
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganic building blocks at inorganic nanomaterial interfaces
Yunping
Huang
 a,
Theodore A.
Cohen
b,
Breena M.
Sperry
a,
Helen
Larson
c,
Hao A.
Nguyen
c,
Micaela K.
Homer
c,
Florence Y.
Dou
c,
Laura M.
Jacoby
c,
Brandi M.
Cossairt
c,
Daniel R.
Gamelin
c and
Christine K.
Luscombe†
*abc
a,
Theodore A.
Cohen
b,
Breena M.
Sperry
a,
Helen
Larson
c,
Hao A.
Nguyen
c,
Micaela K.
Homer
c,
Florence Y.
Dou
c,
Laura M.
Jacoby
c,
Brandi M.
Cossairt
c,
Daniel R.
Gamelin
c and
Christine K.
Luscombe†
*abc
aDepartment of Materials Science & Engineering, University of Washington, Seattle, WA 98195, USA. E-mail: christine.luscombe@oist.jp
bMolecular Engineering & Sciences Institute, University of Washington, Seattle, WA 98195, USA
cDepartment of Chemistry, University of Washington, Seattle, WA 98195, USA
First published on 1st December 2021
Abstract
This tutorial review presents our perspective on designing organic molecules for the functionalization of inorganic nanomaterial surfaces, through the model of an “anchor-functionality” paradigm. This “anchor-functionality” paradigm is a streamlined design strategy developed from a comprehensive range of materials (e.g., lead halide perovskites, II–VI semiconductors, III–V semiconductors, metal oxides, diamonds, carbon dots, silicon, etc.) and applications (e.g., light-emitting diodes, photovoltaics, lasers, photonic cavities, photocatalysis, fluorescence imaging, photo dynamic therapy, drug delivery, etc.). The structure of this organic interface modifier comprises two key components: anchor groups binding to inorganic surfaces and functional groups that optimize their performance in specific applications. To help readers better understand and utilize this approach, the roles of different anchor groups and different functional groups are discussed and explained through their interactions with inorganic materials and external environments.
1. Introduction
Inorganic nanomaterials possess unique physical, chemical, electronic, and optical properties different from their bulk counterparts. Such properties stem from their high surface areas and from quantum confinement effects,1–7 opening new opportunities for innovative technology concepts. For example, the high efficiency and narrow emission of quantum dots (QDs) promote energy efficiency and color quality in TV and cell phone displays, attracting significant attention in the commercial market.8,9 Meanwhile, many other technologies based on inorganic nanomaterials are currently undergoing rapid development towards future commercialization, covering both conventional semiconductor domains (e.g., solar cells10,11 and light-emitting diodes12–15) and newly emergent technology concepts (e.g., photocatalysis,16,17 nanomedicine,18,19 spintronics20–22 and quantum computing23,24).With at least one external dimension reduced to below 100 nm, the surface area to volume ratios of nanomaterials are larger than those of bulk materials by orders of magnitude. Additionally, these terminal surfaces tend to form chemical bonds with or otherwise absorb nearby molecules to reach a thermodynamically stable state. Depending on the size of the incoming molecules, the interfacial layer thickness can be as small as ∼0.1 nm in the case of H-terminated diamonds or oxides, or as large as ∼2 nm when polymers are adsorbed onto a QD surface.25 Thin as they are, such alteration of the surface can induce dramatic changes to the material's properties. For example, a hydrogen mono-layer on the surface of diamond thin films induces band bending extending as deep as 150 nm below the interface,26 and subsequently a ∼105 increase in conductivity.27–29 Similar electronic effects are observed in QDs as well, although now with the crystal's physical dimensions often smaller than the depletion widths. For example, surface dipoles generated by mono-layers of organic molecules on QD surfaces can tune band-edge potentials of PbS QDs by over 1 eV.30,31 The effects of interface engineering are not limited to the manipulation of electronic structures, but also dispersity, molecular interaction, and the stability of the nanomaterials, which will be discussed in the following sections. These examples highlight the efficacy of interface engineering as an attractive complement to conventional methods such as composition and phase engineering to regulate the properties of inorganic nanomaterials.
Controlled-atmosphere annealing and plasma treatment are common conventional methods for the modification of inorganic surfaces. However, these methods give rather limited choices for surface functionalization, terminating the material surface with hydrogen, oxygen, nitrogen, or chlorine atoms.32 Fortunately, this limitation has been alleviated by the collaborative efforts from both inorganic and organic chemists in the past decade. Taking advantage of the structural flexibility and tunability of organic compounds, a variety of organic molecules were introduced to the surfaces of inorganic and related nanomaterials, showing great potential for different material types (e.g., lead-halide perovskites, II–VI semiconductors, III–V semiconductors, metal oxides, diamond, carbon dots, silicon, etc.), and for different applications (e.g., light-emitting diodes (LEDs), photovoltaics (PVs), lasers, photonic cavities, photocatalysis, fluorescent imaging, photodynamic therapy, drug delivery, etc.). Several reviews have comprehensively summarized these surface modification methods regarding one or several specific types of materials or applications.32–35 On the other hand, research in inorganic nanomaterials is quickly evolving, with new materials being developed and applications expanding. To guide and facilitate developments of emerging materials and applications, it is crucial to generalize the principles of interface engineering by summarizing a wide scope of previous examples in both material types and applications.
In this tutorial review, we share a generalized molecular paradigm for organic molecules used for interface modification of inorganic nanomaterials - the “anchor-functionality” design shown in Fig. 1. The anchor group is a building block that binds to nanomaterial surfaces, either through ionic or covalent bonding depending on the target nanomaterial; the functional group is a building block that endows the nanomaterials with targeted functionalities to cater to different requirements depending on the different applications. Once the anchor group and the functional group are selected, a linker (usually inert alkyl chains or polymer backbones) covalently attaches the anchor group and the functional group. The linker, anchor, and functional groups together constitute an interface modifier. The interaction between anchor groups and different types of inorganic materials will be discussed in Section 2. In Section 3, we discuss the mechanism by which functional groups endow new functionalities to nanomaterials. Considering this field is a highly interdisciplinary field with researchers from different backgrounds (including organic chemists, inorganic chemists, physicists, healthcare professionals), the “anchor-functionality” paradigm offers researchers with a non-chemistry background an accessible toolset to understand interface molecules. We hope that this will help bridge the knowledge gap between researchers and facilitate in-depth discussion for future interdisciplinary collaborations, expediting solutions to current challenges leading to future innovations.
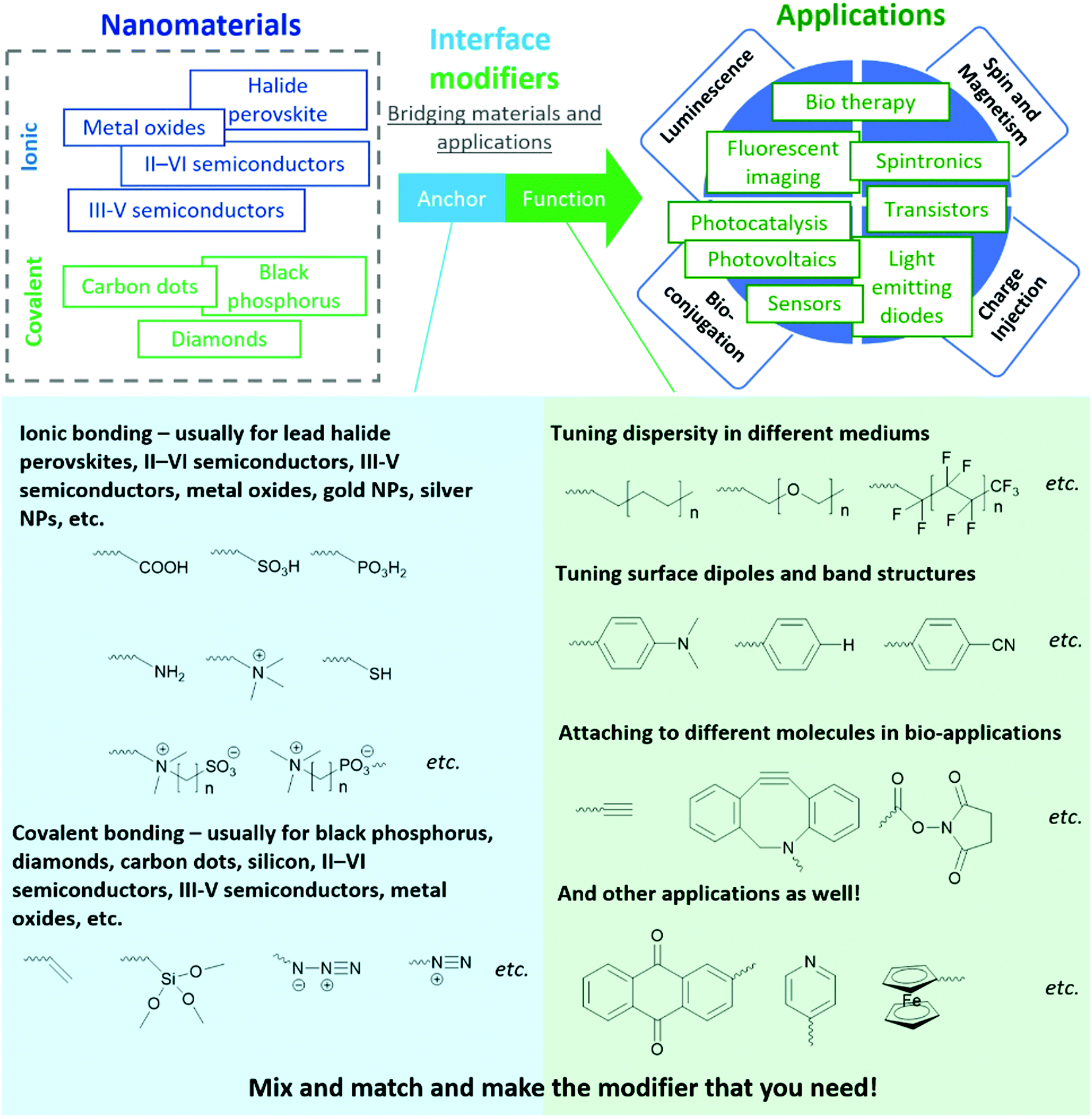 | ||
| Fig. 1 An interface modifier can be seen as molecule comprised of two key components: (i) anchor groups that attaches to inorganic nanomaterials, and (ii) functional groups that tune the properties of inorganic nanomaterials to better suit the target applications. Some examples of anchors and functional groups are shown here. | ||
2. Anchor groups for different interfaces
The binding modes, affinities, and types of capping ligands that can be bound to the surfaces of nanomaterials depend on the nature of the ligand-surface interactions. Nanocrystals (NCs) can be broadly categorized in terms of their lattice ionicity, which spans from purely covalent (diamond) to highly ionic (metal-halide perovskites), and this ionicity frequently dictates the nature of the interface. Such a variety of lattice features requires matching to different anchor groups to form a molecular interaction between ligands and their target nanomaterials. Therefore, in Section 2, we highlight the common inorganic nanomaterial surfaces and explain their interactions with different anchor groups, to elucidate the “anchor” component in the “anchor-functionality” design.The synthesis and processing of metal-halide perovskite NCs is a good example to illustrate the anchor group interactions at the surface of an ionic lattice. Oleylammonium is often used as a ligand to stabilize perovskite NCs and to prevent large aggregate formation during grain growth.36 In this case, the ammonium group is the anchor group of the ligand attaching to the surfaces of the NCs. Due to the high ionicity of the perovskite lattice, the ammonium groups desorb and reattach to the perovskite surface in a dynamic equilibrium, which allows the precursor ions (e.g., Pb2+, Cs+, I−) to freely move into the lattice and grow the NC core structure. Moreover, this ionic interaction allows facile ligand exchange after NC synthesis, offering a convenient method to further tailor the NC properties.
When an ionic core is shelled by a more covalent lattice, the ionic core is shielded, and ligand exchange is inhibited. For example, the core of CdSe QDs will no longer be able to react once the surface is terminated with a silica shell. On the other hand, such strong covalent bonds provide excellent isolation that shields the CdSe core from external molecules, and thus silica shelling is a common method used to enhance the stability of QDs.
In the following sections, we discuss examples of anchor-surface interactions based on these two different lattice types. In the ionic lattice section (Section 2.1), we discuss II–VI semiconductors, III-V semiconductors, metal oxides, I–III–VI nanoparticles, and lead halide perovskites. The covalent lattice surface section (Section 2.2) discusses surface binding for diamond, silicon, and silica. Although we are not able to include all material types, we believe the above examples are sufficient to explain the interactions between anchors and nanomaterials and demonstrate the wide application scope of this “anchor-functionality” paradigm.
2.1 Anchor groups for surfaces of ionic lattices
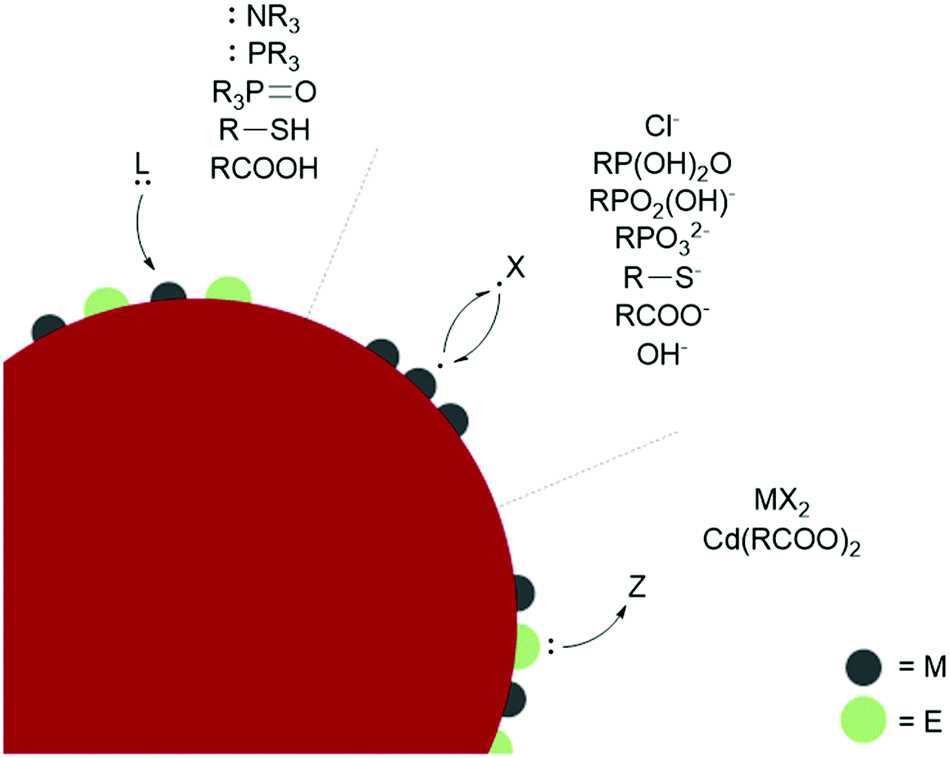 | ||
| Fig. 2 Classification of ligand anchor groups on representative metal chalcogenide (ME) NCs. | ||
L-type ligands coordinate to electron-poor sites on the NC surfaces. The most common L-type NC capping ligands are amines, phosphines, phosphine oxides, thiols, and carboxylic acids (Fig. 2). These ligands are neutral and passivate stoichiometric facets or charge neutral sites of a NC surface. For ligand exchange performed in nonpolar solvents, there is no charge barrier to L-type ligand dissociation. As a result, L-type ligand exchange generally occurs through a dissociative pathway to relieve steric crowding, yielding a dynamic surface of bound and unbound ligands.38 Two materials that can employ L-type species as native ligands during synthesis are CdSe (II–VI) and CuInS2 (I–III–VI). Both provide interesting examples of L-L-type ligand exchange. In an early example, Meijerink and co-workers used the quenching of CdSe photoluminescence to study L-type ligand exchange from native amine, phosphine, and phosphine oxide ligands to thiols.40 The exchange of native amines with thiols was shown to be the most facile, while the exchange with phosphines and phosphine oxides occurred more slowly. Amines, phosphine oxides, and thiols compete to bind to the cadmium surface sites, while the phosphines bound to surface selenides must dissociate to allow the thiol to coordinate to a nearby cadmium. Additionally, the bulkier phosphine and phosphine oxide anchor groups are proposed to slow ligand exchange and prevent nearby surface-ligand interactions.40 More recent studies on CdSe surface chemistry and ligand exchange add additional nuance to these conclusions. For example, it is now understood from NMR spectroscopic investigations that phosphines bind the Lewis acidic metal sites on QD surfaces, rather than perturbing the chalcogenide oxidation states.41 Quantitative 1H and 31P NMR experiments also report contradicting steric and electronic effects on ligand affinity to QD surfaces. The more electron-rich phosphines show a higher affinity for CdSe QD surfaces than isostructural amines, but bulkier ligands have a lower binding affinity despite having more electron-donating substituents due to repulsive interactions between the ligands. Hens and co-workers reported charge-neutral CuInS2 NCs synthesized with octadecylamine and studied ligand exchange with in situ variable temperature NMR experiments.42 Unlike the dynamic surfaces of common L-bound NCs, the amines remained tightly bound at room temperature but became dynamic at higher temperatures. Maintaining charge neutrality at the surface, the amine ligands were shown to exchange with L-type thiols but not with X-type carboxylates during a multi-step ligand exchange in a nonpolar solvent at elevated temperatures. A study of another I–III–VI material, CuInSe2, reported the exchange of L-type amine and phosphine native ligands with thiols or other phosphines assisted by the addition of a charge-neutral Z-type I2 ligand, which is necessary to displace the tightly bound native ligands.43 Z-type ligands were found to exchange with L-type amines in II–VI and IV–VI NCs as well.44–46
Ligand binding is also influenced by facet-dependent surface composition, which is presented here with studies of materials with native X-type ligands. NMR and FTIR techniques have been used to identify chelating and bridging carboxylate X-type ligands passivating the polar {100} and {111} facets of CdSe NCs, while the nonpolar {110} facet is weakly coordinated by modified Z-type cadmium carboxylates.47 PbS NCs showed a similar pattern, where the Pb-rich {111} facets were passivated with X-type oleate and hydroxide ligands, while the stoichiometric {100} facets, which do not require any charge balance, were coordinated by L-type oleic acid.38 Atomically precise semiconductor clusters also provide defined platforms for elucidating ligand binding.45,48 L- and X-type binding modes of carboxylates on InP surfaces were quantified with an analytical 1H NMR method using InP magic sized clusters, and partial X-X-type ligand exchange was shown with thiolates and phosphonates. Phosphonates are known to strongly bind and replace carboxylates in CdSe QDs as well.49 Carboxylic acids passivate metal oxides such as HfO2 and ZrO2 in nonpolar solvents by separating into a carboxylate and a proton, which bind as two separate X-type ligands (sometimes referred to as X2-type) under basic conditions.50 The basicity of surface oxygen ions in metal oxides allows protons to act as ligands, and the resulting dissociative carboxylic acid binding allows for the exchange of the neutral X2-type with L-type ligands.51
The binding affinity of specific anchor groups to different NC surfaces can also be predicted by the hard/soft acid base principle. Within II-VI and metal oxide NCs, surfaces with harder Lewis acidic cations such as Zn2+ in ZnSe or ZnO tend to bind to harder bases such as OH−,52 the softer Cd2+ cations of CdTe bind to softer ligands such as HS−, and CdSe and CdS NCs of intermediate polarizability are ligated by both hard and soft bases.53 The covalency of the lattice affects the classification of the metal sites because the hard acid In3+ behaves as a softer acid in InP and InAs NCs due to the increased sharing of charge density. The strong soft-acid/soft-base bond formed by Au and S means that thiolates are common native ligands for Au NCs. These ligands can be easily exchanged for other soft ligands, but Au NCs are not stabilized by harder anchor groups.53,54 Au, Ag, and other metal NCs are commonly synthesized with capping ligands for stability and in the scope of the ligands discussed above for stabilization in nonpolar solvents. Generally, thiols and phosphines give higher stability for metal NCs than amines, carboxylic acids, and alcohols.55
An exciting recent addition to the nanomaterial surface characterization toolbox is isothermal titration calorimetry (ITC), which can elucidate ligand binding and exchange thermodynamics by quantifying the heat changes upon addition of a ligand titrant.56 ITC can detect ligand binding over a wide range of particle concentrations in their native solvent environment, allowing the results to be compared to both optical and NMR spectroscopy data. As discussed above, NCs often have multiple facets with potentially different binding enthalpies that could be resolved in ITC data. ITC measurements and associated time-trace analyses allowed Greytak and co-workers to identify a two-step ligand exchange and association mechanism for the exchange of native carboxylates with alkylphosphonates on CdSe QDs and to extract thermodynamic parameters.56 The first set of ITC signals included a fast exothermic curve followed by fast endothermic line shape. The authors assigned the exothermic process to the X-type ligand exchange of carboxylates for phosphonates, while the endothermic signal was tentatively attributed to a rapid proton exchange between the incoming and outgoing ligands. Then, after a stoichiometric amount of phosphonate for X-type exchange, the ITC signals changed to a slower exothermic response that was assigned to the L-type association of phosphonic acid to a second set of surface sites. Additional studies of ligand exchange by ITC report the effects of ligand chain length and NC size on the exchange thermodynamics.57–59 ITC measurements of the exchange of native oleate with alkylthiols of various chain lengths on CdSe QDs report an enthalpy–entropy compensation effect that maintains near-constant Gibbs free energy.57 The longer chain ligands have stronger inter-ligand van der Waals interactions that lead to increased exothermicity. This increase is mitigated by a loss of entropy from restricted chain conformations and in this case, an elimination of the disorder caused by the double bond in oleate. The study also investigated the effects of particle size on ligand-exchange thermodynamics and reported a less favourable exchange for larger NCs, an observation that was explained by the ability to more closely pack ligands on the larger, less curved NC surfaces. Alivisatos and co-workers reported an endothermic exchange of native straight chain carboxylates with oleic acid, also attributed to stronger interligand interactions and higher ordering of longer, fully saturated ligand chains.58 Moving beyond semiconductor QDs, ITC measurements of the binding of thiols to gold NC surfaces showed an increase in binding enthalpy with increasing chain length accompanied by a larger loss of entropy, following the previously mentioned enthalpy–entropy compensation effect.59 The Au-thiol system also showed more favourable ligand binding with smaller-sized NCs, understood by the increased ratio of edge sites to facet surface sites.
