
Molecular characterization of COVID-19 therapeutics: luteolin as an allosteric modulator of the spike protein of SARS-CoV-2
Walter
Alvarado†
 a,
Gustavo R.
Perez-Lemus†
b,
Cintia A.
Menéndez†
b,
Fabian
Byléhn†
b and
Juan J.
de Pablo
*b
a,
Gustavo R.
Perez-Lemus†
b,
Cintia A.
Menéndez†
b,
Fabian
Byléhn†
b and
Juan J.
de Pablo
*b
aBiophysical Sciences, University of Chicago, 929 E. 57th Street, Chicago, IL 60637, USA. E-mail: walt@uchicago.edu
bPritzker School of Molecular Engineering, University of Chicago, 5640 S. Ellis Avenue, Chicago, IL 60637, USA
First published on 15th September 2021
Abstract
The interactions between the receptor binding domain (RBD) of SARS-CoV-2 and the angiotensin-converting enzyme 2 (ACE2) are crucial for viral entry and subsequent replication. Given the large and featureless contact surfaces between both proteins, finding a suitable small-molecule drug that can bind and disrupt regulatory pathways has remained a challenge. A promising therapeutic alternative has been the use of small compounds that bind at the protein–protein interface or at distal “hotspots” and induce conformational changes that inhibit or stabilize protein–protein interactions (PPIs). In this work, we conduct large-scale all-atom explicit solvent simulations of the top scoring compounds from a recent large-scale high-throughput docking screening to investigate their interaction with the RBD domain of the spike (S) protein in complex with ACE2. We identify several promising candidates that exhibit a large negative free energy of binding to the RBD/ACE2 complex based on ab initio thermodynamic integration calculations. A systematic structural analysis of two glycosylation profiles of the RBD/ACE2 complex reveals the important role glycans play in modulating the binding of small-molecules. Cross correlation, fluctuation and strain analysis identify several of these compounds as effective PPI modulators that inhibit or stabilize the protein–protein interactions of RBD/ACE2 based on the glycosylation profile. Among them, luteolin, a drug currently approved for asthma, exhibits an intense allosteric effect when it binds to a previously unidentified distal binding site of the RBD domain far from the RBD and ACE2 interface which may serve as a potential target for future drug discovery.
Design, System, ApplicationThe design of small-molecules that target the spike protein of SARS-CoV-2 has been the focus of recent studies searching for an effective treatment strategy against COVID-19. In this work, molecular dynamics simulations are used to study how several previously identified therapeutics alter the structure and dynamics of the spike protein. We analyze the strain and molecular stiffness induced by each small molecule upon binding, and identify a previously uncharacterized distal binding site that induces significant allosteric conformational changes to the protein–protein interaction between the subunits of the spike protein and the active site of the angiotensin-converting enzyme 2 (ACE2), a key binding partner required for host cell infectivity. The fundamental understanding obtained in this study may enable the development and engineering of novel small-molecules that disrupt viral entry, and provides an informed perspective on how allosteric modulators serve as a template for the design of suitable therapeutics. |
1 Introduction
As of February 2021, the human coronavirus referred to as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has infected millions around the world, and is responsible for the COVID-19 disease that has so far led to well over 2.61 million deaths worldwide.1 As vaccinations begin to roll out, viable treatments for COVID-19 are still necessary to treat those who may contract the disease. One promising treatment strategy is the identification of previously approved drugs that could be repurposed to act on different stages of SARS-CoV-2 infection and host response.The surface-anchored viral spike (S) glycoprotein mediates coronavirus entry into host cells. The S protein is a type I transmembrane protein comprising two functionally distinct regions, S1 and S2, that mediate receptor binding and membrane fusion, respectively. Similar to SARS-CoV, responsible for the 2003 SARS outbreak in Asia, the S1 subunit of the SARS-CoV-2 engages the same angiotensin-converting enzyme 2 (ACE2) receptor for host cell entry and utilizes the serine protease TMPRSS2 for S protein priming.2 Of the two regions, the S2 subunit is the most conserved among different coronavirus genera and nine residues involved in the interaction between ACE2 and the receptor binding domain (RBD) of the S1 subunit are conserved between SARS-CoV and SARS-CoV-2.3,4
Specific RBD-receptor binding determines host cell infection and has been the target of several drug design efforts. Recent flow cytometry studies have shown that SARS-CoV-2 RBD exhibits a higher binding affinity to human ACE2 (hACE2) and bat ACE2 (bACE2) receptors than SARS-CoV RBD.5 Deletions in the RBD of closely related S protein sequences have also been shown to inhibit binding to hACE2.6 In addition, several studies indicate that the RBD is an attractive antigen for specific antibody detection.7,8 Taken together, these results suggest that SARS-CoV-2 RBD is a promising target for the development of novel vaccines and antiviral drugs.
Recent studies have identified key surface contacts that can be leveraged to disrupt the RBD/ACE2 protein–protein interaction (PPI).9,10 PPIs are physical contacts of high specificity between proteins that play an important role in cellular function.11 PPIs are characterized by their large, flat, and featureless binding interfaces, as in the case of the RBD/ACE2 complex. One major challenge in PPI inhibition is designing small molecules that can compete with the high binding affinity of a natural protein partner. In addition, the large and flat nature of the protein interaction surface often lacks clear binding pockets or grooves that can act as binding sites for smaller ligands. A promising alternative is the identification or engineering of allosteric modulators that stabilize protein–protein interactions, thereby interfering with the downstream pathways they mediate.12 The key strategy in this approach involves indirectly affecting PPI interfaces by targeting distal binding sites that are structurally distinct. Allosteric modulators induce a conformational change that either inhibits or stabilizes association with another protein and have shown promise in the design of suitable therapeutics.13,14
In this work, we focus on a set of small-molecule drugs that can be repurposed for the therapeutic treatment of COVID-19. Specifically, we focus on the top ten scoring compounds proposed in a recent high-throughput supercomputer-based docking screen that was performed in vacuum and relied on minimization of the potential energy of the ACE/RBD complex.15 Here we perform molecular simulations in explicit water and determine the relative and absolute binding free energies of each drug at various binding locations and with different glycosylation profiles. We find the affinity of the ligands to the complex to be different than previously predicted on the basis of energy minimization, with several of the top candidate drugs unbinding from the protein over the course of nanosecond long trajectories. We study the effect glycans have on binding affinity for several ligands by comparing two glycoforms of the RBD/ACE2 complex: a simple and “Abundant” model. Notably, our free energy results show that for an RBD/ACE2 complex glycosylated with small sugar moieties, high-affinity binding ligands stabilize the binding between RBD and ACE2. An RBD/ACE complex with glycans that were determined as the most abundant from a glycoproteomics study conducted by Zhao et al. was analyzed for comparison.16 The presence of these complex-glycans affect the binding affinity of several drug candidates and their mechanism of action. We then analyze the strain and molecular stiffness induced by each small molecule upon binding and identify a “hotspot”, comprising only a few amino acid residues, as a potential target for drug discovery. Surprisingly, binding of luteolin – one of the drugs considered here, to this distal site, induces a profound allosteric conformational change to residues near the RBD/ACE2 binding interface, which disrupts non-native contacts at the protein–protein interface. We also provide additional quantitative insights into the binding mechanism of luteolin to the RBD/ACE2 complex using cross-correlation analysis and principal component analysis (PCA). We conclude with a discussion of our findings and several suggestions for future experimental studies.
