
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Addressing challenges in the removal of unbound dye from passively labelled extracellular vesicles†
Kaisa
Rautaniemi
 a,
Jacopo
Zini‡
b,
Emilia
Löfman‡
a,
Heikki
Saari
bc,
Iida
Haapalehto
a,
Johanna
Laukka
a,
Sami
Vesamäki
a,
Alexander
Efimov
a,
Marjo
Yliperttula
b,
Timo
Laaksonen
ab,
Elina
Vuorimaa-Laukkanen
a and
Ekaterina S.
Lisitsyna
*a
a,
Jacopo
Zini‡
b,
Emilia
Löfman‡
a,
Heikki
Saari
bc,
Iida
Haapalehto
a,
Johanna
Laukka
a,
Sami
Vesamäki
a,
Alexander
Efimov
a,
Marjo
Yliperttula
b,
Timo
Laaksonen
ab,
Elina
Vuorimaa-Laukkanen
a and
Ekaterina S.
Lisitsyna
*a
aChemistry and Advanced Materials, Faculty of Engineering and Natural Sciences, Tampere University, Korkeakoulunkatu 8, 33720 Tampere, Finland. E-mail: ekaterina.lisitsyna@tuni.fi
bDrug Research Program, Division of Pharmaceutical Biosciences, Faculty of Pharmacy, University of Helsinki, Viikinkaari 5, 00790 Helsinki, Finland
cFinnish Red Cross Blood Services, Kivihaantie 7, 00310 Helsinki, Finland
First published on 23rd November 2021
Abstract
Studies of extracellular vesicles (EVs), their trafficking and characterization often employ fluorescent labelling. Unfortunately, little attention has been paid thus far to a thorough evaluation of the purification of EVs after labelling, although the presence of an unbound dye may severely compromise the results or even lead to wrong conclusions on EV functionality. Here, we systematically studied five dyes for passive EV labelling and meticulously compared five typical purification methods: ultracentrifugation (UC), ultracentrifugation with discontinuous density gradient (UCG), ultrafiltration (UF), size exclusion chromatography (SEC), and anion exchange chromatography (AEC). A general methodology for evaluation of EV purification efficiency after the labelling was developed and tested to select the purification methods for the chosen dyes. Firstly, we found that some methods initially lead to high EV losses even in the absence of the dye. Secondly, the suitable purification method needs to be found for each particular dye and depends on the physical and chemical properties of the dye. Thirdly, we demonstrated that the developed parameter Erp (relative purification efficiency) is a useful tool for the pre-screening of the suitable dye-purification method combinations. Additionally, it was also shown that the labelled EVs properly purified from the unbound dye may show significantly reduced contrast and visibility in the target application, e.g. in the live cell fluorescence lifetime imaging.
1 Introduction
Extracellular vesicles (EVs) have gained significant attention as promising drug carriers for personalized nanomedicine over the recent decade. EVs are structurally heterogenous membrane-bound nanoparticles that are excreted by cells.1 EVs have been associated with immune responses,2 viral pathogenicity,3 central nervous system related diseases,4,5 and cancer progression.6 Because of the natural origin of the EVs and their unique innate properties as natural cargo, the EV research foresees their potential applications as diagnostic or therapeutic tools for various diseases.EV trafficking and their interactions with cells, tissues and in vivo are typically studied with fluorescence-based microscopy methods.4,7–9 Furthermore, fluorescent labelling of EVs can enhance the sensitivity and selectivity of EV characterization methods, i.e., flow cytometry10,11 and nanoparticle tracking analysis.12,13 There are several different approaches for the EV labelling;14,15 the simplest and most commonly used method is the incubation of isolated EVs with lipid-tracer fluorescent dyes, such as long-chain dialkylcarbocyanines, including DiI, DiD8,16–18 and PKH dyes.9,10,17,19 This is referred to as passive loading of the dyes in contrast to active loading methods20 and covalent dye grafting.21,22 The passive labelling is a relatively safe alternative among the other labelling methods due to the least effect on the natural EV structure.23 Since the covalent labels often react with the amine groups of the proteins at the surface of the EV membrane and active labelling requires extra chemical or physical treatments of EVs, the covalent or active labelling may lead to a reduction of EV unique intrinsic features and a deterioration of their potential as nanocarrier.
Being an equilibrium process, the passive labelling usually results in a mixture of the labelled EVs and an unbound dye in an aqueous solvent.21,23–26 Covalent labelling is also often based on equilibrium chemical reactions and thus excess of reactive dye remains in the final labelled sample.21 Consequently, in both cases the unbound dye needs to be removed from the labelled EVs. The methods used for this include the same procedures as used for the initial EV isolation, such as ultracentrifugation,9,19,27 various filtrations,8,17 and size-exclusion chromatography.16,22 A successful removal of the unbound dye from the EVs is a crucial step for most EV studies, as the fluorescent dye not associated with the EVs will likely compromise the research outcomes or even lead to wrong conclusions of the EV functionality.28
Although the challenges in unbound dye removal have been noticed and discussed earlier, there are no clear criteria for selecting a suitable purification method to remove the excess dye from the labelled EVs, and the success of the purification is rarely estimated. In a few studies, the purification success was confirmed by comparison with free dye controls,21,24 while characterization of the labelled EVs was done by quite exotic methods which can hardly be available in all laboratories, e.g. asymmetric-flow field-flow fractionation coupled to a multi-angle light-scattering detector,24 or nanoscale fluorescence analysis and cytometric sorting.21 Although the above methods bring important and convincing information, they can be too laborious for screening of multiple labels, labelling conditions, and suitable purification methods. That is why there is a need for a simpler pre-screening protocol for an assessment of the purification after labelling. In some papers, the EV labelling efficiency was estimated as a relation of the recovered amount of label either to the recovered amount of EVs,20 or to the protein content in the labelled EV preparation.29 They however lacked an evaluation of the purification efficiency after labelling which clearly determines the accuracy of the labelling efficiency estimation. Conclusively, the lack of a standard way to characterize the purification efficiency makes published work difficult to compare. Therefore, there is a clear need for a systematic comparison of the purification methods.15 The present work is our attempt to fulfil this urgent need.
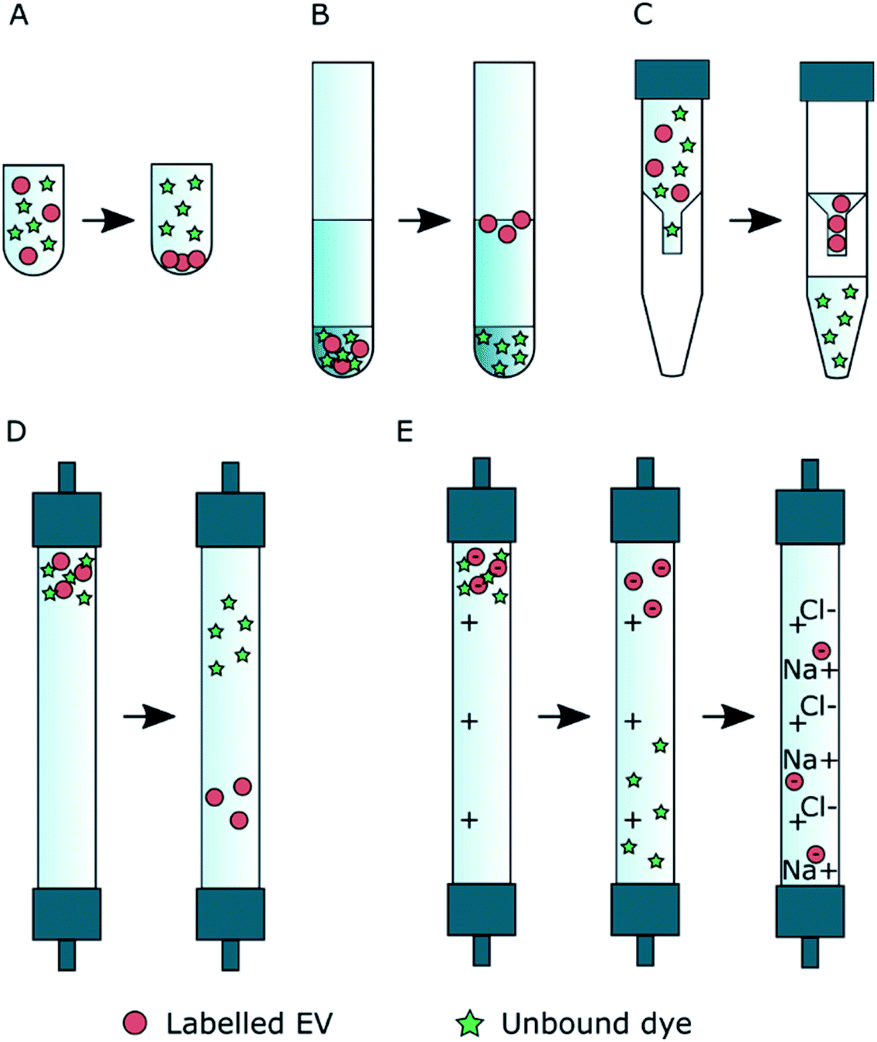
In this study, we focused only on purification method selection and estimation of purification efficiency and did not study EV labelling efficiency. Five widely available purification methods were studied for their ability to separate fluorescently labelled EVs from unbound dye: ultracentrifugation (UC), ultracentrifugation with discontinuous density gradient (UCG), ultrafiltration (UF), size exclusion chromatography (SEC) and anion exchange chromatography (AEC). Five fluorescent dyes were used for the passive EV labelling, resulting in 25 dye – purification method combinations. The overall workflow of the study and the molecular structures of the studied dyes are presented in Scheme 1. First, the behaviour of the dyes and the EVs were studied separately with each of the methods to screen the potential methods offering sufficient separation between the dyes and the EVs. Based on these control results, potential purification methods were chosen for each dye, and they were subsequently applied to the labelled EVs. The relative purification efficiency was estimated based on the EV and dye recovery for each sample, and the results were compared for different purification methods and dyes. Finally, the labelled and purified EVs were applied to cells and imaged by fluorescence lifetime microscopy.
