
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSerum proteomic analysis of differentially expressed proteins and pathways involved in the mechanism of endemic osteoarthritis†
Yan
Zhang‡
,
Qiong
Wang‡
,
Jingqi
Liang
,
Liang
Liu
,
Peilong
Liu
and
Hongmou
Zhao
 *
*
Foot and ankle surgery department, Honghui Hospital of Xi’an Jiaotong University, Xi’an, Shaanxi, China. E-mail: zhao_hongmou@hotmail.com
First published on 29th June 2022
Abstract
Kashin-Beck disease (KBD) is a chronic and endemic osteochondral disease and the etiology and pathogenic mechanism of KBD are still unknown. This study aimed to elucidate and screen KBD-associated proteins, which were differentially expressed between KBD patients and healthy controls. We combined protein fractionation and liquid chromatography-tandem mass spectrometry (LC-MS/MS) with a high-resolution mass spectrometer coupled with tandem mass tags (TMTs) to quantitatively analyze and screen KBD-associated proteins, which were differentially expressed between KBD patients and healthy controls. In addition, we used parallel reaction monitoring (PRM) to quantify proteins in serum from patients with KBD and healthy controls in order to verify the differentially expressed proteins in patients with KBD. We identified 224 differentially expressed proteins, including 11 up-regulated and 213 down-regulated proteins. Catalase (CAT) was observed to be significantly elevated in patients with KBD compared with control patients. Further, the fold difference of CAT is significantly elevated in PRM compared with label-free quantification. The results in this study suggest that CAT may be the reflection of the dynamic nature of KBD and could be considered as a novel pathogenic indicator for patients with KBD.
Introduction
Kashin-Beck disease (KBD), a chronic and endemic osteochondral disease with serious pathological changes and severe clinical signs, has some similarity to osteoarthritis (OA), including chondrocyte necrosis, apoptosis, and degradation, in the extracellular matrix (ECM).1 The clinical signs such as shortened and enlarged fingers always happened at an early stage of the onset of KBD, and then severe clinical changes such as deformed limb joints, limited movement and even dwarfism appeared in the advanced cases.2 With the implementation of comprehensive prevention and control measures, the incidence and prevalence of KBD have been drastically reduced to 0.47% of the general population in 2020 (according to the Health Yearbook of China 2021), but the existing cases of KBD are still more than 0.17 million patients in the endemic areas in China. What needs to be alerted is that there are still some newly diagnosed juvenile KBD patients in endemic areas from Tibet.3 In addition, selenium deficiency and T-2 toxin contamination and the environmental risk factors can still be found in partial endemic areas in China.3 Therefore, the threat of KBD becoming an epidemic in China again still exists. The etiology and pathogenic mechanism of KBD are still unknown, which hinder the development of prevention, early diagnosis and treatments.The proteins in vivo are considered one of the most important and widespread molecules, which regulate the mechanisms that dominate nearly all aspects of cellular functions;4–6 therefore, the expression of proteins definitely reflects some important information regarding the disease states.7 Proteomics is one of the most important techniques for analyzing the expression of proteins, and this method has developed into a crucial protein research area in recent decades. One of the main purposes of clinical proteomics is to identify potential biomarkers, which are important for studying the pathogenesis and therapeutic intervention of diseases by comparatively analyzing proteomic profiles between normal and diseased states and different physiological states. By using two-dimensional electrophoresis coupled with matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry, some differentially expressed proteins were identified,8,9 but the crucial proteins involved in the pathogenesis of KBD were still not determined. Therefore, it is urgent to identify some key proteins related to KBD as new targets for pathogenic mechanism studies of KBD, improving the understanding of the pathology of different stages of KBD, and exploring possible ways to block the pathophysiological process of disease.
In this study, we combine protein fractionation and liquid chromatography-tandem mass spectrometry (LC-MS/MS) with a high-resolution mass spectrometer coupled with tandem mass tags (TMTs)10 to quantitatively analyze and screen KBD-associated proteins that were differentially expressed between KBD patients and healthy controls. 224 proteins were identified to be differentially expressed in KBD serum. Parallel reaction monitoring (PRM) was performed for verification of proteins involved with disease activity and may be implicated in the pathogenesis of KBD.