2 Methods
2.1 Molecular docking
Autodock 4.2 was used for the molecular docking between target proteins and ligands using the Lamarckian genetic algorithm (LGA), and pseudo-Solis and Wets local search method.17 The initial configuration of receptor-binding domain (RBD) of the spike protein of SARS-CoV-2 bound to the ACE2 receptor was taken from crystal structure (PDB: 6M0J) after an NPT relaxation for 100 ns and averaged for the last 20 ns (see section below for simulation details). The search space was centered around the S-protein and spanning the binding interface of the ACE2 receptor. The initial configurations of ligands were randomized before each docking calculation. A total of 200 docking runs were performed and each run was set to terminate after 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 energy calculations. The best pose of each ligand was selected for further analysis in MD simulations.
000000 energy calculations. The best pose of each ligand was selected for further analysis in MD simulations.
2.2 Molecular dynamics
The recently determined crystal structure of the receptor-binding domain (RBD) of the spike protein of SARS-CoV-2 bound to the ACE2 receptor was used as an initial structure (PDB: 6M0J). Crystal waters were removed and force field parameters for the S-protein/ACE2 complex were determined with the antechamber program using the ff14SB and GLYCAM-06j-1 force fields. An octahedron box with 40000 TIP3P water molecules and 23 Na+ ions were added. Energy minimization included 3000 steps which involved 1500 steepest descent steps constrained to heavy atoms, followed by a second minimization of 30000 steps involving 15000 steepest descent steps. Equilibration was performed through a gradual temperature increase from 0 K to 300 K over 400 ps using Langevin thermostat with a temperature coupling constant of 1.0 ps in a constant volume ensemble. Density equilibration and production runs were conducted using a constant pressure ensemble (NPT). All simulations were performed using periodic boundary conditions and 2 fs time step. Long-range electrostatic interactions were modeled using the particle mesh Ewald method with a non-bonded cut-off of 10 Å and the SHAKE algorithm. The ligands included for MD simulations were described by the general Amber force field (GAFF). Partial charges for all small-molecules were generated using the AM1-BCC charge model. Their initial position was selected from the best score in docking calculations.
2.3 MM/GBSA calculations
The relative binding free energy between ACE2 and RBD domains were calculated using the GBSA method implemented in MMPBSA.py with igb2 = 2 and mbondi2 parameter.18 This relative free energy is defined as: ΔΔGBinding = ΔGLig − ΔGN where the superscript Lig denotes the RBD/ACE2 binding free energy with the presence of the ligand in simulations and N denotes the RBD/ACE2 system with no ligands. Three different replicas were used for every ligand/no ligand system, each one with 5000 snapshots sampled every 20 ps from a previously 300 ns equilibrated system.2.4 Thermodynamic integration calculations
The absolute binding free energy for ligands is defined as: ΔGAbsolute = ΔGL − ΔGRL, where ΔGRL is the free energy change of the ligand annihilation in the RBD/ACE2 complex, and ΔGL is the free energy change of ligand annihilation in water. To calculate these free energy changes, we use thermodynamic integration (TI) implemented in PMEMD for Amber20. A one step annihilation protocol with soft core potentials was implemented. Runs were conducted from a starting equilibrated ligand position extracted via K-means clustering from previous 300 ns MD simulations. In this way, three independent replicas for each ligand were considered, as well as three replicas for solvation in pure water. Twelve windows were selected using Gaussian quadrature with 10 ns of simulation time per window. To keep the ligand from wandering in TI calculations, we used a soft restraint of 10 kcal mol−1 Å−2.2.5 Contact maps and RMSF calculations
Contacts maps by residue and root mean squared fluctuations (RMSF) calculations were generated using the native contacts and RMSF functions in CPPTRAJ.19 The native contacts were defined relative to crystal PDB (6M0J) with a cutoff distance of 7 Å. The maps were averaged over 1000 snapshots taken every 100 ps for each ligand. Root mean squared fluctuations calculations were calculated with respect to a 100 ns averaged structure using the mass-weighted average over the CA, N and C atoms and reported by residue. A total of 5000 snapshots were used taken every 20 ps.2.6 Strain analysis
The effects of functional and non-functional fluctuations after ligand binding were performed through the application of the discrete form of the strain formalism from continuum theory.20,21 In this form, derivatives are replaced by differential operators. The local neighborhood for a given central atom, i, is determined by a radius R that contains n number of atoms j. Distances between atoms i and j are related through the deformation matrix F,| xj − xi = F(x0,j − x0,i) | (1) |
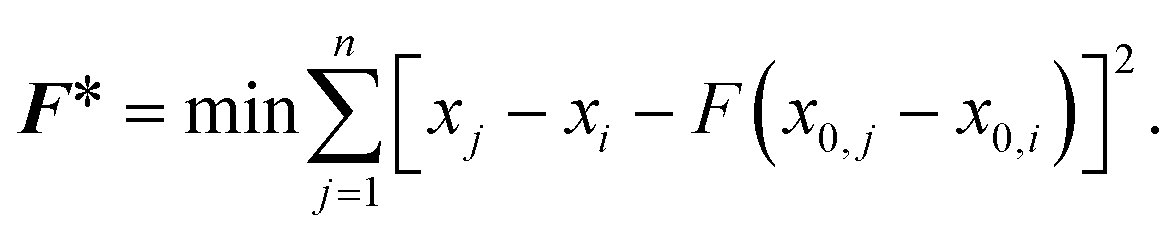 | (2) |
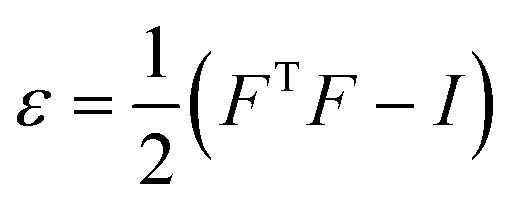 | (3) |
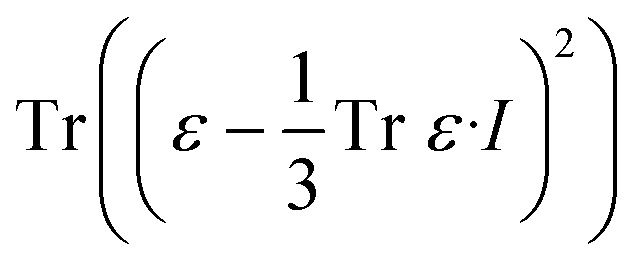 | (4) |
For this study, a 10 Å radius around each Cα atom for our analysis was considered. A simulation of RBD/ACE2 without ligand was used as reference for strain calculations to elucidate the binding of several ligands at different sites.