The first reported synthesis of colloidally stable perovskite NCs used a combination of oleylamine and oleic acid, as shown in Fig. 3a.36 Methylammonium is a key component of thin-film perovskites, and early reports demonstrated that longer chain alkylammonium molecules have high potential as defect passivating groups for perovskite solar cells.108,109 These results suggest that ammonium ions are a critical anchor group for stabilizing the surfaces of perovskite NCs, a hypothesis that was confirmed by NMR studies that show that oleylammonium oleate groups are the main anchor groups that coordinate with perovskite NC surfaces, rather than oleylamine and oleic acid.1 This study and other explorations of ligand-NC binding110,111 show that perovskite NC-ligand interactions are far weaker than those of II–VI and III–V semiconductor NCs,38,42,112 despite these materials having very high photoluminescence quantum yields (PLQYs). These high PLQYs were explained by DFT studies that demonstrate that most perovskite defect structures produce trap states that do not lie within the material's bandgap,94,95 but it was later shown experimentally71 and with DFT113 that undercoordinated lead sites at terminal surfaces or grain boundaries are critical defects that may be responsible for reduced NC PLQY. These studies inspired a large amount of synthetic and ligand chemistry investigations to improve perovskite NC PLQY and increase overall NC stability via passivating such lead sites.
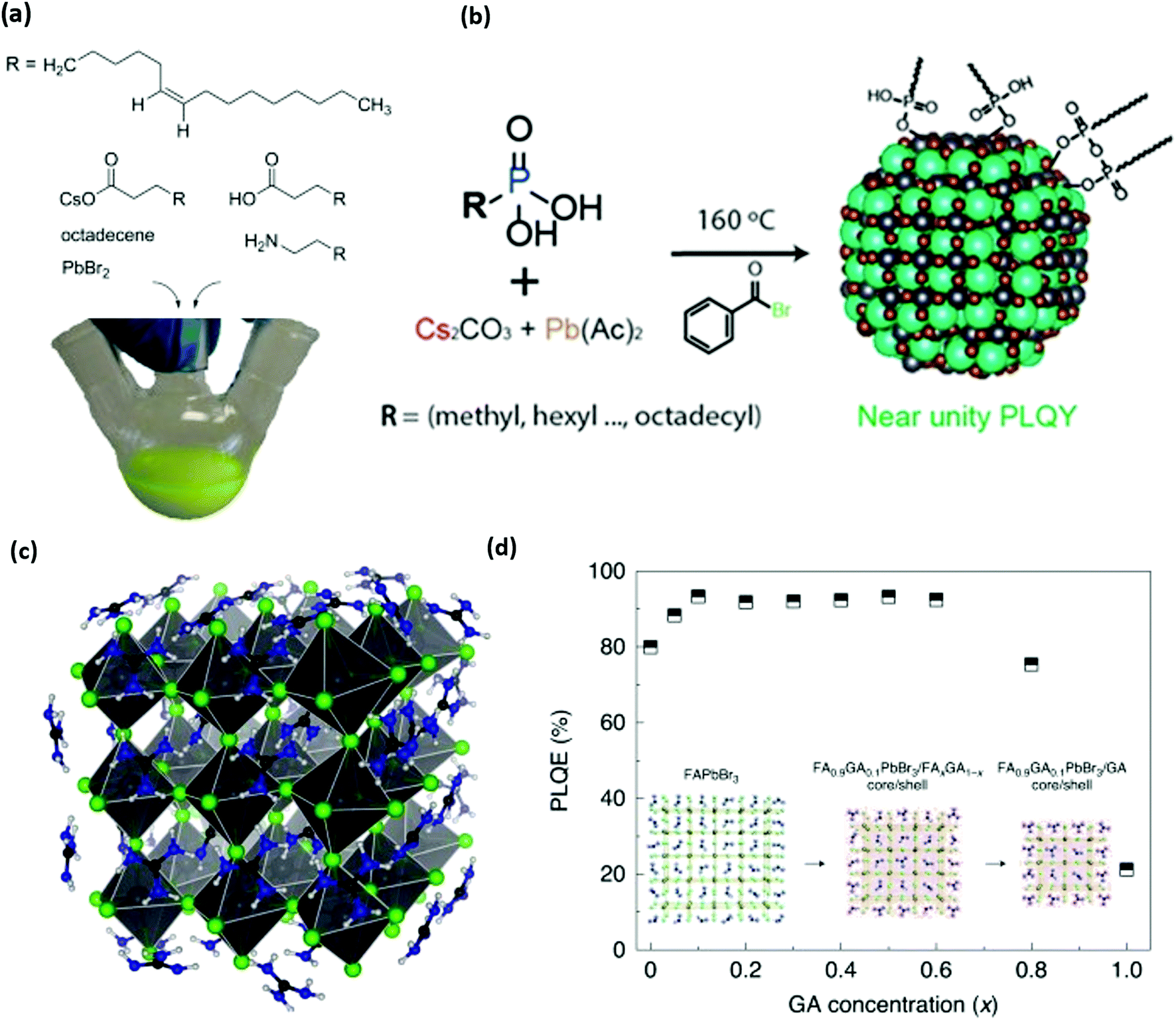 | ||
| Fig. 3 (a) Structure of organic and inorganic precursors typically used to synthesize perovskite NCs. (b) Structure and scheme for the synthesis of perovskite NCs without any cationic ligands. Phosphonic acids are used to dissolve precursors and stabilize NC surfaces. (c and d) Molecular structure of guanidinium (GA) stabilized perovskite NCs and photoluminescence quantum efficiency (PLQY) of synthesized NCs as a function of GA content. Panel a reproduced with permission from ref. 1 and 2, panel b reproduced with permission from ref. 130, and panel c reproduced with permission from ref. 2 and 3. | ||
Early on, it was noted that bromide ions play a critical role in ligand desorption reactions by coordinating with ammonium ions in solution.1 This idea – along with the attribution of low NC PLQY to undercoordinated lead71 – led researchers to explore routes to add additional halide anions to passivate these defects, considering that halides are native lead-coordinating components in perovskite lattices. From the NC synthesis perspective, the original PbBr2, Cs oleate injection synthesis has been supplemented with methods that involve an injection of alternative halide sources, such as benzol halides,98,114 trioctylphosphine-halides,115 and trimethylsilyl halides96,116,117 into solutions of Cs and Pb oleates. Additionally, excess halide can be added in the form of ZnBr2.118 This allows more precise control of ion ratios to improve NC PLQYs from synthesis. Post-synthetic treatment with excess lead bromide,119 and halide containing ligands such as didodecyl dimethylammonium bromide (DDABr)14,120,121 and oleylammonium halide122 also improve the PLQY and stability of perovskite NCs. This treatment with DDABr and other similar ligands has proven effective, and the quaternary ammonium bromide has become a popular perovskite NC passivating group.96,106,116,123–125
Other researchers have focused on alternative anchor groups for perovskite materials. Phosphonate and phosphine groups have exposed lone pair electrons that have shown high potential as passivating groups for thin-film perovskites.126,127 Trioctylphosphine was occasionally used to help dissolve lead precursors for hot injection syntheses,36 but was further explored to show that trioctylphosphine128 and trioctylphosphine oxide129 can be used to improve NC stability and PLQY. It was even shown that NCs with traditional ligands could be recovered by simply treating the NCs with trioctylphosphine.130,131 This recovery is presumably due to an L-type association of electron pairs on phosphine or phosphine oxide moieties with the undercoordinated Pb2+ sites that are typically associated with low NC PLQY.93,132 The hypothesis was verified for perovskite thin films by studying the effect of film treatment with electron pair containing thiophene and pyridine,133 and a thorough study of DFT and experimental results from anionic ligand exchanges showed that softer Lewis base anions such as phosphonates, fluorinated carboxylates, and sulfonates are effective passivating groups for the soft Lewis acid character of undercoordinated lead.93 Such results explain the high performance of phosphonate and sulfonate containing zwitterionic ligands.115,134 More recently, it has been shown that phosphonic acid groups can be used as the sole ligand for synthesizing CsPbBr3 as described in Fig. 3b. The resulting phosphonic capped CsPbBr3 achieved near-unity PLQY confirming excellent passivation of surface traps.135,136 These studies reinforce the idea that soft Lewis bases are effective anchor groups for passivating critical halide vacancies on perovskite NC surfaces.
The cationic ligand anchor group has not received as much research attention as the anionic or charge-neutral anchor groups. Most studies of perovskite materials only investigate variations on primary amines1,36 and quaternary ammonium ions.14,120,121,137 DFT studies suggest that N–H moieties exhibit hydrogen bonding character between the ammonium cation and the halide anions in bulk methylammonium138,139 or formamidinium140 lead halide perovskite materials. Hydrogen bonding has been explored as an important NC stabilization factor for ammonium ligands141 and formamidinium cations,142 but some recent reports now investigate the favourable effect of guanidinium143 and pyrazine144 on the perovskite's properties. Recently, guanidinium was shown to be an important surface ion in the synthesis of perovskite NCs for the highest efficiency green-emitting perovskite LED reported so far, an advance that was enabled by the increased PLQY with guanidinium shown in Fig. 3c.3 These promising results motivate the further investigation of ligand hydrogen bonding character and guanidinium passivation as strategies to understand and enhance the performance of metal-halide perovskites in future applications.
2.2 Anchor groups for covalent lattices
As demonstrated in Section 2.1, the “anchor-functionality” paradigm emphasizes the use of anchors matching the chemical environment of nanomaterials surface to achieve efficient binding and effective surface passivation. In Section 2.2, we discuss covalent lattices, and therefore also the different organic building blocks that can form covalent bonds to these covalent lattice surfaces. Like Section 2.1, there are differences in ionicity in these covalent lattices as well, and they require different bond forming strategies. For example, on silica surfaces, the strong electronegativity of oxygen atoms compared to silicon atoms leads to the formation of polar Si–O bonds, which is significantly different from covalent bonds by atoms of similar electron negativities, such as Si–Si bonds and Si–C bonds. Therefore, in this section, we categorize the examples according to such variance to better illustrate and emphasize the “anchor” section criteria in the “anchor-functionality” paradigm – matching the chemical environment of the nanomaterial surface.The growth of silica shells on NCs has been extensively studied, with two predominant methods emerging: the Stöber method and the reverse microemulsion method, as shown in Fig. 4. In the first method, silica shells are formed directly on the surface of the NC seeds by replacing the stabilizing ligands with silica precursors. In an early example, Mulvaney and coworkers introduced bifunctional (3-aminopropyl)trimethoxysilane to citrate-stabilized gold particles in aqueous solution.149 The –NH2 coordinates to the gold surface and the –Si(OEt)3 groups extend outward, allowing the formation of a silica layer on the gold NCs. This method can also be used to achieve ultra-thick silica shells via seeded-growth using small silica-coated particles as seeds and tetraethyl orthosilicate (TEOS) as a typical precursor of silicon alkoxides.150,151 Water-in-oil (W/O) microemulsion or reverse microemulsion is another technique to encase NCs in silica. In this method, NCs are introduced to stable W/O microemulsions or nanodroplets of organic solvent, water, and surfactants. Upon successive additions of TEOS and ammonia, hydrolyzed TEOS replaces the surfactants on the NC surface and the silanization process is initialized. Compared with the classical Stöber method, the reverse microemulsion method allows more control over silica nucleation, final particle size, and monodispersity since the particles are contained in small, stable, and narrowly distributed nanometer-sized droplets in a continuous phase.152 More detailed discussion on the synthesis of silica-coated NCs, metal oxide NCs, and metal NCs can be found in recent review articles.153–155
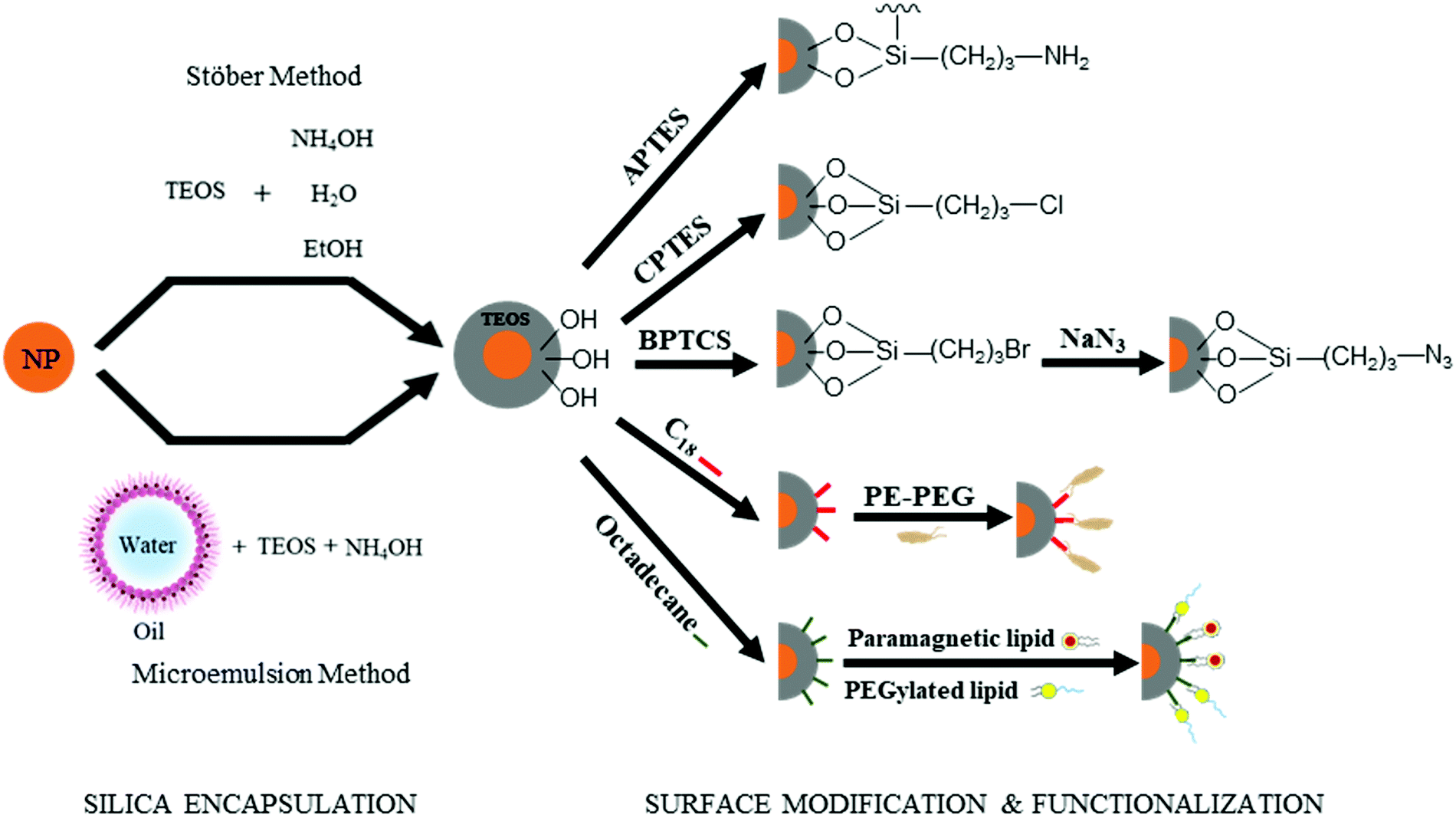 | ||
| Fig. 4 Silica encapsulation of nanoparticles (orange) can be achieved by two main methods: the Stöber method and the microemulsion method. The surface of the silica shell is rich in hydroxyl groups, which can be modified and functionalized by silane coupling agents, polymers, or lipids. APTES = (3-Aminopropyl)triethoxysilane; CPTES = (3-Chloropropyl)triethoxysilane; BPTCS = (3-Bromopropyl)trichlorosilane. | ||
Besides acting as a shield to protect NCs from surface oxidation and ripening, silica shells can also serve as a platform for surface modification and functionalization (Fig. 4). The outer surfaces of silica-coated NCs are rich in silanol, which can act as a handle to covalently attach various functionalized organosilanes to modify the surface with different functional groups. For example, Qin and co-workers prepared superparamagnetic Fe3O4 NCs encapsulated in an amine-functionalized silica shell using (3-aminopropyl) triethoxysilane.156 By using amino groups as reactive handles, polymerization of glycidyl methacrylate was initiated at the surface, and the resulting particles were successfully utilized for lipase immobilization. For thiol-functionalized silica coated NPs, the density of thiol groups on silica shells can be tuned by varying the ratio of methyltriethoxysilane, n-propyltriethoxysilane, and 3-mercaptopropyltriethoxysilane. By coating magnetite NCs with different thiol-functionalized silica shells, Kessler and co-workers demonstrated heavy metal uptake processes for wastewater treatment.157 Carbonyl groups were also attached by modifying amine groups on silica surfaces with succinic anhydride.158 The carbonyl-functionalized silica-encapsulated gold particles subsequently form covalent bonds with oligonucleotides when EDC is present (see mechanism with EDC details in Section 3.3.1) to detect gastric cancer. Furthermore, chloroalkyl alkoxysilanes are widely used to terminate the silica surfaces with chlorine atoms, which serve as leaving groups for the introduction of other functional groups. Utilizing this method, Veisi and co-workers replaced the chlorine atoms on silica surfaces with isoniazide moieties, which immobilize Pd catalysts on the silica surface. The resulting Fe3O4@SiO2/isoniazide/Pd catalysts successfully facilitated Suzuki coupling reactions at room temperature.159
In addition, click chemistry handles can also be attached to silica surfaces (a more detailed discussion of click chemistry is included in Section 3.3.4). Typically, silica-coated Fe3O4 particles are bromine-functionalized with (3-bromopropyl)trichlorosilane and further treated with NaN3 to achieve azide-functionalized nanoparticles.160,161 Some applications of these azide-functionalized magnetic nanoparticles have been demonstrated for click immobilization of mono-alkyne metalloporphyrin to obtain free-base porphyrin conjugate,161 and gold NC modified porous paper working electrode, and magnetic silicon NC with protein.160 Thiol groups are another common click chemistry handle applied as an intermediate to functionalize the surface of silica shells. In the work of Shahsavarifar and coworkers,162 click reactions between thiols and acrylic acid groups were used to form a functional group for immobilizing molybdenum complexes on silica-coated magnetite nanoparticles to prepare a recoverable molybdenum catalyst. The resulting catalyst showed a high catalytic activity and selectivity in the epoxidation of olefins and allylic alcohol. Click chemistry handle-functionalized silica shells can also be achieved by modifying the Stöber method; By using a mixture of TEOS and a click-handle-functionalized triethoxysilane, for example (3-azidopropyl)triethoxysilane or O-(propargyloxy)-N-(triethoxysilylpropyl)urethane in the Stöber method, azido- or alkyne-functionalized silica-coated nanoparticles can be prepared in a one-step procedure.160–162
The silanol surface can also be directly coated by small biological additives or polymers without silane coupling reagents. Riedel et al. coated Au@SiO2 with bovine serum albumin, a blood albumin protein derived from cows, for chemotherapeutic applications.163 The stability and biocompatibility of NCs in harsh chemical treatments including strong acids can also be guaranteed by dual encapsulating with silica and polymers. Hu et al. combined silica and amphiphilic polymer coating techniques to prepare ultra-stable NCs, which can be used for biosensing in a broad range of chemical conditions.164 Mulder and co-workers made bio-applicable and target-specific NCs coated with silica and a dense monolayer of lipids without the use of silane coupling agents.165,166 These particles were suitable for both fluorescence imaging and MRI studies, and showed highly specific uptake by human umbilical vein endothelial cells.
These hydrogenated and oxygenated surfaces are excellent templates for further surface modification, as they can be subjected to a variety of different environments and chemistries to achieve adsorption and covalent anchoring of molecules. This section will provide a mechanistic overview of subsequent techniques employed to further modify these surfaces. These modified surfaces allow for material compatibility within drug delivery,183–186 sensing,184,187 catalysts,188,189 electrochemistry,188,189 and tribology,190,191 amongst other applications.
Instances of the direct adsorption of molecules onto hydrogenated SiNP or ND surfaces are limited. H-terminated silicon and diamond have weak surface dipoles,192,193 making the surfaces averse to hydrogen bonding, dipole interactions, or physisorption. Unlike H-terminated SiNPs and ND, O-terminated surfaces have more pronounced surface dipole moments that facilitate hydrogen bonding and dipole–dipole interactions.194 This is observed through the nanomaterial's stability in aqueous environments; hydrogen bonds form between the partially positively charged hydrogen atom of water (Hδ+) and terminal oxygen atoms with a partial negative charge (Oδ−). While hydrogen bonding and dipole interactions are prevalent, electrostatic interactions via ion exchange reactions with carboxyl functionalized surfaces have also been observed. In neutral environments, carboxyl groups deprotonate to form charged carboxylate anions, which associate with cations in solution, such as protonated amino groups of proteins168 and with cationic polymers.195
However, these electrostatic bindings between organic molecules and inorganic surfaces break down easily due to their weak binding strengths. Therefore, the anchoring of molecules to the surfaces of SiNPs and NDs is more preferably and more commonly achieved through covalent bond formation via the functional groups on the surface (e.g., hydrogen or oxygen moieties such as hydroxyl, ether, carbonyl, and carboxyl groups). Organic molecules are grafted onto the surface of silicon and diamond through a variety of methods such as alkene/alkyne addition,196,197 silanization,191,198 diazonium chemistry,32,199,200 esterification,201 and other reactions. Similar to exchanging hydrogen with oxygen and vice versa, these methods rely on a combination of the following strategies: (1) withdrawing surface atoms to generate dangling bonds, (2) withdrawing ionized electrons from the surface, or (3) chemical reactions with surface moieties. These strategies have been grouped accordingly in Fig. 5.
