
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergic effect of graphene oxide and boron nitride on the mechanical properties of polyimide composite films†
Yi Kai
Cheng
a,
Benoît Denis Louis
Campéon
b,
Seiji
Obata
b and
Yuta
Nishina
 *ab
*ab
aGraduate School of Natural Science and Technology, Okayama University, 3-1-1 Tsushima-naka Kita-ku, Okayama 700-8530, Japan. E-mail: nisina-y@cc.okayama-u.ac.jp
bResearch Core for Interdisciplinary Sciences, Okayama University, 3-1-1 Tsushima-naka Kita-ku, Okayama 700-8530, Japan
First published on 4th May 2022
Abstract
The addition of two-dimensional (2D) materials into polymers can improve their mechanical properties. In particular, graphene oxide (GO) and hexagonal boron nitride (h-BN) are expected to be potential nanoplatelet additives for polymers. Interactions between such nanoplatelets and polymers are effective in improving the above properties. However, no report has investigated the effect of using two types of nanoplatelets that have good interaction with polymers. In this study, we fabricated polyimide (PI) films that contain two types of nanoplatelets, amine-functionalized h-BN (BNNH2) and GO. We have elucidated that the critical ratio and the content of BNNH2 and GO within PI govern the films' mechanical properties. When the BNNH2/GO weight ratio was 52![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and their content was 1 wt% in the PI film, the tensile modulus and tensile strength were increased by 155.2 MPa and 4.2 GPa compared with the pristine PI film.
:1 and their content was 1 wt% in the PI film, the tensile modulus and tensile strength were increased by 155.2 MPa and 4.2 GPa compared with the pristine PI film.
Introduction
Polymer films with molecularly designed structures have been playing an important role in applications such as medical devices,1 separators in batteries,2–4 high-performance audio diaphragms,5 and computer memory.6 Therefore, the fabrication of polymers with targeted properties is of great interest for the development of next-generation technologies. Polymers have advantages such as their ease of processing, light weight, and chemical resistance compared with metallic materials.7,8 Polyimide (PI), which was first synthesized in 1908,9 has extremely high heat and chemical resistances, mechanical properties, and electrical insulation compared to other polymer materials. These properties enable PI to be used in a wide range of applications such as waterproof coatings,10,11 speaker components,12–14 separators for batteries,15,16 gas separations,17–19 and water purification membranes.20,21The introduction of fillers has been investigated to improve the properties of polymer films. Addition of one- and/or two-dimensional (2D) carbon materials has been aimed at forming strong π–π interaction between the aromatic frameworks in PI and carbon materials.22,23 For example, the physical properties of PI can be improved by the addition of low-dimensional carbon materials, such as carbon nanotubes,24–26 graphene oxide (GO),27–31 and amine-functionalized carbon materials.32,33 Another promising filler with a honeycomb structure is hexagonal boron nitride (h-BN), which can interact strongly with 2D carbon materials.34 Therefore, mixing with carbon composites of h-BN would exert a synergic effect, resulting in thermal conductivity, as well as thermoelectric and mechanical properties.35–37 Several methods of modifying h-BN edges with hydroxyl groups or amino (NH2) groups were reported recently.38–40 However, the fabrication of the PI-BN/GO composite film and evaluation of its mechanical properties have not been reported so far.
In this study, we focused on edge functionalization of h-BN with NH2 groups (BNNH2) to ensure a good affinity with GO and PI to improve mechanical strength. The BNNH2 edge can bond with PI through amide bonds, while its basal plane retains a non-reactive honeycomb structure. Interestingly, h-BN and GO show good interaction with each other.41–43 Therefore, we hypothesized that covering the basal plane of BNNH2 with GO results in a filler with superior mechanical strength through interaction with PI at both the basal plane (oxy functional groups on GO) and edge (amino groups of BNNH2) of the 2D structure. In this context, we prepared a PI-BNNH2/GO composite containing both GO and BNNH2 to elucidate the effect of employing multiple types of nanoplatelets in PI films (Fig. 1). As a result, the mechanical properties of PI films were improved by the introduction of BNNH2, and a further improvement was obtained with an additional small amount of GO.