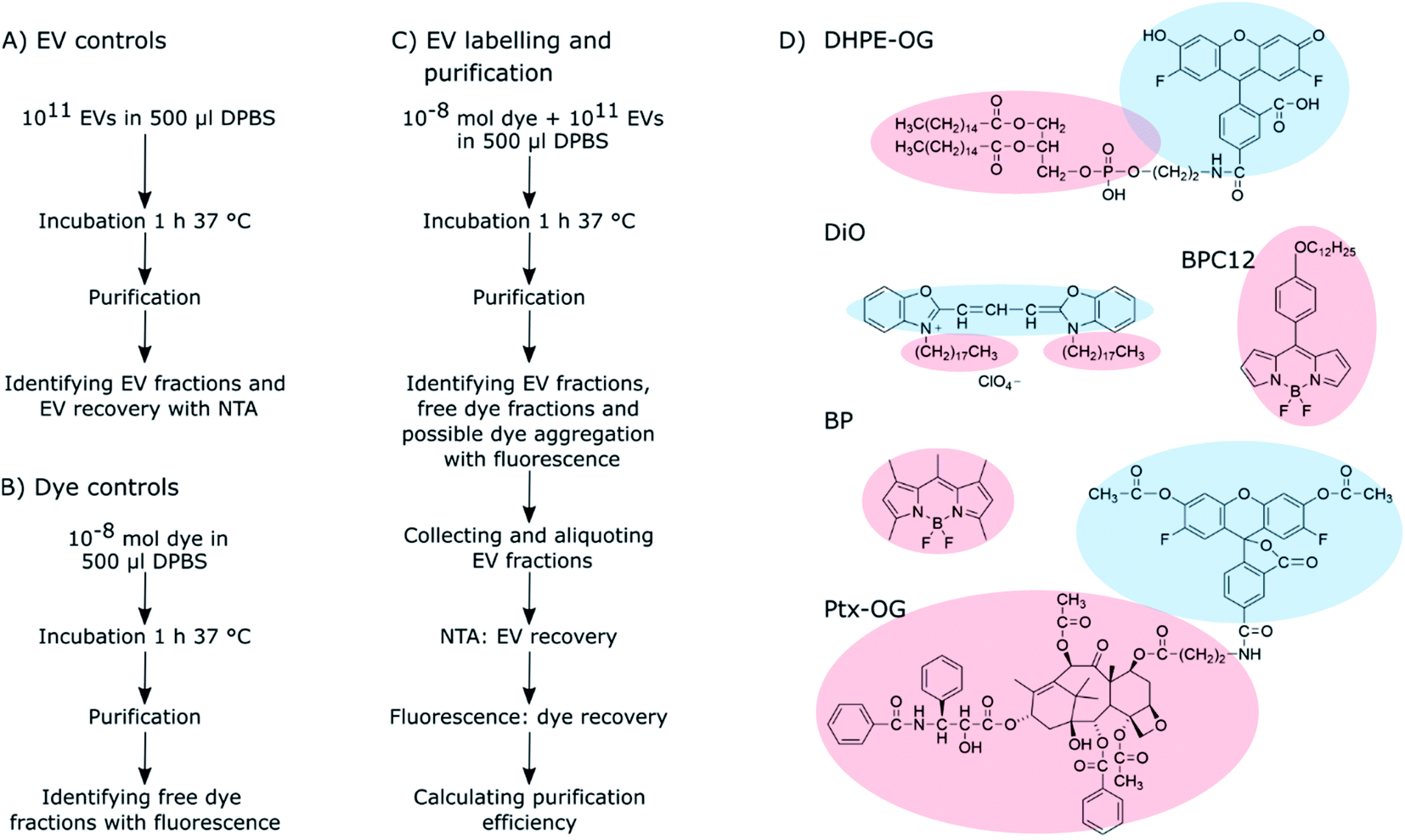 | ||
| Scheme 1 The workflow of the (A) EV control, (B) dye control, and (C) EV labelling and purification experiments. The details of these experiments are described in Sections 2.3–2.6. (D) The molecular structures of the fluorescent dyes used for the EV labelling. Light red clouds point out the hydrophobic parts of the molecules intercalating into the EV lipid membrane, and light blue ones indicate more hydrophilic parts probably located in the outer surface of the EVs exposed to the surroundings. | ||
As clearly seen from the chemical structures of the dyes, they are all significantly hydrophobic and, thus, should intercalate into the EV membrane in aqueous solution (Scheme 1D). DHPE-OG is a fluorescent conjugate of a lipid molecule. Being able to label EVs with lipids would be beneficial since they are natural components of the EV membrane and thus useful e.g., for adding targeting units to EVs.30 DiO was chosen since carbocyanine dyes are commonly used in membrane staining.31 BPC12 is a molecular rotor dye and its viscosity dependent fluorescence could be used to study the integrity of the EVs during cell up take and consequent trafficking inside the cell.32 BP is a neutral, nonpolar lipid stain.33 Both these BP dyes are very hydrophobic and will thus intercalate deep into the EV membrane.34 All the other studied dyes are at least partially at the hydrophilic part of the EV membrane and thus exposed to the surroundings. Hence, they can influence the EV functionality and especially the cell uptake.35 As an example of loading EVs with a biologically active molecule, the labelling was also studied with a Ptx-OG, a tubulin targeting anti-cancer agent Paclitaxel labelled with fluorescent dye OG.7,36 Being somewhat hydrophobic, Ptx part will intercalate into the EV membrane.
2 Materials and methods
2.1 EV isolation
PC-3 cell line was obtained from the American Type Culture Collection (ATCC, USA) and cultured in the CELLine AD 1000 bioreactor (Sigma-Aldrich, USA) at 37 °C and 5% of CO2. The cell culture compartment was filled with 15 ml Advanced DMEM/F-12 glucose (4.5 g ml−1) and L-glutamine (2 mM), while the media compartment was filled with 750 ml Ham's F-12k medium supplemented with 10% fetal bovine serum (FBS) and glucose (4.5 g ml−1). Cell culture compartment and media compartment are devised by a semi-permeable membrane (molecular weight cut-off 10 kDa), which permits a continuous nutrient diffusion and waste elimination. The membrane prevents the diffusion of EVs and large proteins from one compartment to the other. Cell culture media, FBS and L-glutamine were purchased from ThermoFisher Scientific (USA) and the glucose from Sigma-Aldrich (USA).PC-3 cell derived extracellular vesicles were isolated from the cell culture media by differential ultracentrifugation. First, buoyant cells, cell fragments and apoptotic bodies were removed with low-speed centrifugation at 2500×g for 25 min at +4 °C (Eppendorf Centrifuge, rotor FA-45-6-30, Germany). Next, the first EV fraction (20k EVs) was pelleted with centrifugation at +4 °C with 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×g (12741 rpm, k-factor 1287) for 1 h (38.5 ml, Open-Top Thinwall Polypropylene Tube, Optima L-80 XP ultracentrifuge with SW 32 Ti Rotor, Beckman Coulter, USA). A second EV fraction (110k EVs) was collected from the supernatant with centrifugation at +4 °C with 110000×g for 2 h (29881 rpm, k-factor 234).
000×g (12741 rpm, k-factor 1287) for 1 h (38.5 ml, Open-Top Thinwall Polypropylene Tube, Optima L-80 XP ultracentrifuge with SW 32 Ti Rotor, Beckman Coulter, USA). A second EV fraction (110k EVs) was collected from the supernatant with centrifugation at +4 °C with 110000×g for 2 h (29881 rpm, k-factor 234).
Next, the EV pellets were resuspended in DPBS (Dulbecco's phosphate buffered saline, Sigma-Aldrich, USA) buffer and the EVs were further purified with three-layered (0% to 35% to 45%) discontinuous iodixanol density gradient. The EV suspension was mixed with iodixanol (Optiprep™, Alere Technologies AS, Norway) to a final volume of 2 ml and a 45% iodixanol concentration and loaded to the bottom of the density gradient. 4 ml of 35% iodixanol was placed over the bottom layer, and the rest of the tube was filled with DPBS. The gradient was centrifuged at +4 °C with 200000×g (40291 rpm, k-factor 129) for 3.5 h (13.2 ml, Open-Top Thinwall Ultra-Clear Tube, Optima L-80 XP ultracentrifuge with SW 41 Ti Rotor, Beckman Coulter, USA). Upon the centrifugation the EVs move from their original bottom layer (density of >1.215 g ml−1) to the top of the layer with the density of ∼1.195 g ml−1 with a velocity dependent on the cube of their size,37i.e., the EVs concentrate on the 0–35% iodixanol interface according to their buoyant density. DPBS on top of the gradient was discarded, and the EVs were collected from the 0–35% iodixanol interface. Finally, the iodixanol was removed by serial ultrafiltration (Amicon Ultra-15 Centrifugal Filter units, cut-off 10 kDa, Millipore, USA) at 5000×g, 20 min, +4 °C (Eppendorf Centrifuge, 5810 R fixed-angle rotor Hamburg, Germany). The EV suspension was divided to aliquots of 1011 particles and stored at −80 °C until they were used for the experiments. In this study, the EV populations are classified as the 20k EVs and the 110k EVs, according to the centrifugal forces used for their isolation.
2.2 Characterization of the isolated EVs
The isolated EVs were characterized according to MISEV2018 standards38 by western blot, transmission electron microscopy and ATR-FTIR as described in (ESI Section S1†). The particle concentrations and the size distributions of the isolated EV fractions were analysed using the NanoSight LM-14 instrument (LCM14C, 405 nm laser, 60 mW, Nanosight, Salisbury, United Kingdom), equipped with the sCMOS camera (Hamamatsu Photonics K.K., Hamamatsu, Japan). The samples were diluted with DPBS and measured using a camera level 15 and an acquisition time of 90 s. Every sample was measured in triplicates. The resulting videos were analysed using the NanoSight NTA software (NanoSight Ltd., v. 3.0) with a detection threshold set to 5. Noteworthy, the NTA instrument can detect only particles larger than 80 nm. After the NTA analysis, the EV samples were divided to aliquots of 1011 particles and stored frozen in −80 °C until they were used for labelling. Similar NTA analysis was done for all the labelled EVs and non-labelled EV controls after every purification for estimating EV recoveries (Scheme 1A and C).2.3 Passive labelling of EVs
Five different fluorescent dyes were used for the EV labelling: Oregon Green™ 488 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (DHPE-OG), 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO), 4,4-difluoro-1,3,5,7,8-pentamethyl-4-bora-3a,4a-diaza-s-indacene (BP), 4,4′-difluoro-4-bora-3a,4a-diaza-s-indacene meso-substituted with para-dodecylphenyl moiety (BPC12),32 and a tubulin tracer dye Oregon Green 488 taxol, bis-acetate (trade name Tubulin Tracker Green; here, Ptx-OG). BPC12 was synthetized in our group as described in the literature,32 and the other dyes were purchased from ThermoFisher Scientific (USA). The dye stocks were prepared in dimethyl sulfoxide (DMSO, Sigma-Aldrich, Germany). The emission properties of the dyes were studied without EVs in two different solutions: DPBS and solubilized in DPBS by addition of 1% Triton X-100 (Surfact-Amps X-100, ThermoFisher Scientific, USA) with spectrofluorometer (FluoroLog-3, Horiba Scientific, Japan; excitation wavelength 483 nm, emission range 500–800 nm) to find possible aggregation-related emission bands. For the dyes showing clear aggregate emission, the emission spectra were measured also for the purified EVs as one criteria of the purification quality.To be able to study the purification abilities of the chosen methods, the dye-to-EV ratio was chosen to have a significant excess of the fluorescent dye. EV aliquots of 1011 particles were diluted to a final volume of 0.5 ml in DPBS, and 10−8 mol of dye was added to the EVs while mixing with a vortex mixer to prevent immediate aggregation of the hydrophobic dye in aqueous buffer solution. The final DMSO concentration in the EV suspension was less than 2.5%. The labelling mixture was then incubated for 1 h at 37 °C upon shaking while protected from light (Scheme 1C). All the EV labelling was done in triplicates.