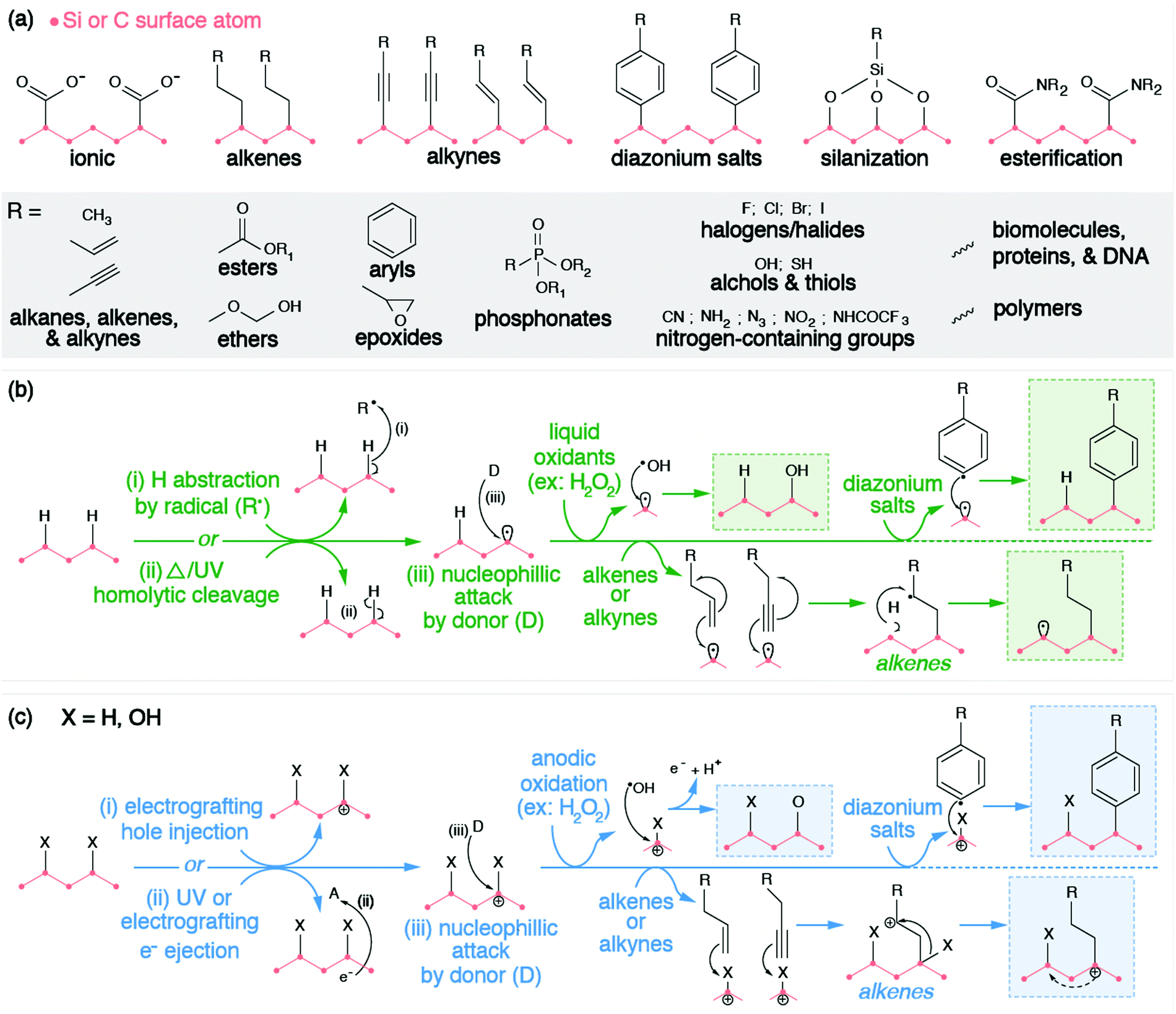 | ||
| Fig. 5 (a) Examples of different functional groups introduced to silicon or diamond surfaces through different surface modification techniques; (b) surface modification of silicon or diamond surface via radical pathway; (c) surface modifications of silicon or diamonds via electrochemical pathways. | ||
The chemical functionalization of the H-terminated surfaces of silicon and diamond can be achieved through thermal, photochemical, or electrochemical grafting of α-olefins.196 Three methods have been reported: radical chain reaction (RCR), photochemical grafting, and electrochemical grafting.
Initiation of RCR requires cleavage between H atoms and C or Si atoms on the surface to form reactive surface dangling bonds (free radicals) that react with the terminal double bond of the incoming organic molecules, covalently grafting the organic molecules to the particle's surface.202 The H–Si or H–C bond cleavage is achieved with a radical initiator (e.g., diacyl peroxide) that abstracts hydrogen from the material's surface. The resulting radical attaches the double bond of the α-olefin and forms an intermediate secondary alkyl radical. Propagation occurs as the secondary alkyl radical abstracts an adjacent hydrogen atom from the silicon or diamond surface, therefore producing a new surface dangling bond and continuing the chain reaction.
Alternatively, the photochemical grafting of α-olefin requires irradiation with UV light instead of a radical initiator.196 It was first applied to functionalize H-terminated silicon, and later translated to H-terminated diamonds.32 The photochemical mechanism of this method starts with an electron transfer from Si or diamonds to incoming α-olefins under UV excitation, forming a positively charge Si or C center at the surface and an alkyl anion. The nucleophilic alkyl anion attacks the positively charged center at the surface and forms a covalent bond.203,204 Similar to photochemical grafting, anodic and cathodic electrochemical grafting through electron transfer and grafting of terminal alkanes and alkynes has been utilized for doped semiconducting SiNPs and ND.197 Such photochemical and electrochemical grafting of α-olefin has been applied to other H-terminated nanomaterials (e.g., Ge and B particles),205,206 metal surfaces,207 and other carbon allotropes.202 Covalent grafting of terminal alkenes introduces various terminal functional groups (e.g., –NHCOCF3, –NHCOO(tert-butyl), –COOCH3, –CH3, –COOH) onto the material's surface to provide new functional properties, or to serve as reactive handles for further modification.205,206,208–210
Diazonium chemistry can be used to functionalize either hydrogenated or oxygenated surfaces with covalently bonded aromatic organic compounds.32,196 The material's surface donates electrons to reduce the electron-withdrawing diazonium salt (Ar–N2+), forming an aryl radical (Ar˙) and molecular nitrogen (N2). Electron-rich donating surfaces are achieved through the injection of electrons (cathodic electrografting) or through electron ionization. The former method requires the material to be conductive and is only applicable to doped SiNPs or NDs. Due to the surface dipole of the H-terminated diamonds, the resulting band bending raises the conduction band of H-terminated diamond above vacuum level. Therefore, when an electron is either injected or excited onto the conduction band, such a high-lying conduction band provides sufficient offset for efficient electron transfer from diamonds to diazonium salts.26 Regardless of the method, the produced aryl radical attacks and displaces the native surface group, forming a surface radical. Radical recombination occurs, covalently grafting the aryl radical to the surface of the SiNP or ND. Similar to other methods, diazonium salts introduce a wide range of terminal functional groups (e.g., –COOH, –Br, –NO2, –CN, –NH2) to the surfaces of these nanomaterials, enabling further chemistries or molecular anchoring.211 Furthermore, covalent attachment via diazonium reactions has also been displayed for other inorganic NCs (e.g., TiO2, Fe2O3, Fe, Au, Cu, Ni)199,200,212–215 and carbon allotropes.216
Chemical reactions with moieties on the O-terminated surfaces are also effective in creating covalent bonds with incoming organic molecules. Hydroxyl groups on surfaces can be modified through silanization chemistry, passivating the material's surface with siloxide. Considering that the surface of O-terminated SiNPs is silica, please refer to Section 2.2.1 for further description of this functionalization method. Alternatively, SiNPs and ND surfaces with carboxyl groups can be subjected to amidation or esterification reactions. Carbodiimide reagents (commonly EDC or DCC) activate the carbonyls to form an intermediate complex that is susceptible to nucleophilic attack by primary amines or hydroxyl groups. This process is also commonly utilized to graft proteins and biomolecules directly to the surfaces of SiNPs or NDs, which will be discussed in the following bio-medical section (Section 3.3), but can also be used to covalently bind NPs to polymers for electrochemical applications.201,217,218 It is worth mentioning that this esterification is also feasible in modifying the surfaces of graphene quantum dots, considering the large number of carboxyl groups on their surfaces.219
3. Functional groups for enhanced performance
In Section 2, the different chemical interactions that anchor groups utilize to attach to a variety of nanomaterials was discussed. In this section, we will elaborate on the roles of the functional group element of the “anchor-functionality” paradigm, with an emphasis on the interactions between functional groups and the original nanomaterials or their surroundings, through which they further enhance the performance of the original nanomaterials.3.1 Tuning dispersity for different applications
Due to their small sizes, most inorganic nanomaterials can be dispersed into solution and form a meta-stable colloidal dispersion. This enables processing with low-cost and efficient solution-based techniques, such as inkjet printing or roll-to-roll printing, to manufacture different electronic devices (e.g., solar cells,11 LEDs,220 photodetectors221) at room temperature, in contrast to the high-temperature and/or high-vacuum techniques used to process traditional inorganic materials. Usually, to accelerate drying of the printed devices, volatile organic solvents are used and the ligands of such nanomaterials are often comprised of long alkyl chains as functional groups to increase their solubility in organic solvents.222 On the other hand, by choosing hydrophilic functional groups, resulting inorganic nanomaterials can be dispersed into aqueous solution and be injected into living organisms as nanomedicines.34 Regarding dispersity, the functional groups in the “anchor-functionality” design offer camouflage for nanomaterials to fit into different environments.The dispersity of a ligand-modified nanomaterial is governed by the interaction between the solvent molecules and the ligand shell, following the similarity–intermiscibility theory, which is more well-known by the “like dissolves like” principle.223,224 Solvent molecules prefer to interact with molecular domains sharing a similar polarity, and thus the solute dissolves better in solvents that are structurally similar.
The interactions between solutes and solvents can be classified into three categories, – non-polar, polar, and protic/ionic, as exemplified in Fig. 6 by the different ligands used in lead halide perovskite NCs to tune their solubility in different solvent mediums. These ligands possess the same zwitterionic anchor groups. Therefore, it is implied that the difference in dispersity originates from the different functional groups. To facilitate the dispersion in non-polar solvent, Kovalenko and co-workers pair a long alkyl chain (–C18H37) with a zwitterionic anchor group for perovskite NCs.115 The long alkyl chain only consists of non-polar hydrocarbon moieties, which is similar to the chemical structure of non-polar solvents such as benzene, toluene and hexane. Therefore, the resulting capped perovskite possesses good solubility in non-polar solvents due to the favorable intermolecular interaction between the alkyl chain and the solvents. When polar groups are introduced into a ligand, for example, the ester groups in ZP3,225 the resulting perovskite NCs can be dispersed in polar solvents such as ethyl acetate and butyl acetate, which are known to be anti-solvents for alkyl-capped perovskite. When the ligand is modified by functional groups that are able to form hydrogen bonds, for example, the polyethylene glycol (PEG) and carboxylate groups in ZW-PIMA-PEG, it enables NC dispersion in ethanol.226 Moreover, there have been examples of aqueous dispersion of perovskite NCs, where much longer PEG chains of more than 100 repeating units were applied to facilitate the solvation of NCs in water.226–228 There are many other examples of functional groups and solvents in each category that are not listed here, and it is always important to be able to identify the interaction mechanism of the functional groups and solvents when designing a ligand for inorganic nanomaterials.
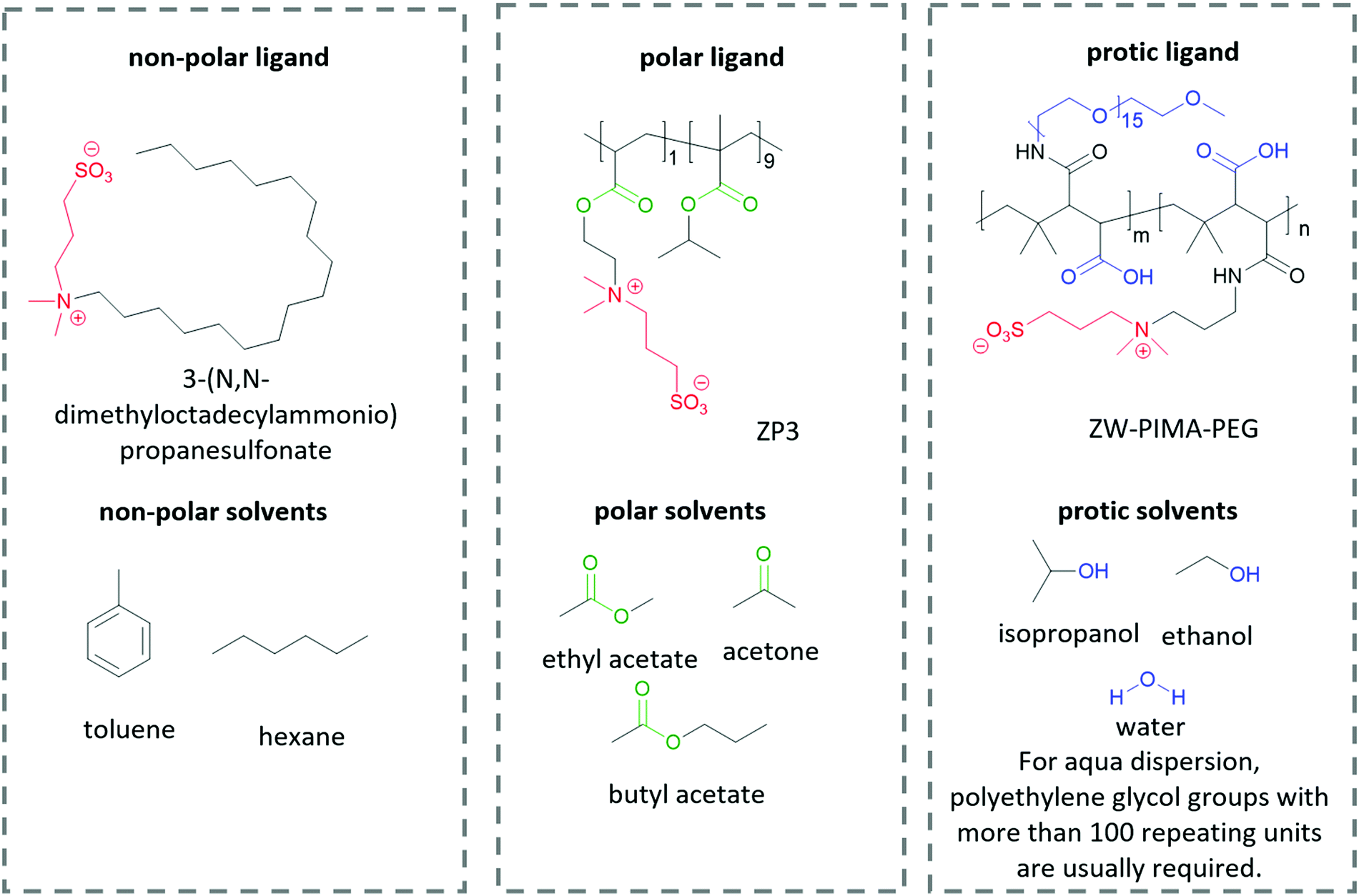 | ||
| Fig. 6 The structure of different ligands for perovskite QDs and the solvents used to disperse the QD-ligand composites. Anchor groups are labeled red, non-polar groups are labeled black, polar groups are labeled green, and H-bonding reactive groups are labeled blue. Due to the increasing interaction intensity in the order of non-polar interaction, to polar interaction and to H-bonding interaction, the stronger interaction dominates when two or more types of interactions are present. | ||
3.2 Optoelectronic applications
Optoelectronics is one of the most studied properties of inorganic nanomaterials. In Section 3.2, we discuss using organic building blocks to tune or enhance the performance of inorganic nanomaterials, by elucidating the steric and electronic interactions between ligands and nanomaterials. Through these examples, we present the basic selection criteria for functional groups in the “anchor-functionality” design.Ligands with dipole-inducing functional groups have now been heavily explored as interfacial modifiers for a variety of inorganic materials. Dipole-induced vacuum level shifting was used to explain improved thin-film perovskite solar cell performance,109 but functional-group-based energy level tuning was critical to the development of PbS photovoltaics. The interfacial dipole's potential as an important electronic tuning tool was best exemplified by a screening of 12 different ligating molecules, which showed that the band energies of PbS could be tuned by a full electron volt by changing the dipole of the surface functional group.30 Further explorations based on solution-state ligand exchanges expanded this range to 2 eV, and explained this change using the dipole of the ligand and the effects of ligand interdigitation on band energies (Fig. 7).31 This motivated research to tune the band energies of PbS and CdSe for organic–inorganic solar cells with functional group engineering.234,235 Additionally, Sargent and co-workers made the impressive innovation of creating a bulk PbS heterojunction solar cell by combining PbS NCs functionalized with different ligands in the same device.236 By respectively using thioglycerol and methylammonium iodide237 as ligands for PbS, the band offsets between these two resulting NCs were engineered to create a bulk heterojunction solar cell with a PCE of 10.7%. This method of device fabrication was further optimized to yield devices with PCEs of 13.3%.238 Such innovations demonstrate the exciting and unique opportunities for functional-group-induced band engineering of inorganic NCs.
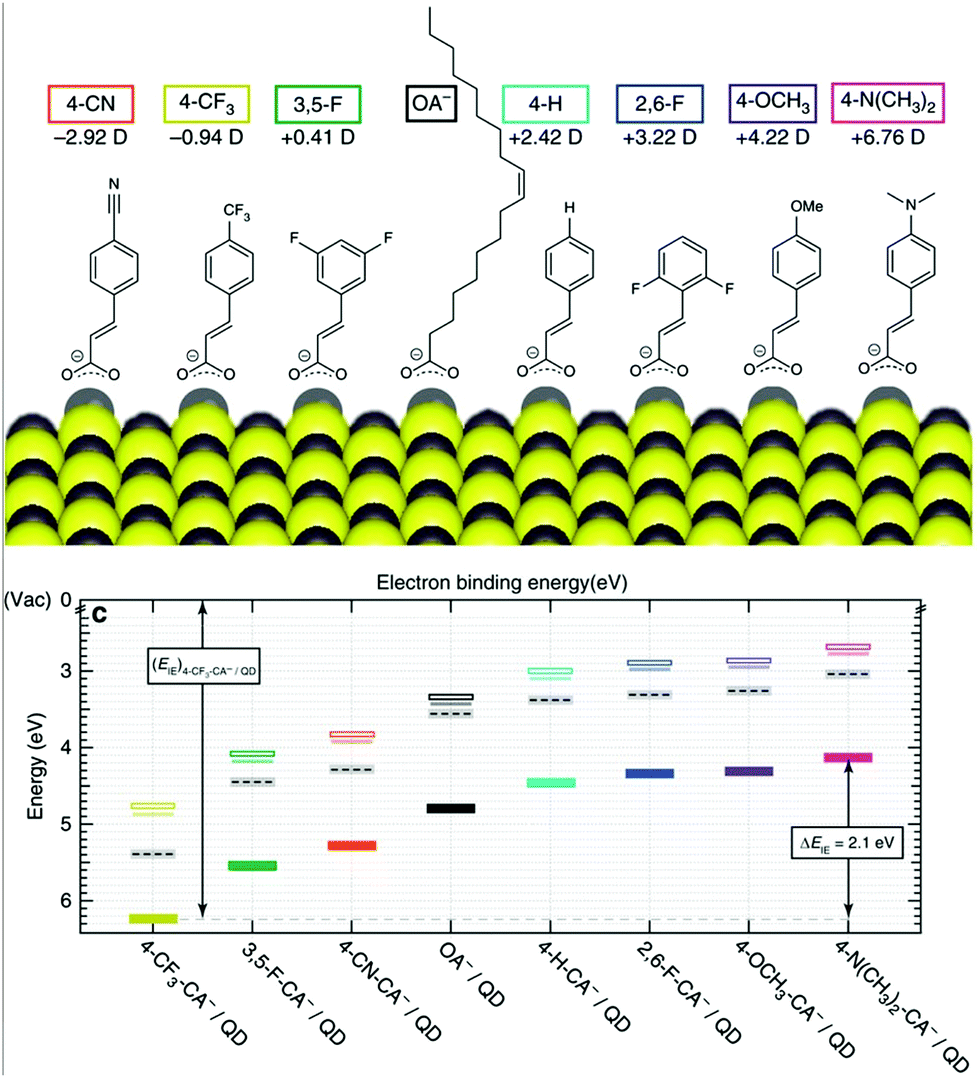 | ||
| Fig. 7 (Top) The molecular structures of organic molecules on PbS QDs and their dipole intensity; (bottom) the shifting of energy levels from the surface treatments with the dipole molecules determined by UPS. Reproduced with permission from ref. 31. | ||
3.2.2.1 Charge transfer. When ligands act as charge acceptors or donors, they should have frontier orbitals energetically aligned to accept or donate electrons to the NP. If fast and efficient charge transfer (CT) is desired, frontier orbitals of the ligand or band edges of the NP (see Section 3.2.1) should be tuned such that there is a driving force for charge transfer (−ΔGo in Fig. 8a). Analysis of the rate of charge transfer, kct, may be framed in the context of Marcus theory.239 A 2013 review by Knowles and co-workers aggregated several studies that vary the driving force for charge transfer.240 Within each data set, kct increases with increased driving force, corresponding to the Marcus normal region.240 At present, the Marcus inverted region has not been observed in NP-ligand CT systems.
