
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile fabrication of conductive MoS2 thin films by sonication in hot water and evaluation of their electrocatalytic performance in the hydrogen evolution reaction†
Dipankar
Saha
 a,
Vinay
Patel
b,
Ponnambalam Ravi
Selvaganapathy
bc and
Peter
Kruse
*a
a,
Vinay
Patel
b,
Ponnambalam Ravi
Selvaganapathy
bc and
Peter
Kruse
*a
aDepartment of Chemistry and Chemical Biology, McMaster University, Hamilton, Ontario L8S 4M1, Canada. E-mail: pkruse@mcmaster.ca
bSchool of Biomedical Engineering, McMaster University, Hamilton, Ontario L8S 4L8, Canada
cDepartment of Mechanical Engineering, McMaster University, Hamilton, Ontario L8S 4L7, Canada
First published on 29th October 2021
Abstract
Molybdenum disulfide (MoS2) has long been used in catalysis and is a promising material for energy conversion devices. In order to utilize MoS2 in electrocatalytic applications, it needs to be sufficiently conductive. Even though a metallic 1T phase of MoS2 exists, its exfoliation process is expensive and difficult to scale because it involves hazardous materials and procedures, limiting its practical applications. We have previously reported an efficient and environmentally friendly procedure to exfoliate conductive MoS2via sonication in very dilute aqueous hydrogen peroxide. Here, we report a new way of exfoliating heavily doped conductive MoS2 by sonication in pure water at 60 °C without additives. Conductivity measurements, Raman spectroscopy and X-ray photoelectron spectroscopy demonstrate that controlling the sonication time and temperature lead to the generation of small quantities of hydrogen peroxide in the water that interact with MoS2 to form a small amount of sub-stoichiometric MoO3−y. This impurity acts as a dopant and is responsible for the increase in conductivity of the MoS2 films without compromising their structural integrity. We also evaluate the performance of the doped MoS2 films as electrocatalysts in the hydrogen evolution reaction. We elucidate the mechanistic origin of the catalytic properties of these materials which may be of future use to develop a family of electrocatalysts based on doped MoS2.
Introduction
Molybdenum disulfide (MoS2) is a 2-dimensional layered transition metal dichalcogenide with a tuneable bandgap, room temperature stability in ambient conditions, and natural abundance.1,2 It is most commonly found in the semiconducting 2H–MoS2 phase and used as a lubricant,3 desulfurization catalyst,4 in gas sensors, or as a channel material for field effect transistors.5,6 However, for applications in batteries,7 supercapacitors,8 electrocatalysts9 and liquid sensors,10 a material with a higher conductivity is required, such as the metallic 1T phase or a heavily doped 2H–MoS2.9,10 The 1T-MoS2 phase does not occur naturally and is generally prepared either via liquid exfoliation with lithium intercalation at elevated temperatures (∼100 °C) for two days,11 or using a hydrothermal synthesis process in an autoclave.12 We have recently reported the synthesis of a conducting, heavily doped 2H–MoS2 phase (c-MoS2) using a liquid exfoliation procedure with dilute aqueous hydrogen peroxide. Doping with hydrogen molybdenum bronze and sub-stoichiometric MoO3−y was identified as the origin of the improved conductivity of the 2H–MoS2 phase while preserving its crystal structure.10 Even though liquid exfoliation methods reduce cost and eliminate safety hazards such as metallic lithium, they commonly utilize toxic solvents such as N-methyl-2-pyrrolidone (NMP).13 Pure water would be a green choice as a dispersion solvent to exfoliate 2H–MoS2, but most two-dimensional material surfaces (including MoS2) are hydrophobic in nature,14,15 requiring an added surfactant.16 Kim et al.15 demonstrated direct exfoliation of semiconducting MoS2 from bulk in pure water by controlling the sonication temperature, but did not report the conductivity of their materials, and accordingly did not optimize their process for conductivity.Over the past few years, MoS2 has garnered attention as an electrocatalyst for the production of renewable energy to help combat climate change. Hydrogen is proposed as a potential alternative energy carrier because it has a high energy density and is easily mass-produced, e.g. via electrolytic water splitting.9 Noble metals such as platinum are the best electrocatalysts currently known for the hydrogen evolution reaction (HER),17–19 but their rarity and cost are impediments to scale-up and motivate the search for alternative catalysts. MoS2 is a promising catalyst candidate due to its high density of active sites in the basal plane or at edge sites and high stability in acidic medium (depending on the exfoliation and synthesis process).19,20 The poor electrical conductivity of 2H–MoS2 is an impediment, however, which is why composites with carbon-based materials such as carbon nanotubes, graphene or graphitic materials have been explored.21–23 The metallic 1T phase has also shown promise for this application but suffers from a lack of stability.24–26 The use of heavily doped 2H–MoS2 phases as HER electrocatalysts remains to be explored.
Here we demonstrate a safe and efficient way to prepare few-layer, nanometer thick c-MoS2 material in pure water at room temperature. The c-MoS2 flakes were first exfoliated at elevated sonication temperatures (60 °C) followed by room temperature sonication. The samples were characterized by conductivity and Hall measurements, Raman spectroscopy and X-ray photoelectron spectroscopy (XPS). Sonication of water at elevated temperature produces small amounts of hydrogen peroxide (H2O2)27 which immediately reacts with MoS2 to form small amounts of sub-stoichiometric MoO3−y which as a dopant is mainly responsible for increasing the conductivity of MoS2.10 We further evaluate the performance of these newly prepared c-MoS2 films as electrocatalysts for HER. We have investigated the active sites for hydrogen evolution in c-MoS2 and established a correlation between the active sites and HER performances of c-MoS2. Our study helps to understand the mechanism of a simple way of preparing doped conductive MoS2 (Fig. 1).
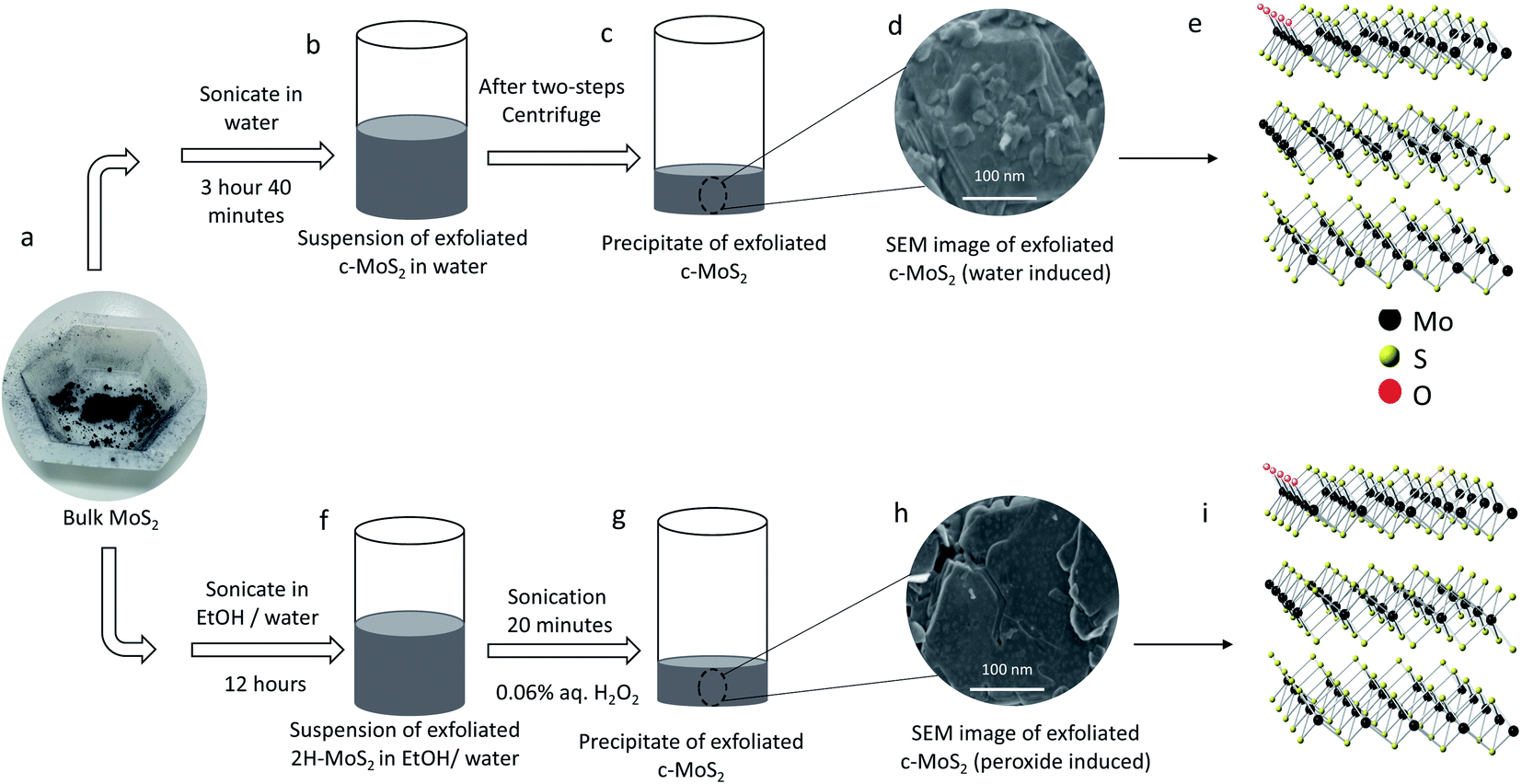 | ||
| Fig. 1 Schematic representation of two different ways of exfoliating conductive MoS2. (a) Bulk MoS2 powder is the starting material for both exfoliation processes. Steps are (b) suspension of water exfoliated conductive MoS2 in water after 3 hours 40 minutes sonication of bulk MoS2; (c) precipitate of conductive MoS2 after two stages of centrifugation; (d) SEM image of water exfoliated conductive MoS2 (Scale bar is 100 nm); (e) structure of water exfoliated conductive MoS2; (f) suspension of semiconducting 2H–MoS2 in an ethanol/water mixture after 12 hours sonication with bulk MoS2; (g) precipitate of 2H–MoS2 after two stages of centrifugation, followed by washing the precipitate with water, adding aqueous 0.06% H2O2 and sonicating for 20 minutes; (h) SEM image of peroxide exfoliated conductive MoS2 (Scale bar is 100 nm); (i) structure of peroxide exfoliated conductive MoS2. | ||
Materials and methods
Materials
Bulk 2H–MoS2 powder (∼6 μm to max. 40 μm, product number 69860, batch number WXBD2352V) and single-walled carbon nanotubes (SWCNTs, 0.78 nm average diameter, product number 773735, batch number MKCJ7287) were purchased from Sigma Aldrich and used without further purification. Ultrapure type I water (18.2 MΩ cm) from a Millipore Simplicity® water purification system was used for all experiments. All organic solvents were HPLC grade and used without further purification. A bath sonicator (Elmasonic P60H ultrasonic cleaner) was used for sonication and an Eppendorf MiniSpin Plus Microcentrifuge was used for centrifugation.Exfoliation of conductive MoS2 (c-MoS2) in hot water
40 mg MoS2 powder were sonicated (37 kHz, 100% power, sweep mode) in 15 mL pure water for 3 hours at 60 °C followed by 40 minutes sonication at 30 °C. The temperature during sonication was controlled using the built-in thermostat and heater of the sonicator. A cooling coil running with tap water was immersed into the sonicator bath for enhanced cooling. The optimized centrifugation process in 2 mL vials consisted of a first step at 3500 rpm (820×g) for 8 minutes, the supernatant of which was centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm (6708×g) for 15 minutes. The supernatant from the second step was discarded using a glass pipette and the precipitate was collected for further use.
