DOI:
10.1039/D1MH01119G
(Communication)
Mater. Horiz., 2022,
9, 444-451
Enantiopure 2-(2-ethylhexyl)dinaphtho[2,3-b:2′,3′-f]thieno[3,2-b]thiophenes: synthesis, single-crystal structure and a surprising lack of influence of stereoisomerism on thin-film structure and electronic properties†
Received
15th July 2021
, Accepted 3rd November 2021
First published on 3rd November 2021
Abstract
Starting from a chiral resolution of 2-ethylhexanoic acid followed by conversions of functional groups without interfering with the enantiopurity, we have successfully introduced an enantiopure 2-ethylhexyl group on to dinaphtho[2,3-b:2′,3′-f]thieno[3,2-b]thiophene (DNTT) via a Negishi-coupling reaction to synthesize 2-(R)-(2-ethylhexyl)- and 2-(S)-(2-ethylhexyl)-DNTT (R- and S-EH-DNTT, respectively). Then, the crystallinities, thin-film structures, and the organic field-effect transistors (OFETs) based on R-, S- and racemic EH-DNTT (rac-EH-DNTT) were studied to elucidate the effect of stereoisomerism in the 2-ethylhexyl group. The crystal structures of the R- and S-EH-DNTTs are classified as herringbone packing and contain two crystallographically independent molecules connected by edge-to-face CH–π intermolecular interactions, and the molecules’ directly opposite directions avoid steric repulsion between the 2-ethylhexyl groups. Thin films of the EH-DNTTs fabricated using both the spin-coating and vacuum-deposition methods were revealed to have similar but slightly different packing structures to that in the single crystal. Intriguingly, the packing structures in the thin-film state depend on the deposition method, and not on the stereoisomers of EH-DNTT. Consistent with the packing structures in the thin-film state, the performance of OFETs based on the thin films of the R-, S- and rac-EH-DNTTs were affected by the deposition method, and not by the stereoisomerism. This means that the stereoisomerism in the alkyl side chain has a marginal effect on the packing structure and electronic properties in the thin-film state. This is endorsed by the theoretical calculations using the functional-group symmetry-adapted perturbation theory (F-SAPT), which indicated that the intermolecular interactions between the DNTT cores are dominant in the total intermolecular interaction energies, and implies that the crystallization process in the thin-film deposition could be governed by intermolecular interactions between the DNTT cores. We conclude that in 2-ethylhexyl-substituted organic semiconductors with a large and highly aggregative π-conjugated core, like EH-DNTT, the enantiopurity in the 2-ethylhexyl group does not significantly affect the thin-film structure and thus the performance of thin-film OFETs.
New concepts
The 2-ethylhexyl group is the most frequently employed branched alkyl group in organic semiconductors based on both polymers and small molecules, in order to enhance the solubility of given semiconducting materials. Despite its usefulness, the 2-ethylhexyl group contains a stereo center that induces stereoisomerism, making the characterization and structure–property relationship of the resulting organic semiconductors ambiguous. In this work, a reliable and scalable method for the preparation of enantiopure (R)- and (S)-2-ethylhexyl reagents including 2-ethylhexyl zincate that can be employed in Negishi-coupling reactions was reported. Furthermore, with this method, the synthesis and characterization of enantiopure (R)- and (S)-2-(2-ethylhexyl)dinaphtho[2,3-b:2',3'-f]thieno[3,2-b]thiophene (R- and S-EH-DNTT) were demonstrated, which enabled us to elucidate the relationship between the stereoisomerism and the solid-state structure of enantiopure and racemic EH-DNTTs.
|
Introduction
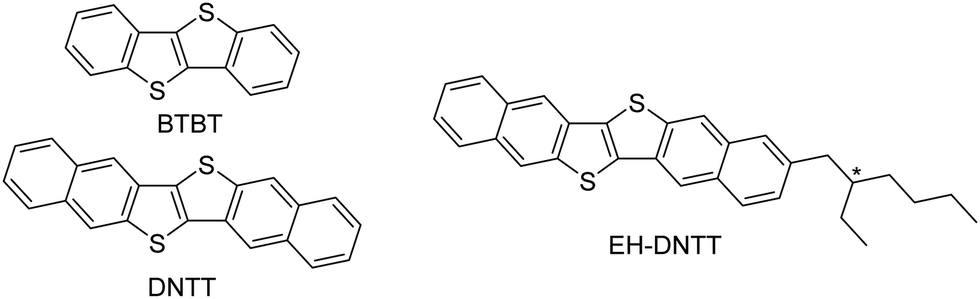
The last three decades have witnessed great improvements in the performance of organic field-effect transistors (OFETs). This is mainly due to optimization of fabrication processes as well as the development of novel, superior active semiconducting materials.1–3 For small-molecule-based organic semiconductors, acenes and thienoacenes have been the workhorse compounds that have shown the record-high field-effect mobilities and have pushed OFETs forwards to practical applications.4,5 Dinaphtho[2,3-b:2′,3′-f]thieno[3,2-b]thiophene (DNTT, Fig. 1) consisting of six fused-aromatic rings in a linear manner has been one of the representative thienoacene-based organic semiconductors6 that affords high-performance, stable OFETs.7 DNTT has contributed to the development of flexible state-of-the-art devices, such as electronic watermarks,8 and those for medical applications9,10 by being integrated into transistor arrays on flexible substrates. The DNTT-based transistors in these devices are fabricated using the vacuum-deposition process. Although vacuum deposition is a reliable method for fabricating devices with high reproducibility, the solution processes are key methods that characterize organic electronics for future technologies. For this reason, soluble organic semiconductors play a pivotal role as the active material in solution-processed OFETs. In this regard, [1]benzothieno[3,2-b][1]benzothiophene (BTBT, Fig. 1) consisting of four fused-aromatic rings has been an ideal platform for developing solution-processable, high-performance organic semiconductors, where the introduction of linear alkyl groups is quite effective in increasing the solubility while retaining high mobility.11–14 In contrast, for the solubilization of DNTT, a higher homologue of BTBT, it turned out that the introduction of linear alkyl groups is not that effective,15 and that thin films of the linear-alkyl-substituted DNTTs were only deposited from their solution in hot high-boiling point solvents such as chlorobenzene.16,17 For solubilizing the π-extended and rigid semiconducting cores, the introduction of branched alkyl groups, such as 2-ethylhexyl groups, has been often examined. In fact, a series of branched-alkyl-substituted DNTTs have been synthesized, and their solubilities were reported to be sufficient for fabricating their OFETs using solution processes such as spin-coating (Fig. 1).18
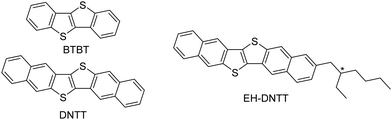 |
| | Fig. 1 Molecular structures of BTBT and DNTT derivatives. | |
Through experimental works on branched-alkyl-substituted DNTT derivatives, we have recognized their potential as solution-processable organic semiconductors, which can be summarized as follows: although the dialkylated derivatives only afford poor OFETs exhibiting mobilities of up to 1.1 × 10−2 cm2 V−1 s−1, devices with mono-alkylated DNTTs as the active layer demonstrated comparable transistor performances (μ ∼2.6 cm2 V−1 s−1) to those of vacuum-deposited DNTT-based OFETs.18,19 Such high mobilities can be rationalized by the formation of crystalline thin films on the substrates using both the solution- and vacuum-deposition methods, even with bulky branched alkyl groups. In particular, the herringbone packing structures in the thin-film state, which are similar to those of other DNTT derivatives, were suggested using thin-film X-ray diffraction (XRD) measurements.18
Although the herringbone structure proposed by previous studies qualitatively explains the comparable OFET performance with those of other DNTT derivatives, the remaining questions on the packing structures of the mono branched-alkylated DNTTs are: (i) their exact packing structures were not elucidated, which means that the actual orientation of the branched alkyl groups to be adapted in the herringbone structure is not clear. In practice, it was difficult to grow single crystals of the racemic mono-alkylated DNTTs suitable for single-crystal X-ray analysis. The racemate is an enantiomeric mixture, which might affect the crystallinity, and we thus suspected that enantiopure mono-alkylated DNTTs may have better crystallinity; (ii) the purity of the active semiconducting material is key in the performance of OFETs. In this sense, the active layer of the racemate always consists of a mixture of enantiomers. We thus wondered how the stereoisomerism affects the molecular ordering in the solid-state and then the performance of OFETs.