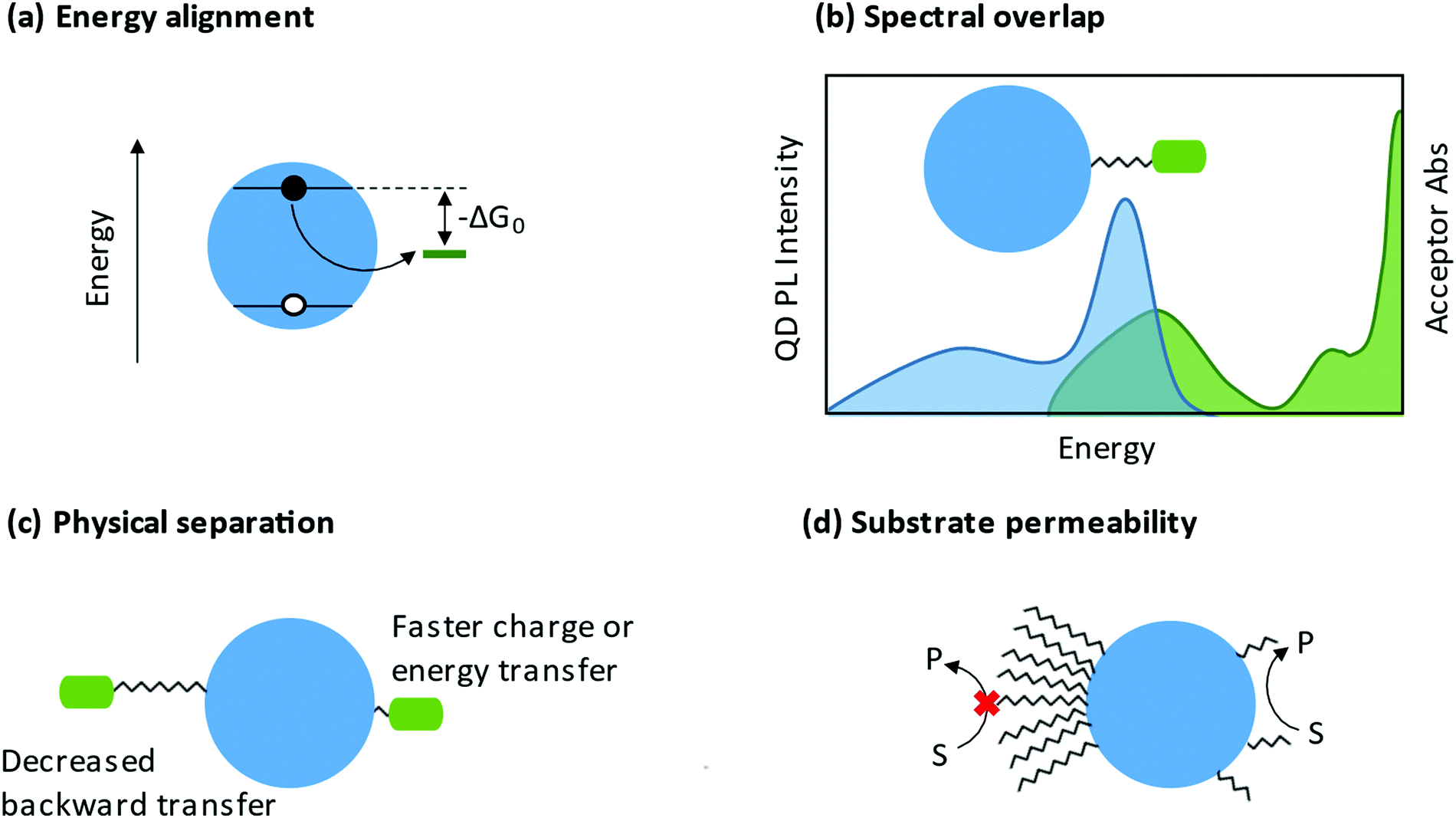 | ||
| Fig. 8 (a) Scheme showing that proper energetic alignment allows a driving force for electron transfer. (b) Overlap between the quantum dot photoluminescence spectrum and the energy accepting ligand absorbance spectrum is required for energy transfer. (c) Physical separation between the nanocrystal core and the energy or charge accepting moiety can control the rate of forward, and by extension, backward transfer. (d) Long, dense ligand shells can prevent substrate access to the nanocrystal. Shorter, less dense ligands allow close interaction between the nanocrystal and substrate, promoting catalysis. | ||
Both organometallic241,242 and conjugated organic moieties243–245 may have their energy levels tuned through substituents to achieve desired energy alignment with the NP. Organometallic complexes that contain an anchor group to bind to the NP may act as charge localizing ligands because of multiple low energy redox states. These include ferrocene derivatives246–249 that act as model charge localizing centers and catalytically competent complexes (see Section 3.2.3). Organometallic ligands are especially interesting because the metal center may have multiple oxidation states, allowing for multiple charges to transfer per ligand.250 Conjugated organic molecules containing an anchor group are also good candidates for charge localization including viologens,251 quinones,252 and fused-ring arenes.253 In such ligands, the conjugated organic moiety acts as a charge acceptor to create long-lived, highly delocalized radicals.
There are additional characteristics that influence charge transfer besides the driving force. An increased distance between the NP and the charge localizing center decreases the rate of charge transfer, as highlighted in Fig. 8c.254–257 The anchor group may also be important to charge transfer. In 2017, Olshansky and co-workers reported on a CT system that was described by surface trap-mediated transfer rather than Marcus two-state direct transfer, highlighting the importance of the NP/ligand interface.248 The head group of the ligand may relax quantum confinement or create charge carrier traps, both of which influence charge transfer to the rest of the ligand or to external substrate.258 By extension, the charge localizing center of the ligand may itself be viewed as an additional charge carrier trap.
3.2.2.2 Energy transfer. The study of energy transfer from NCs began with investigations of Förster resonance energy transfer (FRET) systems. Because of the strong distance- and orientation-dependence of FRET efficiency, FRET can be used as a “spectroscopic ruler”.259 This ability to estimate of distances and orientations of molecules has made FRET a common tool for studying conformational changes in biomolecules.260 In a QD-ligand system, the donor chromophore is photoexcited and transfers energy to an acceptor chromophore (photoresponsive adsorbates such as dye-labeled peptides) through a non-radiative process.261 Important considerations for FRET systems are the spectral overlap of the donor emission and acceptor absorption and the physical separation between the NC and the photoresponsive ligand (Fig. 8b and c). A 2008 perspective from Medintz and Mattoussi outlines the basis for use of NC FRET systems for sensing and biological applications.262
An emerging use of NC energy-transfer systems is photon up-conversion via triplet–triplet annihilation. The NC acts as the photosensitizer, readily forming a spin–forbidden triplet state due to strong spin–orbit coupling. This triplet is transferred in a Dexter-type energy transfer to a triplet transmitter ligand, and then transferred again to an unbound annihilator.263 The 2017 perspective by Huang and Tang proposes that the bottleneck to efficient triplet–triplet annihilation is the efficiency of the triplet energy transfer from the NC to the transmitter ligand.264 Optimization of this step requires energy alignment of the triplet states to the photosensitizer and annihilator, a short distance from the NC to the transmitter core, and strong binding between the NC and transmitter. The triplet transmitter ligand is typically composed of a head-group to bind to the NP and a highly conjugated organic group to act as a stepping-stone between the NC and the annihilator.
The ligand shell is a physical barrier that can be easily manipulated to enhance access of substrates to the NP surface. From a ligand coverage perspective, it has been shown that a less dense ligand shell enhances catalysis, likely explained by increased “permeability” facilitating interaction between substrates and NP surfaces (Fig. 8d). For example, a study by Chen et al. showed that zwitterionic ligand-capped CsPbBr3 QDs with a surface coverage of 3.0 nm−2 (compared to 5.4 nm−2 for oleate- and oleylamine-capped NPs) exhibited an increased rate of stereoselective C–C oxidative homocoupling of α-aryl ketonitriles.277 From a similar permeability argument, the Weiss group has shown that a mixed ligand shells enhance C–C coupling between 1-phenylpyrrolidine (PhPyr) and phenyl trans-styryl sulfone.278 Furthermore, there are numerous reports demonstrating that shorter ligands are better for catalysis.279 Shorter ligands decrease the steric bulk at the NP surface, and if charge transfer proceeds via an electron-tunnelling mechanism, shorter ligands can increase the rate of this transfer.280 In fact, some studies show that a NP passivated with small inorganic ligands,281 or a NP with a completely bare surface,282 is actually better for catalysis. This therefore begs the question, does the presence of ligands ultimately hinder catalysis? Surface ligands have been at the forefront of the long-standing conflict between maintaining colloidal stability and impeding charge transfer, but we argue that they are most certainly necessary and can be designed intentionally to be advantageous in catalysis.
As mentioned in Section 3.2.2, NP surface ligands can be non-innocent in the context of charge separation. Upon photoexcitation, one of the main competing pathways between charge transfer to a substrate is recombination. This pathway can be avoided by prolonging this excited state and promoting charge transfer by engineering a trap state at a ligand site. This can serve the dual purpose of spatially separating the electron and the hole to prevent recombination in the NP and bringing the charge carrier closer in proximity to substrate. This has been shown particularly in the context of hole-quenching ligands,283 which is significant since hole transfer is widely believed to be rate-limiting in photocatalysis.284 For instance, 3-mercaptopropionic acid (MPA) and other thiol capping ligands are often used as ligands on NP photocatalysts because they are able to quench photogenerated holes due to their mid-gap highest occupied molecular orbital (HOMO) levels.285 In another study, a known hole acceptor, phenothiazine, was ligated onto the surface of CdSe QDs to dramatically improve H2 evolution rates.286 Similar effects have also been observed with conductive ligands that facilitate electron transfer to substrate. Beard et al. showed that CsPbBr3 NCs functionalized with short, cinnamate ligands display enhanced electron-donating ability to benzoquinone by up to ∼23x, and an increased activity for α-alkylation of aldehydes.287 Given these findings, we can imagine an idealized Janus particle system wherein one facet is bound by hole-quenchers and the other by electron-acceptors, to achieve precise and controlled separation of charge carriers for a photoredox event.
Ligands can play an even more active role in photocatalysis by directing reaction pathways through interactions with substrates and intermediates, which can ultimately impact product selectivity. In a system where the NP acts as a photosensitizer, metal complex catalysts can be anchored onto the surface. This has been demonstrated by a ruthenium oxygen-evolution reaction catalyst bound to CdS nanorods.288 In two examples of electrostatic immobilization, a [Ni(terpy)2]2+/CsPbBr3 composite was synthesized, where the Ni(II) complex acted as an electron sink for CO2 reduction.289 Similarly, a cationic ruthenium species was self-assembled onto the surface of negatively charged Au NPs to catalyze the transfer hydrogenation of acetophenone.290 Substrates themselves have also been shown to transiently bind to the surface of CdSe QDs and form diastereoselective products.291 In the [2+2] cycloaddition of 4-vinylbenzoic acid derivatives, selectivity for the minor syn-cyclobutane products was achieved by a proposed mechanism of triplet–triplet energy transfer following binding to the surface at the carboxylate functional group.
Finally, ligands can greatly increase the stability of the photocatalyst. Hole-quenching amines have been shown to displace native ligands in situ and confer greater activity and stability to CdS QDs.292 When anisotropic materials are used, ligands can also help maintain the unique morphology of the NPs.293 This is particularly important because speciation of the NP post-catalysis often reveals aggregation and degradation.284 Being able to maintain the catalyst during operation has great implications for benchmarking, fully understanding what the active photocatalyst is, and painting an accurate overall picture of reaction rate and mechanism.294,295
Furthermore, given the promising spin coherence properties of perovskite materials,21,307 researchers have begun exploring the possibilities for tuning perovskite spin properties with chiral ligands. For instance, two-photon absorption upconverted chiral photoluminescence in CsPbBr3 NCs with enantiomeric ligands was observed, where the chirality was attributed to mechanism ii.302 He and co-workers engineered chirality in perovskite NCs by varying the amount of ligand. They found that with high concentration of the functional group, geometric aggregation of the ligands induces chiroptical properties (mechanism iii), while at lower concentrations, surface distortion/defects contribute to chirality (mechanism ii).301 This chiral effect was realized for perovskite devices when α-phenylethylamine groups were used in the synthesis of 2D perovskites to produce spin-selective photodetectors that can differentiate left and right circularly polarized light.302 Similarly, these chiral-functionalized 2D perovskites have been used to fabricate a circularly polarized white LED.308 This device concept was further optimized and chiral 2D perovskites have now been used as spin selective hole injection layers for perovskite NC-based circularly polarized LEDs with a room-temperature electroluminescent efficiency of 2.6%.309 These impressive devices based on chiral ligand functionalization are important for the future of spintronic applications of inorganic nanomaterials, but more research regarding the impact of ligand structure on performance should be conducted to advance this field.
3.3 Bio-medical applications
Different from optoelectronic applications, where functional groups directly enable new functionality in inorganic nanomaterials, in bio-medical applications, functional groups in the “anchor-functionality” design act as linkers that bind nanomaterials to different biomolecules, and it is the incoming biomolecules that enable new properties of the resulting bioconjugates. For example, proteins or peptides on the surface of QDs can be used to control their self-assembly,310,311 or enable selective interaction between QDs and specific small molecules312 or macromolecules,313 which are essential for chemical sensors, target drug delivery and different bioimaging techniques. These new properties of the resulting bioconjugates originate from the inherent structural or special recognition properties of the biomolecules.310,311 Bioconjugation methods are dependent on the functional groups on both the nanomaterial surfaces and the incoming biomolecules, as briefly summarized in Fig. 9. However, biomolecules, such as proteins and peptides, contain complicated structures and a large number of functional groups compatible with different bioconjugation methods. To avoid ambiguity in discussion, in this review, we took a different perspective and categorized the research cases by chemical interactions instead of by applications, to better illustrate the intrinsic and fundamental aspect of bioconjugation reactions – chemical bond formation.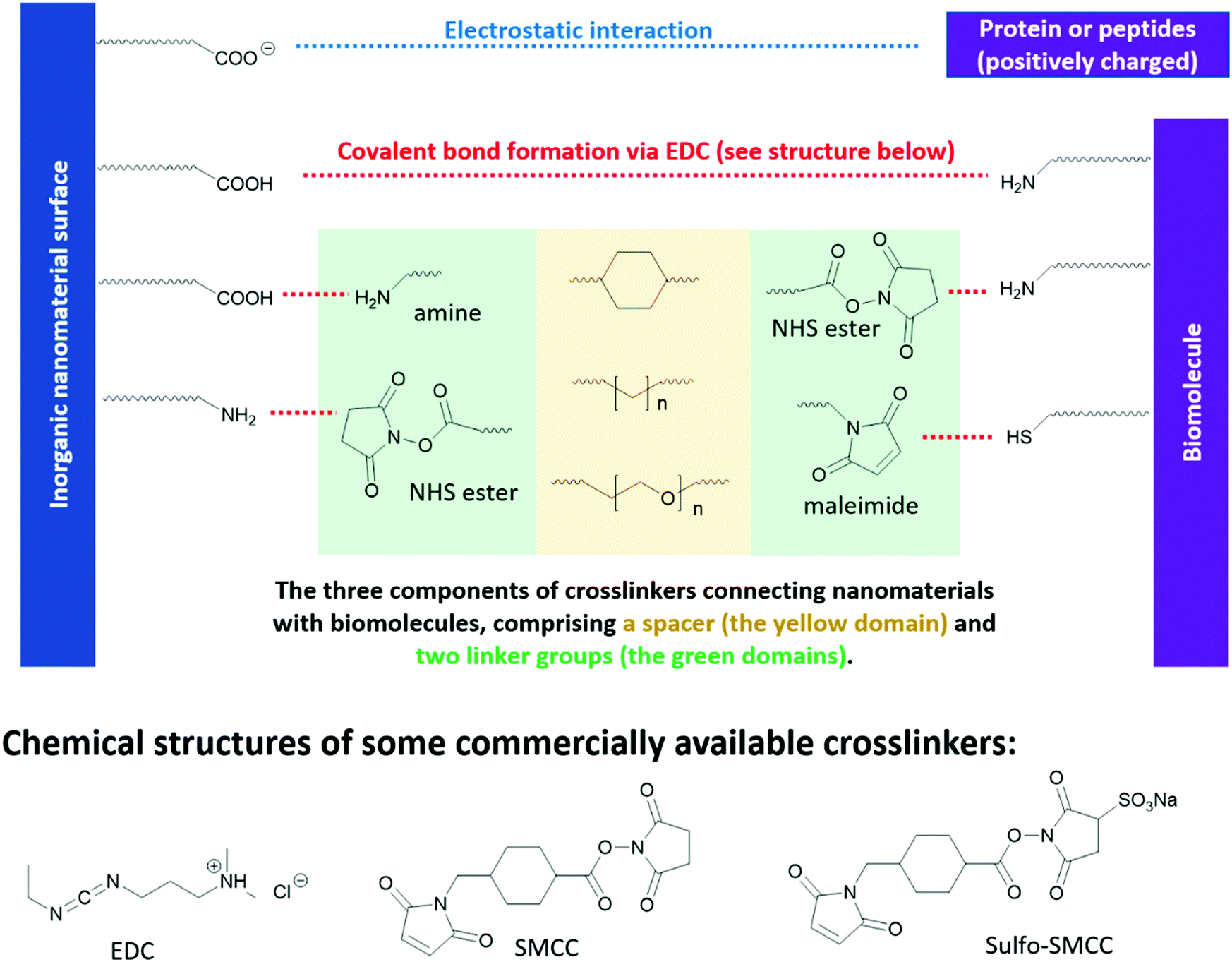 | ||
| Fig. 9 Reaction handles on inorganic nanomaterials and biomolecules commonly used in bioconjugation reactions and the structures of some commercial linkers for bioconjugation. | ||
Among the methods for carboxyl bioconjugation, electrostatic interaction is the simplest approach. Carboxyl groups are negatively charged in aqueous environments, which matches well with typical positively charged proteins. For example, trypsin is a positively charged enzyme in the small intestine that digest proteins. Gole et al. used a carboxylate-rich polymer to coat the surface of gold nanorods, which endowed the gold nanorods with negative charges.315 The bioconjugation between the modified gold nanorods and trypsin was carried out by simple mixing in solution. However, it was observed that the bioactivity of the trypsin was reduced to 19%. Gole et al. ascribed this reduced performance to alteration of the protein secondary structures when it is associated with a charged surface,316 and protein immobilization.317 On the other hand, Gole et al. also applied a zero-space linker (EDC as shown in Fig. 9) to covalently link the carboxylate-rich gold nanorods to trypsin, via the amine groups on the trypsin surface as handle. However, this bioconjugation method with EDC leads to even lower bioactivity of trypsin, which is 13%. This can be ascribed to the acidic nature of EDC, and it requires an excessive amount to achieve a high conversion rate. The resulting low pH environment may lead to irreversible change in the secondary structure of trypsin.
The above results emphasize the importance of maintaining the pH environment in bioconjugation reactions. To better preserve the bioactivity of trypsin, Gole et al. also experimented with “click” chemistry methods to form bioconjugation without altering the pH environment. As a result, the bioactivity of trypsin was better preserved to 53%. A small decrease of the bioactivity after binding to gold nanorod may be inevitable due to immobilization of the protein. It is worth mentioning that “click” reactions are popular in biochemistry research due to their high bio-orthogonality, which is crucial to maintain the bioactivity of biomolecules. A detailed discussion of “click” reactions will be later discussed in Section 3.3.4.
N-Hydroxysuccinimide (NHS) esters have low toxicity319 and are generally considered to be primary-amine-specific,320 making it a good crosslinker for amine groups. The reaction between NHS ester and a primary amine is nucleophilic, where the primary amine is the nucleophile attacking the NHS-activated ester bond. Although thiol and hydroxide groups are nucleophiles as well, it is demonstrated that NHS ester only reacts with the thiol and hydroxide groups in biomolecules when NHS ester is present in excess.320,321 Therefore, one can achieve a high selectivity of amine-NHS-ester reaction by controlling the amount of NHS ester added. Fig. 9 shows a commercial bio-crosslinker functionalized with NHS ester, SMCC, which also has a maleimide handle (see Section 3.3.3) to link to other nanomaterials or biomolecules.
