
Propylene and butylene glycol: new alternatives to ethylene glycol in conjugated polymers for bioelectronic applications†
Maximilian
Moser‡
 *a,
Yazhou
Wang‡
*b,
Tania Cecilia
Hidalgo‡
c,
Hailiang
Liao
b,
Yaping
Yu
b,
Junxin
Chen
b,
Jiayao
Duan
b,
Floriana
Moruzzi
a,
Sophie
Griggs
a,
Adam
Marks
a,
Nicola
Gasparini
d,
Andrew
Wadsworth
a,
Sahika
Inal
*c,
Iain
McCulloch
ae and
Wan
Yue
*b
*a,
Yazhou
Wang‡
*b,
Tania Cecilia
Hidalgo‡
c,
Hailiang
Liao
b,
Yaping
Yu
b,
Junxin
Chen
b,
Jiayao
Duan
b,
Floriana
Moruzzi
a,
Sophie
Griggs
a,
Adam
Marks
a,
Nicola
Gasparini
d,
Andrew
Wadsworth
a,
Sahika
Inal
*c,
Iain
McCulloch
ae and
Wan
Yue
*b
aUniversity of Oxford, Department of Chemistry, Oxford, OX1 3TA, UK
bState Key Laboratory of Optoelectronic Materials and Technologies, Key Laboratory for Polymeric Composite and Functional Materials of the Ministry of Education, School of Materials and Engineering, Guangzhou Key Laboratory of Flexible Electronic Materials and Wearable Devices, Sun Yat-Sen University, Guangzhou, 510275, China. E-mail: wangyzh53@mail2.sysu.edu.cn; yuew5@mail.sysu.edu.cn
cKing Abdullah University of Science and Technology (KAUST), Biological and Environmental Science and Engineering (BESE), Organic Bioelectronics Laboratory, Thuwal, 23955-6900, Saudi Arabia. E-mail: sahika.inal@kaust.edu.sa
dImperial College London, Department of Chemistry and Centre for Plastic Electronics, London, W12 0BZ, UK
eKing Abdullah University of Science and Technology (KAUST), KAUST Solar Center (KSC), Thuwal, 23955-6900, Saudi Arabia
First published on 22nd December 2021
Abstract
To date, many of the high-performance conjugated polymers employed as OECT channel materials make use of ethylene glycol (EG) chains to confer the materials with mixed ionic-electronic conduction properties, with limited emphasis placed on alternative hydrophilic moieties. While a degree of hydrophilicity is required to facilitate some ionic conduction in hydrated channels, an excess results in excessive swelling, with potentially detrimental effects on charge transport. This is therefore a subtle balance that must be optimised to maximise electrical performance. Herein a series of polymers based on a bithiophene–thienothiophene conjugated backbone was synthesised and the conventional EG chains substituted by their propylene and butylene counterparts. Specifically, the use of propylene and butylene chains was found to afford polymers with a more hydrophobic character, thereby reducing excessive water uptake during OECT operation and in turn significantly boosting the polymers’ electronic charge carrier mobility. Despite the polymers’ lower water uptake, the newly developed oligoether chains retained sufficiently high degrees of hydrophilicity to enable bulk volumetric doping, ultimately resulting in the development of polymers with superior OECT performance.
New conceptsIn recent years, several conjugated polymers functionalised with hydrophilic ethylene glycol (EG)-based side chains have been investigated as channel materials for improving the performance of organic electrochemical transistors. In these materials, electronic charge carrier conduction occurs along the conjugated polymer backbone, while ionic conduction is facilitated by the presence of the hydrophilic side chains. While considerable efforts have gone into varying the polymers’ conjugated backbone to maximise their OECT performance, little work has thus far been conducted on using alternative hydrophilic pendant chains besides EG-based ones. Herein, we present the first use of propylene glycol (PG) and butylene glycol (BG) as solubilising chains in conjugated polymers and discuss their impact on the polymers’ electrochemical, structural, and swelling properties. Specifically, we demonstrate how the increased hydrophobicity of the PG and BG functionalities benefits the polymers’ microstructure and mass uptake during device operation, thereby leading to superior OECT performances. We envisage that the proposed molecular design concept will not only be helpful for the development of future high-performance OECT channel materials but will also guide material design in related research areas where mixed ionic-electronic conduction is of paramount importance, including organic thermoelectrics, batteries, and electrochromics. |
Introduction
Organic mixed ionic-electronic conductors (OMIECs) are an emerging class of materials that have shown considerable promise for bioelectronic applications. The ability of OMIECs to conduct both electronic and ionic charge carriers also makes them highly attractive candidates for alternative application areas such as batteries, supercapacitors, and electrochromic devices, all of which rely on mixed transport behaviour.1–6 One bioelectronic device, which has attracted considerable interest recently is the organic electrochemical transistor (OECT).7–9 Analogously to field-effect transistors (FETs), OECTs act as switches and convert a gate voltage (VG) input into a current output (ID). In comparison to their FET counterparts, OECTs can offer superior transduction (i.e. signal amplification) abilities,10,11 while also enabling for operation in aqueous media, promoting their use as biomarker and metabolite sensors,12–15 neuroelectronic recording elements,16–18 transmembrane protein activity monitors,19 amongst many others. The most widely employed OMIEC in OECTs is the commercially available aqueous dispersion of poly(3,4-ethylenedioxythiophene)-poly(styrenesulfonate) (PEDOT:PSS). In PEDOT:PSS, electronic conduction occurs along the conjugated polymer backbone of PEDOT, while the negatively charged sulfonate groups on PSS lead to water uptake and a pathway for efficient ionic conduction.20,21 PEDOT:PSS-based OECTs, however, suffer from: (i) their depletion mode of operation, incurring low on/off ratios