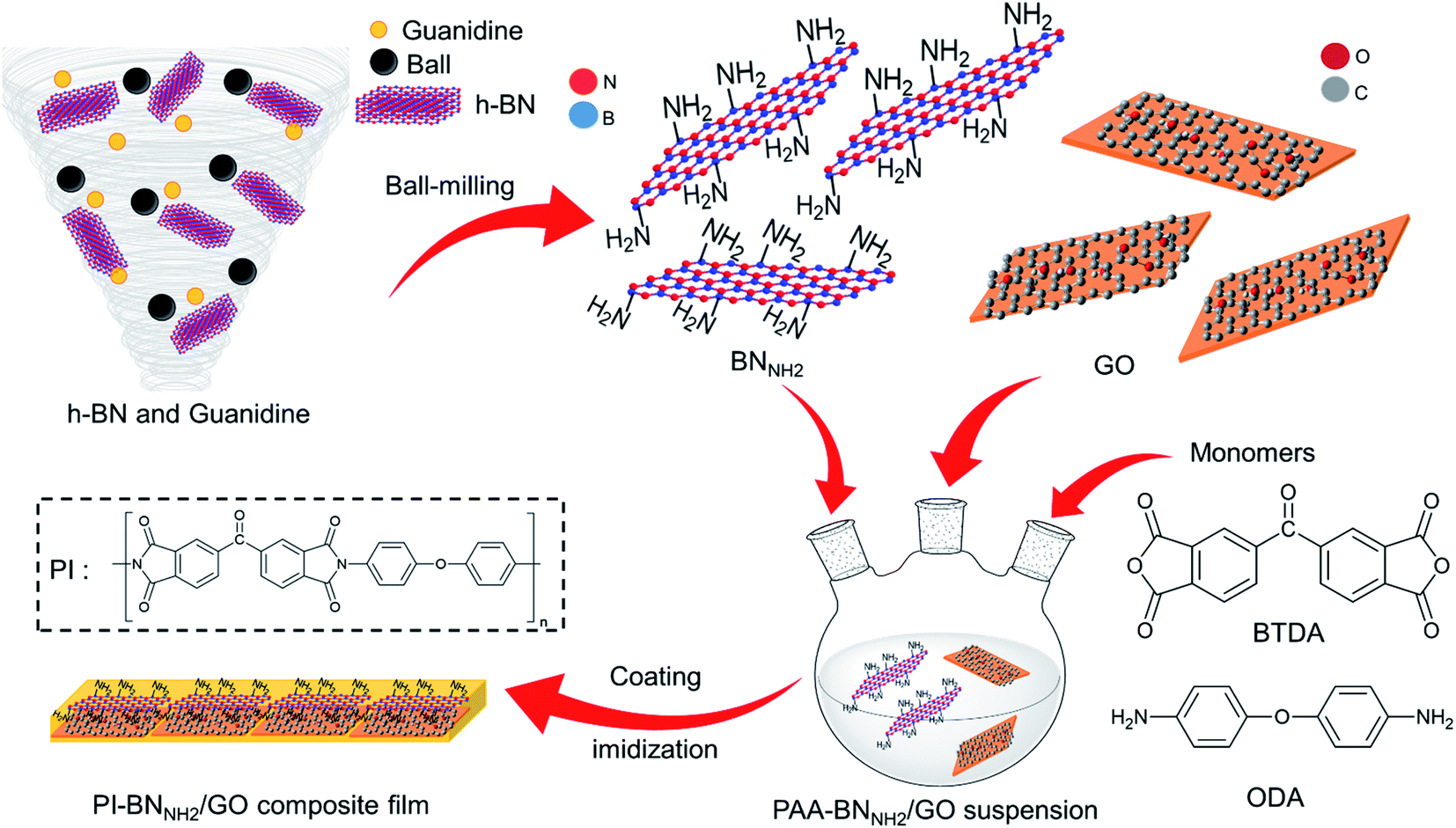 | ||
| Fig. 1 Schematic illustration of the fabrication of the PI-BNNH2/GO composite film. | ||
Results and discussion
Synthesis and characterization of BNNH2
First, we synthesized amine-functionalized h-BN (BNNH2) by a ball milling method (details are given in the Experimental section). We used guanidine or urea as a nitrogen source. Hereafter, we call h-BN treated with urea BNNH2(U) and h-BN treated with guanidine BNNH2(G), respectively. After ball milling, dialysis was carried out to remove unreacted guanidine or urea. The specimens were treated by sonication for 1 h in dimethylacetamide (DMAc), resulting in a homogeneous dispersion (Fig. S1†).The Fourier transform infrared (FTIR) spectra exhibited strong peaks at 780 cm−1 and 1380 cm−1 originating from the in-plane B–N stretching vibration and out-of-plane B–N–B bending vibration (Fig. S2†).44,45 Furthermore, an additional peak appeared at around 3250 cm−1 in BNNH2(G). This peak is assigned to the N–H vibration mode.46,47 Thermal gravimetric analysis (TGA) results are shown in Fig. 2a. According to the TGA results, BNNH2(U) and BNNH2(G) showed 2 wt% and 4 wt% loss, respectively, while pure h-BN did not show weight loss. These results indicate that functionalized h-BN was successfully synthesized and that more amine-functionalization proceeded with guanidine than with urea. Therefore, BNNH2(G) was used for the following experiments. X-ray diffraction (XRD) data are presented in Fig. 2b; BNNH2(G) showed two main characteristic diffraction peaks at 26.2° and 42.8° originating from the h-BN structure. Compared to the pristine h-BN, the (002) and (100) peak intensities of BNNH2(G) decreased. Thus, the FWHM of (002) was changed from 0.25 to 0.34; in contrast, the (002) peak position was not significantly shifted (Fig. S3†). These results suggest that h-BN sheets were exfoliated because of the physical force and/or the chemical functionalization during the ball milling process.