Due to the hydrophobic nature of the NHS ester, crosslinking reactions are usually conducted in organic solvents, which may perturb the bioactivities of biomolecules. To enable crosslinking reactions in physiologic solutions, sulfonated versions of NHS ester crosslinkers are designed and commercially available, for example sulfo-SMCC shown in Fig. 9. Furthermore, the formation of amide bond between NHS ester and the primary amines in biomolecules can be carried out at pH 7–8, demonstrating high biocompatibility. In lower pH environments, protonation of primary amines take place, which reduces their nucleophilicity and thus their reactivity towards NHS esters. In higher pH environments, the stability of NHS ester decreases, due to the relatively higher concentration of the nucleophilic hydroxide group. The half-life of NHS ester drops from 4–5 hours at pH 7, to 1 hour at pH 8, and to 10 mins at pH 8.6.322,323 However, the half-life of the resulting amide bonds is very stable in water, in the range of 7 years,324 rendering it a reliable method in constructing bio-conjugates.322,323
Maleimide is the most popular crosslinker for thiols, due to their exceptionally fast kinetics and high selectivity towards the thiols on cysteine. A variety of maleimide-based crosslinkers available commercially, for example, SMCC and sulfo-SMCC in Fig. 9. The fast kinetics of maleimide towards thiols can be ascribed to its lower lowest unoccupied molecule orbital (LUMO) levels, due to the electron-withdrawing imide group and the strained five-member ring – which facilitates its orbital interaction with the non-bonding orbitals of thiols and therefore bond formation.325
The nucleophilic reaction between thiol and maleimide readily occurs at near-neutral conditions, pH 6.5–7.5.314 At pH above 8, the hydrolysis of the imide group takes place, which leads to the ring-opening of maleimide. This ring-opening reaction is problematic in bioconjugations. First, it destroys the strained five-member ring of maleimide and yield undesired products that are non-reactive toward thiols. Second, the hydrolysis of the newly formed maleimide-thiol adduct eventually leads to the breakdown of the bioconjugation. Although amine is also a nucleophile, its reaction with maleimide is negligible due to its much slower kinetics, only 1/1000 of thiol.314 This explains the high selectivity of maleimide towards cysteine in proteins and peptides. There are other bioconjugation methods for thiol as well. However, they are not as widely applicable as maleimide due to low selectivity and/or slow kinetics and are therefore not included in this tutorial review. For those who are interested, please refer to the reviews by Koniev et al.325 and Ochtrop et al.326
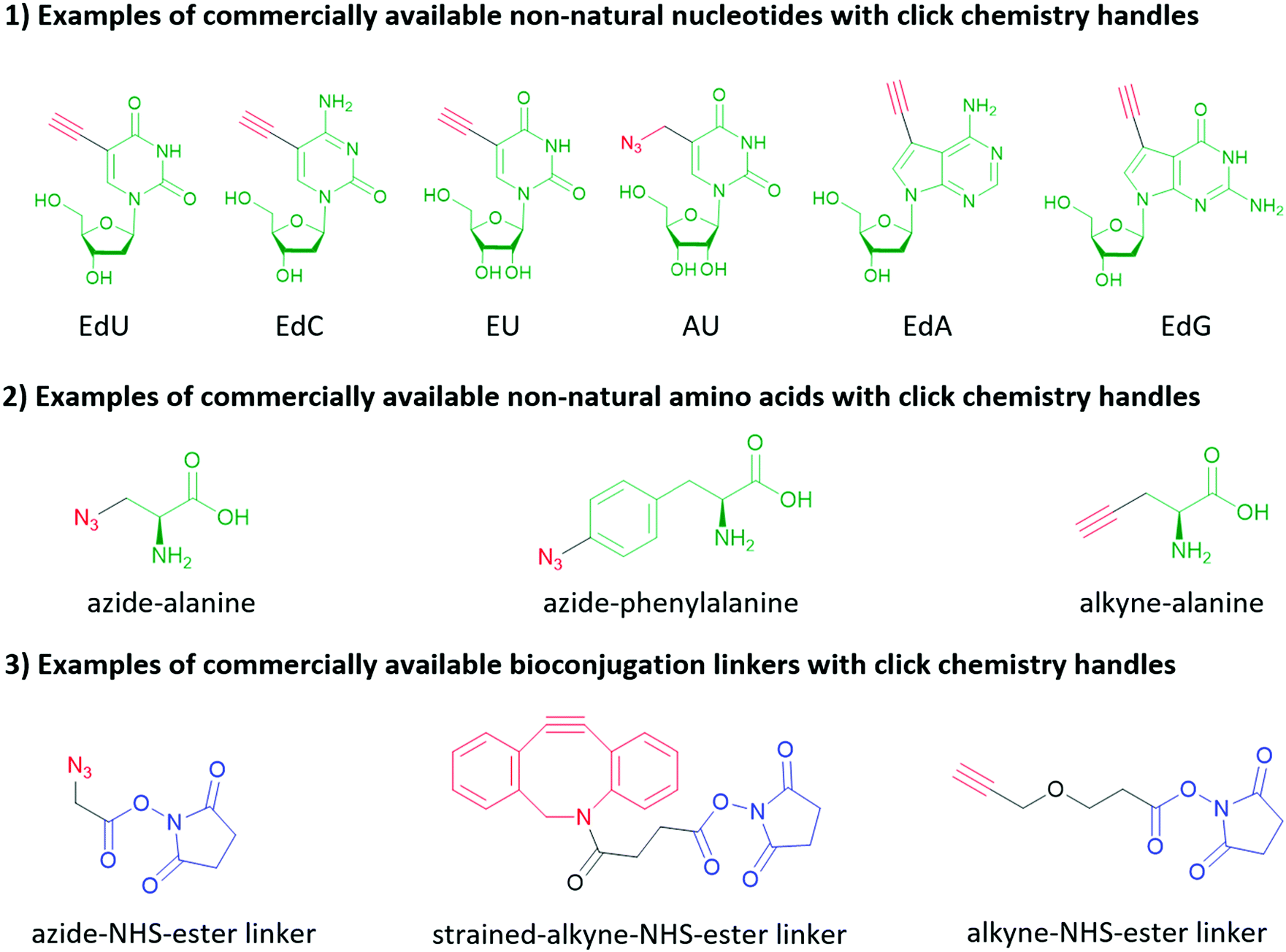 | ||
| Fig. 10 Examples of commercially available non-natural nucleotides, non-natural amino acids and bioconjugation linkers functionalized with click chemistry handles. The click chemistry handles are highlighted red, the structural analogs of natural nucleotides and amino acids are highlighted green, and the handles attaching to amino groups (as discussed in Section 3.3.2) are highlighted blue. | ||
Click chemistry handles (e.g., azide, alkyne, and alkene) are popular in non-natural nucleotides for the following two reasons. First, click chemistry reactions possess the advantages of fast kinetics, high yield, bio-orthogonality, and applicability in aqueous solvents. Second, click chemistry handles do not exist in natural biomolecules and possess high selectivity towards their coupling partners, which is critical to eliminate the side reactions during the synthesis of nucleic acids, and further increase the selectivity of subsequent bioconjugation reactions. For similar reasons, click chemistry is also widely applied in protein and peptide bioconjugation, starting with the non-natural amino acids shown in Fig. 10. It is worth mentioning that in addition to biomolecules, click chemistry handles can also be applied to inorganic nanomaterial surfaces (see Section 2) and crosslinkers as shown in Fig. 10. Two types of click chemistry reactions attract most attention in bioconjugation methods: azide–alkyne cycloaddition and inverse electron-demand Diels–Alder cycloaddition.
3.3.4.1 Click chemistry: azide−alkyne cycloaddition. Copper(I)-catalysed azide–alkyne cycloaddition (CuAAC) has fast kinetic rates in the range of 10–100 M−1 s−1,329 high selectivity and almost quantitative yields,330,331 which are advantageous for bioconjugation. However, due to the biotoxicity of Cu(I) compounds and their insolubility in aqua solution, concerns were raised regarding the biocompatibility of CuAAC. The small amount of Cu(I) required in bioconjugation reactions, which is in the μM range, has demonstrated to impair the functionality of biomolecules by promoting the generation of reactive oxygen species.332,333 Efforts have been focused on reducing the toxicity of Cu(I) complex and increasing their aqueous solubility. With an appropriate ligand design, Bevilacqua et al. demonstrated CuAAC can be performed in living cells.334
Strain promoted azide–alkyne cycloaddition (SPAAC) is a way to bypass the biotoxicity of Cu(I) compounds, as SPAAC does not require catalysts due to the higher activity of the strained alkyne group. However, these reactions have much slower kinetics, in the range of 10−3–1 M−1 s−1,329 depending on the level of strain applied to alkyne group, which is linear when free standing.335Fig. 11 shows the different SPAAC alkyne. Due to the flexible nature of sp3 carbons in the eight-member ring, the alkyne group in OCT is under a low level of strain, which leads to a low reaction rate of 2.4 × 10−3 M−1 s−1. With 4 rigid sp2 carbons atoms in the eight-member ring, the reaction rate of DIBAC is increased to 0.31 M−1 s−1. BARAC possesses one more sp2 carbon compared to DIBAC, by changing the methylene group in DIBAC into carbonyl group, and therefore its reaction rate is further increased to 0.96 M−1 s−1.335 However, it is worth pointing out that the increased strain at the alkyne group also increases its activity towards thiols, reducing the bio-orthogonality of this method.335,336
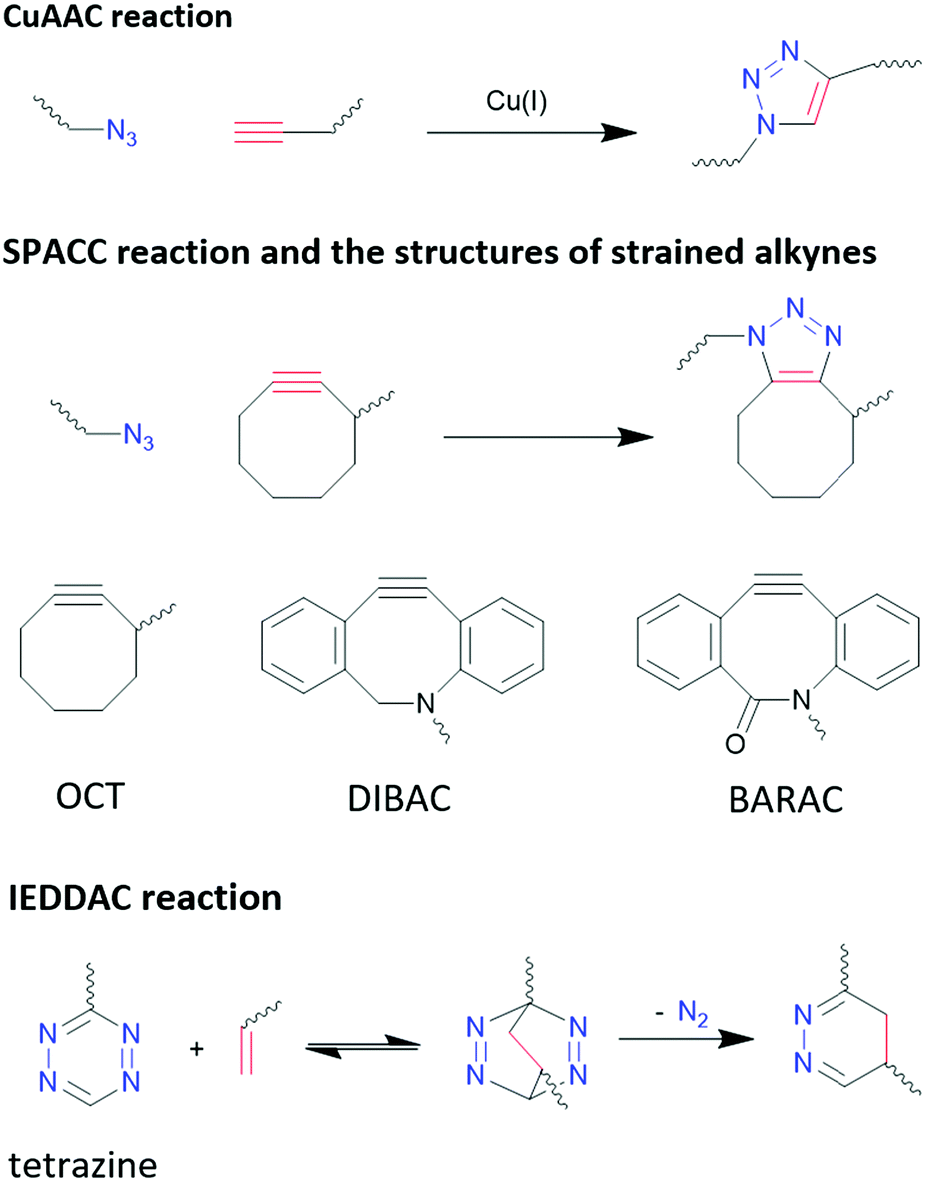 | ||
| Fig. 11 Reaction schemes of different click chemistry reactions. | ||
3.3.4.2 Click chemistry: inverse electron-demand Diels–Alder cycloaddition (IEDDAC). IEDDA reaction stands out from other bio-orthogonal reactions considering its unmatchable kinetics (as much as 106 M−1 s−1)329 and its excellent orthogonality and biocompatibility. IEDDAC is a subcategory of Diels–Alder reaction, characterized by a [4+2]-cycloaddition between a diene (usually tetrazine derivatives) and a dienophile (alkynes or alkenes), and subsequent evolution of 1 equivalent of nitrogen that pushes the equilibrium to one end and eliminates the reversible nature of Diel–Alder reactions, as shown in Fig. 11.
Although strain on alkynes and alkenes can promote the reaction rate of IEDDAC, it is not necessarily required. Oliveria et al. demonstrated a successful bioconjugation between terminal-alkene-functionalized protein and tetrazine, with a kinetic rate of 0.21 M−1 s−1,337 which is comparable to SPAAC reactions. Nevertheless, faster reaction kinetic are preferred in bioconjugations, and strained alkynes and alkenes are often paired with tetrazine for such purposes. Transcyclooctene (TCO) moiety is one of the most common strained alkenes used, and its reaction with tetrazine reaches a rate of more than 1000 M−1 s−1 in organic solvents.338 In addition to strain, electronic effects, steric effects, solvent, and pH have significant impacts on the kinetic of IEDDAC, which have been discussed comprehensively by Oliveria et al.329
4. Conclusion and outlook
In this tutorial review, we coalesce a wide range of examples and concepts regarding inorganic nanomaterial surface functionalization into the streamlined “anchor-functionality” paradigm. To help the reader better understand this idea, the functions of different anchor groups and functional groups were elaborated with examples covering a variety of material types and applications. In Section 2, the different interactions between inorganic surfaces and organic anchors were discussed, and it was demonstrated that the selection of anchor groups is determined by the chemical features of the inorganic surfaces, such as lattice type and bonding motif. In Section 3, the roles of functional groups were discussed focusing their mechanisms on enhancing and/or endowing new functionalities of inorganic nanomaterials to optimize their performances in certain applications.Although the types of materials and applications covered in this review are somewhat specific, the chemistry methods discussed are not limited to the examples mentioned. For example, Section 2 discussed using diazonium chemistry to graft organic molecules to only silicon and diamond. In fact, diazonium chemistry has also been used to modify other inorganic materials with covalent lattices, such as black phosphorus,339 WS2, MoSe2, WSe2,340 and Ni2P.341 The covalent characteristics these materials share allow the use of the same chemistry for surface modification. On a broader scope, the silanization chemistry used to modify O-terminated silicon and diamond surfaces applies to cellulose as well, because all of these material surfaces are functionalized with hydroxyl groups.342 This highlights the importance of identifying the chemical features of a surface prior to modification, and meanwhile provides a useful mindset for modifying other novel materials emerging in the future. Similarly, the functionalities that organic building blocks can introduce are not limited to the categories in Section 3. For example, the ethylene oxide moiety has been successfully introduced to ligands, forming crosslinking ligand shells and providing good oxygen and water insolation for lead halide perovskite NCs.343 (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl, commonly known as TEMPO, is magnetic resonance (MR) active due to its radical nature. When introduced to surfaces of fluorescent QDs, the resulting TEMPO-QD complex makes a bimodal MR/optical imaging reagent providing high signal-to-noise ratios.344 Considering the structural flexibility of organic compounds, the choices of functionality are extensive and leave much room for exploration.
Interdisciplinary collaborations can not only consummate certain existing ideas as stated above, but also establish the foundation for a new research domain. Take the recent surge of chiral spintronics research as an example.309,345,346 Chiral spintronics utilize the connection between the electron spins (a quantum physics concept) and the chirality of organic compounds (a chemistry concept), which is their mirror-reflection asymmetry as shown in Fig. 12. Moving spins can couple to a chiral object via spin–orbit coupling. When the spin possesses the same handedness as the chiral object, it can travel a longer distance. Such hybridization with chiral structures not only removes the requirement of an external magnetic field in traditional spintronics, but also leads to significantly larger spin currents compared to other spin–orbit-coupling-based devices from the achiral counterparts. Such enhancement is crucial to achieve higher signal-to-noise ratios and reduce electricity consumption. Currently one of the topics of interest in chiral spintronics is introducing chiral ligand to QD surfaces, leveraging the quantum confinement effect to obtain robust spin states at room temperature and chiral effect to enhance spin current.309 This case exemplifies the fact that the organic–inorganic interface is a hybrid domain that requires the fusion of knowledge from multiple scientific disciplines via familiarizing a broad range of introductory content regarding organic building blocks to researchers from non-organic-chemistry backgrounds. We hope that by sharing this anchor-functionality paradigm with interdisciplinary researchers, we can spark other novel collaborative ideas in these communities.
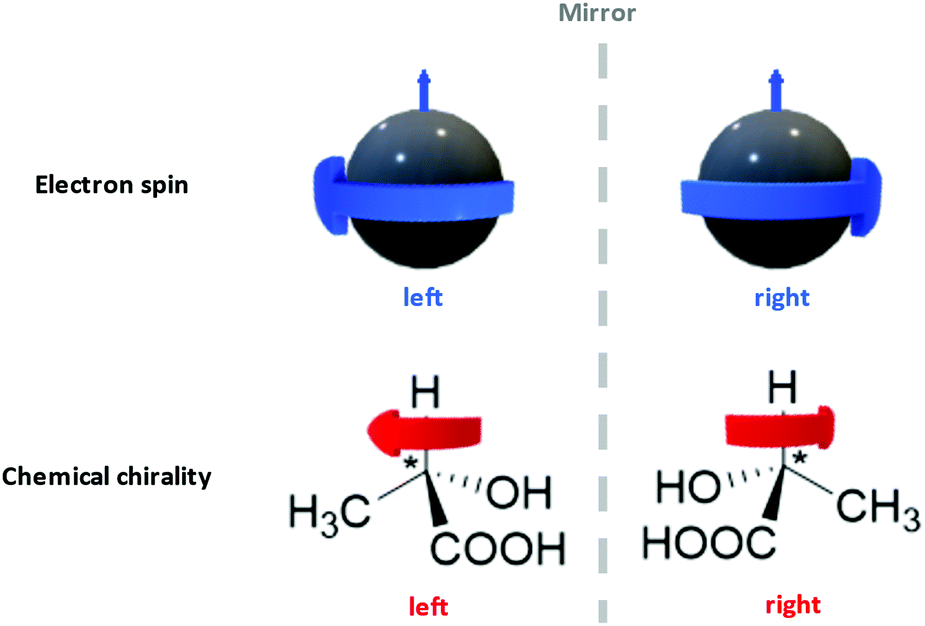 | ||
| Fig. 12 The mirror-reflection asymmetry comparison between electron spins and chirality of chemical compounds. The small blue arrows and the big blue arrows respectively represent the moving directions and spinning directions of electrons. The red arrows indicated the chirality of two lactic acid enantiomers. The direction of the red arrow is defined by the arrangement of the functional groups around the chiral centers (marked with “*”), with the order of –OH > –COOH > –CH3. | ||
Author contributions
Y. H. and C. K. L. led and coordinated the writing of this paper. Y. H. wrote Sections 1, 3.1, 3.3, and 4. T.A.C. wrote Sections 2.1.2, 3.2.1, and 3.2.4. B. M. S. wrote Section 2.2.2. Y. H., T. A. C., and B. M. S. combined, formatted, and proofread the manuscript. H. L. wrote Section 2.1.1. H. A. N. wrote Section 2.2.1. M. K. H. wrote Section 3.2.2. F. Y. D. wrote Section 3.2.3. L.M.J. and T. A. C. wrote Section 3.2.4. B. M. C., D. R. G., and C. K. L. supervised the writing. All authors provided critical feedback and helped shape the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was primarily supported by the National Science Foundation (NSF) through the UW Molecular Engineering Materials Center, a Materials Research Science and Engineering Center (DMR-1719797 to B.M.C., D.R.G., and C.K.L.). The authors would also like to acknowledge the financial support from the Clean Energy Institute (B.M.C., D.R.G., and C.K.L.).Notes and references
- J. De Roo, M. Ibáñez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071 CrossRef CAS.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706 CrossRef CAS.
- Y.-H. Kim, S. Kim, A. Kakekhani, J. Park, J. Park, Y.-H. Lee, H. Xu, S. Nagane, R. B. Wexler, D.-H. Kim, S. H. Jo, L. Martínez-Sarti, P. Tan, A. Sadhanala, G.-S. Park, Y.-W. Kim, B. Hu, H. J. Bolink, S. Yoo, R. H. Friend, A. M. Rappe and T.-W. Lee, Nat. Photonics, 2021, 15, 148 CrossRef CAS.
- A. Ekimov and A. Onushchehko, ZhETF Pis ma Redaktsiiu, 1981, 34, 363 CAS.
- R. Rossetti, S. Nakahara and L. E. Brus, J. Chem. Phys., 1983, 79, 1086 CrossRef CAS.
- J. Zhou, Y. Yang and C.-Y. Zhang, Chem. Rev., 2015, 115, 11669 CrossRef CAS.
- C. Tan, X. Cao, X.-J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han, G.-H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225 CrossRef CAS.
- X. Wang, Z. Bao, Y.-C. Chang and R.-S. Liu, ACS Energy Lett., 2020, 5, 3374 CrossRef CAS.
- E. Jang, S. Jun, H. Jang, J. Lim, B. Kim and Y. Kim, Adv. Mater., 2010, 22, 3076 CrossRef CAS.