and high power requirements,22,23 (ii) their often lower OECT steady-state performance compared to alternative OECT channel materials,22,24,25 (iii) the presence of an insulating PSS phase, which limits volumetric capacitance,24 and (iv) the frequent necessity of extensive pre- and post-deposition film processing to achieve maximum OECT performance.20,26 To address these issues, a novel class of OMIECs has recently been developed, namely ethylene-glycol (EG) functionalised conjugated polymers. Similar to PEDOT:PSS, in this group of OMIECs, electronic conduction occurs along the conjugated polymer backbone. On the other hand, water uptake and in turn ion conduction is facilitated by the grafting of hydrophilic EG side chains onto the conjugated polymer backbone.Several molecular design strategies have been pursued to maximise the OECT performance of EG functionalised conjugated polymers.23,27,28 Broadly speaking these synthetic modifications can be categorised into conjugated polymer backbone engineering22,29–33 and EG-side chain engineering.25,34–39 In this context, one of the highest-performing EG-functionalised polymers reported up to date is poly(2-(3,3-bis(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-[2,2′-bithiophen]-5-yl)thieno[3,2-b]thiophene), p(g2T-TT). p(g2T-TT)'s steady-state OECT performance has, however, been documented to be limited by excessive polymer swelling, i.e. water uptake, during OECT operation.40 In fact, reducing water uptake by 50%, achieved by increasing the NaCl concentration in the external electrolyte from 100 mM to 6 M, was shown to double the recorded electronic charge carrier mobility (μ) and consequently also transconductance (gm).40 Electrolyte concentrations in mammalia and in planta, however, typically span across 100–200 mM with sodium and chloride concentrations lying close to 100 mM in human blood plasma and interstitial fluids.41,42 A more suitable strategy to overcome this OECT performance-biological tissue electrolyte concentration mismatch is thus to reduce p(g2T-TT)'s swelling during OECT operation by chemical modification of the polymer itself, rather than the external electrolyte. With this in consideration, in this work, we have developed two new semiconducting polymers employing the same bithiophene–thienothiophene backbone as p(g2T-TT) but with more hydrophobic oligoether side chains, specifically propylene glycol (PG) in p(p2T-TT) and butylene glycol (BG) in p(b2T-TT), see Fig. 1. To the best of our knowledge, this is the first report disclosing the use of PG and BG chains for conjugated polymers both within the context of OECTs and also in the broader field of organic electronics. From a molecular design point of view, we envisaged PG and BG chains to be a suitable alternative to EG ones, given their similar ion transport abilities,43,44 while simultaneously binding to significantly less water molecules per oligoether repeat unit.45 Moreover, by retaining the same number of oligoether repeat units per solubilising chain (three), good solubility of the polymers and a fair comparison against the reference p(g2T-TT) polymer was ensured.
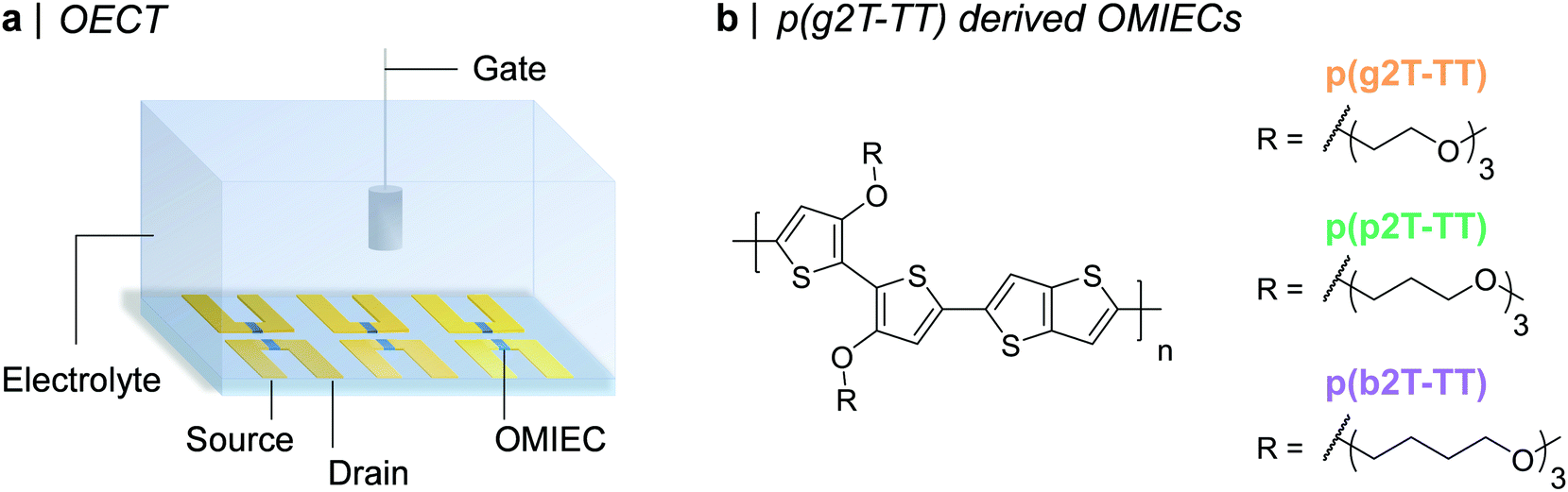 | ||
| Fig. 1 (a) Schematic representation of an OECT array. (b) Chemical structures of the synthesised OMIECs, namely p(g2T-TT), p(p2T-TT), and p(b2T-TT). | ||
Results and discussion
Monomer preparation and polymer synthesis are described extensively in the ESI.† In brief, this involved (i) oligoether chain synthesis, (ii) oligoether chain grafting onto the 3-position of thiophene, (iii) preparation of the oligoether functionalised bithiophene monomers, and (iv) Stille cross-coupling polymerisation. Both of the two new polymers, p(p2T-TT) and p(b2T-TT), and the reference p(g2T-TT) polymer, were obtained in good yields (>85%). Gel permeation chromatography (GPC) was used to quantify the molecular weights of the polymers. Similarly, to previous reports on oligoether functionalised conjugated polymers, bimodal GPC traces were obtained for the polymers, whereby the higher molecular weight peak was attributed to the formation of aggregates.29,39 Thus, to forego molecular weight overestimations, the signal observed for short elution times (high molecular weights) was neglected. Comparable number average molecular weights (Mn) and dispersity values (Đ) were recorded for the three polymers, see Table 1.| Polymer | M n (kDa) | Đ | E ox,org (V vs. Ag/AgCl) | E ox,aq (V vs. Ag/AgCl) | IP (eV) | λ max,soln (nm) | λ max,film (nm) | E g,opt (eV) |
|---|---|---|---|---|---|---|---|---|
| a Determined from GPC measurements (CHCl3, 40 °C) against a set of narrow polystyrene standards. b Using a 0.1 M solution of TBA PF6 in acetonitrile as the supporting electrolyte. c Using a 0.1 M solution of NaCl in distilled water as the supporting electrolyte. d Measured in dilute chloroform solutions. e Recorded while applying a negative bias to ensure complete dedoping of the polymer film. f Calculated from the onset of absorption in thin film. | ||||||||
| p(g2T-TT) | 24.7 | 1.4 | −0.12 | −0.18 | 4.55 | 580 | 606, 647 | 1.74 |
| p(p2T-TT) | 24.7 | 1.4 | −0.14 | −0.02 | 4.53 | 596 | 612, 648 | 1.73 |
| p(b2T-TT) | 19.8 | 1.5 | −0.14 | +0.05 | 4.53 | 584 | 607, 643 | 1.73 |
The optical properties of the polymers were investigated by UV-vis absorption spectroscopy in solution and thin film, see Fig. S27 (ESI†), and are summarised in Table 1. No significant differences were recorded in the polymers’ solution UV-vis spectra with all polymers exhibiting an absorption maximum (λmax,soln) around 585 nm. In comparison to their λmax,soln, the polymers’ thin film absorption maxima (λmax,film) were shifted bathochromically and featured two absorption maxima, one around 610 nm and one around 650 nm, indicating molecular ordering upon thin film formation.