000 rpm (6708×g) for 15 minutes. The supernatant from the second step was discarded using a glass pipette and the precipitate was collected for further use.
Exfoliation of semiconducting 2H–MoS2
2H–MoS2 was exfoliated from bulk powder using 45% (v/v) ethanol in water via sonication (80 kHz frequency, 100% power and sweep mode) for 12 hours at 30 °C (temperature control as above).28 The optimized centrifugation process consisted of a first step at 3500 rpm (820×g) for 15 minutes, the supernatant of which was centrifuged at 4500 rpm (1700×g) for 3 minutes, resulting in a grey precipitate of 2H–MoS2 that was further washed with water and the supernatant discarded.Exfoliation of conductive MoS2 (c-MoS2) in very dilute aqueous H2O2
c-MoS2 was exfoliated from bulk MoS2 in dilute aqueous H2O2 using a previously reported procedure.10 Briefly, semiconducting 2H–MoS2 was exfoliated and the precipitate was then washed with water. Aqueous H2O2 (600 ppm by volume) was added to the precipitate of 2H–MoS2 and sonicated (37 kHz, 100% power, sweep mode) for 20 minutes at 30 °C (temperature control as above). The suspension was then centrifuged at 3500 rpm (820×g) for 8 minutes. The resulting supernatant was centrifuged at 10000 rpm (6708×g) for 15 minutes. The supernatant from the second step was discarded by aspiration and the precipitate was collected for further use.
UV-visible absorption spectroscopy
UV-visible absorption spectra of pure water and samples mixed with ethanol or MoS2 and sonicated under different conditions were recorded using an Orion Aquamate 8000 spectrophotometer.Scanning electron microscopy and transmission electron microscopy
High resolution images were obtained on a JEOL JSM-7000F scanning electron microscope (SEM) at 3 kV. Low resolution images were obtained on a TESCAN VEGA-II LSU SEM at 20 kV. A Talos 200X transmission electron microscope (TEM) was used to obtain images at 300 kV.Optical microscopy
The thickness of the films was measured on a Bruker Alicona Infinite FocusG5 plus 3D optical measurement system using a 10× objective for height measurements (100 nm vertical resolution).X-ray diffraction
The sample structure was analysed by XRD using a Bruker D8 Discover instrument with Cu Kα radiation having a wavelength of 0.154 nm.Raman spectroscopy
A Renishaw inVia Raman spectrometer was used over a range of 100–3000 cm−1, with a spectral resolution of 2 cm−1, using a 20× objective in backscattering configuration. Spectra were obtained from three different spots of each sample using a fully focused 633 nm laser on a spot size of about 50 μm limited to 1% of laser power to avoid sample damage.X-ray photoelectron spectroscopy
The XPS analyses were carried out with a Kratos AXIS Supra X-ray photoelectron spectrometer using a monochromatic Al K(alpha) source (15 mA, 15 kV). XPS can detect all elements except hydrogen and helium, probes the surface of the sample to a depth of 7–10 nm, and has detection limits ranging from 0.1–0.5 at% depending on the element. The instrument work function was calibrated to give a binding energy (BE) of 83.96 eV for the Au 4f7/2 line for metallic gold and the spectrometer dispersion was adjusted to give a BE of 932.62 eV for the Cu 2p3/2 line of metallic copper. The Kratos charge neutralizer system was used on all specimens. Survey scan analyses were carried out with an analysis area of 300 × 700 μm2 and a pass energy of 160 eV. High resolution analyses were carried out with an analysis area of 300 × 700 μm2 and a pass energy of 20 eV. Spectra have been charging corrected to the main line of the carbon 1s spectrum (adventitious carbon) set to 284.8 eV. Spectra were analysed using CasaXPS software. Survey scans and high-resolution spectra of C 1s, O 1s, S 2p and Mo 3d were recorded and analyzed of all doped conductive MoS2 and 2H–MoS2 including hydrogen evolution reaction. 2H–MoS2 was used as a reference for comparison.Device fabrication for bulk resistivity
Bulk resistivity was measured in a four-probe geometry. A 500 nm thick silicon dioxide (SiO2) layer was grown by thermally oxidising silicon wafers (Virginia Semiconductor, orientation 〈111〉±0.1°, boron doped, resistivity: 0.04–0.4 Ω cm) for electrical insulation. The oxidised wafers were cut into pieces of 1 × 1 cm2 using a dicing saw and cleaned first using first acetone, then methanol and DI water and dried with N2 gas. Cr (20 nm)/Au (200 nm) 3 × 3 mm2 pads were sputtered onto the four corners of the oxidised wafers to lower the contact resistance. To define the area for MoS2 deposition, a Kapton mask was used in the centre of the substrate (7 × 7 mm2). Water exfoliated c-MoS2 material was first suspended in a 95% (v/v) ethanol and water mixture and airbrushed onto the masked area as a uniform film using a NEO for Iwata CN Gravity Feed Dual Action Brush #N4500 with 20 psi of N2. The sample was kept at 80 °C during airbrushing to facilitate solvent evaporation. The mask was removed once the film dried. Two replicates were fabricated of each device to ensure reproducibility. Bulk resistivity and Hall mobility of the films were measured on a Nanometrics HL 5500PC Hall effect measurement system.Oxygen plasma treatment
Oxygen plasma treatment on doped MoS2 samples was done in a Harrick plasma PDC-001-HP system with a 45 W power supply at a chamber pressure of 630 mTorr O2.Preparation of SWCNT suspension
2 mg of SWCNTs were suspended in 15 mL of methanol by sonicating for 6 hours, whereas 1 mg conductive MoS2 was added to 0.1 mL of water to prepare the MoS2 suspension. Then the supernatants were mixed in a 1.25:1 ratio by suspension volume (1:1 by suspension weight) corresponding to a 1:75 ratio (SWCNT: MoS2) by material weight.
Electrochemistry
All electrochemical measurements were performed with a three-electrode configuration in a 0.5 M H2SO4 electrolyte on an EC301 electrochemical workstation (Stanford Research Systems; following the American polarity convention of the cathodic current having a positive sign). The electrolyte was purged with dry N2 gas for at least for 15 minutes to remove any dissolved O2 prior to all electrochemical measurements. Typically, 80 μL of MoS2 were drop-cast from aqueous suspension onto a graphite electrode with a working area of 1.13 cm2 (outline was defined by a coat of hot glue). Linear sweep voltammetry (LSV) (+0 V to −1.1 V potential range, binning rate 500 ms, and sampling rate 1024 μs) with a sweep rate of 2 mV s−1 was conducted in 0.5 M H2SO4 using an Ag/AgCl reference electrode, a graphite rod counter electrode, and a graphite electrode (pencil was drawn on the graphite electrode to improve adhesion) coated with MoS2 catalyst as a working electrode. All potentials are reported relative to the reversible hydrogen electrode (RHE). Cyclic voltammetry was carried out using the same three electrode set up after HER using +0.5 V to −1.1 V potential window with 15 mV s−1 scan rate, binning rate 500 ms, and sampling rate 1024 μs. The working electrodes for electrochemical surface area calculation and the chronoamperometry experiments were fabricated in a square geometry on glass slides to be able to carry out XPS, Raman and SEM characterization. Hence, a lower conductivity is observed in all cases compared to the initial electrochemical performances on graphite rod substrates.Results and discussion
Conductivity and morphology of MoS2 in water exfoliation
The conductivity of hot water exfoliated MoS2 was determined using samples that were airbrushed onto a silicon dioxide (SiO2) substrate with four gold contacts to measure the bulk resistivity (Fig. 2a). The measured bulk resistivity (28 Ω cm in a 10 μm thick film) of hot water exfoliated c-MoS2 is significantly lower than that of 2H–MoS2. SEM images (Fig. 2b) show the distribution of multilayer water exfoliated c-MoS2 flakes in the film deposited on the SiO2 substrate. Even though hot water exfoliated MoS2 is more conductive than 2H–MoS2, it is less conductive than peroxide exfoliated MoS2.10 The conductivity difference between water and peroxide exfoliated MoS2 (ref. 10) can at least in part be explained by comparing the surface morphology of both samples (Fig. 2). The hot water exfoliation process starts directly from bulk MoS2, resulting in a lower degree of exfoliation (compare Fig. 2c and d) and a resulting poor distribution of the c-MoS2 flakes in the deposited films the c-MoS2 flakes (Fig. 2b for SEM and TEM images showing film distribution of bulk and water exfoliated MoS2, Fig. 2c and d) compared to the homogenously distributed peroxide exfoliated c-MoS2 flakes10 which were prepared from ethanol/water exfoliated 2H–MoS2. The conductivity of MoS2 was also not improved by water sonication from pre-exfoliated 2H–MoS2 films, due to residual ethanol preventing the in situ formation of H2O2. Nevertheless, both hot water exfoliated (28 Ω cm) and peroxide exfoliated c-MoS2 (0.42 Ω cm) samples are several orders of magnitude more conductive than 2H–MoS2 (77 kΩ cm). Water exfoliation is therefore a simple and reliable way of preparing conductive MoS2. An important objective in this work is sustainability. Both H2O2 and water exfoliated MoS2 showed good conductivities compared to 2H–MoS2. To get conductive MoS2 in 4 hours using water as a solvent is a simpler, cheaper and safer way of preparing conductive MoS2. Furthermore, handling and storage of H2O2 are challenging especially on a larger scale due to its reactivity and facile decomposition. | ||
| Fig. 2 Surface morphology of MoS2 samples. (a) Water exfoliated c-MoS2 (grey area ∼7 × 7 mm2) on SiO2 substrate (1 × 1 cm2) with Au contacts, and (b) SEM image of water exfoliated c-MoS2. The scale bar is 1 μm; (c) TEM image of bulk MoS2; and (d) TEM image of water exfoliated conductive MoS2. Scale bar for both TEM images is 50 nm. | ||
It is important to follow the hot water sonication step with a shorter sonication step at lower temperatures. For a MoS2 sample airbrushed directly after sonication in hot water without the second (cold sonication) step, the bulk resistivity was found to be 27 kΩ cm in a 6 μm thick film, demonstrating the need for the cold sonication step in making MoS2 sufficiently conductive. We explored hot sonication at lower temperatures, e.g. 50 °C, but the conductivity of the resulting material drops off significantly (Fig. S1†). Positive values (150 and 350 cm2 V−1 s−1) were observed for the Hall mobilities at room temperature in both cases, indicating that holes are the majority charge carriers (p-doping).29
Origins of the conductivity
To clarify the nature of the conducting phase in the hot water exfoliated c-MoS2, XRD was carried out on two samples: (a) hot water sonicated conductive MoS2, and (b) hot water sonication followed by cold water sonicated conductive MoS2 (Fig. S2†). A broad peak (002) at 2θ ∼16.7° was found for both samples which is close to the value of 2H–MoS2 thus implying that no phase change had taken place, but rather that the samples were heavily doped in agreement with the Hall measurement data.30 Further, the absence of a (001) peak at 2θ ∼7.3° rules out the metallic 1T phase of MoS2. This is also supported by Raman data and XPS analysis. None of the characteristic Raman peaks were observed at 156, 226 and 333 cm−1 (Fig. S3†), confirming the absence of the 1T phase in our samples. Finally, the XPS binding energies of Mo 3d5/2, Mo 3d3/2 (Fig. 3a, c and d), S 2p3/2 and S 2p 1/2 (Fig. S4†) were identical between the 2H–MoS2 and the hot water exfoliated c-MoS2 samples, further corroborating that our conductive samples do not contain the metallic 1T-MoS2 phase.31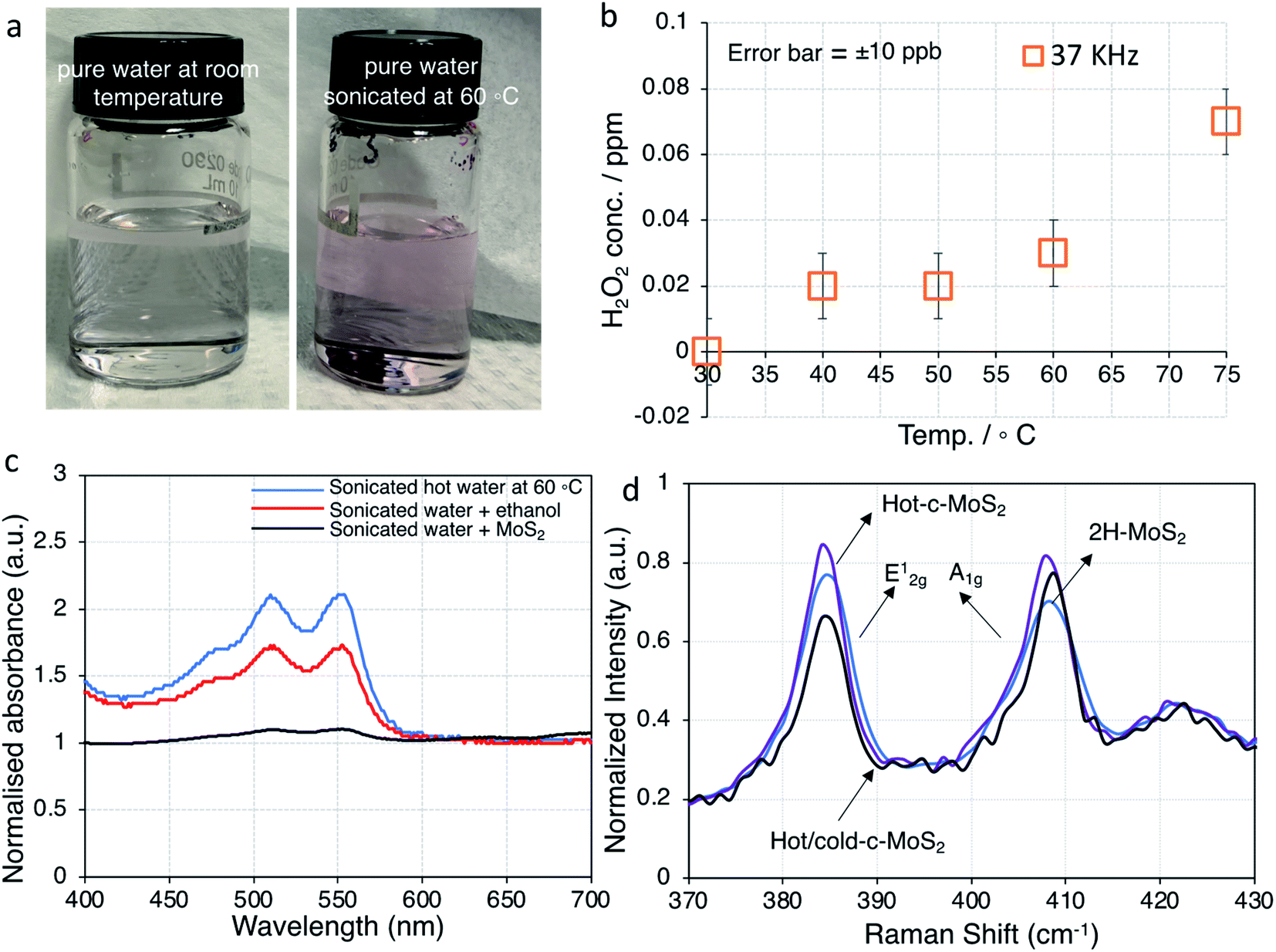 | ||
| Fig. 3 Temperature dependent H2O2 formation in water and properties of MoS2. (a) photographic images of H2O2 formation in pure water at room temperature vs. sonicated hot water at 60 °C based on the colorimetric experiment; (b) amount of H2O2 formation as a function of temperature after sonication for 220 minutes. The error bars are given based on the display resolution of the instrument. (c) UV-visible spectra of samples sonicated at 37 kHz for 220 minutes; (d) Raman spectra of unsonicated, hot water sonicated, and hot & cold water sonicated 2H–MoS2 samples. Spectra are normalized to the ∼466 cm−1 peak. | ||
The electronic properties of MoS2 can be greatly influenced by sulfur vacancies. The sulfur to molybdenum atomic ratios in the samples is expected to be well below the ideal value of 2:1 due to defects in the starting material and sonication damage. Analysis of the S2−/Mo4+ ratios from high resolution XPS data (Table 1) demonstrates that there are no significant changes in the ratio from the bulk 2H phase to the various samples with higher conductivity, therefore the conductivity difference cannot be explained by the introduction of additional sulfur defects.
| Samples | S2−/Mo4+ | Mo4+/Mo | Mo5+/Mo | Mo6+/Mo |
|---|---|---|---|---|
| Semiconducting 2H–MoS2 | 1.31 | 0.670 | 0.110 | 0.218 |
| Water exfoliated (hot + cold sonication) c-MoS2 | 1.30 | 0.764 | 0.055 | 0.180 |
| Water exfoliated (hot sonication only) c-MoS2 | 1.25 | 0.925 | 0.030 | 0.044 |
| Peroxide exfoliated c-MoS2 | 1.29 | 0.805 | 0.060 | 0.131 |
A potential explanation for the conductivity can be derived from our previous finding that the treatment of 2H–MoS2 with very dilute aqueous H2O2 also leads to the formation of a conductive phase (due to doping with hydrogen molybdenum bronze and sub-stoichiometric molybdenum oxide MoO3−y).10 Sonication of water is known to produce small quantities of H2O2in situ, which might react to increase of conductivity of hot water exfoliated c-MoS2 films.27,32,33 The temperature dependence and magnitude of this effect under our conditions therefore needs to be established.
