
Heterogeneous protein co-assemblies with tunable functional domain stoichiometry†
Shaheen A.
Farhadi
,
Antonietta
Restuccia
,
Anthony
Sorrentino
,
Andrés
Cruz-Sánchez
and
Gregory A.
Hudalla
 *
*
J. Crayton Pruitt Family Department of Biomedical Engineering, University of Florida, Biomedical Sciences J293, PO BOX 116131, 1275 Center Drive, Gainesville, FL 32611, USA. E-mail: ghudalla@bme.ufl.edu; Tel: +1 (352) 273 9326
First published on 24th September 2021
Abstract
In nature, the precise heterogeneous co-assembly of different protein domains gives rise to supramolecular machines that perform complex functions through the co-integrated activity of the individual protein subunits. A synthetic approach capable of mimicking this process would afford access to supramolecular machines with new or improved functional capabilities. Here we show that the distinct peptide strands of a heterotrimeric α-helical coiled-coil (i.e., peptides “A”, “B”, and “C”) can be used as fusion tags for heterogeneous co-assembly of proteins into supramolecular structures with tunable subunit stoichiometry. In particular, we demonstrate that recombinant fusion of A with NanoLuc luciferase (NL-A), B with superfolder green fluorescent protein (sfGFP-B), and C with mRuby (mRuby-C) enables formation of ternary complexes capable of simultaneously emitting blue, green, and red light via sequential bioluminescence and fluorescence resonance energy transfer (BRET/FRET). Fusion of galectin-3 onto the C-terminus of NL-A, sfGFP-B, and mRuby-C endows the ternary complexes with lactose-binding affinity that can be tuned by varying the number of galectin-3 domains integrated into the complex from one to three, while maintaining BRET/FRET function. The modular nature of the fusion protein design, the precise control of domain stoichiometry, and the multiplicity afforded by the three-stranded coiled-coil scaffold provides access to a greater range of subunit combinations than what is possible with heterodimeric coiled-coils used previously. We envision that access to this expanded range of co-integrated protein domain diversity will be advantageous for future development of designer supramolecular machines for therapeutic, diagnostic, and biotechnology applications.
Design, System, ApplicationNon-covalent co-association of different proteins can produce supramolecular constructs capable of performing complex functions through the concerted activity of the individual components. Fusing proteins to peptides that form specific supramolecular structures, such as an α-helical coiled-coil, is a widely used synthetic approach for non-covalent protein assembly. However, typical systems only enable incorporation of one or two different proteins. Increasing the number of co-integrated protein types, while maintaining control of their proportions, would enable design of constructs with greater diversity of functional complexity. Here, we demonstrate an approach to create constructs with emergent and synergistic functionality via fusion of proteins to each of three distinct peptide strands that form a heterotrimeric α-helical coiled-coil. Constructs that simultaneously emit blue, green, and red light in response to a chemical stimulus were created via co-assembly of a luciferase enzyme and a pair of fluorescent proteins, highlighting the potential to develop modular diagnostics with this approach. These constructs were also endowed with tunable carbohydrate-binding affinity by co-integrating different numbers of galectin-3 domains, demonstrating the potential to develop targeted drug-delivery vehicles with this approach. This modular protein co-assembly approach is expected to provide access to new constructs with sophisticated functional capabilities for medical and biotechnology applications. |
Introduction
Within living systems, the ordered assembly of proteins gives rise to sophisticated machines that can perform complex functions.1 Broadly, protein assembly can be subdivided into two types, homogeneous and heterogeneous. In homogeneous assembly, the protein domains are identical, and the resultant multivalency often serves as a checkpoint for functional activity (e.g., via allostery or receptor crosslinking) or enables stronger intermolecular interactions (e.g., via avidity). In heterogeneous assembly, two or more different protein molecules co-assemble into a single supramolecular structure. An important additional outcome of heterogeneous assembly is the emergent or synergistic activity that results from co-integrating complementary functional domains. For example, integrin receptors consist of co-assembled α and β chains that work in concert to mediate transmembrane anchoring of the cytoskeleton to the extracellular matrix.2 Likewise, T cell receptors consist of either α and β chain pairs or γ and δ chain pairs that work collaboratively to recognize peptide antigens presented by class I or II major histocompatibility complexes.3,4 Cytokine receptors (e.g., interleukin receptors), which typically consist of a common β and/or γ receptor chain and a distinctive α receptor chain, can form modular docking sites on the cell surface for recognition of cognate soluble cytokines and signaling pathway activation.5 Beyond dimers, the basement membrane glycoprotein laminin is a heterotrimer of α, β, and γ chains, where permutations of these trios provides biomolecular assemblies with unique structural and functional features.6 Further, additional functional complexity is realized in higher-ordered heterogeneous assemblies, such as the eukaryotic proteasome, which mediates regulatory and quality-control protein degradation through the collective activity of nearly 50 protein subunits.7Inspired by these and other examples, synthetic protein co-assemblies are attractive as both tools to interrogate natural heterogeneous protein assemblies and as the basis for creating entirely new molecular machines. Central to these efforts is the development of co-assembly motifs that provide precise control of the stoichiometry of the co-integrated protein domains. One approach relies on sophisticated computational methods that enable the design of protein partners with complementary association interfaces.8,9 Another approach relies on fusing the protein of interest onto a peptide “handle” that directs its assembly into a prescribed supramolecular architecture.10,11 Handles based on peptides that form β-sheets have been shown to provide control of protein stoichiometry within an entire system of molecules;12 however, β-sheet nanofiber chain length is difficult to control, while co-assembly of β-strands is often heterogeneous and stochastic.13 Thus, although system-level control of protein ratio can be achieved, molecular level precision is lacking. In contrast, α-helical coiled-coils are self-limiting, deterministic structures formed from a discrete number of peptide strands, where this number can be tuned through rational design of peptide–peptide interfaces.14,15
Due to their predictable and reproducible behavior, α-helical coiled-coils find extensive use as scaffolding for supramolecular protein assembly. For example, homodimeric and trimeric coiled-coils are used for multivalent display of antibody fragments.16,17 Likewise, a leucine zipper peptide that forms a homodimeric coiled-coil could stabilize the homodimeric quaternary structure of galectin-1.18 Fusing galectin-1 and galectin-3 onto opposing ends of this dimeric leucine zipper peptide provided a heterotetrameric (i.e., ‘dimer of dimers’) construct with increased immunomodulatory activity when compared to either galectin alone.19,20 Heterodimeric protein constructs can also be formed by inserting charged amino acids along the hydrophilic interface residing between two coiled-coil forming peptides (i.e., hydrophobic residues at a and d positions, whereas charged residues at e and g positions of the canonical abcdefg heptad sequence repeat).21,22 In this design, charge complementarity favors the association of two different coiled-coil strands (i.e., A + B peptide pairing), while electrostatic repulsion prevents self-association (i.e., A + A or B + B peptide pairing). This approach has been used to create a soluble T cell receptor analog,23 CD8 heterodimers,24 HLA-DR1:HLA-DM heterodimers,25 multivalent antibody fragments,26–28 probes for high resolution molecular imaging,29 molecular recognition screens and sensors,30,31 mediators of membrane fusion,32 protein purification tags,33 controlled drug release vehicles,34 probes of multivalent cell adhesion,35 stabilized growth factors,36 growth factor immobilization anchors,37–39 and transcription factor immobilization anchors.40 However, although higher-ordered homomeric assemblies have been reported,41–43 examples of higher-ordered heterogeneous assemblies are limited.44 Further, there is presently little understanding of the effect of the number and type of co-integrated protein domains on the co-assembly process or functional capabilities of the resulting heterogeneous constructs.
Here we tested the concept that constructs with modular and tunable protein domain stoichiometry could be created through the use of a trimeric coiled-coil peptide scaffold in which each strand was unique. To test this, we employed an ABC heterotrimeric coiled-coil developed by Alber and colleagues.45 Although each peptide A, B, or C demonstrated some capacity for self-association when alone, the heterotrimer was the preferred state when all three molecules were present in the system. To test the potential of the A, B, and C peptides as handles for protein co-assembly, we created a library of fusion proteins in which strand A was fused to NanoLuc luciferase (NL), B was fused to superfolder green fluorescent protein (sfGFP), and C was fused to mRuby (Fig. 1A). In some instances, the β-galactoside-binding protein galectin-3 (Gal3) was fused onto the opposing terminus of A, B, or C to generate NL-A-Gal3, sfGFP-B-Gal3, and mRuby-C-Gal3 (Fig. 1B). We chose NL, sfGFP, and mRuby because their co-assembly was expected to yield constructs capable of sequential bioluminescence and fluorescence resonance energy transfer (BRET/FRET),46 which provides both an analytical measure of co-assembly and a demonstration of the potential of this approach to create modular diagnostics with emergent activity arising from the co-integrated activity of the co-assembled protein domains. We chose galectin-3 based on a recent report showing that the carbohydrate-binding affinity of homogeneous synthetic galectin-3 constructs can be tuned by varying the number of galectin-3 domains,43 which provides an additional analytical measure of co-assembly and a demonstration of the potential of this approach to create targeted drug delivery vehicles.42Fig. 1C outlines the assortment of heterotrimeric trios that are possible with this protein library, which were evaluated in this report using native PAGE, fluorimetry and luminometry, and lactose affinity chromatography. These studies demonstrate that distinct peptide strands of a heterotrimeric coiled-coil can be used as handles to create heterogeneous protein co-assemblies with tunable functional domain stoichiometry, thereby enabling opportunities to develop modular multi-protein machines with new functional capabilities.