In relation to this, there have been several organic semiconductors with enantiopure 2-ethylhexyl groups reported, and the authors discussed how the stereoisomerism of the 2-ethylhexyl groups can influence the solid-state structure, chiroptical properties, and the performance of their electronic devices such as field-effect transistors and solar cells.20–24 These works have encouraged us to synthesize enantiopure 2-(R)-(2-ethylhexyl)- and 2-(S)-(2-ethylhexyl)-dinaphtho[2,3-b:2′,3′-f]thieno[3,2-b]thiophenes (R- and S-EH-DNTT, respectively) and to investigate the effect of stereoisomerism on the molecular properties and molecular ordering in the solid state. In this work, we report the synthesis and characterization of enantiopure R- and S-EH-DNTT, the crystal structures of R- and S-EH-DNTT, and the thin-film structure and transport properties of thin-film OFETs based on R-, S- and racemic EH-DNTT (rac-EH-DNTT).
Results
Synthesis of enantiopure EH-DNTTs
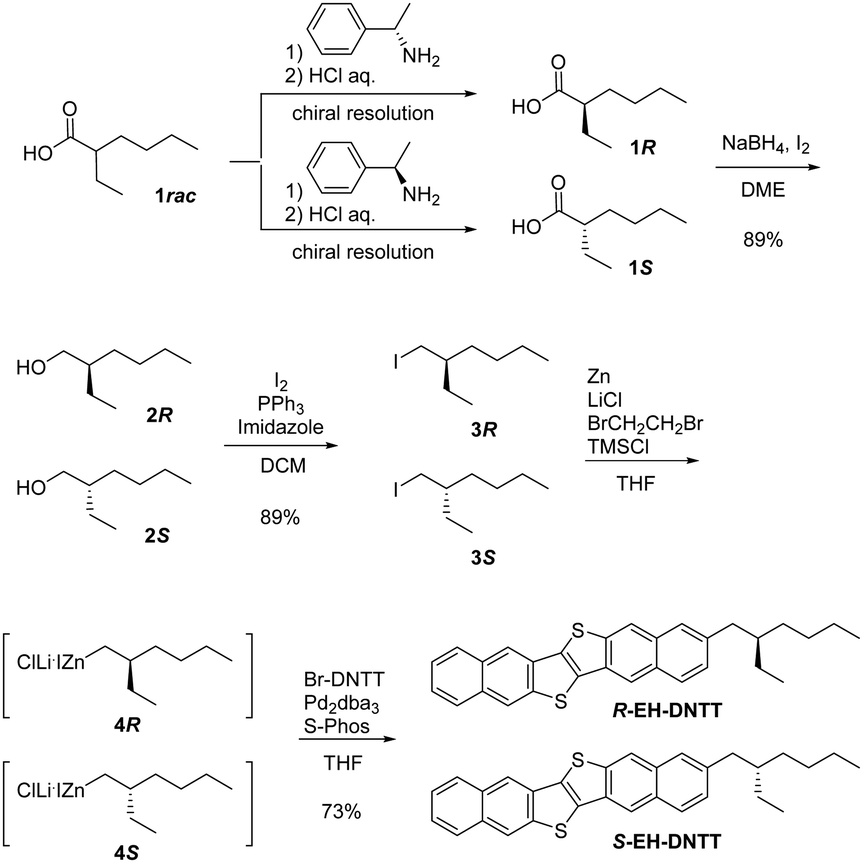
The original synthesis of rac-EH-DNTT from 6-(2-ethylhexyl)-2-methoxynaphthalene was based on the consecutive thiophene-annulation approach25 or the non-selective unsymmetrical coupling approach26 to selectively or unselectively construct the unsymmetrical DNTT derivative, respectively. In theory, these approaches can be applicable to the synthesis of enantiopure EH-DNTTs, but the poor total yield of the target compound makes them unpreferable. Thus, we envisaged that the one-step functionalization of 2-bromodinaphtho[2,3-b:2′,3′-f]thieno[3,2-b]thinenothiophene (Br-DNTT) via the transition metal-catalyzed cross-coupling reaction27 is more suitable for the synthesis of enantiopure EH-DNTTs. Even with this approach, however, a good amount of the enantiopure 2-ethylhexyl precursor is necessary for the preparation of the 2-ethylhexyl organometallic reagents, which is generally carried out on a relatively large scale in practice. We thus pursued an efficient method for the preparation of enantiopure 2-ethylhexyl compounds. In fact, several different ways to obtain enantiopure 2-ethylhexyl compounds can be found in the literature; e.g., enantioselective synthesis,21,28 baker's yeast mediated synthesis,29 and lipase-catalyzed resolution.30 Among these, we chose the chiral resolution of the commercially available racemic 2-ethylhexanoic acid via the formation of diastereomeric salts with (R)- and (S)-1-phenylethylamine,31 since the method seems to be amenable to the large-scale preparation of both (R)- and (S)-2-ethylhexanoic acid.
Following the reported procedure,31 we carried out the chiral resolution of the commercially available racemic 2-ethylhexanoic acid by forming the diastereomeric salt with (R)-1-phenylethylamine. However, the enantiopurity of the acid after the liberation was not as high (ca. 75% ee) as that reported in the literature (91% ee).31 Thus we tried to optimize the chiral resolution by altering the temperature, time, and the number of resolution operations. As a result, a reproducible procedure for the preparation of (S)- (>98% ee, 1S) and (R)-2-ethylhexanoic acid (>97% ee, 1R) was established (Fig. S1, see the ESI†). The enantiopure acids were then reduced to the corresponding alcohols (89% yield, 2R/S),32 which were further converted to the iodides (89% yield, 3R/S) according to the reported procedure.33 Then the iodides were derived into the corresponding organozinc species (4R/S) by the direct insertion of metallic zinc mediated by lithium chloride,34 which were coupled with Br-DNTT to give enantiopure EH-DNTTs (Scheme 1). The enantiopurity of EH-DNTTs could not be directly determined, but the enantiopurity at the acid stage was maintained until the iodide stage (Fig. S2, ESI†). We also confirmed that Negishi-coupling reactions of the enantiopure 2-ethylhexyl zincate did not affect the enantiopurity of the product (Fig. S3, see the ESI†). Furthermore, the absolute structures of R- and S-EH-DNTTs were confirmed using single-crystal X-ray analysis (vide infra).
