
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePAC-FragmentDEL – photoactivated covalent capture of DNA-encoded fragments for hit discovery†
Huiyong
Ma
 b,
James B.
Murray
a,
Huadong
Luo
b,
Xuemin
Cheng
b,
Qiuxia
Chen
b,
Chao
Song
b,
Cong
Duan
b,
Ping
Tan
b,
Lifang
Zhang
b,
Jian
Liu
b,
Barry A.
Morgan
b,
Jin
Li
b,
Jinqiao
Wan
b,
Lisa M.
Baker
a,
William
Finnie
a,
Lucie
Guetzoyan
a,
Richard
Harris
a,
Nicole
Hendrickson
a,
Natalia
Matassova
a,
Heather
Simmonite
a,
Julia
Smith
a,
Roderick E.
Hubbard
*a and
Guansai
Liu
b
b,
James B.
Murray
a,
Huadong
Luo
b,
Xuemin
Cheng
b,
Qiuxia
Chen
b,
Chao
Song
b,
Cong
Duan
b,
Ping
Tan
b,
Lifang
Zhang
b,
Jian
Liu
b,
Barry A.
Morgan
b,
Jin
Li
b,
Jinqiao
Wan
b,
Lisa M.
Baker
a,
William
Finnie
a,
Lucie
Guetzoyan
a,
Richard
Harris
a,
Nicole
Hendrickson
a,
Natalia
Matassova
a,
Heather
Simmonite
a,
Julia
Smith
a,
Roderick E.
Hubbard
*a and
Guansai
Liu
b
aVernalis (R&D) Ltd, Granta Park, Abington, Cambridge, CB21 6GB, UK. E-mail: r.hubbard@vernalis.com; j.murray@vernalis.com; l.baker@vernalis.com; w.finnie@vernalis.com; l.guetzoyan@vernalis.com; r.harris@vernalis.com; n.hendrickson@vernalis.com; natalia_matassova@hotmail.com; h.simmonite@vernalis.com; j.smith@vernalis.com; r.hubbard@vernalis.com
bHitGen Inc., Building 6, No. 8 Huigu First East Road, Tianfu International Bio-Town, Shuangliu District, Chengdu 610000, Sichuan, P. R. China. E-mail: huiyong.ma@hitgen.com; hd.luo@hitgen.com; xm.cheng@hitgen.com; qx.chen@hitgen.com; chao.song@hitgen.com; cong.duan@hitgen.com; ping.tan@hitgen.com; lf.zhang@hitgen.com; jian.liu@hitgen.com; barry.morgan@hitgen.com; jin.li@hitgen.com; jq.wan@hitgen.com; gs.liu@hitgen.com
First published on 26th August 2022
Abstract
We describe a novel approach for screening fragments against a protein that combines the sensitivity of DNA-encoded library technology with the ability of fragments to explore what will bind. Each of the members of the library consists of a fragment which is linked to a photoactivatable diazirine moiety. Split and pool synthesis combines each fragment with a set of linkers with the version of the library reported here containing some 70k different compounds, each with an individual DNA code. Incubation of the library with a protein sample is followed by photoactivation, washing and subsequent PCR and sequencing which allows the individual fragment hits to be identified. We illustrate how the approach allows successful hit fragment identification using only microgram quantities of material for two targets. PAK4 is a kinase for which conventional fragment screening has generated many advance leads. The as yet undrugged target, 2-epimerase, presents a more challenging active site for identification of hit compounds. In both cases, PAC-FragmentDEL identified fragments validated as hits by ligand-observed NMR measurements and crystal structure determination of off-DNA sample binding to the proteins.
Introduction
The screening of small compounds (typically <16 heavy atoms) or fragments is now established as an effective approach for identification of hit compounds in drug discovery.1,2 Fragment hits have been developed into lead compounds3 and clinical candidates4 for many therapeutic targets, with 6 approved fragment-derived drugs now treating patients. There are two essential features of the approach: (1) low molecular weight compounds are sufficiently complex to make detectable interactions with a portion of a binding site but unlike larger compounds (as screened in high throughput screens) are not sterically prevented from binding5 and (2) most screens generate a number of fragment hits, providing choice for identifying suitable hit and lead compounds. Fragments that bind with affinities (KD) as low as 5 mM have successfully been optimised to clinical candidates, and such weak binding is usually detected in a screen against a target using a biophysical approach such as surface-plasmon resonance (SPR), ligand-observed (LO-) or protein-observed (PO-) NMR, thermal shift analysis or X-ray crystallography.6Fragments bind to chemically attractive sites on a protein. A recent development for chemical biology is the use of photoactivated fragments to identify and characterise such sites. This was initially demonstrated by the Cravatt group7 where a small number (14) of fragments were each prepared linked to diazirine and an alkyne handle (Fig. 1(a)). The fragments were incubated with cells, photoactivated and click chemistry used to isolate the proteins which had been covalently labelled. GSK extended the idea with an approach they termed PhABits,8 where a larger number (567) of fragments were similarly linked to diazirine and alkyne and incubated with an isolated protein. In both cases, identification of the protein or binding site used mass spectrometry, which constrains the number of fragments that can be screened and the amount of protein target required.
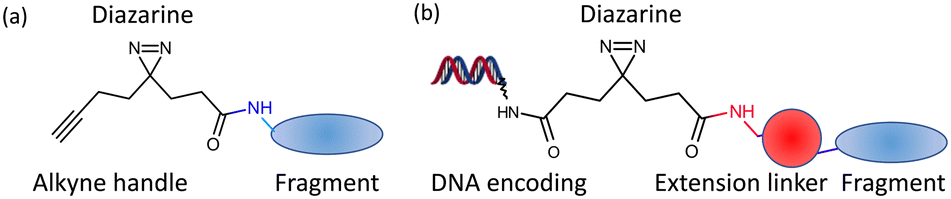 | ||
| Fig. 1 Design of photoactivatable fragments (a). The libraries from Cravatt7 and GSK;8 (b). PAC-FragmentDEL. | ||
DNA-encoded library (DEL) technology emerged 30 years ago9 and has been developed as an effective method for hit identification, where successive synthesis using split and pool combinatorial methods builds a library of many millions (billions) of molecules,10 each of which is labelled with a specific DNA tag. The usual process is to incubate a DEL with the target that can be immobilised onto a bead, then separate the target, wash to remove low affinity or non-binding compounds and then use PCR and sequencing to identify the hits. Provided the DEL hits are retained on the protein during the wash process, the approach provides great sensitivity, allowing many millions of molecules to be screened rapidly against only small amounts of protein material. In this way, DEL screening has successfully identified hit compounds that have been progressed to a number of clinical candidates.11 The stringency of the wash process in DEL screening typically requires a KD for binding of compound to target of less than tens of μM, so non-covalent DELs are unlikely to find many single fragment hits for most targets. There have been reports of DELs constructed with a fragment on each strand of DNA to exploit avidity where the two fragments bind in adjacent sites12 or for a dynamic combinatorial approach where fragments react with each other on the surface of the protein.13–15 These approaches enrich ligands on specific targets by either crosslinking of DELs on a target or affinity enhancement.