2.4 Purification methods
Five widely available purification methods were studied for their ability to separate the labelled EVs from the unbound dye: ultracentrifugation (UC), ultracentrifugation with discontinuous iodixanol density gradient (UCG), ultrafiltration (UF), size exclusion chromatography (SEC), and anion exchange chromatography (AEC). In a successful UC purification, the EVs pellet during the centrifugation while the unbound dye stays mostly in the supernatant and can be carefully aspirated above the EV pellet. In UCG, because of their buoyant density and bigger size compared to fluorescent dye molecules, the EVs float to the higher interface between the layers with smaller densities while the unbound dye is expected to stay mostly at the bottom of the gradient in the highest density layer. Both UF and SEC are based on the size separation: in UF, the EVs concentrate above the filter membrane as most of the unbound dye is washed to the filtrate, and in SEC, the EVs elute from the column before the unbound dye. In AEC, the EVs first bind to the positively charged column and the unbound dye is washed from the column; then, the EVs are released from the column with buffer containing NaCl.000×g (18185 rpm, k-factor 289) for 1 h, and the 110k EVs were pelleted with centrifugation at +4 °C with 110000×g (42647 rpm, k-factor 53.0) for 2 h. The centrifugations were done by an Optima MAX ultracentrifuge equipped with the MLA-130 fixed angle rotor in 1 ml Open-Top Thickwall polycarbonate tubes (Beckman Coulter, USA). The supernatant was aspirated just above the formed pellet to avoid disturbing the rather loose pellet, and the EVs were allowed to disperse in 70 μl of DPBS overnight at +4 °C.
000×g (40291 rpm, k-factor 129.0) for 3.5 h by an Optima XPN ultracentrifuge, equipped with a SW 41 Ti swinging bucket rotor (Beckman Coulter, USA). After collecting 1 ml fractions (a total of 13–14 fractions), the fluorescence of the collected fractions was measured for identifying the EV and the free dye fractions. The fluorescence experiments were done either by a spectrofluorometer (FluoroLog-3, Horiba Scientific, Japan) or by a fluorescence plate reader (Fluorescent Ascent FL, Thermo Scientific, USA). Based on the NTA results at EV isolation step (Section 2.1) and in the EV controls (Section 2.5), the highest particle concentrations were expected to be in the interface between DPBS and 35% iodixanol. Fluorescence intensity registered in 1–2 fractions on this interface verified their choice as the EV fractions. These fractions were either pooled and the iodixanol was removed by serial ultrafiltration as described in Section 2.4.3 (UCG + UF), or the pooled EV fractions were analysed without removing the iodixanol (UCG).
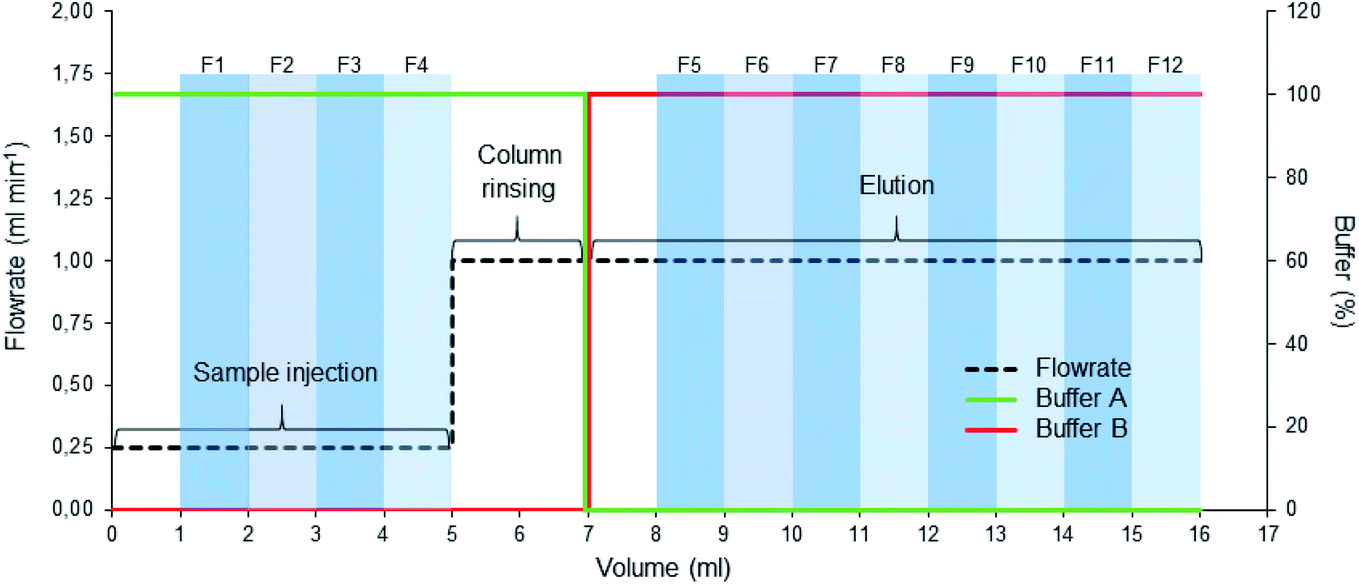 | ||
| Fig. 1 AEC running protocol. After preparing the column, the sample was injected at 0.25 ml min−1 (0–5 ml) and washed with the start buffer (100% A), collecting fractions (1–4) after one column volume (1 ml). The elution was then started with buffer B, collecting fractions (5–12) after one column volume had passed, followed by washing and regeneration of the column. | ||
2.5 Control purifications
Before using any of the purification methods for the EVs incubated with a fluorescent dye, the behaviour of the dye was studied without the EVs (Scheme 1A). Conversely, the EV behaviour and recoveries in all the methods were studied also without the fluorescent dyes (Scheme 1B). The dye and EV controls were used to select the methods with potential to separate the unbound dye from the EVs. Dye controls were prepared by diluting 10−8 mol of the dye to a final volume of 0.5 ml in DPBS; similarly, the non-labelled EV controls were prepared by diluting EV aliquots of 1011 particles to a final volume of 0.5 ml of DPBS (Scheme 1). Both control samples were then incubated and purified in the same way as the labelled EV suspensions. The EV controls were performed in triplicates and the dye controls once.In the EV controls, the expected EV fractions were collected and analysed with NTA for determining the EV recoveries (Section 2.6). In the chromatography methods, the EV containing fractions were identified by analysing fractions 1–10 (SEC) or 1–12 (AEC) with NTA for the first of the EV control replicates. Based on the results for the rest of the replicates only the EV containing fractions were analysed. With UCG, the majority of the unlabelled EVs were in the 0–35% iodixanol interface. In higher-density fractions, the scattering background from iodixanol made the NTA analysis unreliable, and therefore those fractions were not analysed with NTA.
For the dye controls, fluorescence was used for studying whether the dyes behave as expected for a successful purification. For the UC and UF dye controls, the dye recovery Rdye,c was estimated by comparing fluorescence in the expected EV fraction (resuspended UC pellet and UF retentate) to the total fluorescence of the sample, measured with the spectrofluorometer:
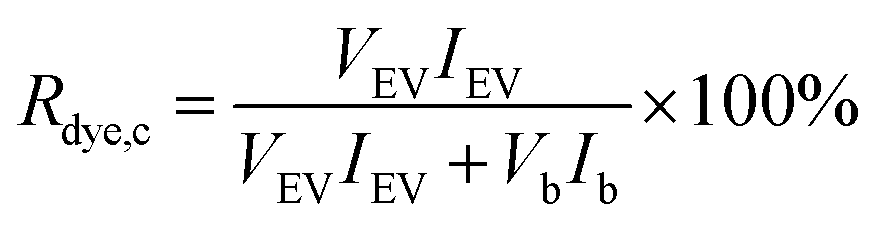 | (1) |
2.6 Characterization after purification
After the purification, all the EV samples were divided into aliquots and stored at −80 °C for the characterization (Scheme 1C). The particle concentrations of the EV suspensions recovered after labelling and purification were measured with NTA to obtain EV recovery (REV):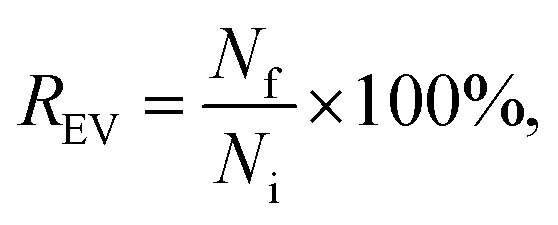 | (2) |
The dye concentration in the purified samples was measured against a dye calibration curve either with a plate reader (Ptx-OG, DHPE-OG, BPC12, DiO, BP UCG and BP AEC) or with a fluorescence spectrophotometer (BP UC and BP UF). The fluorescence of the samples with known amount of a dye were measured to form the calibration curve. The dyes were released from the EVs and solubilized in the buffer by adding 1% Triton X-100 to both the EV and the calibration samples. Ptx-OG was hydrolysed to its fully fluorescent form by adding sodium hydroxide (0.015 M final concentration) together with 1% Triton X-100 to both the EV and the calibration samples and incubating at 37 °C for 1 h before the measurements. Dye recovery Rdye was calculated as
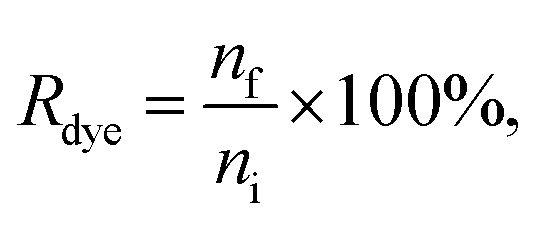 | (3) |
To compare the purification result between the different methods and the different dyes, relative purification efficiency Erp was calculated as
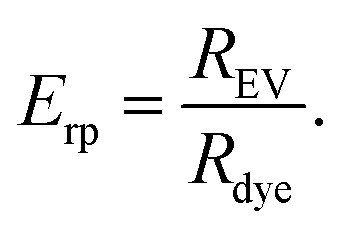 | (4) |
Given that a significant excess of dyes was always used, Erp < 1 indicates that the method concentrates the unbound dye more efficiently than the EVs and therefore it is not suitable for the purification of the EVs after fluorescent labelling. For the methods yielding Erp > 1, the relative purification efficiency was used for comparing the suitability of the purification methods for each dye.