Cyclic voltammetry was conducted on polymer-coated ITO slides employing both an organic (0.1 M solution of tetrabutylammonium hexafluorophosphate in acetonitrile) and an aqueous-based (0.1 M solution of sodium chloride in distilled water) supporting electrolyte, see Fig. S28 and S29 (ESI†). Virtually identical onsets of oxidation in organic media (Eox,org) between −0.14 and −0.12 V were obtained for all polymers, conferring them with similar ionisation potentials (IP) between 4.53 and 4.55 eV. Notably, when employing a water-based electrolyte, significantly different onsets of oxidation (Eox,aq) were recorded for the three polymers, with p(g2T-TT), p(p2T-TT), and p(b2T-TT) incurring values of −0.18, −0.02, and +0.05 V, respectively. This shift in Eox,aq was attributed to side chain substitution impacting both side chain-side chain and side chain-electrolyte interactions, in turn affecting ion penetration. A numerical estimate for the relative ease of ion penetration into the various polymers can in turn be obtained by calculating the difference between the polymers’ Eox,org and Eox,aq.34 Differences of +0.07, −0.12, and −0.19 V were obtained for p(g2T-TT), p(p2T-TT), and p(b2T-TT), respectively. The calculated value of +0.07 V for p(g2T-TT) is similar to the +0.06 V value reported in previous literature.34 Note how the values calculated for p(p2T-TT) and p(b2T-TT) sit exactly between the +0.07 V difference reported for p(g2T-TT) and the −0.42 V difference reported for p(a2T-TT), its fully alkylated counterpart.34 This indicates that the energetics associated with penetration of Cl− ions from an aqueous NaCl solution become increasingly less favourable upon increasing the hydrophobicity of the oligoether side chains, yet remains significantly more favourable compared to their fully alkylated counterpart. Both PG and BG side chains thus confer the polymers with properties that reside between their EG and alkyl counterparts. Importantly, unlike their alkyl counterparts, for which the doping–dedoping process has been reported to be only partially reversible in aqueous media,34 spectroelectrochemistry studies suggested the doping process for both PG and BG functionalised polymers to be reversible, vide infra.
The compatibility of PG and BG side chains to enable bulk reversible electrochemical doping in an aqueous medium were confirmed by spectroelectrochemical measurements of the polymers in a 0.1 M aqueous NaCl solution, see Fig. S30 (ESI†). As can be seen from Fig. S30 (ESI†) the polymers required the application of a negative bias to fully discharge the polymer films. Gradual increments in the applied bias subsequently resulted in a progressive depression of the π–π* absorption feature between 600–650 nm with the concomitant appearance of new absorption features across 700–1100 nm, which were ascribed to the charged polymer species, including the polymers’ polaronic and bipolaronic forms.46,47 Subsequent dedoping of the polymer films by application of a negative applied potential restored their original optical signature therefore highlighting the compatibility of both p(p2T-TT) and p(b2T-TT) to undergo reversible electrochemical doping. This feature also suggests the suitability of these polymers for alternative electrochemical applications, such as batteries, supercapacitors, or electrochromic devices, where reversible electrochemical cycling is a prerequisite for stable device operation.4,48
OECTs were fabricated for the various polymers and their performance evaluated employing a 0.1 M aqueous sodium chloride solution as the supporting electrolyte and an Ag/AgCl pellet as the gate electrode. Note that no pre- or post-processing treatments of the semiconductor films were required to achieve the incurred OECT performances. The polymers’ transfer, transconductance, and ID0.5vs. VG curves are displayed in Fig. 2a–c, while their output curves can be found in Fig. S31 in the ESI.† Finally, the polymers’ steady-state OECT performance is summarised in Table 2.
 | ||
| Fig. 2 (a) Transfer, (b) transconductance, and (c) ID0.5vs. VG curves recorded for p(g2T-TT), p(p2T-TT), and p(b2T-TT). Comparison of the (d) on/off ratios, (e) electronic charge carrier mobilities, and (f) volumetric capacitances measured for the polymers. | ||
| Polymer | d (nm) | V Th (V) | g m (mS) | [μC*]ac (F cm−1 V−1 s−1) | on/off | μ OECT (cm2 V−1 s−1) | C*e (F cm−3) |
|---|---|---|---|---|---|---|---|
a Average values calculated from all operational channels (four p(g2T-TT) and six p(p2T-TT) or p(b2T-TT)) channels.
b Calculated from the intercept of ID0.5 against VG and the abscissa.
c Extracted using  at saturation VD (−0.6 V for p(g2T-TT) and −0.65 V for p(p2T-TT) and p(b2T-TT)).
d Calculated from the slope of ID0.5 against VG.
e Obtained from electrochemical impedance spectra recorded for the doped films. at saturation VD (−0.6 V for p(g2T-TT) and −0.65 V for p(p2T-TT) and p(b2T-TT)).
d Calculated from the slope of ID0.5 against VG.
e Obtained from electrochemical impedance spectra recorded for the doped films.
