones (K13C, D70C, A418C, H446C) and wild type GOx were used for protein expression in 2 L Erlenmeyer flasks with cotton tissue plugs. In accordonace with Weis et al., the cells were added to flasks containing 75 mL of BMD 1% medium, and they were incubated at 30 °C while shaking at 300 rpm for two days.45 On the third day, 75 mL of BMM2 medium was added to induce protein expression. After 12 h, 15 mL of BMM10 medium was added to the flask, and this step was repeated twice. The cell cultures were then centrifuged at 3220 × g for 10 min, and the supernatants were filtered through a 0.2 μm porous filter. The supernatants were kept on ice and concentrated to 30 mL using a Vivaflow 50R 30 kDa MWCO crossflow dialysis device (Sartorius). The solutions were further concentrated in an Amicon Ultra centrifugal filter unit (Merck Millipore) with a 10 kDa MWCO, and the buffer was exchanged with PBS (pH 7.4). Finally, 6 mL of each solution was filtered through a 0.2 μm porous filter and stored at 4 °C.GOx purification
Purification and extraction of GOx from the protein mixture were performed using the AKTA Start setup (GE Healthcare) at 5 °C. A HiPrep 16/60 Sephacryl S-300 high-resolution column (GE Healthcare) was used for size-exclusion protein purification. 5.5 mL of the protein mixtures were loaded onto the column and eluted using a 10 mM PBS at pH 7.4 with 140 mM NaCl. The protein presence in each eluted fraction was confirmed using the colorimetric GOx enzymatic activity assay, and the GOx-containing fractions were collected and concentrated to 1 mL in a 10 kDa MWCO Amicon Ultra centrifugal unit. During concentration, the buffer was exchanged with 10 mM PBS (pH 7.0) with 10 mM ethylenediaminetetraacetic acid (EDTA, Sigma) and 150 mM NaCl (PBS-EDTA). The concentration of GOx in the stock solutions were measured in a NanoDrop 2000 (flavin extinction coefficient at 450 nm is 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1)46,48 and adjusted to 3 mg mL−1 (molecular weight of GOx was determined from SDS-PAGE to be 85 kDa). The final protein yields ranged between 8 to 15 mg L−1. GOx solutions were stored at 4 °C in PBS-EDTA (with EDTA as an antimicrobial agent).49
000 M−1 cm−1)46,48 and adjusted to 3 mg mL−1 (molecular weight of GOx was determined from SDS-PAGE to be 85 kDa). The final protein yields ranged between 8 to 15 mg L−1. GOx solutions were stored at 4 °C in PBS-EDTA (with EDTA as an antimicrobial agent).49
SDS-PAGE analysis
1 μL of each protein solution was diluted in 9 μL of PBS, and the resulting solution was added to 10 μL of a 2× BlueJuice loading buffer (invitrogen) containing SDS and 20 mM 1,4-dithiothreitol (Carl Roth GmbH). A wild type GOx solution was prepared in PBS using commercially available GOx from A. niger (type II, 19440 U g−1, Sigma Aldrich). This solution was also mixed with the loading buffer. The proteins were heated to 95 °C for 6 min while shaking at 500 rpm in 1.5 mL tubes. After denaturation, 15 μL of each solution was loaded in a gel. Gel electrophoresis was performed in a Mini-PROTEAN Tetra cell system (Bio-Rad Laboratories) at 100 V for 10 min and 250 V for 50 min. The gel was stained with a 0.25% Coomassie brilliant blue R-250 (ITW Reagents) solution in 40% ethanol and 10% acetic acid for 2 h at room temperature. The gel was then destained for 4 h in an ethanol:acetic acid:water solution prepared in a 4:1:5 ratio. The gel was imaged in a Fusion Solo S gel imager (Vilber Loumat).