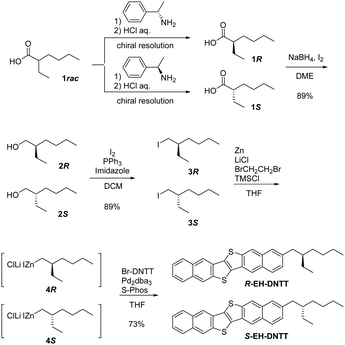 |
| | Scheme 1 Synthesis of enantiopure R- and S-EH-DNTT. | |
Crystal structures of R- and S-EH-DNTT
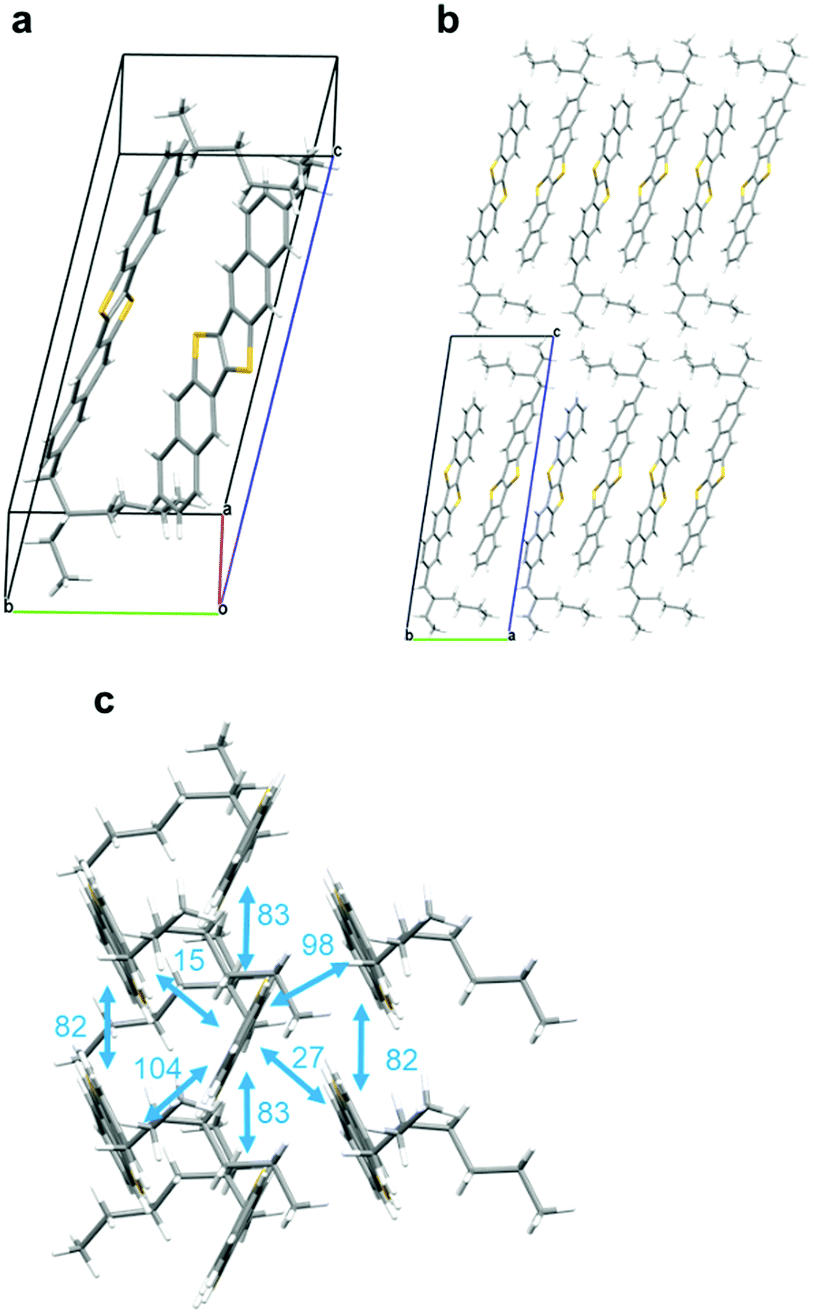
As mentioned above, we could not grow single crystals of rac-EH-DNTT by any means, e.g., recrystallization or sublimation methods. In contrast, the single crystals suitable for the single-crystal X-ray analysis were readily obtained from the enantiopure EH-DNTTs by slow vapor-diffusion of methanol into a toluene solution (Fig. S4–S6, ESI†). Fig. 2 shows a representative crystal structure of S-EH-DNTT, the space group of which is non-centrosymmetric P1. The structure contains two crystallographically independent molecules (Fig. 2a), and both molecules are determined to be the (S)-isomer. Note that the R-factor and Flack parameter were reasonably refined, indicating that the absolute structure of the sample can be determined as the (S)-isomer (see the ESI†). The two crystallographically independent molecules inside the unit cell interact through edge-to-face CH–π contact, and the 2-ethylhexyl group in each molecule is oriented in the opposite direction to avoid causing steric bulk (Fig. 2a). The molecular long-axes are almost along the crystallographic c-axis, and thus the crystallographic ab plane corresponds to the layered structure of the DNTT cores, which results in the lamella structure along the crystallographic c-axis (Fig. 2b). The packing motif in the DNTT layer is classified as herringbone packing and is similar to those of the parent and other DNTT derivatives (Fig. 2c). In the herringbone packing structure, the molecules stack along the a-axis direction, and all these molecules are related by the translation operation, which means that the 2-ethylhexyl groups orient in the same direction. On the other hand, the neighboring molecular stacks consist of the crystallographically different molecules as mentioned, and thus the 2-ethylhexyl moieties alternately exist in the crystallographic b-axis direction. This packing structure can rationally minimize the steric bulk originating in the 2-ethylhexyl groups.
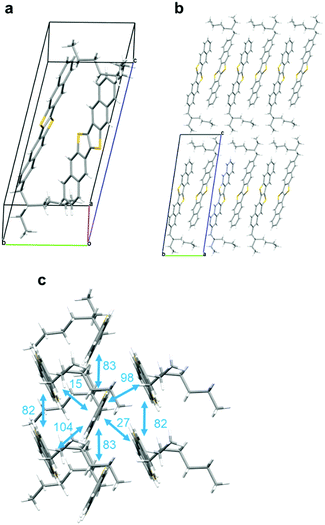 |
| | Fig. 2 Crystal structure of S-EH-DNTT. (a) Molecular structures of two crystallographically-independent molecules, (b) lamella structure, and (c) the herringbone packing structure observed in the lamella layers and the calculated intermolecular electronic coupling (transfer integrals, meV, in cyan) of the HOMO. | |
The intermolecular orbital overlaps of the HOMO (transfer integral: t, Fig. 2c) are calculated with the Amsterdam Density Functional (ADF) program.35 The distribution of ts in the conducting DNTT-layer is similar to that of the parent DNTT (Fig. S7, ESI†),6,36 the features of which can be summarized in the two-dimensional (2D) electronic structure and the large ts. These features rationalize the comparable field-effect mobility reported for the EH-DNTT-based OFETs with those of the parent DNTT-based ones well (vide infra).