|
|||||||
| p(g2T-TT) | 77.2 ± 5.25 | 0.00 ± 0.01 | 15.8 ± 1.0 | 299.42 ± 5.15 | 1.5 × 105 | 1.82 ± 0.09 | 171.4 ± 8.41 |
| p(p2T-TT) | 134.8 ± 18.6 | −0.18 ± 0.01 | 15.1 ± 2.1 | 182.24 ± 16.5 | 6.0 × 105 | 1.76 ± 0.14 | 103.21 ± 8.17 |
| p(b2T-TT) | 106.3 ± 11.67 | −0.27 ± 0.01 | 19.4 ± 2.2 | 342.2 ± 12 | 6.0 × 105 | 3.03 ± 0.38 | 113.93 ± 5.07 |
Threshold voltages (VTh) of the devices were determined by plotting the square root of the source-drain current as a function of the applied gate voltage, see Fig. 2c. As can be seen from Fig. 2c, VTh of 0.00 ± 0.01, −0.18 ± 0.01, and −0.27 ± 0.01 V were obtained for p(g2T-TT), p(p2T-TT), and p(b2T-TT), respectively, whereby the value recorded for p(g2T-TT) is in line with previous literature.40 The recorded trend in VTh was therefore also in full agreement with the Eox,aq obtained from the CV experiments, confirming the increased energetic ion injection requirements upon increasing the hydrophobicity of the oligoether chains. Note the close resemblance of this trend as to increasing the salt concentration in the supporting electrolyte, where a similar shift of VTh towards more negative values was observed.40
The enhancement mode of operation of these devices was envisaged to be a beneficial property for bioelectronic applications, as higher on/off ratios can be achieved compared to their depletion mode counterparts. In fact, good ON/OFF ratios in the order of 105 were achieved for all polymers, see Fig. 2d, which compares favourably compared to those typically afforded by PEDOT:PSS.10,17,49 More careful examination of the on/off ratios incurred by the three polymers revealed that although all of the polymers were able to yield similar ‘on’ currents, p(p2T-TT) and p(b2T-TT) were also able to achieve around one order of magnitude lower ‘off’ currents than p(g2T-TT), thus leading to an equally sized increment in the recorded ON/OFF ratios, see Fig. 2d and Table 2. As outlined in the literature, a reduction in the recorded OECT ‘OFF’ currents is indicative of a reduced propensity of polymers to spontaneously undergo undesirable faradaic oxygen reduction reactions during OECT operation.29 Thus, while leaving the nature of the conjugated polymer backbone fully unaltered, substitution of the EG side chains by more hydrophobic oligoether ones is a simple, yet effective strategy to minimise these parasitic reactions. Note that the origin of this behaviour here is not due to varying the electron density on the conjugated polymer backbone,29,30 but instead due to the use of more hydrophobic oligoether chains.
Transconductance (gm) values of 15.8 ± 1.0, 15.1 ± 2.1, and 19.4 ± 2.2 mS were obtained for p(g2T-TT), p(p2T-TT), and p(b2T-TT), respectively. As follows from the transconductance (gm) equation, gm = (Wd/L) μC* (VTh–VG), gm depends on both device geometry (where W, d, and L are the channel width, thickness, and length, respectively) and material dependent (where μ is the electronic charge carrier mobility and C* the volumetric capacitance) terms.50 Therefore, a more suitable and less device geometry-dependent performance comparison across various channel materials can be achieved by comparison of the individual values recorded for μ and C*.
Electronic charge carrier mobilities of the polymers were calculated using transfer curves at saturation VD. Here, a mobility of 1.82 ± 0.09 cm2 V−1 s−1 was extracted for p(g2T-TT), see Fig. 2e and Table 2, which was higher than the reported value of 0.95 cm2 V−1 s−1, 34 This difference can be ascribed to the higher molecular weight of this particular batch. p(p2T-TT) and p(b2T-TT), on the other hand, afforded mobilities of 1.76 ± 0.17 and 3.03 ± 0.38 cm2 V−1 s−1 respectively. To understand the recorded electronic charge carrier mobility trend, we evaluated the polymers’ microstructure through atomic force microscopy (AFM), see Fig. S34 (ESI†). Here, polymer films were evaluated in their as-cast, passively swollen, and electrochemically doped states. As indicated by Fig. S34 (ESI†) all as cast-films showed relatively smooth surface morphologies, with root-mean square (RMS) roughness values of 1.61, 1.29, and 1.23 nm recorded for p(g2T-TT), p(p2T-TT), and p(b2T-TT), respectively. Nonetheless, compared to the reference polymer p(g2T-TT), p(p2T-TT) and p(b2T-TT) exhibited larger fibrillar structures, which have been shown to be beneficial towards improved electronic charge carrier transport.51 Importantly, these structures were also retained after exposing p(p2T-TT) and p(b2T-TT) to an aqueous electrolyte and following electrochemical doping, see Fig. S34 (ESI†).
Electrochemical impedance spectroscopy (EIS) was conducted to further elucidate differences in the polymers’ OECT performance. We fabricated a microelectrode array with gold patterns of varying sizes that were coated with the various polymers. We subjected these polymer films to an applied bias matching the one of the gate voltage (VG) incurring the maximum steady-state performance.25C* values were estimated by calculating the effective capacitance (Ceff) at 0.1 Hz and normalising this value to the volume of the polymer on the electrode. For all polymers, a good linear fit of capacitance vs. volume was observed, see Fig. S35 (ESI†). As follows from Fig. 2f and Table 2, C* decreased from 171.4 ± 8.41 to 113.93 ± 5.07 to 103.21 ± 8.17 F cm−3 across p(g2T-TT), p(b2T-TT), and p(p2T-TT). The lower C* values obtained for p(p2T-TT) and p(b2T-TT) were attributed to the effective dilution of the redox active conjugated polymer backbone fraction upon increasing the molecular mass of the insulating side chains.35,38 Similarly, the trend in C* can also be understood when describing the polymers volumetric capacitance in terms of sites in which ions can be incorporated from the electrolyte to balance the electronic charges that are injected into the conjugated polymer film from a metal electrode.52 Here, the presence of additional methylene units in the PG and BG side chains effectively increases the volume per site (vide grazing incidence wide-angle X-ray scattering data), thus resulting in an overall lower site density in the conjugated polymer film and lower volumetric capacitance.
Grazing incidence wide angle X-ray scattering (GIWAXS) analysis was conducted on dry as-cast polymer films, see Fig. S36 and S37 (ESI†), to evaluate the polymers’ microstructure. As can be seen, p(g2T-TT) and p(p2T-TT) adopted a predominantly edge-on orientation, while p(b2T-TT) a face-on one. Similar π–π stacking distances around 3.55 ± 0.06 Å were obtained for all the polymers, which are comparable with those previously recorded for p(g2T-TT).34,40 On the other hand increasing the length of the pendant side chains from EG to PG to BG resulted in progressively larger lamellar spacings (100), with p(g2T-TT), p(p2T-TT), and p(b2T-TT) yielding values of 17.4, 20.9, and 25.1 Å, respectively. As mentioned before, this can be traced back to the inclusion of additional methylene units within the OMIEC's side chains, which in turn are also likely responsible for increasing the average site volume and therefore the decreased C* recorded for p(p2T-TT) and p(b2T-TT).
The steady-state OECT performance data was complemented by recording OECT ON (τon) and OFF (τoff) switching times, which were measured by applying square voltage pulses at the gate electrode, see Fig. S32 (ESI†). Low τon values of 456.6 μs and τoff values of 139.55 μs were recorded for p(g2T-TT). However, both p(p2T-TT) and p(b2T-TT) exhibited higher values, of the order of ms, with τon = 1.11 ms for p(p2T-TT) and τon = 30.04 ms for p(b2T-TT), which we attributed to the increased energetics of ion injection. On the other hand, similar τoff values in the order of ms were obtained for the more hydrophobic polymers. We also calculated the cut-off frequency of each OECT using bandwidth vs. frequency plots (Fig. S32 d–f, ESI†), which were in agreement with the values extracted from the measurements relying on using square shaped pulses at the gate electrode. Table S1 (ESI†) reports a summary of the transient characteristics. Overall, the devices became slower when made from polymers with increased hydrophobicity.
Finally, to validate our results and determine whether EG oligoether chain substitution with PG and BG-based ones does indeed lead to reduced water uptake and hence reduced volumetric swelling during electrochemical doping, we conducted electrochemical quartz crystal microbalance with dissipation monitoring (eQCM-D) measurements, see Fig. 3. Polymer-coated gold substrates were first exposed to air to quantify both the mass and thickness of the deposited polymer films (see Fig. S38 and S39 (ESI†) for the raw QCM-D data and the real time mass changes, respectively). Next, their passive swelling (swelling at 0.0 V vs. VOC) was assessed by exposure to an aqueous 0.1 M NaCl electrolyte, hence the same electrolyte that had been employed to assess the polymers’ OECT performance. As illustrated from Fig. 3a, immersion of the polymers into an aqueous electrolyte in the absence of an applied voltage (passive swelling) led to a thickness increase for all polymers, due to the diffusion of water molecules and hydrated ions into the films. The largest passive swelling of 20% was recorded for the EG functionalised p(g2T-TT) polymer. Increasing the hydrophobicity in the polymers’ side chains instead decreased the polymers’ degree of passive swelling, with p(p2T-TT) and p(b2T-TT) only swelling by 5% and 13% respectively, hence suggesting a reduced tendency to take up water molecules. To gain a better insight into the polymers’ interaction with water molecules under more device relevant conditions, an external electrical bias was applied to the polymer-coated quartz crystals, with their volumetric swelling and water uptake as a function of applied potential given in Fig. 3a. Doping pulses were applied against the open-circuit potential (VOC), therefore compensating for any changes in the polymers’ threshold voltage.40 Fig. S40 (ESI†) shows the current versus time traces that were recorded from the films on the QMC-D crystals upon application of these potentials. The increase in the current start as soon as 0.1 V was applied on the films with respect to their VOC measured against the Ag/AgCl reference electrode. As shown in Fig. 3a, application of increasingly positive voltages resulted in additional polymer swelling and corroborating previous findings on EG functionalised conjugated polymers.40,53,54 Specifically, p(g2T-TT), p(p2T-TT), and p(b2T-TT) swelled to 39%, 31%, and 29% of their initial dry volume upon application of a +0.3 V doping potential relative to VOC, respectively. To understand the origin of the polymers’ swelling tendency upon electrochemical biasing, the amount of injected chloride anions and water molecules at each doping pulse was calculated, see Fig. 3b and c. As shown in Fig. 3b, a similar density of Cl− anions is injected into p(p2T-TT) and p(b2T-TT) polymer films when applying doping potentials up to 0.6 V vs. Ag/AgCl. However, for the more hydrophilic p(g2T-TT), the Cl− density is largest at all doping potentials, reaching about 11 × 1020 molecules per cm−3 at the highest doping voltage that the film can withstand. This trend in anion uptake is reflected in our volumetric capacitance measurements, with p(g2T-TT) having the highest C*, while p(p2T-TT) and p(b2T-TT) afforded similar and lower C* than p(g2T-TT). Next, the number of injected water molecules upon electrochemical doping was calculated according to the literature,40 see Fig. 3c. From Fig. 3c it can be seen that while p(g2T-TT) under all biasing conditions takes up more water molecules than the other two polymers. The increase in doping bias, on the other hand, causes p(p2T-TT) to take more water free or solvated water molecules than the most hydrophobic polymer of the series, p(b2T-TT). Specifically, when applying a +0.3 V vs. VOC bias, p(g2T-TT), p(p2T-TT), and p(b2T-TT) took up 1.16 × 1022, 1.04 × 1022, and 9.3 × 1021 H2O molecules per cm3, respectively. Consequently, increasing the hydrophobicity in of the polymers’ pendant oligoether side chains leads to a reduction in the number of water molecules taken up during electrochemical doping. In particular, the reduced water uptake in p(b2T-TT) to a reduced disruption of the structural order compared to p(g2T-TT) during electrochemical doping,25,40 therefore, incurring higher mobilities. However, the low water uptake had no correlation with device stability (see Fig. S33, ESI†) which is mainly controlled by the magnitude of the voltages applied at the gate and drain contacts. Ultimately, our results show that while polymer swelling upon electrolyte exposure and electrochemical biasing is a fundamental requirement to enable OECT operation, polymer swelling must be carefully balanced to minimise structural changes in the polymer films, which can otherwise reduce the electronic charge mobilities, and therefore maximise OECT performance.