2.7 FLIM imaging of cells incubated with EVs
One day before imaging, 10000 PC-3 cells were seeded in 70 μl 2-well inserts (Ibidi, Germany) attached to a glass-bottom 35 mm Petri dish (poly-D-lysine coated, no. 1.5 coverslip, 10 mm glass diameter, MatTek, USA), or 75000 cells were seeded directly on the Petri dish. Cells were incubated with labelled and purified EV sample (30000–400000 particles/seeded cell) for 3 hours, washed once with DPBS and imaged in FluoroBrite DMEM (Gibco, USA) supplemented with 10% (v/v) FBS. Samples for the free dye control were prepared similarly, using free dye instead of the labelled EVs. Similar amount of free dye was added to the cells as would have been added with 30000 particle/cell – ratio.
Fluorescence lifetime images were acquired using the fluorescence lifetime microscope MicroTime-200 (PicoQuant, Germany) coupled to the inverted microscope Olympus IX-71 (Olympus, Japan) equipped with 100× oil objective (NA = 1.4). The pulsed laser diode LDH-P-C483 (PicoQuant, Germany) emitting at 483 nm (time resolution 120 ps) was used for the excitation and the emission was monitored on wavelengths 510–900 nm. The samples were imaged at 37 °C and 5% CO2 using an objective heater (TC-1-1005 Temperature Controller, Bioscience Tools, USA) and a custom-made incubator. The FLIM images were analysed in SymPhoTime 64 software (PicoQuant, Germany). The colours of the FLIM images are based on the mean arrival times of the emitted photons after the excitation pulse (fast lifetime). The intensity-averaged lifetimes τav were obtained by 2- or 3-exponential lifetime fitting7 (DHPE-OG and DiO, respectively) of the decay curves of the selected regions of interest, excluding the cell autofluorescence background.
3 Results
3.1 EV characterization
Two different subpopulations of PC-3 EVs, 20k and 110k, were isolated from conditioned media by differential ultracentrifugation, followed by density gradient centrifugation. The isolated EVs were characterized by NTA, TEM, FTIR and WB analysis. The detailed characterization is presented in ESI Section S1.† Briefly, the isolated EVs were confirmed to be of high purity having consistently similar properties over each sample replicate regarding size, enrichment of the EV-associated proteins and overall biochemical composition according to FTIR spectroscopy. The most apparent differences between 20k and 110k EVs were that 20k EVs were larger on average, which is in line with their consequently faster sedimentation during 20000×g centrifugation, and the more intense peaks in the 1040–1110 cm−1 region in the FTIR spectra for the 110k EVs.
3.2 Control purifications
| Method | 110k, REV (%) | 20k, REV (%) |
|---|---|---|
| UC | 12.2 ± 3.0 | 24.0 ± 11.0 |
| UCG | 82.3 ± 11.6 | 59.5 ± 6.2 |
| UCG + UF | 0.6 ± 0.1 | 1.3 ± 0.3 |
| UF | 4.3 ± 3.2 | 11.1 ± 15.2 |
| SEC | 33.8 ± 16.3 | 10.2 ± 5.9 |
| AEC | 64.7 ± 6.9 | 45.9 ± 9.0 |
 | ||
| Fig. 2 The expected separation principles for a successful EV separation from non-EV-bound dye. (A) Ultracentrifugation: the EVs pellet during the centrifugation while the unbound dye stays in the supernatant; (B) ultracentrifugation with density gradient: the EVs float to the higher interface between the layers with smaller densities, and the unbound dye stays at the bottom of the gradient in the highest density layer; (C) ultrafiltration: the EVs concentrate above the filter membrane as the unbound dye is washed to the filtrate; (D) size exclusion chromatography: the EVs elute from the column before the unbound dye; (E) anion exchange chromatography: first, the EVs are bound to the positively charged column and the unbound dye is washed from the column; then, the EVs are released from the column with buffer containing NaCl. | ||
3.2.2.1 Ultracentrifugation and ultrafiltration (Fig. 2A and C). For the UC and UF dye controls, the dye recoveries in the expected EV fractions (UC pellet and UF retentate) were estimated from the fluorescence intensity of resuspended pellet and supernatant (UC, Fig. 2A), or from the sample collected above the filter membrane and the highest concentration filtrate (UF, Fig. 2C) according to eqn (1). Thus, the optimal Rdye,c-value would be ∼0 for the dye controls. The obtained Rdye,c-values are presented in Table 2.
000×g and 20000×g) and ultrafiltration (UF) dye control results. The dye recoveries in the expected EV fractions Rdye,c were estimated from fluorescence spectra measured at 550 nm (DiO), 515 nm (BP) and 522 nm (DHPE-OG and Ptx-OG)
| Dye | UC 110k, Rdye,c (%) | UC 20k, Rdye,c (%) | UF, Rdye,c (%) |
|---|---|---|---|
| DHPE-OG | 3 | 4 | 9 |
| Ptx-OG | 52 | 59 | 96 |
| BP | <1 | 1 | 2 |
| BPC12 | — | — | — |
| DiO | 80 | 65 | 97 |
DiO and Ptx-OG had virtually no fluorescence in the UC supernatant and UF filtrates, while the emission intensity in the resuspended UC pellet and the UF retentate was high. The result clearly shows that these dyes aggregate and, therefore, they are efficiently pelleted during UC and do not pass the filter membrane in UF. Consequently, these methods are not suitable for removing DiO or Ptx-OG from the labelled EVs.
BP and DHPE-OG gave better control results. DHPE-OG fluorescence was still clearly detected both in the UC pellet and the UF retentate (Rdye,c = 3–9%), while almost no BP fluorescence was observed (Rdye,c = 1–2%). Because of the low dye recoveries, BP was chosen as a model dye for the UC and UF purifications.
BPC12 stuck to the centrifuge tube walls during the ultracentrifugation and attached to the UF filter device membrane showing orange colouring on the tube walls and the filter membrane. The detected fluorescence intensities were low even upon addition of surfactant (1% Triton X) to solubilize the dye, indicating strong surface adsorption. Since UC and UF did not work as expected with BPC12, the dye recoveries could not be reliably estimated. However, the possibility of using these methods to purify EVs from unbound BPC12 based on the dye adsorption on the surfaces was studied further with BPC12-labelled EVs.
3.2.2.2 Ultracentrifugation with density gradient (Fig. 2B). In the UCG controls, most of the Ptx-OG, DHPE-OG and BP stayed in the bottom fractions (45% iodixanol) of the gradient (Fig. 3A). During the centrifugation, DiO formed visible crystals that did not concentrate in 45% iodixanol layer but had migrated further in the 35% iodixanol layer (Fig. 3A, pink). With all these dyes, there was also some emission at the interface between 0% and 35% iodixanol, where also the EVs concentrate according to the unlabelled EV controls. However, as most of the free dye stayed in higher density fractions and the EV recoveries before UF were high (Table 1), UCG purification was studied further for the EVs labelled with Ptx-OG, DHPE-OG, BP and DiO. In contrast, BPC12 emission was observed only in the fractions where EV accumulation is expected (Fig. 3A, yellow), implying that UCG is not suitable method to purify the BPC12 labelled EVs due to similarities in the dye and EV particle density.
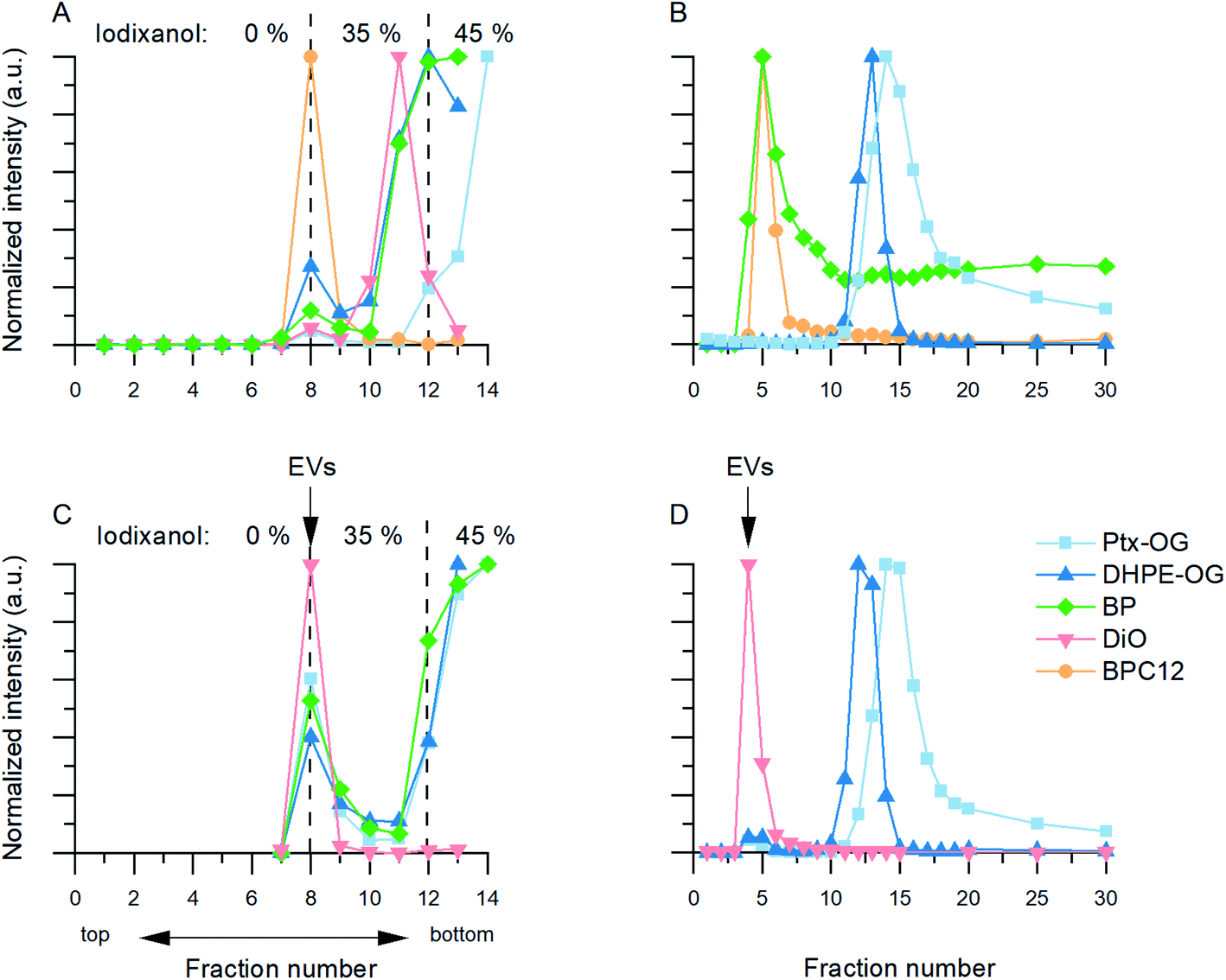 | ||
| Fig. 3 Dye control results and examples of EV fluorescence distributions for the UCG (A and C) and SEC purification (B and D). In all graphs, normalized fluorescence intensity is presented as a function of fraction number. The same colour scale has been used in all graphs. (A) UCG control. The iodixanol concentration is presented in the top of the figure. (B) SEC control. DiO did not pass the SEC column and is therefore not shown here. (C) Fluorescent dye distribution in UCG fractions with labelled 110k EVs. (D) Fluorescent dye distribution in SEC fractions with labelled 110k EVs. | ||
3.2.2.3 Size-exclusion chromatography (Fig. 2D). Three types of dye behaviour were observed in the SEC controls. Both BODIPY dyes eluted in the same fractions as EVs (Fig. 3B, green and yellow). Most of the BPC12 eluted in fractions 4 and 5, indicating that it forms particles large enough for size exclusion to occur in the column. Separate NTA controls confirmed that BPC12 forms particles with about 90 nm diameter in DPBS (ESI Fig. S4†). Although BP did not form particles visible by NTA (ESI Fig. S4†), the highest concentrations of BP eluted in the fractions 4–7, indicating the formation of particles too small for NTA to detect, but large enough for the size exclusion effect, followed by a high background level in later fractions. Instead, both Oregon Green dyes had good separation from EVs and started eluting first after fraction 10 (Fig. 3B, blue and light blue). DiO, being the least water soluble of the dyes in this study, did not pass the column at all: instead, it crystallized and stayed on top of the column. Based on the control results, SEC purification was studied further with Ptx-OG, DHPE-OG and DiO.