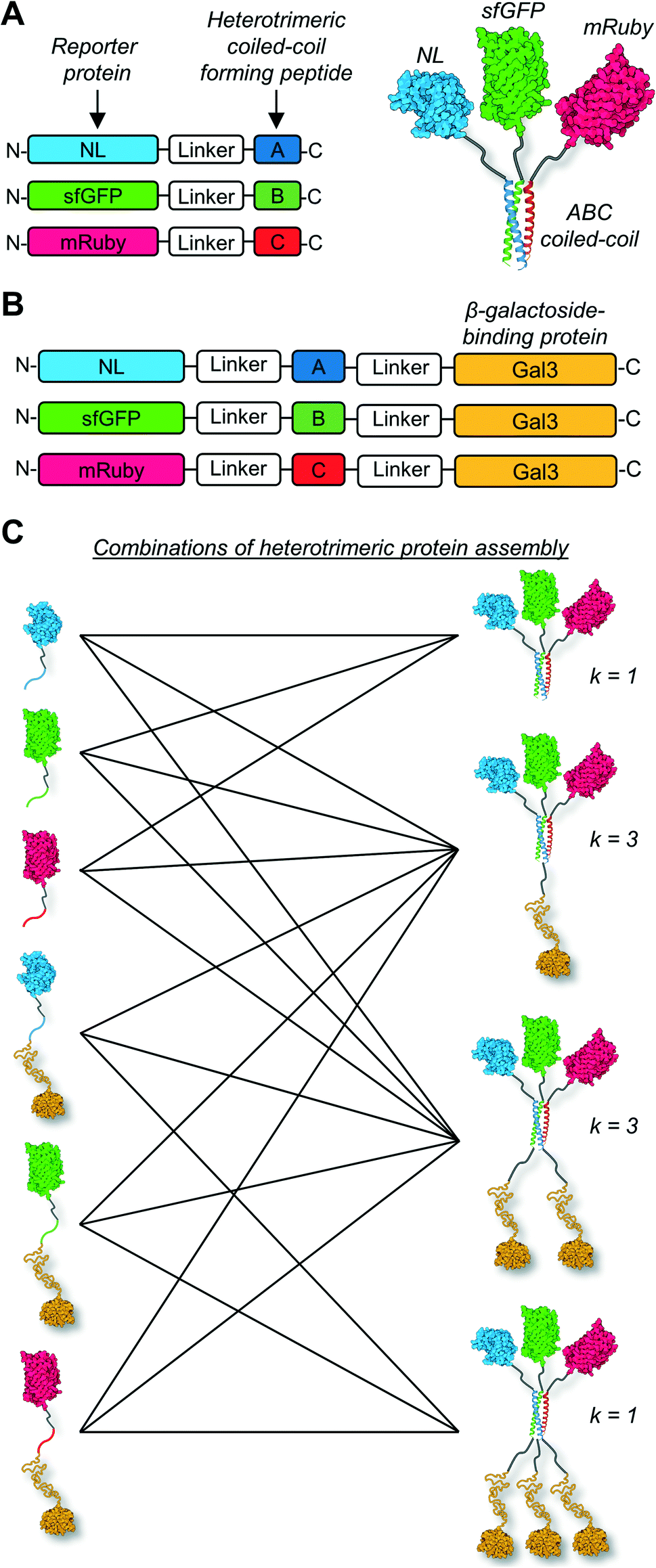 | ||
| Fig. 1 Schematic of heterogeneous protein co-assemblies with tunable functional domain stoichiometry. (A) Design of α-helical coiled-coil fusion protein sequences and structural illustration of functional protein co-assembly via the heterotrimeric coiled-coil scaffold. (B) An expansion to the library of fusion proteins incorporating the β-galactoside-binding protein galectin-3 (Gal3) at the C-terminus. (C) The different number of combinations of heterotrimeric co-assemblies (k) that can be generated using the six fusion proteins. | ||
Methods
Expression and purification of fusion proteins
Methods were adapted from prior reports.19,20,42,43 Genes encoding fusion proteins were inserted into pET-21d(+) vectors between NcoI and XhoI restriction sites. Genetic sequences, along with amino acid sequences, are provided in the ESI.† Plasmids were transformed into Origami™ B (DE3) E. coli (Novagen®). Positive clones were grown on ampicillin (100 μg mL−1) and kanamycin B (15 μg mL−1) doped lysogeny broth (LB)/agar plates overnight at 37 °C. Positive colonies were selected to inoculate 5 mL of LB containing 100 μg mL−1 ampicillin and 15 μg mL−1 kanamycin B. Cultures were grown overnight at 37 °C, 225 rpm on an orbital shaker and then sub-cultured into 1 L of 2xTY growth medium (16 g tryptone, 10 g yeast extract, 5 g NaCl) containing 100 μg mL−1 ampicillin and 15 μg mL−1 kanamycin B. Cultures continued to grow at 37 °C, 225 rpm until reaching an optical density of 0.6–0.8 at 600 nm wavelength, at which point cultures were supplemented with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) (ThermoFisher) to induce protein expression for 18 h at 18 °C, 225 rpm. Bacteria were then pelleted and washed with phosphate-buffered saline (1× PBS, pH 7.4) via centrifugation (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 × g at 4 °C for 10 min) in a Sorvall™ RC 6 Plus Superspeed Centrifuge (ThermoFisher). Pelleted bacteria were resuspended and lysed in B-PER bacterial protein extraction reagent (ThermoFisher) supplemented with a Pierce protease inhibitor tablet (ThermoFisher), 2400 units mL−1 DNAse I (ThermoFisher), and 50 mg mL−1 lysozyme (ThermoFisher) for 20 min at room temperature. Bacterial lysates were centrifuged (42600 × g at 4 °C for 15 min) to separate the soluble protein fraction into the supernatant. Supernatant was applied to HisTrap™ FF crude prepacked columns (GE Healthcare) connected to an ÄKTA™ Pure FPLC system (GE Healthcare) where His-tagged proteins were purified via immobilized metal affinity chromatography (0–250 mM imidazole gradient for protein elution). Amicon® Ultra centrifugal filters (MilliporeSigma) with 10 kDa cut-off were used to concentrate purified proteins to 5 mL for further purification and removal of imidazole via size-exclusion chromatography on a HiLoad™ 26/600 Superdex™ 200 column (GE Healthcare) connected to an ÄKTA™ pure FPLC system. Molar concentration of purified proteins was determined by Beer–Lambert law. Absorbance (λ = 280 nm) was measured on a NanoDrop spectrophotometer (ThermoFisher) and extinction coefficient of each protein was calculated based on amino acid content using ExPASy ProtParam tool (available at https://web.expasy.org/protparam/). Extinction coefficients are as follows: 2842.0 M−1 mm−1 for NL-A; 2201.5 M−1 mm−1 for sfGFP-B; 2602.5 M−1 mm−1 for mRuby-C; 6441.5 M−1 mm−1 for NL-A-Gal3; 5788.5 M−1 mm−1 for sfGFP-B-Gal3; 6053.0 M−1 mm−1 for mRuby-C-Gal3.
300 × g at 4 °C for 10 min) in a Sorvall™ RC 6 Plus Superspeed Centrifuge (ThermoFisher). Pelleted bacteria were resuspended and lysed in B-PER bacterial protein extraction reagent (ThermoFisher) supplemented with a Pierce protease inhibitor tablet (ThermoFisher), 2400 units mL−1 DNAse I (ThermoFisher), and 50 mg mL−1 lysozyme (ThermoFisher) for 20 min at room temperature. Bacterial lysates were centrifuged (42600 × g at 4 °C for 15 min) to separate the soluble protein fraction into the supernatant. Supernatant was applied to HisTrap™ FF crude prepacked columns (GE Healthcare) connected to an ÄKTA™ Pure FPLC system (GE Healthcare) where His-tagged proteins were purified via immobilized metal affinity chromatography (0–250 mM imidazole gradient for protein elution). Amicon® Ultra centrifugal filters (MilliporeSigma) with 10 kDa cut-off were used to concentrate purified proteins to 5 mL for further purification and removal of imidazole via size-exclusion chromatography on a HiLoad™ 26/600 Superdex™ 200 column (GE Healthcare) connected to an ÄKTA™ pure FPLC system. Molar concentration of purified proteins was determined by Beer–Lambert law. Absorbance (λ = 280 nm) was measured on a NanoDrop spectrophotometer (ThermoFisher) and extinction coefficient of each protein was calculated based on amino acid content using ExPASy ProtParam tool (available at https://web.expasy.org/protparam/). Extinction coefficients are as follows: 2842.0 M−1 mm−1 for NL-A; 2201.5 M−1 mm−1 for sfGFP-B; 2602.5 M−1 mm−1 for mRuby-C; 6441.5 M−1 mm−1 for NL-A-Gal3; 5788.5 M−1 mm−1 for sfGFP-B-Gal3; 6053.0 M−1 mm−1 for mRuby-C-Gal3.
Gel electrophoresis
Protein molecular weight under denaturing conditions was determined by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie staining alongside a protein ladder (BP3602500, ThermoFisher) (ESI† S1). Protein size and co-assembly under native conditions were assessed by native PAGE and Coomassie staining or visualization of protein activity within gels. Prior to native PAGE experiments, proteins fused to A, B, or C peptides were mixed at equal molar concentration and volume (e.g., 10 μL, 20 μM A + 10 μL, 20 μM B + 10 μl, 20 μM C) or diluted to 20 μM alone in 1× PBS. Mixed proteins and proteins alone were incubated for 1 h at room temperature. Samples were then mixed with an equal volume of native sample buffer (1610738, Bio-Rad) (e.g., 10 μL sample + 10 μL native sample buffer), which contains 62.5 mM Tris–HCl, pH = 6.8, 40% glycerol, 0.01% bromophenol blue. Samples were loaded into wells of 4–20% Mini-PROTEAN® TGX™ precast protein gels (4561094, Bio-Rad). Electrophoresis was performed at 150 V for 1 h in ice-cold native PAGE running buffer (192 mM glycine, 25 mM Tris base, pH = 8.3). After electrophoresis, gels were carefully placed on a blue light transilluminator and photographic images showing sfGFP and mRuby fluorescence of protein bands were taken. Next, photographic images showing NL luminescence of protein bands were taken on a white background with 2 μL droplet of stock furimazine (Nano-Glo™ substrate, PRN1120, Promega) carefully pipetted above each protein band. Finally, photographic images of gels were taken after Coomassie staining and destaining. A control native PAGE experiment was also performed wherein wild-type (WT) variants of the fusion proteins were mixed at equal molar concentration and volume (e.g., 5 μL, 40 μM WT-NL + 5 μL, 40 μM WT-sfGFP + 5 μL, 40 μM mRuby-C + 5 μL, 40 μM WT-Gal3) and evaluated as described above to determine the extent of protein co-assembly independent of the heterotrimeric coiled-coil (ESI† S8).Protein size analysis
Native size of heterotrimer (NL-A-Gal3 + sfGFP-B + mRuby-C) was determined by size-exclusion chromatography and dynamic light scattering (ESI† S4). Generally, heterotrimer was prepared by mixing 15 μM NL-A-Gal3, 15 μM sfGFP-B, and 15 μM mRuby-C at equal volumes to total of 400 μL 1× PBS. The protein mixture was loaded onto a Superdex™ 200 10/30 GL column (GE Healthcare) connected to an ÄKTA™ Pure FPLC system. Protein eluting from the column was detected at 280 nm wavelength. Raw signal was normalized based on maximum signal intensity and then plotted. Native protein molecular weight was estimated via extrapolation from a size-exclusion chromatography calibration curve, which was prepared from protein standard markers (Bio-Rad, GE Healthcare, ThermoFisher). Prior to taking dynamic light scattering measurements, the protein mixture described above was filtered through a 0.2-micron syringe filter (ESI† S4B). To ensure no significant amount of protein was lost due to aggregation, the molar concentration of heterotrimer was measured before and after filtration. Measurements were taken on a NanoBrook 90Plus particle size analyzer with BIC Particle Sizing Software (Brookhaven Instruments) in technical triplicate after ten 30 second runs. Hydrodynamic diameter ± standard deviation was normalized based on number-weighted size distribution.Lactose affinity chromatography
Prior to lactose binding experiments, proteins fused to A, B, or C peptides were mixed at equal molar concentration and volume (e.g., 167 μL, 45 μM A + 167 μL, 45 μM B + 167 μl, 45 μM C) in 1× PBS. Protein mixtures were incubated for 1 h at room temperature. Lactose binding affinity of protein assemblies was evaluated via lactose affinity chromatography on an ÄKTA pure FPLC system (GE Healthcare).19,20,42,43 Specifically, a Tricorn™ 5/50 column (GE Healthcare) packed with α-lactose-agarose resin (L7634, MilliporeSigma) was loaded with protein samples described above and then washed with 1× PBS until unbound protein was removed. Protein bound to the α-lactose-agarose column was eluted using a linear gradient of 0–100 mM soluble β-lactose (AAH54447A1, ThermoFisher) in 1× PBS. Eluted proteins were detected at 280 nm wavelength and chromatograms were normalized based on maximum signal intensity.Fluorimetry and luminometry assays