- J. Y. Kim, J.-W. Lee, H. S. Jung, H. Shin and N.-G. Park, Chem. Rev., 2020, 120, 7867 CrossRef CAS.
- G. H. Carey, A. L. Abdelhady, Z. Ning, S. M. Thon, O. M. Bakr and E. H. Sargent, Chem. Rev., 2015, 115, 12732 CrossRef CAS.
- H. Guan, S. Zhao, H. Wang, D. Yan, M. Wang and Z. Zang, Nano Energy, 2020, 67, 104279 CrossRef CAS.
- Z. Wang, Y. Liu, S. Zhen, X. Li, W. Zhang, X. Sun, B. Xu, X. Wang, Z. Gao and X. Meng, Adv. Sci., 2020, 7, 1902688 CrossRef CAS.
- J. Pan, L. N. Quan, Y. Zhao, W. Peng, B. Murali, S. P. Sarmah, M. Yuan, L. Sinatra, N. M. Alyami, J. Liu, E. Yassitepe, Z. Yang, O. Voznyy, R. Comin, M. N. Hedhili, O. F. Mohammed, Z. H. Lu, D. H. Kim, E. H. Sargent and O. M. Bakr, Adv. Mater., 2016, 28, 8718 CrossRef CAS.
- H. Moon, C. Lee, W. Lee, J. Kim and H. Chae, Adv. Mater., 2019, 31, 1804294 CrossRef.
- K. Wu and T. Lian, Chem. Soc. Rev., 2016, 45, 3781 RSC.
- G. Liu, C. Zhen, Y. Kang, L. Wang and H.-M. Cheng, Chem. Soc. Rev., 2018, 47, 6410 RSC.
- V. Biju, Chem. Soc. Rev., 2014, 43, 744 RSC.
- G. Xu, S. Zeng, B. Zhang, M. T. Swihart, K.-T. Yong and P. N. Prasad, Chem. Rev., 2016, 116, 12234 CrossRef CAS.
- A. Privitera, M. Righetto, F. Cacialli and M. K. Riede, Adv. Opt. Mater., 2021, 9, 2100215 CrossRef CAS.
- M. J. Crane, L. M. Jacoby, T. A. Cohen, Y. Huang, C. K. Luscombe and D. R. Gamelin, Nano Lett., 2020, 20, 8626 CrossRef CAS.
- Z. Wu, Y. Zhang, R. Hu, M. Jiang, P. Liang, Q. Yang, L. Deng, T. Jia, Z. Sun and D. Feng, J. Phys. Chem. Lett., 2021, 12, 2126 CrossRef CAS.
- Z. Luo, S. Sun, A. Karasahin, A. S. Bracker, S. G. Carter, M. K. Yakes, D. Gammon and E. Waks, Nano Lett., 2019, 19, 7072 CrossRef CAS.
- Y.-J. Wei, Y.-M. He, M.-C. Chen, Y.-N. Hu, Y. He, D. Wu, C. Schneider, M. Kamp, S. Höfling, C.-Y. Lu and J.-W. Pan, Nano Lett., 2014, 14, 6515 CrossRef CAS.
- G. Zorn, S. R. Dave, T. Weidner, X. Gao and D. G. Castner, Surf. Sci., 2016, 648, 339 CrossRef CAS.
- K. G. Crawford, I. Maini, D. A. Macdonald and D. A. J. Moran, Prog. Surf. Sci., 2021, 96, 100613 CrossRef CAS.
- M. I. Landstrass and K. V. Ravi, Appl. Phys. Lett., 1989, 55, 975 CrossRef CAS.
- J. A. Garrido, S. Nowy, A. Härtl and M. Stutzmann, Langmuir, 2008, 24, 3897 CrossRef CAS.
- M. V. Hauf, B. Grotz, B. Naydenov, M. Dankerl, S. Pezzagna, J. Meijer, F. Jelezko, J. Wrachtrup, M. Stutzmann, F. Reinhard and J. A. Garrido, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 081304 CrossRef.
- P. R. Brown, D. Kim, R. R. Lunt, N. Zhao, M. G. Bawendi, J. C. Grossman and V. Bulović, ACS Nano, 2014, 8, 5863 CrossRef CAS.
- D. M. Kroupa, M. Vörös, N. P. Brawand, B. W. McNichols, E. M. Miller, J. Gu, A. J. Nozik, A. Sellinger, G. Galli and M. C. Beard, Nat. Commun., 2017, 8, 15257 CrossRef CAS.
- J. Raymakers, K. Haenen and W. Maes, J. Mater. Chem. C, 2019, 7, 10134 RSC.
- K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904 CrossRef CAS.
- G. Palui, F. Aldeek, W. Wang and H. Mattoussi, Chem. Soc. Rev., 2015, 44, 193 RSC.
- A. Heuer-Jungemann, N. Feliu, I. Bakaimi, M. Hamaly, A. Alkilany, I. Chakraborty, A. Masood, M. F. Casula, A. Kostopoulou, E. Oh, K. Susumu, M. H. Stewart, I. L. Medintz, E. Stratakis, W. J. Parak and A. G. Kanaras, Chem. Rev., 2019, 119, 4819 CrossRef CAS.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692 CrossRef CAS.
- A. I. Ekimov, A. L. Efros and A. A. Onushchenko, Solid State Commun., 1985, 56, 921 CrossRef CAS.
- M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, Nat. Mater., 2016, 15, 141 CrossRef CAS.
- M. L. H. Green and G. Parkin, J. Chem. Educ., 2014, 91, 807 CrossRef CAS.
- R. Koole, P. Schapotschnikow, C. de Mello Donegá, T. J. H. Vlugt and A. Meijerink, ACS Nano, 2008, 2, 1703 CrossRef CAS.
- N. C. Anderson, P. E. Chen, A. K. Buckley, J. D. Roo and J. S. Owen, J. Am. Chem. Soc., 2018, 140, 7199 CrossRef CAS.
- R. Dierick, F. Van den Broeck, K. De Nolf, Q. Zhao, A. Vantomme, J. C. Martins and Z. Hens, Chem. Mater., 2014, 26, 5950 CrossRef CAS.
- D. W. Houck and B. A. Korgel, Chem. Mater., 2018, 30, 8359 CrossRef CAS.
- Y. Zhou, F. Wang and W. E. Buhro, J. Am. Chem. Soc., 2015, 137, 15198 CrossRef CAS.
- K. N. Lawrence, P. Dutta, M. Nagaraju, M. B. Teunis, B. B. Muhoberac and R. Sardar, J. Am. Chem. Soc., 2016, 138, 12813 CrossRef CAS.
- N. C. Anderson, M. P. Hendricks, J. J. Choi and J. S. Owen, J. Am. Chem. Soc., 2013, 135, 18536 CrossRef CAS.
- J. Zhang, H. Zhang, W. Cao, Z. Pang, J. Li, Y. Shu, C. Zhu, X. Kong, L. Wang and X. Peng, J. Am. Chem. Soc., 2019, 141, 15675 CrossRef CAS.
- A. Ritchhart and B. M. Cossairt, Inorg. Chem., 2019, 58, 2840 CrossRef CAS.
- R. Gomes, A. Hassinen, A. Szczygiel, Q. Zhao, A. Vantomme, J. C. Martins and Z. Hens, J. Phys. Chem. Lett., 2011, 2, 145 CrossRef CAS.
- J. De Roo, F. Van den Broeck, K. De Keukeleere, J. C. Martins, I. Van Driessche and Z. Hens, J. Am. Chem. Soc., 2014, 136, 9650 CrossRef CAS.
- J. De Roo, Y. Justo, K. De Keukeleere, F. Van den Broeck, J. C. Martins, I. Van Driessche and Z. Hens, Angew. Chem., Int. Ed., 2015, 54, 6488 CrossRef CAS.
- C. Sun and J. C. Berg, Adv. Colloid Interface Sci., 2003, 105, 151 CrossRef CAS.
- A. Nag, M. V. Kovalenko, J.-S. Lee, W. Liu, B. Spokoyny and D. V. Talapin, J. Am. Chem. Soc., 2011, 133, 10612 CrossRef CAS.
- H. Häkkinen, Nat. Chem., 2012, 4, 443 CrossRef.
- L. M. Rossi, J. L. Fiorio, M. A. S. Garcia and C. P. Ferraz, Dalton Trans., 2018, 47, 5889 RSC.
- M. Y. Gee, Y. Shen and A. B. Greytak, J. Phys. Chem. C, 2020, 124, 23964 CrossRef CAS.
- O. Elimelech, O. Aviv, M. Oded and U. Banin, Nano Lett., 2020, 20, 6396 CrossRef CAS.
- J. J. Calvin, E. A. O’Brien, A. B. Sedlak, A. D. Balan and A. P. Alivisatos, ACS Nano, 2021, 15, 1407 CrossRef CAS.
- V. Ravi, J. M. Binz and R. M. Rioux, Nano Lett., 2013, 13, 4442 CrossRef CAS.
- C. K. Møller, Nature, 1958, 182, 1436 CrossRef.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050 CrossRef CAS.
- L. Etgar, P. Gao, Z. Xue, Q. Peng, A. K. Chandiran, B. Liu, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2012, 134, 17396 CrossRef CAS.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643 CrossRef CAS.
- D. P. McMeekin, Z. Wang, W. Rehman, F. Pulvirenti, J. B. Patel, N. K. Noel, M. B. Johnston, S. R. Marder, L. M. Herz and H. J. Snaith, Adv. Mater., 2017, 29, 1607039 CrossRef.
- J. Berry, T. Buonassisi, D. A. Egger, G. Hodes, L. Kronik, Y.-L. Loo, I. Lubomirsky, S. R. Marder, Y. Mastai, J. S. Miller, D. B. Mitzi, Y. Paz, A. M. Rappe, I. Riess, B. Rybtchinski, O. Stafsudd, V. Stevanovic, M. F. Toney, D. Zitoun, A. Kahn, D. Ginley and D. Cahen, Adv. Mater., 2015, 27, 5102 CrossRef CAS.
- S. Song, R. Hill, K. Choi, K. Wojciechowski, S. Barlow, J. Leisen, H. J. Snaith, S. R. Marder and T. Park, Nano Energy, 2018, 49, 324 CrossRef CAS.
- D. A. Valverde-Chávez, E. Rojas-Gatjens, J. Williamson, S. Jariwala, Y. Shi, D. P. McCarthy, S. Barlow, S. R. Marder, D. S. Ginger and C. Silva-Acuña, J. Mater. Chem. C, 2021, 9, 8204 RSC.
- J. Jeong, M. Kim, J. Seo, H. Lu, P. Ahlawat, A. Mishra, Y. Yang, M. A. Hope, F. T. Eickemeyer, M. Kim, Y. J. Yoon, I. W. Choi, B. P. Darwich, S. J. Choi, Y. Jo, J. H. Lee, B. Walker, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, A. Hagfeldt, D. S. Kim, M. Grätzel and J. Y. Kim, Nature, 2021, 592, 381 CrossRef CAS.
- R. Lin, K. Xiao, Z. Qin, Q. Han, C. Zhang, M. Wei, M. I. Saidaminov, Y. Gao, J. Xu, M. Xiao, A. Li, J. Zhu, E. H. Sargent and H. Tan, Nat. Energy, 2019, 4, 864 CrossRef CAS.
- A. Al-Ashouri, E. Köhnen, B. Li, A. Magomedov, H. Hempel, P. Caprioglio, J. A. Márquez, A. B. M. Vilches, E. Kasparavicius, J. A. Smith, N. Phung, D. Menzel, M. Grischek, L. Kegelmann, D. Skroblin, C. Gollwitzer, T. Malinauskas, M. Jošt, G. Matič, B. Rech, R. Schlatmann, M. Topič, L. Korte, A. Abate, B. Stannowski, D. Neher, M. Stolterfoht, T. Unold, V. Getautis and S. Albrecht, Science, 2020, 370, 1300 CrossRef CAS.
- B. A. Koscher, J. K. Swabeck, N. D. Bronstein and A. P. Alivisatos, J. Am. Chem. Soc., 2017, 139, 6566 CrossRef CAS.
- Y. Zhang, T. D. Siegler, C. J. Thomas, M. K. Abney, T. Shah, A. De Gorostiza, R. M. Greene and B. A. Korgel, Chem. Mater., 2020, 32, 5410 CrossRef CAS.
- N. Pradhan, ACS Energy Lett., 2019, 4, 1634 CrossRef CAS.
- H. Utzat, K. E. Shulenberger, O. B. Achorn, M. Nasilowski, T. S. Sinclair and M. G. Bawendi, Nano Lett., 2017, 17, 6838 CrossRef CAS.
- Z. Chen, H. Li, Y. Tang, X. Huang, D. Ho and C.-S. Lee, Mater. Res. Express, 2014, 1, 015034 CrossRef CAS.
- G. Li, Z.-K. Tan, D. Di, M. L. Lai, L. Jiang, J. H.-W. Lim, R. H. Friend and N. C. Greenham, Nano Lett., 2015, 15, 2640 CrossRef CAS.
- L. Zhao, K. Roh, S. Kacmoli, K. Al Kurdi, S. Jhulki, S. Barlow, S. R. Marder, C. Gmachl and B. P. Rand, Adv. Mater., 2020, 32, 2000752 CrossRef CAS.
- S. Yakunin, L. Protesescu, F. Krieg, M. I. Bodnarchuk, G. Nedelcu, M. Humer, G. D. Luca, M. Fiebig, W. Heiss and M. V. Kovalenko, Nat. Commun., 2015, 6, 8056 CrossRef CAS.
- Y. Jia, R. A. Kerner, A. J. Grede, A. N. Brigeman, B. P. Rand and N. C. Giebink, Nano Lett., 2016, 16, 4624 CrossRef CAS.
- Y. Jia, R. A. Kerner, A. J. Grede, B. P. Rand and N. C. Giebink, Nat. Photonics, 2017, 11, 784 CrossRef CAS.
- H. Kim, K. Roh, J. P. Murphy, L. Zhao, W. B. Gunnarsson, E. Longhi, S. Barlow, S. R. Marder, B. P. Rand and N. C. Giebink, Adv. Opt. Mater., 2020, 8, 1901297 CrossRef CAS.
- H. Wei, Y. Fang, P. Mulligan, W. Chuirazzi, H.-H. Fang, C. Wang, B. R. Ecker, Y. Gao, M. A. Loi, L. Cao and J. Huang, Nat. Photonics, 2016, 10, 333 CrossRef CAS.
- J. Liu, B. Shabbir, C. Wang, T. Wan, Q. Ou, P. Yu, A. Tadich, X. Jiao, D. Chu, D. Qi, D. Li, R. Kan, Y. Huang, Y. Dong, J. Jasieniak, Y. Zhang and Q. Bao, Adv. Mater., 2019, 31, 1901644 CrossRef.
- T. A. Cohen, T. J. Milstein, D. M. Kroupa, J. D. MacKenzie, C. K. Luscombe and D. R. Gamelin, J. Mater. Chem. A, 2019, 7, 9279 RSC.
- X. Luo, T. Ding, X. Liu, Y. Liu and K. Wu, Nano Lett., 2019, 19, 338 CrossRef CAS.
- K. Nikolaidou, S. Sarang, C. Hoffman, B. Mendewala, H. Ishihara, J. Q. Lu, B. Ilan, V. Tung and S. Ghosh, Adv. Opt. Mater., 2016, 4, 2126 CrossRef CAS.
- J. Tong, J. Luo, L. Shi, J. Wu, L. Xu, J. Song, P. Wang, H. Li and Z. Deng, J. Mater. Chem. A, 2019, 7, 4872 RSC.
- F. Meinardi, Q. A. Akkerman, F. Bruni, S. Park, M. Mauri, Z. Dang, L. Manna and S. Brovelli, ACS Energy Lett., 2017, 2, 2368 CrossRef CAS.
- G. Rainò, G. Nedelcu, L. Protesescu, M. I. Bodnarchuk, M. V. Kovalenko, R. F. Mahrt and T. Stöferle, ACS Nano, 2016, 10, 2485 CrossRef.
- Y.-S. Park, S. Guo, N. S. Makarov and V. I. Klimov, ACS Nano, 2015, 9, 10386 CrossRef CAS.
- H. Utzat, W. Sun, A. E. K. Kaplan, F. Krieg, M. Ginterseder, B. Spokoyny, N. D. Klein, K. E. Shulenberger, C. F. Perkinson, M. V. Kovalenko and M. G. Bawendi, Science, 2019, 363, 1068 CrossRef CAS.
- Q. A. Akkerman, G. Rainò, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394 CrossRef CAS.
- D. P. Nenon, K. Pressler, J. Kang, B. A. Koscher, J. H. Olshansky, W. T. Osowiecki, M. A. Koc, L.-W. Wang and A. P. Alivisatos, J. Am. Chem. Soc., 2018, 140, 17760 CrossRef CAS.
- J. Kang and L.-W. Wang, J. Phys. Chem. Lett., 2017, 8, 489 CrossRef CAS.
- S. Ten Brinck, F. Zaccaria and I. Infante, ACS Energy Lett., 2019, 4, 2739 CrossRef CAS.
- S. E. Creutz, E. N. Crites, M. C. De Siena and D. R. Gamelin, Chem. Mater., 2018, 30, 4887 CrossRef CAS.
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635 CrossRef CAS.
- M. Imran, V. Caligiuri, M. Wang, L. Goldoni, M. Prato, R. Krahne, L. De Trizio and L. Manna, J. Am. Chem. Soc., 2018, 140, 2656 CrossRef CAS.
- T. J. Milstein, D. M. Kroupa and D. R. Gamelin, Nano Lett., 2018, 18, 3792 CrossRef CAS.
- W. Liu, Q. Lin, H. Li, K. Wu, I. Robel, J. M. Pietryga and V. I. Klimov, J. Am. Chem. Soc., 2016, 138, 14954 CrossRef CAS.
- G. Pan, X. Bai, D. Yang, X. Chen, P. Jing, S. Qu, L. Zhang, D. Zhou, J. Zhu, W. Xu, B. Dong and H. Song, Nano Lett., 2017, 17, 8005 CrossRef CAS.
- D. Zhou, D. Liu, G. Pan, X. Chen, D. Li, W. Xu, X. Bai and H. Song, Adv. Mater., 2017, 29, 1704149 CrossRef.
- M. C. De Siena, D. E. Sommer, S. E. Creutz, S. T. Dunham and D. R. Gamelin, Chem. Mater., 2019, 31, 7711 CrossRef CAS.
- B. Conings, J. Drijkoningen, N. Gauquelin, A. Babayigit, J. D'Haen, L. D'Olieslaeger, A. Ethirajan, J. Verbeeck, J. Manca, E. Mosconi, F. D. Angelis and H.-G. Boyen, Adv. Energy Mater., 2015, 5, 1500477 CrossRef.
- C. Manspeaker, S. Venkatesan, A. Zakhidov and K. S. Martirosyan, Curr. Opin. Chem. Eng., 2017, 15, 1 CrossRef.
- G. Rainò, A. Landuyt, F. Krieg, C. Bernasconi, S. T. Ochsenbein, D. N. Dirin, M. I. Bodnarchuk and M. V. Kovalenko, Nano Lett., 2019, 19, 3648 CrossRef.
- G. Yuan, C. Ritchie, M. Ritter, S. Murphy, D. E. Gómez and P. Mulvaney, J. Phys. Chem. C, 2018, 122, 13407 CrossRef CAS.
- Y. Ogomi, A. Morita, S. Tsukamoto, T. Saitho, Q. Shen, T. Toyoda, K. Yoshino, S. S. Pandey, T. Ma and S. Hayase, J. Phys. Chem. C, 2014, 118, 16651 CrossRef CAS.
- L. Zuo, Z. Gu, T. Ye, W. Fu, G. Wu, H. Li and H. Chen, J. Am. Chem. Soc., 2015, 137, 2674 CrossRef CAS.
- A. Pan, B. He, X. Fan, Z. Liu, J. J. Urban, A. P. Alivisatos, L. He and Y. Liu, ACS Nano, 2016, 10, 7943 CrossRef CAS.
- V. K. Ravi, P. K. Santra, N. Joshi, J. Chugh, S. K. Singh, H. Rensmo, P. Ghosh and A. Nag, J. Phys. Chem. Lett., 2017, 8, 4988 CrossRef CAS.
- Z. Hens and J. C. Martins, Chem. Mater., 2013, 25, 1211 CrossRef CAS.
- S. ten Brinck and I. Infante, ACS Energy Lett., 2016, 1, 1266 CrossRef CAS.
- M. Imran, P. Ijaz, D. Baranov, L. Goldoni, U. Petralanda, Q. Akkerman, A. L. Abdelhady, M. Prato, P. Bianchini, I. Infante and L. Manna, Nano Lett., 2018, 18, 7822 CrossRef CAS.
- F. Krieg, S. T. Ochsenbein, S. Yakunin, S. ten Brinck, P. Aellen, A. Süess, B. Clerc, D. Guggisberg, O. Nazarenko, Y. Shynkarenko, S. Kumar, C.-J. Shih, I. Infante and M. V. Kovalenko, ACS Energy Lett., 2018, 3, 641 CrossRef CAS.
- Y. Cai, H. Wang, Y. Li, L. Wang, Y. Lv, X. Yang and R.-J. Xie, Chem. Mater., 2019, 31, 881 CrossRef CAS.
- S. E. Creutz, E. N. Crites, M. C. De Siena and D. R. Gamelin, Nano Lett., 2018, 18, 1118 CrossRef CAS.
- D. Yoo, J. Y. Woo, Y. Kim, S. W. Kim, S.-H. Wei, S. Jeong and Y.-H. Kim, J. Phys. Chem. Lett., 2020, 11, 652 CrossRef CAS.
- F. Di Stasio, S. Christodoulou, N. Huo and G. Konstantatos, Chem. Mater., 2017, 29, 7663 CrossRef.
- M. Imran, P. Ijaz, L. Goldoni, D. Maggioni, U. Petralanda, M. Prato, G. Almeida, I. Infante and L. Manna, ACS Energy Lett., 2019, 4, 819 CrossRef CAS.
- M. I. Bodnarchuk, S. C. Boehme, S. ten Brinck, C. Bernasconi, Y. Shynkarenko, F. Krieg, R. Widmer, B. Aeschlimann, D. Günther, M. V. Kovalenko and I. Infante, ACS Energy Lett., 2019, 4, 63 CrossRef CAS.
- A. Dutta, R. K. Behera, S. K. Dutta, S. Das Adhikari and N. Pradhan, J. Phys. Chem. Lett., 2018, 9, 6599 CrossRef CAS.
- H. Sun, Z. Yang, M. Wei, W. Sun, X. Li, S. Ye, Y. Zhao, H. Tan, E. L. Kynaston, T. B. Schon, H. Yan, Z.-H. Lu, G. A. Ozin, E. H. Sargent and D. S. Seferos, Adv. Mater., 2017, 29, 1701153 CrossRef.
- S. Yakunin, J. Chaaban, B. M. Benin, I. Cherniukh, C. Bernasconi, A. Landuyt, Y. Shynkarenko, S. Bolat, C. Hofer, Y. E. Romanyuk, S. Cattaneo, S. I. Pokutnyi, R. D. Schaller, M. I. Bodnarchuk, D. Poulikakos and M. V. Kovalenko, Nat. Commun., 2021, 12, 981 CrossRef CAS.
- I. Cherniukh, G. Rainò, T. Stöferle, M. Burian, A. Travesset, D. Naumenko, H. Amenitsch, R. Erni, R. F. Mahrt, M. I. Bodnarchuk and M. V. Kovalenko, Nature, 2021, 593, 535 CrossRef CAS.
- X. Li, M. Ibrahim Dar, C. Yi, J. Luo, M. Tschumi, S. M. Zakeeruddin, M. K. Nazeeruddin, H. Han and M. Grätzel, Nat. Chem., 2015, 7, 703 CrossRef CAS.
- D. W. deQuilettes, S. Koch, S. Burke, R. K. Paranji, A. J. Shropshire, M. E. Ziffer and D. S. Ginger, ACS Energy Lett., 2016, 1, 438 CrossRef CAS.
- F. Liu, Y. Zhang, C. Ding, S. Kobayashi, T. Izuishi, N. Nakazawa, T. Toyoda, T. Ohta, S. Hayase, T. Minemoto, K. Yoshino, S. Dai and Q. Shen, ACS Nano, 2017, 11, 10373 CrossRef CAS.
- L. Wu, Q. Zhong, D. Yang, M. Chen, H. Hu, Q. Pan, H. Liu, M. Cao, Y. Xu, B. Sun and Q. Zhang, Langmuir, 2017, 33, 12689 CrossRef CAS.
- H. Wang, N. Sui, X. Bai, Y. Zhang, Q. Rice, F. J. Seo, Q. Zhang, V. L. Colvin and W. W. Yu, J. Phys. Chem. Lett., 2018, 9, 4166 CrossRef CAS.
- H. Wang, X. Zhang, N. Sui, Y. Hu, V. L. Colvin, W. W. Yu and Y. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 11769 CrossRef CAS.
- Y. Tan, Y. Zou, L. Wu, Q. Huang, D. Yang, M. Chen, M. Ban, C. Wu, T. Wu, S. Bai, T. Song, Q. Zhang and B. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 3784 CrossRef CAS.
- N. K. Noel, A. Abate, S. D. Stranks, E. S. Parrott, V. M. Burlakov, A. Goriely and H. J. Snaith, ACS Nano, 2014, 8, 9815 CrossRef CAS.
- F. Krieg, Q. K. Ong, M. Burian, G. Rainò, D. Naumenko, H. Amenitsch, A. Süess, M. J. Grotevent, F. Krumeich, M. I. Bodnarchuk, I. Shorubalko, F. Stellacci and M. V. Kovalenko, J. Am. Chem. Soc., 2019, 141, 19839 CrossRef CAS.
- B. Zhang, L. Goldoni, J. Zito, Z. Dang, G. Almeida, F. Zaccaria, J. de Wit, I. Infante, L. De Trizio and L. Manna, Chem. Mater., 2019, 31, 9140 CrossRef CAS.
- B. Zhang, L. Goldoni, C. Lambruschini, L. Moni, M. Imran, A. Pianetti, V. Pinchetti, S. Brovelli, L. De Trizio and L. Manna, Nano Lett., 2020, 20, 8847 CrossRef CAS.
- Y. Shynkarenko, M. I. Bodnarchuk, C. Bernasconi, Y. Berezovska, V. Verteletskyi, S. T. Ochsenbein and M. V. Kovalenko, ACS Energy Lett., 2019, 4, 2703 CrossRef CAS.
- E. Mosconi, C. Quarti, T. Ivanovska, G. Ruani and F. D. Angelis, Phys. Chem. Chem. Phys., 2014, 16, 16137 RSC.
- S. Liu, F. Zheng, N. Z. Koocher, H. Takenaka, F. Wang and A. M. Rappe, J. Phys. Chem. Lett., 2015, 6, 693 CrossRef CAS.
- A. Amat, E. Mosconi, E. Ronca, C. Quarti, P. Umari, M. K. Nazeeruddin, M. Grätzel and F. De Angelis, Nano Lett., 2014, 14, 3608 CrossRef CAS.
- S. Toyoshima, K. Kuwabara, T. Sakurai, T. Taima, K. Saito, H. Kato and K. Akimoto, Jpn. J. Appl. Phys., 2007, 46, 2692 CrossRef CAS.
- Y. R. Park, S. Eom, H. H. Kim, W. K. Choi and Y. Kang, Sci. Rep., 2020, 10, 14758 CrossRef CAS.
- M. Li, Y. Zhang, H. Gao, Y. Peng, S. Tang, L. Yu, R. Chen and W. Huang, J. Phys. Chem. C, 2021, 125, 2866 CrossRef CAS.
- M.-S. Lee, S. Sarwar, S. Park, U. Asmat, D. T. Thuy, C.-H. Han, S. Ahn, I. Jeong and S. Hong, Sustainable Energy Fuels, 2020, 4, 3318 RSC.
- A. Bitar, N. M. Ahmad, H. Fessi and A. Elaissari, Drug Discovery Today, 2012, 17, 1147 CrossRef CAS.
- S. T. Selvan, T. T. Tan and J. Y. Ying, Adv. Mater., 2005, 17, 1620 CrossRef CAS.
- J. Fokkema, J. Fermie, N. Liv, D. J. van den Heuvel, T. O. M. Konings, G. A. Blab, A. Meijerink, J. Klumperman and H. C. Gerritsen, Sci. Rep., 2018, 8, 13625 CrossRef.
- Y. Xu, Y. Lv, R. Wu, J. Li, H. Shen, H. Yang, H. Zhang and L. S. Li, Inorg. Chem., 2021, 60, 6503 CrossRef CAS.
- L. M. Liz-Marzán, M. Giersig and P. Mulvaney, Langmuir, 1996, 12, 4329 CrossRef.
- Y. Han, Z. Lu, Z. Teng, J. Liang, Z. Guo, D. Wang, M.-Y. Han and W. Yang, Langmuir, 2017, 33, 5879 CrossRef CAS.
- Y. Yang, L. Jing, X. Yu, D. Yan and M. Gao, Chem. Mater., 2007, 19, 4123 CrossRef CAS.
- Y. Han, J. Jiang, S. S. Lee and J. Y. Ying, Langmuir, 2008, 24, 5842 CrossRef CAS.
- A. Guerrero-Martínez, J. Pérez-Juste and L. M. Liz-Marzán, Adv. Mater., 2010, 22, 1182 CrossRef.
- S. Liu and M.-Y. Han, Chem. – Asian J., 2010, 5, 36 CAS.
- S. T. Selvan, Biointerphases, 2010, 5, FA110 CrossRef.
- L. Lei, X. Liu, Y. Li, Y. Cui, Y. Yang and G. Qin, Mater. Chem. Phys., 2011, 125, 866 CrossRef CAS.
- I. V. Melnyk, R. P. Pogorilyi, Y. L. Zub, M. Vaclavikova, K. Gdula, A. Dąbrowski, G. A. Seisenbaeva and V. G. Kessler, Sci. Rep., 2018, 8, 8592 CrossRef.
- M. J. Marchant, L. Guzmán, A. H. Corvalán and M. J. Kogan, Nanomaterials, 2019, 9, 1333 CrossRef CAS.
- F. Heidari, M. Hekmati and H. Veisi, J. Colloid Interface Sci., 2017, 501, 175 CrossRef CAS.
- K.-M. Hsieh, K.-C. Lan, W.-L. Hu, M.-K. Chen, L.-S. Jang and M.-H. Wang, Biosens. Bioelectron., 2013, 49, 450 CrossRef CAS.
- J. V. Hollingsworth, N. V. S. D. K. Bhupathiraju, J. Sun, E. Lochner, M. G. H. Vicente and P. S. Russo, ACS Appl. Mater. Interfaces, 2016, 8, 792 CrossRef CAS.
- M. Masteri-Farahani and S. Shahsavarifar, Appl. Organomet. Chem., 2018, 32, 4064 CrossRef.
- R. Riedel, N. Mahr, C. Yao, A. Wu, F. Yang and N. Hampp, Nanoscale, 2020, 12, 3007 RSC.
- X. Hu and X. Gao, ACS Nano, 2010, 4, 6080 CrossRef CAS.
- R. Koole, M. M. van Schooneveld, J. Hilhorst, K. Castermans, D. P. Cormode, G. J. Strijkers, C. de Mello Donegá, D. Vanmaekelbergh, A. W. Griffioen, K. Nicolay, Z. A. Fayad, A. Meijerink and W. J. M. Mulder, Bioconjugate Chem., 2008, 19, 2471 CrossRef CAS.
- M. M. van Schooneveld, A. Gloter, O. Stephan, L. F. Zagonel, R. Koole, A. Meijerink, W. J. M. Mulder and F. M. F. de Groot, Nat. Nanotechnol., 2010, 5, 538 CrossRef CAS.
- L. Mangolini, J. Vac. Sci. Technol., B, 2013, 31, 020801 CrossRef.
- V. N. Mochalin, O. Shenderova, D. Ho and Y. Gogotsi, Nat. Nanotechnol., 2012, 7, 11 CrossRef CAS.
- T.-W. Kim, C.-H. Cho, B.-H. Kim and S.-J. Park, Appl. Phys. Lett., 2006, 88, 123102 CrossRef.
- S. P. Pujari, H. Driss, F. Bannani, B. van Lagen and H. Zuilhof, Chem. Mater., 2018, 30, 6503 CrossRef CAS.
- X. Zhang, D. Neiner, S. Wang, A. Y. Louie and S. M. Kauzlarich, Nanotechnology, 2007, 18, 095601 CrossRef.
- S. Xun, X. Song, L. Wang, M. E. Grass, Z. Liu, V. S. Battaglia and G. Liu, J. Electrochem. Soc., 2011, 158, A1260 CrossRef CAS.
- B. J. Winters, J. Holm and J. T. Roberts, J. Nanopart. Res., 2011, 13, 5473 CrossRef CAS.
- O. O. Mykhaylyk, Y. M. Solonin, D. N. Batchelder and R. Brydson, J. Appl. Phys., 2005, 97, 074302 CrossRef.
- J. Xiao, G. Ouyang, P. Liu, C. X. Wang and G. W. Yang, Nano Lett., 2014, 14, 3645 CrossRef CAS.
- K. Iakoubovskii, M. V. Baidakova, B. H. Wouters, A. Stesmans, G. J. Adriaenssens, A. Y. Vul and P. J. Grobet, Diamond Relat. Mater., 2000, 9, 861 CrossRef CAS.
- K. M. Song, J. Y. Shim and H. K. Baik, Diamond Relat. Mater., 2002, 11, 185 CrossRef CAS.
- Z. Q. Li and Y. Zhou, Phys. B, 2010, 405, 1004 CrossRef CAS.
- K. Niwase, T. Tanaka, Y. Kakimoto, K. N. Ishihara and P. H. Shingu, Mater. Trans., JIM, 1995, 36, 282 CrossRef CAS.
- V. Pichot, M. Comet, E. Fousson, C. Baras, A. Senger, F. Le Normand and D. Spitzer, Diamond Relat. Mater., 2008, 17, 13 CrossRef CAS.
- C. L. Cheng, H. C. Chang, J. C. Lin, K. J. Song and J. K. Wang, Phys. Rev. Lett., 1997, 78, 3713 CrossRef CAS.
- P. Thissen, O. Seitz and Y. J. Chabal, Prog. Surf. Sci., 2012, 87, 272 CrossRef CAS.
- T. J. Barnes, K. L. Jarvis and C. A. Prestidge, Ther. Delivery, 2013, 4, 811 CrossRef CAS.
- D. H. Jariwala, D. Patel and S. Wairkar, Mater. Sci. Eng., C, 2020, 113, 110996 CrossRef CAS.
- N. Aissaoui, L. Bergaoui, J. Landoulsi, J.-F. Lambert and S. Boujday, Langmuir, 2012, 28, 656 CrossRef CAS.
- X. Li, J. Shao, Y. Qin, C. Shao, T. Zheng and L. Ye, J. Mater. Chem., 2011, 21, 7966 RSC.
- J. Hwang, M. P. Hwang, M. Choi, Y. Seo, Y. Jo, J. Son, J. Hong and J. Choi, Sci. Rep., 2016, 6, 35565 CrossRef CAS.
- I. S. Curtis, R. J. Wills and M. Dasog, Nanoscale, 2021, 13, 2685 RSC.
- Z. Kang, Y. Liu and S.-T. Lee, Nanoscale, 2011, 3, 777 RSC.
- J. B. Zang, Y. H. Wang, S. Z. Zhao, L. Y. Bian and J. Lu, Diamond Relat. Mater., 2007, 16, 16 CrossRef CAS.
- S. Jiang, B. Hu, R. Sahore, L. Zhang, H. Liu, L. Zhang, W. Lu, B. Zhao and Z. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 44924 CrossRef CAS.
- M. J. Rutter and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 9241 CrossRef CAS.
- D. Mizuno, K. Yamasue and Y. Cho, Appl. Phys. Lett., 2013, 103, 101601 CrossRef.
- V. Puddu and C. C. Perry, ACS Nano, 2012, 6, 6356 CrossRef CAS.
- H. A. Girard, S. Perruchas, C. Gesset, M. Chaigneau, L. Vieille, J.-C. Arnault, P. Bergonzo, J.-P. Boilot and T. Gacoin, ACS Appl. Mater. Interfaces, 2009, 1, 2738 CrossRef CAS.
- S. Ciampi, J. B. Harper and J. J. Gooding, Chem. Soc. Rev., 2010, 39, 2158 RSC.
- E. G. Robins, M. P. Stewart and J. M. Buriak, Chem. Commun., 1999, 2479 RSC.
- B. Arkles, J. R. Steinmetz, J. Zazyczny and P. Mehta, J. Adhes. Sci. Technol., 1992, 6, 193 CrossRef CAS.
- A. Mesnage, X. Lefèvre, P. Jégou, G. Deniau and S. Palacin, Langmuir, 2012, 28, 11767 CrossRef CAS.
- M. Busson, A. Berisha, C. Combellas, F. Kanoufi and J. Pinson, Chem. Commun., 2011, 47, 12631 RSC.
- C.-H. Jung, K.-H. Kim and S.-H. Hong, ACS Appl. Mater. Interfaces, 2019, 11, 26753 CrossRef CAS.
- Y. Zhang, A. A. Tamijani, M. E. Taylor, B. Zhi, C. L. Haynes, S. E. Mason and R. J. Hamers, J. Am. Chem. Soc., 2019, 141, 8277 CrossRef CAS.
- L. Diederich, O. M. Küttel, P. Aebi and L. Schlapbach, Surf. Sci., 1998, 418, 219 CrossRef CAS.
- L. A. Huck and J. M. Buriak, Langmuir, 2012, 28, 16285 CrossRef CAS.
- J. P. L. Perez, J. Yu, A. J. Sheppard, S. D. Chambreau, G. L. Vaghjiani and S. L. Anderson, ACS Appl. Mater. Interfaces, 2015, 7, 9991 CrossRef CAS.
- E. Fok, M. Shih, A. Meldrum and J. G. C. Veinot, Chem. Commun., 2004, 386 RSC.
- S. Gabriel, R. Jérôme and C. Jérôme, Prog. Polym. Sci., 2010, 35, 113 CrossRef CAS.
- E. V. Rogozhina, D. A. Eckhoff, E. Gratton and P. V. Braun, J. Mater. Chem., 2006, 16, 1421 RSC.
- C. Stavis, T. L. Clare, J. E. Butler, A. D. Radadia, R. Carr, H. Zeng, W. P. King, J. A. Carlisle, A. Aksimentiev, R. Bashir and R. J. Hamers, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 983 CrossRef CAS.
- P. E. Colavita, B. Sun, K.-Y. Tse and R. J. Hamers, J. Am. Chem. Soc., 2007, 129, 13554 CrossRef CAS.
- C. Gautier, I. López and T. Breton, Mater. Adv., 2021, 2, 2773 RSC.
- K. Brymora, J. Fouineau, A. Eddarir, F. Chau, N. Yaacoub, J.-M. Grenèche, J. Pinson, S. Ammar and F. Calvayrac, J. Nanopart. Res., 2015, 17, 438 CrossRef.
- S. Betelu, I. Tijunelyte, L. Boubekeur-Lecaque, I. Ignatiadis, J. Ibrahim, S. Gaboreau, C. Berho, T. Toury, E. Guenin, N. Lidgi-Guigui, N. Félidj, E. Rinnert and M. L. D. L. Chapelle, J. Phys. Chem. C, 2016, 120, 18158 CrossRef CAS.
- L. Laurentius, S. R. Stoyanov, S. Gusarov, A. Kovalenko, R. Du, G. P. Lopinski and M. T. McDermott, ACS Nano, 2011, 5, 4219 CrossRef CAS.
- F. Mousli, A. Chaouchi, S. Hocine, A. Lamouri, M. Rei Vilar, A. Kadri and M. M. Chehimi, Appl. Surf. Sci., 2019, 465, 1078 CrossRef CAS.
- D. Hetemi, V. Noël and J. Pinson, Biosensors, 2020, 10, 4 CrossRef CAS.
- N. Bondon, L. Raehm, C. Charnay, R. Boukherroub and J.-O. Durand, J. Mater. Chem. B, 2020, 8, 10878 RSC.
- A. Azioune, A. Ben Slimane, L. Ait Hamou, A. Pleuvy, M. M. Chehimi, C. Perruchot and S. P. Armes, Langmuir, 2004, 20, 3350 CrossRef CAS.
- H.-H. Cho, H. Yang, D. J. Kang and B. J. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 8615 CrossRef CAS.
- L. Qian, Y. Zheng, J. Xue and P. H. Holloway, Nat. Photonics, 2011, 5, 543 CrossRef CAS.
- W. Zhou, Y. Shang, F. P. García de Arquer, K. Xu, R. Wang, S. Luo, X. Xiao, X. Zhou, R. Huang, E. H. Sargent and Z. Ning, Nat. Electron., 2020, 3, 251 CrossRef CAS.
- A. Kiani, B. R. Sutherland, Y. Kim, O. Ouellette, L. Levina, G. Walters, C.-T. Dinh, M. Liu, O. Voznyy, X. Lan, A. J. Labelle, A. H. Ip, A. Proppe, G. H. Ahmed, O. F. Mohammed, S. Hoogland and E. H. Sargent, Appl. Phys. Lett., 2016, 109, 183105 CrossRef.
- R. Schmid, Monatsh. Chem., 2001, 132, 1295 CrossRef CAS.
- Q. Lu, C. Wu, D. Liu, H. Wang, W. Su, H. Li, Y. Zhang and S. Yao, Green Chem., 2017, 19, 900 RSC.
- T. A. Cohen, Y. Huang, N. A. Bricker, C. S. Juhl, T. J. Milstein, J. D. MacKenzie, C. K. Luscombe and D. R. Gamelin, Chem. Mater., 2021, 33, 3779 CrossRef CAS.
- S. Wang, L. Du, Z. Jin, Y. Xin and H. Mattoussi, J. Am. Chem. Soc., 2020, 142, 12669 CrossRef CAS.
- B. Shi, J. Lü, Y. Liu, Y. Xiao and C. Lü, Mater. Chem. Front., 2021, 5, 4343 RSC.
- S. M. Lee, H. Jung, W. I. Park, Y. Lee, E. Koo and J. Bang, ChemistrySelect, 2018, 3, 11320 CrossRef CAS.
- J. Liu, T. Tanaka, K. Sivula, A. P. Alivisatos and J. M. J. Fréchet, J. Am. Chem. Soc., 2004, 126, 6550 CrossRef CAS.
- E. T. Vickers, T. A. Graham, A. H. Chowdhury, B. Bahrami, B. W. Dreskin, S. Lindley, S. B. Naghadeh, Q. Qiao and J. Z. Zhang, ACS Energy Lett., 2018, 3, 2931 CrossRef CAS.
- D. Cui, J. Xu, T. Zhu, G. Paradee, S. Ashok and M. Gerhold, Appl. Phys. Lett., 2006, 88, 183111 CrossRef.
- Y. Liu, S. R. Scully, M. D. McGehee, J. Liu, C. K. Luscombe, J. M. J. Fréchet, S. E. Shaheen and D. S. Ginley, J. Phys. Chem. B, 2006, 110, 3257 CrossRef CAS.