2.7 Principal component analysis
Principal component analysis (PCA) calculations were performed using the Bio3D package for R.23 PCA is a dimensionality reduction technique that is effective in identifying correlated motions in atomic simulations of proteins from experimental structures or MD trajectories. Essential correlated conformational changes between structures can be represented in this low-dimensional subspace spanned by the first few principal components (PCs).Mathematically, PCs are evaluated by diagonalizing the correlation matrix Cij,
| RTCR = Λ | (5) |
| Cij = 〈(xi − 〈xi〉)(xj − 〈xj〉)〉 | (6) |
3 Results and discussion
3.1 Calculating binding free energies
The configuration of the complex was derived from a recently published crystal structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor (PDB: 6M0J), whose conformation noticeably differed from the homology modeled structure used by Smith et al.15 The glycosylation profile of the RBD/ACE2 protein was based on a simple glycan model and a glycomics-informed glycoproteomics structure (“Abundant” model) generated by Zhao et al.16 which included one glycan group on the RBD domain and six on ACE2. Molecular dynamics simulations were carried out using the Amber20 simulation package.25 To increase sampling, ensure convergence, and extract an equilibrated representative structure, K-means clustering was applied to three independent 100 ns molecular dynamics replicas sans ligand. To identify potential binding sites and validate the results of the previous docking study, candidates were docked using the Autodock4 software.17 Three binding sites were identified for the simple glycan model: the binding interface between the S protein and the ACE2 receptor, a distal RBD region, and a site inside the ACE2 receptor (Fig. 1). For the “Abundant” model, the interface and distal binding sites were identified. The structure with the highest binding affinity, as identified through docking, was used as the starting structure for production runs. Three 100 ns replicas for each ligand in complex with RBD/ACE2 were conducted to determine whether ligands would remain bound. Six ligands remained bound to the simple glycan model, and only three for the “Abundant” form. A representative structure with the ligand bound was extracted and used as a starting structure for three additional MD replicas totaling 300 ns for each ligand bound to the RBD/ACE2 complex and used for analysis.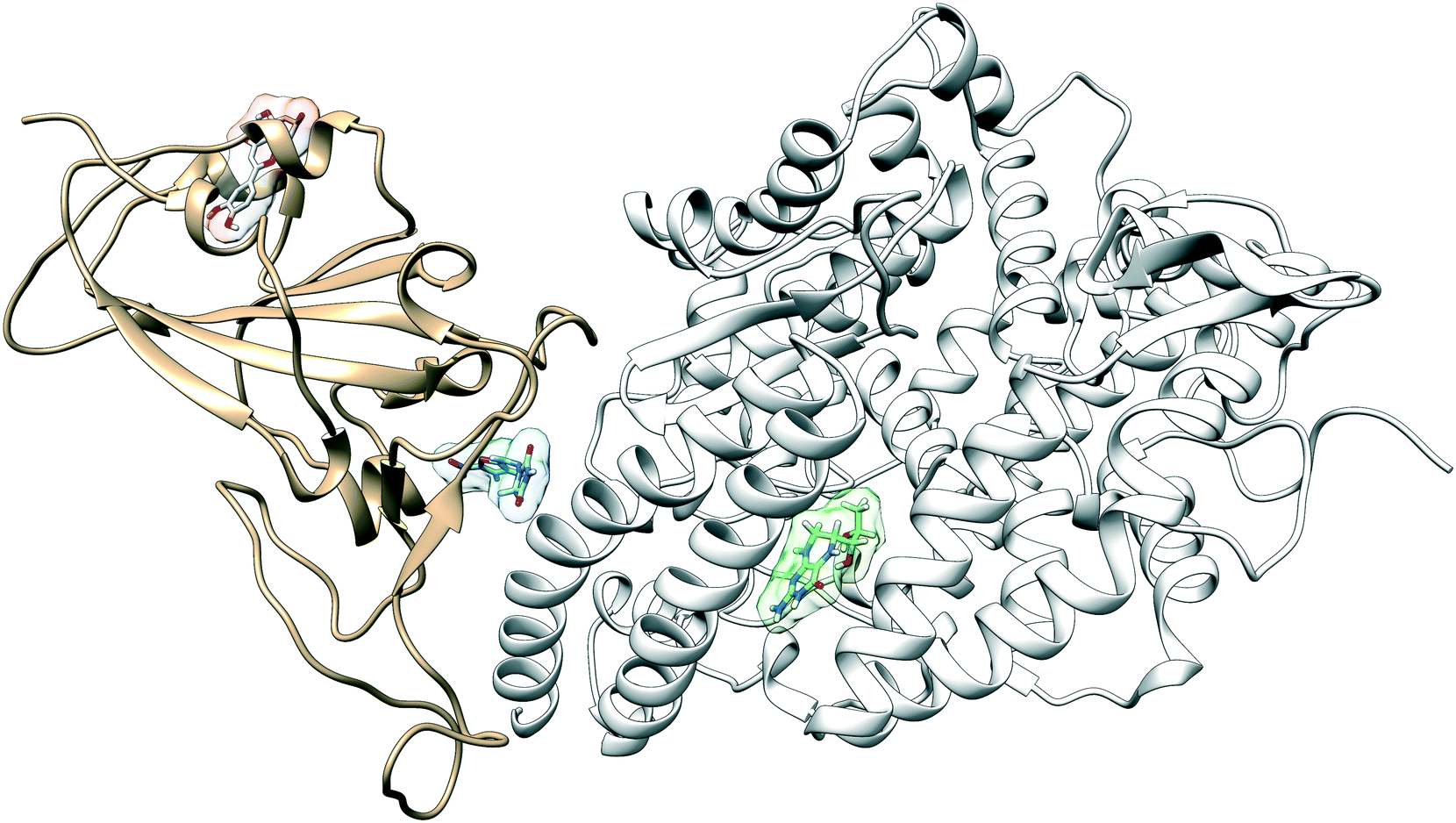 | ||
| Fig. 1 The RBD domain (tan) of SARS-CoV-2 recognizes ACE2 (white) as its receptor. We identify and characterize the interactions at three potential small-drug binding sites located at the binding interface between the RBD and the ACE2 receptor, inside the ACE2 protein, and a new previously unidentified distal site to which the drug luteolin has a high binding affinity (red). Nitrofurantoin is shown in blue and sapropterin is shown in green. | ||
Multiple binding free energy calculations were conducted to determine the binding affinity between the RBD/ACE2 complex and each small ligand (Table 1). The results were compared to those of the high-throughput screening.15 We observed standard deviations ranging from 0.60–0.91 and 0.16–0.53 between our docking binding energies to that of the previous screening study for the simple and “Abundant” models, respectively. This could be due to the differences in the structures used (homology modeled vs. crystal structure) and the fact that important disulfide bonds and glycans groups were accounted for in our structure but were omitted in the Smith et al. analysis.15 The absolute binding free energies between ligand and protein reported here were determined using thermodynamic integration (TI) in the presence of explicit water. In our TI protocol, each ligand atom is treated as a softcore atom and is subsequently “removed” in a one-step alchemical cycle both in solution and in complex. This alchemical method, also referred to as free energy perturbation (FEP), allows for the calculation of absolute binding free energies (ABFE). We observed larger binding energies with standard deviations as high as 10.79 for the simple glycan structure and 5.04 for the “Abundant” glycoform in our free energy calculations than the docking binding energies. Of the three ligands studied, luteolin showed the highest binding affinity for both glycoforms of the RBD/ACE2 complex. Luteolin was also the only ligand to bind at the distal site compared to the other candidates which were found to have a stronger binding affinity for the binding interface of RBD and ACE2 (Fig. 1).
| Molecule | Oak ridge ΔG (kcal mol−1) | Docking ΔG (kcal mol−1) | TI ΔG (kcal mol−1) | RBD/ACE2 ΔΔG (kcal mol−1) |
|---|---|---|---|---|
| Luteolin | −7.4 | −6.19 | −11.31 ± 0.62 | −13.93 ± 1.63 |
| Protirelin | −7.3 | −8.45 | −1.22 ± 5.79 | −11.22 ± 5.10 |
| Nitrofurantoin | −7.2 | −9.22 | −28.98 ± 4.83 | −13.96 ± 2.84 |
| Sapropterin | −7.1 | −5.67 | −6.10 ± 1.21 | −18.89 ± 3.64 |
| Vidarabine | −7.1 | −6.24 | −3.39 ± 1.70 | −17.77 ± 1.66 |
| Eriodictyol | −7.1 | −7.86 | −9.36 ± 2.41 | −15.99 ± 3.53 |
To determine how each compound affects S protein and ACE2 binding, we calculated the free energy of binding between the RBD domain of the S-protein and the ACE2 receptor in the presence of each ligand using the molecular mechanics/generalized Born surface area (MM/GBSA) method. Tables 1 and 2 show the difference in binding free energy with ligand present versus without (ΔΔGBinding) for the simple and “Abundant” glycan models of the complex. In the case of the simple glycan model, our results show that every ligand enhances the binding free energy between the RBD domain and ACE2, acting as a binding stabilizer for the complex. Sapropterin, which binds inside ACE2 (Fig. 1), appears to be the most potent stabilizer of the analyzed group. For the “Abundant” glycan model, our results show that Isoniazid acts as an allosteric inhibitor as indicated by the positive binding energy between the RBD domain and ACE2 (ΔΔGBinding) when the ligand is present. In contrast, eriodictyol was observed to increase the binding affinity of the RBD domain to ACE2 thereby acting more as an allosteric stabilizer. Luteolin was observed to have negligible effect on the binding energy between RBD and ACE2. We hypothesize that inhibition/stabilization of the complex induced by these small molecules may serve to disrupt downstream events such as S protein priming by TMPRSS2, an essential step for cellular entry by the virus.
| Molecule | Oak ridge ΔG (kcal mol−1) | Docking ΔG (kcal mol−1) | TI ΔG (kcal mol−1) | RBD/ACE2 ΔΔG (kcal mol−1) |
|---|---|---|---|---|
| Luteolin | −7.4 | −7.71 | −15.04 ± 5.04 | 0.0027 ± 8.94 |
| Isoniazid | −7.3 | −6.57 | −7.29 ± 4.54 | 11.87 ± 7.26 |
| Eriodictyol | −7.1 | −8.16 | −12.65 ± 2.36 | −8.06 ± 9.92 |
3.2 Structural analysis of luteolin binding
Fig. 2 provides a closer look at the interactions between ligand and protein for each identified binding region. For both glycoprofiles, several of the potential binding sites were observed to localize to the same regions. Given its higher affinity for the RBD/ACE2 complex, we focus on the interactions of nitrofurantoin (Fig. 2A) within the binding interface. In this region, hydrogen bonds are the dominant interactions responsible for stabilizing nitrofurantoin. These hydrogen bonds are formed between the carbonyl oxygens of nitrofurantoin and ACE2 residue Lys353 and RBD residue Gln493. Fig. 2B shows a representative configuration for luteolin and the distal RBD binding site. Luteolin is stabilized by a hydrogen bond between its carbonyl oxygen and Tyr369 and pi-alkyl stacking between its aromatic group and Phe377. For comparison, sapropterin, which was found to bind to only ACE2, is stabilized by hydrogen bonds formed with residues of the ACE2 protein (Fig. 2C).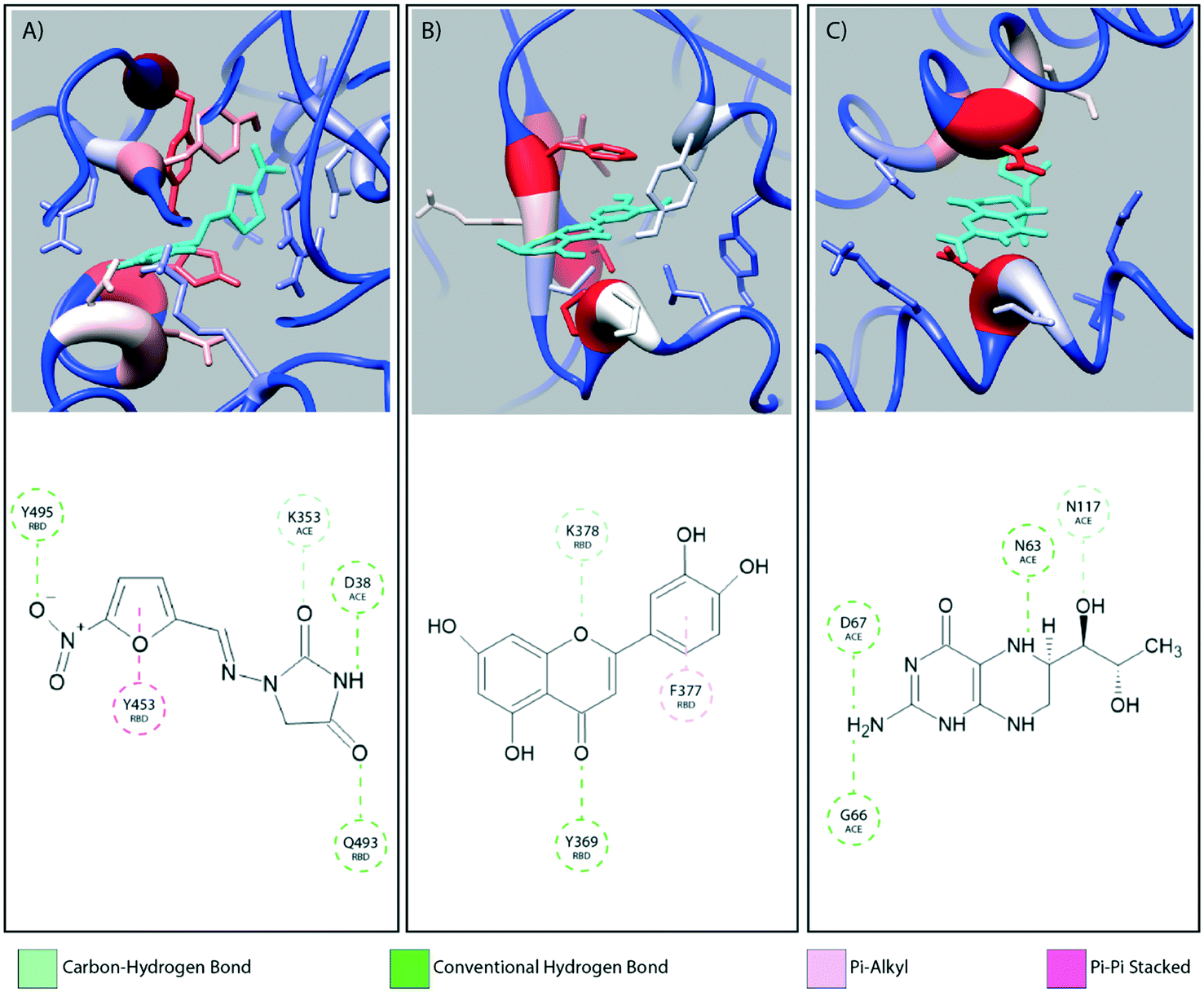 | ||
| Fig. 2 The interaction diagrams for equilibrated configurations of ligands at the interface (A, nitrofurantoin), in the RBD domain (B, luteolin) and in ACE2 region (C, sapropterin) are shown. Hydrogen bonds act as the dominating interactions responsible for stabilizing nitrofurantoin which are formed between the carbonyl oxygens of nitrofurantoin and the Lys353 residue of ACE2 and RBD residue Gln493. Luteolin is stabilized by a hydrogen bond between its carbonyl oxygen and Tyr369 and pi-alkyl stacking between its aromatic group and Phe377 of the RBD domain. Sapropterin, which is found buried in the ACE2 cavity is stabilized by hydrogen bonds. | ||
We study the changes of non-native contacts between the S protein and ACE2 receptor upon ligand binding by comparing the non-native contact maps of each compound (Fig. 3). Similar changes in non-native contacts are observed for compounds eriodictyol and nitrofurantoin, which bind at the RBD/ACE2 interface (Fig. 3A and B), and sapropterin, which binds inside ACE2 (Fig. 3C). Interestingly, binding of luteolin (Fig. 3D) to the distal binding region shows a clear difference in the number of contacts between the S protein and ACE2 receptor. These results suggest that an allosteric effect is induced when luteolin binds to the distal binding site.
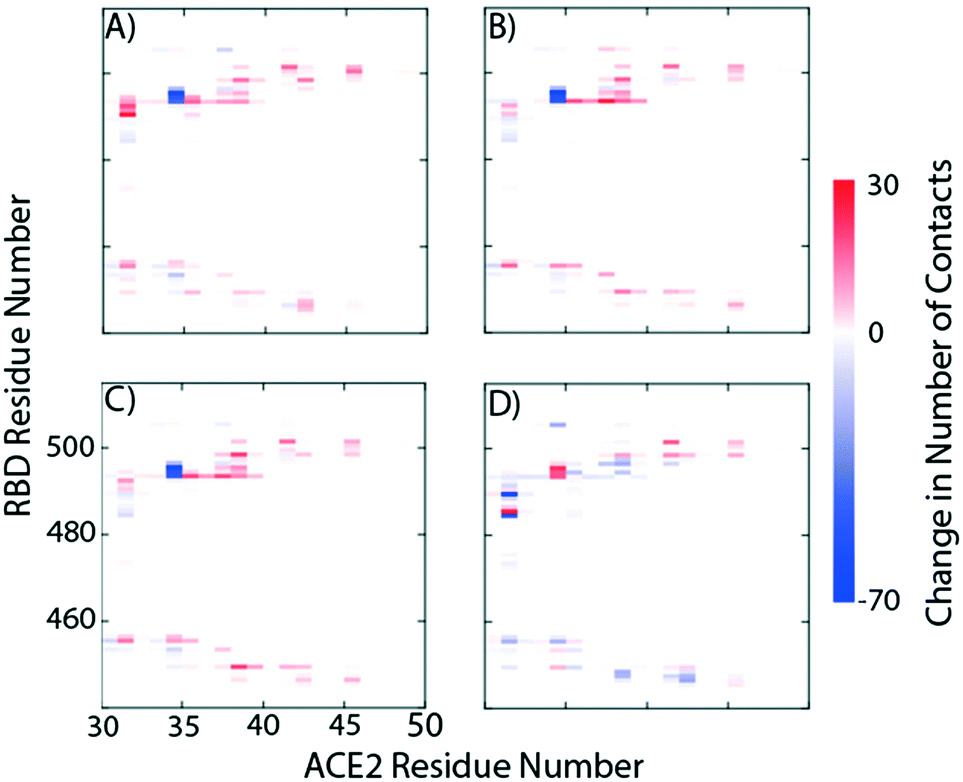 | ||
| Fig. 3 Relative protein–protein non-native contact maps in the presence of A) eriodictyol, B) nitrofurantoin, C) sapropterin, and D) luteolin. The relative non-native contact maps measure the change in contacts relative to the complex with no ligands (red more contacts, blue less contacts). From the graphs we see that the first three panels have identical contact profiles compared to D. | ||
Fig. 4 shows the estimated root mean squared fluctuations and the shear strain on each residue of the RBD/ACE2 complex for several ligands. Strain is a natural quantity to study local protein deformations, which are a good indicator of mechanical allosteric coupling between two binding events on the same protein. This deformation upon ligand binding is calculated as the shear strain measured relative to the average conformation from all frames of the RBD/ACE2 sans ligand. Average root mean square fluctuations (RMSF), a measure of the displacement of a residue relative to the initial crystal structure (PDB: 6M0J), suggest high levels of deviations for RBD residues Asn481, Gly482 and Val483 near the binding interface of ACE2, particularly for the luteolin ligand (Fig. 4A and 5A). Strain analysis also shows a significant peak at these residues (Fig. 4B and 5B). Consistent with the results of Fig. 3D, this profound allosteric effect is induced when luteolin is bound to the distal RBD binding site for both the simple and “Abundant” glycan forms of the RBD/ACE2 complex.
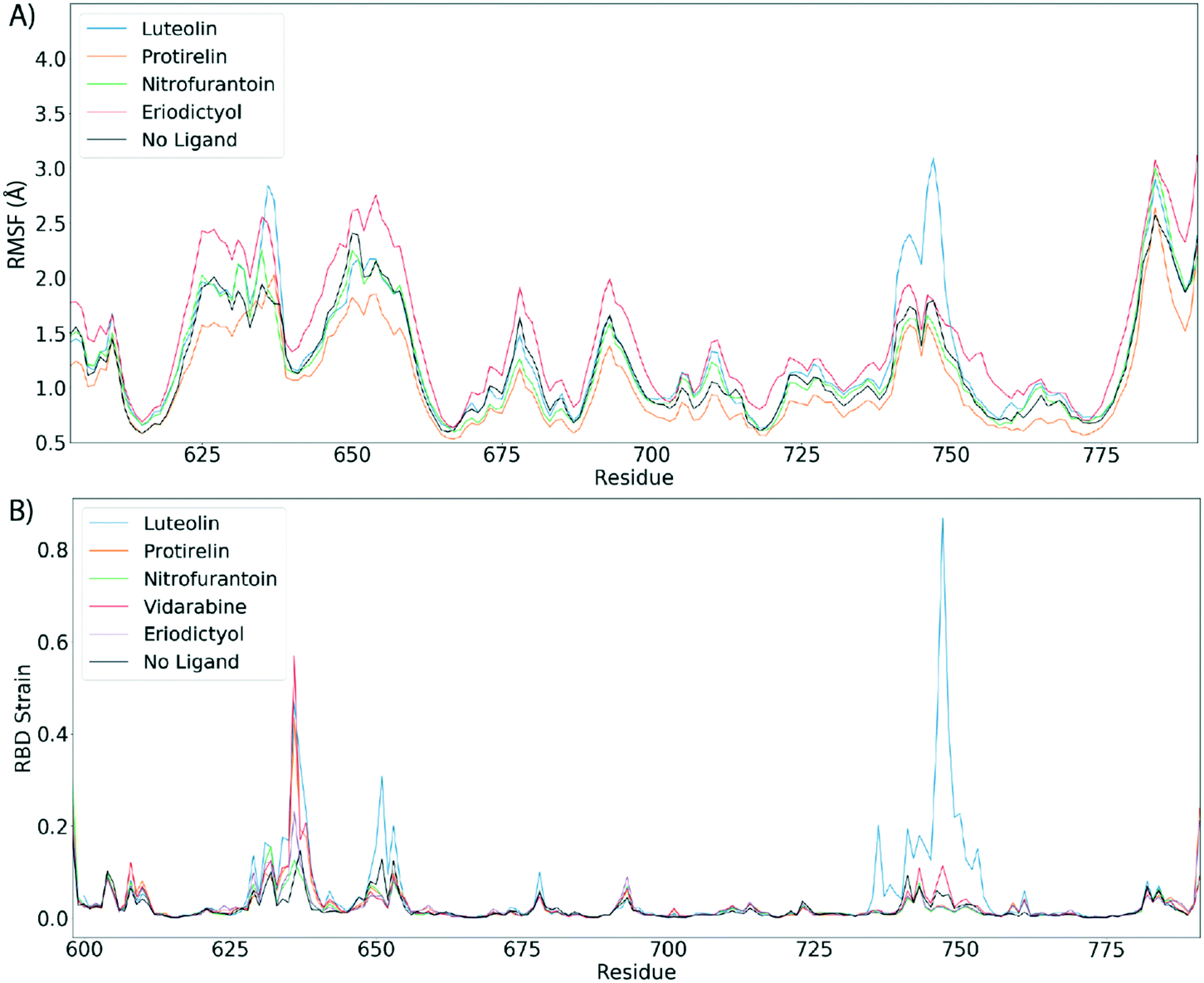 | ||
| Fig. 4 Luteolin induces large allosteric strain when RBD domain is bound to ACE2. A) Root mean squared fluctuations (RMSF) between the RBD/ACE2 complex with top scoring ligand. B) Shear strains mapped. For shear strain calculations, only Cα atoms are included. Strain analysis suggest a strong allosteric strain to the ACE2 binding region of the RBD domain when RBD is in complex with ACE2 and luteolin. | ||
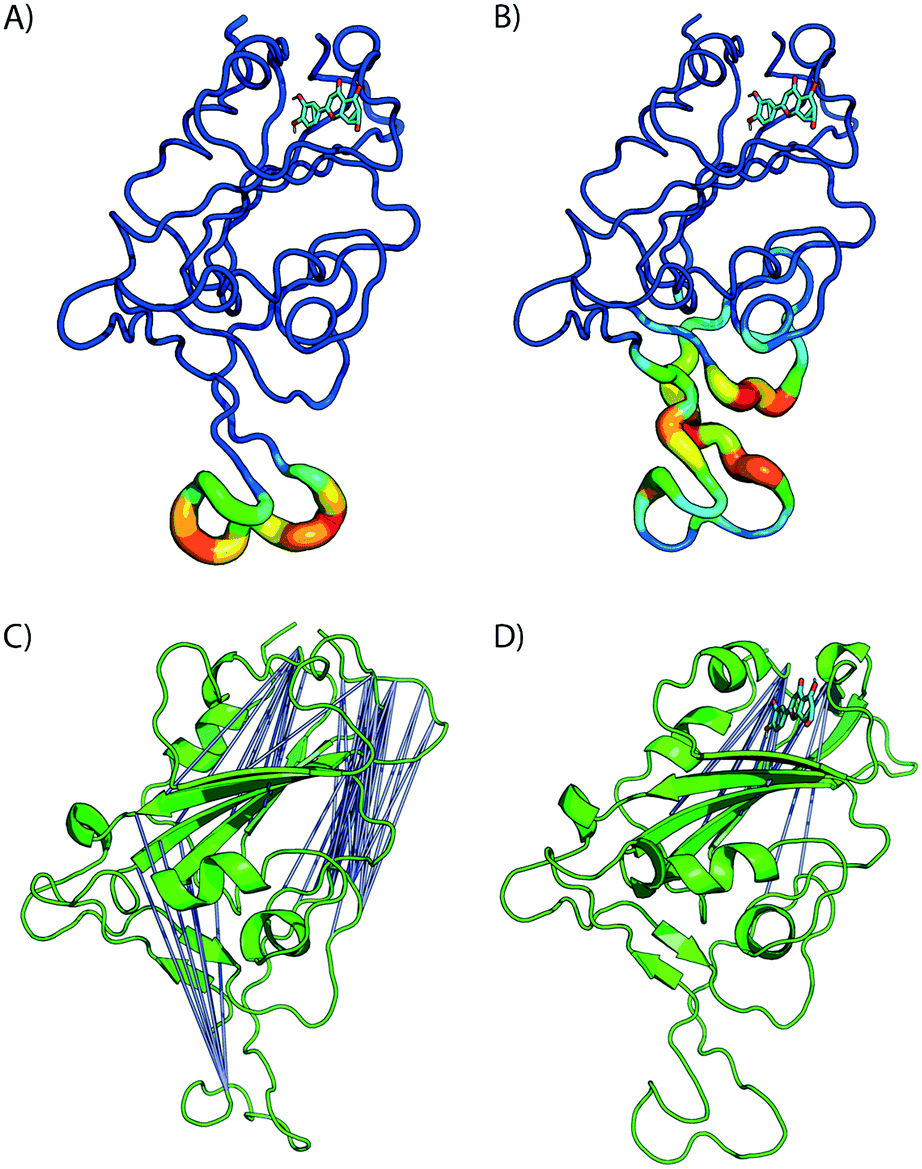 | ||
| Fig. 5 Bound luteolin induces large strain at RBD/ACE binding region. A) Difference in estimated root mean squared fluctuations (RMSF) between the RBD domain and luteolin in complex are shown. (B) Shear strains mapped onto RBD/LUT complex. Regions flanking disulfide bonds have the highest atomic fluctuations that contribute to the deformation energy of the ACE2 binding region. C) and D) Distal site binding disrupts intramolecular RBD interactions inducing conformational changes at ACE2 binding interface. Visualization of residue–residue cross correlations. Blue lines indicate anti-correlation motions with values between −0.4 and −0.6. Higher correlations between distal sites sans ligand (C) and in the presence of luteolin (D). | ||
The formation of cross-correlation networks allows the transmission of information when the binding of a molecule at one site of the protein induces a change in local structure elsewhere in the protein. From our structural analysis, luteolin was the only ligand to induce a conformational change to the RBD protein after binding to a distal site. To validate the extent by which the atomic fluctuations of the complex are correlated with one another in the presence of luteolin, a dynamic cross-correlation network analysis was conducted using the Bio3D software.23 Anti-correlations between −0.40 and −0.60 are represented as blue lines between several distant regions of the RBD, as seen in Fig. 5C and D (ACE2 has been omitted for clarity). A comparison of simulations of the RBD/ACE2 with and without luteolin shows the disruption of the dynamic cross-correlation network by this drug. The disappearance of elements of the network, which include anti-correlated sites between this distal binding site and the RBD/ACE2 interface, may be critical in mediating allosteric transitions and ACE2 binding.
Principal component analysis (PCA) was used to gain quantitative insights into the binding of luteolin to the RBD/ACE2 complex. The orthogonal eigenvectors of the resulting principal components (PCs) describe the maximal variance of the distributions of structures. Details of the data processing are described in the Methods section. The two dominant principal components (PCs) were sufficient to describe the observed conformational changes at the RBD/ACE2 interface. The first principal component (PC1) corresponds to the rotational motion of the RBD/ACE2 complex. The second principal component (PC2) captures the conformational change observed at the RBD/ACE2 interface induced by Luteolin binding (Fig. 6). As previously discussed, residues near the binding site were observed to be anti-correlated to residues near the ACE2 binding site. Binding of luteolin disrupts this cross-correlation network, thereby inducing a conformational shift near the ACE2 binding site. Conformational changes to the RBD protein, as captured by PCA, confirm not only correlated motions of the RBD protein but suggest that the “hotspot” identified here may be a useful target in the discovery and design of new therapeutics that modulate the RBD/ACE2 protein–protein interface. Taken together, our results suggest that luteolin may serve as an important small-molecule allosteric modulator that affects RBD/ACE2 protein dynamics and may provide an alternative therapeutic approach.
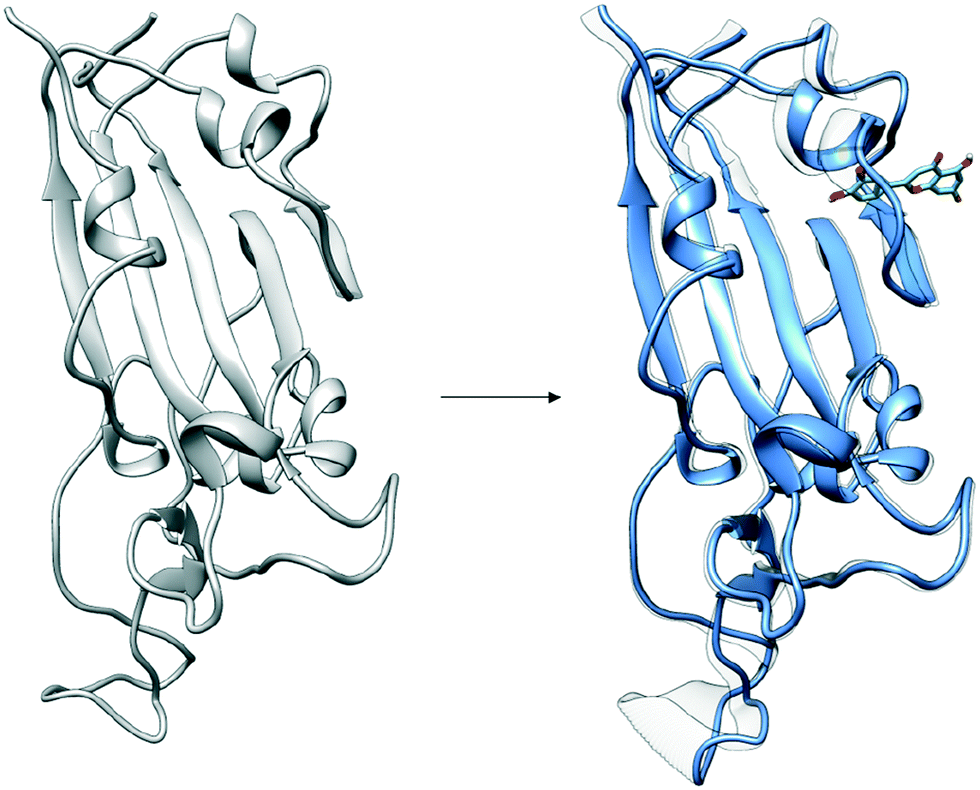 | ||
| Fig. 6 Distal binding by luteolin induces conformational changes at ACE2 binding site. Induced conformational changes at loop binding regions are visualized as captured by the second dominant principal component. Images of important conformational changes are superimposed to emphasize conformations changes introduced after luteolin binding to distal binding site. | ||
4 Conclusion
The repurposing of approved drug candidates provides an alternative strategy to identify lead candidates for viral infections. The time and expense of bringing a drug to market are significantly reduced when the safety and pharmacokinetic profiles of existing drugs are already known. Drug repositioning is guided by a rational approach that requires detailed knowledge of the target structure, the spatial arrangement, interactions and structural conformation of the compound, and the mechanism of action.26 For protein–protein complexes, the full binding inhibition is hard to achieve using small molecules, and novel approaches based on the concept of interfacial inhibition are needed, where macromolecules are trapped in dead-end complexes that cannot fulfill their biological function.27 In this sense, allosteric modulation of PPIs of the RBD/ACE2 complex may serve as an approach in drug discovery. While current high-throughput virtual screenings have identified several potential therapeutic candidates, the mechanism by which they affect protein–protein interactions remains unclear.In this study, we have focused on determining the binding mechanism and associated conformational changes in the presence of the top compounds from a docking screen recently conducted by Smith et al.15 After filtering compounds by their residence time in complex through MD simulations, rigorous binding free energy calculations were conducted in explicit water using thermodynamic integration. Our results have revealed a range of free energies and binding positions that were not anticipated in the original docking studies. The majority of such positions lie far from the interface, and the free energy extrema correspond to the shallow interface region of binding (Tables 1 and 2). The relative RBD/ACE2 binding free energy changes induced by the top ligands considered here indicate a binding enhancement of the RBD/ACE2 complex, even for drugs bound in the interface region. This enhancement is accompanied by a modification of the non-native contact maps of the protein interface, which lead to changes in the manner in which the proteins interact. Taken together, our results suggest that these ligands could not act as ordinary competitive inhibitors for RBD, since there is no binding disruption, but, given the contact modification and the energy differences with respect to the pure RBD/ACE2 complex, we propose that they may inhibit the system by serving as allosteric or direct PPI stabilizers.12,28
PPIs with large, flat and featureless surfaces, as in the case in the RBD/ACE2 complex, lack good drug-binding pockets for ligands. Recent approaches have shown that allosteric modulators provide an alternative strategy to target PPIs such as the RBD/ACE2 complex. Our results have uncovered pronounced effects at the RBD/ACE2 interface upon ligand binding at a distal site. In particular, upon luteolin binding, RMSF and strain analysis unveil significant levels of fluctuations and strain in RBD regions away from the binding site. In addition, cross-correlational analysis reveals the disruption of anti-correlated motions between the luteolin binding site and the binding interface of RBD/ACE2. Future research will require investigating the possible binding motifs for luteolin and its effects on S protein binding to the ACE2 receptor. Beyond the finding that luteolin might serve to inhibit viral entry, the discovery of this distal binding site offers potential for the design and engineering of future therapeutics for COVID-19.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The study of protein−DNA complexes in epigenetics is supported by the NSF, under grant EFRI CEE 1830969, and the development of new DNA models is supported by BIO/ MCB 1818328. The study of allostery and its role in triggerable materials is supported by MURI: W911NF-15-1-0568. The simulations reported here were carried out on the GPU cluster supported by the NSF through grant DMR- 1828629. The authors are grateful to the Research Computing Center of the University of Chicago for additional computational resources.Notes and references
- E. Dong, H. Du and L. Gardner, Lancet Infect. Dis., 2020, 20, 533–534 CrossRef CAS.