Here we describe and demonstrate PAC-FragmentDEL, an approach where photoactivated covalent capture of DNA-encoded fragments can identify hits binding to a target. There have been reports16,17 of DELs of non-fragment sized molecules incorporating an electrophile, where forming a covalent bond with a protein requires reaction with an available cysteine side chain. In contrast, photoactivation generates a carbene which rapidly inserts into nearby bonds.18 The PAC-FragmentDEL approach is demonstrated with two examples. The serine/threonine protein kinase PAK4 (p21 activated kinase 4) was chosen as a positive control.13 The bacterial enzyme 2-epimerase (UDP-N-acetylglucosamine 2-epimerase) was chosen as a challenging protein for which no validated inhibitors have been reported. 2-Epimerase catalyses the conversion of UDP-GlcNAc to UDP-ManNAc for biosynthesis of glycans that maintain antibacterial cell wall integrity and is a potential target for antimicrobial development.19
The binding to PAK4 and 2-epimerase of off-DNA samples of fragment hits identified by PAC-FragmentDEL screens have been validated by ligand observed NMR (LO-NMR) and X-ray structure determination.
Results and discussion
Library design and synthesis
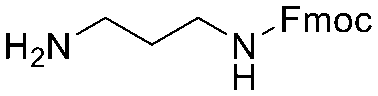
Each molecule in a PAC-FragmentDEL library is composed of three parts: DNA encoding system, linker, and fragment (Fig. 1(b)), where the DNA encoding system records the chemical matter installed at a specific step. The linker consists of a fixed linker containing the diazirine and an extension of variable length of 0 to 13 atoms between diazirine and fragment to probe different depths of the binding site. The work reported here used two types of library as summarised in Fig. 2. Type 1 consists of sub-library 1, made from 2634 amines and sub-library 2 from 4783 fragments containing amine capping reagents (acids, aldehydes, sulfonyl chlorides), where such functional groups are required to be compatible with DEL chemistry. The Type 2 library consists of sub-libraries 3 and 4, where DEL chemistry was used to merge two smaller fragments, but without an extension linker.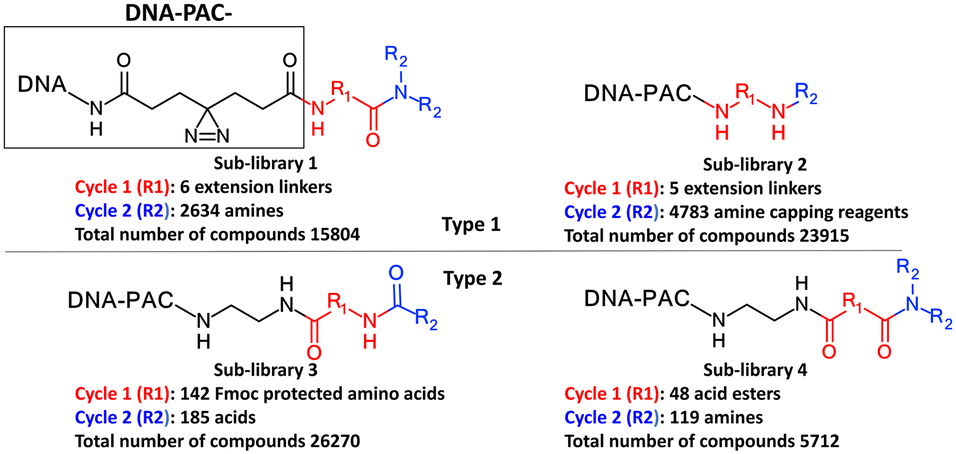 | ||
| Fig. 2 Design of the PAC-FragmentDEL libraries. The same DNA and photoactivation group is present in all sub-libraries. The DNA encodes the library and the groups added in cycles 1 and 2. | ||
The same headpiece-conjugated diazirine linker is present in all library members. The library was synthesized in a “split & pool” fashion in two cycles. The synthesis of the Type 1 sub-libraries is outlined in Scheme S1.† For sub-library 1, the conjugated diazirine linker was split into 6 wells, each ligated to a unique code, followed by installation of the amino ester extension linker shown in Table 1. Upon completion of the reaction, as monitored by LCMS, the 6 wells were pooled and purified; hydrolysis of ester to free carboxylic acid afforded the cycle 1 mixture. This cycle 1 mixture was split into 2636 wells (2634 fragments and 2 nulls) and each was encoded with unique code 2, followed by installation of a fragment by amide condensation. The final step was ligation with the library ID DNA and purification. Sub-library 2 was synthesised in a similar way, with reactions differing according to the nature of the 5 extension linkers (Table 1) and electrophilic handles on the fragments. This resulted in 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 804 members of sub-library 1 and 23905 members of sub-library 2.
804 members of sub-library 1 and 23905 members of sub-library 2.
| Item | Structure of extension linkers | Used in |
|---|---|---|
| 1 | Null | Sub-library 1 |
| 2 |
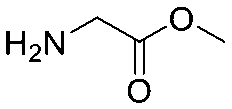
|
Sub-library 1 |
| 3 |
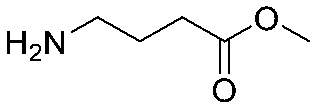
|
Sub-library 1 |
| 4 |
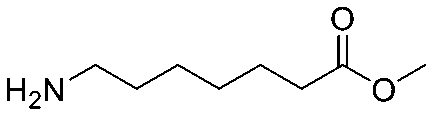
|
Sub-library 1 |
| 5 |
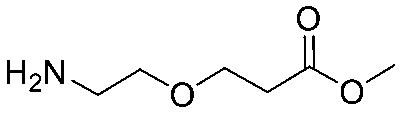
|
Sub-library 1 |
| 6 |
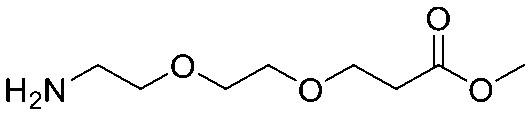
|
Sub-library 1 |
| 7 |
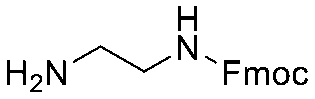
|
Sub-library 2 |
| 8 |
|
Sub-library 2 |
| 9 |
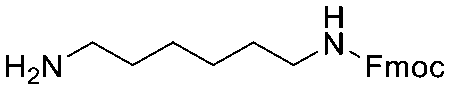
|
Sub-library 2 |
| 10 |
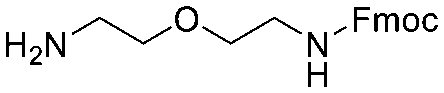
|
Sub-library 2 |
| 11 |
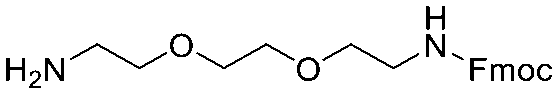
|
Sub-library 2 |
The Markush structure of the Type 2 sub-libraries 3 and 4 is also shown in Fig. 2 which were synthesised as outlined in Scheme S2† in a similar fashion to the Type 1 library but without an extension linker. Fig. S1† is a summary of the properties of the fragment component of the compounds in the two types of library. Although these properties will be affected by the DNA code, diazirine and linker, the main recognition moiety of the Type 1 libraries is well within the usual property space of most fragment libraries.20,21 Most Type 1 fragments contain less than 18 heavy atoms, aLogP less than 2, less than 3 hydrogen bond acceptors and 2 hydrogen bond donors. The two cycles of synthesis of Type 2 sub-libraries mostly generates fragments with between 18 to 26 heavy atoms, MW of 260 to 350, slightly lower aLogP and small increase in the number of hydrogen bonding groups. Although this larger molecular weight means the coverage of potential chemical space is not as great for these Type 2 compounds compared to the smaller Type 1 library compounds,22 the larger compounds may be more suitable for screening against binding sites which will bind with lower ligand efficiency.23 For many of the compounds in the Type 2 library, the cycle 1 fragment forms a structured linker, positioning the cycle 2 fragment for binding. This construction of the overall PAC-FragmentDEL library means that the same fragment is present a number of times with different extended linkers in Type 1 sub-libraries and in some cases is also present in the Type 2 sub-libraries.
Performing a PAC-FragmentDEL screen
The process for performing a screen is outlined schematically in Fig. 3. The combined Type 1 and Type 2 PAC-FragmentDEL (notionally 107 copy number per individual DEL molecule, estimated by qPCR) is incubated with 250 pmol of the his-tagged protein for 1 h at room temperature and then exposed to UV light of 365 nm for 10 minutes on ice. The sample is then incubated with Ni-magnetic beads and washed 10 times with buffer to remove non-binding PAC-FragmentDEL members and then heat washed for 10 minutes with buffer at 75 °C and once at 95 °C to denature the protein and remove non-covalent binders. An equivalent sample is taken through the process without exposure to UV light. Two further pairs (with and without UV light exposure) are also prepared similarly; one pair contains 10 μM of a competitor compound and the other pair are control samples with blank beads (i.e. no protein).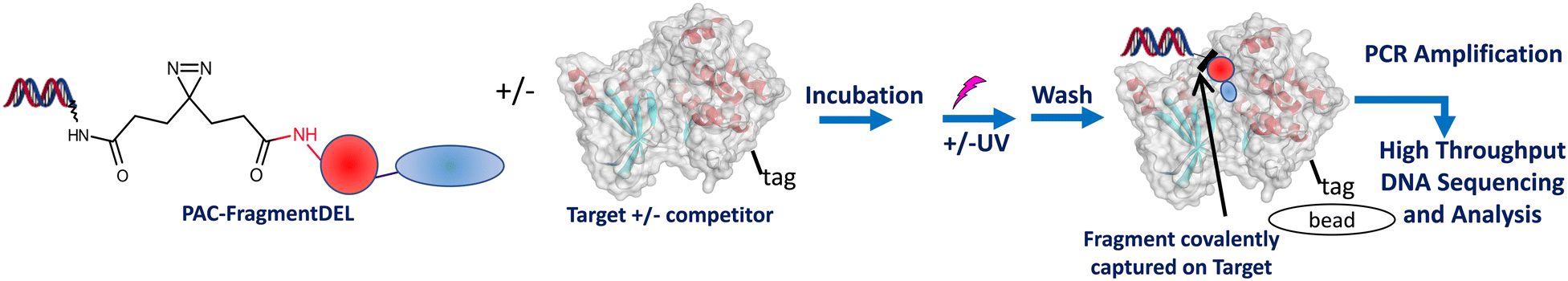 | ||
| Fig. 3 The PAC-FragmentDEL process. Typically a total of 3 pairs of samples (+/− UV exposure) are prepared with the library incubated with blank beads, and for the target +/− competitor. | ||
The DNA remaining in the prepared samples is then subjected to PCR amplification, sequenced and the number of sequence counts for each fragment in each sample recorded. In brief, the analysis consists of subtraction of signal from the controls to remove fragments that have bound strongly without UV irradiation to either beads or protein or to remove fragments which show higher sequence count in the presence of the competitor (either artefacts or compounds that bind more strongly when the competitor is bound to the protein). The selected fragments are those that show enrichment compared to controls and with enhanced sequence count in the absence of the competitor. Off-DNA samples of the selected fragments (see Fig. S3† for the mapping between fragment and corresponding PAC-FragmentDEL member) are directly available for Type 1 or require synthesis for Type 2 library members.
Initial proof of concept experiment with PAK4
An initial set of experiments were performed on the enzyme PAK4 for individual PAC-FragmentDEL compounds encoding fragments known to bind to PAK4: 1 (with null linker) and 2 (with linker 7 from Table 1) where the KD measured by SPR for the isolated fragments was measured as 510 μM and 300 μM respectively. The results of incubation with the isolated PAC-FragmentDEL members (Fig. 4) demonstrate significant enrichment of the conjugated fragments binding to bead-immobilised PAK4 compared to the blank bead control when exposed to UV light.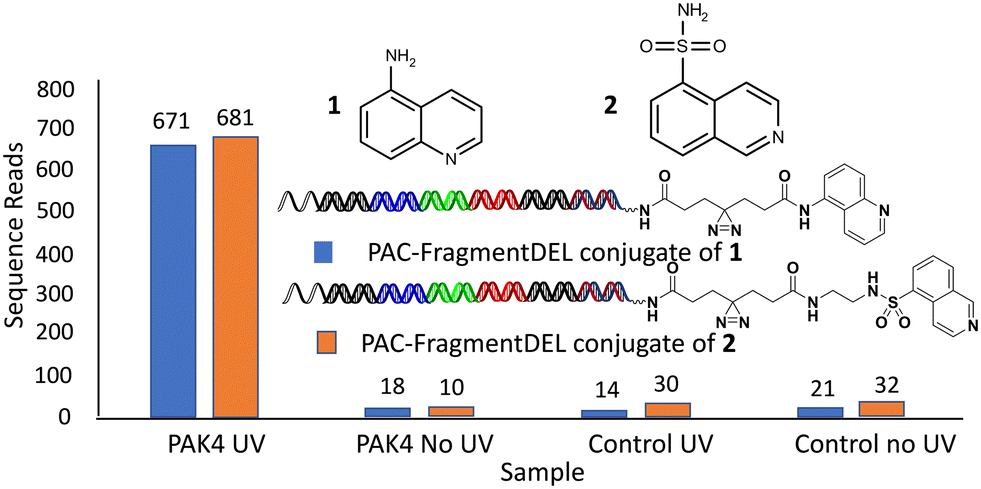 | ||
| Fig. 4 Individual PAC-FragmentDEL compounds bind to PAK4. | ||
PAC-FragmentDEL screen for PAK4 and 2-epimerase
For PAK4, the screen was performed in the presence and absence of 10 μM of compound 3 which binds to the ATP-binding site of PAK4 with a KD measured by SPR of 50 nM. Analysis of the sequence counts obtained for the competitive hits showed that none of these PAC-FragmentDEL compounds bound to the beads (Fig. 5(a)), but some bound weakly to PAK4 in the absence of UV irradiation. In total, 301 fragments were identified that bound competitively, some with different linkers (hence more than 301 dots in the PAK4 plots in Fig. 5). 11 of the fragments were chosen for further validation based on signal strength and structural diversity.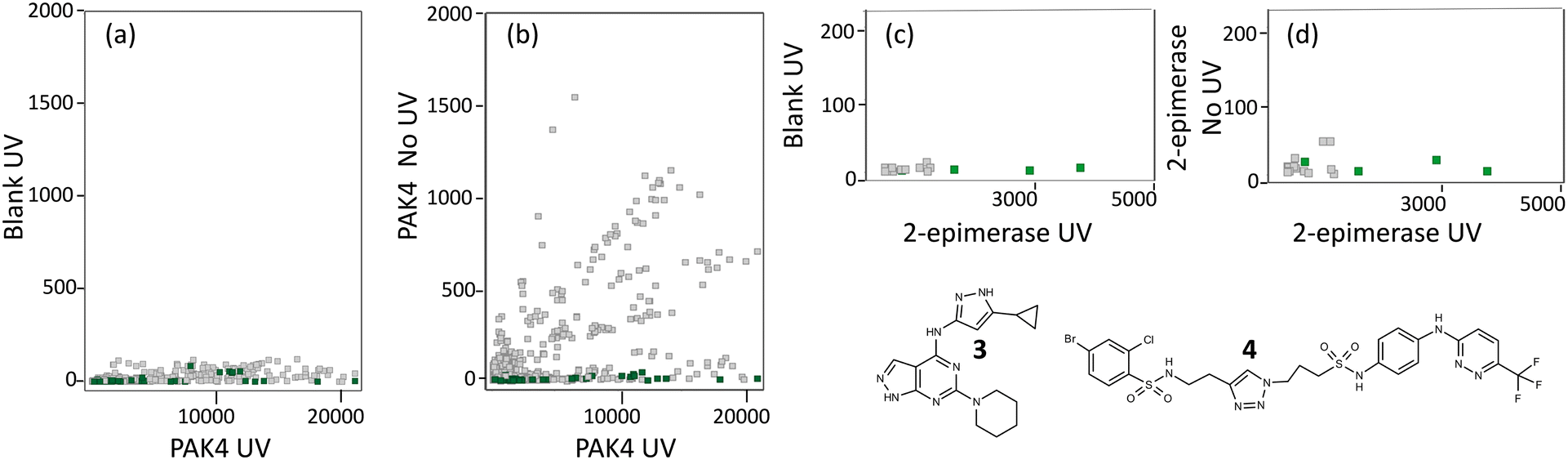 | ||
| Fig. 5 Identifying PAC-FragmentDEL hits. Each dot represents a particular fragment which was identified as binding to the target but not binding in the presence of the competitor. The x-axis is the sequence count for that fragment in the sample isolated after UV photoactivation of the PAC-FragmentDEL incubated with the indicated protein, labelled UV (a) and (b) for PAK4 and (c) and (d) for 2-epimerase. The y-axis of each plot is the count for each fragment in the control samples: (b) and (d) where the sample with protein was not irradiated, labelled no UV and (a) and (c) where the sample without protein was irradiated, labelled blank UV. The plot of sequence count for blank UV vs. protein UV show that the binding is specific to the protein and the plots of protein UV vs. blank UV show the extent of fragment binding without photoactivation, treated as non-specific. The fragments selected for validation are shown in green. | ||
The 301 hits contained several different linkers and at least in the case of PAK4, there was no preference for a specific linker.
For 2-epimerase, there is a reported inhibitor (Epimerox24) which in our hands did not show binding by isothermal titration calorimetry (ITC). We therefore generated compound 4 (manuscript in preparation) which binds to 2-epimerase with a KD of 100 nM as measured by ITC and was competitive with UDP-GlcNAc as confirmed by LO-NMR (see Fig. S2†). The PAC-FragmentDEL screen for 2-epimerase was performed in the presence and absence of 4 and as can be seen in Fig. 5(c), the sequence count of the PAC-FragmentDEL hits is low. The sequence count for a particular library member is affected by a number of factors, in particular the success of individual reactions and thus number of compound copies. However, the overall low sequence count for 2-epimerase probably reflects weak affinity for this binding site and only 21 competitive fragments could be identified, 9 of which were selected for experiments to validate binding.
Hit validation by NMR and X-ray crystallography
The fragments within the selected hits from the two PAC-FragmentDEL screens were sourced or synthesised. These isolated off-DNA fragments were then validated as binding by measurement of competitive LO-NMR spectra and by determination of crystal structures of off-DNA compounds bound to the protein. All of the hits were confirmed as binding by LO-NMR, crystal structures were obtained for 10 of the 11 PAK4 hits and 5 of the 9 2-epimerase hits. The hits for which data is presented are shown in Fig. 6.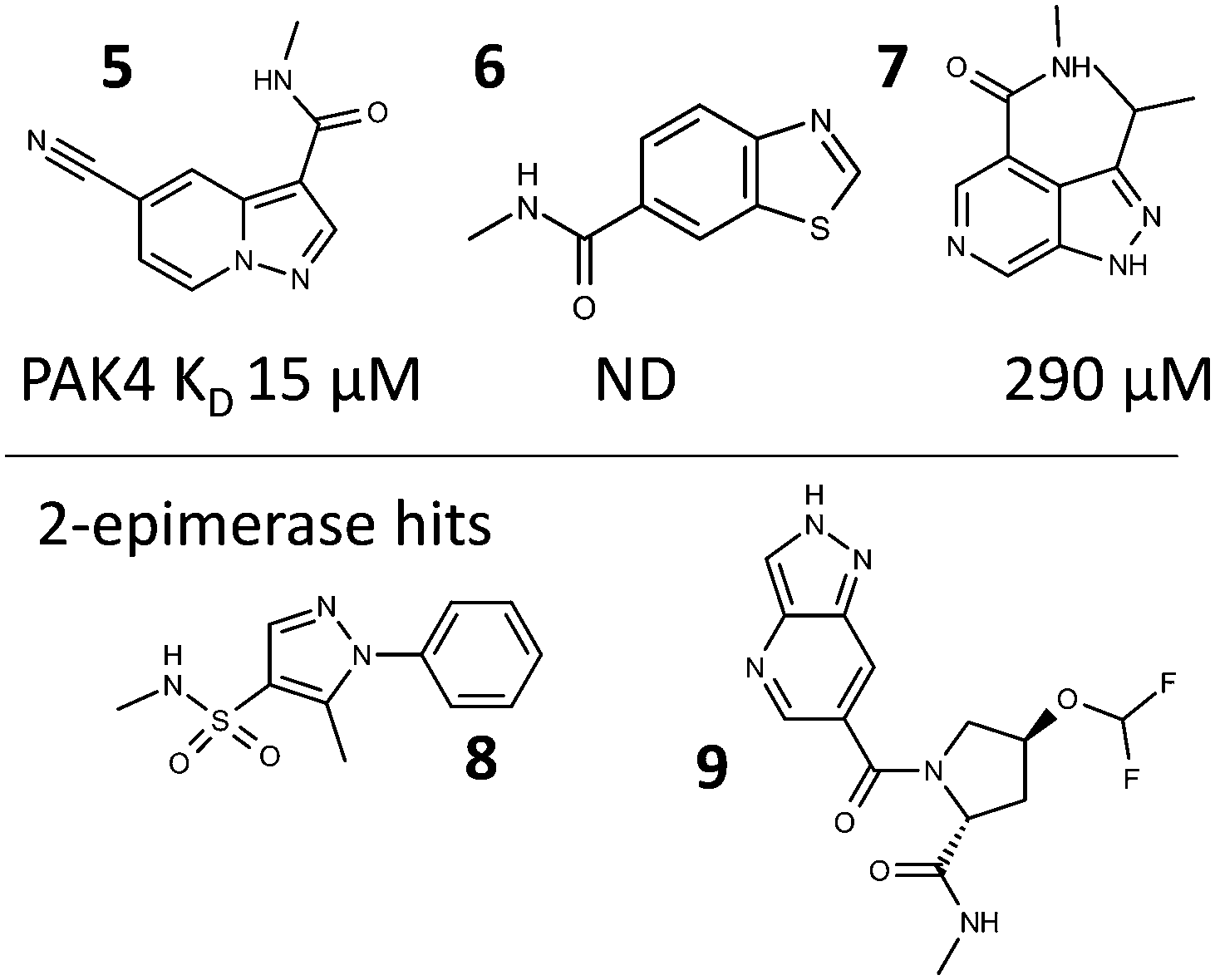 | ||
| Fig. 6 Fragment hits for which binding information is presented; with the KD for 5, 6 and 7 as measured by SPR. | ||
A representative set of spectra for binding to PAK4 is shown for 5 in Fig. 7, where the changes in the NMR spectra in the presence and absence of staurosporine are consistent with binding to the ATP-binding site. Fig. 8 shows detail of the crystal structures of the ATP-binding site of PAK4 with each of 5, 6 and 7 bound. Each of these hits are similar to sub-structures in known kinase hinge binding compounds, but none have been reported previously as binding to PAK4. As explained earlier, each individual fragment can be present in both Type 1 and Type 2 PAC-FragmentDELs. 5 was a hit several times with different extended linkers in Type 1, 6 was a hit only in Type 2 library and 7 was found from both Type 1 and Type 2 libraries.
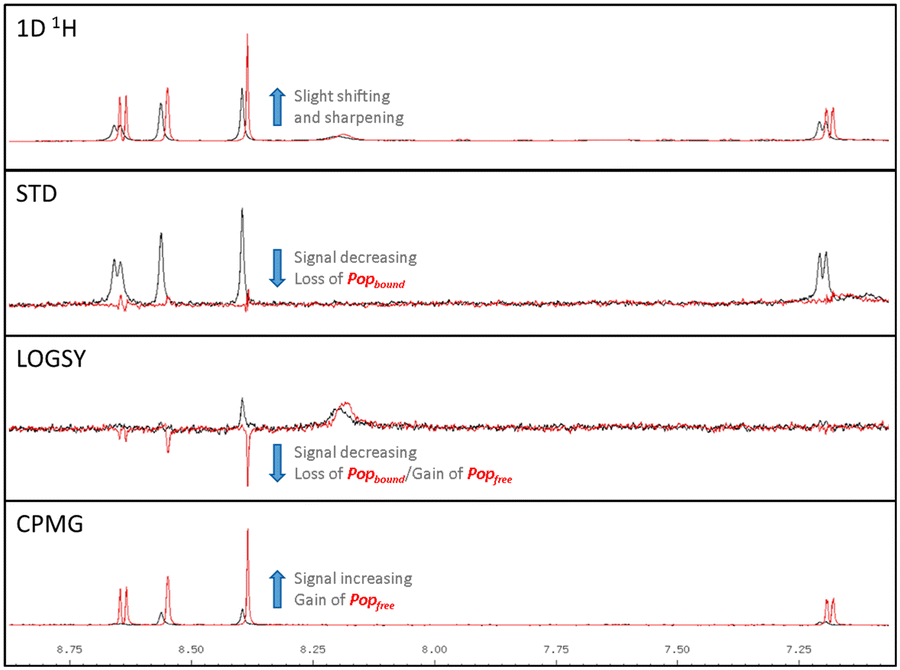 | ||
| Fig. 7 LO-NMR spectra for 5 binding to PAK4 in the presence (red) and absence (black) of 10 μM 3, indicating the shifts observed, annotated how they reflect differences in the bound (Popbound) and free (Popfree) ligand population. | ||
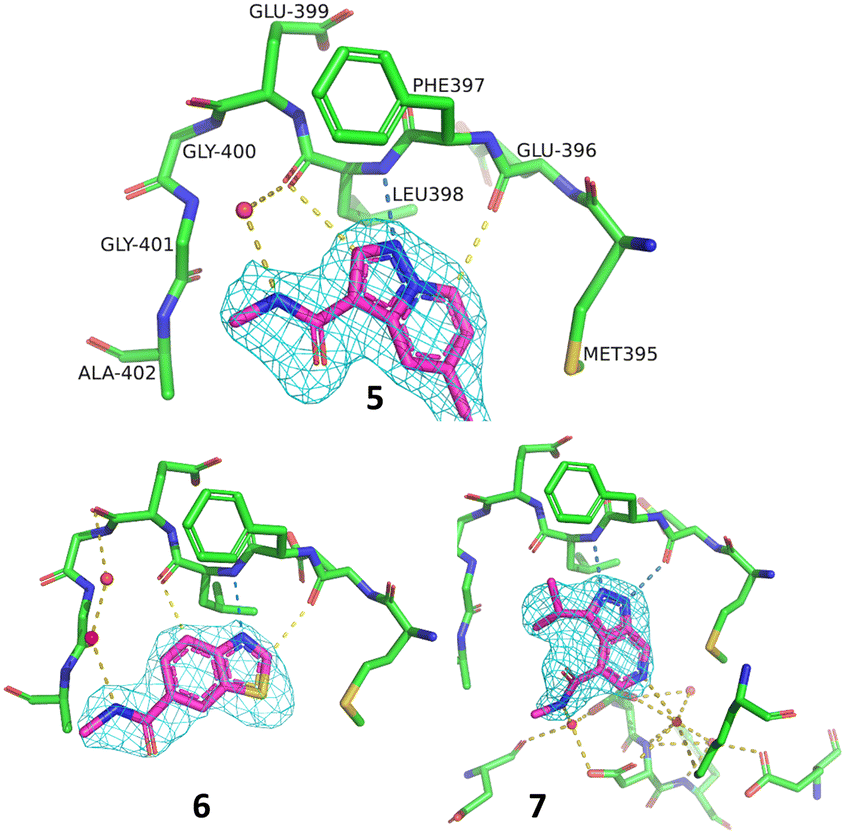 | ||
| Fig. 8 Details of the ATP-binding site in the crystal structure of 5 (PDB code: 8AHH), 6 (8AHI) and 7 (8AHG) bound to PAK4 all in the same orientation, amino acids labelled for 5, with hydrogen bonding contacts and 2Fo–Fc maps on the compound contoured at 1σ. | ||
Fig. 9(a) shows the crystal structure of 2-epimerase (PDB code: 3BEO) with UDP and UDP-GlcNAc bound. A comparison of the molecular surface of this structure (Fig. 9(b)) with the structure with the hits 8 and 9 (Fig. 9(c) and (d)) emphasises the conformational change to an active form associated with substrate and cofactor binding.25 The fragment hits both bind in the UDP cofactor site, with the enzyme in an open, deactivated conformation. Fragment 8 was from Type 1 and 9 from Type 2 library.
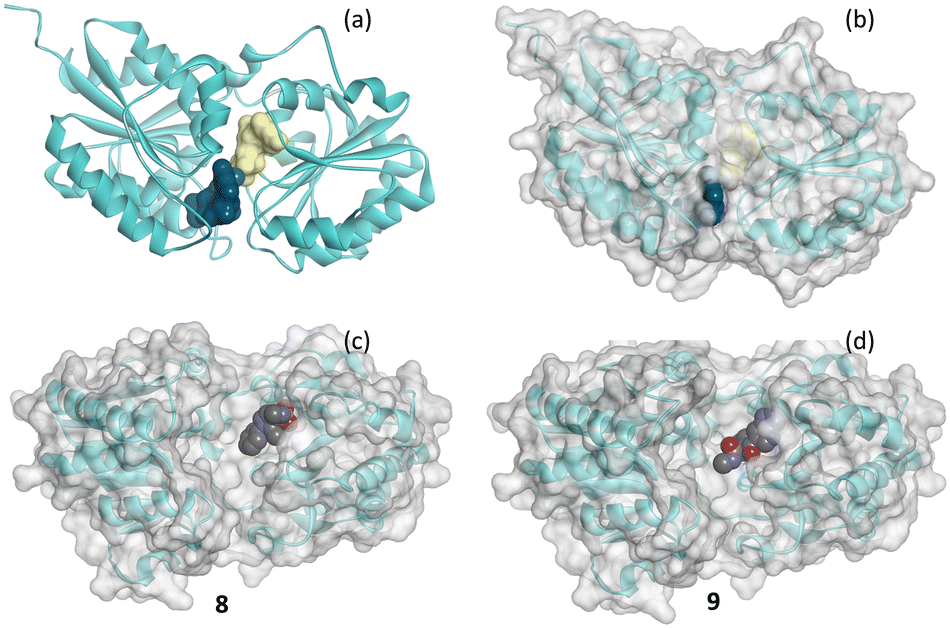 | ||
| Fig. 9 Schematic of the secondary structure of 2-epimerase showing (a) structure of pdb code 3BEO with a molecular surface on each of UDP (yellow) and UDP-GlcNAc (blue) and with (b) transparent molecule surface on the protein and (c) and (d) crystal structures of 8 (8AHE) and 9 (8AHF) shown in CPK bound to 2-epimerase. | ||
Perspectives and conclusions
There are several advantages of the PAC-FragmentDEL approach. Only a small amount of tagged protein (250 pmol per sample) is required, and it is relatively rapid to identify hits, with selection from the DEL library and sequencing and analysis taking only a few weeks to perform. The solubility of the fragments that are incorporated into the library can be lower than required for most fragment screening approaches. Typically, biophysical screens (such as NMR or SPR) require aqueous solubility for the fragment in the high 100 s of μM to enable detection of weak binding. Conjugation with DNA removes this solubility requirement, at least for the PAC-FragmentDEL screen, broadening the chemical space of fragments that can be included in the library. Finally, confirmation of hits depends on which library they are from. The fragments that are conjugated in the Type 1 sub-libraries are mostly readily available; for Type 2, synthesis of off-DNA samples is required.In the examples described here, the selection of PAC-FragmentDEL hits is by comparison of the hits in the presence and absence of a competitor ligand that binds to the site of interest. If such a competitor is not available (and it could be a peptide or protein partner), then an alternate approach would be to block the active site by mutation(s) and compare the hits with those obtained for the wild type protein. Similarly, comparative screens of orthologous proteins could be used to identify selective fragments.
There are many interesting features of the PAC-FragmentDEL approach which are yet to be investigated, such as which types of targets and binding sites are appropriate for the approach? The examples described here demonstrate a low false positive rate for the fragments selected for characterisation, but will this hold for all targets? The false negative rate (which is related to the cut-offs applied in hit analysis) is of less interest – the priority in early hit discovery is to find some chemically tractable hits for progression and as with any screening, it is accepted that compounds will be missed. Another area for analysis is the impact on whether a particular fragment is found to bind of different extension linkers in Type 1 sub-libraries or the coupling of two fragments in Type 2 libraries. We have also not investigated scale up of individual PAC-FragmentDEL hits for incubation with the target protein to characterise by mass spectrometry or crystallography, where the covalent bond is formed on photoactivation. The carbene formed by photoactivation inserts into proximal H–heteroatom and H–C bonds.18 A limited analysis of the hits in the PhABits8 study showed covalent bonding to acidic side chains. It is likely that the high temperature washing step in our protocol would hydrolyse at least a proportion of any ester bonds formed but any C–C bonds formed would survive. It remains to be seen whether the PAC-FragmentDEL approach can successfully identify new binding sites. This will depend on whether such specific binding is masked by a large amount of non-specific attachment from hits that are not affected by competitor.
In conclusion, the PAC-FragmentDEL approach successfully identified fragments that were validated as binding to the two targets presented here. PAK4 is from the protein kinase class of proteins for which the ATP binding groove is well established as a highly druggable26–28 site for small molecules to bind. This target was selected as a demonstrator for the approach, with known fragments included in the screen as a positive control. 2-Epimerase has a more open and polar active site which is less ligandable and for which no small molecules have been reported to bind. The hit rate from the PAC-FragmentDEL screen confirmed the different ligandability28 of the two proteins and selected hits were validated as binding in solution (NMR) and by crystallography.
Experimental
Protein production
Synthesis of PAC-FragmentDEL and fragments that are not commercially available
See ESI.†Screening, sequencing, and decoding
Selection was carried out using a KingFisher Duo Prime Purification System (ThermoFisher) in a 96 well plate. 2.5 μM PAK4 (or 2-epimerase) was incubated with a mixture of the Type 1 and Type 2 sub-libraries at room temperature for 60 min in 100 μL selection buffer (25 mM HEPES, 150 mM NaCl, 0.01% Triton X-100, 10 mM imidazole, 0.3 mg ml−1 ssDNA, pH 7.4). The protein–PAC-FragmentDEL mixture was incubated on ice and subjected to UV irradiation at 365 nm for 10 min. The control without UV treatment was included in parallel. Protein was immobilized on Ni-Charged Magbeads at RT for 30 min. The weak or non-specific binders were removed by 1 min wash in 500 μL selection buffer 10 times, followed by two 10 min heat washes at 75 °C and one at 95 °C in 50 mM HEPES, 300 mM NaCl, pH 7.4 buffer to remove the non-covalent binders. The DNA in the sample was quantified by qPCR and samples amplified by PCR and sequenced on an Illumina NovaSeq 6000.The selection output was quantified by qPCR using PowerUp SYBR Green Master Mix (Thermo, A25778) and amplified by PCR using Q5 Hot Start High-Fidelity 2X Master Mix (NEB, M0494L). PCR cycle was determined by qPCR result. The resulting PCR amplicons were purified by using QIAGEN-MinElute PCR Purification Kit (QIAGEN, 28006) and quantified using a Qubit DNA High-Sensitivity Kit (Invitrogen, 32854) before library construction and sequencing. The library preparation was performed by Nextflex Rapid DNA-Seq Kit (BI00 Scientific, 5144-08) following the manufacturer's operational manual. Specifically, 10 ng DNA was used as input. DNA library was constructed through end-repair, dA-tailing, adaptor-ligation and 13 cycles PCR amplification followed a final purification by AMPure XP beads (Beckman, A63882). Subsequently, the resulting libraries were denatured and neutralized and subjected to 116 bp single-end sequencing on an Illumina NovaSeq platform (Illumina, USA) by Chengdu HitGen Pharmaceuticals Inc., China.
After decoding, the DNA sequences enriched in selection were translated into chemical structures and visualized by DataWarrior files (Openmolecules). The enrichment of fragments can be determined by the sequence count which is the copy number of the corresponding fragment's DNA barcode. The difference between groups was compared by enrichment ratio which is calculated by sequence count in different selection conditions, and background subtraction was performed by removing high signals that appeared in the blank control.
Ligand-observed NMR (LO-NMR) validation of competitive compound binding
1D 1H, STD,29 the water ligand observed via gradient spectroscopy (waterLOGSY),30 and relaxation-filtered31 spectra were acquired at 298 K using excitation sculpting to suppress the solvent peak on a 600 MHz Bruker Avance III spectrometer fitted with a cryoprobe. Samples contained 10 μM protein with either 100 μM or 500 μM compound in 20 mM Tris pH 7.5 (PAK4) or 20 mM Tris pH 7.5, 50 mM NaCl (2-epimerase). Competition was determined by the addition of 30 μM staurosporine (PAK4) or 1 mM UDP-GlcNAc (2-epimerase).SPR to measure KD for binding to PAK4
SPR measurements were performed on a BIAcore T200 instrument (BIAcore GE Healthcare) at 25 °C with double His8-tagged PAK4 prepared in running buffer of 20 mM HEPES pH 7.5, 150 mM NaCl, 5 mM MgCl2, 0.01% P-20, 0.025 mM EDTA and 1% DMSO. Series S NTA chips were prepared by injection of 0.5 mM NiCl2 at 10 μL min−1 until the sensor showed an RU of 80–160. A solution of 200 nM PAK4 was injected at 10 μl min−1 until a stable surface was achieved (usually 2000–5000 RU). The reference surface on the chip was without Ni2+ to control for non-specific binding and refractive index changes. For most measurements, the immobilised protein was flushed with running buffer for each concentration in titration series. If needed, the sensor surface can be regenerated between experiments by consecutive injections of 0.35 M EDTA and 0.1 M imidazole (all for 120 s at a flow rate of 10 μl min−1) to eliminate any carry-over of the protein and/or analyte. Measurement of fragment binding was conducted in dose response titrations of nine two-fold diluted experimental points with the top concentration of 500–2000 μM. Each measurement reported is the average of at least three determinations. Data processing was performed using BIAevaluation 2.1 (BIAcore GE Healthcare Bio-SciencesCorp) software. Sensorgrams were double referenced prior to global fitting of the concentration series to the Steady State Affinity or 1:1 Kinetic models.
ITC to measure KD for binding of 4 to 2-epimerase
ITC measurements were performed using an iTC200 instrument (Microcal, GE Healthcare). The feedback mode was ‘low’ with reference power setting of 4 μCals−1. The cell was stirred at 1000 rpm and thermostated at 25 °C. All experiments were performed using the dialysis buffer (see below) with 1% DMSO (v/v). Both experiments were conducted with 6 μM ligand in the cell and 60 μM protein in the syringe. The experiments were conducted with 12 injections of 3.05 μL and 240 s spacing. The first ‘waste’ injection of 0.5 μL was discarded in each case. All data were fitted to a one site model using the provided software.Protein (7.2 mg ml−1) was dialysed with stirring for four hours 20 °C, in 25 mM HEPES pH 7.4, 150 mM NaCl. Upon recovery the protein was filtered (0.22 μM spin filter). The protein concentration was determined by UV absorbance spectroscopy at 280 nM using an extinction coefficient of 17420. The dialysis buffer was degassed, this was then used for subsequent preparation of protein and ligand solutions for the titration experiments. For each experiment the ligand was freshly prepared from a 20 mM DMSO stock solution.
X-ray crystal structures of protein–ligand complexes
Data deposition
The refined crystal structures of 5, 6 and 7 bound to PAK4 and of 8 and 9 bound to 2-epimerase are deposited in the PDB with codes 8AHH, 8AHI, 8AHG, 8AHE and 8AHF respectively.Author contributions
GL, JW, HM, and HL designed the PAC-FragmentDEL approach and wrote the initial draft of the manuscript, CS and CD performed the chemistry of DEL, QC contributed on DEL screening, Jian Liu performed the sequencing, XC and LZ analysed the DEL screening data, PT resynthesized the hits. REH oversaw the initial concept of the application of PAC-FragmentDEL to these targets and wrote the final version of the manuscript. JBM designed the details of the experiments on these targets and made ITC measurements and analysed the data. NM made SPR measurements. LMB conducted all the crystallographic structure determination and RH and HS conducted all NMR experiments. WF, JS and NH produced the protein samples. BAM and Jin Li oversaw the PAC-FragmentDEL design and manuscript writing.Conflicts of interest
There are no conflicts to declare. All authors are employees of either Vernalis (R&D) Ltd or HitGen who funded the research.References
- D. A. Erlanson, S. W. Fesik, R. E. Hubbard, W. Jahnke and H. Jhoti, Twenty years on: The impact of fragments on drug discovery, Nat. Rev. Drug Discovery, 2016, 15, 605–619 CrossRef CAS PubMed.
- M. N. Schulz and R. E. Hubbard, Recent progress in fragment-based lead discovery, Curr. Opin. Pharmacol., 2009, 9, 615–621 CrossRef CAS PubMed.
- I. J. P. de Esch, D. A. Erlanson, W. Jahnke, C. N. Johnson and L. Walsh, Fragment-to-lead medicinal chemistry publications in 2020, J. Med. Chem., 2022, 65, 84–99 CrossRef CAS PubMed.
- D. A. Erlanson, Practical fragments blog – fragments in the clinic, 2021 Search PubMed.
- A. R. Leach and M. M. Hann, Molecular complexity and fragment-based drug discovery: Ten years on, Curr. Opin. Chem. Biol., 2011, 15, 489–496 CrossRef CAS PubMed.
- J.-P. Renaud, C.-W. Chung, U. H. Danielson, U. Egner, M. Hennig, R. E. Hubbard and H. Nar, Biophysics in drug discovery: Impact, challenges and opportunities, Nat. Rev. Drug Discovery, 2016, 15, 679–698 CrossRef CAS PubMed.
- C. G. Parker, A. Galmozzi, Y. Wang, B. E. Correia, K. Sasaki, C. M. Joslyn, A. S. Kim, C. L. Cavallaro, R. M. Lawrence, S. R. Johnson, I. Narvaiza, E. Saez and B. F. Cravatt, Ligand and target discovery by fragment-based screening in human cells, Cell, 2017, 168, 527–541 CrossRef CAS PubMed.
- E. K. Grant, D. J. Fallon, M. M. Hann, K. G. M. Fantom, C. Quinn, F. Zappacosta, R. S. Annan, C. W. Chung, P. Bamborough, D. P. Dixon, P. Stacey, D. House, V. K. Patel, N. C. O. Tomkinson and J. T. Bush, A photoaffinity-based fragment-screening platform for efficient identification of protein ligands, Angew. Chem., Int. Ed., 2020, 59, 21096–21105 CrossRef CAS PubMed.
- S. Brenner and R. A. Lerner, Encoded combinatorial chemistry, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 5381–5383 CrossRef CAS PubMed.
- M. A. Clark, R. A. Acharya, C. C. Arico-Muendel, S. L. Belyanskaya, D. R. Benjamin, N. R. Carlson, P. A. Centrella, C. H. Chiu, S. P. Creaser, J. W. Cuozzo, C. P. Davie, Y. Ding, G. J. Franklin, K. D. Franzen, M. L. Gefter, S. P. Hale, N. J. Hansen, D. I. Israel, J. Jiang, M. J. Kavarana, M. S. Kelley, C. S. Kollmann, F. Li, K. Lind, S. Mataruse, P. F. Medeiros, J. A. Messer, P. Myers, H. O'Keefe, M. C. Oliff, C. E. Rise, A. L. Satz, S. R. Skinner, J. L. Svendsen, L. Tang, K. van Vloten, R. W. Wagner, G. Yao, B. Zhao and B. A. Morgan, Design, synthesis and selection of DNA-encoded small-molecule libraries, Nat. Chem. Biol., 2009, 5, 647–654 CrossRef CAS PubMed.
- A. Gironda-Martinez, E. J. Donckele, F. Samain and D. Neri, DNA-encoded chemical libraries: A comprehensive review with succesful stories and future challenges, ACS Pharmacol. Transl. Sci., 2021, 4, 1265–1279 CrossRef CAS PubMed.
- M. Wichert, N. Krall, W. Decurtins, R. M. Franzini, F. Pretto, P. Schneider, D. Neri and J. Scheuermann, Dual-display of small molecules enables the discovery of ligand pairs and facilitates affinity maturation, Nat. Chem., 2015, 7, 241–249 CrossRef CAS PubMed.
- F. V. Reddavide, M. Cui, W. Lin, N. Fu, S. Heiden, H. Andrade, M. Thompson and Y. Zhang, Second generation DNA-encoded dynamic combinatorial chemical libraries, Chem. Commun., 2019, 55, 3753–3756 RSC.
- S. Melkko, J. Scheuermann, C. E. Dumelin and D. Neri, Encoded self-assembling chemical libraries, Nat. Biotechnol., 2004, 22, 568–574 CrossRef CAS PubMed.
- K. E. Denton and C. J. Krusemark, Crosslinking of DNA-linked ligands to target proteins for enrichment from DNA-encoded libraries, MedChemComm, 2016, 7, 2020–2027 RSC.
- C. Zambaldo, J. P. Daguer, J. Saarbach, S. Barluenga and N. Winssinger, Screening for covalent inhibitors using DNA-display of small molecule libraries functionalized with cysteine reactive moieties, MedChemComm, 2016, 7, 1340–1351 RSC.
- J. P. Guilinger, A. Archna, M. Augustin, A. Bergmann, P. A. Centrella, M. A. Clark, J. W. Cuozzo, M. Däther, M.-A. Guié, S. Habeshian, R. Kiefersauer, S. Krapp, A. Lammens, L. Lercher, J. Liu, Y. Liu, K. Maskos, M. Mrosek, K. Pflügler, M. Siegert, H. A. Thomson, X. Tian, Y. Zhang, D. L. Konz Makino and A. D. Keefe, Novel irreversible covalent btk inhibitors discovered using DNA-encoded chemistry, Bioorg. Med. Chem., 2021, 42, 116223 CrossRef CAS PubMed.
- J. R. Hill and A. A. B. Robertson, Fishing for drug targets: A focus on diazirine photoaffinity probe synthesis, J. Med. Chem., 2018, 61, 6945–6963 CrossRef CAS PubMed.
- R. Schuch, A. J. Pelzek, A. Raz, C. W. Euler, P. A. Ryan, B. Y. Winer, A. Farnsworth, S. S. Bhaskaran, C. E. Stebbins, Y. Xu, A. Clifford, D. J. Bearss, H. Vankayalapati, A. R. Goldberg and V. A. Fischetti, Use of a bacteriophage lysin to identify a novel target for antimicrobial development, PLoS One, 2013, 8, e60754 CrossRef CAS PubMed.
- I. J. Chen and R. E. Hubbard, Lessons for fragment library design: Analysis of output from multiple screening campaigns, J. Comput.-Aided Mol. Des., 2009, 23, 603–620 CrossRef CAS PubMed.
- G. M. Keseru, D. A. Erlanson, G. G. Ferenczy, M. M. Hann, C. W. Murray and S. D. Pickett, Design principles for fragment libraries: Maximizing the value of learnings from pharma fragment-based drug discovery (fbdd) programs for use in academia, J. Med. Chem., 2016, 59, 8189–8206 CrossRef CAS PubMed.
- J.-L. Reymond, The chemical space project, Acc. Chem. Res., 2015, 48, 722–730 CrossRef CAS PubMed.
- A. L. Hopkins, C. R. Groom and A. Alex, Ligand efficiency: A useful metric for lead selection, Drug Discovery Today, 2004, 9, 430–431 CrossRef PubMed.
- Y. Xu, B. Brenning, A. Clifford, D. Vollmer, J. Bearss, C. Jones, V. McCarthy, C. Shi, B. Wolfe, B. Aavula, S. Warner, D. J. Bearss, M. V. McCullar, R. Schuch, A. Pelzek, S. S. Bhaskaran, C. E. Stebbins, A. R. Goldberg, V. A. Fischetti and H. Vankayalapati, Discovery of novel putative inhibitors of udp-glcnac 2-epimerase as potent antibacterial agents, ACS Med. Chem. Lett., 2013, 4, 1142–1147 CrossRef CAS PubMed.
- L. M. Velloso, S. S. Bhaskaran, R. Schuch, V. A. Fischetti and C. E. Stebbins, A structural basis for the allosteric regulation of non-hydrolysing udp-glcnac 2-epimerases, EMBO Rep., 2008, 9, 199–205 CrossRef CAS PubMed.
- P. J. Hajduk, J. R. Huth and C. Tse, Predicting protein druggability, Drug Discovery Today, 2005, 10, 1675–1682 CrossRef CAS PubMed.
- T. A. Halgren, Identifying and characterizing binding sites and assessing druggability, J. Chem. Inf. Model., 2009, 49, 377–389 CrossRef CAS PubMed.
- F. N. Edfeldt, R. H. Folmer and A. L. Breeze, Fragment screening to predict druggability (ligandability) and lead discovery success, Drug Discovery Today, 2011, 16, 284–287 CrossRef CAS PubMed.
- M. Mayer and B. Meyer, Characterization of ligand binding by saturation transfer difference nmr spectroscopy, Angew. Chem., Int. Ed., 1999, 38, 1784–1788 CrossRef CAS PubMed.
- C. Dalvit, G. Fogliatto, A. Stewart, M. Veronesi and B. Stockman, Waterlogsy as a method for primary nmr screening: Practical aspects and range of applicability, J. Biomol. NMR, 2001, 21, 349–359 CrossRef CAS PubMed.
- P. J. Hajduk, E. T. Olejniczak and S. W. Fesik, One-dimensional relaxation- and diffusion-edited nmr methods for screening compounds that bind to macromolecules, J. Am. Chem. Soc., 1997, 119, 12257–12261 CrossRef CAS.
- W. Kabsch, Xds, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Overview of the ccp4 suite and current developments, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- F. Long, R. A. Nicholls, P. Emsley, S. Gra
 ulis, A. Merkys, A. Vaitkus and G. N. Murshudov, Acedrg: A stereochemical description generator for ligands, Acta Crystallogr., Sect. D: Struct. Biol., 2017, 73, 112–122 CrossRef CAS PubMed.
ulis, A. Merkys, A. Vaitkus and G. N. Murshudov, Acedrg: A stereochemical description generator for ligands, Acta Crystallogr., Sect. D: Struct. Biol., 2017, 73, 112–122 CrossRef CAS PubMed. - G. N. Murshudov, A. A. Vagin and E. J. Dodson, Refinement of macromolecular structures by the maximum-likelihood method, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 240–255 CrossRef CAS PubMed.
- P. Emsley and K. Cowtan, Coot: Model-building tools for molecular graphics, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00197g |
| This journal is © The Royal Society of Chemistry 2022 |