Thin-film deposition of enantiopure and racemic EH-DNTTs
To characterize the molecular properties of the enantiopure and racemic EH-DNTTs, the racemate in this study was prepared by mixing equimolar amounts of enantiopure R- and S-EH-DNTTs. First, the solubility of both the enantiopure and racemic forms were determined by obtaining the 1H NMR spectra of their saturated solutions in CDCl3 at 20 °C. The solubility of rac-EH-DNTT was 0.86 g L−1, which qualitatively agrees with the reported value in chloroform (1.0 g L−1) determined by the slow addition of the solvent into an exactly weighed sample.18 On the other hand, the solubility of the enantiopure R- and S-EH-DNTTs in CDCl3 were determined to be 0.52 and 0.53 g L−1, respectively (Fig. S8, ESI†); the solubility of the racemate is higher than that of the enantiomers by about 1.5 times, which implies that the crystallinity is affected by the enantiopurity. Although the solubilities are different, the thin films of the racemate and enantiomers were fabricated by spin-coating of the chloroform solution, and the resulting thin films have similar surface textures (Fig. S9 and S10, ESI†). Similarly, the vacuum deposition of the racemate and enantiomers afforded uniform thin films on the substrate, and no clear difference between the racemate and enantiomers was observed in the optical microscopy and atomic force microscopy (AFM) images (Fig. S9 and S11, ESI†), or in the UV-vis spectra (Fig. S12, ESI†), which implies that the thin films were virtually identical independent of the enantiopurity (vide infra).
X-ray diffraction patterns of the thin films
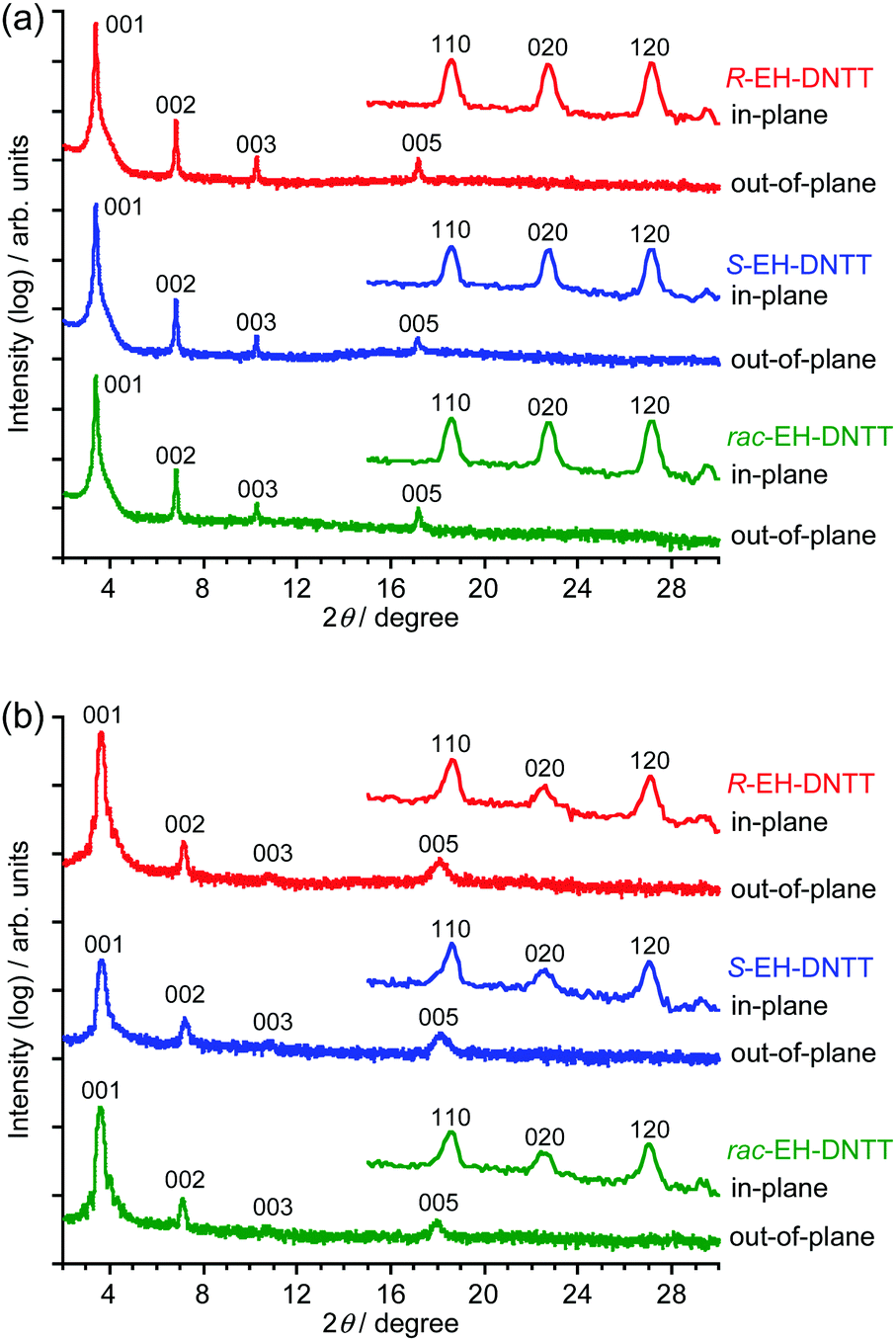
Fig. 3 shows the XRD patterns of the spin-coated and vacuum-deposited thin films on the octyltrichlorosilane (OTS)-treated SiO2/Si substrate. The out-of-plane XRD patterns of both thin films show a series of 00l diffractions assignable to the lamella structure along the normal to the substrate. Interestingly, the 2θ values of the out-of-plane peaks depend on the deposition method, but not on the compounds; the extracted d-spacings are about 25.9 Å for the spin-coated thin films and about 24.2–24.5 Å for the vacuum-deposited thin films. It is noticeable that the d-spacings in the thin-film state are different from the one expected from the single-crystal X-ray analysis of S-EH-DNTT (d-spacing is about 24.0 Å, Fig. S13 (ESI†), see also Table 1). In particular, the difference in the d-spacing is pronounced for the spin-coated thin films (25.9 Å), which implies that the conformation of the 2-ethylhexyl group could be different in the spin-coated and the vacuum-deposited thin films. In the single crystal of S-EH-DNTT, the branch part, i.e., the ethyl part, of the 2-ethylhexyl group orients in the molecular long axis direction (Fig. 2a). It is thus expected that the trunk part (i.e., the hexyl part) of the alkyl group could orient along the molecular long axis in the spin-coated thin films. This consideration can be endorsed by the theoretically optimized molecular structures; the molecular length of EH-DNTT with the trunk-stretched 2-ethylhexyl group is about 23.0 Å, whereas the one with the branch-stretched 2-ethylhexyl group is about 20.5 Å, which indicates that the conformation of the 2-ethylhexyl group can largely affect the d-spacing of the thin film (Fig. S14, ESI†). The d-spacings of the vacuum-deposited thin films are between these two, which implies that the conformation of the 2-ethylhexyl groups is sensitive to the fabrication methods (vide supra).