Materials and methods
Blood samples were collected under approval from the human ethics committee of Xi’an Jiaotong University, and all subjects properly consented before samples were collected. All methods were carried out in accordance with relevant guidelines and regulations.Patient recruitment
A total of 17 patients with KBD and 15 healthy controls from Linyou County, one of the endemic areas for KBD in China, were recruited for this study. The patients with KBD were diagnosed strictly according to the national diagnostic criteria of KBD in China [WS/T 207-2010]. All subjects were diagnosed with KBD when manifested with X-ray alterations, such as defects and sclerosis on the bone end of phalanges combined with compression changes of the calcaneus and talus, and enlarged/deformed limb joints (hand, elbow, knee, ankle, etc.). Subjects were excluded for the following reasons: they were suffering or had previously suffered from any other osteoarticular diseases (such as osteoarthritis, rheumatoid arthritis, gout, or skeletal fluorosis) or any other type of macrosomia, osteochondrodysplasia, or chronic disease (such as hypertension, diabetes, or coronary heart disease). All the healthy controls did not have any musculoskeletal pathologies or recent injuries. All subjects signed a written informed consent form. Their general clinical data, including age, sex, educational background, and body mass index (BMI), were recorded. All patients and healthy controls were Shaanxi Han Chinese from similar geographic areas. Four KBD patients and 4 healthy controls were included to perform proteomics. And the other 10 KBD patients and 8 healthy controls were performed to verification of proteins specific to patients with KBD using parallel reaction monitoring (PRM) in this study, three KBD patients and three normal subjects for immunohistochemistry verification (Table S1, ESI†).Sample collection and protein extraction
5–7 mL veinal blood was collected from each subject, and stood for 5–10 min. Firstly, the cellular debris from the serum sample was removed by centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g at 4 °C for 10 min. Then, the supernatant was transferred to a new centrifuge tube. The top 14 high abundance proteins were removed using Pierce™ Top 12 Abundant Protein Depletion Spin Columns Kit (Thermo Fisher). Finally, the protein concentration was determined with BCA kit according to the manufacturer's instructions.
000 g at 4 °C for 10 min. Then, the supernatant was transferred to a new centrifuge tube. The top 14 high abundance proteins were removed using Pierce™ Top 12 Abundant Protein Depletion Spin Columns Kit (Thermo Fisher). Finally, the protein concentration was determined with BCA kit according to the manufacturer's instructions.
Trypsin digestion, TMT labeling and HPLC fractionation
The protein solution was reduced with 5 mM dithiothreitol for 30 min at 56 °C and alkylated with 11 mM iodoacetamide for 15 min at room temperature in darkness. The alkylated samples were transferred to ultrafiltration tubes for FASP digestion. The samples were firstly replaced by 8 M urea 3 times at 12000 g at room temperature for 20 min, and then replaced by 100 mM TEAB 3 times. Trypsin was added at 1:50 trypsin-to-protein mass ratio for digestion overnight. The peptide was recovered by centrifugation at 12000 g for 10 min at room temperature, and repeated two times. Finally, the combined peptides were desalted using a C18 SPE column.
Tryptic peptides were firstly dissolved in 0.5 M TEAB. Each channel of peptide was labeled with their respective TMT reagent (based on the manufacturer's protocol, Thermo Scientific), and incubated for 2 hours at room temperature. Five microliters of each sample were pooled, desalted and analyzed by MS to check labeling efficiency. After labeling efficiency check, samples were quenched by adding 5% hydroxylamine. The pooled samples were then desalted with a Strata X C18 SPE column (Phenomenex) and dried by vacuum centrifugation.
The sample was fractionated into fractions by high pH reverse-phase HPLC using an Agilent 300 Extend C18 column (5 μm particles, 4.6 mm ID, 250 mm length). Briefly, peptides were separated with a gradient of 2% to 60% acetonitrile in 10 mM ammonium bicarbonate pH 10 over 80 min into 80 fractions. Then, the peptides were combined into 9 fractions and dried by vacuum centrifugation.
LC-MS/MS analysis
The tryptic peptides were dissolved in solvent A (0.1% formic acid, 2% acetonitrile/in water), and directly loaded onto a home-made reversed-phase analytical column (25 cm length, 100 μm i.d.). Peptides were separated with a gradient from 5% to 16% solvent B (0.1% formic acid in 90% acetonitrile) over 65 min, 16% to 26% in 20 min and climbing to 70% in 2 min then holding at 70% for the last 3 min, all at a constant flow rate of 300 nL min−1 on an EASY-nLC 1000 UPLC system (Thermo Fisher Scientific).The separated peptides were analyzed in an Orbitrap FusionTM LumosTM (Thermo Fisher Scientific) with a nano-electrospray ion source. The electrospray voltage applied was 2.4 kV. The full MS scan resolution was set to 60000 for a scan range of 350–1550 m/z. Up to 10 most abundant precursors were then selected for further MS/MS analyses with 30 s dynamic exclusion. The HCD fragmentation was performed at a normalized collision energy (NCE) of 35%. The fragments were detected in the Orbitrap at a resolution of 30000. Fixed first mass was set as 100 m/z. Automatic gain control (AGC) target was set at 5E4, with an intensity threshold of 5E4 and a maximum injection time of 60 ms.