- C. Goh, S. R. Scully and M. D. McGehee, J. Appl. Phys., 2007, 101, 114503 CrossRef.
- A. E. Colbert, W. Wu, E. M. Janke, F. Ma and D. S. Ginger, J. Phys. Chem. C, 2015, 119, 24733 CrossRef CAS.
- J. Park, J. H. Lee, H. Doh, S. W. Heo, K. Cho, S. Kim, S. Cho and J. Bang, J. Phys. Chem. C, 2020, 124, 25775 CrossRef CAS.
- Z. Yang, J. Z. Fan, A. H. Proppe, F. P. G. D. Arquer, D. Rossouw, O. Voznyy, X. Lan, M. Liu, G. Walters, R. Quintero-Bermudez, B. Sun, S. Hoogland, G. A. Botton, S. O. Kelley and E. H. Sargent, Nat. Commun., 2017, 8, 1325 CrossRef PubMed.
- H. Zhang, J. Jang, W. Liu and D. V. Talapin, ACS Nano, 2014, 8, 7359 CrossRef CAS PubMed.
- M.-J. Choi, F. P. García de Arquer, A. H. Proppe, A. Seifitokaldani, J. Choi, J. Kim, S.-W. Baek, M. Liu, B. Sun, M. Biondi, B. Scheffel, G. Walters, D.-H. Nam, J. W. Jo, O. Ouellette, O. Voznyy, S. Hoogland, S. O. Kelley, Y. S. Jung and E. H. Sargent, Nat. Commun., 2020, 11, 103 CrossRef CAS PubMed.
- R. A. Marcus, J. Chem. Phys., 1956, 24, 966 CrossRef CAS.
- K. E. Knowles, M. D. Peterson, M. R. McPhail and E. A. Weiss, J. Phys. Chem. C, 2013, 117, 10229 CrossRef CAS.
- C. Yun, J. Lee, J. Lee, B. Lüssem, F. Ventsch, K. Leo and M. C. Gather, Appl. Phys. Lett., 2012, 101, 243303 CrossRef.
- B. Minaev, G. Baryshnikov and H. Agren, Phys. Chem. Chem. Phys., 2014, 16, 1719 RSC.
- Y. Huang, D. L. Elder, A. L. Kwiram, S. A. Jenekhe, A. K. Y. Jen, L. R. Dalton and C. K. Luscombe, Adv. Mater., 2021, 33, 1904239 CrossRef CAS.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868 CrossRef CAS PubMed.
- Y. Huang, T. A. Cohen, P. J. W. Sommerville and C. K. Luscombe, J. Mater. Chem. C, 2021, 9, 7274 RSC.
- Y. Hassan, T. Janes, R. D. Pensack, S. Rafiq, P. M. Brodersen, M. A. Winnik, D. Song and G. D. Scholes, Chem. Mater., 2016, 28, 4953 CrossRef CAS.
- K. Tarafder, Y. Surendranath, J. H. Olshansky, A. P. Alivisatos and L.-W. Wang, J. Am. Chem. Soc., 2014, 136, 5121 CrossRef CAS.
- J. H. Olshansky, A. D. Balan, T. X. Ding, X. Fu, Y. V. Lee and A. P. Alivisatos, ACS Nano, 2017, 11, 8346 CrossRef CAS PubMed.
- J. H. Olshansky, T. X. Ding, Y. V. Lee, S. R. Leone and A. P. Alivisatos, J. Am. Chem. Soc., 2015, 137, 15567 CrossRef CAS PubMed.
- J. Huang, K. L. Mulfort, P. Du and L. X. Chen, J. Am. Chem. Soc., 2012, 134, 16472 CrossRef CAS.
- A. J. Morris-Cohen, M. T. Frederick, L. C. Cass and E. A. Weiss, J. Am. Chem. Soc., 2011, 133, 10146 CrossRef CAS.
- A. J. Harvie, C. T. Smith, R. Ahumada-Lazo, L. J. C. Jeuken, M. Califano, R. S. Bon, S. J. O. Hardman, D. J. Binks and K. Critchley, J. Phys. Chem. C, 2018, 122, 10173 CrossRef CAS.
- H. Zhao, C. Sun, H. Yin, Y. Li, J. Gao, Y. Shi and M. Sun, Sci. Rep., 2019, 9, 7756 CrossRef PubMed.
- D. A. Henckel, M. J. Enright, N. Panahpour Eslami, D. M. Kroupa, D. R. Gamelin and B. M. Cossairt, Nano Lett., 2020, 20, 2620 CrossRef CAS PubMed.
- E. P. A. M. Bakkers, A. W. Marsman, L. W. Jenneskens and D. Vanmaekelbergh, Angew. Chem., Int. Ed., 2000, 39, 2297 CrossRef CAS.
- R. S. Dibbell and D. F. Watson, J. Phys. Chem. C, 2009, 113, 3139 CrossRef CAS.
- A. J. Morris-Cohen, M. D. Peterson, M. T. Frederick, J. M. Kamm and E. A. Weiss, J. Phys. Chem. Lett., 2012, 3, 2840 CrossRef CAS.
- J. J. Buckley, E. Couderc, M. J. Greaney, J. Munteanu, C. T. Riche, S. E. Bradforth and R. L. Brutchey, ACS Nano, 2014, 8, 2512 CrossRef CAS PubMed.
- L. Stryer and R. P. Haugland, Proc. Natl. Acad. Sci. U. S. A., 1967, 58, 719 CrossRef CAS.
- B. Schuler, Chem. Phys. Chem., 2005, 6, 1206 CrossRef CAS.
- T. Förster, Ann. Phys., 1948, 437, 55 CrossRef.
- I. L. Medintz and H. Mattoussi, Phys. Chem. Chem. Phys., 2009, 11, 17 RSC.
- C. Mongin, S. Garakyaraghi, N. Razgoniaeva, M. Zamkov and F. N. Castellano, Science, 2016, 351, 369 CrossRef CAS.
- Z. Huang and M. L. Tang, J. Am. Chem. Soc., 2017, 139, 9412 CrossRef CAS.
- Z.-W. Xi, L. Yang, D.-Y. Wang, C.-W. Feng, Y. Qin, Y.-M. Shen, C. Pu and X. Peng, J. Org. Chem., 2021, 86, 2474 CrossRef CAS.
- M. J. Enright, K. Gilbert-Bass, H. Sarsito and B. M. Cossairt, Chem. Mater., 2019, 31, 2677 CrossRef CAS.
- J. A. Caputo, L. C. Frenette, N. Zhao, K. L. Sowers, T. D. Krauss and D. J. Weix, J. Am. Chem. Soc., 2017, 139, 4250 CrossRef CAS.
- M. Zhang, Z. Li, X. Xin, J. Zhang, Y. Feng and H. Lv, ACS Catal., 2020, 10, 14793 CrossRef CAS.
- Y. Sahin, A. T. Sika-Nartey, K. E. Ercan, Y. Kocak, S. Senol, E. Ozensoy and Y. E. Türkmen, ACS Appl. Mater. Interfaces, 2021, 13, 5099 CrossRef CAS PubMed.
- X. Zhu, Y. Lin, Y. Sun, M. C. Beard and Y. Yan, J. Am. Chem. Soc., 2019, 141, 733 CrossRef CAS PubMed.
- H. Xu, S. Ouyang, L. Liu, P. Reunchan, N. Umezawa and J. Ye, J. Mater. Chem. A, 2014, 2, 12642 RSC.
- C. B. Ong, L. Y. Ng and A. W. Mohammad, Renewable Sustainable Energy Rev., 2018, 81, 536 CrossRef CAS.
- Y. Huang, Z. Liu, G. Gao, Q. Xiao, W. Martens, A. Du, S. Sarina, C. Guo and H. Zhu, Catal. Sci. Technol., 2018, 8, 726 RSC.
- U. Aslam, V. G. Rao, S. Chavez and S. Linic, Nat. Catal., 2018, 1, 656 CrossRef.
- J. Bang, S. Das, E.-J. Yu, K. Kim, H. Lim, S. Kim and J. W. Hong, Nano Lett., 2020, 20, 6263 CrossRef CAS.
- A. Meng, B. Zhu, B. Zhong, L. Zhang and B. Cheng, Appl. Surf. Sci., 2017, 422, 518 CrossRef CAS.
- Y. Yuan, H. Zhu, K. Hills-Kimball, T. Cai, W. Shi, Z. Wei, H. Yang, Y. Candler, P. Wang, J. He and O. Chen, Angew. Chem., Int. Ed., 2020, 59, 22563 CrossRef CAS PubMed.
- Z. Zhang, K. Edme, S. Lian and E. A. Weiss, J. Am. Chem. Soc., 2017, 139, 4246 CrossRef CAS PubMed.
- X. Wu, S. Xie, C. Liu, C. Zhou, J. Lin, J. Kang, Q. Zhang, Z. Wang and Y. Wang, ACS Catal., 2019, 9, 8443 CrossRef CAS.
- M. B. Wilker, J. K. Utterback, S. Greene, K. A. Brown, D. W. Mulder, P. W. King and G. Dukovic, J. Phys. Chem. C, 2018, 122, 741 CrossRef CAS.
- S. Yu, X.-B. Fan, X. Wang, J. Li, Q. Zhang, A. Xia, S. Wei, L.-Z. Wu, Y. Zhou and G. R. Patzke, Nat. Commun., 2018, 9, 4009 CrossRef PubMed.
- J. G. Smith and P. K. Jain, J. Am. Chem. Soc., 2016, 138, 6765 CrossRef CAS PubMed.
- E. H. Edwards, A. A. Fertig, K. P. McClelland, M. T. Meidenbauer, S. Chakraborty, T. D. Krauss, K. L. Bren and E. M. Matson, Chem. Commun., 2020, 56, 8762 RSC.
- M. J. Berr, P. Wagner, S. Fischbach, A. Vaneski, J. Schneider, A. S. Susha, A. L. Rogach, F. Jäckel and J. Feldmann, Appl. Phys. Lett., 2012, 100, 223903 CrossRef.
- R. Abargues, J. Navarro, P. J. Rodríguez-Cantó, A. Maulu, J. F. Sánchez-Royo and J. P. Martínez-Pastor, Nanoscale, 2019, 11, 1978 RSC.
- X.-B. Li, B. Liu, M. Wen, Y.-J. Gao, H.-L. Wu, M.-Y. Huang, Z.-J. Li, B. Chen, C.-H. Tung and L.-Z. Wu, Adv. Sci., 2016, 3, 1500282 CrossRef.
- H. Lu, X. Zhu, C. Miller, J. San Martin, X. Chen, E. M. Miller, Y. Yan and M. C. Beard, J. Chem. Phys., 2019, 151, 204305 CrossRef PubMed.
- C. M. Wolff, P. D. Frischmann, M. Schulze, B. J. Bohn, R. Wein, P. Livadas, M. T. Carlson, F. Jäckel, J. Feldmann, F. Würthner and J. K. Stolarczyk, Nat. Energy, 2018, 3, 862 CrossRef CAS.
- Z. Chen, Y. Hu, J. Wang, Q. Shen, Y. Zhang, C. Ding, Y. Bai, G. Jiang, Z. Li and N. Gaponik, Chem. Mater., 2020, 32, 1517 CrossRef CAS.
- L. C. M. Souza, T. A. Santos, C. R. A. Do Prado, B. A. V. Lima, R. S. Corrêa, A. A. Batista, L. Otubo, J. Ellena, L. T. Ueno, L. R. Dinelli and A. L. Bogado, RSC Adv., 2016, 6, 53130 RSC.
- Y. Jiang, C. Wang, C. R. Rogers, M. S. Kodaimati and E. A. Weiss, Nat. Chem., 2019, 11, 1034 CrossRef CAS.
- W. Li, J. R. Lee and F. Jäckel, ACS Appl. Mater. Interfaces, 2016, 8, 29434 CrossRef CAS.
- K. P. Acharya, R. S. Khnayzer, T. O’Connor, G. Diederich, M. Kirsanova, A. Klinkova, D. Roth, E. Kinder, M. Imboden and M. Zamkov, Nano Lett., 2011, 11, 2919 CrossRef CAS PubMed.
- M. Melchionna and P. Fornasiero, ACS Catal., 2020, 10, 5493 CrossRef.
- P. Christopher, S. Jin, K. Sivula and P. V. Kamat, ACS Energy Lett., 2021, 6, 707 CrossRef CAS.
- V. Kuznetsova, Y. Gromova, M. Martinez-Carmona, F. Purcell-Milton, E. Ushakova, S. Cherevkov, V. Maslov and Y. K. Gun’ko, Nanophotonics, 2021, 10, 797 CrossRef CAS.
- A. Ben Moshe, D. Szwarcman and G. Markovich, ACS Nano, 2011, 5, 9034 CrossRef CAS PubMed.
- U. Tohgha, K. K. Deol, A. G. Porter, S. G. Bartko, J. K. Choi, B. M. Leonard, K. Varga, J. Kubelka, G. Muller and M. Balaz, ACS Nano, 2013, 7, 11094 CrossRef CAS.
- J. K. Choi, B. E. Haynie, U. Tohgha, L. Pap, K. W. Elliott, B. M. Leonard, S. V. Dzyuba, K. Varga, J. Kubelka and M. Balaz, ACS Nano, 2016, 10, 3809 CrossRef CAS.
- N. Suzuki, Y. Wang, P. Elvati, Z.-B. Qu, K. Kim, S. Jiang, E. Baumeister, J. Lee, B. Yeom, J. H. Bahng, J. Lee, A. Violi and N. A. Kotov, ACS Nano, 2016, 10, 1744 CrossRef CAS.
- T. He, J. Li, X. Li, C. Ren, Y. Luo, F. Zhao, R. Chen, X. Lin and J. Zhang, Appl. Phys. Lett., 2017, 111, 151102 CrossRef.
- C. Chen, L. Gao, W. Gao, C. Ge, X. Du, Z. Li, Y. Yang, G. Niu and J. Tang, Nat. Commun., 2019, 10, 1927 CrossRef.
- A. Ben-Moshe, A. Teitelboim, D. Oron and G. Markovich, Nano Lett., 2016, 16, 7467 CrossRef CAS.
- H. Zhang, H. He, X. Jiang, Z. Xia and W. Wei, ACS Appl. Mater. Interfaces, 2018, 10, 30680 CrossRef CAS.
- W. Ma, L. Xu, L. Wang, C. Xu and H. Kuang, Adv. Funct. Mater., 2019, 29, 1805512 CrossRef.
- C. Hao, R. Gao, Y. Li, L. Xu, M. Sun, C. Xu and H. Kuang, Angew. Chem., Int. Ed., 2019, 58, 7371 CrossRef CAS PubMed.
- V. V. Belykh, D. R. Yakovlev, M. M. Glazov, P. S. Grigoryev, M. Hussain, J. Rautert, D. N. Dirin, M. V. Kovalenko and M. Bayer, Nat. Commun., 2019, 10, 1 CrossRef CAS.
- K. Trujillo-Hernández, G. Rodríguez-López, A. Espinosa-Roa, J. González-Roque, A. P. Gómora-Figueroa, W. Zhang, P. S. Halasyamani, V. Jancik, M. Gembicky, G. Pirruccio and D. Solis-Ibarra, J. Mater. Chem. C, 2020, 8, 9602 RSC.
- Y.-H. Kim, Y. Zhai, H. Lu, X. Pan, C. Xiao, E. A. Gaulding, S. P. Harvey, J. J. Berry, Z. V. Vardeny, J. M. Luther and M. C. Beard, Science, 2021, 371, 1129 CrossRef CAS PubMed.
- K. E. Sapsford, T. Pons, I. L. Medintz, S. Higashiya, F. M. Brunel, P. E. Dawson and H. Mattoussi, J. Phys. Chem. C, 2007, 111, 11528 CrossRef CAS.
- J. Wang, P. Jiang, Z. Han, L. Qiu, C. Wang, B. Zheng and J. Xia, Langmuir, 2012, 28, 7962 CrossRef CAS PubMed.
- Y. Cui, S. N. Kim, S. E. Jones, L. L. Wissler, R. R. Naik and M. C. McAlpine, Nano Lett., 2010, 10, 4559 CrossRef CAS.
- D. S. Seferos, D. A. Giljohann, H. D. Hill, A. E. Prigodich and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 15477 CrossRef CAS.
- A. Foubert, N. V. Beloglazova, A. Rajkovic, B. Sas, A. Madder, I. Y. Goryacheva and S. De Saeger, TRAC, 2016, 83, 31 CAS.
- A. Gole and C. J. Murphy, Langmuir, 2008, 24, 266 CrossRef CAS PubMed.
- R. Bilan, F. Fleury, I. Nabiev and A. Sukhanova, Bioconjugate Chem., 2015, 26, 609 CrossRef CAS.
- K. Kang, C. Kan, A. Yeung and D. Liu, Mater. Sci. Eng., C, 2006, 26, 664 CrossRef CAS.
- N. V. Beloglazova, A. Foubert, A. Gordienko, M. D. Tessier, T. Aubert, E. Drijvers, I. Goryacheva, Z. Hens and S. De Saeger, Talanta, 2016, 160, 66 CrossRef CAS.
- O. Klykov and M. G. Weller, Anal. Methods, 2015, 7, 6443 RSC.
- N. Abello, H. A. M. Kerstjens, D. S. Postma and R. Bischoff, J. Proteome Res., 2007, 6, 4770 CrossRef CAS.
- D. R. Dempsey, H. Jiang, J. H. Kalin, Z. Chen and P. A. Cole, J. Am. Chem. Soc., 2018, 140, 9374 CrossRef CAS PubMed.
- P. Cuatrecasas and I. Parikh, Biochemistry, 1972, 11, 2291 CrossRef CAS.
- A. J. Lomant and G. Fairbanks, J. Mol. Biol., 1976, 104, 243 CrossRef CAS.
- D. Kahne and W. C. Still, J. Am. Chem. Soc., 1988, 110, 7529 CrossRef CAS.
- O. Koniev and A. Wagner, Chem. Soc. Rev., 2015, 44, 5495 RSC.
- P. Ochtrop and C. P. R. Hackenberger, Curr. Opin. Chem. Biol., 2020, 58, 28 CrossRef CAS.
- N. Venkatesan and B. H. Kim, Chem. Rev., 2006, 106, 3712 CrossRef CAS PubMed.
- M. Amblard, J.-A. Fehrentz, J. Martinez and G. Subra, Mol. Biotechnol., 2006, 33, 239 CrossRef CAS.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895 RSC.
- T. Machida and N. Winssinger, ChemBioChem, 2016, 17, 811 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef.
- B. Cao, Y. Zheng, T. Xi, C. Zhang, W. Song, K. Burugapalli, H. Yang and Y. Ma, Biomed. Microdevices, 2012, 14, 709 CrossRef CAS.
- D. C. Kennedy, C. S. McKay, M. C. B. Legault, D. C. Danielson, J. A. Blake, A. F. Pegoraro, A. Stolow, Z. Mester and J. P. Pezacki, J. Am. Chem. Soc., 2011, 133, 17993 CrossRef CAS PubMed.
- V. Bevilacqua, M. King, M. Chaumontet, M. Nothisen, S. Gabillet, D. Buisson, C. Puente, A. Wagner and F. Taran, Angew. Chem., Int. Ed., 2014, 53, 5872 CrossRef CAS PubMed.
- J. Dommerholt, F. P. J. T. Rutjes and F. L. van Delft, Top. Curr. Chem., 2016, 374, 16 CrossRef PubMed.
- B. D. Fairbanks, E. A. Sims, K. S. Anseth and C. N. Bowman, Macromolecules, 2010, 43, 4113 CrossRef CAS.
- B. L. Oliveira, Z. Guo, O. Boutureira, A. Guerreiro, G. Jiménez-Osés and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2016, 55, 14683 CrossRef CAS PubMed.
- I. Nikić, T. Plass, O. Schraidt, J. Szymański, J. A. G. Briggs, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2014, 53, 2245 CrossRef.
- Z. Liu, F. Gan, N. Dong, B. Zhang, J. Wang and Y. Chen, J. Mater. Chem. C, 2019, 7, 10789 RSC.
- D. O. Li, M. S. Gilliam, X. S. Chu, A. Yousaf, Y. Guo, A. A. Green and Q. H. Wang, Mol. Syst. Des. Eng., 2019, 4, 962 RSC.
- I. Murphy, P. Rice, M. Monahan, L. Zasada, E. Miller, S. Raugei and B. Cossairt, 2021 DOI:10.33774/chemrxiv-2021-3p4pp.
- N. H. Mohd, N. F. H. Ismail, J. I. Zahari, W. F. B. Wan Fathilah, H. Kargarzadeh, S. Ramli, I. Ahmad, M. A. Yarmo and R. Othaman, J. Nanomater., 2016, 2016, 4804271 CrossRef.
- H. Yu, Y. Lu, Z. Feng, Y. Wu, Z. Liu, P. Xia, J. Qian, Y. Chen, L. Liu, K. Cao, S. Chen and W. Huang, Nanoscale, 2019, 11, 9103 RSC.
- X. Lu, Z. Zhang, Q. Xia, M. Hou, C. Yan, Z. Chen, Y. Xu and R. Liu, Mater. Sci. Eng., C, 2018, 82, 190 CrossRef CAS.
- S.-H. Yang, Appl. Phys. Lett., 2020, 116, 120502 CrossRef CAS.
- R. Naaman, Y. Paltiel and D. H. Waldeck, Nat. Rev. Chem., 2019, 3, 250 CrossRef CAS.
Footnote |
| † Current address: Pi-Conjugated Polymers Unit, Okinawa Institute of Science & Technology Graduate University, Onna-son, Okinawa, Japan. |
| This journal is © The Royal Society of Chemistry 2022 |