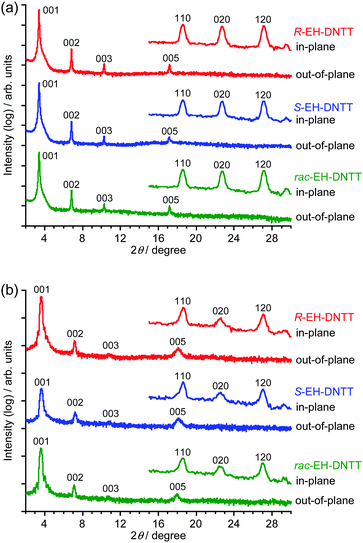 |
| | Fig. 3 Out-of-plane and in-plane XRD patterns of spin-coated (a) and vacuum-deposited (b) thin films on OTS-treated substrates. | |
Table 1 Summary of thin-film XRD measurements
| Deposition method |
Isomer |
Out-of-plane/° |
In-plane/° |
| 001 (d-spacing/Å) |
110 |
020 |
120 |
| Vacuum deposition |
R
|
3.64 (24.3) |
18.6 |
22.6 |
27.1 |
|
S
|
3.65 (24.2) |
18.6 |
22.6 |
27.0 |
|
rac
|
3.61 (24.5) |
18.6 |
22.6 |
27.0 |
| Spin-coating |
R
|
3.41 (25.9) |
18.6 |
22.7 |
27.1 |
|
S
|
3.41 (25.9) |
18.6 |
22.8 |
27.1 |
|
rac
|
3.42 (25.9) |
18.6 |
22.8 |
27.1 |
| Single crystal |
S
|
3.68 (24.0) |
19.1 |
22.2 |
27.5 |
In all the in-plane XRDs (Fig. 3), three peaks assignable to the 110, 020, and 120 diffractions, which are characteristic of herringbone packing, are observed. Independent of the deposition method and the stereoisomers, each of the three peaks appear at almost the same position, which indicates that the in-plane structures in the thin-film state that correspond to the herringbone ab-cells are very close to one another. On the other hand, the in-plane structures of the thin films can be different from that in the single crystal, as the three peaks estimated from the single-crystal structural analysis appear at different 2θ values (Table 1 and Fig. S13, ESI†). Comparison of the 2θ values between the thin-film state and the single-crystal state implies that the crystallographic a-axis in the thin-film state tends to be longer, whereas the b-axis tends to be shorter than that in the structure of the single crystal, provided that the crystallographic angles are the same. Qualitatively speaking, this corresponds to the fact that the dihedral angle between the DNTT cores in the herringbone dimer, often called the “herringbone angle” (Fig. 2a), is smaller in the thin-film state than that in the single crystal, which makes the molecules in the stacking direction (i.e., the crystallographic a-axis direction) far apart, whereas the molecules in the “herringbone” direction (i.e., the crystallographic b-axis direction) are close. Such structural changes can be understood by considering the less ordered structure of the 2-ethylhexyl group in the thin-film state; the disorder of the 2-ethylhexyl group can induce larger steric hindrance in the crystallographic a-axis direction, as the molecules in this direction are in the relationship of translation operation, and thus the 2-ethylhexyl groups are close to one another. On the other hand, as the 2-ethylhexyl groups exist alternately in the crystallographic b-axis direction, the steric bulk cannot be significantly affected by the disorder. As a result, the crystallographic ab cell of the thin-film state tends to resemble that of the parent DNTT (a = 6.187 Å, b = 7.662 Å, S-EH-DNTT: a = 6.002 Å, b = 8.108 Å).
Characteristics of thin-film transistors with enantiopure and racemic EH-DNTTs
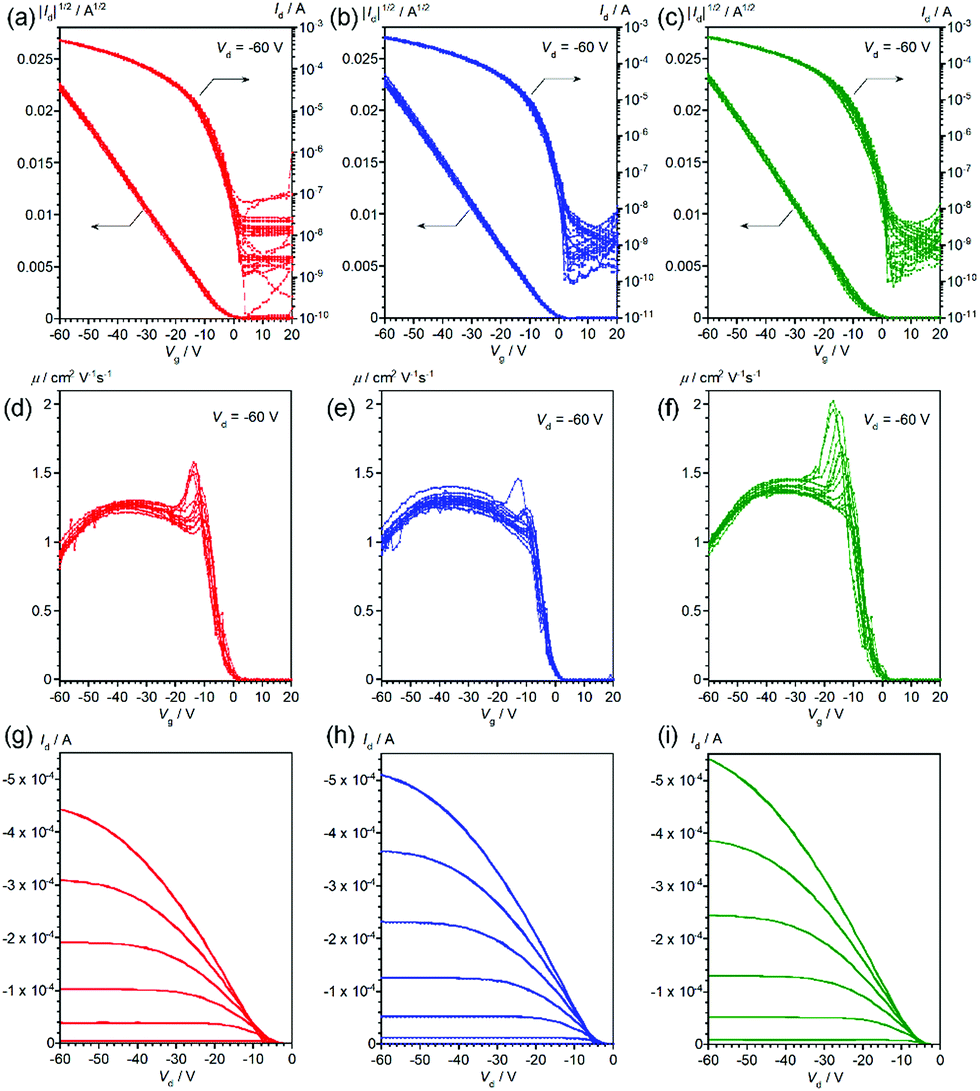
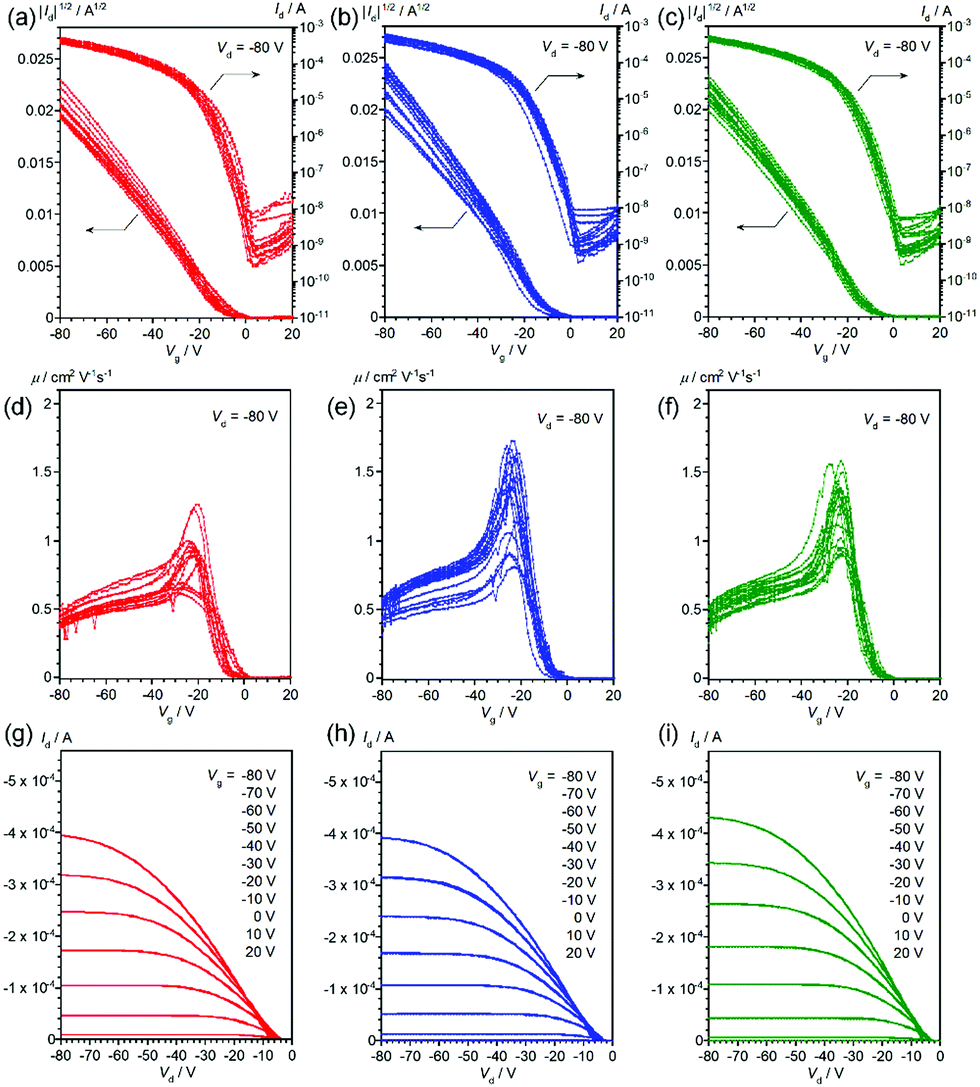
Thin-film OFETs with a bottom-gate-top-contact configuration were fabricated using vapor-deposition of the source and drain electrodes on top of the thin films on the SiO2/Si substrates. All the devices showed typical transistor behaviors (Fig. 4 and 5), and the mobilities extracted from the saturation regime are about 1.2–1.3 cm2 V−1 s−1 for the devices with vapor-deposited thin films and 0.57–0.68 cm2 V−1 s−1 for the ones with spin-coated thin films (Table 2). As can be clearly seen from Fig. 4, 5 and Table 2, the mobility does depend on the deposition method, but not on the stereoisomers in the 2-ethylhexyl moiety.22 This is consistent with the thin-film XRDs, where no clear differences between the stereoisomers are observed. Improved device performance for vapor-deposited thin films has been already reported in previous work with racemic EH-DNTT,18 and it can be thus concluded that the method for thin-film deposition rather than the stereoisomers is critical for device performance.
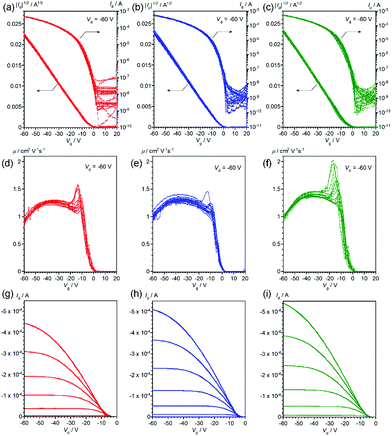 |
| | Fig. 4 Transfer curves (a–c), gate-voltage-dependence of mobility (d–f), and output curves (g–i) of OFET devices based on vacuum-deposited R- (red), S- (blue), and rac- (green) EH-DNTT thin films, respectively. Transfer and gate-voltage-dependent mobility curves are overlaid for more than twelve devices. | |
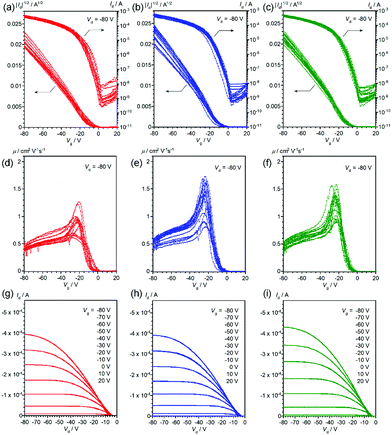 |
| | Fig. 5 Transfer curves (a–c), gate-voltage-dependence of mobility (d–f), and output curves (g–i) of OFET devices based on spin-coated R- (red), S- (blue), and rac- (green) EH-DNTT thin films, respectively. Transfer and gate-voltage-dependent mobility curves are overlaid for more than twelve devices. | |
Table 2 Summary of the FET characteristics of EH-DNTT-based thin-film transistors
| Deposition method |
Isomer |
μ/cm2 V−1 s−1 |
V
th/V |
I
on/Ioff |
|
Mobilities were extracted from the Vg range of −30 to −60 V, and the values from more than twelve devices are averaged.
Mobilities were extracted from the Vg range of −50 to −80 V, and the values from more than sixteen devices are averaged.
|
| Vacuum deposition |
R
|
1.17 ± 0.02a |
−2.8 ± 0.6 |
105 |
|
S
|
1.23 ± 0.04a |
−2.5 ± 0.6 |
105 |
|
rac
|
1.28 ± 0.02a |
−3.1 ± 0.7 |
105 |
| Spin-coating |
R
|
0.51 ± 0.05b |
−0.4 ± 1.7 |
105 |
|
S
|
0.64 ± 0.08b |
−0.2 ± 2.0 |
105 |
|
rac
|
0.58 ± 0.06b |
−0.5 ± 2.1 |
105 |
Discussion
Formation of a single crystal and thin films of EH-DNTTs
The fact that the performance of the thin-film transistors was not susceptible to the stereoisomerism of the 2-ethylhexyl groups is consistent with the thin-film structure elucidated using the in-plane XRDs, which are mostly the same as one another.22 Interestingly, and as discussed above, the packing structures in the thin-film state are at least different from that in the single crystal of the R- and S-EH-DNTTs, which means that polymorphs exist in the packing structures of the R- and S-EH-DNTTs in the solid state.
The packing structure of organic molecules is in general affected by several factors in the crystallization process, which is often discussed in terms of thermodynamic or kinetic control. The formation of crystals via slow recrystallization from solution is said to be a thermodynamically controlled process, where equilibrium between the nucleation and crystal growth in the supersaturation state is the key.37 With this consideration, the crystal structures of the R- and S-EH-DNTTs elucidated using X-ray single crystal structure analysis of the crystals grown via slow recrystallization can be regarded as the thermodynamically stable phase. In contrast, the deposition of thin films on the substrate, regardless of spin-coating or the vacuum deposition, is in general regarded as a kinetically controlled process, where rapid nucleation followed by crystallization takes place to form crystallites. We thus believe that the difference in the crystallization mechanism is the primary reason for the two polymorphs and depends on the method used for the crystallization.