Database search and bioinformatics analysis
The resulting MS/MS data were processed using the MaxQuant search engine (v.1.5.2.8). Tandem mass spectra were searched against the human SwissProt database concatenated with a reverse decoy database. Trypsin/P was specified as a cleavage enzyme allowing up to 2 missing cleavages. The mass tolerance for precursor ions was set as 20 ppm in First search and 5 ppm in Main search, and the mass tolerance for fragment ions was set as 0.02 Da. Carbamidomethyl on Cys was specified as fixed modification. Acetylation on protein N-terminal and oxidation on Met were specified as variable modifications. TMT-10plex quantification was performed. FDR was adjusted to <1% and minimum score for peptides was set >40.In this study, the quantitative values of proteins in multiple replicates were obtained by the protein quantification experiment. The first step is to get the expression ratio of each protein between two samples; we calculate the average value in multiple replicates, and then use the ratio of the average values between two samples as the final expression ratio. The second step is to calculate the significant p value of differentially expressed proteins between two samples. First, log2 transformation was performed on the relative quantitative value of each sample in multiple replicates (making the data conform to the normal distribution), and then p value was calculated by the two-sample two-tailed T-test method. Proteins with p value <0.05 and expression ratio >2.0 were regarded as up-regulated, while proteins with p value <0.05 and expression ratio <1/2.0 were regarded as down-regulated.
GO annotation
Gene Ontology (GO) annotation proteome was derived from the UniProt-GOA database (https://www.ebi.ac.uk/GOA/). Firstly, identified protein ID was converted to UniProt ID and then mapped to GO IDs by protein ID. If some identified proteins were not annotated by the UniProt-GOA database, the InterProScan soft would be used to annotate protein's GO functional based on the protein sequence alignment method. Then proteins were classified by Gene Ontology annotation based on three categories: biological process, cellular component and molecular function.KEGG pathway annotation
The Kyoto Encyclopedia of Genes and Genomes (KEGG) database was used to annotate protein pathways. Firstly, using KEGG online service tools KAAS to annotated protein's KEGG database description. Then mapping the annotation result on the KEGG pathway database using KEGG online service tools KEGG mapper.Enrichment of gene ontology analysis
Proteins were classified by GO annotation into three categories: biological process, cellular compartment and molecular function. For each category, a two-tailed Fisher's exact test was employed to test the enrichment of the differentially expressed protein against all identified proteins. The GO with a corrected p-value <0.05 is considered significant.Enrichment of pathway analysis
The Encyclopedia of Genes and Genomes (KEGG) database was used to identify enriched pathways by a two-tailed Fisher's exact test to test the enrichment of the differentially expressed protein against all identified proteins. The pathway with a corrected p-value <0.05 was considered significant. These pathways were classified into hierarchical categories according to the KEGG website.Enrichment of protein domain analysis
For each category proteins, the InterPro (a resource that provides functional analysis of protein sequences by classifying them into families and predicting the presence of domains and important sites) database was researched and a two-tailed Fisher's exact test was employed to test the enrichment of the differentially expressed protein against all identified proteins. Protein domains with a p-value <0.05 were considered significant.Transcriptomes and proteomics crosstalk analysis
In the study, the crosstalk analysis from quantification of proteomics and transcriptomics was performed. The transcriptomics data were gene expression profiles in peripheral blood mononuclear cells from normal controls and patients with KBD, which has been published before.11Data analysis of parallel reaction monitoring
The resulting MS data were processed using Skyline (v.3.6). Peptide settings: enzyme was set as Trypsin [KR/P], and Max missed cleavage was set as 2. The peptide length was set as 8–25, Variable modification was set as Carbamidomethyl on Cys and oxidation on Met, and max variable modifications was set as 3. Transition settings: precursor charges were set as 2, 3, ion charges were set as 1, 2, and ion types were set as b, y, p. The product ions were set as from ion 3 to last ion, and the ion match tolerance was set as 0.02 Da.Immunohistochemical verification in cartilage
Cartilage tissues were fixed with 4% (w/v) paraformaldehyde for 24 h immediately after acquisition and decalcified in 10% (w/v) ethylenediaminetetraacetic acid disodium salt (EDTA-Na2) for 4 weeks. The samples were dehydrated in an alcohol series, cleared in xylene, and embedded in paraffin wax. Paraffin sections were cut into 5 μm sections, mounted on slides, and stored at room temperature until use for staining. The paraffin-embedded sections were baked at 65 °C for 1 h, deparaffinized with xylene and then rehydrated in decreasing concentrations of ethanol. Endogenous peroxidase activity was blocked by 0.3% (w/v) hydrogen peroxide for 10 min at room temperature, and then the sections were washed with 1 × PBS. Then, the sections were incubated in 10 mol L−1 urea solution diluted with water at 37 °C for 20 min and washed with 1 × PBS. The sections were incubated in 0.1% trypsin/CaCl2 at 37 °C and digested for another 20 min for antigen retrieval. After blocking with 5% (w/v) goat serum for 20 min at room temperature, anti-CAT (1:200 dilution, bs-6874R, Bioss) and IgG as a negative control were applied on the sections, and the samples were further incubated overnight at 4 °C. After washing with 1 × PBS, sections were incubated using a human serum amyloid P (SAP) kit (solution B contains a goat anti-rabbit secondary antibody; Zhongshan, Jinqiao, Guangzhou, China) according to the manufacturer's instructions. The substrate 3,3′-diaminobenzidine was added to stain the sections, and haematoxylin counterstaining was performed. Finally, the sections were dehydrated and mounted under alcohol-cleaned coverslips. Each zone was identified based on previously reported characteristics11–14 and articular cartilage was divided into superficial, middle, and deep zones, according to light microscopy observation. The percentage of positive cells was calculated using the number of positively stained cells divided by the total number of cells (positively and negatively stained cells) in the chosen fields of view.
Results
Protein identification and quantification
An LC-MS analysis produced 324149 spectra, corresponding to 6558 unique peptides representing 1305 proteins (Table S2, ESI†), including 940 quantifiable proteins (Table S3, ESI†). Proteins with an abundance difference ratio ≥2.0 and p < 0.05 (t-test) were considered as up-regulated proteins, and those with ratio ≤0.5 as down-regulated proteins. Following this criterion, we identified 224 differentially expressed proteins, including 11 upregulated and 213 downregulated proteins in patients with KBD (Table 1 and Table S4, ESI†).
| Gene aymbol | Accession | Description | Fold change | P-value |
|---|---|---|---|---|
| CFHR1 | Q03591 | Complement factor H-related protein 1 | 3.30 | 0.037 |
| KRT1 | P04264 | “Keratin, type II cytoskeletal 1” | 2.72 | 0.041 |
| CAT | P04040 | Catalase | 2.68 | 0.009 |
| SPP1 | P10451 | Osteopontin | 2.24 | 0.002 |
| LRG1 | P02750 | Leucine-rich alpha-2-glycoprotein | 2.20 | 0.004 |
| CALML5 | Q9NZT1 | Calmodulin-like protein 5 | 2.13 | 0.020 |
| APCS | P02743 | Serum amyloid P-component | 2.12 | 0.002 |
| FCN2 | Q15485 | Ficolin-2 | 2.11 | 0.003 |
| THBS4 | P35443 | Thrombospondin-4 | 2.07 | 0.029 |
| COL1A2 | P08123 | Collagen alpha-2(I) chain | 2.06 | 0.041 |
| MCM3 | P25205 | DNA replication licensing factor MCM3 | 0.05 | 0.0006 |
| RPL14 | P50914 | 60S ribosomal protein L14 | 0.08 | 0.0008 |
| EIF4B | P23588 | Eukaryotic translation initiation factor 4B | 0.09 | 0.0005 |
| SRSF1 | Q07955 | Serine/arginine-rich splicing factor 1 | 0.10 | 0.0004 |
| HNRNPC | P07910 | Heterogeneous nuclear ribonucleoproteins C1/C2 | 0.10 | 0.0002 |
| RPL5 | P46777 | 60S ribosomal protein L5 | 0.10 | 0.0002 |
| RPS3A | P61247 | 40S ribosomal protein S3a | 0.11 | 0.00001 |
| RPL17 | P18621 | 60S ribosomal protein L17 | 0.11 | 0.000007 |
| RPL28 | P46779 | 60S ribosomal protein L28 | 0.11 | 0.00001 |
| NOLC1 | Q14978 | Nucleolar and coiled-body phosphoprotein 1 | 0.12 | 0.0002 |
GO terms and GO enrichment analysis of differentially expressed proteins
The gene ontology (GO) term and GO enrichment analysis were performed, in order to understand the functions and biological processes of the differentially abundant proteins involved in the KBD group. GO terms for biological processes showed that all differentially expressed proteins (DEPs) participated in response to a cellular process, biological regulation, metabolic process and single-organism process. The GO terms for molecular function indicated that DEPs were mainly enriched in the binding, catalytic activity and structural molecule activity; the GO terms for cellular component enriched in the cell, organelle and macromolecular complex (Fig. 1 and Table S5, ESI†). The GO enrichment analysis for the molecular function and cellular component showed that most of DEPs were enriched in the nucleic acid binding, RNA binding and heterocyclic compound binding; non-membrane-bounded organelle, intracellular non-membrane-bounded organelle and nucleus (Fig. 2A and B and Table S6, ESI†). The GO enrichment analysis for biological processes showed that most of the DEPs participated in the nucleobase-containing compound metabolic process, regulation of gene expression and heterocycle metabolic process (Fig. 2C and Table S6, ESI†). The protein domain enrichment analysis showed that upregulated DEPs were involved in the nucleotide-binding alpha–beta plait domain and RNA recognition motif domain (Fig. 2D and Table S7, ESI†). | ||
| Fig. 1 The GO terms for biological process, cellular component and molecular function enriched by differentially expressed proteins. | ||
 | ||
| Fig. 2 The GO enrichment analysis of differentially expressed proteins. (A) The GO enrichment analysis for molecular function, (B) the GO enrichment analysis for cellular component, (C) the GO enrichment analysis for biological processes, and (D) the GO enrichment analysis for protein domain. | ||
KEGG enrichment analysis
To investigate the functions and signaling pathways, which the differentially expressed proteins were involved in, Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis and multiple corrections were conducted. The upregulated DEPs from the KEGG pathway analysis were involved in the ECM-receptor interaction, human papillomavirus infection, focal adhesion, apelin signaling pathway and PI3K–Akt signaling pathway (Fig. 3A and Table S8, ESI†). Downregulated DEPs from the KEGG pathway analysis were involved in ribosome, Epstein-Barr virus infection, spliceosome, legionellosis, mismatch repair, DNA replication, RNA degradation, Huntington disease and alcoholism (Fig. 3B and Table S9, ESI†). | ||
| Fig. 3 The kyoto encyclopedia of genes and genomes (KEGG) enrichment analysis. (A) The upregulated differentially expressed proteins from the KEGG pathway analysis, and (B) the downregulated differentially expressed proteins from the KEGG pathway analysis. | ||
Verification of proteins specific to patients with KBD using parallel reaction monitoring (PRM) and immunohistochemistry
To further screen the proteins specific to patients with KBD, quantitative assays based on PRM were performed on the thirty selected differentially expressed proteins with serum samples from 10 patients with KBD and 8 healthy controls. Finally, eight proteins were quantified; however, only four proteins were consistent with the quantitative proteomic results and the relative abundance data of proteins from four individual proteins are presented as a linear box-and-whisker plot (Fig. 4). As shown in the figure, catalase (CAT), thrombospondin 4 (THBS4), and keratin 1 (KRT1) were observed to be consistent in patients with KBD compared with those in control patients between PRM and label-free quantification. However, only the fold difference of CAT is significantly elevated in PRM compared with label-free quantification. The expression of CAT in cartilage tissues was also investigated by using immunohistochemistry assays. The results showed that CAT was upregulated in the superficial and middle zone of KBD cartilage compared to the normal control (Fig. 5).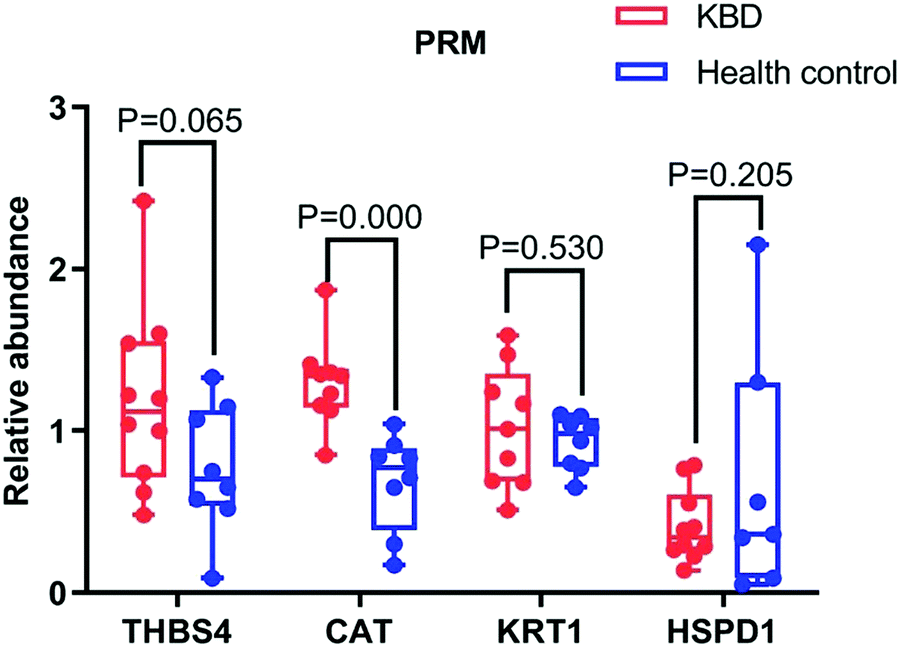 | ||