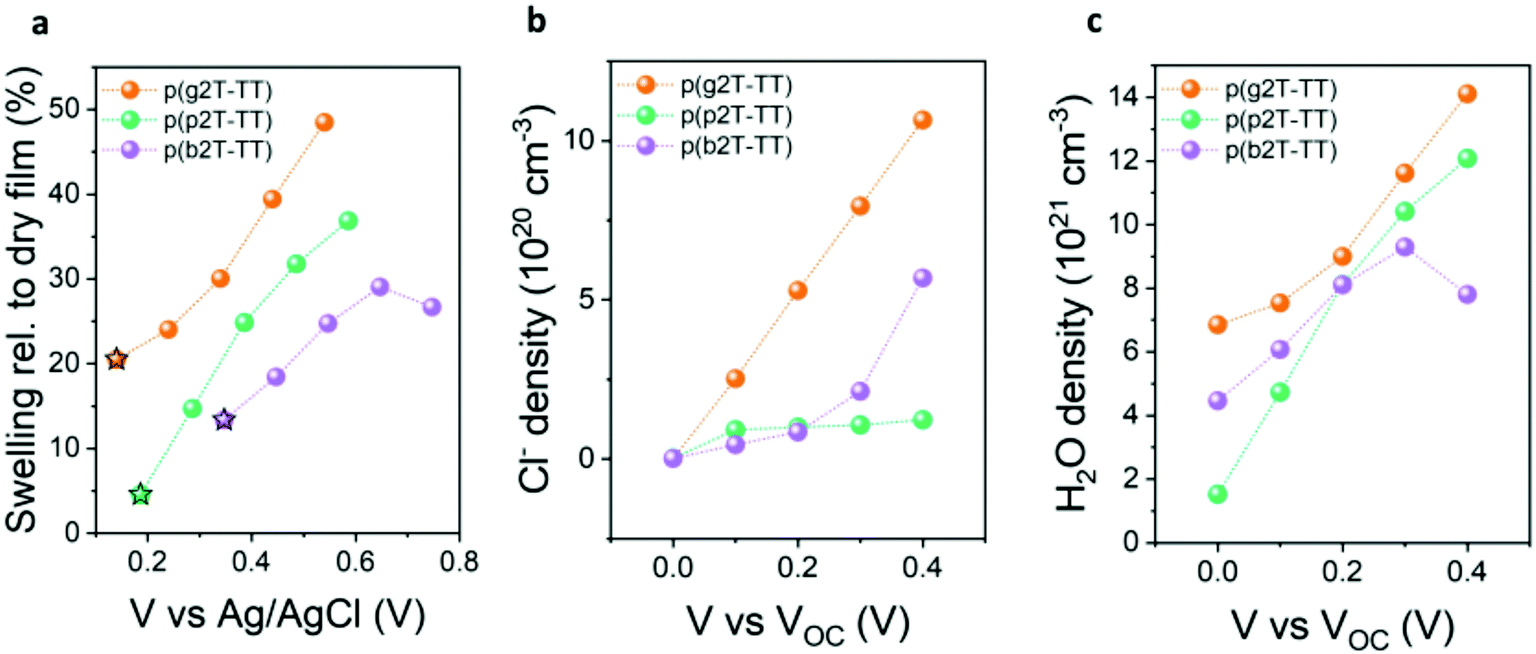 | ||
| Fig. 3 (a) Polymer swelling relative to the corresponding dry state. Star symbols correspond to the swelling values of the film at VOC conditions (i.e., passive swelling). The voltage values are plotted vs. Ag/AgCl. (b) Chloride ion and (c) water molecule density in the conjugated polymer film following electrochemical doping at various potentials against VOC. VOC values are 0.14 V for p(g2T-TT), 0.19 V for p(p2T-TT), and 0.35 V for p(b2T-TT). | ||
Conclusion
In summary we synthesised a series of organic mixed ionic-electronic conductors based on an oligoether functionalised bithiophene–thienothiophene conjugated backbone for OECT applications. Copolymers were either functionalised with the traditionally employed ethylene glycol side chains as the reference p(g2T-TT) polymer or with their more hydrophobic propylene and butylene glycol counterparts to afford p(p2T-TT) and p(b2T-TT), respectively. In OECTs, substitution of the EG side chains in p(g2T-TT) with BG in p(b2T-TT) resulted in an improvement in the electronic charge carrier mobility, while preserving high volumetric capacitance values, therefore leading to an overall superior OECT performance compared to the p(g2T-TT) benchmark. The improved OECT performance of p(b2T-TT) is suggested to arise due to this polymer's incurring larger and more fibrillar microstructures that are preserved during electrochemical doping in water, thereby aiding charge carrier transport. In parallel, the polymers’ more hydrophobic side chains were also able to minimise the amount of water molecules taken up per chloride counterion, thereby also reducing the degree of polymer swelling and the structural changes that impair hole mobility. Ultimately, this work presents a new approach to augment the signal transduction performance of organic mixed ionic-electronic conductors in bioelectronic devices through a synthetic approach, thus foregoing the necessity to manipulate the electrolyte of the biological entity under study to incur optimum performances.Author contributions
Maximilian Moser – synthesised the polymers and wrote the manuscript. Yazhou Wang – synthesised the monomers and polymers, did all the characterisation of the monomers and polymers, including the 1HNMR, 13C NMR and MS, measured and analysised the GIWAX data. Tania Cecilia Hidalgo fabricated the OECTs, collected the device data, performed EIS and EQCM-D measurements, did data analysis, and assisted in writing the manuscript. Hailiang Liao, did the UV-vis absorption spectra. Yaping Yu, did the CV in organic phase and aqueous. Junxin Chen, helped to analysis the GIWAX data. Jiayao Duan, assisted in the measurement and analysis of the data. Floriana Moruzzi, synthesised propylene glycol side chains used in the polymers. Sophie Griggs, synthesised the butylene glycol side chains used in the polymers. Adam Marks, synthesised the thienothiophene monomers. Nicola Gasparini, assisted in the measurement and analysis of the data. Andrew Wadsworth, managed the synthetic plan. Sahika Inal managed the device fabrication and measurement and electrochemical characterization of the films. Iain McCulloch managed the project, designed the polymers and co-wrote the manuscript. Wan Yue, co-managed the project, planned the experiments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from KAUST, including Office of Sponsored Research (OSR) awards no. OSR-2018-CRG/CCF-3079, OSR-2019-CRG8-4086 and OSR-2018-CRG7-3749. The authors acknowledge funding from National Research Foundation of China (21875291 and 21702240) and China Postdoctoral Foundation (Grant No. 2021M693580) for the financial support, ERC Synergy Grant SC2 (610115), the European Union's Horizon 2020 research and innovation program under grant agreement no. 952911, project BOOSTER and grant agreement no. 862474, project RoLAFLEX, as well as EPSRC Project EP/T026219/1.References
- A. Malti, J. Edberg, H. Granberg, Z. U. Khan, J. W. Andreasen, X. Liu, D. Zhao, H. Zhang, Y. Yao, J. W. Brill, I. Engquist, M. Fahlman, L. Wågberg, X. Crispin and M. Berggren, Adv. Sci., 2016, 3, 1500305–1500313 CrossRef PubMed.