Intermolecular interaction energy in the S-EH-DNTT single crystal
The initial stage of nucleation in the crystallization process can be largely affected by intermolecular interaction, and thus it could be informative to estimate the intermolecular interaction energy (Eint), which can be calculated using the symmetry-adapted perturbation theory (SAPT) method.38 Among the various SAPT methods, the functional-group SAPT (F-SAPT) that allows for an effective two-body partition of the various SAPT terms to localized chemical functional groups was employed in the present analyses,39 in order to understand how the “functional groups” in EH-DNTT, i.e., the DNTT core and the 2-ethylhexyl group, contribute to the intermolecular interaction in the solid state. We thus picked up six molecular pairs in the ab-plane in the crystal structure of S-EH-DNTT to calculate the Eint values between the DNTT cores, the 2-ethylhexyl groups, and the DNTT core and the 2-ethylhexyl groups (Table 3 and Tables S1–S6, ESI†). As can be clearly seen in Table 3, the Eint values between the DNTT cores are always dominant in all the molecular pairs, which indicates that the intermolecular interactions involving the 2-ethylhexyl group play a marginal role. It is thus rationally considered that in the thin-film deposition of EH-DNTT using spin-coating and vacuum deposition, the intermolecular interactions between the DNTT cores are dominant in contributing to the nucleation: in other words, the stereoisomerism and the conformation of the 2-ethylhexyl groups cannot be dominant factors in the thin-film deposition. These theoretical studies explain the experimental results well, that is, there are no observable differences between the thin films of the different EH-DNTT samples. Therefore, with these experimental as well as theoretical studies on the enantiopure and racemic EH-DNTTs, we can conclude that the enantiopurity of EH-DNTT is not critical for the performance of the thin-film OFETs.
Table 3 Partitioned intermolecular interaction energies (Eints/kcal mol−1) of various molecular pairs in the single crystal of S-EH-DNTTa
|
|
Molecular pairs |
| 1 |
2 |
3 |
4 |
5 |
6 |
|
Calculated with the F-SAPT method using the Psi4 program with the jun-cc-pVDZ basis set.
Sum of Eint values calculated for two possible combinations.
Sum of all the partitioned Eint values, which are consistent with the Eint values calculated using SAPT0.
|
| DNTT-DNTT |
−10.77 |
−16.07 |
−15.15 |
−15.27 |
−14.92 |
−10.98 |
| EH-EH |
−1.75 |
−2.08 |
−1.61 |
−2.25 |
−2.60 |
−1.26 |
| DNTT-EHb |
−1.28 |
0.01 |
0.02 |
0.02 |
0.01 |
−1.29 |
| Totalc |
−13.79 |
−18.14 |
−16.73 |
−17.50 |
−17.50 |
−13.53 |
To further confirm this consideration experimentally, we synthesized racemic and enantiopure 2-(2-ethylhexyl)[1]benzothieno[3,2-b][1]benzothiophene (EH-BTBT), a lower homologue of EH-DNTT, and checked its packing structure in the thin-film state. Fig. S15 (ESI†) shows the out-of-plane and in-plane XRD patterns of the spin-coated thin-films of rac-EH-BTBT and S-EH-BTBT. As can be clearly observed, both the out-of-plane and in-plane XRDs are identical for rac-EH-BTBT and S-EH-BTBT, which indicates that no noticeable structural differences exist in the thin-film state. This result is consistent with the EH-DNTT case. Although the poor crystallinity of S-EH-BTBT did not allow us to elucidate the exact crystal structure, the result on the EH-BTBT thin-films supports that the intermolecular interaction between the largely π-extended cores, such as BTBT and DNTT, dictates the packing structure in the thin-film state.
Conclusions
We have successfully established a method for introducing an enantiopure 2-ethylhexyl group on to the DNTT core, which consists of the initial chiral resolution of 2-ethylhexanoic acid followed by a series of chemical conversions without interfering with the enantiopurity of 2-ethylhexyl zincate utilized in the Negishi-coupling reaction. This synthetic protocol represents a versatile way to introduce both enantiopure R- and S-2-ethylhexyl group(s) directly on to the sp2 carbon atom in π-conjugated systems, which can be a complementary method for synthesizing organic semiconductors with enantiopure 2-ethylhexyl group(s) to the previously reported ones that consist of nucleophilic substitution on 2-ethylhexyl-bromide or -p-toluenesulfonate. The enantiopure and racemic EH-DNTTs were characterized in terms of their crystallinities, thin-film structures, and OFET properties to elucidate the effect of stereoisomerism in the 2-ethylhexyl group. As expected, the enantiopure R- and S-EH-DNTTs enabled us to elucidate the exact packing structures of a mono-functionalized DNTT derivative, for the first time. The packing structures can be classified into a herringbone packing, which is similar to those of other DNTT derivatives, and the feature of the structure is an alternating molecular-orientation in the edge-to-face direction (crystallographic b-axis direction), so as to avoid steric repulsion between the 2-ethylhexyl groups. On the other hand, the packing structures in the thin-film state were similar to, but different from the ones in the single crystals. Interestingly, the stereoisomerism in the 2-ethylhexyl groups did not affect the packing structures in the thin-film state; the thin films of the R-, S- and rac-EH-DNTTs were identical and accordingly the performances of their OFETs were well within experimental error. The insensitivity of the stereoisomerism to the packing structures in the thin-film state could be explained by the relative strength of the intermolecular interactions caused by the DNTT core and the 2-ethylhexyl groups. The partitioned intermolecular interactions were calculated for the crystal structure of S-EH-DNTT using F-SAPT, which confirmed that the intermolecular interactions between the DNTT cores are much larger than the ones involving the 2-ethylhexyl groups, and strongly implies that the intermolecular interactions between the DNTT cores predominantly contributes to the crystallization process during thin-film deposition, which can be regarded as a kinetically controlled process. We thus conclude that in 2-ethylhexyl-substituted organic semiconductors with a large and highly aggregative π-conjugated core, like EH-DNTT, the stereoisomerism in the 2-ethylhexyl group does not significantly affect the thin-film structure and thus the performances of the OFETs. This knowledge of enantiopure EH-DNTT in comparison with its racemic counterpart gives us inspiration to design 2-ethylhexyl-substituted organic semiconductor systems.
Author contributions
K. Sumitomo carried out most parts of the experiments including the synthesis and device studies. Y. Sudo and K. Kawabata analysed and established the enantiopurity of the materials. K. Kanazawa carried out the theoretical calculations. K. Kawabata and K. Takimiya supervised the project. All authors contributed to the discussion of the results and the writing of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge the Supercomputer System in the Advanced Centre for Computing and Communication (ACCC) of RIKEN for support in the theoretical calculations. We also thank the Centre for Computational Materials Science, Institute for Materials Research, Tohoku University for the use of MASAMUNE-IMR (MAterials science Supercomputing system for Advanced MUlti-scale simulations towards NExt-generation-Institute for Materials Research). This work was financially supported by JSPS KAKENHI Grant Numbers JP19H00906 (Grant-in-Aid for Scientific Research (A)) and JP20H05865 (Grant-in-Aid for Transformative Research Areas (A) “Condensed Conjugation”), and the Mitsubishi Foundation.
Notes and references
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724–6746 CrossRef CAS PubMed.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS.