| Fig. 4 Three potential markers were validated in 10 patients with KBD and eight healthy individuals using PRM. One potential marker CAT showed significant difference (P < 0.05) in patients with KBD compared with healthy controls. | ||
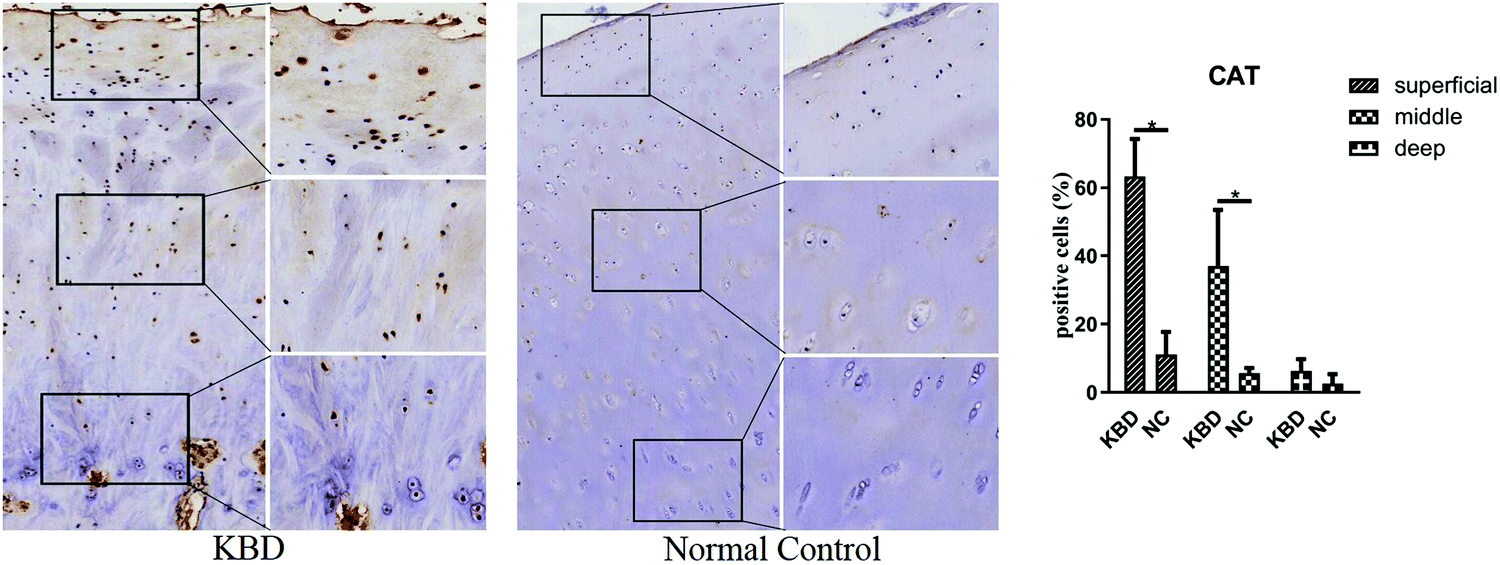 | ||
| Fig. 5 Representative immunohistochemistry staining of CAT in KBD and healthy cartilage tissues. Scale bar, left, 500 μm; right, 50 μm, and comparative quantification of positive cells of different areas (superficial, middle, and deep) between KBD and healthy cartilage tissues displayed by a box plot (n = 3). *p < 0.05. | ||
Transcriptomes and proteomics crosstalk analysis
A previous study15 has performed transcriptomics research of blood from patients with KBD, and 20381 transcriptions were identified, among which 62 and 74 genes exhibited more than a 2-fold decrease or increase in mRNA expression, respectively. We compared the protein expression in this study with mRNA expression in KBD patients. We observed remarkable concordance between the proteomics and transcriptomics datasets (Fig. 6B). Most of the upregulated genes revealed by transcriptomics research exhibited an increased protein expression, whereas downregulated genes were associated with a reduced protein expression. In addition, 1091 proteins or transcriptions were identified in both proteomics and transcriptomics studies (Fig. 6A).
 | ||
| Fig. 6 (A) The Venn diagram of transcriptomics and proteomics identified of the patients with KBD. (B) The correlation analysis of transcriptomics and proteomics of the patients with KBD. | ||
Discussion
Recent developments in proteomic technologies for massive protein analyses have facilitated the identification of novel biomarkers for the early diagnosis, clinical management, and severity evaluation of osteochondral diseases10,16–19 and provided new clues to elucidate underlying pathogenesis and to identify therapeutic targets.In 2010, Du et al.8 identified potential biomarkers of KBD by using mass spectrometry (MS)-based proteomic profiling, identified correlative mass points and obtained a discriminative pattern with 90.91% sensitivity and 82.61% specificity. The results of this study suggested that further proteomics studies may be useful with a larger number of appropriate specimens, careful experimental manipulation and improved MS techniques. In another study, MALDI-TOF/TOF analysis was performed to investigate the protein changes in KBD cartilage and to identify candidate proteins that can improve the understanding of the pathogenesis of KBD. A total of 27 proteins from KBD chondrocyte cultures, which consisted of 17 upregulated and ten downregulated proteins, were identified that were involved in cellular redox homeostasis and stress response (MnSOD, Hsp27, peroxiredoxin-1, and cofilin-1), glycolysis (PGK-1, PGM-1, a-enolase), and cell motility and cytoskeletal organization (actin, calponin-2, and keratin).9 All these studies showed that proteomic analyses were effective in exploiting pathogenic target candidates for KBD.
Catalase is the key antioxidant enzyme for the human body to resist oxidative stress. It is a heme enzyme that exists in peroxisome of almost all aerobic cells. Catalase transforms the active oxygen hydrogen peroxide into water and oxygen, thus reducing the toxic effect of hydrogen peroxide. Oxidative stress is thought to play a key role in the development of many chronic diseases, such as rheumatoid arthritis.20 In a rat KBD model study, catalase in the serum and cartilage from rats in the KBD group was significantly lower than that in the rats in the normal diet group,21 but this protein was observed to be significantly elevated in patients with KBD compared with control patients in our study, and is worth further verification. In other studies, it has been proven that CAT was significantly increased in OA chondrocytes compared with human normal chondrocytes and serum.22,23 In this study, CAT was observed to be significantly elevated in patients with KBD compared with control patients by PRM. Therefore, CAT may reflect the dynamic nature of KBD, and could be considered an important pathogenic indicator of patients with KBD.
In this study, GO and KEGG enrichment analyses indicated that most of the upregulated DEPs participated in the regulation of responses to a stimulus and in the immune response. Upregulated DEPs from the KEGG pathway analysis were involved in the ECM-receptor interaction and the PI3K–Akt signaling pathway. In addition, most of the downregulated DEPs participated in nucleic acid metabolic processes, and downregulated DEPs in the KEGG pathway analysis were involved in ribosomes, DNA replication, and RNA degradation. These results showed that these proteins might contribute to the pathogenesis of KBD, and some of them were consistent with the findings from previous research. Zhao's study found that 124 miRNAs had lower expression levels in the subchondral bone sampled from KBD patients in comparison with OA patients, and the predicted targets demonstrated numerous significantly enriched GO terms and signal pathways, such as PI3K–Akt, which were essential for bone development and integrity.24 In another study, the protein expression levels of PI3K/Akt signaling molecules in whole blood and chondrocytes were detected, suggesting that apoptosis induced by tBHP in chondrocytes might be mediated via upregulation of PI3K/Akt, and Na2SeO3 has an anti-apoptotic effect by downregulating the PI3K/Akt signaling pathway.25
The limitation of this study is the lack of the large sample size to determine the sensitivity and specificity of serum CAT in distinguishing KBD from the healthy control. Firstly, the dramatically decreased incidence of KBD and gradual death of elderly patients in the past decade led to the drop in the number of patients with KBD. Secondly, the pathological changes of many adult KBD have been changed or merged with OA, making the collection of a typical KBD case extremely difficult. Therefore, it is very difficult to collect a large number of biological samples from patients with KBD to perform the verification study.
Conclusion
In summary, LC-MS/MS analysis was performed to screen differentially expressed proteins between patients with KBD and healthy controls. A total of 224 differentially expressed proteins, including 11 upregulated and 213 downregulated proteins with a threshold value of expression fold change over 2 and a P value <0.05, were identified. To validate the differential proteins of potential markers in patients with KBD, CAT in serum from patients with KBD was observed to be significantly elevated by using PRM. Therefore, CAT may reflect the dynamic nature of KBD and may be considered a novel pathogenic indicator for patients with KBD.Data availability
The datasets supporting this article have been uploaded as part of the ESI† material.Ethics approval and consent to participate
All samples were collected under approval from the human ethics committee of Xi’an Jiaotong University, and all subjects properly consented before samples were collected.Author contributions
Yan Zhang, Qiong Wang and Hongmou Zhao: writing – original draft, conceptualization and methodology. Jingqi Liang, Liang Liu and Peilong Lliu: investigation and formal analysis. Hongmou Zhao: project administration and funding acquisition.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This study was financially supported by the Key R & D plan of Shaanxi Province (No. 2021SF-025) and the Natural Science Foundation of Shaanxi Province (No. 2020JQ940).References
- Y. Ning, X. Wang, M. J. Lammi and X. Guo, Exp. Cell Res., 2019, 379, 140–149 CrossRef CAS PubMed.
- X. Wang, Y. Ning, L. Yang, F. Yu and X. Guo, Biol. Trace Elem. Res., 2017, 179, 178–184 CrossRef CAS PubMed.
- R. Lei, N. Jiang, Q. Zhang, S. Hu, B. S. Dennis, S. He and X. Guo, Biol. Trace Elem. Res., 2016, 171, 34–40 CrossRef CAS PubMed.
- T. Hunter, Cell, 2000, 100, 113–127 CrossRef CAS PubMed.
- Y. Kabuyama, K. A. Resing and N. G. Ahn, Curr. Opin. Genet. Dev., 2004, 14, 492–498 CrossRef CAS PubMed.
- I. H. Chen, L. Xue, C. C. Hsu, J. S. Paez, L. Pan, H. Andaluz, M. K. Wendt, A. B. Iliuk, J. K. Zhu and W. A. Tao, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3175–3180 CrossRef CAS PubMed.
- A. B. Iliuk, J. V. Arrington and W. A. Tao, Electrophoresis, 2014, 35, 3430–3440 CrossRef CAS PubMed.
- J. Du, X. Wu, H. Zhang, S. Wang, W. Tan and X. Guo, Mol. Med. Rep., 2010, 3, 821–824 CAS.
- W. J. Ma, X. Guo, J. T. Liu, R. Y. Liu, J. W. Hu, A. G. Sun, Y. X. Yu and M. J. Lammi, Proteomics, 2011, 11, 2881–2890 CrossRef CAS PubMed.
- Y. Cheng, Y. Chen, X. Sun, Y. Li, C. Huang, H. Deng and Z. Li, Inflammation, 2014, 37, 1459–1467 CrossRef CAS PubMed.
- T. Chen, M. J. Hilton, E. B. Brown, M. J. Zuscik and H. A. Awad, Biotechnol. Bioeng., 2013, 110, 1476–1486 CrossRef CAS PubMed.
- C. Karlsson and A. Lindahl, Arthritis Res. Ther., 2009, 11, 121 CrossRef PubMed.
- B. L. Schumacher, J. A. Block, T. M. Schmid, M. B. Aydelotte and K. E. Kuettner, Arch. Biochem. Biophys., 1994, 311, 144–152 CrossRef CAS PubMed.
- P. Lorenzo, M. T. Bayliss and D. Heinegard, J. Biol. Chem., 1998, 273, 23463–23468 CrossRef CAS PubMed.
- S. Wang, X. Guo, X. M. Wu and M. J. Lammi, PLoS One, 2012, 7, e28439 CrossRef CAS PubMed.
- M. Camacho-Encina, V. Balboa-Barreiro, I. Rego-Perez, F. Picchi, J. VanDuin, J. Qiu, M. Fuentes, N. Oreiro, J. LaBaer, C. Ruiz-Romero and F. J. Blanco, Ann. Rheum. Dis., 2019, 1699–1705 CrossRef CAS PubMed.
- S. Hosseininia, P. Onnerfjord and L. E. Dahlberg, J. Orthop. Res., 2019, 37, 131–135 CrossRef CAS PubMed.
- C. S. Thudium, H. Lofvall, M. A. Karsdal, A. C. Bay-Jensen and A. R. Bihlet, J. Proteomics, 2019, 190, 55–66 CrossRef CAS PubMed.
- L. Lourido, B. Ayoglu, J. Fernandez-Tajes, N. Oreiro, F. Henjes, C. Hellstrom, J. M. Schwenk, C. Ruiz-Romero, P. Nilsson and F. J. Blanco, Sci. Rep., 2017, 7, 137 CrossRef PubMed.
- W. Z. Wang, X. Guo, C. Duan, W. J. Ma, Y. G. Zhang, P. Xu, Z. Q. Gao, Z. F. Wang, H. Yan, Y. F. Zhang, Y. X. Yu, J. C. Chen and M. J. Lammi, Osteoarthr. Cartil., 2009, 17, 83–90 CrossRef CAS PubMed.
- J. H. Chen, S. Xue, S. Li, Z. L. Wang, H. Yang, W. Wang, D. Song, X. Zhou and C. Chen, J. Orthop. Res., 2012, 30, 1229–1237 CrossRef CAS PubMed.
- C. Zhuang, Y. Wang, Y. Zhang and N. Xu, Int. J. Biol. Macromol., 2018, 115, 281–286 CrossRef CAS PubMed.
- M. Fernandez-Moreno, A. Soto-Hermida, S. Pertega, N. Oreiro, C. Fernandez-Lopez, I. Rego-Perez and F. J. Blanco, BMC Musculoskeletal Disord., 2011, 12, 264 CrossRef CAS PubMed.
- G. H. Zhao, L. Yang, M. J. Lammi and X. Guo, Clin. Rheumatol., 2019, 38, 2637–2645 CrossRef PubMed.
- X. A. Du, H. M. Wang, X. X. Dai, Y. Kou, R. P. Wu, Q. Chen, J. L. Cao, X. Y. Mo and Y. M. Xiong, Osteoarthr. Cartil., 2015, 23, 210–216 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2mo00154c |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2022 |