3.2.2.4 Anion exchange chromatography (Fig. 2E). Since both OG dyes (Ptx-OG after hydrolysation) contain a negatively charged carboxylic acid group, their separation with AEC was expected to be challenging. Neutral dyes (BODIPY dyes) and positively charged dyes (DiO) were expected to elute in the sample injection step. Unfortunately, the AEC principle turned out to be unsuitable for the studied dyes. Based on the dye controls, all of the OG and BP dyes elute in the same fractions as the EVs (ESI Fig. S5†). DiO had the most promising result, giving only negligible fluorescence in the expected EV fractions. Although it was not removed in the sample injection step as expected, DiO crystallized similar to the SEC DiO control and did not pass the column at all. Consequently, IEC was studied only for the purification of the DiO-labelled EVs.
3.3 EV purification results
The chosen purification methods for each dye were applied to the labelled EVs. The EVs were incubated with a constant concentration of fluorescent dye in DPBS as described in Section 2.3 and Scheme 1C. The EV and dye recoveries (eqn (2) and (3)) as well as the relative purification efficiencies (eqn (4)) were used to estimate the quality of the purification (Table 3). In general, labelling decreased the EV recoveries (Table 3) compared to the unlabelled controls (Table 1). DiO- and Ptx-OG-labelling reduced the recoveries the most with all the used purification methods. On the other hand, BP-labelled EVs had almost the same REV after UCG as the unlabelled EVs.| Dye | Method | 110k EVs | 20k EVs | ||||
|---|---|---|---|---|---|---|---|
| R EV (%) | R dye (%) | E rp , | R EV (%) | R dye (%) | E rp , | ||
| a Colour code: green – acceptably high; red – unacceptably low; black – acceptable with caution. b E rp > 1 indicates successful separation of the labelled EVs from the unbound dye: the greater Erp, the better separation; conversely, Erp < 1 indicates unsuccessful removal of the dye. | |||||||
| DHPE-OG | UCG | 43.0 ± 2.8 | 44.6 ± 4.2 | 1.0 | 52.9 ± 7.5 | 39.6 ± 3.3 | 1.3 |
| SEC | 12.2 ± 1.6 | 8.7 ± 1.4 | 1.4 | 8.2 ± 0.9 | 9.3 ± 5.0 | 0.9 | |
| Ptx-OG | UCG | 10.3 ± 0.4 | 67.8 ± 11.5 | 0.2 | 6.5 ± 3.3 | 41.3 ± 38.4 | 0.2 |
| SEC | 3.8 ± 1.6 | 2.9 ± 1.3 | 1.3 | 3.7 ± 0.9 | 1.3 ± 0.3 | 2.8 | |
| BP | UC | 7.6 ± 4.8 | 16.6 ± 1.3 | 0.5 | <1 | 7.0 ± 0.8 | — |
| UF | 1.2 ± 0.7 | 1.8 ± 0.9 | 0.7 | 2.3 ± 1.0 | 4.8 ± 3.8 | 0.5 | |
| UCG | 78.6 ± 10.3 | 6.2 ± 0.9 | 12.7 | 54.0 ± 6.0 | 15.5 ± 4.8 | 3.5 | |
| BPC12 | UC | 12.5 ± 6.8 | 35.5 ± 37.6 | 0.4 | 5.9 ± 6.2 | 17.2 ± 15.3 | 0.3 |
| UF | 3.9 ± 2.8 | 7.9 ± 3.4 | 0.5 | 8.4 ± 11.2 | 9,5 ± 15,4 | 0.9 | |
| DiO | UCG | n.d. | n.d. | — | n.d. | n.d. | — |
| SEC | 1.1 ± 0.2 | n.d. | — | <1 | n.d. | — | |
| AEC | 10.1 ± 12.3 | 2.2 ± 2.0 | 4.6 | 6.4 ± 0.3 | 1.8 ± 0.3 | 3.5 | |
The dyes in this study were relatively hydrophobic, and thus they were expected to locate mainly in the EV membrane after successful labelling. About 60000 dye molecules were added per each EV in the initial labelling suspension. By estimating the EV membrane area and how many lipid molecules it can accumulate,39,40 the dye-to-lipid molar ratio in the labelled EV suspension before purification was at least 1:3. As an example, for paclitaxel-loaded liposomes, drug-to-lipid ratios of 1:20–1:33 have been reported.41,42 Such high dye-to-lipid ratios are unlikely reached by passive labelling, and it can be considered that a large excess of dye related to the EVs was used in the present experiments. Therefore, a successful purification process would remove most of the initial dye and a minimum requirement for the purification is that Rdye < REV, yielding Erp > 1. In other cases, the dye-to-EV ratio would actually increase during the purification.
Although the purification efficiencies were similar for both methods, the EV yields were higher after UCG (>40%) than SEC purification (about 10%).
In contrast to DHPE-OG, UCG did not separate the unbound dye from the Ptx-OG-labelled EVs. Only 10% or less of the EVs were recovered, while these fractions contained 40–70% of the fluorescent dye. Instead, the SEC purification gave promising and even reproducible results: although REV was less than 4% for both EV types, Erp was 1.3 (110k EVs) and 2.8 (20k EVs).
For the UC and UF purifications, BP dye control results were the most promising (Table 2). Unfortunately, UC did not remove the unbound BP from the labelled EVs efficiently enough during one centrifugation round: 17% (110k EVs) or 7% (20k EVs) of the dye was recovered while only 8% or less than 1% (respectively) of the EVs were collected after the centrifugation. UF gave slightly better results in terms of Erp; however, Erp < 1 suggests a poor separation of the labelled EVs from the unbound dye, and the EV recoveries were very low (1–2%). For BP, the best purification method was UCG. The BP-labelling did not reduce the EV recoveries compared to the unlabelled controls, while majority of the fluorescent dye added to the EVs was removed during the purification. The UCG-purified BP EVs had the highest relative purification efficiencies of the studied samples, 12.7 (110k EVs) and 3.5 (20k EVs).
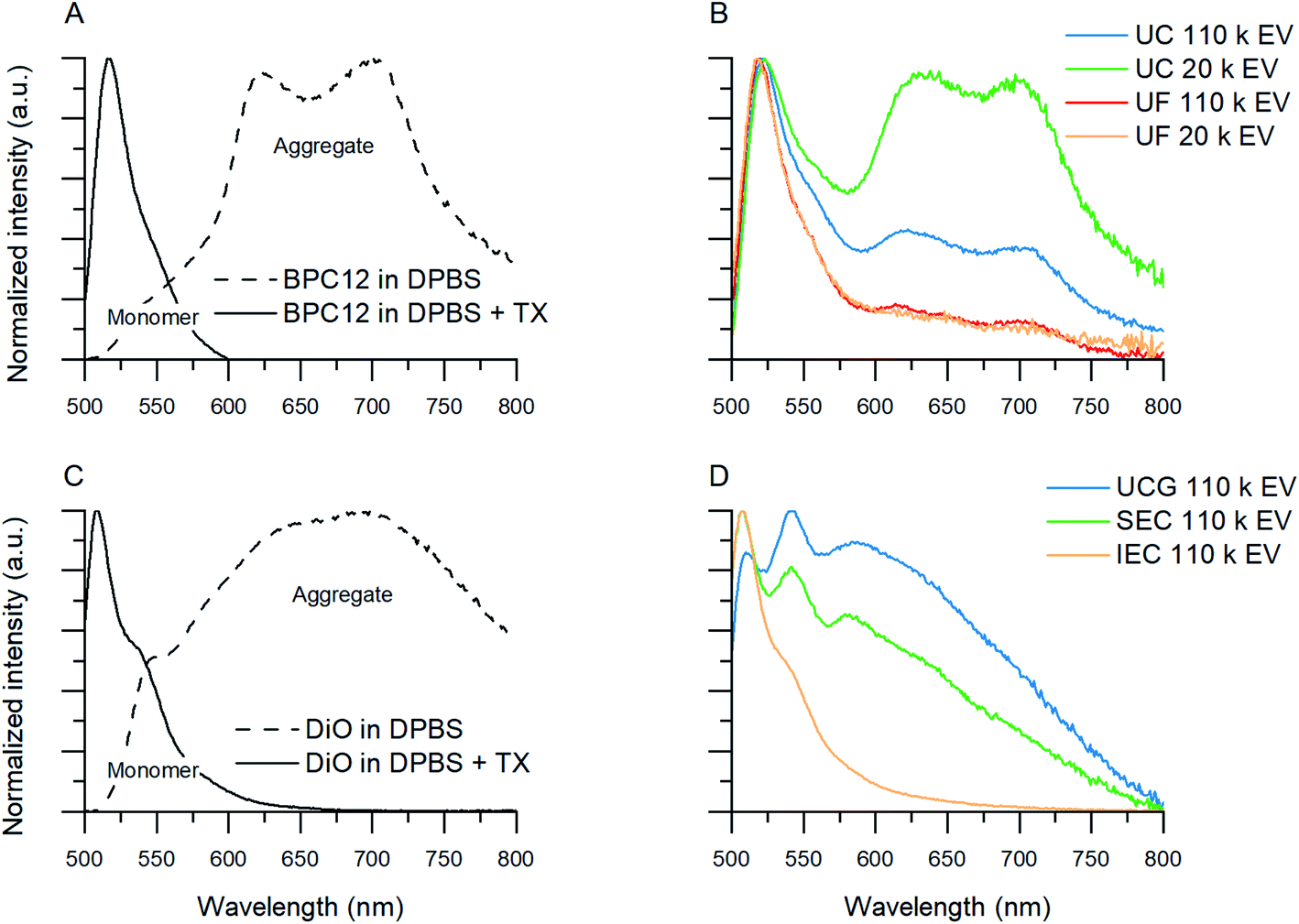 | ||
| Fig. 4 Normalized fluorescence spectra showing aggregated and monomeric BPC12 and DiO emission in solvent samples and purified EV samples. (A) Aggregated BPC12 (maxima at 630 nm and 700 nm) in DPBS and monomeric BPC12 (520 nm) in DPBS with Triton X (TX). (B) Examples of emission spectra of BPC12-labelled EVs after UC and UF purifications. (C) Aggregated DiO (broad band above 550 nm) in DPBS and monomeric DiO (maximum at 510 nm) in DPBS with TX. (D) Examples of emission spectra of DiO-labelled EVs after UCG, SEC and AEC purifications. All the spectra were measured with excitation at 483 nm. | ||
As the unbound BPC12 forms aggregates in DPBS that have similar size as the EVs (Section 3.2.2, ESI Fig. S4†), some of the particles detected by NTA are probably not EVs, and consequently REV is not reliable for the BPC12-labelled EVs leading to a low Erp showing a failure of the purification. For the UC purified BPC12 EVs, a strong aggregate fluorescence was observed (Fig. 4B, green and blue). The 520 nm emission peak relates to the monomeric dye in the EV membrane, while the longer wavelength emission (bands at 630 nm and 700 nm) relates to the dye aggregates in the aqueous buffer. The relative intensity of the aggregate peaks compared to monomer (EV-related) peak is higher for 110k than 20k EVs, indicating higher concentration of the unbound BPC12 in 110k EV sample than in 20k EVs. The fluorescence spectra of the UF-purified BPC12-labelled EVs (Fig. 4B, red and yellow) do not have as pronounced aggregate emission peaks as the UC-purified EVs, indicating more efficient removal of the unbound dye especially for the 20k EVs. However, the UF purification results had high variation, which is seen in the fluorescence spectra (ESI Fig. S6†) and is reflected also in REV and Rdye (Table 3), suggesting that the fluorescence spectra should always be checked after the UF purification.
For DiO, UCG, SEC and AEC were studied to purify the labelled EVs. In UCG, the DiO control result showed only slight accumulation of the dye in the expected EV-fractions, while with the EVs, the dye accumulated almost exclusively in the EV-fractions (Fig. 3C, pink). This could designate strong binding of DiO to the EVs. However, given the high excess of the dye used for the labelling, there must be unbound dye present in the EV fraction. The result indicates that the EVs have affected the DiO aggregation or the diffusion of the DiO aggregates in the gradient, leading to no separation between the unbound dye aggregates and the EVs. Indeed, the fluorescence spectra of the UCG-purified DiO-labelled EVs (Fig. 4D, blue) confirms the presence of DiO aggregates in the EV fractions: monomer DiO peak at 510 nm is weak, and the spectrum is dominated by an aggregated DiO emission (bands above 550 nm). Compared to UCG, the SEC purification removed the unbound dye more efficiently. In the emission spectra of DiO EVs after SEC purification (Fig. 4D, green) the most intensive emission peak at 510 nm corresponds to the monomeric, EV-bound DiO. Nevertheless, there is still clearly a visible aggregate emission band above 550 nm. Additionally, the DiO aggregation seemed to lead to very low EV recoveries which varied from non-detectable to slightly over 1% (Table 3, ESI Table S2†).
The third method studied for purification of the DiO-labelled EVs was AEC. Similar to the SEC dye control, DiO did not pass the AEC column. However, the DiO-labelled EVs passed the AEC column better than the SEC column: the average EV recoveries were 10% for 110k EVs and 6% for 20k EVs. The fluorescence spectrum (Fig. 4D, yellow) shows only a monomeric EV-related emission peak at 510 nm, which is in line with Erp ≥ 3.5 for both EV types, indicating AEC as the best purification method for DiO-labelled EVs.
3.4 FLIM imaging of purified EVs with cells
A selection of labelled and purified EVs and corresponding free dyes were applied to PC-3 cells and imaged with FLIM to confirm the visibility and examine the distribution of the labelled EVs in the cells, and to compare the fluorescence staining patterns to those of the free dyes. Examples of the FLIM images are presented in Fig. 5.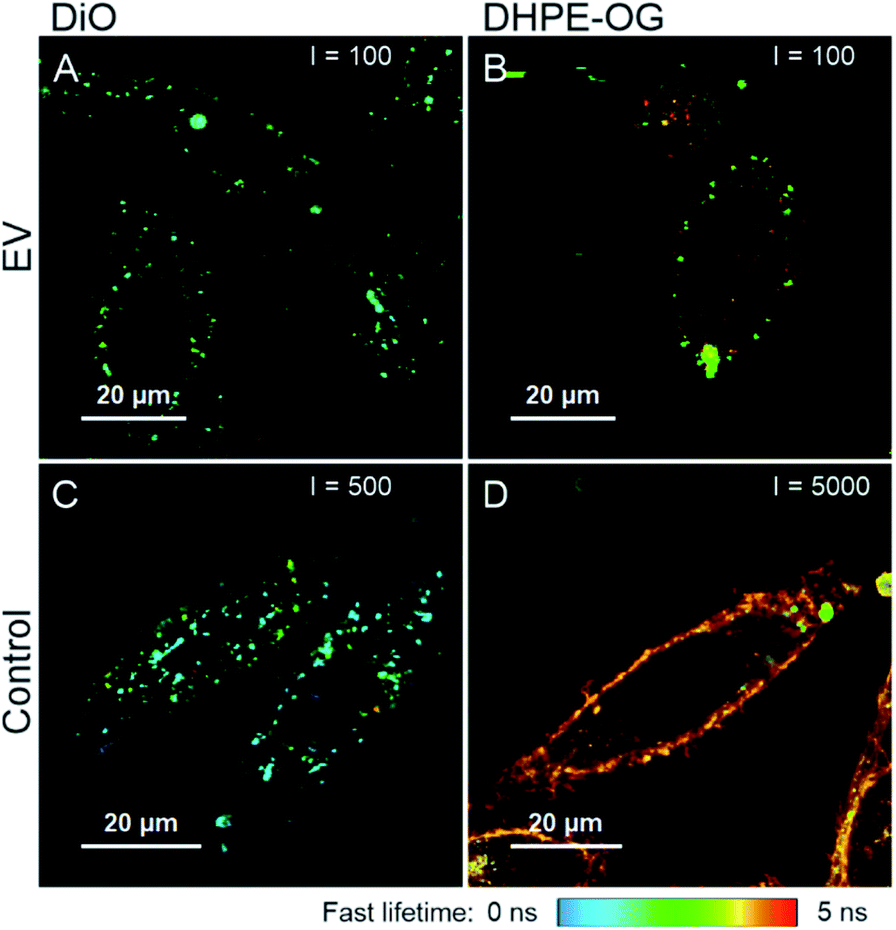 | ||
| Fig. 5 FLIM images of labelled and purified EVs (A, B) and corresponding free dyes (C and D) incubated with PC-3 cells for 3 hours. (A) AEC DiO 110k EVs, 80000 EVs/cell; (B) DHPE-OG SEC 110k EVs, 80000 EVs/cell; (C) DiO; and (D) DHPE-OG. The dye concentration in the free dye controls is the same as would be added with 30000 of similar EVs/cell. The maximum intensity threshold (events) is presented in the upper corner of each figure. The figures are presented in the same fluorescence lifetime scale presented below the figure. | ||
The FLIM imaging results demonstrate the importance of the purification step and underlines the difficulties in separating the free dye background from the fluorescence signal originating from the EVs. In general, the fluorescence intensities of the samples incubated with labelled and successfully purified EVs (Fig. 5A and B) were lower than the free dye controls (Fig. 5C and D), although the dye concentrations were higher in the EV samples, indicating slower dye internalization via EVs. The average fluorescence lifetime of DiO is similar in both samples (τav = 1.17 ns in the EV sample and τav = 1.16 ns in the control), while there is a clear difference in the DHPE-OG lifetimes (τav = 2.63 in the EV sample and τav = 3.99 ns in the control).
Based on Erp (Table 3), purification of DiO and DHPE-OG 110k EVs succeeded with AEC and SEC, correspondingly. The staining patterns of the EVs labelled with these dyes are spot-like inside the cells (Fig. 5A and B), which could be interpreted as the internalization of EVs into the endosomal pathway. However, the free DiO staining pattern and fluorescence lifetime is very similar to the DiO-labelled EVs (Fig. 5A and C); consequently, it is very easy to misinterpret the free DiO background as labelled EVs. For DHPE-OG, the difference in the staining patterns and fluorescence lifetime is clearer (Fig. 5B and D), indicating different uptake mechanisms of the free dye and EV-bound dye. Free DHPE-OG seems to stay attached to the cell membrane at least for 3 h of incubation with cells while EV-bound dye enters the cell confirming the EV mediated dye internalisation. However, even if the labelling and purification of the EVs from the unbound dye would have been successful, the lipophilic dyes may leach from the EVs and stain other cellular membranes.28,45
Our results also demonstrate that with a successful removal of the unbound dye, the EVs might not be detectable when applied to the cells. The emission intensity of the cells incubated with SEC-purified Ptx-OG EVs (30000 EVs/cell) was close to the cell autofluorescence, and due to low EV recoveries, intensity could not be increased by adding more EVs. The UCG-purified BP EVs gave best Erp values but were not visible even with 400000 EVs/cell, suggesting that either the labelling efficiency has not been high enough for detecting the EVs or the iodixanol has a negative effect on the EV-cell interactions.
4 Discussion
The FLIM images of the labelled EVs (Fig. 5) clearly demonstrate that although fluorescence signal is observed, it is not necessarily related to the EVs. Thus, for further reliable applications, e.g. in microscopy, it is extremely important to ensure both successful labelling and purification from the unbound dye with a dedicated method. To evaluate the purity of labelled EVs, we relied on a simple parameter, Erp to describe the approximate success of each purification protocol. The approach is simple and applies to the cases where the unbound dye is present in the system after labelling or the dye-to-EV ratio is high enough. As described earlier, this is very often the case for passive and covalent labelling. Moreover, it is clear that using a dye control solely is not sufficient to validate the purification success. The unbound dye behaviour may be affected by the presence of EVs as, for example, in the case of DIO in ultracentrifugation with gradient (Fig. 3 A and C).As the EV samples were initially isolated with a differential ultracentrifugation protocol prior to a density gradient, it may have also affected the EVs morphologically causing EV aggregation and shape distortion. The effect on the results presented in this study is difficult to estimate. However, the study is focused on screening purification methods, and the important point is that the initial EVs have been treated in the same way for all experiments.
Nonlabelled EV controls of a purification method are important as some of the methods themselves may result in almost complete EV loss. This makes the method uninteresting for labelled EV purification as in the case of consequent UCG and UF (Table 1). The comparison of nonlabelled and labelled EV recoveries also gives important information about the possible effects of the dye on EV recovery. Significant reduction of EV recovery due to labelling questions the probe's suitability for EVs. Lastly, the parameter Erp, compares EV and dye recoveries after purification. If the percentage of EV losses is higher than that of dye removal (Erp < 1) the method obviously does not perform as desired and indicates a significant amount of unbound dye in the final labelled EVs. The need to compare the EV recovery with the dye recovery becomes clear upon taking a look at Table 3, where a relatively good amount of recovered particles in EV samples does not always match with efficient dye removal. Thus, it turns out that only the combination of the controls together with the Erp metric leads to a comprehensive assessment of the tested purification method.
In this study, we relied on NTA for evaluating the EV recovery, assuming that labelling does not significantly change the refractive index or size of the labelled EVs, excluding EV aggregation. Due to NTA's limits, not all the smallest EVs can be detected, and the method does not discriminate between EVs and other nanoparticles. However, the use of NTA in our attempt to suggest a protocol for evaluation of purification efficiency can be justified as we compare initially well-purified EVs using the same NTA settings and the same instrument prior and after labelling and purification. Thus, the relative values, such as ratios of the concentrations measured by NTA, can be reasonable estimates of the EV recoveries. Moreover, Erp parameter represents a criterium of a purification success, i.e. can reflect both successful (>1) or non-successful (<1) purification and should not be considered as absolute value in contrast to usually used labelling efficiency. The latter is meant to show a particular ratio of labels attached to a labelling target and is limited by 100% over which it becomes irrelevant.
Based on the above methodology (Scheme 1A–C), the purification methods were classified into three categories for each dye: good, unknown, and poor, (“+”, “?”, or “−“) as presented in Table 4. Most of the methods were directly classified based on the negative dye control results, and the rest were classified according to the relative purification efficiencies (Erp) of the labelled EVs. The methods which could reproducibly concentrate labelled EVs and remove unbound dye efficiently are marked with “+”. According to the same logic, the methods that can recover more dye than EVs, and therefore cannot provide good separation of the labelled EVs from the unbound dye, are marked with “−“. The third category, marked with “?”, had high variation in the purification results or yielded Erp close to 1. These methods may be considered for removing these dyes, provided that an appropriate verification of the purification result is done separately for each sample. For the samples applied for live cell imaging, also the visibility of the EVs with cells is summarized in Table 4. As can be seen, Table 4 is quite empty and most attempted purification protocols failed. But encouragingly, it was possible to find a method that at least partly worked for each dye applying the approach proposed in our study.
| Method | Ptx-OG | DHPE-OG | DiO | BPC12 | BP |
|---|---|---|---|---|---|
| UC | − | − | − | − | − |
| UCG | − | ? | − | − | + (−) |
| UF | − | − | − | ? | − |
| SEC | + (−) | ? (+) | − | − | − |
| AEC | − | − | + (+) | − | − |
Interestingly, the molecular structure of the dyes has a clear effect on the purification results. Unbound BP and DiO were effectively isolated from the EVs: BP has a rigid molecular skeleton and DiO two hydrocarbon chains anchoring it to the EV membrane during the labelling. On the other hand, the dyes with freely rotating units, OG-chromophore in DHPE-OG as well as the BP-chromophore in BPC12, showed either poor purification or high result variation. Furthermore, the dyes do not always act according to the expected purification principle. Ptx-OG and DiO (Table 2) form particle-like aggregates in aqueous environments that are large enough to be collected by centrifugation and BPC12 precipitates into the tube walls. Thus UC, the most used protocol for the EV purification,46 is not a suitable purification method for these dyes. BPC12 is also retained in the polyethersulfone-based filter membrane used in UF instead of passing it. In UCG, DiO and BPC12 gathered into the same layer as the EVs, probably due to aggregate formation. For SEC and AEC, DiO did not pass the columns at all.
From the studied methods, UC and UF were not efficient methods for purification of unbound dye from the labelled EVs. For UC, even with the dyes that stay mostly in the supernatant during the centrifugation, several UC concentration cycles would be necessary for efficient removal of the dye. However, since in most cases over 90% of the initial EVs were lost during a single UC run, no EVs would be left after few centrifugations. According to our results, also UF leads to very low EV recoveries and is, thus, not suitable for the unbound dye removal. The recoveries after UF might be improved by choosing different filter devices,47 but a comprehensive study of these matters is missing and there is no univocal consensus on the best way to purify EVs with UF.24,47–51 The use of UF for removal of the iodixanol proved to be the limiting step of UCG as well. In our work, we observed high initial UCG EV yields of 60–80% before gradient removal, while using the common ultrafiltration protocol for removing the iodixanol from the EVs, we observed a heavy loss of EVs down to ∼1% yield (Table 1). Despite the wide usage of UCG for EV purification, methods to remove the iodixanol are not yet fully addressed. As iodixanol is a non-charged small-sized molecule, both AEC and SEC are promising candidate methods for the iodixanol removal.
The use of chromatographic methods for EV isolation and purification has raised a lot of interest, because they can be highly automatized and scaled up.52,53 From the two chromatographic methods studied here, SEC showed more potential for removing the unbound dye from the labelled EVs, and is already a commonly used technique in EV field with different ready to use solutions available on the market.54 On the other hand, AEC was incompatible with the studied dyes, although the unlabelled EV recoveries were high. DiO-labelled EVs were successfully purified with AEC; however, the purification principle was rather related to the dye aggregation. With both chromatographic methods, the recoveries of labelled EVs were smaller than those of non-labelled EVs, suggesting that the dye may partly clog the column or increase EV binding to the column. The AEC method provided an interesting comparison for SEC as the EV losses were smaller in AEC than in SEC. This can be at least partially explained by non-specific binding of EVs to the column: the SEC bed size was more than 10-fold larger than the AEC bed size, therefore containing much more surface area for binding. Furthermore, saturating the AEC column with BSA significantly increased the EV recoveries. The results indicate that the EV yields could be increased in the SEC by reducing the column volume and if feasible, by blocking the column with BSA.
Based on this study, it is difficult to say whether the dye is actually located in the EV membrane. Still, Erp is a useful tool for the pre-screening of the suitable dye-purification method combinations. As some dyes, such as PKH dyes24 or BPC12 used here, tend to form dye aggregates with similar sizes as the EVs, detecting fluorescent EV-sized particles is not sufficient proof for the successful EV labelling and purification and there is clearly a need for a method for separating the EVs from possible dye aggregates. Generally, EV marker antibodies are recommended for separating EVs from other particles.38 Consequently, quantitative particle detection methods such as NTA12,13 and flow cytometry10,11 could offer a solution for identifying the labelled EVs.
5 Conclusions
Five common purification methods were tested with five dyes for their ability to separate the passively labelled EVs from the unbound dye. None of the studied purification methods was suitable for all the studied dyes. Most dyes could be successfully purified with only one of the methods tested, suggesting that the successful purification method is related to the physical and chemical properties of the dye. Notably, in many cases the expected purification principle did not work. The unbound dye might form aggregates, bind to the purification matrices or gather to the same location with the EVs for some other reason. Thus, we suggest the following steps when working with a new dye to ensure the successful removal of the unbound dye: (1) studying the behaviour of free dye and unlabelled EVs separately with the chosen method to determine whether the method can effectively separate those from each other, (2) ensuring the separation still exists with the labelled EVs, and (3) evaluating the purification quality by Erp. Importantly, the labelled EVs need to have high enough fluorescence intensity to be visible in the target application that is not always the case after successful purification.The most promising methods for the used dyes were SEC and UCG, and the highest recoveries were obtained by UCG before removing the density gradient. With SEC, the purification is faster to perform involving only one separation step, making the SEC purification favourable over the UCG when there is a possibility to choose between the methods. However, both methods should be further developed: the gradient removal step after UCG causes high EV losses and SEC purification should be still improved to yield higher EV recoveries. Sometimes also an unexpected purification principle may provide good purification results, as we demonstrated with AEC.
In this study, we used only EVs from a single cell line: the purification results with the same dyes but EVs from different sources might be different. We admit that the Erp values used in this study to evaluate the success of the purification of labelled EVs from unbound dye is an approximate method. However, the parameter in combination with the proposed controls allows to assess the purification outcome in a more comprehensive way compared to using only dye controls or labelling efficiency values. The proposed approach represents an easy methodology for initial pre-screening of multiple labelling conditions and purification methods.
Author contributions
Conceptualization: KR, EV-L, ESL; methodology: KR, HS, EV-L, ESL; investigation: KR, JZ, EL, HS, IH, JL, SV, AE, ESL; writing – original draft: KR, JZ, HS, AE, MY, TL, EV-L, ESL; supervision: MY, TL, EV-L, ESL.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Academy of Finland under Grants 311362 (KR, ESL, EV-L), 316893 (TL), 314406 (JZ), 315409 (HS), and 323669 (ESL); and Business Finland EVE ecosystem under Grants 1842/31/2019 (KR, JZ, EL, IH, SV, ESL, EV-L) and 2581/31/2018 (HS).References
- R. Kalluri and V. S. LeBleu, The biology, function, and biomedical applications of exosomes, Science, 2020, 367, 6478 CrossRef PubMed.
- Z. Chen, A. T. Larregina and A. E. Morelli, Impact of extracellular vesicles on innate immunity, Curr. Opin. Organ Transplant., 2019, 24(6), 670–678 CrossRef CAS.
- H. Saari, et al., Extracellular vesicles provide a capsid-free vector for oncolytic adenoviral DNA delivery, J. Extracell. Vesicles, 2020, 9(1), 1747206 CrossRef CAS PubMed.
- Z. Ruan, et al., Alzheimer's disease brain-derived extracellular vesicles spread tau pathology in interneurons, Brain, 2021, 144(1), 288–309 CrossRef PubMed.
- A. F. Hill, Extracellular Vesicles and Neurodegenerative Diseases, J. Neurosci., 2019, 39(47), 9269–9273 CrossRef PubMed.
- A. Becker, B. K. Thakur, J. M. Weiss, H. S. Kim, H. Peinado and D. Lyden, Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis, Cancer Cell, 2016, 30(6), 836–848 CrossRef CAS PubMed.
- H. Saari, et al., FLIM reveals alternative EV-mediated cellular up-take pathways of paclitaxel, J. Controlled Release, 2018, 284, 133–143 CrossRef CAS.
- T. Tian, et al., Exosome uptake through clathrin-mediated endocytosis and macropinocytosis and mediating miR-21 delivery, J. Biol. Chem., 2014, 289(32), 22258–22267 CrossRef CAS PubMed.
- S. Kamerkar, et al., Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer, Nature, 2017, 546(7659), 498–503 CrossRef CAS PubMed.
- E. J. van der Vlist, E. N. M. Nolte-'t Hoen, W. Stoorvogel, G. J. A. Arkesteijn and M. H. M. Wauben, Fluorescent labeling of nano-sized vesicles released by cells and subsequent quantitative and qualitative analysis by high-resolution flow cytometry, Nat. Protoc., 2012, 7(7), 1311–1326 CrossRef.
- S. A. Stoner, et al., High sensitivity flow cytometry of membrane vesicles, Cytometry, Part A, 2016, 89(2), 196–206 CrossRef.
- A. Desgeorges, J. Hollerweger, T. Lassacher, E. Rohde, C. Helmbrecht and M. Gimona, Differential fluorescence nanoparticle tracking analysis for enumeration of the extracellular vesicle content in mixed particulate solutions, Methods, 2020, 177, 67–73 CrossRef CAS.
- K. E. Thane, A. M. Davis and A. M. Hoffman, Improved methods for fluorescent labeling and detection of single extracellular vesicles using nanoparticle tracking analysis, Sci. Rep., 2019, 9(1), 1–13 CAS.
- S. Salunkhe, S. Dheeraj, M. Basak, D. Chitkara and A. Mittal, Surface functionalization of exosomes for target-specific delivery and in vivo imaging & tracking: Strategies and significance, J. Controlled Release, 2020, 326, 599–614 CrossRef CAS.
- M. Dehghani and T. R. Gaborski, Fluorescent Labeling of Extracellular Vesicles, Elsevier Inc., 1st edn, vol. 645, 2020 Search PubMed.
- M. Mehanny, M. Koch, C. M. Lehr and G. Fuhrmann, Streptococcal Extracellular Membrane Vesicles Are Rapidly Internalized by Immune Cells and Alter Their Cytokine Release, Front. Immunol., 2020, 11, 80 CrossRef CAS PubMed.
- K. Takov, D. M. Yellon and S. M. Davidson, Confounding factors in vesicle uptake studies using fluorescent lipophilic membrane dyes, J. Extracell. Vesicles, 2017, 6(1), 1388731 CrossRef.
- V. Fonsato, et al., Human liver stem cell-derived extracellular vesicles enhance cancer stem cell sensitivity to tyrosine kinase inhibitors through Akt/mTOR/PTEN combined modulation, Oncotarget, 2018, 9(90), 36151–36165 CrossRef PubMed.
- W. Ying, et al., Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate in Vivo and in Vitro Insulin Sensitivity, Cell, 2017, 171(2), 372–384 CrossRef CAS PubMed.
- G. Fuhrmann, A. Serio, M. Mazo, R. Nair and M. M. Stevens, Active loading into extracellular vesicles significantly improves the cellular uptake and photodynamic effect of porphyrins, J. Controlled Release, 2015, 205, 35–44 CrossRef CAS PubMed.
- A. Morales-Kastresana, et al., Labeling extracellular vesicles for nanoscale flow cytometry, Sci. Rep., 2017, 7(1), 1–10 CrossRef CAS.
- S. A. A. Kooijmans, J. J. J. M. Gitz-Francois, R. M. Schiffelers and P. Vader, Recombinant phosphatidylserine-binding nanobodies for targeting of extracellular vesicles to tumor cells: a plug-and-play approach, Nanoscale, 2018, 10(5), 2413–2426 RSC.
- M. Wehbe, et al., A simple passive equilibration method for loading carboplatin into pre-formed liposomes incubated with ethanol as a temperature dependent permeability enhancer, J. Controlled Release, 2017, 252, 50–61 CrossRef CAS PubMed.
- P. Pužar Dominkuš, et al., PKH26 labeling of extracellular vesicles: characterization and cellular internalization of contaminating PKH26 nanoparticles, Biochim. Biophys. Acta, Bioenerg., 2018, 1860(6), 1350–1361 CrossRef PubMed.
- M. Dehghani, S. M. Gulvin, J. Flax and T. R. Gaborski, Systematic Evaluation of PKH Labelling on Extracellular Vesicle Size by Nanoparticle Tracking Analysis, Sci. Rep., 2020, 10(1), 1–10 CrossRef PubMed.
- B. Kneidl, M. Peller, G. Winter, L. H. Lindner and M. Hossann, Thermosensitive liposomal drug delivery systems: state of the art review, Int. J. Nanomed., 2014, 9, 4387–4398 Search PubMed.
- A. Hoshino, et al., Tumour exosome integrins determine organotropic metastasis, Nature, 2015, 527(7578), 329–335 CrossRef CAS PubMed.
- J. B. Simonsen, Pitfalls associated with lipophilic fluorophore staining of extracellular vesicles for uptake studies, J. Extracell. Vesicles, 2019, 8(1), 1582237 CrossRef CAS PubMed.
- S. A. A. Kooijmans, C. G. Aleza, S. R. Roffler, W. W. van Solinge, P. Vader and R. M. Schiffelers, Display of GPI-anchored anti-EGFR nanobodies on extracellular vesicles promotes tumour cell targeting, J. Extracell. Vesicles, 2016, 5(1), 31053 CrossRef PubMed.
- V. Agrahari, V. Agrahari, P. A. Burnouf, C. H. Chew and T. Burnouf, Extracellular Microvesicles as New Industrial Therapeutic Frontiers, Trends in Biotechnology, 2019, 37(7), 707–729 CrossRef CAS.
- F. Progatzky, M. J. Dallman and C. Lo Celso, From seeing to believing: labelling strategies for in vivo cell-tracking experiments, Interface Focus, 2013, 3(3), 20130001 CrossRef.
- J. A. Levitt, M. K. Kuimova, G. Yahioglu, P. H. Chung, K. Suhling and D. Phillips, Membrane-bound molecular rotors measure viscosity in live cells via fluorescence lifetime imaging, J. Phys. Chem. C, 2009, 113(27), 11634–11642 CrossRef CAS.
- J. Karolin, L. B.-A. Johansson, L. Strandberg, and T. Ny, Fluorescence and Absorption Spectroscopic Properties of Dipyrrometheneboron Difluoride (BODIPY) Derivatives in Liquids, Lipid Membranes, and Proteins, UTC, 1994 Search PubMed.
- H. Wang, W. Zhao, X. Liu, S. Wang and Y. Wang, BODIPY-Based Fluorescent Surfactant for Cell Membrane Imaging and Photodynamic Therapy, ACS Appl. Bio Mater., 2020, 3(1), 593–601 CrossRef CAS.
- C. Charoenviriyakul, Y. Takahashi, M. Morishita, M. Nishikawa and Y. Takakura, Role of Extracellular Vesicle Surface Proteins in the Pharmacokinetics of Extracellular Vesicles, Mol. Pharm., 2018, 15(3), 1073–1080 CrossRef CAS.
- H. Saari, E. Lázaro-Ibáñez, T. Viitala, E. Vuorimaa-Laukkanen, P. Siljander and M. Yliperttula, Microvesicle- and exosome-mediated drug delivery enhances the cytotoxicity of Paclitaxel in autologous prostate cancer cells, J. Controlled Release, 2015, 220, 727–737 CrossRef CAS PubMed.
- E. G. Eikenberry, Analytical Centrifugation in Density Gradients, in Centrifugation in Density Gradients, ed. C. A. Prixe, Academic Press Inc., 1982 Search PubMed.
- C. Théry, et al., Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines, J. Extracell. Vesicles, 2018, 7(1), 1535750 CrossRef.
- S. Takamori, et al., Molecular Anatomy of a Trafficking Organelle, Cell, 2006, 127(4), 831–846 CrossRef CAS PubMed.
- J. F. Nagle and S. Tristram-Nagle, Structure of lipid bilayers, Biochim. Biophys. Acta, Rev. Biomembr., 2000, 1469(3), 159–195 CrossRef CAS.
- S. Strieth, et al., Phase I/II clinical study on safety and antivascular effects of paclitaxel encapsulated in cationic liposomes for targeted therapy in advanced head and neck cancer, Head Neck Oncol., 2014, 36(7), 976–984 CrossRef PubMed.
- M. Slingerland, et al., Bioequivalence of Liposome-Entrapped Paclitaxel Easy-To-Use (LEP-ETU) Formulation and Paclitaxel in Polyethoxylated Castor Oil: A Randomized, Two-Period Crossover Study in Patients With Advanced Cancer, Clin. Ther., 2013, 35(12), 1946–1954 CrossRef CAS PubMed.
- D. A. Merkushev, et al., BODIPY associates in organic matrices: spectral properties, photostability and evaluation as OLED emitters, Mater. Chem. Phys., 2017, 187, 104–111 CrossRef CAS.
- R. Hu, et al., Twisted intramolecular charge transfer and aggregation-induced emission of BODIPY derivatives, J. Phys. Chem. C, 2009, 113(36), 15845–15853 CrossRef CAS.
- R. Münter, K. Kristensen, D. Pedersbæk, J. B. Larsen, J. B. Simonsen and T. L. Andresen, Dissociation of fluorescently labeled lipids from liposomes in biological environments challenges the interpretation of uptake studies, Nanoscale, 2018, 10(48), 22720–22724 RSC.
- C. Gardiner, et al., Techniques used for the isolation and characterization of extracellular vesicles: Results of a worldwide survey, J. Extracell. Vesicles, 2016, 5(1), 32945 CrossRef PubMed.
- R. J. Lobb, et al., Optimized exosome isolation protocol for cell culture supernatant and human plasma, J. Extracell. Vesicles, 2015, 4(1), 27031 CrossRef PubMed.
- C. Lässer, et al., Human saliva, plasma and breast milk exosomes contain RNA: uptake by macrophages, J. Transl. Med., 2011, 9(1), 9 CrossRef PubMed.
- B. Wu, et al., Separation and characterization of extracellular vesicles from human plasma by asymmetrical flow field-flow fractionation, Anal. Chim. Acta, 2020, 1127, 234–245 CrossRef CAS PubMed.
- B. J. Benedikter, et al., Ultrafiltration combined with size exclusion chromatography efficiently isolates extracellular vesicles from cell culture media for compositional and functional studies, Sci. Rep., 2017, 7(1), 1–13 CrossRef CAS PubMed.
- R. Xu, D. W. Greening, A. Rai, H. Ji and R. J. Simpson, Highly-purified exosomes and shed microvesicles isolated from the human colon cancer cell line LIM1863 by sequential centrifugal ultrafiltration are biochemically and functionally distinct, Methods, 2015, 87, 11–25 CrossRef CAS PubMed.
- J. Z. Nordin, et al., Ultrafiltration with size-exclusion liquid chromatography for high yield isolation of extracellular vesicles preserving intact biophysical and functional properties, Nanomedicine, 2015, 11(4), 879–883 CrossRef CAS PubMed.
- A. N. Böing, E. van der Pol, A. E. Grootemaat, F. A. W. Coumans, A. Sturk and R. Nieuwland, Single-step isolation of extracellular vesicles by size-exclusion chromatography, J. Extracell. Vesicles, 2014, 3(1), 1–11 Search PubMed.
- M. Monguió-Tortajada, C. Gálvez-Montón, A. Bayes-Genis, S. Roura and F. E. Borràs, Extracellular vesicle isolation methods: rising impact of size-exclusion chromatography, Cell. Mol. Life Sci., 2019, 76(12), 2369–2382 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00755f |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2022 |