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS.
- J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452–483 CrossRef CAS.
- K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347–4370 CrossRef CAS PubMed.
- T. Yamamoto and K. Takimiya, J. Am. Chem. Soc., 2007, 129, 2224–2225 CrossRef CAS.
- U. Zschieschang, T. Yamamoto, K. Takimiya, H. Kuwabara, M. Ikeda, T. Sekitani, T. Someya and H. Klauk, Adv. Mater., 2011, 23, 654–658 CrossRef CAS.
- K. Kuribara, H. Wang, N. Uchiyama, K. Fukuda, T. Yokota, U. Zschieschang, C. Jaye, D. Fischer, H. Klauk, T. Yamamoto, K. Takimiya, M. Ikeda, H. Kuwabara, T. Sekitani, Y.-L. Loo and T. Someya, Nat. Commun., 2012, 3, 723 CrossRef.
- M. Kaltenbrunner, T. Sekitani, J. Reeder, T. Yokota, K. Kuribara, T. Tokuhara, M. Drack, R. Schwödiauer, I. Graz, S. Bauer-Gogonea, S. Bauer and T. Someya, Nature, 2013, 499, 458–463 CrossRef CAS PubMed.
- H. Ebata, T. Izawa, E. Miyazaki, K. Takimiya, M. Ikeda, H. Kuwabara and T. Yui, J. Am. Chem. Soc., 2007, 129, 15732–15733 CrossRef CAS PubMed.
- H. Minemawari, T. Yamada, H. Matsui, J. Y. Tsutsumi, S. Haas, R. Chiba, R. Kumai and T. Hasegawa, Nature, 2011, 475, 364–367 CrossRef CAS.
- H. Iino, T. Usui and J.-I. Hanna, Nat. Commun., 2015, 6, 6828 CrossRef CAS.
- S. Inoue, H. Minemawari, J. Y. Tsutsumi, M. Chikamatsu, T. Yamada, S. Horiuchi, M. Tanaka, R. Kumai, M. Yoneya and T. Hasegawa, Chem. Mater., 2015, 27, 3809–3812 CrossRef CAS.
- M. J. Kang, I. Doi, H. Mori, E. Miyazaki, K. Takimiya, M. Ikeda and H. Kuwabara, Adv. Mater., 2011, 23, 1222–1225 CrossRef CAS PubMed.
- K. Nakayama, Y. Hirose, J. Soeda, M. Yoshizumi, T. Uemura, M. Uno, W. Li, M. J. Kang, M. Yamagishi, Y. Okada, E. Miyazaki, Y. Nakazawa, A. Nakao, K. Takimiya and J. Takeya, Adv. Mater., 2011, 23, 1626–1629 CrossRef CAS.
- R. Hofmockel, U. Zschieschang, U. Kraft, R. Rödel, N. H. Hansen, M. Stolte, F. Würthner, K. Takimiya, K. Kern, J. Pflaum and H. Klauk, Org. Electron., 2013, 14, 3213–3221 CrossRef CAS.
- M. Sawamoto, M. J. Kang, E. Miyazaki, H. Sugino, I. Osaka and K. Takimiya, ACS Appl. Mater. Interfaces, 2016, 8, 3810–3824 CrossRef CAS PubMed.
- M. Sawamoto, H. Sugino, M. Nakano and K. Takimiya, Org. Electron., 2017, 46, 68–76 CrossRef CAS.
- J. Liu, Y. Zhang, H. Phan, A. Sharenko, P. Moonsin, B. Walker, V. Promarak and T.-Q. Nguyen, Adv. Mater., 2013, 25, 3645–3650 CrossRef CAS.
- R. B. Zerdan, N. T. Shewmon, Y. Zhu, J. P. Mudrick, K. J. Chesney, J. Xue and R. K. Castellano, Adv. Funct. Mater., 2014, 24, 5993–6004 CrossRef CAS.
- T. He, P. Leowanawat, C. Burschka, V. Stepanenko, M. Stolte and F. Würthner, Adv. Mater., 2018, 30, 1804032 CrossRef.
- T. Ikai, R. Kojima, S. Katori, T. Yamamoto, T. Kuwabara, K. Maeda, K. Takahashi and S. Kanoh, Polymer, 2015, 56, 171–177 CrossRef CAS.
- S. L. Fronk, Y. Shi, M. Siefrid, C.-K. Mai, C. McDowell and G. C. Bazan, Macromolecules, 2016, 49, 9301–9308 CrossRef CAS.
- T. Mori, T. Nishimura, T. Yamamoto, I. Doi, E. Miyazaki, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2013, 135, 13900–13913 CrossRef CAS PubMed.
- K. Niimi, M. J. Kang, E. Miyazaki, I. Osaka and K. Takimiya, Org. Lett., 2011, 13, 3430–3433 CrossRef CAS PubMed.
- K. Kawabata, S. Usui and K. Takimiya, J. Org. Chem., 2020, 85, 195–206 CrossRef CAS PubMed.
- C. R. G. Grenier, S. J. George, T. J. Joncheray, E. W. Meijer and J. R. Reynolds, J. Am. Chem. Soc., 2007, 129, 10694–10699 CrossRef CAS PubMed.
- Y. Huang, F. Zhang and Y. Gong, Tetrahedron Lett., 2005, 46, 7217–7219 CrossRef CAS.
- K. Baczko and C. Larpent, J. Chem. Soc., Perkin Trans. 2, 2000, 521–526, 10.1039/A907987D.
- Z. Xu, Y. Zhang, H. Fu, H. Zhong, K. Hong and W. Zhu, Bioorg. Med. Chem. Lett., 2011, 21, 4005–4007 CrossRef CAS.
- J. V. B. Kanth and M. Periasamy, J. Org. Chem., 1991, 56, 5964–5965 CrossRef CAS.
- L. Jones, J. S. Schumm and J. M. Tour, J. Org. Chem., 1997, 62, 1388–1410 CrossRef CAS.
- A. Krasovskiy, V. Malakhov, A. Gavryushin and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 6040–6044 CrossRef CAS.
- ADF: powerful DFT code for modeling molecules; Scien-tific Computing and Modeling: Amsterdam; (http://www.scm.com/ADF/).
- K. Takimiya, I. Osaka, T. Mori and M. Nakano, Acc. Chem. Res., 2014, 47, 1493–1502 CrossRef CAS PubMed.
- N. Uyeda, J. Jpn. Soc. Colour Mater., 1970, 43, 612–621 CrossRef CAS.
- R. M. Parrish, L. A. Burns, D. G. A. Smith, A. C. Simmonett, A. E. DePrince, E. G. Hohenstein, U. Bozkaya, A. Y. Sokolov, R. Di Remigio, R. M. Richard, J. F. Gonthier, A. M. James, H. R. McAlexander, A. Kumar, M. Saitow, X. Wang, B. P. Pritchard, P. Verma, H. F. Schaefer, K. Patkowski, R. A. King, E. F. Valeev, F. A. Evangelista, J. M. Turney, T. D. Crawford and C. D. Sherrill, J. Chem. Theory Comput., 2017, 13, 3185–3197 CrossRef CAS.
- R. M. Parrish, T. M. Parker and C. D. Sherrill, J. Chem. Theory Comput., 2014, 10, 4417–4431 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2096392 and 2092243. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1mh01119g |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Kohsuke
Kawabata
ab,
Kohsuke
Kawabata