The varying amounts of H2O2 formed during sonication at different temperatures were quantified colorimetrically for a series of pure DI water samples after sonication for 220 minutes without added MoS2 (Fig. 3b and S5†). Since our ultrasonic bath is capable of operating at either of two frequencies (37 kHz and 80 kHz) for the purpose of tuning sample damage according to application, we conducted these experiments at both frequencies. Even though some differences were observed in the generated H2O2 concentrations (Fig. 3b and S5†), all following work is carried out at 37 kHz, which is more commonly found in basic sonicator devices.
It was observed that up to 0.07 ppm H2O2 was formed at 75 °C sonication temperature (37 kHz sonication frequency), and correspondingly less at lower sonication temperatures (Fig. 3a, b and S4†). While 75 °C was the maximum temperature we could achieve, running our ultrasonic bath at 60 °C was more reliable, which is why that temperature was chosen for our further work. The UV-visible spectra of the same samples are dominated by peaks with maxima at 511 nm and 555 nm for pure water samples after sonication at 60 °C. Those peaks decreased in samples with added MoS2, indicating that during the sonication process MoS2 completely reacted with any H2O2 that would have formed during sonication. Ultrasonic formation of H2O2 involves radical intermediates such as ˙OH, which can be quenched by alcohols.27 Accordingly, the spectral features of H2O2 were suppressed in water samples with added ethanol (Fig. 3c). Since the normal exfoliation process for 2H–MoS2 involves sonication in an ethanol/water mixture, peroxide formation is suppressed in that case, and traces of ethanol in the precipitate are sufficient to prevent peroxide-induced doping during a subsequent hot water sonication step. Even for subsequent explicit treatment with peroxide, a thorough washing step is required in order to yield a conducting material.10 We therefore conclude that the c-MoS2 phases from hot water sonication and from sonication in very dilute aqueous H2O2 are formed via the same mechanism. Both procedures result in the formation of small amounts of sub-stoichiometric MoO3−y and hydrogen molybdenum bronze that eventually make the material conductive. This is further confirmed by XPS analysis.
As resolved in the high resolution XPS data in Fig. 4, a Mo 3d5/2 binding energy of 229.7 eV represents Mo4+ in 2H–MoS2, and a Mo 3d5/2 binding energy of 233.0 eV is characteristic of Mo6+ such as in molybdenum trioxide (MoO3).34,35 Additionally, Mo 3d5/2 peaks at 232.1 eV attributable to the formation of Mo5+ were observed in all samples. The presence of small amounts of Mo in the +5 and +6 oxidation states in all samples is consistent with the known propensity of MoS2 to undergo edge oxidation compared to its resistance to attacks on its basal plane in water. XPS results further indicates (Fig. 4a and c) oxidation of bulk-MoS2 (Fig. 5) due to the interaction with H2O2 which formed during water sonication at elevated temperatures. H2O2 can decompose and form atomic hydrogen, or hydroxyl radicals32 which may react with edge Mo without impacting the bulk. This interaction with edge MoS2 can proceed through several intermediate MoOxSy species where Mo may be in the +4, +5, or +6 oxidation states. This leads to the formation of hydrogen molybdenum bronze HxMoO3 and sub-stoichiometric MoO3−y, where an increase in y is correlated with a decrease of the electronic bandgap of MoS2, making the material more conductive.36 The oxidation state of molybdenum in the bronze is +5, as observed by XPS. HxMoO3 is significantly more conductive than 2H–MoS2 and MoO3.37,38 Hence, we propose the conductivity of the hot and cold water exfoliated c-MoS2 and hot water exfoliated c-MoS2 samples to be due to the presence of HxMoO3 and MoO2. The ratios of Mo5+/Mo and Mo6+/Mo are higher for samples that were both hot and cold sonicated (Fig. 4b), compared to samples that underwent only hot sonication (Table 1, Fig. 4a). The percentages of hydrogen molybdenum bronze and sub-stoichiometric oxide in hot water exfoliated c-MoS2 are found to be lower when the room temperature sonication step was omitted. This is also supported by our conductivity data, as hot/cold sonicated c-MoS2 samples are more conductive than only hot water sonicated c-MoS2. In addition to the increase of the Mo5+/Mo ratio, the ratios of Mo6+/Mo and Mo4+/Mo are found to decrease from hot/cold water exfoliated c-MoS2 to only hot water exfoliated c-MoS2 upon exposure to H2O (Table 1). The ratios of Mo5+ to Mo in peroxide sonicated MoS2 samples are higher compared to water exfoliated samples, whereas Mo4+ to Mo and Mo6+ to Mo are higher in hot water and hot/cold water exfoliated samples (Table 1, Fig. 4b). The procedure of fabricating peroxide exfoliated c-MoS2 is slightly different from that of hot water exfoliation, as the exfoliation of c-MoS2 in peroxide was preceded by an exfoliating step in an ethanol/water mixture, followed by reaction with 0.06% aq. H2O2. This results in the direct interaction of hydroxyl radicals or hydrogen atoms with the edges of the exfoliated material, and the formation of more bronze and sub-stoichiometric oxides.
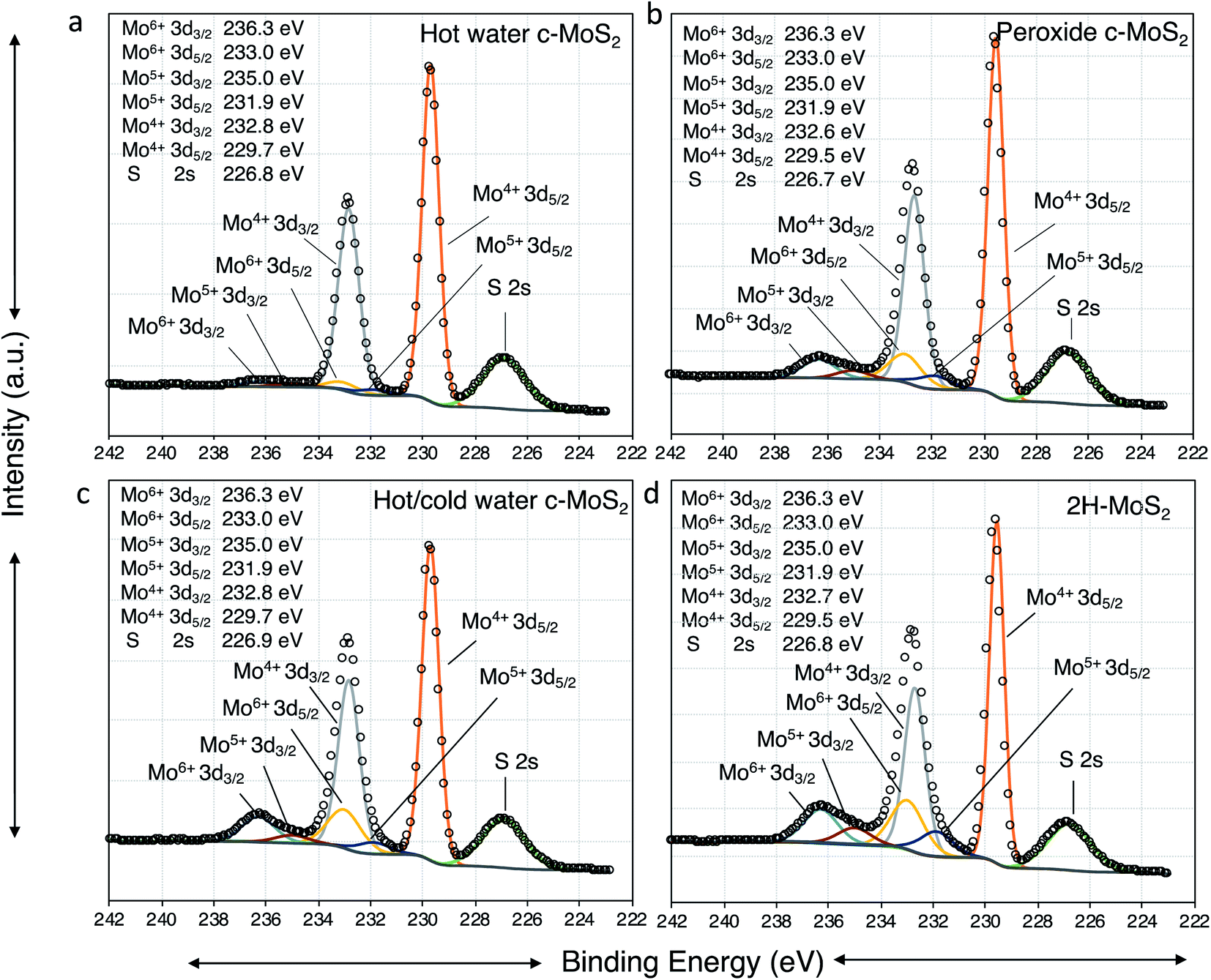 | ||
| Fig. 4 XPS high resolution spectra of Mo 3d for (a) hot water exfoliated conductive MoS2; (b) peroxide exfoliated conductive MoS2; (c) hot and cold water exfoliated conductive MoS2; and (d) exfoliated semiconducting 2H–MoS2. | ||
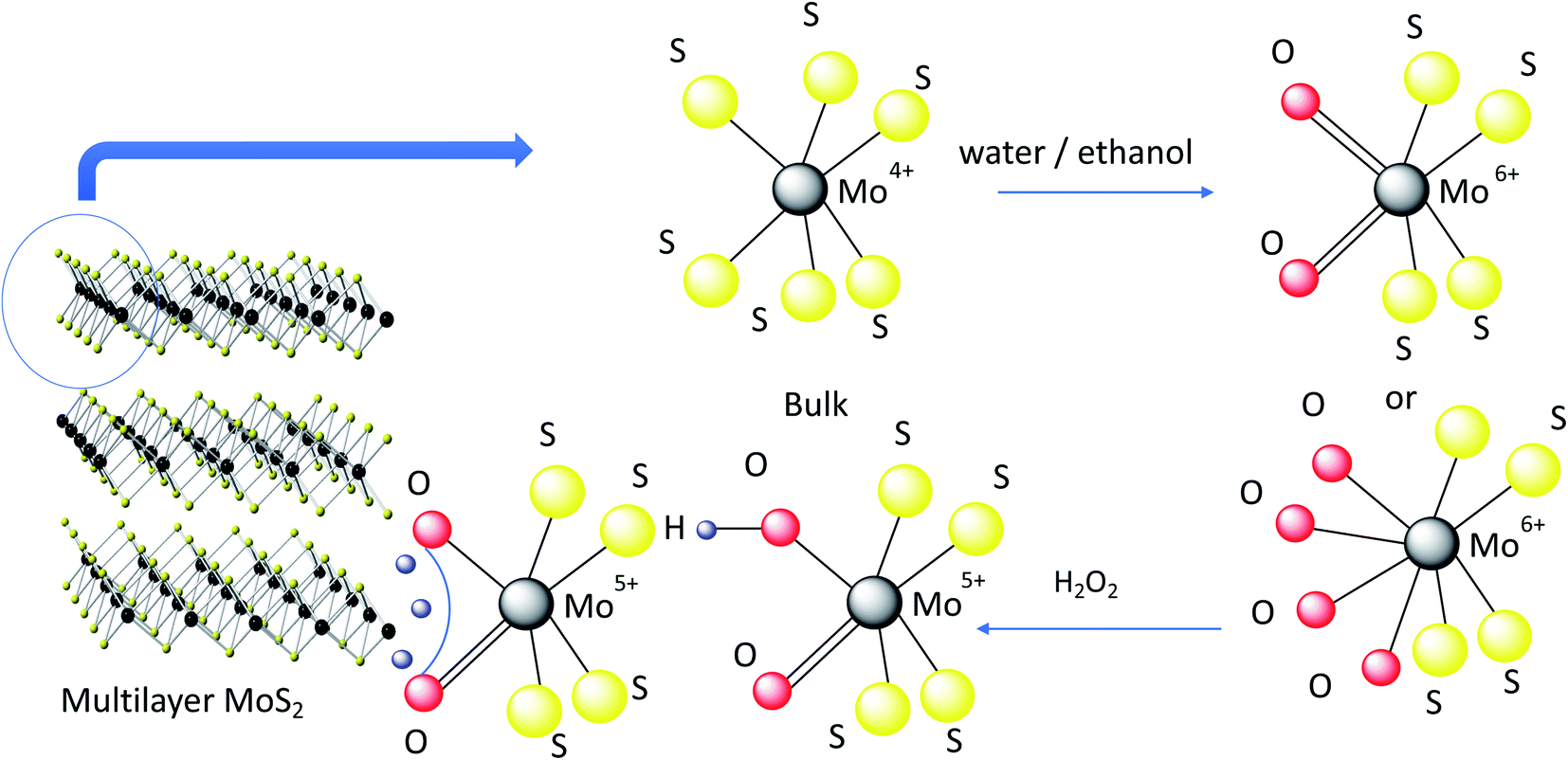 | ||
| Fig. 5 Schematic representation of the mechanism of formation of conductive MoS2. | ||
Water exfoliation differs from peroxide exfoliation in two major ways: (a) the starting material is not partially oxidized, and (b) H2O2 is not used directly, but instead formed in situ during the sonication in hot water, hence the maximum concentration of peroxide is lower. Therefore, availability of H2O2 is limited during water sonication and less interaction is expected between in situ formed H2O2 and MoS2 in hot water exfoliated samples compared to the interaction between the directly added H2O2 and MoS2 in peroxide exfoliated samples. This results in a lower conductivity of hot water exfoliated samples compared to peroxide exfoliated c-MoS2. During the hot water exfoliation process, the H2O2 concentration is lower by 4 orders of magnitude (0.07 ppm, Fig. 3) which explains the lower conductivity of hot water sonicated c-MoS2 compared to peroxide exfoliated c-MoS2. Due to the preceding exfoliation step in the ethanol/water mixture, peroxide treated samples are more exfoliated than the hot water sonicated samples, thereby changing the surface to bulk ratio. Since the reaction can only happen at the surface of the flakes, a higher degree of exfoliation will not only improve the connectivity of the flakes in the film, but also increase the number of available sites for doping. On the other hand, 2H–MoS2 has a higher Mo6+/Mo ratio (Table 1) compared to all other samples, indicating MoO3 to be the major product. In pure form, MoO3 is an insulator and does not contribute to the conductivity of the material, which is supported by our conductivity data as well.
Raman spectroscopy was carried out to further probe the bulk structure and properties of water exfoliated MoS2 samples, as XPS only probes the top 10 nm at the surface of the material. The main characteristic peaks for MoS2 are the E12g and A1g peaks at 384 and 408 cm−1 (Fig. 3d).39 A combination of a very small red shift of the E12g mode and a blue shift of the A1g modes (Fig. 3d) are indicative of the formation of multilayer films (consistent with TEM data in Fig. 2). Resonant Raman scattering peaks at 178, 423, 466, 526, 600, and 644 cm−1 are observed as a result of illumination with a 633 nm laser (Fig. S3†).39 Characteristic peaks of MoO2 at 570 cm−1 and 738 cm−1 are vibrational modes, and 230 cm−1 and 492 cm−1 are phonon modes of MoO2.40 Both the hydrogen molybdenum bronze and sub-stoichiometric MoO3−y are unstable intermediate species and can be reduced to MoO2, which is conductive. The presence of MoO2 in a sample should result in a broad Mo4+ feature in the Mo 3d XPS spectrum, much broader than the Mo4+ peak from MoS2. While there is no clear evidence of MoO2 in XPS in any of the samples, a small amount Mo4+ for MoO2 might be obscured by the large Mo4+ signal of MoS2. Hydrogen molybdenum bronze at 204 cm−1 was not observed in any of the samples, instead a broad feature of sub-stoichiometric MoO3−y peak at 780 cm−1 was detected in all the samples. MoO3 is present in all the samples including semiconducting 2H–MoS2 but small features due to MoO2 and MoO3−y are also detected as impurities in the 2H–MoS2 Raman spectra. The most prominent characteristic peak for MoO3 is 820 cm−1, but since MoO3 is an insulator it does not have any effect on bulk or surface conductivity. No oxysulfide peaks (440 cm−1) were detected in the Raman spectra in agreement with the high resolution XPS spectra of S 2p (Fig. S4†). Raman spectroscopy of freshly prepared and three weeks old water exfoliated conductive MoS2 samples did not yield any differences in peak positions or intensities, leading us to conclude that water exfoliated conductive MoS2 can be stable in air for at least 21 days (Fig. S6†).
Hydrogen evolution reaction and identifying the active sites
Different surface properties and morphology of water exfoliated c-MoS2 and 2H–MoS2 can lead to differences in the performance as an electrocatalyst for the hydrogen evolution reaction (HER). Hence, hydrogen production analysis was done using a graphite rod as a working electrode with a three-electrode system in a nitrogen purged 0.5 M H2SO4 electrolyte. A significant catalytic activity difference for hydrogen generation was observed between semiconducting 2H–MoS2, peroxide exfoliated c-MoS2 and water exfoliated c-MoS2 (Fig. 6). The Tafel slopes for water exfoliated c-MoS2 at 204 mV per decade (overpotential of 780 mV) and peroxide exfoliated c-MoS2 at 242 mV per decade (overpotential of 632 mV) were lower than that of semiconducting 2H–MoS2 at 333 mV per decade (overpotential of 773 mV), both at 10 mA cm−2 current density. The Tafel slope is a key parameter to quantify catalytic performance and gather information about the mechanistic pathway of the HER. According to the Tafel equation,| η = alog|current density| + b | (1) |
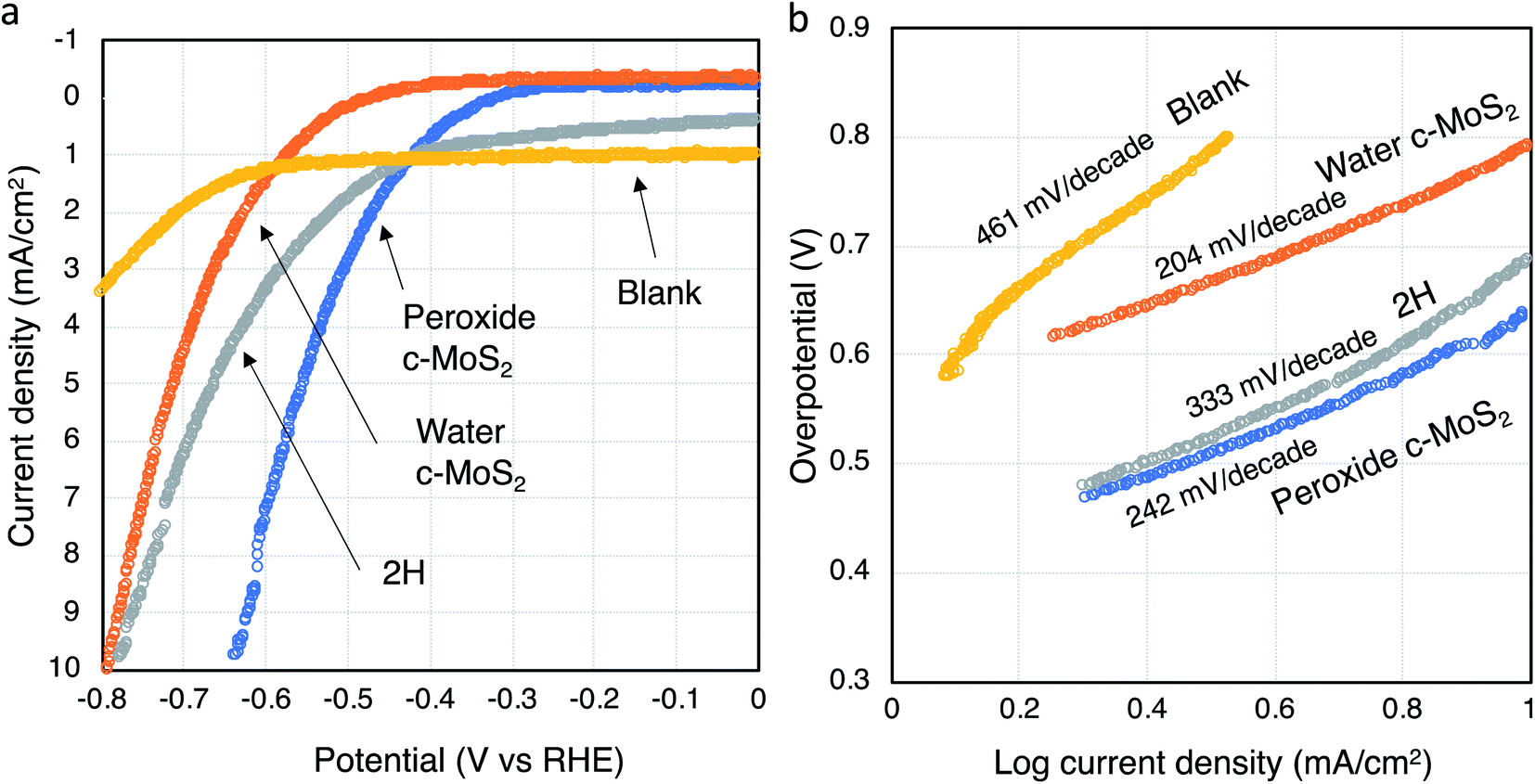 | ||
| Fig. 6 HER activity of MoS2 catalysts. (a) Linear sweep voltammograms of all catalysts, normalized by geometric surface area, and (b) corresponding Tafel plots. An uncoated graphite rod (‘blank’) is used as reference for comparison. | ||
In order to properly scale the HER activity by electrochemical surface area (ECSA), electrochemical double layer capacitances (Cdl) were determined for all samples since they are proportional to the ECSA (Fig. S7†).41,42 Capacitance current densities are measured for all the doped conductive MoS2 materials along with 2H–MoS2 for certain potential windows (0.1 to 0.25 V vs. RHE) at different scan rates and then plotted and linearly fitted. The Cdl were found to vary for all three MoS2 materials, with higher Cdl's corresponding to larger surface areas. Peroxide and water exfoliated MoS2 samples were found to have higher Cdl values (and thus higher surface areas) compared to 2H–MoS2. This result is consistent with our calculated Tafel value being higher for 2H–MoS2 compared to the conductive MoS2 samples doped via the two different pathways.
To further identify the active sites, semiconducting 2H–MoS2, water exfoliated c-MoS2 and peroxide exfoliated c-MoS2 samples were oxidised using oxygen plasma for 1 minute and characterized using XPS (Table 2). Partial oxidation occurred for all samples (Fig. S8†). Linear sweep voltammetry was performed to determine the electrochemical performance and calculate the Tafel slope from the Tafel plot. The Tafel slope for water exfoliated c-MoS2 increased from 204 to 238 mV per decade due to oxidation, whereas the Tafel slope for semiconducting 2H–MoS2 slightly decreased to 278 mV per decade and to 206 mV per decade for peroxide exfoliated c-MoS2 (Fig. S9†). A higher Tafel slope is expected because the MoS2 samples are oxide doped which already limits the active sites. Oxygen plasma oxidation further blocked those limited active sites in the edges due to oxidation as the edge sides of MoS2 are more prone to oxidise, causing a decrease in the rate of hydrogen evolution. It has been reported that oxygen plasma can lead to erosion of the basal plane and create holes in the structure to increase the number of active edge sites at the same time as passivating existing catalytically active edge side.19 Hence, the slight decrease in the Tafel slope for peroxide exfoliated c-MoS2 and semiconducting 2H–MoS2 might be the result of a small net increase in the number of active sites. It is important to note that a defect-free basal plane in 2H–MoS2 would be inert during HER.43
| Sample | Mo4+/Mo | Mo5+/Mo | Mo6+/Mo | Stoichiometric amount of MoO3 |
|---|---|---|---|---|
| 2H–MoS2 | 0.505 | 0.136 | 0.358 | 13.530 |
| Water exfoliated c-MoS2 | 0.656 | 0.063 | 0.273 | 8.793 |
| Peroxide exfoliated c-MoS2 | 0.773 | 0.094 | 0.130 | 8.448 |
In order to separate the impact of conductivity difference between water exfoliated c-MoS2, peroxide exfoliated c-MoS2, and semiconducting 2H–MoS2 from the impact of structural effects on the catalytic activity of the materials, SWCNTs were combined with the respective MoS2 catalyst materials (1:75 ratio by weight as described) for measurement of the electrocatalytic properties. This material ratio did not undergo further optimization for electrocatalytic activity since the sole focus was on elucidating the impact of conductivity of the material on its electrocatalytic properties. SEM images of the CNT and MoS2 mixture are shown in Fig. S10.† SWCNTs by themselves are known to not to be good electrocatalysts for HER.44 The overpotential indeed decreases for water exfoliated c-MoS2 (759 mV), and 2H–MoS2 (755 mV) materials but slightly increased by 84 mV for peroxide exfoliated c-MoS2 due to the addition of SWCNT (Table S1†). However, the Tafel slopes (Table S1†) were higher (304 mV per decade) in water exfoliated c-MoS2 and peroxide exfoliated c-MoS2 (282 mV per decade) but slightly lower in 2H–MoS2 (323 mV per decade) with added SWCNTs than without, further indicating that the active sites are present in the edge site and no other active sites are present to increase rate of the hydrogen production (otherwise a decrease in the Tafel slope would have been observed). Adding SWCNTs reduces the active site density by diluting the active material, decreasing the rate of hydrogen generation.
Hydrogen evolution reaction mechanism and stability
The HER activity is directly related to the availability of active sites on the surface of the catalyst (Fig. 7). In MoS2, the sulfur atoms in edge sites are the major active sites whereas the basal plane of 2H–MoS2 is inert in the absence of defects. Sulfur vacancies may also play a role in the evolution of hydrogen gas. In c-MoS2, the partially oxidation of the material may lead to the blocking of the reactive S sites, as reflected in the Tafel values (Fig. 5b) being higher than for previously reported MoS2-based catalysts.44,45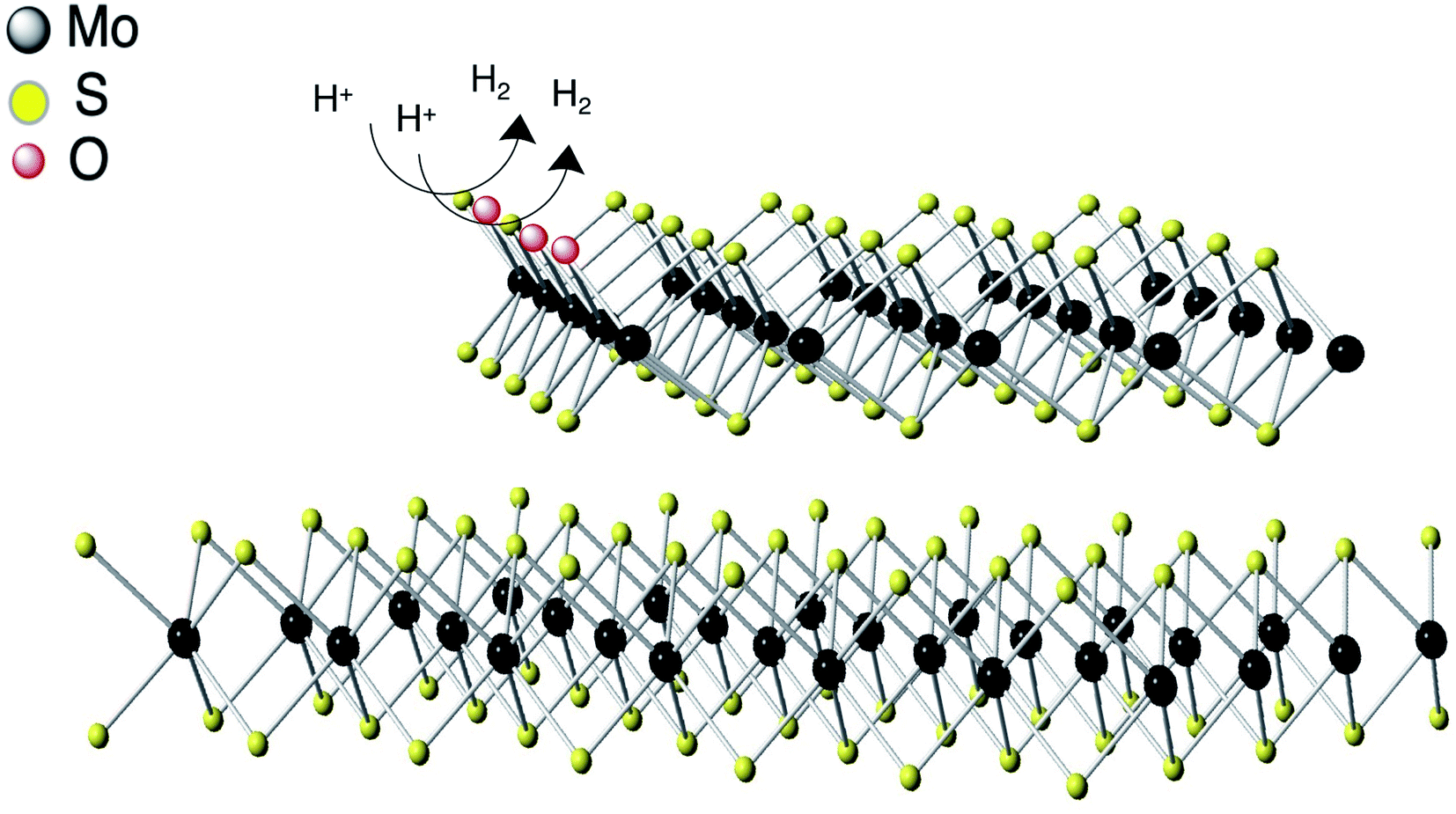 | ||
| Fig. 7 Schematic representation of HER mechanism of conductive MoS2. Hydrogen evolution reaction process at the edges of conductive MoS2. | ||
XPS analysis of the catalysts after operation gives further insight into the fate of the active sites. According to the high resolution Mo 3d and S 2p spectra (Fig. S11†), a higher S2−/Mo4+ ratio was observed compared to the pristine material (Table 3) after HER. The increase in the ratio may have two possible explanations: either the S vacancies were repaired with added sulfur atoms during the electrocatalytic process, or the Mo composition may have changed during the HER process. From the XPS data, we found that total the S atomic percentage was not changing significantly compared to the pristine material during HER, but the total Mo atomic percentage was lower than its initial amount (Table 3). This can be explained by the sub-stoichiometric oxide dissolving into the electrolyte throughout the electrocatalyst process, as reflected in both the S2−/Mo4+ ratio and the overpotential of the catalysts. This also explains why the overpotentials of water exfoliated c-MoS2 and peroxide exfoliated c-MoS2 are the range of 630–780 mV (Fig. 6a) as the sub-stoichiometric oxide was mainly responsible for the conductivity of those two materials. On the other hand, the Tafel slopes for all the conductive MoS2 catalysts was lower than for semiconducting 2H–MoS2, because the active site accessibility increases as the sub-stoichiometric oxide starts to dissolve. We also observed that the leaching rate of sub oxide MoO3−y into the solution was higher for water exfoliated c-MoS2 compared to peroxide exfoliated c-MoS2 (Table 3), which is also consistent with the electrochemical performance with an overpotential of 750 mV for water exfoliated c-MoS2 compared to 650 mV for peroxide exfoliated c-MoS2. In the case of semiconducting 2H–MoS2, the main contribution was from MoO3 which started dissolving during the HER, resulting in a higher overpotential. High resolution SEM images of the catalysts after HER performance (Fig. S12†) reveal no significant changes in the surface morphologies of water exfoliated c-MoS2, peroxide exfoliated c-MoS2 and 2H–MoS2, implying that the production of hydrogen gas did not have any effect on the film surfaces.
| Samples | Total Mo% (before HER) | Total Mo% (after HER) | Total S% (before HER) | Total S% (after HER) | S2−/Mo4+ (before HER) | S2−/Mo4+ (after HER) |
|---|---|---|---|---|---|---|
| 2H–MoS2 | 14.80 | 7.90 | 15.60 | 14.10 | 1.31 | 1.64 |
| Water exfoliated c-MoS2 | 13.80 | 8.90 | 16.90 | 16.20 | 1.30 | 1.71 |
| Peroxide exfoliated c-MoS2 | 16.60 | 14.80 | 19.40 | 21.50 | 1.29 | 1.42 |
Stability of all the catalysts was further verified in the acidic electrolyte solution. We measured the electrocatalytic activity using linear sweep voltammetry (Table S2†) after 250 cycles of cyclic voltammetry (Fig. S13†). We found a decrease in the Tafel slope after 250 cycles, while the overpotential gradually increases over 250 cycles for water exfoliated c-MoS2 and peroxide exfoliated c-MoS2. This is consistent with the dissolving sub oxide during the HER process making more sites available for the evolution of hydrogen gas. However, we did not observe any significant changes in the Tafel slope or the overpotential for the semiconducting 2H–MoS2 after 250 cycles, confirming that 2H–MoS2 was less catalytically active compared to the other two catalysts. We also verified the catalytic stability of water exfoliated c-MoS2 and peroxide exfoliated c-MoS2 up to 950 cycles (183 mV per decade at 800 mV overpotential for water exfoliated c-MoS2 and 160 mV per decade at 653 mV overpotential for peroxide exfoliated c-MoS2 after 950 cycles) where slight decreases in the Tafel slope were observed after each set of 250 cycles with a similar change in the overpotential. This further confirms that the sub oxides were dissolved during the HER process, but after 250 cycles the rate of sub oxide leaching gradually decreases. Even though this resulted in a lower Tafel slope for the HER, stability was an issue since the conductivity decreases at the same time, resulting in a higher overpotential. We also performed long-term measurements on all catalysts using chronoamperometry at a fixed overpotential for the duration of 14 hours (Fig. S14†). The current density for all materials is lower than during the previous HER performance tests due to the use of a different geometry that facilitates spectroscopic and microscopic analysis at the end of the run. The current continues to increase over 14 hours for peroxide and water exfoliated conductive MoS2 because of the gradual increase in the number of active sites (Tables 3 and S3†). In the case of 2H–MoS2, the initial current is higher, but within three hours of the measurement, the current started decreasing due to decreasing availability of active sites for HER. This further supports that 2H–MoS2 is less catalytically active for HER compared to the two doped conductive materials. We further characterized those materials with Raman and XPS after 14 hours in the chronoamperometry experiment. We did not observe any changes of Raman peak position and intensity compared to the pristine materials prior to undergoing chronoamperometry (Fig. S15†). However, according to the high resolution XPS data, the S2−/Mo4+ ratio increased for all doped materials and also for 2H–MoS2 after 14 hours of electrochemical testing (Table S3†). This further supports our hypothesis that the suboxide gradually dissolves in the electrolyte throughout the electrocatalytic process. No differences in surface morphology were observed after 14 hours stability test for all the samples using high resolution SEM (Fig. S16†). A comparison between our synthesised catalyst with other reported MoS2 catalyst shows that even though our oxide doped water and peroxide exfoliated conductive MoS2 have competitive Tafel values, their overpotentials are higher that what has been reported for other MoS2 electrocatalysts.19,46,47 Nevertheless, a correlation between the active sites of oxide doped conductive MoS2 and the rate of hydrogen evolution was established.
Conclusion
We have demonstrated exfoliation of p-doped conductive MoS2 in pure water by controlling the sonication temperature and time. SEM images shown sufficient overlap between the multilayer conductive MoS2 flakes to enable good conductivity of the overall film. Sonication of water leads to in situ formation of trace amounts of H2O2 which interact with MoS2 to form small amounts of hydrogen molybdenum bronze and sub-stoichiometric MoO3−y to significantly dope the bulk 2H–MoS2. Electrocatalytic activity for the hydrogen evolution reaction was demonstrated for conductive MoS2 phases produced by both hot water sonication and sonication in dilute aqueous hydrogen peroxide. An overpotential range of 630–780 mV was observed for water and peroxide exfoliated c-MoS2 with Tafel slopes of 204 and 242 mV per decade respectively. The higher Tafel slopes indicate that the active sites are present only at the sheet edges, and accessibility of those edge sites is limited for hydrogen production in all doped MoS2 materials due to suboxide doping. Stability is a concern for the doped conductive MoS2 materials, since a decrease in current density was observed over 950 CV cycles due to dissolution of the sub-oxides. While further work will be required to optimize conducting MoS2 materials as electrocatalysts, the synthesis of these materials can now be accomplished in a scalable, safe, sustainable, and cheap manner, opening up a wide range of applications.Author contributions
D. S. and P. K. conceived and designed the experiments. D. S. conducted all experimental work. V. P. assisted in electrochemical measurements and calculation. D. S. analyzed the experimental data with help from P. K. The results were discussed by all authors. D. S. wrote the manuscript with feedback from P. K., V. P., and P. R. S.Conflicts of interest
D. S., P. R. S., and P. K. have filed for a U.S. patent covering the exfoliation process described in this manuscript.Acknowledgements
We are grateful to Dr Mark Biesinger (Surface Science Western, Canada) for help with XPS; Ali Aliakbari Khoei (McMaster Manufacturing Research Institute, McMaster University) for help with optical microscopy; Prof. Alex Adronov (Chemistry and Chemical Biology, McMaster University) for use of the Raman spectrometer; Dr James F. Britten (McMaster Analytical X-Ray Diffraction Facility, McMaster University) for help with XRD measurement; Dr Carmen Andrei (Canadian Centre for Electron Microscopy (CCEM)-McMaster University) for help with TEM; Chris Butcher (CCEM-McMaster University) for help with high resolution SEM; Md. Ali Akbar (Kruse Lab, McMaster University) for help with preparing SWCNTs; Ramis Arbi (Turak Lab, McMaster University) for help with oxygen plasma treatment; Dr Richa Pandey, Sudarshan Sharma, Jayasree Biswas, Marcia Reid, Shayan Angizi, Maryam Darestani-Farahani, and Johnson Dalmieda (all McMaster University) for fruitful discussions. Electron microscopy was carried out at the CCEM-McMaster University, a national facility supported by NSERC Canada and the Electron Microscopy Facility of the Faculty of Health Science. Sputtering, sheet resistance and Hall measurements were carried out at the Centre for Emerging Device Technologies (CEDT) at McMaster University. The work was financially supported by the Natural Sciences and Engineering Research Council of Canada through the Discovery Grant Program, as well as the Canada First Research Excellence Fund project “Global Water Future”.References
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef.
- D. J. Late, B. Liu, H. S. S. R. Matte, V. P. Dravid and C. N. R. Rao, ACS Nano, 2012, 6, 5635–5641 CrossRef CAS PubMed.
- W. O. Winer, Wear, 1967, 10, 422–452 CrossRef CAS.
- R. V. Mom, J. N. Louwen, J. W. M. Frenken and I. M. N. Groot, Nat. Commun., 2019, 10, 2546 CrossRef.
- D. Kiriya, M. Tosun, P. Zhao, J. S. Kang and A. Javey, J. Am. Chem. Soc., 2014, 136, 7853–7856 CrossRef CAS PubMed.
- D. Sarkar, W. Liu, X. Xie, A. C. Anselmo, S. Mitragotri and K. Banerjee, ACS Nano, 2014, 8, 3992–4003 CrossRef CAS PubMed.
- T. Stephenson, Z. Li, B. Olsen and D. Mitlin, Energy Environ. Sci., 2014, 7, 209–231 RSC.
- L. Cao, S. Yang, W. Gao, Z. Liu, Y. Gong, L. Ma, G. Shi, S. Lei, Y. Zhang, S. Zhang, R. Vajtai and P. M. Ajayan, Small, 2013, 9, 2905–2910 CrossRef CAS.
- D. Saha and P. Kruse, J. Electrochem. Soc., 2020, 167, 126517 CrossRef CAS.
- D. Saha, P. R. Selvaganapathy and P. Kruse, ACS Appl. Nano Mater., 2020, 3, 10864–10877 CrossRef CAS.
- G. Eda, H. Yamaguchi, D. Voiry, T. Fujita, M. Chen and M. Chhowalla, Nano Lett., 2011, 11, 5111–5116 CrossRef CAS PubMed.
- X. Geng, W. Sun, W. Wu, B. Chen, A. Al-Hilo, M. Benamara, H. Zhu, F. Watanabe, J. Cui and T.-p. Chen, Nat. Commun., 2016, 7, 10672 CrossRef CAS PubMed.
- S. P. Ogilvie, M. J. Large, G. Fratta, M. Meloni, R. Canton-Vitoria, N. Tagmatarchis, F. Massuyeau, C. P. Ewels, A. A. K. King and A. B. Dalton, Sci. Rep., 2017, 7, 16706 CrossRef.
- A. Ciesielski and P. Samorì, Chem. Soc. Rev., 2014, 43, 381–398 RSC.
- J. Kim, S. Kwon, D.-H. Cho, B. Kang, H. Kwon, Y. Kim, S. O. Park, G. Y. Jung, E. Shin, W.-G. Kim, H. Lee, G. H. Ryu, M. Choi, T. H. Kim, J. Oh, S. Park, S. K. Kwak, S. W. Yoon, D. Byun, Z. Lee and C. Lee, Nat. Commun., 2015, 6, 8294 CrossRef CAS PubMed.
- K. R. Paton, E. Varrla, C. Backes, R. J. Smith, U. Khan, A. O'Neill, C. Boland, M. Lotya, O. M. Istrate, P. King, T. Higgins, S. Barwich, P. May, P. Puczkarski, I. Ahmed, M. Moebius, H. Pettersson, E. Long, J. Coelho, S. E. O'Brien, E. K. McGuire, B. M. Sanchez, G. S. Duesberg, N. McEvoy, T. J. Pennycook, C. Downing, A. Crossley, V. Nicolosi and J. N. Coleman, Nat. Mater., 2014, 13, 624–630 CrossRef CAS PubMed.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS PubMed.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- G. Ye, Y. Gong, J. Lin, B. Li, Y. He, S. T. Pantelides, W. Zhou, R. Vajtai and P. M. Ajayan, Nano Lett., 2016, 16, 1097–1103 CrossRef CAS PubMed.
- J. Benson, M. Li, S. Wang, P. Wang and P. Papakonstantinou, ACS Appl. Mater. Interfaces, 2015, 7, 14113–14122 CrossRef CAS PubMed.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed.
- H. Huang, W. Huang, Z. Yang, J. Huang, J. Lin, W. Liu and Y. Liu, J. Mater. Chem. A, 2017, 5, 1558–1566 RSC.
- J. Joyner, E. F. Oliveira, H. Yamaguchi, K. Kato, S. Vinod, D. S. Galvao, D. Salpekar, S. Roy, U. Martinez, C. S. Tiwary, S. Ozden and P. M. Ajayan, ACS Appl. Mater. Interfaces, 2020, 12, 12629–12638 CrossRef CAS PubMed.
- Y. Li, L. Wang, S. Zhang, X. Dong, Y. Song, T. Cai and Y. Liu, Catal. Sci. Technol., 2017, 7, 718–724 RSC.
- Q. Tang and D.-e. Jiang, ACS Catal., 2016, 6, 4953–4961 CrossRef CAS.
- D. Voiry, A. Mohite and M. Chhowalla, Chem. Soc. Rev., 2015, 44, 2702–2712 RSC.
- K. R. Moonoosawmy and P. Kruse, J. Am. Chem. Soc., 2010, 132, 1572–1577 CrossRef CAS PubMed.
- K.-G. Zhou, N.-N. Mao, H.-X. Wang, Y. Peng and H.-L. Zhang, Angew. Chem., Int. Ed., 2011, 50, 10839–10842 CrossRef CAS PubMed.
- F. Werner, J. Appl. Phys., 2017, 122, 135306 CrossRef.
- M. Acerce, D. Voiry and M. Chhowalla, Nat. Nanotechnol., 2015, 10, 313–318 CrossRef CAS PubMed.
- N. H. Attanayake, A. C. Thenuwara, A. Patra, Y. V. Aulin, T. M. Tran, H. Chakraborty, E. Borguet, M. L. Klein, J. P. Perdew and D. R. Strongin, ACS Energy Lett., 2018, 3, 7–13 CrossRef CAS.
- P. Riesz and T. Kondo, Free Radicals Biol. Med., 1992, 13, 247–270 CrossRef CAS PubMed.
- S. Ziembowicz, M. Kida and P. Koszelnik, Proceedings, 2018, 2, 188 CrossRef.
- D. O. Scanlon, G. W. Watson, D. J. Payne, G. R. Atkinson, R. G. Egdell and D. S. L. Law, J. Phys. Chem. C, 2010, 114, 4636–4645 CrossRef CAS.
- P. Afanasiev and C. Lorentz, J. Phys. Chem. C, 2019, 123, 7486–7494 CrossRef CAS.
- S. Balendhran, J. Deng, J. Z. Ou, S. Walia, J. Scott, J. Tang, K. L. Wang, M. R. Field, S. Russo, S. Zhuiykov, M. S. Strano, N. Medhekar, S. Sriram, M. Bhaskaran and K. Kalantar-zadeh, Adv. Mater., 2013, 25, 109–114 CrossRef CAS PubMed.
- X. K. Hu, Y. T. Qian, Z. T. Song, J. R. Huang, R. Cao and J. Q. Xiao, Chem. Mater., 2008, 20, 1527–1533 CrossRef CAS.
- J. Z. Ou, J. L. Campbell, D. Yao, W. Wlodarski and K. Kalantar-zadeh, J. Phys. Chem. C, 2011, 115, 10757–10763 CrossRef CAS.
- H. Li, Q. Zhang, C. C. R. Yap, B. K. Tay, T. H. T. Edwin, A. Olivier and D. Baillargeat, Adv. Funct. Mater., 2012, 22, 1385–1390 CrossRef CAS.
- M. Dieterle and G. Mestl, Phys. Chem. Chem. Phys., 2002, 4, 822–826 RSC.
- P. Salarizadeh, M. B. Askari, M. Seifi and S. M. Rozati, J. Electroanal. Chem., 2019, 847, 113198 CrossRef CAS.
- W. Li, Z. Zhang, W. Zhang and S. Zou, ACS Omega, 2017, 2, 5087–5094 CrossRef CAS PubMed.
- Y. Yin, J. Han, Y. Zhang, X. Zhang, P. Xu, Q. Yuan, L. Samad, X. Wang, Y. Wang, Z. Zhang, P. Zhang, X. Cao, B. Song and S. Jin, J. Am. Chem. Soc., 2016, 138, 7965–7972 CrossRef CAS PubMed.
- D. Voiry, M. Salehi, R. Silva, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nano Lett., 2013, 13, 6222–6227 CrossRef CAS PubMed.
- S. Jayabal, G. Saranya, J. Wu, Y. Liu, D. Geng and X. Meng, J. Mater. Chem. A, 2017, 5, 24540–24563 RSC.
- J. Liang, C. Ding, J. Liu, T. Chen, W. Peng, Y. Li, F. Zhang and X. Fan, Nanoscale, 2019, 11, 10992–11000 RSC.
- L. Ma, L. R. L. Ting, V. Molinari, C. Giordano and B. S. Yeo, J. Mater. Chem. A, 2015, 3, 8361–8368 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00456e |
| This journal is © The Royal Society of Chemistry 2022 |