- A. M. Österholm, J. F. Ponder, J. A. Kerszulis and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2016, 8, 13492–13498 CrossRef PubMed.
- G. Hernández, N. Casado, A. M. Zamarayeva, J. K. Duey, M. Armand, A. C. Arias and D. Mecerreyes, ACS Appl. Energy Mater., 2018, 1, 7199–7205 CrossRef.
- D. Moia, A. Giovannitti, A. A. Szumska, I. P. Maria, E. Rezasoltani, M. Sachs, M. Schnurr, P. R. F. Barnes, I. McCulloch and J. Nelson, Energy Environ. Sci., 2019, 12, 1349–1357 RSC.
- A. L. Dyer, E. J. Thompson and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2011, 3, 1787–1795 CrossRef CAS PubMed.
- J. F. Ponder, A. M. Österholm and J. R. Reynolds, Chem. Mater., 2017, 29, 4385–4392 CrossRef CAS.
- J. Rivnay, R. M. Owens and G. G. Malliaras, Chem. Mater., 2014, 26, 679–685 CrossRef CAS.
- D. T. Simon, E. O. Gabrielsson, K. Tybrandt and M. Berggren, Chem. Rev., 2016, 116, 13009–13041 CrossRef CAS PubMed.
- J. Rivnay, S. Inal, A. Salleo, R. M. Owens, M. Berggren and G. G. Malliaras, Nat. Rev. Mater., 2018, 3, 17086 CrossRef CAS.
- D. Khodagholy, J. Rivnay, M. Sessolo, M. Gurfinkel, P. Leleux, L. H. Jimison, E. Stavrinidou, T. Herve, S. Sanaur, R. M. Owens and G. G. Malliaras, Nat. Commun., 2013, 4, 2133 CrossRef PubMed.
- S. T. M. Tan, A. Giovannitti, A. Melianas, M. Moser, B. L. Cotts, D. Singh, I. McCulloch and A. Salleo, Adv. Funct. Mater., 2021, 2010868 CrossRef CAS.
- X. Strakosas, M. Bongo and R. M. Owens, J. Appl. Polym. Sci., 2015, 132, 41735–41748 CrossRef.
- A.-M. Pappa, V. F. Curto, M. Braendlein, X. Strakosas, M. J. Donahue, M. Fiocchi, G. G. Malliaras and R. M. Owens, Adv. Healthcare Mater., 2016, 5, 2295–2302 CrossRef CAS PubMed.
- D. Ohayon, G. Nikiforidis, A. Savva, A. Giugni, S. Wustoni, T. Palanisamy, X. Chen, I. P. Maria, E. Di Fabrizio, P. M. F. J. Costa, I. McCulloch and S. Inal, Nat. Mater., 2020, 19, 456–463 CrossRef CAS.
- A. Koklu, S. Wustoni, V.-E. Musteata, D. Ohayon, M. Moser, I. McCulloch, S. P. Nunes and S. Inal, ACS Nano, 2021, 15, 8130–8141 CrossRef CAS PubMed.
- D. Khodagholy, T. Doublet, P. Quilichini, M. Gurfinkel, P. Leleux, A. Ghestem, E. Ismailova, T. Hervé, S. Sanaur, C. Bernard and G. G. Malliaras, Nat. Commun., 2013, 4, 1575–1581 CrossRef PubMed.
- J. Rivnay, P. Leleux, M. Ferro, M. Sessolo, A. Williamson, D. A. Koutsouras, D. Khodagholy, M. Ramuz, X. Strakosas, R. M. Owens, C. Benar, J. M. Badier, C. Bernard and G. G. Malliaras, Sci. Adv., 2015, 1, e1400251 CrossRef.
- W. Lee, D. Kim, N. Matsuhisa, M. Nagase, M. Sekino, G. G. Malliaras, T. Yokota and T. Someya, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10554–10559 CrossRef CAS.
- A.-M. Pappa, H.-Y. Liu, W. Traberg-Christensen, Q. Thiburce, A. Savva, A. Pavia, A. Salleo, S. Daniel and R. M. Owens, ACS Nano, 2020, 14, 12538–12545 CrossRef CAS.
- J. Rivnay, S. Inal, B. A. Collins, M. Sessolo, E. Stavrinidou, X. Strakosas, C. Tassone, D. M. Delongchamp and G. G. Malliaras, Nat. Commun., 2016, 7, 11287–11295 CrossRef.
- E. Stavrinidou, P. Leleux, H. Rajaona, D. Khodagholy, J. Rivnay, M. Lindau, S. Sanaur and G. G. Malliaras, Adv. Mater., 2013, 25, 4488–4493 CrossRef CAS.
- C. B. Nielsen, A. Giovannitti, D.-T. Sbircea, E. Bandiello, M. R. Niazi, D. A. Hanifi, M. Sessolo, A. Amassian, G. G. Malliaras, J. Rivnay and I. McCulloch, J. Am. Chem. Soc., 2016, 138, 10252–10259 CrossRef CAS.
- M. Moser, J. F. Ponder, A. Wadsworth, A. Giovannitti and I. McCulloch, Adv. Funct. Mater., 2019, 29, 1807033 CrossRef.
- S. Inal, G. G. Malliaras and J. Rivnay, Nat. Commun., 2017, 8, 1–7 CrossRef CAS PubMed.
- M. Moser, T. C. Hidalgo, J. Surgailis, J. Gladisch, S. Ghosh, R. Sheelamanthula, Q. Thiburce, A. Giovannitti, A. Salleo, N. Gasparini, A. Wadsworth, I. Zozoulenko, M. Berggren, E. Stavrinidou, S. Inal and I. McCulloch, Adv. Mater., 2020, 32, 2002748 CrossRef CAS PubMed.
- S.-M. Kim, C.-H. Kim, Y. Kim, N. Kim, W.-J. Lee, E.-H. Lee, D. Kim, S. Park, K. Lee, J. Rivnay and M.-H. Yoon, Nat. Commun., 2018, 9, 3858 CrossRef PubMed.
- H. Sun, J. Gerasimov, M. Berggren and S. Fabiano, J. Mater. Chem. C, 2018, 6, 11778–11784 RSC.
- E. Zeglio and O. Inganäs, Adv. Mater., 2018, 30, 1800941 CrossRef PubMed.