CD spectroscopy
UV-CD spectra of GOx was measured between 195 and 250 nm with a J-810 CD spectropolarimeter (Jasco). The reference cuvette was filled with PBS-EDTA. Spectra were smoothed using a convolution filter kernel from the “convolve” function. The spectra were analyzed using BeStSel software to identify differences in the folding of the proteins.50,51Thiol reactivity assay
The GOx variants were reduced by 10 mM TCEP (abcr) for 1 h at 5 °C while shaking at 500 rpm. The TCEP was removed using a PD midiTrap G-25 desalting column (GE Healthcare) and eluted with PBS-EDTA to minimize the formation of GOx oligomers. The concentrations of GOx were adjusted to 0.87 ± 0.04 mg mL−1 using a NanoDrop 2000 spectrometer. 195 μL from each protein solution was transferred to a 96-well plate and absorption spectra were measured in the plate reader. Next, 5 μL of freshly prepared 20 mM SPDP (abcam) in DMSO (Sigma) were mixed with the solutions. The absorbance spectra were repeatedly measured 1, 8, 30, 60, and 120 minutes after addition of SPDP (Fig. S2).† The difference between the final absorbance and absorbance before SPDP addition at 343 nm was compared among the proteins. The number of moles of SPDP per mole of GOx was calculated according to the equation, (ΔA × Mw)/(cGOx × ε343 nm), where ΔA is the absorbance difference at 343 nm, Mw is a molecular weight of GOx, cGOx is concentration of GOx in mg mL−1, and ε343 nm is the extinction coefficient for pyridine-2-thione at 343 nm (ε343 nm = 8080 M−1 cm−1).52GOx crosslinking with PM
The GOx(D70C) stock solution was reduced with TCEP (as described above) and removed using a PD midiTrap G-25 desalting column. Next, 5 μL of freshly prepared 30 mM PM in DMSO (abcr) was added to 995 μL of the freshly reduced protein solution (∼1 mg mL−1); the PM was added in approximately 10 times excess. The reaction was performed at pH 7.0, where the maleimide can predominantly react with free thiols.53 The sample was incubated overnight at 4 °C and shaken at 500 rpm. The free PM was removed from the GOx(70C)-PM solution using a PD midiTrap G-25 desalting column with PBS (pH 7.4) (Gibco, Life Technologies) equilibration buffer (Fig. S3†). The samples were stored at 4 °C.Fluorescence spectrometry
GOx and GOx(70C)-PM fluorescence spectra were measured in a Varioskan LUX plate reader (ThermoFisher Scientific) in bottom-scanning plate-reading mode. The samples were excited at 345 ± 2.5 nm.Preparation of GOx(70C)-PM-SWCNTs
50 mg of SWCNTs (CoMoCAT (7,6)-enriched carbon nanotubes, Sigma Aldrich) were mixed with 50 mL of 2 wt% SC (Sigma Aldrich) and sonicated for 60 min using a tip sonicator (1/4 in. tip, QSonica Q700) at 1% amplitude in an ice bath. The SC-SWCNT suspension was ultracentrifuged at 164000 × g for 3 h (Beckman Optima XPN-80). Approximately 80% of the supernatant was collected and stored at room temperature. 0.5 mL of the 25 mg L−1 SC-SWCNT stock solution was mixed with 0.5 mL of ∼3 mg mL−1 GOx(70C)-PM (or GOx). The suspension was subsequently dialyzed at 5 °C in a 14 kDa MWCO dialysis tube (D9777, Sigma Aldrich) in 2 L of PBS for 4 h. The dialyzed mixture was transferred to a 300 kDa MWCO dialysis device (Spectra/Por Float-A-Lyzer, Spectrum Laboratories) and dialyzed GOx(70C)-PM-SWCNTs were stored at 4 °C.
Absorbance spectroscopy
Spectra were acquired between 200 and 1350 nm using a UV-Vis-NIR spectrophotometer (UV-3600 Plus, SHIMADZU) (Fig. S4†). All measurements were performed in a quartz cuvette (10 mm, Quartz SUPRASIL, Hellma Analytics). The SWCNT concentration was calculated using the extinction coefficient at 739 nm (ε739 nm = 25.3 mL mg−1 cm−1).54NIR fluorescence spectroscopy
Individual wells of a 96-well plate (costar 3590, Croning Incorporated) were each filled with 49 μL of GOx(70C)-PM-SWCNT and GOx-SWCNT suspensions in PBS. NIR fluorescence spectra were acquired between 950 and 1400 nm (75 L mm−1 grating) using the NIR micro-spectrometer described previously.5 The samples were illuminated at 660 ± 5 nm (SuperK Extreme EXR-15 and SuperK Varia, NKT Photonics). Fluorescence spectra were continuously acquired while 1 μL of 1 M glucose in PBS (pH 7.4) was added to the well. The fluorescence of the GOx(70C)-PM-SWCNT sensor in response to the addition and removal of a 20 mM glucose solution in PBS was measured in a glass-bottom device with a 14 kDa MWCO cellulose membrane (Sigma Aldrich) on top, as described in our previous work.5Miniaturized sensor device
The application-specific design of the sensor device was developed at CSEM. The printed electronic board includes a 660 nm LED (Osram), InGaAs photodiode (Hamamatsu), visible photodiode (Osram), and a photometric front end (Analog Devices). An optical 850 nm long-pass filter is installed in front of the InGaAs photodiode. The optoelectronic components of the device are embedded in optically transparent epoxy resin. The SWCNT-based sensor is applied on the top of the resin and covered with a semipermeable membrane. The optoelectronic components are driven by a microcontroller unit STM32 (STMicroelectronics), which is linked to the device by a cable. The measured data are transferred to a PC and recorded using custom software.Author contributions
V. Z., N. S., and A. A. B. designed research. N. S. engineered plasmids. A. W. and A. G. contributed to protein expression. H. W. contributed to sensor preparation. V. Z. and S. C. contributed to the development of the sensor reader. V. Z. and N. S. performed experiments. All authors contributed to the writing and reviewing of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful for support from the Swiss National Science Foundation Assistant Professor (AP) Energy Grant (Project No. PYAPP2_154269) and Swiss National Science Foundation Project No. 200021_184822. This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No 853005). The authors acknowledge E. Ahunbay and M. Moradel-Casellas, who helped establish protocols for glucose oxidase expression and purification, as well as S. Y. Rahnamaee, who helped to express and purify the proteins. G. Orawez developed the electronics design of the miniaturized device and its firmware, while T. Tatar developed the device software. E. Vuille-Dit-Bille performed sensor monitoring experiments.References
- G. Hong, S. Diao, A. L. Antaris and H. Dai, Chem. Rev., 2015, 115, 10816–10906 CrossRef CAS PubMed.
- N. M. Iverson, P. W. Barone, M. Shandell, L. J. Trudel, S. Sen, F. Sen, V. Ivanov, E. Atolia, E. Farias, T. P. McNicholas, N. Reuel, N. M. A. Parry, G. N. Wogan and M. S. Strano, Nat. Nanotechnol., 2013, 8, 873–880 CrossRef CAS PubMed.
- J. D. Harvey, P. V. Jena, H. A. Baker, G. H. Zerze, R. M. Williams, T. V. Galassi, D. Roxbury, J. Mittal and D. A. Heller, Nat. Biomed. Eng., 2017, 1, 1–11 CrossRef.
- W. Feng and P. Ji, Biotechnol. Adv., 2011, 29, 889–895 CrossRef CAS PubMed.
- V. Zubkovs, N. Schuergers, B. Lambert, E. Ahunbay and A. A. Boghossian, Small, 2017, 1701654 CrossRef PubMed , 1–10..
- H. Yoon, J. H. Ahn, P. W. Barone, K. Yum, R. Sharma, A. a. Boghossian, J. H. Han and M. S. Strano, Angew. Chem., Int. Ed., 2011, 50, 1828–1831 CrossRef CAS PubMed.
- S. F. Oliveira, G. Bisker, N. A. Bakh, S. L. Gibbs, M. P. Landry and M. S. Strano, Carbon, 2015, 95, 767–779 CrossRef CAS.
- H. Muguruma, S. Yoshida, M. Urata, K. Fujisawa and Y. Matsui, Electrochemistry, 2008, 76, 545–548 CrossRef CAS.
- S. Ferri, K. Kojima and K. Sode, J. Diabetes Sci. Technol., 2011, 5, 1068–1076 CrossRef PubMed.
- A. Antonucci, J. Kupis-Rozmyslowicz and A. A. Boghossian, ACS Appl. Mater. Interfaces, 2017, 9, 11321–11331 CrossRef CAS PubMed.
- V. Sanz, H. M. Coley, S. R. P. Silva and J. McFadden, J. Nanopart. Res., 2012, 14, 1–13 Search PubMed.
- C. Ge, J. Du, L. Zhao, L. Wang, Y. Liu, D. Li, Y. Yang, R. Zhou, Y. Zhao, Z. Chai and C. Chen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16968–16973 CrossRef CAS PubMed.
- X. Xu, B. J. Bowen, R. E. A. Gwyther, M. Freeley, B. Grigorenko, A. V. Nemukhin, J. Eklöf-Österberg, K. Moth-Poulsen, D. D. Jones and M. Palma, Angew. Chem., Int. Ed., 2021, 60, 20184–20189 CrossRef CAS PubMed.
- V. Zubkovs, S.-J. Wu, S. Y. Rahnamaee, N. Schuergers and A. A. Boghossian, Chem. Mater., 2020, 32, 8798–8807 CrossRef CAS.
- R. M. Williams, C. Lee, T. V. Galassi, J. D. Harvey, R. Leicher, M. Sirenko, M. A. Dorso, J. Shah, N. Olvera, F. Dao, D. A. Levine and D. A. Heller, Sci. Adv., 2018, 4, 1–11 Search PubMed.
- G. Bisker, J. Dong, H. D. Park, N. M. Iverson, J. Ahn, J. T. Nelson, M. P. Landry, S. Kruss and M. S. Strano, Nat. Commun., 2016, 7, 1–14 Search PubMed.
- G. Wohlfahrt, S. Witt, J. Hendle, D. Schomburg, H. M. Kalisz and H. J. Hecht, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 969–977 CrossRef CAS PubMed.
- M. D. Gouda, S. A. Singh, A. G. A. Rao, M. S. Thakur and N. G. Karanth, J. Biol. Chem., 2003, 278, 24324–24333 CrossRef CAS PubMed.
- N. Filla, R. Ramasamy and X. Wang, Phys. Chem. Chem. Phys., 2018, 20, 11327–11335 RSC.
- Z. Yang, Z. Wang, X. Tian, P. Xiu and R. Zhou, J. Chem. Phys., 2012, 136, 1–10 Search PubMed.
- Z. He and J. Zhou, Carbon, 2014, 78, 500–509 CrossRef CAS.
- P. Kommoju, Z. Chen, R. C. Bruckner, F. S. Mathews and M. S. Jorns, Biochemistry, 2011, 50, 5521–5534 CrossRef CAS PubMed.
- V. Looser, B. Bruhlmann, F. Bumbak, C. Stenger, M. Costa, A. Camattari, D. Fotiadis and K. Kovar, Biotechnol. Adv., 2014, 33, 1177–1193 CrossRef PubMed.
- S. Witt, M. Singh and H. M. Kalisz, Appl. Environ. Microbiol., 1998, 64, 1405–1411 CrossRef CAS PubMed.
- V. Leskovac, S. Trivić, G. Wohlfahrt, J. Kandrač and D. Peričin, Int. J. Biochem. Cell Biol., 2005, 37, 731–750 CrossRef CAS PubMed.
- N. J. Kavimandan, E. Losix, J. J. Wilson, J. S. Brodbelt and N. A. Peppas, Bioconjugate Chem., 2006, 17, 1376–1384 CrossRef CAS PubMed.
- C. W. Wu, L. R. Yarbrough and F. Y. H. Wu, Biochemistry, 1976, 15, 2863–2868 CrossRef CAS PubMed.
- G. Bains, A. B. Patel and V. Narayanaswami, Molecules, 2011, 16, 7909–7935 CrossRef CAS PubMed.
- E. S. Jeng, A. E. Moll, A. C. Roy, J. B. Gastala and M. S. Strano, Nano Lett., 2006, 6, 371–375 CrossRef CAS PubMed.
- J. H. Choi and M. S. Strano, Appl. Phys. Lett., 2007, 90, 1–3 Search PubMed.
- S. Goldrick, K. Lee, C. Spencer, W. Holmes, M. Kuiper, R. Turner and S. S. Farid, Biotechnol. J., 2018, 13, 29247603 Search PubMed.
- T. W. Tsai, G. Heckert, L. F. Neves, Y. Tan, D. Y. Kao, R. G. Harrison, D. E. Resasco and D. W. Schmidtke, Anal. Chem., 2009, 81, 7917–7925 CrossRef CAS PubMed.
- M. Raicopol, A. Prună, C. Damian and L. Pilan, Nanoscale Res. Lett., 2013, 8, 1–8 CrossRef PubMed.
- S. Yabuki, M. Iwamoto and Y. Hirata, Materials, 2014, 7, 899–905 CrossRef PubMed.
- P. W. Barone, R. S. Parker and M. S. Strano, Anal. Chem., 2005, 77, 7556–7562 CrossRef CAS PubMed.
- Y. J. Heo, H. Shibata, T. Okitsu, T. Kawanishi and S. Takeuchi, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2140–2143 Search PubMed.
- Y. J. Heo and S. Takeuchi, Adv. Healthcare Mater., 2012, 2, 43–56 CrossRef PubMed.
- E. Hofferber, J. Meier, N. Herrera, J. Stapleton, K. Ney, B. Francis, C. Calkins and N. Iverson, Methods Appl. Fluoresc., 2021, 9, 025005 CrossRef CAS PubMed.
- E. M. Hofferber, J. A. Stapleton, J. Adams, M. Kuss, B. Duan and N. M. Iverson, Macromol. Biosci., 2019, 19, 1–8 CrossRef PubMed.
- B. L. Allen, G. P. Kotchey, Y. Chen, N. V. K. Yanamala, J. Klein-Seetharaman, V. E. Kagan and A. Star, J. Am. Chem. Soc., 2009, 131, 17194–17205 CrossRef CAS PubMed.
- C. F. Chiu, H. H. Dar, A. A. Kapralov, R. A. S. Robinson, V. E. Kagan and A. Star, Nanoscale, 2017, 9, 5948–5956 RSC.
- H. Ashkenazy, E. Erez, E. Martz, T. Pupko and N. Ben-Tal, Nucleic Acids Res., 2010, 38, 529–533 CrossRef PubMed.
- O. Edelheit, A. Hanukoglu and I. Hanukoglu, BMC Biotechnol., 2009, 9, 1–8 CrossRef PubMed.
- J. Lin-Cereghino, W. W. Wong, S. Xiong, W. Giang, L. T. Luong, J. Vu, S. D. Johnson and G. P. Lin-Cereghino, Biotechniques, 2005, 38, 44–48 CrossRef CAS PubMed.
- R. Weis, R. Luiten, W. Skranc, H. Schwab, M. Wubbolts and A. Glieder, FEMS Yeast Res., 2004, 5, 179–189 CrossRef CAS PubMed.
- R. C. Bateman and J. A. Evans, J. Chem. Educ., 1995, 72, 240–241 CrossRef.
- L. Gu, J. Zhang, B. Liu, G. Du and J. Chen, Appl. Biochem. Biotechnol., 2015, 175, 1429–1447 CrossRef CAS PubMed.
- B. E. P. Swoboda and V. Massey, J. Biol. Chem., 1965, 240, 2209–2215 CrossRef CAS PubMed.
- S. Finnegan and S. L. Percival, Adv. Wound Care, 2015, 4, 415–421 CrossRef PubMed.
- A. Micsonai, F. Wien, É. Bulyáki, J. Kun, É. Moussong, Y. H. Lee, Y. Goto, M. Réfrégiers and J. Kardos, Nucleic Acids Res., 2018, 46, W315–W322 CrossRef CAS PubMed.
- A. Micsonai, F. Wien, L. Kernya, Y.-H. Lee, Y. Goto, M. Réfrégiers and J. Kardos, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3095–E3103 CrossRef CAS PubMed.
- G. T. Hermanson, Bioconjugate Techniques, Elsevier Inc., 3rd edn, 2013 Search PubMed.
- A. Mukhortava and M. Schlierf, Bioconjugate Chem., 2016, 27, 1559–1563 CrossRef CAS PubMed.
- J. Yang, Z. Zhang, D. Zhang and Y. Li, Mater. Chem. Phys., 2013, 139, 233–240 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: GOx gene sequence, primer sequences, cell culture media compositions, CD spectra, and absorbance spectra. See https://doi.org/10.1039/d2na00092j |
| This journal is © The Royal Society of Chemistry 2022 |