Prior to BRET/FRET experiments, a control protein mixture (1 μM WT-NL + 1 μM WT-sfGFP + 1 μM mRuby-C) and a heterotrimer mixture of proteins without Gal3 fused (1 μM NL-A + 1 μM sfGFP-B + 1 μM mRuby-C) were prepared in 1× PBS, followed by 1 h incubation at room temperature. BRET/FRET was then measured immediately thereafter. For mixtures including one or more proteins fused to Gal3, the heterotrimeric assemblies were prepared by mixing the three proteins at equal concentration in equal volumes, these mixtures were subjected to lactose affinity chromatography, and then BRET/FRET was measured in the collected eluent released by the lactose gradient (i.e., “bound fraction”). Specifically, 100 μL of each sample was pipetted into wells of a black, clear plastic bottom, 96-well microplate for detection of sfGFP and mRuby fluorescence on a SpectraMax® M3 multi-mode microplate reader (Molecular Devices). For detection of NL luminescence, 50 μL of each protein assembly was pipetted into a white plastic bottom 96-well microplate followed by the addition of 50 μL diluted furimazine solution (49 μL Nano-Glo™ buffer + 1 μL stock furimazine) (PRN1120, Promega) immediately prior to spectrophotometric measurements. Wells were excited at 485 nm and 558 nm wavelengths to measure fluorescence emission of protein assemblies over a 400–700 nm wavelength sweep at 1 nm increments. Cut-offs were set at 495 nm and 570 nm wavelengths for sfGFP and mRuby detection, respectively. NL luminescence of protein assemblies after adding furimazine solution or Nano-Glo™ buffer only (control) was measured over a 400–700 nm wavelength sweep at 1 nm increments and 500 ms integration time. Data were plotted as either raw signal or normalized signal by dividing every data point across the spectrum by the data point for the highest signal within the spectrum. To assess background signal during BRET/FRET measurements, the same procedures that are described above were performed without the addition of furimazine to the protein mixtures (ESI† S9).Results and discussion
To test the hypothesis that individual A, B, and C strands of a heterotrimeric α-helical coiled-coil can be used as handles to co-assemble three different protein domains, we expressed and purified recombinant fusions of strand A linked to NL (NL-A), strand B linked to sfGFP (sfGFP-B), and strand C linked to mRuby (mRuby-C) using microbial hosts. The fusions were recovered from the soluble phase in high yield and demonstrated the respective luminescence or fluorescence activity associated with each functional protein domain.To characterize the solution behavior of the fusions and their co-assembly into a heterotrimer, we first measured the hydrodynamic size of each alone and in equimolar combination using size-exclusion chromatography (ESI† S10). When alone, all of the proteins eluted from the SEC column at a volume corresponding to a molecular weight that was significantly greater than the theoretical value. In particular, NL-A eluted with a sharp peak corresponding to an empirical molecular weight (MW) of 99.5 kDa, as well as smaller peaks at higher and lower MW (theoretical MW = 25.1 kDa) (ESI† S10A). sfGFP-B eluted with a sharp peak corresponding to an empirical MW of ∼164 kDa, as well as a broad peak centered at an empirical MW of ∼89 kDa (theoretical MW = 34.5 kDa) (ESI† S10B). mRuby-C eluted with a sharp peak corresponding to an empirical MW of 98 kDa, as well as a smaller peak at a lower MW (theoretical MW = 32.5 kDa) (ESI† S10C). The observed discrepancies in the empirical and theoretical MWs of NL-A, sfGFP-B, and mRuby-C may suggest a tendency for the fusion proteins to self-associate. Indeed, in the paper describing the development and characterization of the A, B, and C peptides, Alber and colleagues reported that the peptides had some tendency to self-assemble when alone, but favored heterotrimer formation when present in combination.45 Here, though, it is also important to note that the estimation of molecular weight from elution volume via SEC assumes that the proteins can be approximated as a globular hard sphere, and have similar hydrodynamic properties as the standards used for calibration. It is reasonable to expect that the linker domain and the A, B, or C peptide domain, which are roughly 50 amino acids long and correspond to 10% or more of the protein molecular weight, are likely to be unstructured. Intrinsically disordered proteins, as well as other non-globular fusions, are known to elute at volumes that correspond to much higher MWs than expected due to their larger Stokes radii.47,48
When combined at an equimolar ratio, the heterotrimeric mixture of NL-A, sfGFP-B, and mRuby-C eluted as a relatively narrow peak at an elution volume that corresponded to an empirical MW of 108 kDa, which was slightly higher than the theoretical value of 92.1 kDa (ESI† S10D). As was noted above for the analysis of NL-A, sfGFP-B, and mRuby-C elution, discrepancy in the empirical and theoretical MW likely reflects the non-globular structural features of the assembly. However, the observation that the proteins eluted at unique volume when combined as compared to their elution when alone suggested that they were in a different physical state (e.g., co-assembled into a heterotrimer) when combined at an equimolar ratio. When compared to SEC traces reported previously for homotrimeric protein complexes,42,43 the empirical molecular weight estimated from elution volume suggested that NL-A, sfGFP-B, and mRuby-C formed a complex with a size comparable to the predicted heterotrimer.
Due to the limitations of SEC, we evaluated the migration of NL-A, sfGFP-B, and mRuby-C alone and in an equimolar mixture using native polyacrylamide gel electrophoresis (“native PAGE”) to assess their co-assembly into supramolecular complexes. In native PAGE, protein samples are loaded into a gel and then subjected to an electric field that will induce their mobility through the gel pores. Under native conditions, the distance that a protein will migrate through the gel depends on its size, shape, net charge, and the properties of the gel used. Charge-to-mass ratio is a useful predictor for migration distance, where molecules (or assemblies) with a greater negative charge-to-mass ratio are expected to migrate further (i.e., farther away from the negative pole). This contrasts with sodium dodecylsulfate PAGE (SDS-PAGE) wherein migration distance correlates directly with molecular weight, irrespective of charge, because the proteins are denatured and charge is equilibrated by way of the anionic detergent. Based on the properties of the fusion proteins, we would expect sfGFP-B to migrate a moderate distance (charge-to-mass = −0.377); NL-A to migrate the longest distance because it is smaller than sfGFP-B and has a slightly more negative charge-to-mass ratio (−0.398); and mRuby-C to migrate the shortest distance because it has the smallest charge-to-mass ratio of all of the proteins (−0.09). Consistent with this, we observed that NL-A migrated a slightly longer distance than sfGFP-B, whereas mRuby-C migrated a shorter distance than either of the other proteins (Fig. 2A). When the NL-A, sfGFP-B, and mRuby-C fusion proteins were combined at an equimolar ratio prior to electrophoresis, only one band was identified on the native PAGE gel following Coomassie staining (Fig. 2A). This band migrated a unique distance when compared to the migration of NL-A, sfGFP-B, or mRuby-C alone, and this intermediate distance was consistent with the relative migration distance expected based on its charge, size, and charge-to-mass ratio (∼92 kDa, −26 net charge, −0.282). We also note that these native PAGE experiments employed gradient gels, which will retard the mobility of a larger-sized protein complex that carries a high net negative charge as it progresses further along the length of the gel toward the positive pole of the electric field where the pore size continues to decrease. Thus, some variability in migration distance as a function of charge-to-mass ratio is expected. Collectively, the presence of a single Coomassie band at a unique migration distance relative to the individual proteins suggested that NL-A, sfGFP-B, and mRuby-C co-assembled into a supramolecular complex. Further, the location of the bands of the individual proteins relative to the band for the mixture suggested that the proteins likely did not undergo significant self-association, and thus the discrepancies in empirical and theoretical MW as predicted by SEC were likely due to improper assumptions of protein hydrodynamic shape.
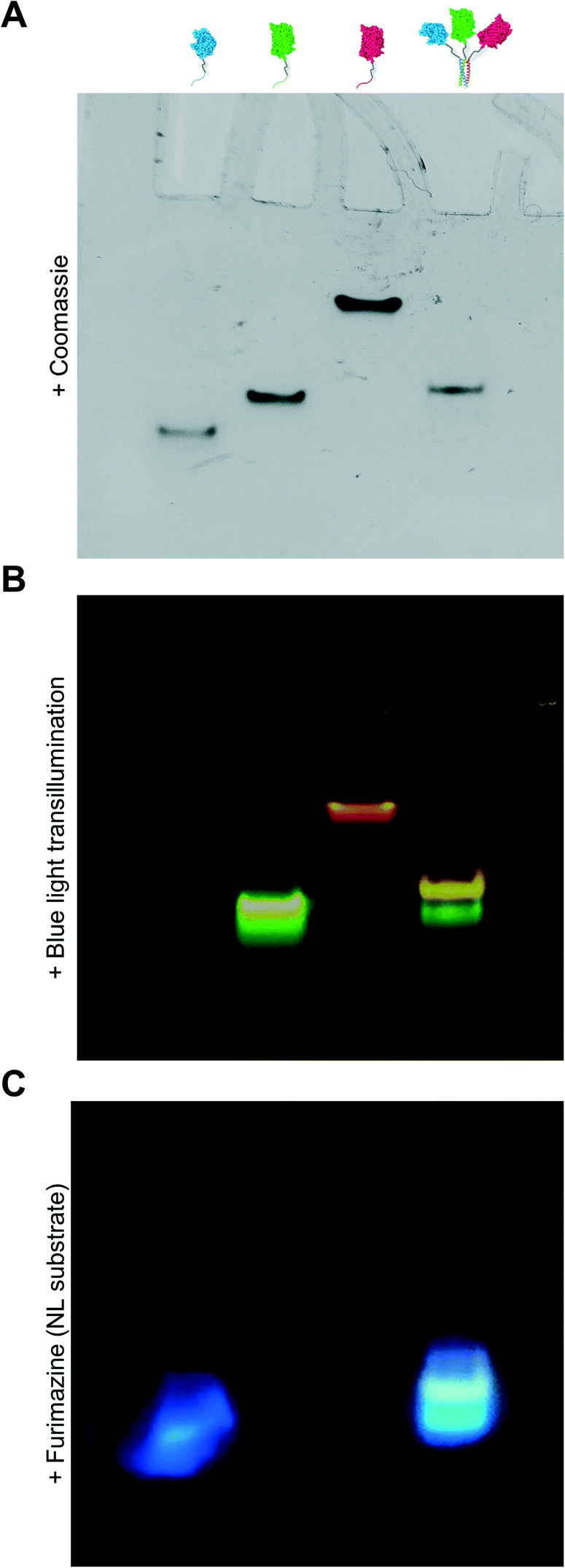 | ||
| Fig. 2 Native PAGE experiment where bands corresponding with NL-A, sfGFP-B, mRuby-C, and all three fusion proteins mixed were detected by (A) Coomassie staining, (B) blue light transillumination, and (C) treatment with furimazine (NL substrate). | ||
Under native PAGE conditions, NL-A, sfGFP-B, and mRuby-C are expected to maintain their folded-state functions (i.e., luminescence, green fluorescence, and red fluorescence, respectively). To determine if all three proteins were co-localized at the site of the single Coomassie band identified after electrophoresis of the ternary mixture, we subjected the gel to blue light transillumination to induce green and red fluorescence emission, and then treated it with the NL substrate furimazine to evaluate luminescence. When subjected to blue light transillumination, we observed a yellow band at the same location as the Coomassie band, which is the expected output for overlapping green and red fluorescence emission (Fig. 2B). Likewise, we observed luminescence from the gel treated with furimazine at the same location as the Coomassie band (Fig. 2C). Collectively, these observations indicated that NL-A, sfGFP-B, and mRuby-C were co-localized at the site of the single band observed with Coomassie stain after subjecting the ternary mixture to electrophoresis. However, we note that a weak red fluorescent band was also observed at the same location as mRuby-C when alone, while stronger green and blue bands were observed at the same locations as sfGFP-B and NL-A, respectively. These bands indicate that some fraction of the proteins were unassembled in the ternary mixture. We note that the weakened signal from mRuby-C is likely due to its weak excitation upon blue light transillumination, while the absence of the NL-A, sfGFP-B, and mRuby-C bands in the Coomassie stained gel is likely due to the detection limit of the dye. Observing some fraction of proteins in the unassembled state would be expected here because this is an equilibrium system, where the ratio of assembled to unassembled proteins would be governed by the dissociation constant of the coiled-coil. We note that for a binary system, which is considerably easier to model quantitatively, 99% assembly on a 1 micromolar protein basis would require a dissociation constant in the low nanomolar range. In a ternary system, which is expected to be thermodynamically less favorable than a binary system due to the lower statistical likelihood of A + B + C collision, the dissociation constant required for 99% assembly on a 1 micromolar protein basis would also be on the order of nanomolar or lower. Most coiled-coil complexes reported to date have dissociation constants that are in the nano- to micromolar range. Thus, quantitative or near-quantitative conversion should not be expected in this system. However, because this method is not quantitative as performed, the excess green fluorescence and blue luminescence relative to red fluorescence in the unassembled fraction could be also due to errors arising from practical limitations of measuring protein concentration.
Due to overlap of their emission and excitation spectra, respectively, proximal NL and sfGFP domains can demonstrate bioluminescence energy transfer, or BRET, wherein photons emitted from NL can mediate excitation and, in turn, emission of green light from sfGFP in the absence of discrete sfGFP excitation.49 Likewise, proximal sfGFP and mRuby domains can demonstrate fluorescence resonance energy transfer, or FRET, wherein photons emitted from sfGFP can mediate excitation and, in turn, emission of red light from mRuby.50 If NL, sfGFP, and mRuby are all co-localized, NL excitation can induce emission of green light from sfGFP, which in turn induces emission of red light from mRuby via sequential BRET/FRET.46 Heterodimeric α-helical coiled-coil tags have previously been shown to enable FRET because they place fused fluorescent protein domains sufficiently close to enable efficient photon transfer.33 Here, we used sequential BRET/FRET to evaluate the co-assembly of NL-A, sfGFP-B, and mRuby-C into a heterotrimer (Fig. 3A). When furimazine was added to a solution containing an equimolar ratio of NL-A, sfGFP-B, and mRuby-C, a peak associated with NL emission was observed along with two additional emission peaks at λ = 515 nm and λ = 585 nm (Fig. 3B, solid and dashed black traces), which correspond to the emission maxima of sfGFP and mRuby, respectively. In contrast, when furimazine was added to a control solution containing an equimolar ratio of NL lacking A (“WT-NL”), sfGFP lacking B (“WT-sfGFP”), and mRuby-C only blue light emission was detected (Fig. 3B, blue trace). Comparison of the raw emission spectra for the heterotrimer and control groups indicated that the NL signal intensity in the heterotrimer group was diminished by ∼20% relative to the control group (ESI† S3). Some decrease in NL “donor” emission should be expected in the heterotrimer group when compared to the control because photons from NL would be absorbed by the acceptor sfGFP during BRET. Although not performed here, donor decay can be used to measure resonance energy transfer efficiency and provide some sense of intermolecular distance.
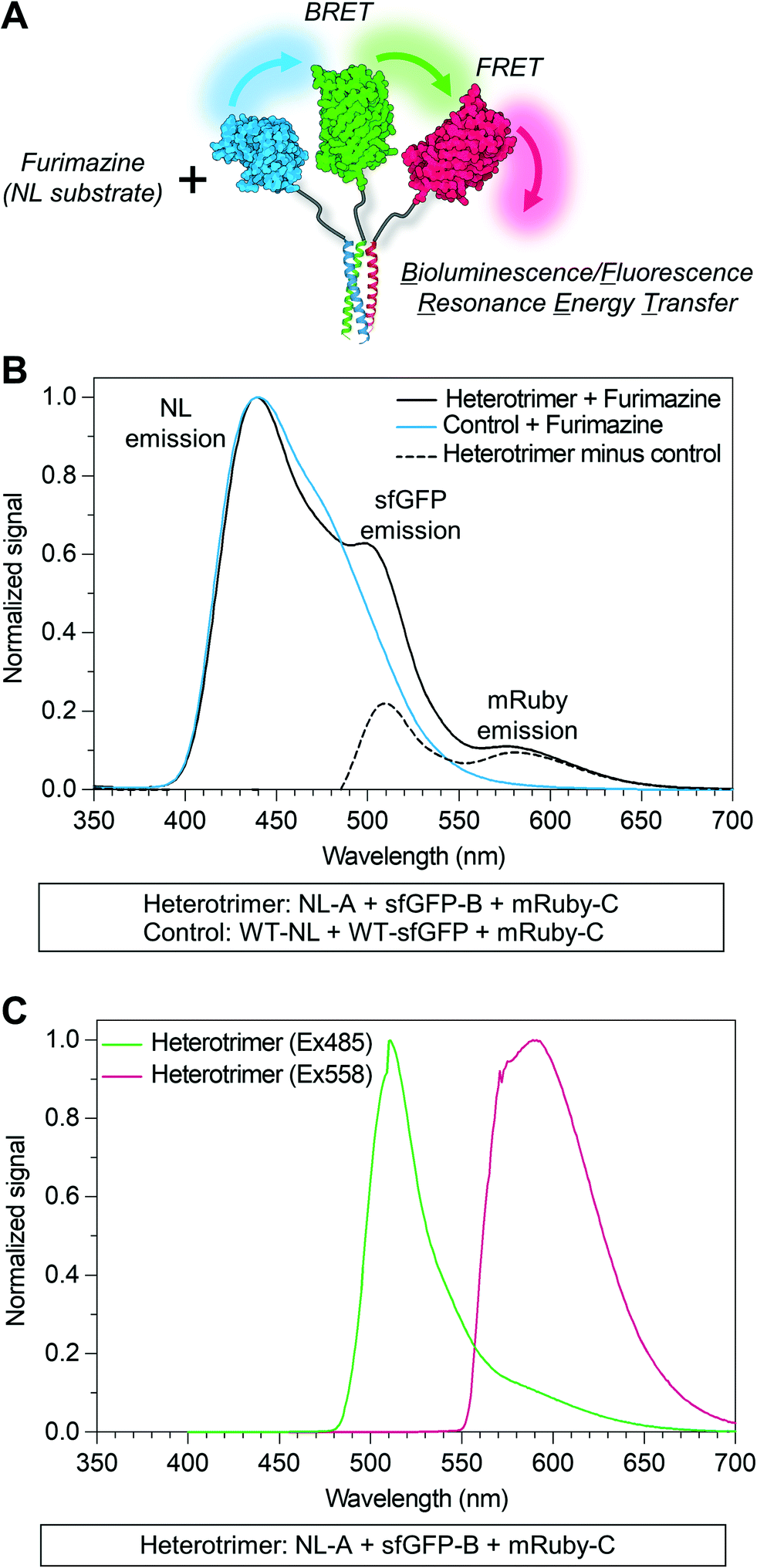 | ||
| Fig. 3 Sequential bioluminescence-to-fluorescence resonance energy transfer (BRET/FRET) properties of the heterotrimeric co-assembly. (A) Schematic illustration of sequential BRET/FRET between reporter proteins within the heterotrimeric co-assembly. In this instance, BRET/FRET is initiated by an interaction between the catalytic site of NL and the substrate furimazine. (B) Emission spectra of the heterotrimer and control protein mixture upon addition of furimazine (black trace and blue trace, respectively). The difference between the emission spectra of the heterotrimer and control is plotted (black dashed trace) to highlight the contribution of superfolder green fluorescent protein (sfGFP) and mRuby excitation and emission, via BRET/FRET, to the emission spectra of the heterotrimer. (C) Emission spectra of the heterotrimer upon excitation at λ = 485 nm (green trace) and λ = 558 nm (red trace), which correspond to the excitation maxima of sfGFP and mRuby, respectively. | ||
As an additional control for FRET within the homotrimer, we also excited the ternary mixture of NL-A, sfGFP-B, and mRuby-C with light at 485 nm (i.e., sfGFP excitation maximum) and 558 nm (i.e., mRuby excitation maximum). When the heterotrimer mixture was excited with 558 nm light, we observed a strong peak corresponding to the emission of mRuby, without any significant emission at the wavelengths corresponding to sfGFP or NL emission (Fig. 3C, red trace). When the heterotrimer mixture was excited with 485 nm light, we observed a strong peak corresponding to the emission of sfGFP-B as well as a small shoulder in the wavelength region that overlaps with mRuby emission (Fig. 3C, green trace), suggesting weak FRET between sfGFP-B and mRuby-C in this sample. When a control mixture was excited with 485 nm light, we observed a strong peak corresponding to the emission of sfGFP but the shoulder overlapping with mRuby emission was absent (ESI† S2), indicating that the weak FRET observed in the heterotrimer sample was enabled by fusion of sfGFP and mRuby to the B and C peptides, respectively. We note that the weakened FRET induced when the ternary mixture of NL-A + sfGFP-B + mRuby-C was excited with 485 nm light (Fig. 3C) as compared to the stronger FRET observed when furimazine was added to the mixture (Fig. 3B) could be due to direct BRET between NL-A and mRuby-C. Red-shifted BRET has been reported before,51 and the NL-A control spectrum (Fig. 3B, blue trace) indicates that photons are emitted with a wavelength that falls within the excitation spectrum of mRuby (558 nm). Collectively, when taken together with the native PAGE data in Fig. 2, these observations demonstrated that A, B, and C mediate co-assembly of NL, sfGFP, and mRuby into a ternary construct with emergent function (i.e., sequential BRET/FRET) attributed to the co-integrated activity of each protein domain.
An inherent benefit of coiled-coil scaffolds is that the N- and C-termini could, in principle, each be fused to a different protein domain to create assemblies with an even greater range of functional capabilities. For example, homomeric coiled-coils have been used as scaffolding to create fluorescent probes to study the emergent signaling function of galectin-3 or constructs that combine the signaling activity of galectin-1 and galectin-3.19,20,42,43 Here, we tested whether an additional protein domain could be co-integrated into assemblies of NL-A, sfGFP-B, and mRuby-C by replacing NL-A with a new fusion protein wherein galectin-3 was fused onto the C-terminus of the A domain (“NL-A-Gal3”) (Fig. 4A). We first subjected each protein and an equimolar ternary mixture to native PAGE to evaluate protein co-assembly qualitatively (Fig. 4B). A single band was observed in each lane corresponding to the individual proteins following Coomassie staining, whereas two bands were observed in the lane corresponding to the ternary mixture. Subjecting the gel to blue light transillumination and furimazine demonstrated that the lower band in the ternary mixture lane emitted yellow fluorescence (indicative of sfGFP and mRuby co-localization), as well as blue luminescence (indicative of NL co-localization). In contrast, the upper band emitted red fluorescence with a consistent hue and at the same location as the band for mRuby-C alone. We note that some unassembled sfGFP-B (green fluorescence) and NL-A-Gal3 (blue luminescence) were also detected in the lane corresponding to the ternary mixture via transillumination and furimazine, although not with Coomassie. These results demonstrate that NL-A-Gal3, sfGFP-B, and mRuby-C can co-assemble, although the assembly efficiency does not approach 100% and the integration of mRuby-C may be hindered by the presence of the Gal3 domain.
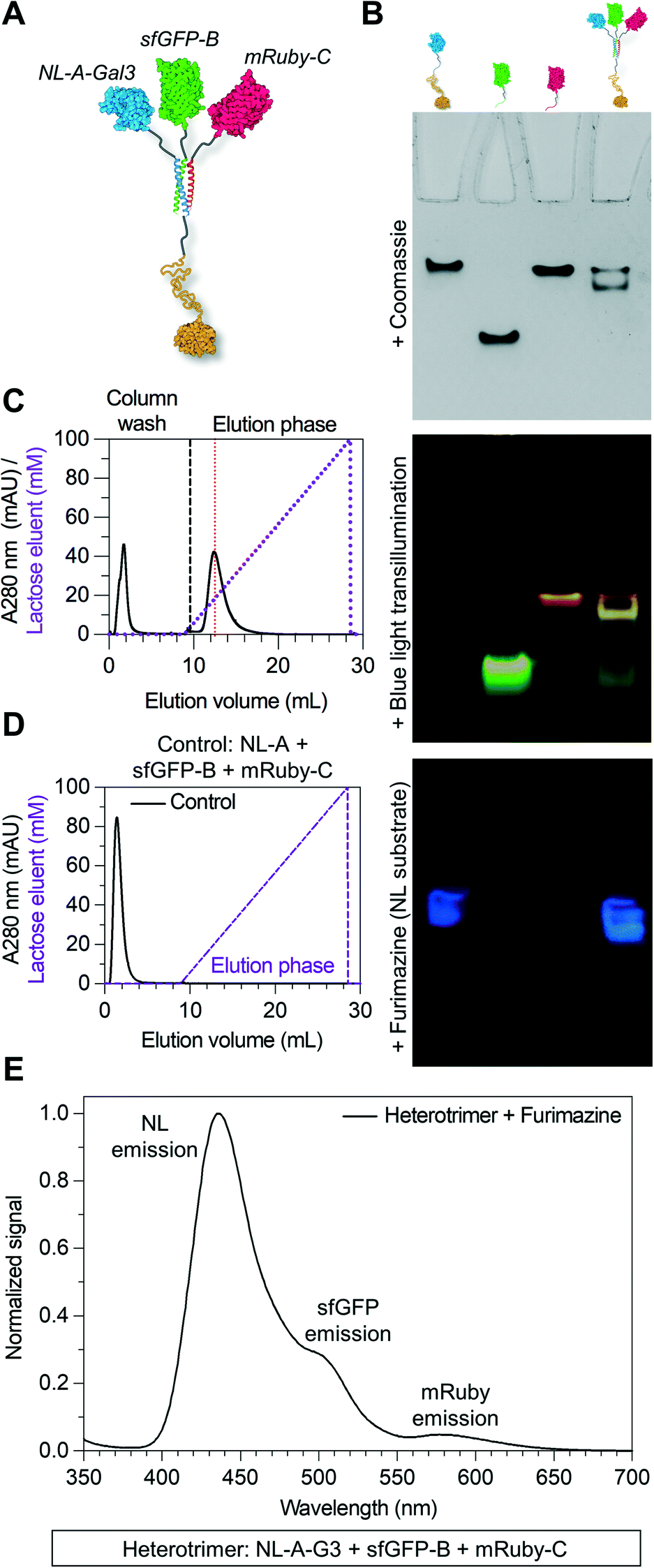 | ||
| Fig. 4 Endowing the heterotrimeric co-assembly with carbohydrate-binding affinity by fusing it to Gal3. (A) Structural illustration of the heterotrimer with a Gal3 domain fused to NL-A within the protein co-assembly. (B) Native PAGE experiment where bands corresponding with NL-A-Gal3 (or “NL-A-G3”), sfGFP-B, mRuby-C, and all three fusion proteins mixed were detected by Coomassie staining, blue light transillumination, and furimazine treatment. (C) Immobilized lactose affinity chromatography experiment performed on the heterotrimer with a Gal3 domain. For comparison, the vertical red dashed line indicates the concentration of lactose required to elute wild-type Gal3. (D) Immobilized lactose affinity chromatography experiment performed on the heterotrimer without a Gal3 domain (control). (E) Sequential BRET/FRET analysis of heterotrimer with a Gal3 domain. | ||
Physical characterization by size-exclusion chromatography and dynamic light scattering suggested that the heterotrimeric assembly of NL-A-Gal3, sfGFP-B, and mRuby-C has a molecular weight and hydrodynamic size in a range comparable to that of previously reported homotrimeric assemblies fused to Gal3 (ESI† S4).42,43 The lower molecular weight shoulder in the SEC trace may be due to unassembled NL-A-Gal3, sfGFP-B, and mRuby-C. The higher molecular weight shoulders in the SEC trace may reflect differences in the hydrodynamic shape of the heteroassemblies, and not the formation of larger (e.g., non-specific aggregates) or smaller (e.g., dimer) constructs. This is supported by the DLS measurements, which did not identify larger aggregates, which would be expected to be detected with greater sensitivity because they will diffract more light, as well as filtration studies which showed that the protein concentration did not change considerably before and after being passed through a 0.2 micron filter (ESI† S4B). Caution should also be taken with the assumption that NL-A-Gal3 + sfGFP-B + mRuby-C can be approximated as a globular hard sphere, given that each reporter protein is linked to the 35 amino acid coiled-coil strand by a flexible 16 amino acid linker, while galectin-3 has an ∼130 amino acid globular C-terminal domain linked to an ∼110 amino acid long, intrinsically disordered N-terminal domain.52 Under the present conditions, we do not know and cannot adequately predict if the unstructured domain of galectin-3 is in a compact or extended conformation in these assemblies. As noted above for the SEC analysis of NL-A, sfGFP-B, and mRuby-C, an extended conformation of the galectin-3 domain would be expected to decrease the elution volume of the construct from the SEC column, which would result in observing a higher-than-expected molecular weight for the assembly.
Galectin-3 can bind to lactose immobilized on agarose chromatography beads and be eluted via a soluble lactose gradient.42,43 In contrast, neither sfGFP nor mRuby demonstrate lactose binding affinity. Thus, the co-assembly of sfGFP-B and mRuby-C with NL-A-Gal3 would be expected to endow sfGFP-B and mRuby-C with lactose binding affinity. For the ternary mixture of NL-A-Gal3, sfGFP-B, and mRuby-C, immobilized lactose affinity chromatography identified two elution peaks measured via UV absorbance (λ = 280 nm), one in the non-binding “void” fraction and the other at a soluble lactose concentration that was similar to the concentration of lactose required to elute wild-type Gal3 (red dashed line) (Fig. 4C compared to ESI† S5). The correlation of the ternary mixture bound fraction elution with that of wild-type Gal3 suggested that assembly did not significantly alter Gal3 carbohydrate-binding affinity, consistent with other previously reported Gal3 fusions.42,43 Calculation of the area under the curve indicated that ∼66% of the protein was in the bound fraction. In contrast, more than 98% of wild-type galectin-3 was in the bound fraction. Consistent with the native PAGE analyses, this suggested that some of the proteins were in the unassembled state in the ternary mixture, as would be expected for an equilibrium system. It is assumed that the proteins eluting in the void fraction were primarily sfGFP-B and mRuby-C, which lack carbohydrate-binding affinity.
To confirm that the observed lactose-dependent elution of the mixture of NL-A-Gal3, sfGFP-B, and mRuby-C was due to specific Gal3:lactose interactions, we also characterized the elution of the control mixture of NL-A, sfGFP-B, and mRuby-C. 100% of the protein was identified in the void fraction (Fig. 4D), confirming that the observed bound fraction in the elution profile of the ternary mixture of NL-A-Gal3 + sfGFP-B + mRuby-C was due to the presence of the NL-A-Gal3 fusion protein.
Although lactose affinity chromatography demonstrated that the majority of the protein in the ternary NL-A-Gal3 + sfGFP-B + mRuby-C mixture eluted in the bound fraction, A280 measurements cannot reliably determine if NL, sfGFP, and mRuby are present in this fraction. To determine if NL-A-Gal3, sfGFP-B, and mRuby-C were co-assembled in the bound fraction, we subjected the eluent to furimazine and measured BRET/FRET. We observed a strong peak corresponding to NL luminescence emission, as well as shoulders indicative of sfGFP emission (via BRET) and mRuby emission (via FRET) (Fig. 4E). This result demonstrates that NL-A-Gal3, sfGFP-B, and mRuby-C co-assembled into a heterogeneous complex that endowed sfGFP-B and mRuby-C with lactose-binding affinity while also maintaining BRET/FRET capability between the reporter domains. When taken in conjunction with the native PAGE (Fig. 4B) and SEC (ESI† S4) for this mixture (Fig. 4B), these analyses suggest that NL-A-Gal3, sfGFP-B, and mRuby-C co-assembled into a heterotrimeric construct demonstrating the functional properties of each co-integrated protein domain.
Comparison of the spectra in Fig. 4E to that in Fig. 3B indicated that the relative sfGFP emission in the NL-A-Gal3 + sfGFP-B + mRuby-C sample was lower than that in the NL-A + sfGFP-B + mRuby-C sample. This could be due to a variety of factors, including a change in the intermolecular distance between NL and sfGFP in the fusion construct with the Gal3 domain which would be expected to decrease BRET efficiency. Additionally, a decrease in BRET could be due to a loss of NL in the NL-A-Gal3 + sfGFP-B + mRuby-C bound fraction eluent. Recall that the NL-A + sfGFP-B + mRuby-C mixture was not subjected to chromatography before BRET/FRET measurements, whereas nearly a third of the protein was eluted in the void fraction of the NL-A-Gal3 + sfGFP-B + mRuby-C mixture. The protein content of the void fraction was not analyzed, and although one would expect sfGFP-B and mRuby-C that lack lactose-binding affinity to dominate this fraction, it would not be unreasonable to assume some NL-A-Gal3 was also present therein. Finally, we would expect that NL-A-Gal3 and the heterotrimer of NL-A-Gal3 + sfGFP-B + mRuby-C would have similar affinity for immobilized lactose and, as such, a similar elution profile in response to the soluble lactose gradient. In contrast, sfGFP-B and mRuby-C would be eliminated in the void fraction. Thus, the bound fraction eluent of NL-A-Gal3 + sfGFP-B + mRuby-C would be expected to have excess NL-A-Gal3 that is in the unassembled state and would not contribute to BRET/FRET. Consistent with this, native PAGE demonstrated that some unassembled NL-A-Gal3 was present in the ternary mixture.
Informed by these data, we next tested whether the Gal3 domain could be fused to either the B or C tag by replacing sfGFP-B with sfGFP-B-Gal3 or mRuby-C with mRuby-C-Gal3 (Fig. 5A). Three bands were observed in the native PAGE lane corresponding to the mixture of NL-A + sfGFP-B-Gal3 + mRuby-C following Coomassie staining, whereas only one band was observed in the lane corresponding to the mixture of NL-A + sfGFP-B + mRuby-C-Gal3 (ESI† S6, top row). The location of two bands in the mixture of NL-A + sfGFP-B-Gal3 + mRuby-C were at the same location as the bands for sfGFP-B-Gal3 and mRuby-C alone, albeit at lower staining intensity, suggesting that some fraction of these proteins was in the unassembled state in the ternary mixture. We note that the staining intensity of sfGFP-B-Gal3 and mRuby-C alone was significantly higher than that of NL-A, suggesting that the former may have been in molar excess of NL-A in this experiment. The third band was in a unique location suggesting formation of a heteroassembly in this mixture. Likewise, the single band in the lane for the mixture of NL-A + sfGFP-B + mRuby-C-Gal3 was in a unique location relative to the bands for the proteins alone suggesting that this trio also formed a heteroassembly.
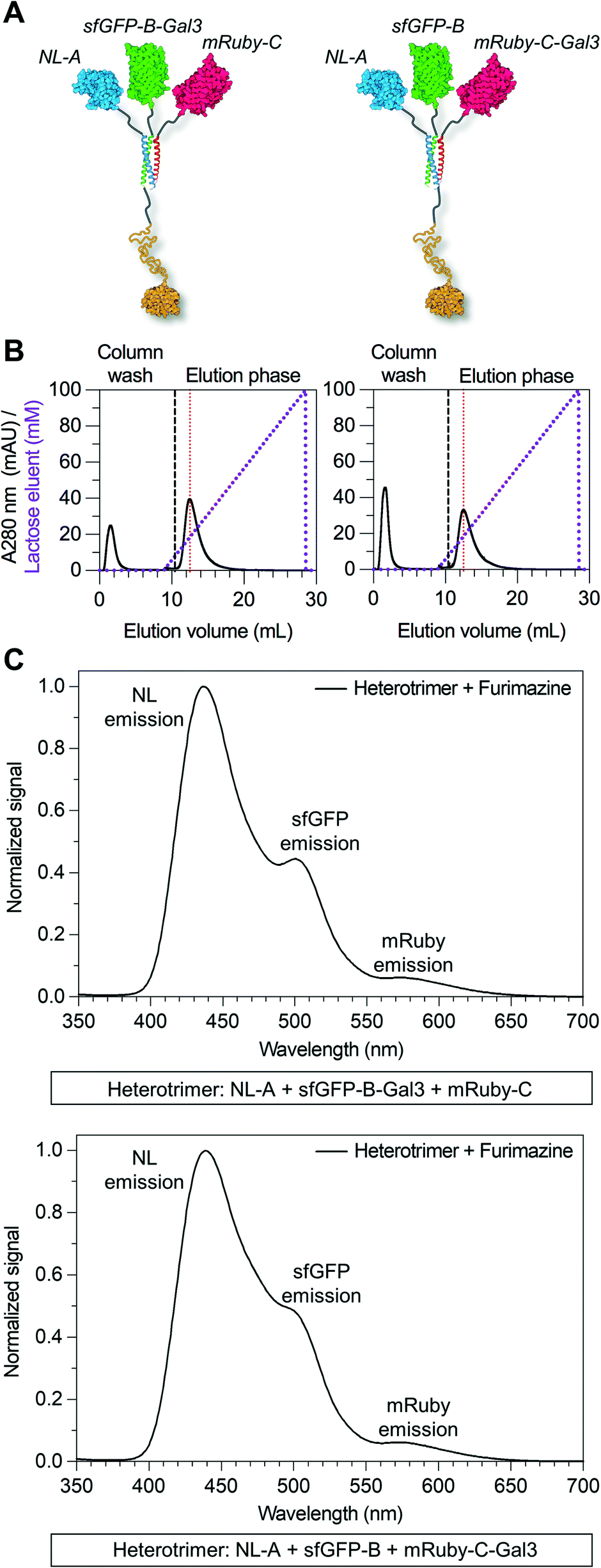 | ||
| Fig. 5 Modular placement of the Gal3 domain within the heterotrimeric co-assembly. (A) Structural illustration of the heterotrimer with a Gal3 domain fused to either sfGFP-B or mRuby-C. (B) Immobilized lactose affinity chromatography traces of the two different heterotrimers fused with Gal3. For comparison, the vertical red dashed line indicates the concentration of lactose required to elute wild-type Gal3. (C) Sequential BRET/FRET analysis of the two different heterotrimers fused with Gal3. | ||
To evaluate the co-localization of the NL, sfGFP, and mRuby fusion proteins, we subjected the native PAGE gel to blue light transillumination and furimazine (ESI† S6, middle and bottom row). Yellow fluorescence and blue luminescence were co-localized at the site of the unique Coomassie bands in the lane for each ternary mixture. Consistent with the Coomassie staining, sfGFP-B-Gal3 and mRuby-C were detected alone in the mixture of NL-A + sfGFP-B-Gal3 + mRuby-C, whereas no NL-A was detected. Notably, this suggested that sfGFP-B-Gal3 and mRuby-C did not form a dimer in the absence of A. Both unassembled sfGFP-B and NL-A were detected in the lane for the mixture of NL-A + sfGFP-B + mRuby-C-Gal3. Unexpectedly, we also observed an intermediate band in the lane corresponding to the ternary mixture of NL-A + sfGFP-B + mRuby-C-Gal3 that emitted yellow fluorescence (indicative of red and green fluorescence co-emission) and blue luminescence (indicative of NL co-localization), which was not detected with Coomassie staining. This could be indicative of a different physical state of the heterotrimer, for example, if the Gal3 domain was in a compact state versus an extended state during electrophoresis, where the former would correlate to a larger hydrodynamic size and, in turn, shorter migration distance.
To further evaluate the co-assembly of these fusion proteins into a heterotrimer, we subjected the ternary mixtures of NL-A + sfGFP-B-Gal3 + mRuby-C and NL-A + sfGFP-B + mRuby-C-Gal3 to lactose affinity chromatography. Protein eluted in both the unbound void fraction and as a bound fraction released by a soluble lactose competitor (Fig. 5B). Both heterotrimers eluted from the lactose chromatography column at a similar soluble lactose concentration as wild-type Gal3 and the NL-A-Gal3 + sfGFP-B + mRuby-C heterotrimer, suggesting that the carbohydrate-binding affinity of Gal3 was not affected by fusion to either the B or C peptide. Quantification of the area under the curve indicated that ∼75% of the protein was in the bound fraction in the NL-A + sfGFP-B-Gal3 + mRuby-C mixture, whereas only ∼61% of the protein was in the bound fraction for the NL-A + sfGFP-B + mRuby-C-Gal3 mixture. When compared alongside the analysis of the NL-A-Gal3 + sfGFP-B + mRuby-C mixture, this suggested that the extent of heterotrimer assembly was highest when Gal3 was fused to sfGFP-B and lowest when Gal3 was fused to mRuby-C.
Finally, we measured BRET/FRET in the bound fraction eluent collected from the ternary mixtures of NL-A + sfGFP-B-Gal3 + mRuby-C and NL-A + sfGFP-B + mRuby-C-Gal3 to determine if the three proteins co-eluted. A strong NL emission peak, as well as weaker sfGFP and mRuby emission peaks, were observed when furimazine was added to the bound eluent fraction collected from both ternary mixtures (Fig. 5C). However, the mixture of NL-A + sfGFP-B-Gal3 + mRuby-C (Fig. 5C, top) yielded more pronounced green fluorescence emission than the mixtures of NL-A-Gal3 + sfGFP-B + mRuby-C (Fig. 4E) and NL-A + sfGFP-B + mRuby-C-Gal3 (Fig. 5C, bottom), which generally correlated with the trend for the percentage of protein in the bound fraction of each mixture. We also note that in contrast to the mixture analyzed in Fig. 4E, any unassembled NL-A was likely to be eliminated in the void fraction of the NL-A + sfGFP-B-Gal3 + mRuby-C and NL-A + sfGFP-B + mRuby-C-Gal3 mixtures. Thus, the BRET/FRET analysis in Fig. 5C is only assessing the activity of proteins in the assembled state. Taken together, the results presented in Fig. 4 and 5 demonstrate that an additional functional domain can be appended onto the C-terminus of the coiled-coil scaffold, increasing the total types of functionalities in the assembly from 3 to 4. However, the co-assembly of A, B, and C may be affected by attaching protein cargoes onto both the N- and C-termini and, although not demonstrated here, the extent of this effect is likely to depend on the physical properties of the protein.
To further extend the functional capabilities of this platform, we asked whether the lactose-binding affinity conferred upon NL-A, sfGFP-B, and mRuby-C via their heterotrimeric co-assembly could be tuned by varying the number of Gal3 domains, including dimeric and trimeric Gal3 (Fig. 6A). We recently demonstrated that the lactose-binding affinity of synthetic Gal3 homo-oligomers could be tuned by varying the number of domains from one to six.43 To test the tunability of heterotrimer carbohydrate-binding affinity, we evaluated different mixtures of NL-A, sfGFP-B, and mRuby-C with and without the Gal3 domain. Native PAGE suggested that any combination of A, B, and C fusion proteins with or without Gal3 domains could co-assemble, as indicated by unique Coomassie and co-localized luminescent/fluorescent bands in the mixtures when compared to the proteins alone (Fig. 6B and ESI† S7). As with the other ternary mixtures studied above, excess NL and sfGFP fusions were detected in the lanes corresponding to each ternary mixture in the native PAGE gels, suggesting that co-assembly does not approach completion under any condition tested.
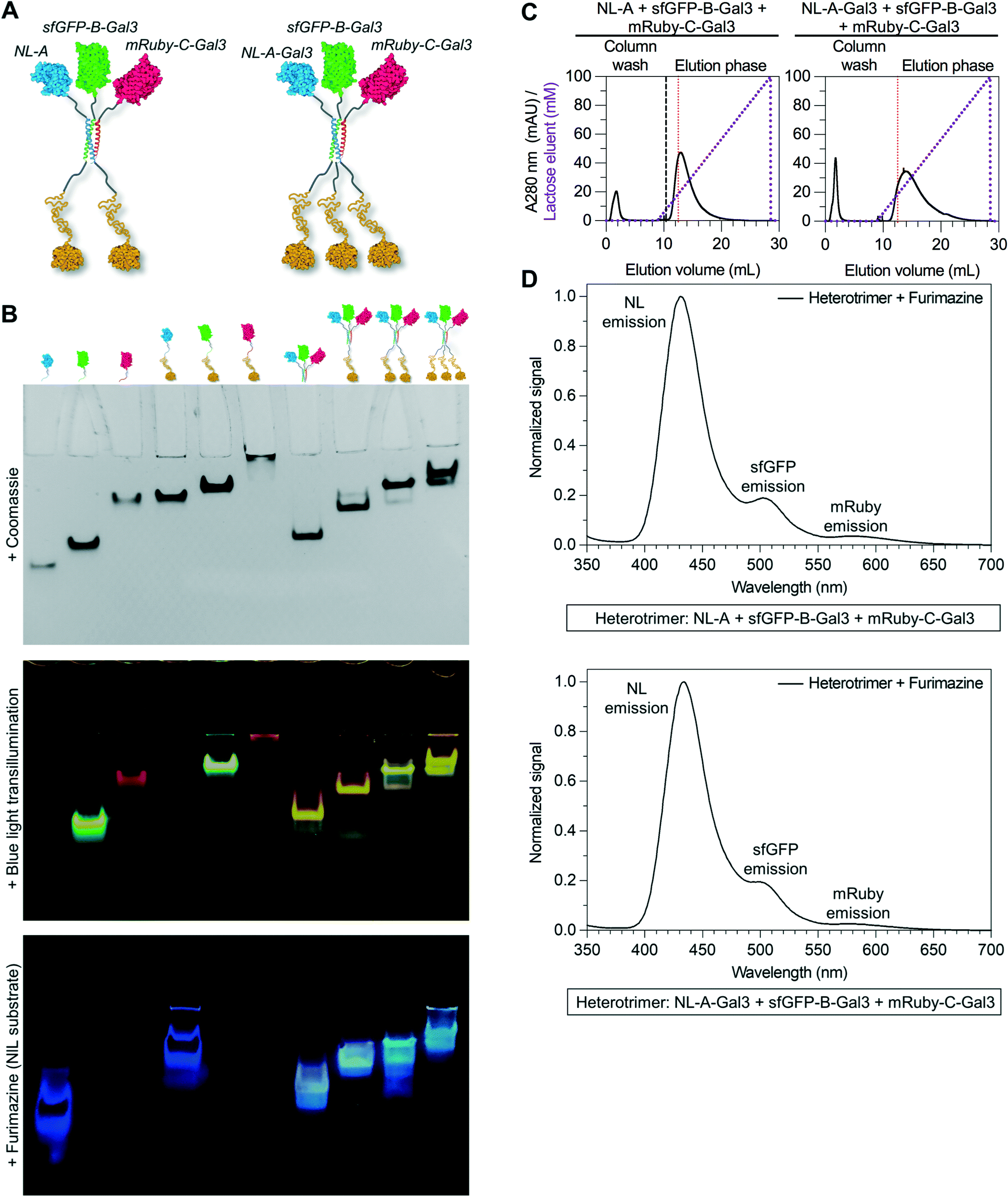 | ||
| Fig. 6 Tuning the stoichiometry of Gal3 within the heterotrimeric co-assembly. (A) Structural illustration of the heterotrimer fused to either two or three Gal3 domains. (B) Native PAGE analysis of the six different fusion proteins and four different heterotrimers integrating up to three Gal3 domains. Bands within the gel were detected by Coomassie staining, blue light transillumination, and furimazine treatment. (C) Immobilized lactose affinity chromatography traces of the heterotrimer fused to two and three Gal3 domains (left and right, respectively). For comparison, the vertical red dashed line indicates the concentration of lactose required to elute wild-type Gal3. (D) Sequential BRET/FRET analysis of the heterotrimer fused to two and three Gal3 domains (top and bottom, respectively). | ||
We used lactose-affinity chromatography to further characterize the co-assembly of the mixtures of fusion proteins with different numbers of Gal3 domains. Here, we expected that the relative binding affinity, which correlates with the soluble lactose competitor concentration, would increase when two proteins with Gal3 domains were co-assembled together. When a mixture of NL-A + sfGFP-B-Gal3 + mRuby-C-Gal3 was passed through a lactose-agarose column, the elution profile shifted to the right relative to the elution for wild-type Gal3 (Fig. 6C, left), as well as to the heterotrimers with one Gal3 domain studied above (Fig. 4c and 5b). The rightward shift of this elution profile was similar to the elution profile for an sfGFP-Gal3 homodimer reported previously,43 suggesting that two Gal3 domains were co-integrated into the construct that eluted in the bound fraction from the ternary mixture studied here. Quantification of the area under the curve demonstrated that ∼71% of the protein was in the bound fraction, indicating that the presence of the second Gal3 domain did not adversely affect A + B + C co-assembly when compared to the relative efficiency of assembly of constructs with one Gal3 domain.
Likewise, when a mixture of NL-A-Gal3 + sfGFP-B-Gal3 + mRuby-C-Gal3 was passed through a lactose-agarose column, the elution profile shifted even further to the right, suggesting a further increase in apparent lactose-binding affinity (Fig. 6C, right). The rightward shift of this elution profile was similar to the elution profile for a sfGFP-Gal3 homotrimer reported previously,43 suggesting that three Gal3 domains were co-integrated into the eluted construct. Quantification of the area under the curve demonstrated that ∼84% of the protein was in the bound fraction, indicating that the presence of the third Gal3 domain did not adversely affect A + B + C co-assembly when compared to the relative efficiency of assembly of constructs with one or two Gal3 domains. Notably, this was the highest percentage of protein recovered from any bound fraction; the low level of detectable protein at the concentration of lactose required to release wild-type Gal3 suggested that the majority of proteins in this ternary mixture were either co-assembled and bound immobilized lactose, or were eluted in the void fraction. We did not characterize the state of the proteins in the void fraction, but suggest that their inability to bind lactose could either be due to the large size of the construct preventing efficient access to the immobilized lactose, or that the neighboring domains imposed a steric impediment that prevented Gal3 interaction with the immobilized lactose.
Finally, we measured BRET/FRET to determine if all three proteins were present and spatially co-localized in the bound fraction eluent. Recall that in control samples containing NL, sfGFP, and mRuby, which cannot co-assemble, no BRET/FRET was observed because the domains were not in close enough proximity (Fig. 3B). Thus, any BRET/FRET observed here would be expected to be due to co-assembly of NL-A-Gal3, sfGFP-B-Gal3, and mRuby-C-Gal3. A strong NL emission peak, as well as weaker sfGFP and mRuby emission peaks, were observed when furimazine was added to the bound fraction eluents collected from the Gal3x2 and Gal3x3 mixtures (Fig. 6D). This indicated that the three different proteins were co-assembled in a manner that placed the NL, sfGFP, and mRuby domains sufficiently close for BRET and FRET. However, the efficiency of BRET was lower in these heterotrimers when compared to heterotrimers formed with either zero or one Gal3 domain. This could be due to an increase in the intermolecular distance between NL and sfGFP in assemblies with an increasing number of Gal3 domains, or other physical features that we have not yet considered.
Conclusions
The results reported here demonstrate that the distinct peptide strands of a heterotrimeric coiled-coil can be used as fusion tags to mediate the heterogeneous co-assembly of proteins into multifunctional supramolecular constructs. The A, B, C peptide trio used here was previously developed and characterized by Alber and colleagues, and was shown to preferentially form a heterotrimeric coiled-coil when the molecules were combined, although some tendency for self-association was noted when the molecules were alone.45 Here we used this trio of peptides as a model to test the hypothesis that heterogeneous coiled-coils can be used to create modular protein assemblies with up to four different protein domains at precisely defined stoichiometries. This represents a significant advance beyond the mono- and bi-functional protein assemblies built using coiled-coil scaffolds that have been reported previously. Collectively, the data presented here demonstrate that predictably heterogenous protein co-assembly is possible using a heterotrimeric coiled-coil scaffold. Co-integrating a luciferase enzyme and green/red fluorescent protein pair that demonstrate sequential BRET/FRET due to close spatial proximity upon co-assembly highlights the potential of this approach to create sophisticated diagnostics. Further, co-integrating a carbohydrate-binding galectin-3 domain highlights the potential of this approach to create targeted drug delivery vehicles. The modular nature of the fusion protein design, coupled with the multiplicity afforded by heterogeneous ternary co-assembly, provides a significantly greater range of possible interchangeable subunit combinations than what is achievable through heterodimeric coiled-coils used previously (Fig. 1). Further, the defined strand number afforded by the self-limiting, deterministic nature of α-helical coiled-coils provides a molecular level stoichiometric precision of domain composition that would be difficult to achieve in other supramolecular systems.Nonetheless, there remains room for improvement with this approach. In particular, the results presented here demonstrate that protein co-assembly is not quantitative or near-quantitative under the employed conditions. Rather, some unassembled protein was detected in each ternary mixture studied, where the unassembled protein concentration was likely determined by the dissociation (or association) constant of the coiled-coil. Future efforts to design a peptide trio demonstrating higher heterogeneous co-assembly affinity and minimal off-pathway homogeneous self-association could lead to more stable constructs with greater extent of formation. Further, the results presented here suggest that appending proteins onto the termini of the A, B, or C peptides may affect their co-association, although the dissociation constants of the heterotrimers were not measured on a case-by-case basis here. Future efforts to optimize the peptide affinity and/or the construct design (e.g., linker length) could yield improvements in co-assembly irrespective of the appended protein cargo. Finally, BRET/FRET efficiency depends on intermolecular distance. Here, BRET/FRET was used as an analytical reporter and no effort was made to optimize these events. However, future efforts to optimize the construct design (e.g., linker length and protein domain orientation) could enable opportunities to create constructs demonstrating highly efficient coupled or synergistic chemical reactions.
Author contributions
S. A. F. designed experiments, conducted experiments, analysed data, and wrote the paper. A. R. conceived of the project and conducted experiments. A. S. contributed to optimization of fusion protein expression and purification. A. C.-S. conducted experiments. G. A. H. conceived of the project, analysed data, and wrote the paper.Conflicts of interest
G. A. H. is a founder and shareholder of Anchor Biologics, Inc. Patents have been applied for by the University of Florida that name S. A. F., A. R., and G. A. H., as co-inventors.Acknowledgements
This work was supported by the National Institutes of Health (NIBIB, R03-EB019684-02; NIDCR, R01-DE027301-01) and in part by the NIH/NCATS Clinical and Translational Science Awards to the University of Florida UL1TR001427 and TL1TR001428.Notes and references
- B. Alberts, Cell, 1998, 92, 291–294 CrossRef CAS PubMed.
- Y. Takada, X. Ye and S. Simon, Genome Biol., 2007, 8, 215 CrossRef PubMed.
- S. C. Meuer, O. Acuto, R. E. Hussey, J. C. Hodgdon, K. A. Fitzgerald, S. F. Schlossman and E. L. Reinherz, Nature, 1983, 303, 808–810 CrossRef CAS PubMed.
- C. A. Janeway, Jr., B. Jones and A. Hayday, Immunol. Today, 1988, 9, 73–76 CrossRef.
- K. Ozaki and W. J. Leonard, J. Biol. Chem., 2002, 277, 29355–29358 CrossRef CAS PubMed.
- H. Colognato and P. D. Yurchenco, Dev. Dyn., 2000, 218, 213–234 CrossRef CAS PubMed.
- J. A. M. Bard, E. A. Goodall, E. R. Greene, E. Jonsson, K. C. Dong and A. Martin, Annu. Rev. Biochem., 2018, 87, 697–724 CrossRef CAS PubMed.
- N. P. King, J. B. Bale, W. Sheffler, D. E. McNamara, S. Gonen, T. Gonen, T. O. Yeates and D. Baker, Nature, 2014, 510, 103–108 CrossRef CAS.
- J. B. Bale, S. Gonen, Y. Liu, W. Sheffler, D. Ellis, C. Thomas, D. Cascio, T. O. Yeates, T. Gonen, N. P. King and D. Baker, Science, 2016, 353, 389–394 CrossRef CAS PubMed.
- K. M. Muller, K. M. Arndt and T. Alber, Methods Enzymol., 2000, 328, 261–282 CAS.
- R. Liu and G. A. Hudalla, Molecules, 2019, 24, 1450 CrossRef CAS.
- G. A. Hudalla, T. Sun, J. Z. Gasiorowski, H. Han, Y. F. Tian, A. S. Chong and J. H. Collier, Nat. Mater., 2014, 13, 829–836 CrossRef CAS.
- Q. Shao, K. M. Wong, D. T. Seroski, Y. Wang, R. Liu, A. K. Paravastu, G. A. Hudalla and C. K. Hall, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 4710–4717 CrossRef CAS PubMed.
- D. N. Woolfson, Adv. Protein Chem., 2005, 70, 79–112 CrossRef CAS.
- D. N. Woolfson, Subcell. Biochem., 2017, 82, 35–61 CAS.
- S. H. Lee, D. W. Park, E. S. Sung, H. R. Park, J. K. Kim and Y. S. Kim, Mol. Immunol., 2010, 47, 816–824 CrossRef CAS PubMed.
- C. Y. Fan, C. C. Huang, W. C. Chiu, C. C. Lai, G. G. Liou, H. C. Li and M. Y. Chou, FASEB J., 2008, 22, 3795–3804 CrossRef CAS PubMed.
- J. van der Leij, A. van den Berg, G. Harms, H. Eschbach, H. Vos, P. Zwiers, R. van Weeghel, H. Groen, S. Poppema and L. Visser, Mol. Immunol., 2007, 44, 506–513 CrossRef CAS PubMed.
- M. M. Fettis, S. A. Farhadi and G. A. Hudalla, Biomater. Sci., 2019, 7, 1852–1862 RSC.
- S. A. Farhadi, M. M. Fettis, R. Liu and G. A. Hudalla, Front. Chem., 2019, 7, 898 CrossRef CAS.
- E. K. O'Shea, K. J. Lumb and P. S. Kim, Curr. Biol., 1993, 3, 658–667 CrossRef.
- A. W. Reinke, R. A. Grant and A. E. Keating, J. Am. Chem. Soc., 2010, 132, 6025–6031 CrossRef CAS.
- H. C. Chang, Z. Bao, Y. Yao, A. G. Tse, E. C. Goyarts, M. Madsen, E. Kawasaki, P. P. Brauer, J. C. Sacchettini and S. G. Nathenson, et al. , Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 11408–11412 CrossRef CAS.
- P. Kern, R. E. Hussey, R. Spoerl, E. L. Reinherz and H. C. Chang, J. Biol. Chem., 1999, 274, 27237–27243 CrossRef CAS.
- R. Busch, A. Pashine, K. C. Garcia and E. D. Mellins, J. Immunol. Methods, 2002, 263, 111–121 CrossRef CAS PubMed.
- T. Ojima-Kato, K. Fukui, H. Yamamoto, D. Hashimura, S. Miyake, Y. Hirakawa, T. Yamasaki, T. Kojima and H. Nakano, Protein Eng., Des. Sel., 2016, 29, 149–157 CrossRef CAS PubMed.
- K. M. Arndt, K. M. Muller and A. Pluckthun, J. Mol. Biol., 2001, 312, 221–228 CrossRef CAS.
- X. Wang, P. Zhong, P. P. Luo and K. C. Wang, PLoS One, 2011, 6, e19023 CrossRef CAS PubMed.
- J. K. Doh, J. D. White, H. K. Zane, Y. H. Chang, C. S. Lopez, C. A. Enns and K. E. Beatty, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 12961–12966 CrossRef CAS PubMed.
- T. J. Magliery, C. G. Wilson, W. Pan, D. Mishler, I. Ghosh, A. D. Hamilton and L. Regan, J. Am. Chem. Soc., 2005, 127, 146–157 CrossRef CAS PubMed.
- T. Plaper, J. Aupic, P. Dekleva, F. Lapenta, M. M. Keber, R. Jerala and M. Bencina, Sci. Rep., 2021, 11, 9136 CrossRef CAS PubMed.
- H. Robson Marsden, N. A. Elbers, P. H. Bomans, N. A. Sommerdijk and A. Kros, Angew. Chem., Int. Ed., 2009, 48, 2330–2333 CrossRef PubMed.
- J. Fernandez-Rodriguez and T. C. Marlovits, Protein Sci., 2012, 21, 511–519 CrossRef CAS PubMed.
- F. Murschel, C. Fortier, M. Jolicoeur, R. S. Hodges and G. De Crescenzo, Biomacromolecules, 2017, 18, 965–975 CrossRef CAS PubMed.
- C. D. Jun, M. Shimaoka, C. V. Carman, J. Takagi and T. A. Springer, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 6830–6835 CrossRef CAS PubMed.
- P. U. Le, A. E. Lenferink, M. Pinard, J. Baardsnes, B. Massie and M. D. O'Connor-McCourt, Protein Expression Purif., 2009, 64, 108–117 CrossRef CAS.
- Y. Assal, Y. Mizuguchi, M. Mie and E. Kobatake, Bioconjugate Chem., 2015, 26, 1672–1677 CrossRef CAS PubMed.
- Y. Assal, M. Mie and E. Kobatake, Biomaterials, 2013, 34, 3315–3323 CrossRef CAS.
- P. Lequoy, F. Murschel, B. Liberelle, S. Lerouge and G. De Crescenzo, Acta Biomater., 2016, 29, 239–247 CrossRef CAS.
- S. Siew, M. Kaneko, M. Mie and E. Kobatake, J. Mater. Chem. B, 2016, 4, 2512–2518 RSC.
- W. Wang, W. He, L. Wang, G. Zhang and B. Gao, Immunol. Cell Biol., 2013, 91, 360–367 CrossRef CAS PubMed.
- S. A. Farhadi, E. Bracho-Sanchez, M. M. Fettis, D. T. Seroski, S. L. Freeman, A. Restuccia, B. G. Keselowsky and G. A. Hudalla, Nat. Commun., 2018, 9, 4943 CrossRef PubMed.
- S. A. Farhadi, R. Liu, M. W. Becker, E. A. Phelps and G. A. Hudalla, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2024117118 CrossRef CAS PubMed.
- Z. Wu, K. W. Johnson, B. Goldstein, Y. Choi, S. F. Eaton, T. M. Laue and T. L. Ciardelli, J. Biol. Chem., 1995, 270, 16039–16044 CrossRef CAS PubMed.
- S. Nautiyal, D. N. Woolfson, D. S. King and T. Alber, Biochemistry, 1995, 34, 11645–11651 CrossRef CAS PubMed.
- P. Carriba, G. Navarro, F. Ciruela, S. Ferre, V. Casado, L. Agnati, A. Cortes, J. Mallol, K. Fuxe, E. I. Canela, C. Lluis and R. Franco, Nat. Methods, 2008, 5, 727–733 CrossRef CAS PubMed.
- A. K. Dunker, I. Silman, V. N. Uversky and J. L. Sussman, Curr. Opin. Struct. Biol., 2008, 18, 756–764 CrossRef CAS PubMed.
- E. Folta-Stogniew, Methods Mol. Biol., 2006, 328, 97–112 CAS.
- Y. Xu, D. W. Piston and C. H. Johnson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 151–156 CrossRef CAS PubMed.
- R. Heim and R. Y. Tsien, Curr. Biol., 1996, 6, 178–182 CrossRef CAS PubMed.
- A. M. Loening, A. Dragulescu-Andrasi and S. S. Gambhir, Nat. Methods, 2010, 7, 5–6 CrossRef CAS.
- A. Flores-Ibarra, S. Vertesy, F. J. Medrano, H. J. Gabius and A. Romero, Sci. Rep., 2018, 8, 9835 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1me00083g |
| This journal is © The Royal Society of Chemistry 2022 |