 | ||
| Fig. 2 (a) TGA curves of h-BN, BNNH2(U), and BNNH2(G) with a heating rate of 10 °C min−1. (b) XRD patterns of BNNH2(G), BNNH2(U), and pristine h-BN. | ||
Reaction of BNNH2 with an acid anhydride
To confirm the presence of NH2 groups in BNNH2, we treated BNNH2(G) with 3,3′,4,4′-benzophenonetetracarboxylic dianhydride (BTDA) to form amide bonds. The detailed synthesis procedure is given in the Experimental section. TGA data of h-BN, BNNH2(G), and BNNH2(G) treated with BTDA (BNNH2(G)/BTDA) are presented in Fig. 3. For comparison, TGA data of h-BN treated with BTDA (h-BN/BTDA) are also presented in Fig. 3. The h-BN/BTDA sample showed two mass losses of 1.7 wt% and 2.2 wt% at 225 °C and 320 °C. On the other hand, the BNNH2(G)/BTDA sample showed two mass losses of 5.6 wt% and 4.2 wt% at 150 °C and 500 °C, respectively, corresponding to the release of water during the thermal imidization and the carbonization reactions.48–50 After TGA analysis, only the surface of BNNH2(G)/BTDA turned black (Fig. 3b–e). Furthermore, FTIR spectra of BNNH2(G)/BTDA (Fig. S4a and b†) show an additional peak at 1720 cm−1 derived from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration,51,52 and the NH stretching vibration at 3520 cm−1 has slightly decreased (Fig. S4c†). The above description suggests that the NH2 group on h-BN can bond with BTDA through an amide bond. In the case of h-BN/BTDA, CO vibration at 1720 cm−1 was not observed (Fig. S4d†). The TGA and FTIR results confirm that NH2 groups are present on h-BN, and the NH2 groups can form amide bonds.
O vibration,51,52 and the NH stretching vibration at 3520 cm−1 has slightly decreased (Fig. S4c†). The above description suggests that the NH2 group on h-BN can bond with BTDA through an amide bond. In the case of h-BN/BTDA, CO vibration at 1720 cm−1 was not observed (Fig. S4d†). The TGA and FTIR results confirm that NH2 groups are present on h-BN, and the NH2 groups can form amide bonds.
 | ||
| Fig. 3 (a) TGA curves of h-BN, h-BN/BTDA, BNNH2(G), and BNNH2(G)/BTDA with a heating rate of 10 °C min−1 under a N2 atmosphere. (b) h-BN after TGA. (c) h-BN/BTDA after TGA. (d) BNNH2(G) after TGA. (e) BNNH2(G)/BTDA after TGA. | ||
Fabrication of PI-BNNH2(G) and PI-BNNH2(G)/GO composite films
DMAc is generally used as a solvent for the preparation of polyamic acid (PAA),32,33 a precursor of PI. Therefore, the dispersibility of BNNH2(G) and BNNH2(G)/GO was investigated in DMAc by sonication. The ratio of BNNH2(G):GO was 10:1 and 100:1, and the obtained samples were termed BNNH2(G)/GO(10:1) and BNNH2(G)/GO(100:1), respectively. The suspensions were stable for more than one month at room temperature without visible precipitation (Fig. S1†). The particle size analysis (Fig. S5†) showed a mean size of 273 nm, 234 nm, and 223 nm for BNNH2(G), BNNH2(G)/GO(100:1), and BNNH2(G)/GO(10:1), respectively.
Next, homogeneous suspensions of PAA composites were prepared in DMAc by a two-step process. Initially, sonication was employed to disperse BNNH2(G) or BNNH2(G)/GO with 4,4′-diaminodiphenyl ether (ODA) and BTDA. Then, the as-obtained dispersion was mixed for 12 hours to allow the polymerization and formation of PAA composite suspensions, which are termed as PAA-BNNH2(G), PAA-BNNH2(G)/GO(10:1), and PAA-BNNH2(G)/GO(100:1). These PAA composite suspensions were coated on glass and heated at 300 °C, resulting in PI composite films, termed as PI-BNNH2(G), PI-BNNH2(G)/GO(10:1), and PI-BNNH2(G)/GO(100:1) (details are given in the Experimental section). A conventional method for the synthesis of PI films consists of heating PAA films at elevated temperatures within 200 to 300 °C.53,54 The heating rate was kept as low as 5 °C min−1 to avoid the formation of bubbles within the PI film during the solvent evaporation.55
Mechanical properties of composite films
First, we investigated the effect of BNNH2(G) and GO on PI film. According to Fig. 4 and Table 1, the tensile modulus of PI containing a low quantity (1 wt%) of GO showed a 22% increase over the tensile modulus of the pristine PI, although the tensile strength of the PI–GO was slightly decreased (Table 1, entries 1 and 2). The addition of only GO does not significantly improve the mechanical properties. A previous study also pointed out that GO decreased the tensile strength because the interaction between GO and PI is not strong.31 | ||
| Fig. 4 (a) Stress–strain curves of the pristine PI film and PI-BNNH2(G) films. (b) Stress–strain curves of the PI-BNNH2(G) (100:1) and PI-BNNH2(G) (10:1) films. | ||
| Entry | Samplea | Tensile strength (MPa) | Tensile modulus (GPa) | Elongation at break (%) |
|---|---|---|---|---|
| a 1 wt% sample was used in the PI matrix. | ||||
| 1 | Pristine PI | 93.9 ± 3.4 | 2.2 ± 0.2 | 10.6 ± 3.6 |
| 2 | PI–GO | 90.2 ± 6.3 | 2.7 ± 0.1 | 4.4 ± 1.3 |
| 3 | PI–hBN | 110.9 ± 7.0 | 2.7 ± 0.1 | 6.8 ± 1.4 |
| 4 | PI-BNNH2(G) | 135.7 ± 5.4 | 3.1 ± 0.5 | 10.3 ± 1.7 |
| 5 | PI–hBN/GO(100:1) |
127.4 ± 6.8 | 3.1 ± 0.2 | 9.5 ± 3.1 |
| 6 | PI-BNNH2(G)/GO(100:1) |
154.8 ± 11.1 | 3.8 ± 0.4 | 7.8 ± 1.6 |
| 7 |
PI-BN
NH
2
(G)/GO(52![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1) :1)
|
155.2 ± 2.1 | 4.2 ± 0.1 | 7.1 ± 0.7 |
Next, we investigated the effect of the addition of h-BN, which improved the mechanical properties (Table 1, entry 3). Due to the surface roughness and wrinkles in h-BN (Fig. S6a and b†), the PI–hBN composite film has better mechanical properties than the pristine PI film, as reported previously.56 Then, we investigated the composite with BNNH2(G) to elucidate the effect of the functionalization. The tensile strength of PI containing a low quantity (0.5 wt%) of BNNH2(G) showed a 23% increase over the tensile strength of the pristine PI (Table S1, entry 1†). Increasing the amount of BNNH2(G) from 0.5 to 1 wt% improved the tensile strength by 44% (Table 1, entry 4). However, further increase of BNNH2(G) to 3 wt% decreased both the tensile strength and tensile modulus (Table S1, entry 2†). These results indicate that an excess amount of BNNH2(G) leads to reducing the positive effects of BNNH2(G) in the PI matrix, probably caused by the stacking of BNNH2(G) and void formation. Therefore, the optimal amount of BNNH2(G) in PI is determined to be 1 wt%.
Finally, the combined use of GO and BN in the PI films was evaluated. The results demonstrated that PI films incorporating h-BN, BNNH2(G) and GO showed superior mechanical properties than those with a single component (GO, h-BN, or BNNH2(G)) (Table 1, entries 2–6). This suggests that there is a good interaction between GO and BN, which enhances the mechanical properties of PI. According to Table 1 (entries 5 and 6), the tensile strength and tensile modulus of the BNNH2(G)/GO composite film are 22% and 23% higher than those of PI–hBN/GO. This proves that the edge-functionalization of h-BN improves the mechanical properties of the composite film. In addition, the PI-BNNH2(G)/GO composite film has a tensile strength of approximately 154.89 MPa, which is 65% higher than that of the original PI, and the tensile modulus was increased to 3.83 GPa, which is 69% higher than that of the original PI (Table 1, entry 1). Upon reducing the amount of BNNH2(G)/GO to 0.5 wt% BNNH2(G), the tensile strength and tensile modulus of PI were also reduced to 118.96 MPa and 2.84 GPa (Table S1, entry 4†). When the BNNH2(G)/GO ratio increased to 10:1, the tensile strength dropped to 137.33 MPa and the tensile modulus also dropped to 3.30 GPa (Table S1, entry 7†). Similar to only h-BN, there is a critical BNNH2(G)/GO amount for the improvement of the PI film's mechanical properties. Furthermore, these data also revealed that the ratio of BNNH2(G) to GO strongly affects the PI film's mechanical properties.
Based on our experimental results, we propose a model structure of PI-BNNH2(G)/GO. As reported previously, GO sheets are easily aggregated in the composite, resulting in lower tensile strength (Fig. S7a†).31 In contrast, GO has a strong affinity with h-BN (Fig. S7c and d†).41–43 The attachment of GO on the BNNH2(G) surface facilitates the interaction with PI because of the following synergic effect: (1) amide bond formation with BTDA at the edge of BNNH2(G),57 and (2) amination of GO at the basal plane with ODA through an epoxy ring-opening reaction.27 Due to these effects, the BNNH2(G)/GO composite would successfully enhance the mechanical properties of PI.
Ideally, BNNH2(G)/GO should be composed of single BNNH2(G) flakes covered by single-layer GO on both sides. According to AFM measurements, the average thickness of BNNH2(G) was 36 nm (Fig. S8†). Considering that the thickness of a single layer h-BN is 0.33 nm,58 the prepared BNNH2(G) is composed of about 109 layers. As GO sheets are adsorbed only on the surface of BNNH2(G), the ideal ratio to fully cover the BNNH2(G) surface with GO is 109 layers of BNNH2(G) and 2 layers of GO. Considering the unit weight of BN and GO (the elemental ratio of C to O is 2:1), the ideal ratio of BNNH2(G)/GO is 52:1. To prove our speculation, we measured the mechanical properties of BNNH2(G)/GO that has a ratio of 52:1 (BNNH2(G)/GO(52:1)); its tensile modulus increased by 86%, and the tensile strength increased by 65% in comparison to the neat PI film (Table 1, entry 6). This specimen shows the highest tensile strength and tensile modulus among all specimens, which supports our hypothesis that surface coating of h-BN with GO is desirable for the strong interaction with PI.
Conclusions
In this research, we have demonstrated the improvement of the mechanical properties of PI films using a mixture of two types of materials, BNNH2(G) and GO. Improvement of the mechanical properties of composite films, including modulus, strength, and failure strain, has attracted tremendous interest in the field of materials science. Although the improvement of the modulus and/or strength has been realized in current studies,59,60 the improvement of failure strain along with modulus and strength is rare in previous studies. Changing the ratio of BN and GO and their ratio to PI, we elucidated that there is a critical value at which the PI composite film showed the highest tensile modulus, tensile strength, and failure strain at the same time. This enhances the toughness and potential application of PI.In the case of 1 wt% PI-BNNH2(G)/GO(52:1) composite film, the tensile modulus increased by 86%, and the tensile strength increased by 65% in comparison to the neat PI film. According to our results, we proposed and experimentally confirmed a model for the improvement of PI mechanical properties via the addition of BNNH2(G) and GO. In the case of BNNH2(G), only NH2 groups at the edges interact with the raw materials of PI because the BNNH2(G) surface is inert. On the other hand, covering the BNNH2(G) surface with GO creates additional interaction between the BNNH2(G)/GO surface and the raw material of PI. Therefore, PI has a good affinity with it on the surfaces and edges, which enhances the mechanical properties of the PI composite film. In contrast, the excess amount of GO that is not attached to the h-BN surface causes the mechanical properties of the composite to deteriorate. Our results and the proposed mechanism will be useful guidelines for fabricating 2D composite materials in polymer matrixes.
Experimental section
Materials
Hexagonal boron nitride (h-BN) (average size 1–2 μm) was purchased from Showa Denko K.K., Japan. Guanidine hydrochloride (purity 99.0+%), 3,3′,4,4′-benzophenonetetracarboxylic dianhydride (BTDA, purity 96.0+%), 4,4′-diaminodiphenyl ether (ODA, purity 99.0+%), dimethylacetamide (DMAc, purity 98.0+%), sulfuric acid (H2SO4, purity 95.0+%), and potassium permanganate (KMnO4, purity 99.3+%) were purchased from FUJIFILM Wako Pure Chemical Corporation, Japan.Characterization instruments
The particle size analyzer (PSA) used in this work was ELSZ-2000N (Photal Otsuka Electronics, Japan). The samples for the PSA were dispersed in DMAc. Fourier transform infrared spectroscopy (FTIR) spectra of the samples were recorded between 400 and 4000 cm−1 with an IRTracer-100 (Shimadzu Corporation, Japan). Scanning electron microscope (SEM) measurements were performed with an S-5200 (Hitachi Limited, Japan) with an accelerating voltage of 30 kV. X-ray diffraction (XRD) measurements were performed on an AERIS equipped with single crystalline silicon (Panalytical, Netherlands). Thermal gravimetric analyses (TGA) were performed with a DTG-60AH (Shimadzu Corporation, Japan) from room temperature to 800 °C in nitrogen (N2) at a heating rate of 10 °C min−1. To measure the tensile strength of PI films, sample sheets were cut into 10 mm × 60 mm and were tested using an AG-Xplus Universal Testing Machine (Shimadzu, Japan) at a tensile testing rate of 1 mm min−1.Preparation of graphene oxide (GO)
GO was prepared using a modified Hummers method.61,62 Graphite (3.0 g) was stirred in 95% H2SO4 (75 mL). KMnO4 (9.0 g) was gradually added to the solution while keeping the temperature below 10 °C using an ice bath. The mixture was then stirred at 35 °C for 2 h. The resulting mixture was diluted with water (75 mL) under vigorous stirring and cooling so that the temperature did not exceed 50 °C. The suspension was further treated with 30% aq. H2O2 (7.5 mL). The resulting graphite oxide suspension was purified by centrifugation with water until neutralization, and freeze-dried.Preparation of amine-functionalized hexagonal boron nitride (BNNH2(G))
BNNH2(G) was prepared using a Pulverisette 7 Classic Line ball mill (Fritsch, Germany).39,40 h-BN (0.5 g) and guanidine hydrochloride (10 g) were mixed using a ball mill at a rotation speed of 750 rpm for 16 h at ambient temperature. Water (ca. 50 mL) was added to the solid product. The resulting BNNH2(G) suspension was purified by dialysis (membrane cutoff: 12000–14000 Da) in deionized (DI) water for 24 h (DI water was changed every 3 h, which was repeated 3 times, then left to stand for 16 h) to remove the guanidine hydrochloride. A 1 week dialysis treatment was also investigated (Fig. S9†), but no difference was detected from the 24 h treated sample. Therefore, the 24 h dialysis treatment was selected. Finally, the sample was ultrasonicated for 10 min in DI water, and freeze-dried.
Preparation of PI-BNNH2(G) composite films
To prepare PAA-BNNH2(G) suspensions of different concentrations, BNNH2(G) (0.026 g, 0.053 g, 0.161 g, or 0.274 g) was dispersed in DMAc (20 mL) by sonication for 1 h. ODA (2.0 g, 10 mmol) was added to the BNNH2(G) suspension by sonication for 10 min. Then, BTDA (3.2 g, 10 mmol) was added into the suspension, sonicated for 10 min, and cooled in a freezer at about −18 °C for 12 h.A viscous PAA-BNNH2(G) suspension was coated on glass using an MSK-AFA-III coater (MTI Corporation, USA) with a coating rate of 20 mm s−1. The coating was heated at 90 °C for 1 h (heating speed 1 °C min−1 under nitrogen) to evaporate the solvent. Finally, PI was formed by heat-treatment from room temperature to 300 °C for 1 h (room temperature to 300 °C for 5 h under nitrogen).
Preparation of PI-BNNH2(G)/GO composite films
BNNH2(G)/GO(100:1) or BNNH2(G)/GO(10:1) (5.2 g) was dispersed in DMAc (20 mL) with sonication for 1 h. ODA (2.0 g, 10 mmol) was added to the BNNH2(G)/GO suspension with sonication for 10 min. BTDA (3.2 g, 10 mmol) was then added into the suspension and sonicated for 10 min, furnishing a PAA-BNNH2(G)/GO suspension. The mixture was coated on glass and heated at 90 °C for 1 h (heating speed: 1 °C min−1 under nitrogen) to evaporate the solvent. Next, heat-treatment was performed from room temperature to 300 °C over 5 h under a nitrogen atmosphere, followed by keeping at 300 °C for 1 h. PI-BNNH2(G)/GO composite films containing 0.5 wt%, 1 wt%, and 3 wt% BNNH2(G)/GO were prepared following the same procedures.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Professor Mitsuhiro Okayasu is acknowledged for the mechanical properties measurements.Notes and references
- M. E. Alf, A. Asatekin, M. C. Barr, S. H. Baxamusa, H. Chelawat, G. Ozaydin-lnce, C. D. Petruczok, R. Sreenivasan, W. E. Tenhaeff, N. J. Trujillo, S. Vaddiraju, J. Xu and K. K. Gleason, Adv. Mater., 2010, 22, 1993–2027 CrossRef CAS PubMed.
- C. Wang, W. Zheng, Z. Yue, C. O. Too and G. G. Wallace, Adv. Mater., 2011, 23, 3580–3584 CrossRef CAS PubMed.
- Z. Chen, P. C. Hsu, J. Lopez, Y. Li, J. W. F. To, N. Liu, C. Wang, S. C. Andrews, J. Liu, Y. Cui and Z. Bao, Nat. Energy, 2016, 1, 15009 CrossRef CAS.
- J. K. Kim, J. Scheers, J. H. Ahn, P. Johansson, A. Matic and P. Jacobsson, J. Mater. Chem. A, 2013, 1, 2426–2430 RSC.
- S. Kang, S. Cho, R. Shanker, H. Lee, J. Park, D. S. Um, Y. Lee and H. Ko, Sci. Adv., 2018, 4, 1–12 CAS.
- Y. J. Park, I. S. Bae, S. Ju Kang, J. Chang and C. Park, IEEE Trans. Dielectr. Electr. Insul., 2010, 17, 1135–1163 CAS.
- D. A. Bernards and T. A. Desai, Soft Matter, 2010, 6, 1621–1631 RSC.
- W. Ogieglo, H. Wormeester, K. J. Eichhorn, M. Wessling and N. E. Benes, Prog. Polym. Sci., 2015, 42, 42–78 CrossRef CAS.
- M. T. Bogert and R. R. Renshaw, J. Am. Chem. Soc., 1908, 30, 1135–1144 CrossRef CAS.
- Y. Ma, L. Xu, Z. He, J. Xie, L. Shi, M. Zhang, W. Zhang and W. Cui, J. Mater. Chem. C, 2019, 7, 7360–7370 RSC.
- T. Komamura, K. Okuhara, S. Horiuchi, Y. Nabae and T. Hayakawa, ACS Appl. Polym. Mater., 2019, 1, 1209–1219 CrossRef CAS.
- N. Hosoya, S. Baba and S. Maeda, J. Acoust. Soc. Am., 2015, 138, EL424–EL428 CrossRef CAS PubMed.
- K. M. Kim and K. Park, J. Mech. Sci. Technol., 2013, 27, 2923–2928 CrossRef.
- M. Sharifzadeh Mirshekarloo, C. Y. Tan, X. Yu, L. Zhang, S. Chen, K. Yao, F. Cui, S. M. Pandit, S. H. Chong and S. T. Tan, Appl. Acoust., 2018, 137, 90–97 CrossRef.
- H. G. Wang, S. Yuan, D. L. Ma, X. L. Huang, F. L. Meng and X. B. Zhang, Adv. Energy Mater., 2014, 4, 1–7 CAS.
- Y. E. Miao, G. N. Zhu, H. Hou, Y. Y. Xia and T. Liu, J. Power Sources, 2013, 226, 82–86 CrossRef CAS.
- T. H. Kim, W. J. Koros, G. R. Husk and K. C. O'Brien, J. Membr. Sci., 1988, 37, 45–62 CrossRef CAS.
- M. R. Coleman and W. J. Koros, J. Membr. Sci., 1990, 50, 285–297 CrossRef CAS.
- Y. Wang, Z. Low, S. Kim, H. Zhang, X. Chen, J. Hou, J. G. Seong, Y. M. Lee, G. P. Simon, C. H. J. Davies and H. Wang, Angew. Chem., 2018, 130, 16288–16293 CrossRef.
- G. Q. Chen, C. A. Scholes, G. G. Qiao and S. E. Kentish, J. Membr. Sci., 2011, 379, 479–487 CrossRef CAS.
- M. Minelli, G. Cocchi, L. Ansaloni, M. G. Baschetti, M. G. De Angelis and F. Doghieri, Ind. Eng. Chem. Res., 2013, 52, 8936–8945 CrossRef CAS.
- M. Okamoto, T. Fujigaya and N. Nakashima, Adv. Funct. Mater., 2008, 18, 1776–1782 CrossRef CAS.
- S. Qin, C. Chen, M. Cui, A. Zhang, H. Zhao and L. Wang, RSC Adv., 2017, 7, 3003–3011 RSC.
- M. Lebrón-Colón, M. A. Meador, J. R. Gaier, F. Solá, D. A. Scheiman and L. S. McCorkle, ACS Appl. Mater. Interfaces, 2010, 2, 669–676 CrossRef PubMed.
- C. Cui, W. Qian, M. Zhao, F. Ding, X. Jia and F. Wei, Carbon, 2013, 60, 102–108 CrossRef CAS.
- W. Ning, Z. Wang, P. Liu, D. Zhou, S. Yang, J. Wang, Q. Li, S. Fan and K. Jiang, Carbon, 2018, 139, 1136–1143 CrossRef CAS.
- D. Chen, H. Zhu and T. Liu, ACS Appl. Mater. Interfaces, 2010, 2, 3702–3708 CrossRef CAS PubMed.
- N. D. Luong, U. Hippi, J. T. Korhonen, A. J. Soininen, J. Ruokolainen, L. S. Johansson, J. Do Nam, L. H. Sinh and J. Seppälä, Polymer, 2011, 52, 5237–5242 CrossRef.
- J. Y. Kong, M. C. Choi, G. Y. Kim, J. J. Park, M. Selvaraj, M. Han and C. S. Ha, Eur. Polym. J., 2012, 48, 1394–1405 CrossRef CAS.
- K. Kim, K. H. Nam, J. Lee, H. J. Kim, M. Goh, B. C. Ku and N. H. You, Carbon, 2017, 122, 614–621 CrossRef CAS.
- Z. Chen, F. Tong, D. Zhu, X. Lu and Q. Lu, ACS Appl. Polym. Mater., 2019, 1, 914–923 CrossRef CAS.
- W. H. Liao, S. Y. Yang, J. Y. Wang, H. W. Tien, S. T. Hsiao, Y. S. Wang, S. M. Li, C. C. M. Ma and Y. F. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 869–877 CrossRef CAS PubMed.
- J. Y. Wang, S. Y. Yang, Y. L. Huang, H. W. Tien, W. K. Chin and C. C. M. Ma, J. Mater. Chem., 2011, 21, 13569–13575 RSC.
- T. Chu, D. Liu, Y. Tian, Y. Li, W. Liu, G. Li, Z. Song, Z. Jian and X. Cai, ACS Appl. Nano Mater., 2020, 3, 5327–5334 CrossRef CAS.
- H. Li, R. Y. Tay, S. H. Tsang, W. Liu and E. H. T. Teo, Electrochim. Acta, 2015, 166, 197–205 CrossRef CAS.
- H. Li, L. Jing, R. Y. Tay, S. H. Tsang, J. Lin, M. Zhu, F. N. Leong and E. H. T. Teo, Chem. Eng. J., 2017, 328, 825–833 CrossRef CAS.
- G. Yang, L. Zhao, C. Shen, Z. Mao, H. Xu, X. Feng, B. Wang and X. Sui, Sol. Energy Mater. Sol. Cells, 2020, 209, 110441 CrossRef CAS.
- J. Wang, D. Liu, Q. Li, C. Chen, Z. Chen, P. Song, J. Hao, Y. Li, S. Fakhrhoseini, M. Naebe, X. Wang and W. Lei, ACS Nano, 2019, 13, 7860–7870 CrossRef CAS PubMed.
- W. Lei, V. N. Mochalin, D. Liu, S. Qin, Y. Gogotsi and Y. Chen, Nat. Commun., 2015, 6, 8849 CrossRef CAS.
- C. Chen, J. Wang, D. Liu, C. Yang, Y. Liu, R. S. Ruoff and W. Lei, Nat. Commun., 2018, 9, 1902 CrossRef PubMed.
- T. Huang, X. Zeng, Y. Yao, R. Sun, F. Meng, J. Xu and C. Wong, RSC Adv., 2016, 6, 35847–35854 RSC.
- S. Byun, J. H. Kim, S. H. Song, M. Lee, J. J. Park, G. Lee, S. H. Hong and D. Lee, Chem. Mater., 2016, 28, 7750–7756 CrossRef CAS.
- X. He and Y. Wang, Ind. Eng. Chem. Res., 2020, 59, 1925–1933 CrossRef CAS.
- Y. Lin, T. V. Williams, T. B. Xu, W. Cao, H. E. Elsayed-Ali and J. W. Connell, J. Phys. Chem. C, 2011, 115, 2679–2685 CrossRef CAS.
- Q. Weng, B. Wang, X. Wang, N. Hanagata, X. Li, D. Liu, X. Wang, X. Jiang, Y. Bando and D. Golberg, ACS Nano, 2014, 8, 6123–6130 CrossRef CAS PubMed.
- R. Geick, C. H. Perry and G. Rupprecht, Phys. Rev., 1966, 146, 543–547 CrossRef CAS.
- Y. Shi, C. Hamsen, X. Jia, K. K. Kim, A. Reina, M. Hofmann, A. L. Hsu, K. Zhang, H. Li, Z. Y. Juang, M. S. Dresselhaus, L. J. Li and J. Kong, Nano Lett., 2010, 10, 4134–4139 CrossRef CAS PubMed.
- J. H. Chang and K. M. Park, Eur. Polym. J., 2000, 36, 2185–2191 CrossRef CAS.
- D. J. Liaw, K. L. Wang, Y. C. Huang, K. R. Lee, J. Y. Lai and C. S. Ha, Prog. Polym. Sci., 2012, 37, 907–974 CrossRef CAS.
- A. Mochizuki, T. Teranishi and M. Ueda, Polym. J., 1994, 26, 315–323 CrossRef CAS.
- S. Shin, J. Jang, S. H. Yoon and I. Mochida, Carbon, 1997, 35, 1739–1743 CrossRef CAS.
- O. Gershevitz and C. N. Sukenik, J. Am. Chem. Soc., 2004, 126, 482–483 CrossRef CAS PubMed.
- A. Henglein, Chem. Rev., 1989, 89, 1861–1873 CrossRef CAS.
- M.-J. Brekner and C. Feger, J. Polym. Sci., Part A: Polym. Chem., 1987, 25, 2479–2491 CrossRef CAS.
- J. M. Lavin, D. M. Keicher, S. R. Whetten, P. B. Moore and S. S. Mani, Solid Freeform Fabr., 2016, 2016, 729–737 Search PubMed.
- C. Zhi, Y. Bando, C. Tang, H. Kuwahara and D. Golberg, Adv. Mater., 2009, 21, 2889–2893 CrossRef CAS.
- X. Zheng, J. Liu, K. Wang, R. Liu, Y. Yuan and X. Liu, Prog. Org. Coat., 2018, 124, 122–128 CrossRef CAS.
- M. Topsakal, E. Aktürk and S. Ciraci, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 115442 CrossRef.
- L. Chen, S. Chai, K. Liu, N. Ning, J. Gao, Q. Liu, F. Chen and Q. Fu, ACS Appl. Mater. Interfaces, 2012, 4, 4398–4404 CrossRef CAS PubMed.
- C. Rodrigues, J. M. M. de Mello, F. Dalcanton, D. L. P. Macuvele, N. Padoin, M. A. Fiori, C. Soares and H. G. Riella, J. Polym. Environ., 2020, 28, 1216–1236 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- N. Morimoto, H. Suzuki, Y. Takeuchi, S. Kawaguchi, M. Kunisu, C. W. Bielawski and Y. Nishina, Chem. Mater., 2017, 29, 2150–2156 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2na00078d |
| This journal is © The Royal Society of Chemistry 2022 |