- A. Giovannitti, R. B. Rashid, Q. Thiburce, B. D. Paulsen, C. Cendra, K. Thorley, D. Moia, J. T. Mefford, D. Hanifi, D. Weiyuan, M. Moser, A. Salleo, J. Nelson, I. McCulloch and J. Rivnay, Adv. Mater., 2020, 32, 1908047 CrossRef CAS PubMed.
- M. Moser, A. Savva, K. Thorley, B. D. Paulsen, T. C. Hidalgo, D. Ohayon, H. Chen, A. Giovannitti, A. Marks, N. Gasparini, A. Wadsworth, J. Rivnay, S. Inal and I. McCulloch, Angew. Chem., Int. Ed., 2021, 60, 7777–7785 CrossRef CAS PubMed.
- X. Chen, A. Marks, B. D. Paulsen, R. Wu, R. B. Rashid, H. Chen, M. Alsufyani, J. Rivnay and I. McCulloch, Angew. Chem., Int. Ed., 2021, 60, 9368–9373 CrossRef CAS PubMed.
- H. Jia, Z. Huang, P. Li, S. Zhang, Y. Wang, J.-Y. Wang, X. Gu and T. Lei, J. Mater. Chem. C, 2021, 9, 4927–4934 RSC.
- G. Krauss, F. Meichsner, A. Hochgesang, J. Mohanraj, S. Salehi, P. Schmode and M. Thelakkat, Adv. Funct. Mater., 2021, 2010048 CrossRef CAS.
- A. Giovannitti, D. T. Sbircea, S. Inal, C. B. Nielsen, E. Bandiello, D. A. Hanifi, M. Sessolo, G. G. Malliaras, I. McCulloch and J. Rivnay, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12017–12022 CrossRef CAS PubMed.
- M. Moser, L. R. Savagian, A. Savva, M. Matta, J. F. Ponder, T. C. Hidalgo, D. Ohayon, R. Hallani, M. Reisjalali, A. Troisi, A. Wadsworth, J. R. Reynolds, S. Inal and I. McCulloch, Chem. Mater., 2020, 32, 6618–6628 CrossRef CAS.
- Y. Wang, E. Zeglio, H. Liao, J. Xu, F. Liu, Z. Li, I. P. Maria, D. Mawad, A. Herland, I. McCulloch and W. Yue, Chem. Mater., 2019, 31, 9797–9806 CrossRef CAS.
- P. Schmode, A. Savva, R. Kahl, D. Ohayon, F. Meichsner, O. Dolynchuk, T. Thurn-Albrecht, S. Inal and M. Thelakkat, ACS Appl. Mater. Interfaces, 2020, 12, 13029–13039 CrossRef CAS PubMed.
- A. Savva, R. Hallani, C. Cendra, J. Surgailis, T. C. Hidalgo, S. Wustoni, R. Sheelamanthula, X. Chen, M. Kirkus, A. Giovannitti, A. Salleo, I. McCulloch and S. Inal, Adv. Funct. Mater., 2020, 30, 1907657 CrossRef CAS.
- A. Giovannitti, I. P. Maria, D. Hanifi, M. J. Donahue, D. Bryant, K. J. Barth, B. E. Makdah, A. Savva, D. Moia, M. Zetek, P. R. F. Barnes, O. G. Reid, S. Inal, G. Rumbles, G. G. Malliaras, J. Nelson, J. Rivnay and I. McCulloch, Chem. Mater., 2018, 30, 2945–2953 CrossRef CAS.
- A. Savva, C. Cendra, A. Giugni, B. Torre, J. Surgailis, D. Ohayon, A. Giovannitti, I. McCulloch, E. Di Fabrizio, A. Salleo, J. Rivnay and S. Inal, Chem. Mater., 2019, 31, 927–937 CrossRef CAS.
- N. Yunos, R. Bellomo, D. Story and J. Kellum, Crit. Care, 2010, 14, 226 CrossRef PubMed.
- X. Niu, R. A. Bressan, P. M. Hasegawa and J. M. Pardo, Plant Physiol., 1995, 109, 735–742 CrossRef CAS PubMed.
- M. Watanabe, S. Nagano, K. Sanui and N. Ogata, Solid State Ionics, 1986, 18–19, 338–342 CrossRef CAS.
- Z. Xue, D. He and X. Xie, J. Mater. Chem. A, 2015, 3, 19218–19253 RSC.
- S. L. Hager and T. B. Macrury, J. Appl. Polym. Sci., 1980, 25, 1559–1571 CrossRef CAS.
- I. Zozoulenko, A. Singh, S. K. Singh, V. Gueskine, X. Crispin and M. Berggren, ACS Appl. Polym. Mater., 2019, 1, 83–94 CrossRef CAS.
- C. Enengl, S. Enengl, S. Pluczyk, M. Havlicek, M. Lapkowski, H. Neugebauer and E. Ehrenfreund, Chem. Phys. Chem., 2016, 17, 3830 CrossRef CAS.
- L. R. Savagian, A. M. Österholm, J. F. Ponder, K. J. Barth, J. Rivnay and J. R. Reynolds, Adv. Mater., 2018, 30, 1804647 CrossRef PubMed.
- X. Wu, A. Surendran, M. Moser, S. Chen, B. T. Muhammad, I. P. Maria, I. Mcculloch and W. L. Leong, ACS Appl. Mater. Interfaces, 2020, 12, 20757–20764 CrossRef CAS PubMed.
- D. A. Bernards and G. G. Malliaras, Adv. Funct. Mater., 2007, 17, 3538–3544 CrossRef CAS.
- X. Wu, A. Surendran, J. Ko, O. Filonik, E. M. Herzig, P. Müller-Buschbaum and W. L. Leong, Adv. Mater., 2019, 31, 1805544 CrossRef PubMed.
- C. M. Proctor, J. Rivnay and G. G. Malliaras, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 1433–1436 CrossRef CAS.
- J. Gladisch, E. Stavrinidou, S. Ghosh, A. Giovannitti, M. Moser, I. Zozoulenko, I. McCulloch and M. Berggren, Adv. Sci., 2020, 7, 1901144 CrossRef CAS PubMed.
- M. Moser, J. Gladisch, S. Ghosh, T. C. Hidalgo, J. F. Ponder, R. Sheelamanthula, Q. Thiburce, N. Gasparini, A. Wadsworth, A. Salleo, S. Inal, M. Berggren, I. Zozoulenko, E. Stavrinidou and I. McCulloch, Adv. Funct. Mater., 2021, 31, 2100723 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1mh01889b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |