
Unveiling the role of structure–property correlation and its validation towards engineering the application potential of sol–gel derived mesoporous gamma-alumina†
Savan Kumar
Sharma
 ,
Paromita
Das‡
and
Barun K.
Sanfui
*
,
Paromita
Das‡
and
Barun K.
Sanfui
*
Department of Ceramic Technology, Government College of Engineering and Ceramic Technology (Autonomous Post Graduate) NAAC Accredited Grade A, 73, A.C.Banerjee Lane, Kolkata-700010, West Bengal, India. E-mail: mailme.drbks@gmail.com
First published on 13th October 2021
Abstract
In the present study, a comprehensive investigation has been carried out on the meticulously designed surfactant-free glycerol-activated sol–gel derived mesoporous γ-alumina powder, specifically with the aim to establish the excellent prospect of the structure–property relationship towards efficiently modifying and conceptually emphasizing the correlative outlook of the application perspective. Herein, the systematic engineering of the selective synthetic parameters such as peptizing acid type and its associated solvent create great strategic influence towards methodically tuning and judiciously constructing the prime controlling facet of the overall process, which successively maintains the distinctive individuality of the sol–gel derived mesoporous gamma-alumina structure. Interestingly, the initiation of discrepancy, which was recorded at the chemical nature site of the molecular bond structure of two distinctive boehmite sols, conspicuously maintains their individuality even while experiencing the sequential advancement of the dehydroxylation process, the consequential rearrangement of the hydroxyl group, alongside the concurrent progressive diffusion of Al3+ ion amongst the available co-ordination sites as a function of increasing temperature. Eventually, while dictating the individual identity of the evolved gamma-alumina's structure, the study aimed at acquiring identical distinctive characteristics even at their corresponding atomic scale, bulk structure, and surface textural features in terms of revealing the Rietveld, NMR (27Al and 1H), pyridine-FTIR, TEM, and XPS spectral analysis with the exploration of comparatively higher crystallinity, greater AlO4/AlO6 ratio, lower Lewis and Brønsted acid sites, lesser amount of hydroxyl groups, and lower extent of agglomeration tendency in inorganic acid peptized gamma-alumina as compared to organic acid peptized gamma-alumina, which thereafter showed concrete interconnectivity by concluding with the proper demonstration of the surface area, pore morphology, and band gap study. Finally, the study presented a detailed mechanistic view with the aim to further exemplify the real-time prospect of the structure–property relationship of the two differently synthesized gamma-alumina samples in terms of categorizing the application potential, particularly in identical dye adsorption and photocatalyst fields, and consecutively building a robust scientific and technological perspective of the structure–property-performance inter-connective linkage beyond the existing limiting factor.
Design, System, ApplicationThe investigation mainly addresses the excellent prospect of the structure–property relationship aspect of the surfactant-free glycerol-activated sol–gel derived mesoporous gamma-alumina structure, wherein the conceptual design and strategic amendment of the synthesis parameters play a decisive role in attaining tunable structural and morphological features of the native gamma-alumina structure, which successively proves its synergistic importance toward engineering the application potential, particularly in the field of water effluent treatment. The atomic as well as surface structural analysis of the sol–gel derived gamma alumina provides a new comprehensive outlook for identifying the origin of disparity arising at their nascent position, categorizing the changes appearing on their transformation pathway, along with exploring the correlating factors toward achieving the ultimate properties and final application performance of the gamma-alumina structure. Therefore, the scientific perspective of the present investigation provides a direct insight into the structure–property-application relationship, which conclusively validates its potential toward engineering the present and future applications of the gamma-alumina structure. |
1. Introduction
Amongst the various available alumina polymorphs, gamma-alumina (γ-alumina) with a specific mesoporous configuration has received enormous scientific and technological interest worldwide in the field of materials science and engineering, especially as a promising candidate in the form of adsorbent, membrane, catalyst, and catalyst support system.1 In particular, the unique combination of the inherent catalytic, electronic, and adsorption properties of γ-alumina can only be extracted and proficiently redirected towards the path of endless opportunities while preserving its competent structural asset of high surface area, controllable pore size and its distribution, adjustable acid–base properties, etc., in an expertise knowledge constructed way.2,3 Over the last few decades, materials scientists and technologists have already witnessed an emergent tailored characteristic feature of nano-structured γ-alumina by simply altering its structural and designing aspect, which directly authenticates the consequence of its structure–property inter-relationship.4,5 In the above context, an utmost requirement is to shed the light primarily at the atomic scale crystallographic position, followed by unlocking its true dependability towards governing the actual structural identity of the γ-alumina nanostructure.Specifically, the crystal structure of γ-alumina with Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space groups, which is comparable with the defective cubic spinel lattice, exhibits an admirable characteristic distribution of tetrahedral and octahedral aluminum (Al) lattice sites accompanied with well-maintained vacant sites.6 It is worth mentioning herein that the slight alteration in the particular crystallographic facets of the γ-alumina structure creates a gigantic influence on its individual structural identity. In general, the definite acquaintance of both the coordinatively saturated and unsaturated aluminum concentration and its collective distribution are considered to be mainly responsible for exerting the actual defective nature within the γ-alumina structure.7 To be specific, the significant alteration in the chemical nature and surface active sites has been noticeably observed while experiencing a huge variation precisely on generating Lewis and Brønsted acidic sites along with preserving the definite concentration of surface hydroxyl groups at the respective particle surface.8 In addition to the above, the atomic-scale arrangement in the γ-alumina local structure also creates synergistic influence towards amending both the surface texture and pore morphological feature of the oxide powder.9 Furthermore, C. Lee et al. elucidated another peculiar but significant aspect of atomic site occupancy, wherein they demonstrated its powerful influence especially towards controlling the band gap structure of γ-alumina.10
m space groups, which is comparable with the defective cubic spinel lattice, exhibits an admirable characteristic distribution of tetrahedral and octahedral aluminum (Al) lattice sites accompanied with well-maintained vacant sites.6 It is worth mentioning herein that the slight alteration in the particular crystallographic facets of the γ-alumina structure creates a gigantic influence on its individual structural identity. In general, the definite acquaintance of both the coordinatively saturated and unsaturated aluminum concentration and its collective distribution are considered to be mainly responsible for exerting the actual defective nature within the γ-alumina structure.7 To be specific, the significant alteration in the chemical nature and surface active sites has been noticeably observed while experiencing a huge variation precisely on generating Lewis and Brønsted acidic sites along with preserving the definite concentration of surface hydroxyl groups at the respective particle surface.8 In addition to the above, the atomic-scale arrangement in the γ-alumina local structure also creates synergistic influence towards amending both the surface texture and pore morphological feature of the oxide powder.9 Furthermore, C. Lee et al. elucidated another peculiar but significant aspect of atomic site occupancy, wherein they demonstrated its powerful influence especially towards controlling the band gap structure of γ-alumina.10
Nonetheless, on a key note, the crystallographic identity, which indeed determines the surface activity of the γ-alumina structure, is intrinsically connected to a large extent on the respective synthetic route and its associated processing parameters.11,12 Therefore, for instance, the application specific construction of γ-alumina primarily demands a comprehensive understanding of the complex structure–property relationship; the essence of the overall consecutive connectivity of which can only be recognized while maintaining an intense control on the synthetic parameters along with reviewing its associated underlying transformation sequence, which in turn provide high degree of command in freezing the target specific surface and atomic scale structural integrity of the desired end product. Recently, some of the well-established synthetic pathways with clearly distinctive protocols are already in use such as combustion synthesis, precipitation, sol–gel, thermal decomposition, template route, spray drying, and combination thereof, all of which show excellent proficiency in producing γ-alumina with subsidiary controlled morphological, structural, and physicochemical characteristics.13,14 However, among these various processing techniques, the soft chemistry-based sol–gel route provides relatively good control over others especially due to a clear sense of flexibility in altering the structure at the atomic scale.15 On a closer look, the sol–gel based synthetic strategy of nano-structured γ-alumina generally involves the preliminary sequential growth of boehmite or aluminum oxy-hydroxide phase, followed by experiencing its controllable progressive transformation into the oxide γ-alumina phase by employing an appropriate dehydration schedule.15 Herein, it is important to note that the fine-tuning of the sol–gel based processing parameters, including the nature and concentration of the selected precursor, solvent, peptizing agent, temperature, and processing pH, generates the first dominating influence at the atomic scale structural level of the precursor stage, which in turn shows extraordinary proficiency in diverging the crystallographic growth of γ-alumina and thereby broadening the range of surface activity in terms of the acidity, hydrophilicity, surface structure, and pore morphology of the final product.16
In the above context, the literature, which highlights the distinctive scientific protocols of the sol–gel science toward deriving the variable γ-alumina structure is worth addressing herein. Yoldas et al. studied the peptization, hydrolysis, and gelling behavior of the alkoxide precursor species with respect to different type of acid content, which authenticate its interdependence toward altering the particle size and their respective crystallinity of the oxide phase.17 In addition, X. Chen et al. and others utilize the different types of acids for synthesizing boehmite sols at nearly constant pH where a significant difference in the particle size of the sol as well as a considerable divergence in the crystallinity, surface area, pore size and its distribution of the calcined γ-alumina powders have been reported.18 In relation to the above, our previous study also evidently denotes that altering the type of peptizing agent along with the solvent effectively changes the particle size of the boehmite sol, which shows promising influence in tuning the microstructure and pore morphology of the γ-alumina unsupported membrane layer, thereby authorizing its application prospect for the fabrication of alumina-based graded membrane support system specialized for CO2/N2 binary gas separation.19
However, in spite of recognizing such decent prospects, there is still scarcity in the availability of literatures demonstrating the versatile significance of the structure–property relationship, especially towards highlighting the appropriateness of selecting the application scenario of the γ-alumina end product.1,5,20 Henceforth, in a generalized approach, the experimental design continuing with a widely accepted sol–gel synthesized γ-alumina structure for the exploration of flexible construction of the structure–property relationship always believed to offer a fascinating scientific prospect in the ray of the application perspective. Herein, it is worth noting that the individual properties of the application directive γ-alumina were found to be hard to correlate or remained unexplained with respect to the structural level, especially while imposing a minor change in the synthetic parameters. In particular, amongst the widespread application prospect of mesoporous γ-alumina, the photocatalytic degradation and adsorption phenomenon, which indigenously act as a key promising aspirant of waste water treatment, shows great dependence on the structural perspective, thereby extends great potential for the effective exploration of the actual influential facet of the characteristic structure–property inter-relationship aspect of the nanomaterials.21–23 In the above context, while experiencing the designing aspect, it is important to describe the results of Piña-Pérez et al., wherein the oxide sample possessing a low band gap not only results in high photochemical stability but also ensures the complete degradation of the organic contaminants, whereas the critically low band gap of the native semiconducting oxides experience high recombination rate of the electrons and holes at their surface, which ultimately hampers the photocatalytic activity.22 In addition, few authors have also highlighted the effective contribution of hydroxyl radicals, Lewis acid sites, and surface morphology in determining the photocatalytic activity of the system.21,24 Similarly, while cultivating the structural dependency of nanostructured materials in determining the adsorption characteristic features, it shows direct interconnectivity with the coordination sites, the association of hydrogen bonding with respect to the surface active sites, and the presence of OH− groups, which can sense the conventional abundant factors of the complicated defective spinel-like γ-alumina structure. Raj et al.25 and Zafar et al.26 showed that the morphology of the oxides materials also imposes an important feature for facilitating dye adsorption, whereas the overlapping and critical inter-pore structure acts as a limiting factor. Furthermore, according to Cai et al.27 and Lahiri et al.,28 controlled surface chemistry along with tunable porous structure at the surface of the oxides also seems to plays a synergetic role in the dye adsorption system. The significant influence of the surface morphological features on the diverse application potentials has already been proven for mesoporous hybrid materials.29 Importantly, the constructive literature study defining the overall scenario of the γ-alumina indicates the fact that the utilization of a basic environment along with some tactical steps in the synthetic route has the possibility to give higher active sites for γ-alumina oxide structure, which in turn assist in extracting its effective application potential.
Importantly, since the presently adopted sol–gel based synthetic route has received its own unique significance via encountering the selective utilization of glycerol as an electric double layer (EDL) stabilizing agent for the effective elimination of the employed assistance of the complex and costlier surfactant-based synthetic technique; thus, considering the state of science, understanding the strategic factors must provide a distinctive protocol for the selective evolution of the γ-alumina structure. Henceforth, in the present investigation, a facile attempt has been undertaken with an integrated scientific decorum to indigenously identify the structure–property relationship of the surfactant-free glycerol-modified sol–gel synthesized mesoporous γ-alumina entities, in concurrence with the additional employment of a distinctive dispersion medium and peptizing agents, followed by the consequent enlightening of the suitable application prospective in order to establish the robust interconnected bridging network between the structure–properties-performance entities, which will exemplify the new scientific perspectives for nano-structured spinel material. The influential aspect of the processing parameters such as the peptizing agent type and its associative solvent towards tailoring the structure–property relationship facet of the selective gamma-alumina has been meticulously investigated with a logical flow, advancing from the initial stage of processing step to the final anticipated network structure, followed by outlining the direct mechanistic view to deepen the scientific understanding for tuning the practical application perspective of the ordinance structure.
2. Experimental
2.1 Materials
Aluminium-tri-sec-butoxide (ATSB, 99.99%, Sigma Aldrich, Germany), ethanol (C2H5OH ≥99.8%, Merck, India), 2-propanol (C3H8O3, 85%, Merck, India), glacial acetic acid (CH3COOH, 99.8–100%, Merck, India), nitric acid (HNO3, 68.5–69.5%, Merck, India), glycerol (C3H8O3, 85%, Merck, India), pyridine (C5H5N, ≥99.0%, Merck, India), Congo red (C32H22N6Na2O6S2, 98%, Merck, Germany), and phenol (C6H5OH, 99.0–100%, Merck, India) were purchased and used as received for the current investigation.2.2 Synthesis of Al based powder precursor
Aluminium-tri-sec-butoxide (ATSB) was firstly hydrolyzed in pre-defined mixtures of ethanol–water and iso-propanol–water separately at 80 °C under vigorous stirring under refluxing conditions. The respective mixtures of aluminum hydroxides with propanol and ethanol were peptized using glacial acetic acid and nitric acid, respectively, in order to synthesize the transparent sol of boehmite having diverse structural behavior. Finally, glycerol was added to stabilize the sol and to avoid the long aging procedure, which is generally used in order to synthesize the stable boehmite sol. Herein, the molar ratio of ATSB![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :solvent:peptizing agent for colloidal boehmite sol synthesized using isopropanol as a solvent and acetic acid as the peptizing agent (denoted as AAI) is 1:20.559:5.24, while that for another sol where ethanol was a solvent and nitric acid was a peptizing agent (denoted as ANE) was 1:27.128:0.5549. The resultant sols were converted to gel under natural conditions, and dried in a hot air oven at 110 °C for 24 h and heat-treated at 600 and 800 °C for 2 h in order to study the structural transformation and their respective properties. The overall schematic diagram of the procedure has been presented in Fig. 1.
:solvent:peptizing agent for colloidal boehmite sol synthesized using isopropanol as a solvent and acetic acid as the peptizing agent (denoted as AAI) is 1:20.559:5.24, while that for another sol where ethanol was a solvent and nitric acid was a peptizing agent (denoted as ANE) was 1:27.128:0.5549. The resultant sols were converted to gel under natural conditions, and dried in a hot air oven at 110 °C for 24 h and heat-treated at 600 and 800 °C for 2 h in order to study the structural transformation and their respective properties. The overall schematic diagram of the procedure has been presented in Fig. 1.
 | ||
| Fig. 1 Schematic diagram of the experimental procedure. | ||
2.3 Characterization
The two synthesized Al-based different powder precursors have been characterized thoroughly. Firstly, the phase transformation and structural parameter of the dried gel and the heat-treated samples were studied by X-ray diffraction measurement (PANalytical X-PERT Pro) with Cu Kα radiation, having a wavelength of λ = 1.54 Å and 2θ angles in the 5–80° range and 2° continuous scanning mode at the rate of 2° min−1. The Rietveld refinement of the acquired data (for γ-alumina) was carried out in order to get quantitative information on the structural data.30 The graphical representations of the modeled structural data have been visualized by the VESTA software.31 The morphological evolution and details of the sample surface were analyzed by FESEM (Zeiss, Germany, Model Sigma) and TEM (JEOL JEM 2100HR). The crystallographic structure of the gamma samples was acquired from a high-resolution transmission electron microscopy (HRTEM) and selected area electron diffraction (SAED) analysis. The d-spacing from the IFFT (inverse fast Fourier transformation) profile as well as from the SAED pattern has been calculated using the GATAN Digital Micrograph software (version 2.1). To analyze the thermal decomposition behavior of the respective dried gel, TGA-DTA (Pyris diamond, PerkinElmer) analyses was performed. BET (Quantachrome ASiQwin-Automated gas adsorption data, ver. 3.0) and BJH analysis were carried out from the N2 adsorption–desorption method (77 K) to determine the surface area and pore size distribution. The bond characteristics and crystalline structure of the respective sol and powder were characterized by Fourier transform infrared spectroscopy (IRAffinity-1S WL Shimadzu, Japan). Furthermore, the relative amounts of Brønsted and Lewis acid sites were characterized by FTIR spectroscopy of the self-supported pyridine-absorbed γ-alumina pellets (dried under vacuum at 75 °C for 12 h) with a resolution of 2 cm−1 and 75 cumulative scans using self-supported pure γ-alumina as the reference pellet. Solid-state 27Al and liquid state 1H nuclear magnetic resonance spectra were measured on an NMR spectrophotometer using 400 MHz, JEOL ECX400 and 400 MHz, Bruker AV-400, respectively. The UV-vis spectra of the γ-alumina samples were acquired from a double beam UV-vis spectrophotometer (UV-2600, Shimadzu, Japan) using an integrating sphere reflectance unit.2.4 Methodology
where C0 is the initial Congo red dye concentration (mg L−1), Cf is the Congo red dye concentration at equilibrium (after adsorption mg L−1), V is the volume of Congo red dye solution (L), and m is the adsorbent mass (g).

3. Results and discussion
3.1 Analysis of AAI and ANE sols and its corresponding gels
The dominating influence of the alteration of the acid type (organic and inorganic acid) and its respective solvent (two distinct organic solvents) in the presence of glycerol as a common electric double layer (EDL) stabilizing agent that has been presented in our previous study for the synthesis of two distinctive boehmite sols preliminarily evidenced tunable characteristic particle size distribution, showing promising prospect in regulating the ultimate surface morphological feature of the resultant γ-alumina structures at the relevant membrane application site.19 Based on these key exploratory features, the present investigation has been further intended to reveal both the atomic scale and surface structure interdependency of the evolved γ-alumina structure towards defining its tangible characteristics, whose critical and systematic correlation help to build up a direct protocol for establishing the versatile significance of the structure–property inter-relationship aspect on dictating its ultimate end-use applicability. Henceforth, in order to probe the genuine influence of the processing parameters on the evolution of the γ-alumina structure, the presently adopted experimental design has been meticulously subjected to reveal the specific origin site from where the actual structural discrepancy has been taking place as well as monitoring the consecutive changes appearing at their intermediate atomic scale and surface structural position until the desired structural asset of γ-alumina is obtained.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Al–(OH) bond, bending of the (OH)–AlO bond, and consequent stretching of the Al–O–Al bond at the respective distorted AlO6 site of the boehmite structure. Interestingly, few available literatures dictated the 1072 cm−1 transmission band as the most considerate band, which depicts the extent of boehmite phase formation. On the contrary, some researchers also presented an exactly opposite interference for the 1072 cm−1 peak, wherein it is strictly indicated as a peak for the nitrate ion.34 Notably, while concentrating specifically on the discrepancy of the hydroxyl group arrangement, a weak but prominent peak observed at 3741 cm−1 of the AAI sol, which might signify either the presence of an isolated OH− ions related to the pre-adsorption of acetic acid or related to the doubly bridging type II hydroxyl group of (Al5c)2OH.35–37 The close comparison of the characteristic bands of the boehmite structure at two respective sols reveals more or less similar intensity bands unless considering the band at 744 cm−1, which shows comparatively high intensity in the case of ANE sol. As a consequence, the presence of a higher AlO6 coordination in the ANE sol stipulates the possibility of presence of higher active sites along with highly crystalline arrangement of the boehmite structure as compared to the AAI sol.
Al–(OH) bond, bending of the (OH)–AlO bond, and consequent stretching of the Al–O–Al bond at the respective distorted AlO6 site of the boehmite structure. Interestingly, few available literatures dictated the 1072 cm−1 transmission band as the most considerate band, which depicts the extent of boehmite phase formation. On the contrary, some researchers also presented an exactly opposite interference for the 1072 cm−1 peak, wherein it is strictly indicated as a peak for the nitrate ion.34 Notably, while concentrating specifically on the discrepancy of the hydroxyl group arrangement, a weak but prominent peak observed at 3741 cm−1 of the AAI sol, which might signify either the presence of an isolated OH− ions related to the pre-adsorption of acetic acid or related to the doubly bridging type II hydroxyl group of (Al5c)2OH.35–37 The close comparison of the characteristic bands of the boehmite structure at two respective sols reveals more or less similar intensity bands unless considering the band at 744 cm−1, which shows comparatively high intensity in the case of ANE sol. As a consequence, the presence of a higher AlO6 coordination in the ANE sol stipulates the possibility of presence of higher active sites along with highly crystalline arrangement of the boehmite structure as compared to the AAI sol.
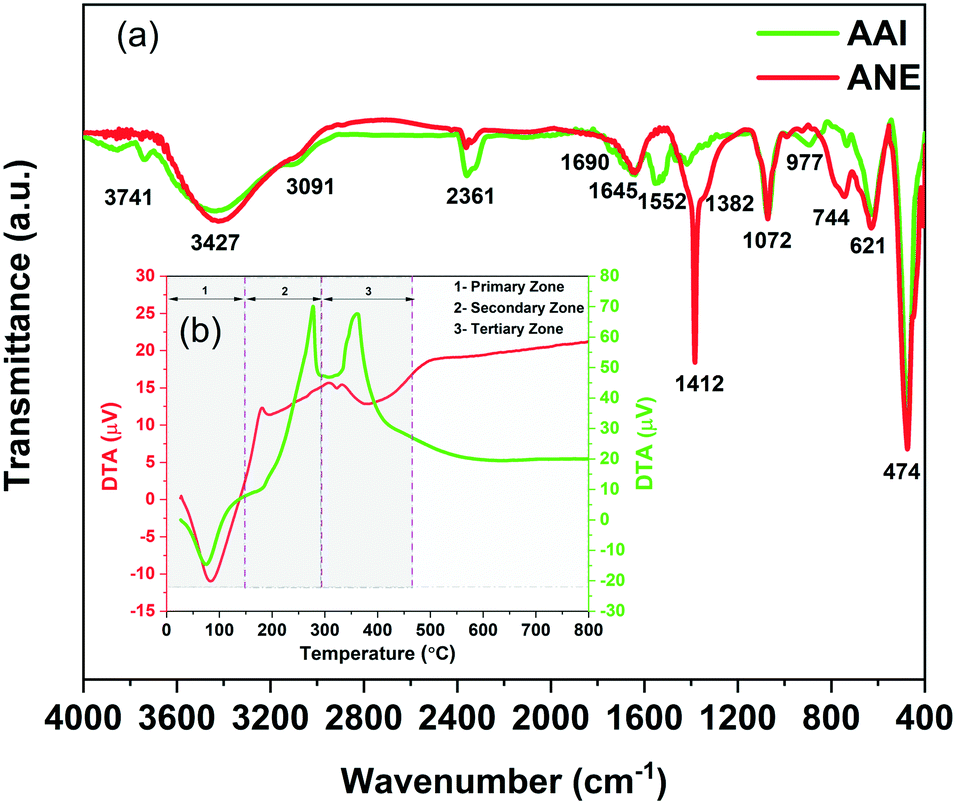 | ||
| Fig. 2 (a) FTIR spectrum of the as-synthesized AAI and ANE sol, (b) DTA curve of the as-synthesized AAI and ANE gel dried at 110 °C. | ||
 | ||
| Fig. 3 XRD (a and b) and FTIR (c and d) spectra of AAI and ANE gel heat treated at 110, 600, and 800 °C. | ||
 | ||
| Fig. 4 Adsorption–desorption N2 isotherms (a and b) and BJH pore size distribution curves (c and d) of AAI and ANE gel heat treated at 110, 600, and 800 °C. | ||
Herein, two differently synthesized gel powders dried at 110 °C exhibit low N2 adsorption behavior, thereby suggesting lower surface area and pore volume of the boehmite structure as compared to the high temperature thermally-treated samples. As confirmed by DTA, FTIR, and XRD analyses, the resultant dried boehmite samples shows a high amount of surface and structural OH groups, which might block the pore in a complicated manner, consequently imposing a strict obstacle for the adsorption of N2 gas.44 In addition, this particular effect becomes more prominent while depicting a detailed comparative analysis of the BET results, which readily demonstrated that the low hydroxyl group and volatile/organic matters containing boehmite synthesized (ANE) from nitric acid shows the lowest surface area and negligible pore volume as compared to the high organic and hydroxyl group comprising acetic acid-peptized samples (AAI). As depicted from the XRD pattern and FTIR spectrum [Fig. 3(a–d)], the associative presence of higher crystallinity might be another significant reason for the lower N2 adsorption and surface area of the respective ANE gel. The Barrett–Joyner–Halenda (BJH) pore size distribution curve [Fig. 4(c and d)] was calculated from the adsorption data of the isotherms, which clearly shows a weak peak at 4.37 nm and 4.44 nm for AAI and ANE powder samples dried at 110 °C, which become centered at 12.45 nm and 4.47 nm at 600 °C, respectively. Surprisingly, the pore size, pore volume, and surface area of the dried precursor powder increases with increasing temperature up to 600 °C, which might arise due to the phase transformation of boehmite to the crystalline γ-alumina phase with individual morphologies. In general, at this particular stage, the continuous progressive disintegration of the hydrogen bonds took place in terms of dehydroxylation, which in turn favors the formation of a morphological specific γ-alumina with increased specific surface area and controllable pore volume in the meso-range. The evaporation of physically- and chemically-bonded water molecules influences the external and internal stresses, which encourages cracking and surface entanglement, leading to micronization. The continuous rise in the temperature tends to form stresses along with the generation of secondary cracks of critical length, which promotes the formation of new cracks via critical crack propagation and ultimately ends up causing particle fracture.45 This unique physical phenomenon combined with major structural transformation sequence will expose new surfaces, hence causing a simultaneous increase in both the surface area, pore size, and its associated pore volume. However, importantly, with the further increase of the thermal treatment up to 800 °C, both the samples experience a sudden decrease in the surface area, pore size, and volume. In addition to the above, the particle coarsening effect is also believed to play a key role in this regard, which involves the pore enlargement and volume contractions, resulting from the combined contribution of grain growth and reduction of the associated surface energy of the agglomerated particles.46
Therefore, the close comparison of the above results clearly confirms the preferential selection of γ-alumina heat-treated at 600 °C as the most suitable material, especially for unlocking its structure-dominating application perspective. Moreover, as the meticulous alteration of the synthetic parameters of the sol–gel process creates a profound influence on the structural evolution of the boehmite phase, herein, in the next proceeding section, it is of utmost requirement to reveal the differential structural characteristic features, which continued to persist even after the thermal treatment at 600 °C at the two distinctive γ-alumina structures.
3.2 Comparative structural analysis of γ-AAI and γ-ANE powders
In the above context, a detailed comparative analysis of the surface and atomic scale structural characteristics of the two evolved γ-alumina structures denoted as γ-AAI and γ-ANE have been systematically demonstrated in terms of FTIR, XRD, FESEM, BET, TEM, NMR, XPS, and band gap studies. Herein, it is worth noting that the FTIR spectra (Fig. 5a) of both the AAI and ANE powder samples calcined at 600 °C possess differential characteristics of Al–O modes in the range of 500–750 cm−1 and 750–850 cm−1, signifying the AlO6 octahedra and AlO4 tetrahedra, respectively. Interestingly, the results are in good agreement with the DTA patterns of the two differently synthesized sols, suggesting an obvious discrepancy at the thermally-induced relative movement of the Al ion from the respective octahedral to tetrahedral coordination sites, specially accounting for the differential extent of the organic content. | ||
| Fig. 5 Comparative FTIR (a) and XRD (b) spectra of γ-AAI and γ-ANE powders. | ||
Similarly, the XRD results (Fig. 5b) also identified a clear difference in the crystallographic orientation of the evolved γ-alumina structure, which is completely in line with the FTIR results, indicating the differential movement of aluminum ions in between the tetrahedral and octahedral coordination sites, thereby experiencing a variation in the structural ordering of the two evolved γ-alumina phases. The results corroborate the fact that, as the ANE sol shows lower average particle size and lesser extent of surface coverage after peptization, it experiences high surface energy, easy movement of Al ion at the respective co-ordination sites, and higher crystal growth rate than that of the AAI sol, thereby extending strong grounds for attaining the comparatively higher level of crystallinity at 600 °C for ANE.47 On the other side, interestingly, it is worth mentioning that since the transformation of boehmite to the gamma-alumina structure generally follows a complex reaction mechanism, comprising of the initial formation of the active nucleation site for the eventual covering of the boehmite surface with the gamma-alumina layer; therefore, the next consecutive step of the dehydration sequence must encounter the diffusion process proceeding through the respective evolved oxide layer. The above sequence makes it clear that the crystallinity of boehmite should play a significant role in the formation of the gamma-alumina structure.48 Precisely, in case of low crystalline boehmite, the formation mechanism of gamma-alumina is restricted by the diffusion-controlled process, whereas for high crystalline boehmite, the similar transformation sequence actively shows the dominating influence of the nucleation step.48 In the above context, it is quite justified to express the preferential aspect of the γ-ANE structure towards providing the relatively high crystalline and well-defined gamma-alumina phase in comparison to the γ-AAI structure. Meanwhile, the diffusion of the aluminum ion from the octahedral to the tetrahedral position at 600 °C is quite efficiently related with the peak intensity of the (220), (440), and (400) crystalline phases. Specifically, herein, the intensity ratio of the (220)/(400) and (400)/(440) peaks may play a conclusive role while dictating the comparison of the developed γ-alumina crystalline phase.49 In particular, as described by the XRD analysis, the intensity ratio of the (220)/(400) and (400)/(440) planes for the γ-ANE powder is higher than that for the γ-AAI powder, which signifies the presence of a higher concentration of an octahedral aluminum ion in γ-AAI powder.50 In addition, it is also important to note that the comparatively higher peak intensity of the characteristic (220) and (440) planes of γ-ANE favors the much stronger orientation in the direction of the (110) plane, which is thereby believed to exhibit more plate-like morphology.
m symmetry of gamma-alumina, we have followed the best fitted model proposed by Zhou et al., wherein the cation occupancy at the highly distorted Wyckoff 32e site has been reported, which is later on defined as the Al(V)-coordinated aluminum ion, thereby eventually designating as a defective spinel structure of γ-alumina.51 However, it is important to mention herein that the non-spinel model for γ-alumina has already been proven to be less considerable for the specific aluminum ion distribution.52,53
Though, the comparative refinement patterns (Fig. S1†) and the respective data (Table 1) reveals the three distinctive Al coordination sites for both the gamma-alumina structures but the more precise one has readily demonstrated the differential distribution fraction of Al ions at the octahedral, tetrahedral, and pentahedral coordination sites of 62.74%, 29.41%, 7.84% and 66.06%, 32.12%, 1.80% for γ-AAI and γ-ANE powders, respectively. In addition, the lattice parameter (a) and unit cell volume were calculated to be 7.879 Å and 489.11 Å3 for γ-AAI, whereas they were 7.884 Å and 490.04 Å3 for γ-ANE powder, respectively. The graphical representations of the respective structural parameters and the modelled data have been shown in Fig. 6a and b. Since the comparative refinement study exhibits the dominating nature of Al ion at the 32e site occupancy for the γ-AAI structure with lower lattice parameter and cell volume, it confirms the presence of a higher amount of abnormally coordinated AlV ions in the γ-AAI structure with respect to the γ-ANE structure, which as a whole is also indicative of the differential characteristic aspects of surface morphology and pore structure, thereby dictating the strict demand to further explore its detailed surface and atomic structural analysis.2
| Structural parameter | Sample γ-AAI | Sample γ-ANE |
|---|---|---|
| Site (space group: Fdm) |
Occupancy | |
| Al1 16(b) | 0.64 | 0.73 |
| Al2 8(a) | 0.60 | 0.71 |
| Al3 32(e) | 0.04 | 0.01 |
| O 32(e) | 1.00 | 1.00 |
| GOF | 2.41 | 2.70 |
| Weighted R profile | 6.69% | 6.97% |
| Site | Aluminium coordination (%) | |
| Octahedral | 62.74 | 66.06 |
| Tetrahedral | 29.41 | 32.12 |
| Pentahedral | 7.84 | 1.80 |
| Lattice parameters | ||
| a (Å) | 7.879 | 7.884 |
| Unit cell volume (Å3) | 489.11 | 490.04 |
 | ||
| Fig. 6 Graphical representations of the crystal structure for γ-AAI (a) and γ-ANE (b) powders modelled by Rietveld refinement. | ||
 | ||
| Fig. 7 TEM, HRTEM, IFFT patterns, IFFT profile, and SAED patterns of the γ-AAI (a–e) and γ-ANE (f– j) powders. | ||
 | ||
| Fig. 8 (a) Comparative 27Al NMR spectra of γ-AAI and γ-ANE powders, (b) de-convoluted NMR spectra of γ-AAI powder, (c) comparative 1H NMR spectra of γ-AAI and γ-ANE powders, comparative pyridine-FTIR spectra, (d) of γ-AAI powder and γ-ANE powders. | ||
Interestingly, as predicted above, the simple employment of the de-convolution study of the respective spectra of the γ-AAI powder sample (Fig. 8b) clearly illustrated the additional findings of Al(V) with more wide range distribution of Al ions, i.e., Al(VI):Al(V):Al(IV) as 76.337:7.05:16.43, whereas in the case of γ-ANE, the distribution has been restricted only to Al(VI):Al(IV) as 78.763:21.237. The chemical shift at 31.42 ppm, in case of γ-AAI, signifies the fivefold-coordinated Al atoms, wherein an aluminum nucleus is surrounded by higher electron density atoms with respect to an oxygen tetrahedron but smaller than that of an oxygen octahedron.58
This particular observation along with the data recorded with the deconvoluted study, as a whole, designated γ-AAI powder as a more defective structure especially due to its restricted movement of aluminum ions from AlO6 to AlO4 coordination sites, which clearly show good agreement with delayed decomposition nature of the AAI gel in the DTA result.59 Another important fact to mention herein is that as indicated by the FTIR and DTA analyses of the native gels, the comparatively higher amount of chemically-bonded hydroxyl groups, which is present in the structure of the γ-AAI powder, replace some of the oxygen atoms surrounding the aluminum atom at octahedral sites, generating an aluminum nucleus with a lower electron density. However, the two distinctive gamma-alumina structure show similar differential trend in the distribution nature of the AlV coordination site while depicting the NMR and the Rietveld refinement analysis but the actual source of the discrepancy might arise due to the presence of surface Al cations and the nearby polarized oxygen anions, which creates a high quadrupole coupling constant, which in turn produces very broad NMR signal, thereby increasing a difference in the measurement of the Al3+ ions.51,53
In addition, for revealing the actual potential of the thermal treatment on monitoring the differential rearrangement of hydroxyl ion vis-a-vis identifying its significant effect towards tuning the local atomic structural aspect and ultimate reactivity of the end product, the study has taken an assistance of the 1H MAS NMR study (Fig. 8c). Herein, the spectrum of the γ-AAI powder sample reveals the chemical shift peaks at 0.9, 1.32, 2.18, and 3.62 ppm, which refers to the distinctive chemical environment of the Al ion in association with different hydroxyl species on the surface of γ-alumina.60 In particular, the chemical shift signal present at 2.18 and 0.9 ppm represents the formation of double bridging hydroxyl groups (μ2-OH), whereas the frequency resonance at 1.32 and 3.62 ppm is designated to an isolated hydroxyl groups (μ1-OH).61 The peak at 2.18 ppm is generally classified as more acidic nature, which originates from the combined contribution of OH2Alo and OHAloAlt, where Alo and Alt denote the octahedral and tetrahedral contribution of the aluminum ions, whereas the peak at 1.32 ppm originates due to the contribution of the μ1-Alt OH group. The signal at 3.62 ppm represents the most acidic nature of the γ-alumina structure. The comparative analysis of the 1H NMR spectrum represents the sharp relative intensities at 0.9, 2.18, and 3.62 in the γ-AAI powder, which suggested the highly reactive nature of the particular γ-alumina surface in comparison to the ANE powder,62 which again provides a concrete support to the FTIR, DTA, and XRD results of the respective sol and their corresponding gel structure via experiencing the restricted progressive advancement of the dehydroxylation sequence of the higher organic acid containing poorly crystalline AAI boehmite gel as a function of the incremental thermal treatment. Henceforth, the NMR results reveal the existence of relatively higher intensity of the coordinatively unsaturated 27Al sites along with a sharp indicative association of more acidic 1H sites with AAI-derived γ-alumina samples, giving a clear sense of indication of its relatively higher defective nature on account the large extent of Lewis and Brønsted acidic sites, which is further intended to be confirmed by the FTIR profile of pyridine adsorption.61
 | ||
| Fig. 9 Deconvoluted Al 2p and O 1s XPS spectra of γ-AAI (a and b) and γ-ANE (c and d) powders and Tauc plots of γ-AAI (e) and (f) γ-ANE powders. | ||
3.3 Application of the synthesized γ-AAI and γ-ANE powders
| γ-alumina + hv → e− + h+ (γ-alumina) |
| h+ + H2O/OH− → ˙OH |
| Phenol + h+/˙OH → CO2 + H2O |
3.3.1(a) Effect of phenol concentration and irradiation time. The photodegradation efficiencies of the synthesized γ-AAI and γ-ANE powders were examined by maintaining a fixed dosage of 0.2 g L−1 with variable phenol concentrations of 20, 40, and 80 ppm. The resultant concentration of phenol has been recorded after a fix time span of 2 h with the help of a UV-vis spectrophotometer. Fig. 10(a) and (b) denotes the initial and final concentration ratio of the phenol pollutant initially after the immediate exposure under dark conditions for 2 h, followed by calculating its degradation efficiency up to the time period of 8 h (while maintaining the time span of 2 h) under UV light. Although the results depict that under dark conditions, lower amount of adsorption has been recorded for both the powders but in a closer sense, the calculated adsorption capacity of the γ-AAI powders is slightly greater than that of the γ-ANE powders. The maximum extent of the degradation efficiency at a particular phenol concentration of 20, 40, and 80 ppm recorded after the effective UV light exposure of 8 h was 19.32%, 49.93%, and 20.43% for γ-AAI and 33.81%, 69.77%, and 49.23% for the γ-ANE powder samples, respectively. The results clearly corroborate the increasing trend in the degradation efficiency of both the powders with an increase in the amount of phenol concentration from 20 ppm to 40 ppm but as the concentration of the phenol increased at 80 ppm, the degradation efficiency suddenly decreases. The reason for the above contradictory results might signify the fact that, especially at lower concentration of phenol (20 ppm), the powder concentration in the system is quite high with respect to the targeted degradation species itself, which may be the cause for the significant hindrance of the effective UV-mediated phenol–catalyst interaction.24 As the concentration of the target pollutant, i.e., phenol, increases the number of adsorbed molecules at the surface of the powder readily increases, thereby causing an enhancement in the pollutant degrading reactive species or radicals, which in turn increases the degradation capacity of the respective powders at 40 ppm concentration of the pollutant. However, the presence of active sites and the generation of reactive species in γ-alumina, which is mainly responsible for higher degradation of phenol remains limited for each structure at similar intensity of light, catalyst amount, and duration.
 | ||
| Fig. 10 Effect of phenol concentration on the photocatalytic degradation, (a) γ-AAI and (b) γ-ANE powder at 0.2 g L−1 loading of the catalyst, effect of catalyst loading on the photocatalytic degradation of (c) γ-AAI and (d) γ-ANE powders for 40 ppm of phenol solution, and comparative analysis of photocatalytic degradation (e) for γ-AAI and γ-ANE. | ||
These structural features act as a rate-limiting factor for the system having high phenol concentration, i.e., 80 ppm. Furthermore, the saturation of the catalyst's surface by the coating of comparatively the largest dimensional form of phenol may also work as a limiting factor. It can be seen that the highest photodegradation efficiencies of the phenol for both the powders have been noted at 40 ppm concentration of the phenol. Therefore, 40 ppm concentration has been selected for the further study in order to explain the effect of catalyst loading under fixed concentration of the pollutant.
3.3.1(b) Effect of the catalyst loading and irradiation time. To find out the optimum dosage limit of the photocatalyst, the effect of catalyst loading on phenol degradation was studied by increasing the concentration of γ-AAI and γ-ANE powders from 0.1 g L−1 to 0.6 g L−1 under constant 40 ppm concentration of phenol. As the degradation time increases, likewise, in the previous section, the linear relation of time vs. degradation has been recorded and showed in Fig. 10(c) and (d). It is evident from the figures that as the contact time increases, the photodegradation of the respective pollutant also increases up to 16 h with 68.67% and 97.92% degradation efficiency at 0.2 g L−1 of the catalyst concentration for the γ-AAI and γ-ANE powders, respectively, which signifies the higher photodegradation potential of both the powders. However, the important fact to reveal herein, although the continuous increase in the irradiation time readily increases the phenol degradation efficiency of the γ-AAI and γ-ANE powders but at the same time, the systematic evaluation also interestingly identified a sudden drop in the rate of degradation efficiency immediately after crossing the initial irradiation time of 4 h at all the loading concentrations. This reduction rate may be explained by the effect of generation of intermediate species, which may adsorb on the surface of the catalyst and result in the deactivation of the surface active sites.24,78 Interestingly, the catalyst loading concentration also plays a decisive role while representing the phenol degradation behavior. Herein, with the increase in the catalyst loading concentration up to 0.2 g L−1, the degradation efficiency increases for both the powders but above that, at 0.4 and 0.6 g L−1 catalyst loading concentration, the degradation efficiency significantly shows diminishing tends with no further improvement in the performance. At lower concentration of the photocatalyst (0.1 g L−1), the possible existence of lower concentration of the active site may be the reason for showing lower photodegradation efficiency of both the powder. As the concentration of the photocatalyst increases to 0.2 g L−1, the active sites of the γ-alumina powder also increase, which ultimately leads to increased photodegradation efficiency. The further increase in the photocatalyst dosage leads to a decrease in the photodegradation efficiency of both the powders. The resultant decreasing efficiency may emerge due to the key factors, which include a reduction in the surface area as well as upsurge in non-uniform light scattering effect. The aggregation of the nanoparticle reduces the efficiency of light harvesting by excessive light scattering, which potentially blocks the passage of incoming photons (shadowing effect) and ultimately reduces the illuminated active sites on the surface of the particles.79 The above explanation confirms that the balance should be maintained between these opposing effects to get the optimum loading of the catalyst at the optimum concentration of the pollutant for getting fascinating degradation efficiency while utilizing the decisive fraction of photons.
3.3.2(a) Effect of dye concentrations and contact time. The dye removal efficiency of both the γ-AAI and γ-ANE powders has been studied at a fixed adsorbent concentration of 0.2 g L−1 with increasing initial dye concentration from 20 ppm to 60 ppm. Fig. 11(a) and (b) clearly exhibits rapid dye removal efficiency after initial exposure of 20 min time period but beyond that period, the adsorption rate of the respective powders gradually decreases and almost tends to attain a flatten curve. Initially, the gradual and similar trends of adsorption behavior of γ-AAI powder with respect to the increasing dye concentration clearly indicate that the 120 min of contact time can be considered as optimum equilibrium time for achieving the maximum extent of dye adsorption in the systems. Herein, the smooth, continuous, and saturation leading curve profiles suggest the monolayer coverage of Congo red on the surface of the sol–gel synthesized γ-alumina particles during the dye removal process.80 The dye removal capacity of the respective γ-alumina particle was noted as 50.00 mg g−1, 92.70 mg g−1, and 133.36 mg g−1 for γ-AAI powder and 24.80 mg g−1, 43.96 mg g−1, and 23.63 mg g−1 for γ-ANE powder under the dye concentration of 20, 40, and 60 ppm, respectively, at a fixed adsorbent concentration of 0.2 g L−1. The data of the respective powders clearly denote that the dye removal capacity of the respective powders also increases with increasing concentration of the dye. This increased capacity may be ascribed to the adsorption of higher extent of the dye molecules at the respective adsorbent's surface, which might arise due to the employment of weak electrostatic attraction forces at the specific active sites. Herein, the decreasing adsorption efficiency of the γ-ANE powder with respect to increased dye concentration, particularly at 60 ppm, can be related to the presence of relatively higher extent of dye molecules at the vicinity of the invariable surface active sites of the γ-alumina powder particles, which would hinder the availability of surface hydroxyl groups of the adsorbents.81 It is quite clear from the figures that the proficient combination of adsorption efficiency and the capacity of Congo red dye can be achieved at 40 ppm dye concentration under constant adsorbent loading of 0.2 ppm. Therefore, in the next section, this dye concentration has been chosen for further evaluating the influence of the adsorbent loading for controlling both the adsorption efficiency and capacity of the respective γ-alumina powders.
 | ||
| Fig. 11 Effect of dye concentration on the relative adsorption capacity of (a) γ-AAI and (b) γ-ANE powders at 0.2 g L−1 loading of the adsorbent, effect of adsorbent loading on the adsorption capacity of (c) γ-AAI and (d) γ-ANE powders for 40 ppm of dye solution, and comparative analysis of adsorption capacity (e) for γ-AAI and γ-ANE. | ||
3.3.2(b) Effect of adsorbent loading and contact time. The effect of variable dosages of γ-AAI and γ-ANE powder adsorbents on the removal extent of Congo red dye has been shown in Fig. 11(c) and (d). The experiments were carried out using the fixed dye concentration of 40 ppm along with variable adsorbent dosages from 0.1 g L−1 to 0.6 g L−1. The result clearly shows that the adsorbent efficiency of the gamma powders increases from 13.49% to 100% and from 5.13% to 52.04% for γ-AAI and γ-ANE powder, respectively, with respect to increasing adsorbent doses, whereas the viable equilibrium time for γ-AAI also tends to reduce. Thus, this closer insight for both the γ-alumina powders ensures a wide variance in their adsorption behavior under the identical experimental conditions. In this regard, both the samples show higher adsorption efficiency at 0.6 g L−1 but the detailed investigation demonstrates 100% removal efficiency of Congo red dye only at the surface of the γ-AAI powder specifically after maintaining 80 min of minimum exposure time. On the contrary, γ-ANE does not reach 100% removal efficiency even after attaining the extreme conditions of the present experimental design for the adsorption study. Furthermore, the comparative analysis of the γ-AAI and γ-ANE powder (Fig. 11(c and d)) shows that the maximum adsorption capacity of 108.77 mg g−1 and 86.18 mg g−1 was noted for the γ-AAI and γ-ANE powder, respectively, under 20 ppm dye concentration with 0.1 g L−1 adsorbent loading at 160 min time. The effective adsorbent capacity as well as the efficiency of γ-AAI powder may be described in terms of the existence of a wide pore size distribution range along with a larger number of surface active sites, which indeed increase the adsorption as well as diffusion of dye molecules in the widely open morphology of the respective γ-alumina particles, whereas limited number of available surface active sites with the narrow pore size distribution range of γ-ANE may act as the rate limiting steps of pore diffusion, which would result in limited adsorption properties.82
3.4 Detail mechanistic view of the photocatalytic and dye removal application directive perspective of distinctive γ-alumina powders
Thus, the present study successfully demonstrated a clear sense of variability in the gamma-alumina structure, which originates from the differential nature of the basic peptized boehmite particles while emphasizing the major difference in dictating the application perspective of two differently synthesized sol–gel processed γ-alumina powders. The comparative analysis of γ-AAI and γ-ANE powders reveals the higher degradation capacity of phenol on the surface of the γ-ANE powder but on the other hand, the γ-AAI powder shows better dye adsorption properties. Herein, it is clear from the above study that two types of waste water treatment application, namely, photocatalytic degradation and dye adsorption, noticeably improve the actual reliability of the various structural aspects of the selected materials, which actively participated especially towards controlling the removal efficiency of both the pollutants but certainly in a different way. While emphasizing these typical influential features such as defect chemistry, surface area, agglomeration tendency, band gap, and Lewis and Brønsted acid sites are considered as key characteristic features of the materials, which directly come in the frontline, specifically for governing the mechanistic study of effluent treatment.In the above context, it is worth noting that though under dark condition, the phenol adsorption capacity of the γ-AAI powders is relatively higher than that of γ-ANE powder (Fig. 11e), especially owing to the presence of higher hydroxyl groups at the γ-AAI surface (confirmed by 1H NMR), which eventually decreases the active sites and consequently reduces the free radicals on the surface, the effect of which is conspicuously reflected while explaining the results of the photodegradation study, wherein it represents completely differential statistics after the immediate exposure of the system under UV condition.83 In general, the intrinsic semiconductor properties are reported to have the most significant influence on the photocatalytic properties of the materials. Herein, the band gaps of γ-alumina act as a driving force for the generation of the OH radical and therefore act as an immediate parameter for identifying the photocatalytic properties. Notably, in most of the cases, the band gap of γ-alumina has been estimated to be 7.0–8.7 eV but the study shows relatively inferior recorded band gap for the γ-ANE powder, which is 3.72 eV and 4.54 eV, whereas for the γ-AAI powder, it is 3.74 eV and 4.67 eV, as calculated from Tauc plots. The γ-ANE powder exhibits comparatively lower band gap values than that of γ-ANE powder due to relatively higher and controlled defective nature of the respective γ-alumina phases as confirmed by NMR, Rietveld refinement, and XPS study.84 Therefore, the comparatively lower band gap of the γ-ANE powder may extend the light adsorption by helping in the excitation of the electron from the valence band to the conduction band at the UV spectrum range, thereby possessing higher degradation capacity.85 Moreover, certain additional factors, which also extend great assistance for the enhancement of the phenol degradation efficiency, are mainly concentrated herein at their appropriate surface texture position. In this regard, on a key note, the higher aggregation/agglomeration tendency of the γ-AAI particle, as seen in the TEM micrographs, lends a stimulated shadowing effect, which causes a substantial decrease in the degradation efficiency of this particles, whereas, there are no such visual effects of agglomeration that have been noticed at γ-ANE powders, which might significantly contribute to the effective utilization of the decisive fraction of photons on the surface.24 In addition, the relatively larger surface area with good light adsorption properties of the γ-ANE powders might also contribute towards increasing the degradation capacity.
On the other hand, from Fig. 11(e), it is quite clear that the adsorption capacity and its corresponding efficiency of the γ-AAI powder is relatively higher than that of the γ-ANE powder. The high dye adsorption capacity of the γ-AAI powder may be explained by electrostatic interaction and surface complexation due to hydrogen bonding characteristics between the surface hydroxyl groups of the oxide powder and the terminal amine groups of Congo red dye. As shown in the 1H NMR study, the qualitative amount of surface hydroxyl groups in γ-AAI powder is relatively higher than that of the γ-ANE powder. Therefore, the large number of structural hydroxyl groups of γ-AAI powder show strong affinity with the polar molecules of Congo red dye, which may promisingly promote both the adsorption capacity and efficiency of the oxide powders. In addition, the coordinatively unsaturated Al3+ ion, which are mainly termed as Lewis acid centers along with the oxygen center of Congo red dye in the γ-alumina structure (as acidic centers) can readily form bonding.79 Henceforth, the combined contributions of higher amount of Brønsted and Lewis acid sites (observed from Rietveld analysis, 27Al–1H NMR, and pyridine-FTIR spectral analysis) in the presence of relatively highly defective and porous structural feature observed at γ-AAI powder have shown synergistic influence, particularly towards enhancing the adsorption performance of the material itself. Interestingly, the highly compacted (low pore volume) sheet-like layered morphology may cause the unilateral adsorption restrictions and/or thereby play the rate limiting role of pore diffusion in γ-ANE powder. The combination of the above factors forces the γ-ANE powder for showing low adsorption capacity in spite of with the high surface area of 223.925 m2 g−1 and uniform particle size. Furthermore, as shown in the BET and BJH results, the increased pore size and higher pore volume in combination with viable surface area might also promote the adsorption of Congo red dye via stronger hydrogen bonding and coordination effect in the γ-AAI powder.25,29
3.5 Comparative study
The comparative study of photocatalyst degradation and dye adsorption behavior of the synthesized γ-alumina observed from the present investigation (Table 2) will give further insight in the field of materials science and engineering. Importantly, there are numerous literatures available that have highlighted the potential aspect of structurally-tuned sol–gel derived γ-alumina, specifically on controlling the high-end adsorbent and photocatalytic application behavior.| Material, synthetic route and effective parameter | Organic pollutant | Optimum pollutant concentration | Optimum sample dosage | Outcome | Ref. | Structure–property-application relationship |
|---|---|---|---|---|---|---|
| Hydroxylated (highly amorphous) sol–gel Al2O3 calcined 400, 500, 600, and 700 °C for 12 h | Phenol | 80 ppm | 3.0 g L−1 | 64% photocatalytic degradation under UV-vis for 6 h | 21 | The structural defects, i.e., aluminum point defects as well as hydroxyl groups are considered a major factor to induce the scavenging of the electron–hole pair by the adsorbed molecular oxygen and thereby considered as a major factor for photocatalytic process initiators |
| Sol–gel processed Al2O3 precursor calcined at 400 °C for 12 h | Phenol | 40 ppm | 0.8 g L−1 | ∼78% photocatalytic degradation under UV-vis for 6 h | 22 | The surface actives site of the synthesized alumina (capturing of electron) specifically help towards regulating its optical as well as semiconducting properties and assist in improving the photocatalytic decomposition of targeted phenol compounds |
| Template assisted γ-alumina calcined at 600 °C for 6 h | Phenol | 100 ppm | 1 g L−1 | 37% phenol degradation for 2 h in the presence of H2O2 and UV light; no further decomposition after that | 12 | The highly defective mesoporous γ-alumina structure with relatively high surface area, appropriate pore morphology, and tunable formation of hydroxyl radical at its defective sites helps in controlling the photocatalytic degradation of phenol |
| Commercial; γ-alumina (procured from Versal) | Phenol | 40 ppm | 0.8 g L−1 | 60% phenol degradation after 180 min of reaction | 67 | The active surface sites, i.e., specific tetrahedral and octahedral coordination of Al ions of crystalline γ-alumina structure, help in promoting the degradation capacity of the organic effluent phenol |
| γ-ANE | Phenol | 40 ppm | 0.2 g L −1 | 99.42% photocatalytic degradation under UV-vis for 16 h | This work | The moderate Lewis acid strength with high surface area, specific crystallographic orientation, and viable band gap lead to the generation of synergic influence on the control of the phenol degradation efficiency |
| γ-Al2O3 calcined at 600 °C for 4 h | Congo red dye | 100 ppm | 200 and 400 mg L−1 | 100.0 mg g−1 adsorption capacity | 68 | The synergistic effect of pore morphology and the quantity of surface active sites play a decisive role for the surface adsorption phenomenon of γ-alumina structure, which is considered to be associated with the electrostatic attraction and surface complexation ability of surface hydroxyl groups and terminal amine groups of Congo red dye |
| γ-Al2O3 400 °C for 4 h | Congo red dye | 6.25–100 ppm | 1 g L−1 | 99.56 mg g−1 adsorption capacity | 23 | The high surface area and highly open mesoporous morphology eventually shows good control towards governing the adsorption capacity of the organic effluent, i.e., Congo red dye |
| Hierarchical spindle-like γ-alumina calcined at 550 °C for 4 h | Congo red dye | 45–720 ppm | 0.8 g L−1 | 176.70 mg g−1 adsorption capacity | 26 | The tunable porous structure as well as adjustable chemical properties and characteristic active site of γ-alumina show a dominating influence towards controlling the rate of adsorption of Congo red dye molecules |
| γ-AAI | Congo red dye | 40 ppm | 0.2 g L −1 | 133.36 mg g −1 adsorption capacity | This work | The higher Lewis as well as Brønsted acid strength with high hydroxyl concentration, relatively defective atomic configurations, open pore structure, elevated surface area, and unique granular surface morphological feature provide assistance in attaining good adsorption capacity |
Specifically, in spite of having convenient thermal stability, high surface area, and well regulated acid–base properties of the γ-alumina structure, it is still demonstrated as the least studied material as the photocatalyst due to its highly insulating nature. However, the electronic properties of γ-alumina are well-governed by precisely controlling the process parameters in the sol–gel based synthetic route. In this regard, F. Tzompantzi has studied the feasibility of sol–gel synthesized hydroxylated Al2O3 as the photocatalyst for the degradation of phenol with a band gap of 1.99 eV, which shows high photodegradation efficiency by exhibiting full degradation of 80 ppm phenol in 7 h with 3 g L−1 catalyst concentration.21 The authors prove that the high concentrations of hydroxyl groups with highly defective γ-alumina structure are responsible for this high photoactivity. Furthermore, the comparative study of various doped and undoped γ-alumina strived by Piña-Pérez et al. approves the synergic impact of surface active sites of the sol–gel synthesized alumina calcined at 400 °C for 12 h towards altering its optical as well as semiconducting properties wherein the presence of 0.8 g L−1 of pure γ-alumina catalyst assist in optimistically regulating the photocatalytic degradation of 40 ppm phenol to ∼78% after effective exposure under UV light for 6 h.22 In addition, Parida et al. conducted a photocatalytic degradation study of 100 ppm phenol, utilizing 1 g L−1 of template-assisted synthesized γ-alumina powder calcined at 600 °C for 6 h, wherein the utilization of hydrogen peroxide extends auxiliary support for the effective tuning of its surface defective sites via the specialized formation of hydroxyl radicals, thereby inducing a robust support for the degradation of ∼37% phenol after 2 h exposure under UV light.12 Another attempt made by Piña-Pérez et al. focuses on explaining the photocatalytic degradation study of 40 ppm phenol with 0.8 g L−1 of commercial γ-alumina powder, which again largely validate the decisive influence of tetrahedral and octahedral coordination of Al ions, especially towards altering the photocatalytic degradation efficiency of phenol, which eventually arrested its value at 60% after an effective exposure under UV light for the duration of 180 min.86 In our study, glycerol-modified sol–gel derived γ-ANE powder possessing relatively less defective structural configuration has shown moderate Lewis acid strength with high specific surface area and viable band gap, which lead to synergic phenol degradation efficiency of 99.42% for 40 ppm of phenol with the lowest catalytic amount of 0.2 g L−1 under UV light.
On the other hand, while considering the dye adsorption study, very few reported literatures are available, which mainly give preference to reveal the systematic influence of the sol–gel based γ-alumina adsorbent specifically without involving any surfactant usage. In this context, the investigation of Cai et al. shows the synergistic influence of pore morphology and the quantity of surface active sites of γ-alumina calcined at 600 °C for 4 h towards monitoring the adsorption efficiency of Congo red dye, wherein the effective exposure of 100 ppm dye in contact with variable adsorbent dosages of 200 and 400 mg L−1 shows an optimized Congo red dye adsorption of 100 mg g−1, thereby verifying its significance towards altering the electrostatic attraction and surface complexation ability of the surface hydroxyl groups and terminal amine groups of Congo red dye.27 S. Ghosh et al. utilized nanofiber-like mesoporous γ-alumina synthesized with triblock copolymer (P123) using evaporation-induced self-assembly (EISA) process that after calcination at 400 °C for 4 h achieved a surface area of 175.5 m2 g−1 and wherein the contact of 1 g L−1 of adsorbent with variable dye concentration of 100 ppm shows maximum Congo red dye adsorption capacity of 99.56 mg g−1, thereby favoring the effectiveness of the surface area and open mesoporous morphology as an effective tool towards governing the adsorption capacity of the organic effluent, i.e., Congo red dye.23 In addition, W. Cai et al. also utilized the hierarchical spindle-like γ-alumina synthesized by the hydrothermal route under highly basic environment, which reveal surface area and average pore volume of 149 m2 g−1 and 0.36 cm3 g−1, respectively.82 Herein, the effective utilization of 0.8 g L−1 of adsorbent in contact with variable dye content from 45 to 720 ppm shows maximum adsorption capacity of 176.7 mg g−1. The synthesized hierarchical spindle-like nanosized γ-alumina assemblies with tunable porous structure as well as adjustable chemical properties and characteristics active site shows dominating influence towards controlling the rate of adsorption of the Congo red dye molecules. In this study, sol–gel derived γ-alumina (γ-AAI powder) synthesized under acidic environment possesses high Lewis acid and Brønsted acid strength, high hydroxyl concentration with a highly open pore structure, and granular morphology, which provides assistance towards obtaining good adsorption capacity. Hence, it is clearly evidenced that not only the surface area but also the combined contribution of surface hydroxyl groups, crystallographic configuration, pore volume, chemical properties, and morphology as a whole play synergetic effect towards governing the adsorption characteristics of Congo red dye.
Therefore, the comparative study will significantly give a more reliable and augmented realistic view of our results, which extends a basic but solid robust ground for validating the unique essence of the structure–property relationship of the defective γ-alumina structure towards dictating an ideological awareness of its application perspective. Herein, the unique process of two surfactant-free glycerol-activated distinctive sol–gel derived γ-alumina, which have already proven their strong candidature in gas separation membrane, provides further promising advancement in the water treatment application field wherein the true potential of the wide variance in the structural, morphological, and designing individualities are noticeably revealed.19 A detailed view of the differential aspect of synthetic parameters and the consequent enlightenment of its wide range of structural variability show promising impact, especially for the flexible engineering of its designing aspect, thereby providing a better present and future application perspective in the waste effluent treatment platform.
4. Conclusions
In summary, the present investigation mainly involves the synthesis of two distinctive surfactant-free glycerol-activated dispersion medium and peptizing agent-mediated sol–gel derived γ-alumina especially with an aim to reveal the conclusive interference of the structure–property relationship towards stimulating and meticulously recognizing its indigenous application-specific perspective. The changes appearing at the distinctive structural position of the two sol–gel mediated γ-alumina precursor sol maintains its individuality during the accompanying thermal treatment, which indeed noticeably exerts promising influence on the final properties of both the oxide phases. Herein, the utilization of higher dosages of organic acetic acid in combination with high viscous solvent resulted in higher concentration of hydroxyl group and lower crystallinity in boehmite sol and its corresponding gel structure, which dictated high density of the pore amount, enhanced hydroxyl group, along with the co-existence of a notable persistence of Lewis and Brønsted acid sites, which imposes higher tendency for the adsorption of Congo red dye at the γ-AAI structure. On the other hand, γ-alumina synthesized using nitric acid peptizing agent and low viscous solvent medium exhibits relatively lower hydroxyl group concentration and higher crystallinity in the associated sol and its corresponding boehmite gel structure, which eventually attained an elevated one dimensional crystalline growth (platy morphology) in combination with a relatively higher surface area, lower band gap, lesser amount of surface acidic sites, and noticeably lower extent of agglomeration tendency, thereby causing relatively higher photoactivity on behalf of the decomposition of phenol. Importantly, the comparative application prospective of our synthesized γ-alumina structure with respect to other literature data approves the effectiveness of the presently synthesized γ-alumina, wherein the nominal extent of the material usage shows significant influence on both the opposite but substantial waste water effluent treatment applications such as the adsorbent and photocatalyst. Finally, the present findings illustrated a detailed mechanistic view that promisingly provided nascent insights for deliberately controlling the structure–property-application relationship aspect of the γ-alumina structure in relation to the synthesis patterns, which can be useful for controlling the present and future specific concern of the current as well as other application perspectives.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Council of Scientific & Industrial Research (CSIR), (file no. 08/709(0002)2K19 EMR-I) and Science and Engineering Research Board (SERB-DST) (file no. EEQ/2016/000762/2017-2018 and EEQ/2020/000218), Government of India for financial support. We are also thankful to the Government College of Engineering and Ceramic Technology, Kolkata, for providing the infrastructural and various testing facilities.References
- S. Said, S. Mikhail and M. Riad, Mater. Sci. Energy Technol., 2020, 3, 344–363 CAS.
- M. Trueba and S. Trasatti, Eur. J. Inorg. Chem., 2005, 2005, 3393–3403 CrossRef.
- G. I. N. Waterhouse, W.-T. Chen, A. Chan, H. Jin, D. Sun-Waterhouse and B. C. C. Cowie, J. Phys. Chem. C, 2015, 119, 6647–6659 CrossRef CAS.
- S. Lamouri, M. Hamidouche, N. Bouaouadja, H. Belhouchet, V. Garnier, G. Fantozzi and J. F. Trelkat, Bol. Soc. Esp. Ceram. Vidrio, 2017, 56, 47–54 CrossRef CAS.
- M. Acikgoz, J. Harrell and M. Pavanello, J. Phys. Chem. C, 2018, 122, 25314–25330 CrossRef CAS.
- M. H. Lee, C.-F. Cheng, V. Heine and J. Klinowski, Chem. Phys. Lett., 1997, 265, 673–676 CrossRef CAS.
- J. Gu, J. Wang and J. Leszczynski, ACS Omega, 2018, 3, 1881–1888 CrossRef CAS PubMed.
- R. Feng, S. Liu, P. Bai, K. Qiao, Y. Wang, H. A. Al-Megren, M. J. Rood and Z. Yan, J. Phys. Chem. C, 2014, 118, 6226–6234 CrossRef CAS.
- J. B. Peri, J. Phys. Chem., 1965, 69, 220–230 CrossRef CAS.
- C.-K. Lee, E. Cho, H.-S. Lee, K. Seol and S. Han, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 245110 CrossRef.
- T. M. Moghadam, P. Alizadeh, M. Ghamari and M. Mousavi, Int. Nano Lett., 2021, 11, 141–147 CrossRef CAS.
- K. M. Parida, A. C. Pradhan, J. Das and N. Sahu, Mater. Chem. Phys., 2009, 113, 244–248 CrossRef CAS.
- P. S. Behera, R. Sarkar and S. Bhattacharyya, Interceram, 2016, 65, 10–16 CAS.
- L. Samain, A. Jaworski, M. Edén, D. M. Ladd, D.-K. Seo, F. Javier Garcia-Garcia and U. Häussermann, J. Solid State Chem., 2014, 217, 1–8 CrossRef CAS.
- M. Nguefack, A. Popa, S. Rossignol and C. Kappenstein, Phys. Chem. Chem. Phys., 2003, 5, 4279–4289 RSC.
- S. Dervin and S. Pillai, in An Introduction to Sol-Gel Processing for Aerogels, ed. S. Pillai and S. Hehir, Springer, Cambridge, 2017, ch. 1, vol. 6, pp. 1–12 Search PubMed.
- B. E. Yoldas, J. Mater. Sci., 1975, 10, 1856–1860 CrossRef CAS.
- X. Chen, W. Zhang, Y. Lin, Y. Cai, M. Qiu and Y. Fan, Microporous Mesoporous Mater., 2015, 214, 195–203 CrossRef CAS.
- S. K. Sharma, B. K. Sanfui, A. Katare and B. Mandal, ACS Appl. Mater. Interfaces, 2020, 12, 40269–40284 CrossRef CAS PubMed.
- M. Fuji, M. T. Takashi Shirai and H. Watanabe, Mater. Sci., 2009, 9, 23–31 Search PubMed.
- F. Tzompantzi, Y. Piña, A. Mantilla, O. Aguilar-Martínez, F. Galindo-Hernández, X. Bokhimi and A. Barrera, Catal. Today, 2014, 220–222, 49–55 CrossRef CAS.
- Y. Piña-Pérez, F. Tzompantzi-Morales, R. Pérez-Hernández, R. Arroyo-Murillo, P. Acevedo-Peña and R. Gómez-Romero, Fuel, 2017, 198, 11–21 CrossRef.
- S. Ghosh, K. P. Dey and M. K. Naskar, J. Am. Ceram. Soc., 2013, 96, 28–31 CrossRef CAS.
- H. Ling, K. Kim, Z. Liu, J. Shi, X. Zhu and J. Huang, Catal. Today, 2015, 258, 96–102 CrossRef CAS.
- S. I. Raj, A. Jaiswal and I. Uddin, RSC Adv., 2019, 9, 11212–11219 RSC.
- M. N. Zafar, Q. Dar, F. Nawaz, M. N. Zafar, M. Iqbal and M. F. Nazar, J. Mater. Res. Technol., 2019, 8, 713–725 CrossRef CAS.
- W. Cai, Y. Hu, J. Chen, G. Zhang and T. Xia, CrystEngComm, 2012, 14, 972–977 RSC.
- S. K. Lahiri and L. Liu, Langmuir, 2021, 37, 8753–8764 CrossRef CAS PubMed.
- S. Banerjee, T. K. Paira, A. Kotal and T. K. Mandal, Adv. Funct. Mater., 2012, 22, 4751–4762 CrossRef CAS.
- Z. Wang, G. M. Kale, Q. Yuan and M. Ghadiri, RSC Adv., 2012, 2, 9993–9997 RSC.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- G. Krishna Priya, P. Padmaja, K. G. K. Warrier, A. D. Damodaran and G. Aruldhas, J. Mater. Sci. Lett., 1997, 16, 1584–1587 CrossRef.
- Y. Zhu, Z. Jiang, L. Zhang, J. Shi and D. Yang, Ind. Eng. Chem. Res., 2012, 51, 255–261 CrossRef CAS.
- M. Mahinroosta and A. Allahverdi, Int. Nano Lett., 2018, 8, 255–261 CrossRef CAS.
- M. Jafar Tafreshi and Z. Masoomi, Mater. Sci., 2015, 21, 28–31 CrossRef.
- H. Barzegar-Bafrooei and T. Ebadzadeh, Adv. Powder Technol., 2011, 22, 366–369 CrossRef CAS.
- V. Franceti and P. Bukovec, Acta Chim. Slov., 2008, 55, 904–908 Search PubMed.
- F. Hosseinzadeh and H. Sarpoolaky, J. Inorg. Organomet. Polym. Mater., 2019, 29, 1956–1971 CrossRef CAS.
- M. S. Ghamsari, Z. A. S. Mahzar, S. Radiman, A. M. A. Hamid and S. R. Khalilabad, Mater. Lett., 2012, 72, 32–35 CrossRef CAS.
- R. Prins, J. Catal., 2020, 392, 336–346 CrossRef CAS.
- J. Gangwar, B. K. Gupta, S. K. Tripathi and A. K. Srivastava, Nanoscale, 2015, 7, 13313–13344 RSC.
- R. Wischert, C. Copéret, F. Delbecq and P. Sautet, Chem. Commun., 2011, 47, 4890–4892 RSC.
- F. M. D. Segal, M. F. Correa, R. Bacani, B. Castanheira, M. J. Politi, S. Brochsztain and E. R. Triboni, Mater. Res., 2018, 21, 1–8 Search PubMed.
- X. Zhang, M. Honkanen, E. Levänen and T. Mäntylä, J. Cryst. Growth, 2008, 310, 3674–3679 CrossRef CAS.
- V. Joshi, S. Dwivedi and G. H. Ward, Pharm. Res., 2002, 19, 7–12 CrossRef CAS PubMed.
- P. P. Nampi, S. Ghosh and K. G. Warrier, Ceram. Int., 2011, 37, 3329–3334 CrossRef CAS.
- R. Bouza, C. Marco, M. Naffakh, L. Barral and G. Ellis, Composites, Part A, 2011, 42, 935–949 CrossRef.
- T. Tsuchida, R. Furuichi and T. Ishii, Thermochim. Acta, 1980, 39, 103–115 CrossRef CAS.
- V. Jayaram and C. G. Levi, Acta Metall., 1989, 37, 569–578 CrossRef CAS.
- C. Jagadeeshwaran, K. Madhan and R. Murugaraj, J. Mater. Sci.: Mater. Electron., 2018, 29, 18923–18934 CrossRef CAS.
- R.-S. Zhou and R. L. Snyder, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 617–630 CrossRef.
- H. O. Ayoola, S. D. House, C. S. Bonifacio, K. Kisslinger, W. A. Saidi and J. C. Yang, Acta Mater., 2020, 182, 257–266 CrossRef CAS.
- R. Prins, Angew. Chem., Int. Ed., 2019, 58, 15548–15552 CrossRef CAS PubMed.
- M. Urbonavicius, Š. Varnagiris, L. Pranevicius and D. Milcius, Materials, 2020, 13, 1300 CrossRef CAS PubMed.
- G. Castruita, Y. A. Perera-Mercado and E. M. Saucedo-Salazar, J. Inorg. Organomet. Polym. Mater., 2013, 23, 1145–1152 CrossRef CAS.
- K. Frikha, L. Limousy, J. Bouaziz, K. Chaari and S. Bennici, Materials, 2020, 13, 1–15 CrossRef PubMed.
- X. Zhang, M. Honkanen, Y. Ge, S.-P. Hannula, E. Levänen and T. Mäntylä, Nanocrystalline γ-alumina and α-alumina from boehmite nanoflakes, 2008 Search PubMed.
- P. Kaur, A. Khanna, N. Kaur, P. Nayar and B. Chen, Phase Transitions, 2020, 93, 596–605 CrossRef CAS.
- L. Zeng, D. T. Tran, C.-W. Tai, G. Svensson and E. Olsson, Sci. Rep., 2016, 6, 29679 CrossRef CAS PubMed.
- L. Shen, Y. Wang, J.-H. Du, K. Chen, Z. Lin, Y. Wen, I. Hung, Z. Gan and L. Peng, ChemCatChem, 2020, 12, 1569–1574 CrossRef CAS PubMed.
- N. Huittinen, P. Sarv and J. Lehto, J. Colloid Interface Sci., 2011, 361, 252–258 CrossRef CAS PubMed.
- E. C. Decanio, J. C. Edwards and J. W. Bruno, J. Catal., 1994, 148, 76–83 CrossRef CAS.
- P. Bai, M. Xie, U. J. Etim, W. Xing, P. Wu, Y. Zhang, B. Liu, Y. Wang, K. Qiao and Z. Yan, Ind. Eng. Chem. Res., 2018, 57, 1389–1398 CrossRef CAS.
- X. Liu, J. Phys. Chem. C, 2008, 112, 5066–5073 CrossRef CAS.
- E. M. Moroz, D. A. Zyuzin, V. Y. Tregubenko, I. E. Udras, A. S. Belyi and V. A. Likholobov, React. Kinet., Mech. Catal., 2013, 110, 459–470 CrossRef CAS.
- N. M. Figueiredo, N. J. M. Carvalho and A. Cavaleiro, Appl. Surf. Sci., 2011, 257, 5793–5798 CrossRef CAS.
- A. Malki, Z. Mekhalif, S. Detriche, G. Fonder, A. Boumaza and A. Djelloul, J. Solid State Chem., 2014, 215, 8–15 CrossRef CAS.
- X. Zhang, P. L. Huestis, C. I. Pearce, J. Z. Hu, K. Page, L. M. Anovitz, A. B. Aleksandrov, M. P. Prange, S. Kerisit, M. E. Bowden, W. Cui, Z. Wang, N. R. Jaegers, T. R. Graham, M. Dembowski, H.-W. Wang, J. Liu, A. T. N'Diaye, M. Bleuel, D. F. R. Mildner, T. M. Orlando, G. A. Kimmel, J. A. La Verne, S. B. Clark and K. M. Rosso, ACS Appl. Nano Mater., 2018, 1, 7115–7128 CrossRef CAS.
- L. Gao, Y. Li, Q. Li, Z. Song and F. Ma, Nanotechnology, 2017, 28, 215201 CrossRef PubMed.
- S. J. Wilson and M. H. Stacey, J. Colloid Interface Sci., 1981, 82, 507–517 CrossRef CAS.
- D. Maarisetty and S. S. Baral, J. Mater. Chem. A, 2020, 8, 18560–18604 RSC.
- Y. Zhu, S. Lin, W. Gao, M. Zhang, C. Yang, P. Feng, C. Xu and Z. L. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 35795–35803 CrossRef CAS PubMed.
- D. Maarisetty, S. Mahanta, A. K. Sahoo, P. Mohapatra and S. S. Baral, ACS Appl. Mater. Interfaces, 2020, 12, 11679–11692 CrossRef CAS PubMed.
- X. Qian, B. Li, H.-Y. Mu, J. Ren, Y. Liu, Y.-J. Hao and F.-T. Li, Inorg. Chem. Front., 2017, 4, 1832–1840 RSC.
- B. Ealet, M. H. Elyakhloufi, E. Gillet and M. Ricci, Thin Solid Films, 1994, 250, 92–100 CrossRef CAS.
- B. D. Evans and M. Stapelbroek, Phys. Rev. B: Condens. Matter Mater. Phys., 1978, 18, 7089–7098 CrossRef CAS.
- A. F. Lotus, R. K. Feaver, L. A. Britton, E. T. Bender, D. A. Perhay, N. Stojilovic, R. D. Ramsier and G. G. Chase, Mater. Sci. Eng., B, 2010, 167, 55–59 CrossRef CAS.
- A. Sobczyński, Ł. Duczmal and W. Zmudziński, J. Mol. Catal. A: Chem., 2004, 213, 225–230 CrossRef.
- M. M. Khan, S. A. Ansari, D. Pradhan, M. O. Ansari, D. H. Han, J. Lee and M. H. Cho, J. Mater. Chem. A, 2014, 2, 637–644 RSC.
- D. Maiti, S. Mukhopadhyay and P. S. Devi, ACS Sustainable Chem. Eng., 2017, 5, 11255–11267 CrossRef CAS.
- S. MiarAlipour, D. Friedmann, J. Scott and R. Amal, J. Hazard. Mater., 2018, 341, 404–423 CrossRef PubMed.
- W. Cai, J. Yu and M. Jaroniec, J. Mater. Chem., 2010, 20, 4587–4594 RSC.
- D. Maarisetty, J. Komandur, S. Sharma, S. S. Baral and P. Mohapatra, J. Environ. Chem. Eng., 2020, 8, 103938 CrossRef CAS.
- E. Filatova and A. S. Konashuk, J. Phys. Chem. C, 2015, 119, 20755–20761 CrossRef CAS.
- A. Nickheslat, M. M. Amin, H. Izanloo, A. Fatehizadeh and S. M. Mousavi, J. Environ. Public Health, 2013, 2013, 815310 Search PubMed.
- Y. Piña-Pérez, O. Aguilar-Martínez, S. Oros-Ruíz, R. Gómez and F. Tzompantzi, J. Photochem. Photobiol., A, 2018, 353, 409–415 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1me00102g |
| ‡ Currently working at Refractory and Traditional Ceramics Division, CSIR-CG&CRI, Jadavpur, Kolkata-700032, West Bengal, India. |
| This journal is © The Royal Society of Chemistry 2022 |