- M. Hoffmann, H. Kleine-Weber, S. Schroeder, N. Krüger, T. Herrler, S. Erichsen, T. S. Schiergens, G. Herrler, N.-H. Wu and A. Nitsche, et al. , Cell, 2020, 181, 271–280 CrossRef CAS PubMed.
- P. Zhou, X.-L. Yang, X.-G. Wang, B. Hu, L. Zhang, W. Zhang, H.-R. Si, Y. Zhu, B. Li and C.-L. Huang, et al. , Nature, 2020, 579, 270–273 CrossRef CAS PubMed.
- Y. Huang, C. Yang, X.-f. Xu, W. Xu and S.-w. Liu, Acta Pharmacol. Sin., 2020, 41, 1141–1149 CrossRef PubMed.
- W. Tai, L. He, X. Zhang, J. Pu, D. Voronin, S. Jiang, Y. Zhou and L. Du, Cell. Mol. Immunol., 2020, 17, 613–620 CrossRef CAS PubMed.
- Q. Wang, Y. Zhang, L. Wu, S. Niu, C. Song, Z. Zhang, G. Lu, C. Qiao, Y. Hu and K.-Y. Yuen, et al. , Cell, 2020, 181, 894–904 CrossRef CAS PubMed.
- L. Premkumar, B. Segovia-Chumbez, R. Jadi, D. R. Martinez, R. Raut, A. Markmann, C. Cornaby, L. Bartelt, S. Weiss and Y. Park, et al. , Sci. Immunol., 2020, 5, 48 Search PubMed.
- N. M. Okba, M. A. Müller, W. Li, C. Wang, C. H. GeurtsvanKessel, V. M. Corman, M. M. Lamers, R. S. Sikkema, E. de Bruin and F. D. Chandler, et al. , Emerging Infect. Dis., 2020, 26, 1478–1488 CrossRef CAS PubMed.
- D. Wrapp, N. Wang, K. S. Corbett, J. A. Goldsmith, C.-L. Hsieh, O. Abiona, B. S. Graham and J. S. McLellan, Science, 2020, 367, 1260–1263 CrossRef CAS.
- R. Yan, Y. Zhang, Y. Li, L. Xia, Y. Guo and Q. Zhou, Science, 2020, 367, 1444–1448 CrossRef CAS PubMed.
- J. Westermarck, J. Ivaska and G. L. Corthals, Mol. Cell. Proteomics, 2013, 12, 1752–1763 CrossRef CAS.
- D. Ni, S. Lu and J. Zhang, Med. Res. Rev., 2019, 39, 2314–2342 CrossRef PubMed.
- M. J. Gorczynski, J. Grembecka, Y. Zhou, Y. Kong, L. Roudaia, M. G. Douvas, M. Newman, I. Bielnicka, G. Baber and T. Corpora, et al. , Chem. Biol., 2007, 14, 1186–1197 CrossRef CAS PubMed.
- J. M. Ostrem, U. Peters, M. L. Sos, J. A. Wells and K. M. Shokat, Nature, 2013, 503, 548–551 CrossRef CAS.
- M. Smith and J. C. Smith, 2020, chemRxiv.11871402.v4.
- P. Zhao, J. L. Praissman, O. C. Grant, Y. Cai, T. Xiao, K. E. Rosenbalm, K. Aoki, B. P. Kellman, R. Bridger and D. H. Barouch, et al. , Cell Host Microbe, 2020, 28, 586–601 CrossRef CAS.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- B. R. Miller III, T. D. McGee Jr, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, J. Chem. Theory Comput., 2012, 8, 3314–3321 CrossRef PubMed.
- D. R. Roe and T. E. Cheatham III, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
- M. R. Mitchell, T. Tlusty and S. Leibler, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E5847–E5855 CrossRef CAS PubMed.
- P. Gullett, M. Horstemeyer, M. Baskes and H. Fang, Modell. Simul. Mater. Sci. Eng., 2007, 16, 015001 CrossRef.
- J.-P. Eckmann, J. Rougemont and T. Tlusty, Rev. Mod. Phys., 2019, 91, 031001 CrossRef CAS.
- B. J. Grant, A. P. Rodrigues, K. M. ElSawy, J. A. McCammon and L. S. Caves, Bioinformatics, 2006, 22, 2695–2696 CrossRef CAS PubMed.
- A. Amadei, A. B. Linssen and H. J. Berendsen, Proteins: Struct., Funct., Bioinf., 1993, 17, 412–425 CrossRef CAS PubMed.
- R. Salomon-Ferrer, D. A. Case and R. C. Walker, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 198–210 CAS.
- W. H. Prusoff, T. Lin, E. M. August, T. G. Wood and M. E. Marongiu, Yale J. Biol. Med., 1989, 62, 215 CAS.
- Y. Ferrandez, W. Zhang, F. Peurois, L. Akendengué, A. Blangy, M. Zeghouf and J. Cherfils, Sci. Rep., 2017, 7, 1–13 CrossRef CAS PubMed.
- P. Thiel, M. Kaiser and C. Ottmann, Angew. Chem., Int. Ed., 2012, 51, 2012–2018 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |