
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, biological evaluation and computational studies of pyrazole derivatives as Mycobacterium tuberculosis CYP121A1 inhibitors†
Lama A.
Alshabani
a,
Amit
Kumar
 b,
Sam J.
Willcocks
cd,
Gayathri
Srithiran
d,
Sanjib
Bhakta
d,
D. Fernando
Estrada
b and
Claire
Simons
*a
b,
Sam J.
Willcocks
cd,
Gayathri
Srithiran
d,
Sanjib
Bhakta
d,
D. Fernando
Estrada
b and
Claire
Simons
*a
aSchool of Pharmacy & Pharmaceutical Sciences, Cardiff University, King Edward VII Avenue, Cardiff, CF10 3NB UK. E-mail: simonsc@cardiff.ac.uk
bDepartment of Biochemistry, Jacobs School of Medicine and Biomedical Science, University at Buffalo, Buffalo, New York-14203, USA
cDepartment of Infection Biology, The London School of Hygiene and Tropical Medicine, London, WC1E 7HT UK
dMycobacteria Research Laboratory, Institute of Structural and Molecular Biology, Department of Biological Sciences, Birkbeck, University of London, London WC1E 7HX, UK
First published on 16th August 2022
Abstract
A series of imidazole and triazole diarylpyrazole derivatives were prepared using an efficient 5-step synthetic scheme and evaluated for binding affinity with Mycobacterium tuberculosis (Mtb) CYP121A1 and antimycobacterial activity against Mtb H37Rv. Antimycobacterial susceptibility was measured using the spot-culture growth inhibition assay (SPOTi): the imidazoles displayed minimum inhibitory concentration (MIC90) in the range of 3.95–12.03 μg mL−1 (10.07–33.19 μM) with 11f the most active, while the triazoles displayed MIC90 in the range of 4.35–25.63 μg mL−1 (11.88–70.53 μM) with 12b the most active. Assessment of binding affinity using UV-vis spectroscopy showed that for the imidazole series, the propyloxy (11f) and isopropyloxy (11h) derivatives of the 4-chloroaryl pyrazoles displayed Mtb CYP121A1 type II binding affinity with Kd 11.73 and 17.72 μM respectively compared with the natural substrate cYY (Kd 12.28 μM), while in the triazole series, only the methoxy substitution with the 4-chloroaryl pyrazole (12b) showed good type II Mtb CYP121A1 binding affinity (Kd 5.13 μM). Protein-detected 1D 19F-NMR spectroscopy as an orthogonal strategy was used to evaluate ligand binding independent of perturbations at the haem. For imidazole and triazole compounds, perturbations were more intense than cYY indicating tighter binding and confirming that ligand coordination occurs in the substrate-binding pocket despite very modest changes in UV-vis absorbance, consistent with computational studies and the demonstrated potential anti-tuberculosis properties of these compounds.
Introduction
Tuberculosis (TB) was, prior to COVID-19, the leading cause of death from a single infectious agent. The impact of COVID-19 on TB has been substantial, most notably with TB resources, funding and facilities being redirected for COVID-19 and also a reduction in reporting data for newly diagnosed TB patients.1 Reduced access to testing facilities has resulted in an increase in TB related deaths in 2020 with estimates of 1.3 million deaths in HIV-negative people and 214![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths in HIV-positive people.1 The rise in drug-resistant strains of Mycobacterium tuberculosis (Mtb), the main causative agent of TB, continues to be of concern with rifampicin resistant TB (RR-TB), multidrug resistant TB (MDR-TB) which is resistant to both first line anti-TB drugs rifampicin and isoniazid, and extremely drug resistant TB (XDR-TB) which is resistant against rifampicin and any second line fluoroquinolone plus one second line injectable anti-TB drug, exacerbating the challenge of effectively treating TB.2 Therefore, research looking at alternative Mtb targets, for which no resistance is already present in the population, is of interest to provide additional therapeutic drugs as part of the combinatorial drug approach used for TB treatment.
000 deaths in HIV-positive people.1 The rise in drug-resistant strains of Mycobacterium tuberculosis (Mtb), the main causative agent of TB, continues to be of concern with rifampicin resistant TB (RR-TB), multidrug resistant TB (MDR-TB) which is resistant to both first line anti-TB drugs rifampicin and isoniazid, and extremely drug resistant TB (XDR-TB) which is resistant against rifampicin and any second line fluoroquinolone plus one second line injectable anti-TB drug, exacerbating the challenge of effectively treating TB.2 Therefore, research looking at alternative Mtb targets, for which no resistance is already present in the population, is of interest to provide additional therapeutic drugs as part of the combinatorial drug approach used for TB treatment.
A target of interest is cytochrome P450 121A1 (CYP121A1, mycocyclosin synthase), an essential enzyme for Mtb viability,3 which catalyses the biotransformation of the tyrosine dipeptide cyclo-L-Tyr-L-Tyr (cYY) to mycocyclosin (Fig. 1).4 The exact role of mycocyclosin is not known, however it has been suggested that it may have an essential role in cell growth or structural stability, alternatively inhibition of CYP121A1 could result in an accumulation of cYY, which may be toxic to Mtb.4,5
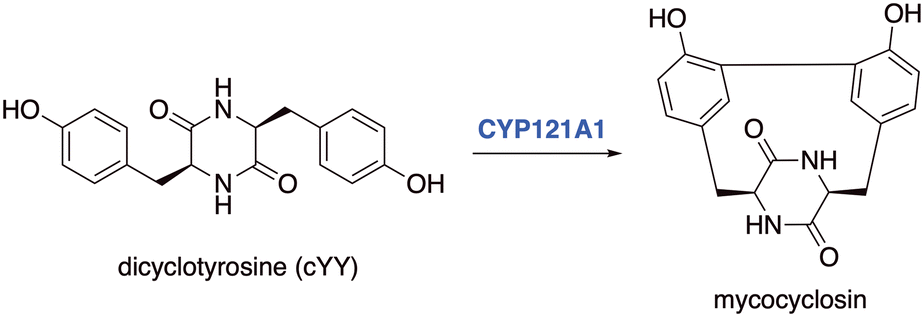 | ||
| Fig. 1 Mycocyclosin formed through a CYP121A1 mediated C–C bond formation between carbon atoms in the ortho-positions of two tyrosine groups in the cyclodipeptide dicyclotyrosine (cYY). | ||
CYP121A1 inhibitors with a range of structural scaffolds have been described by us6–9 and others.10–12 In all cases a basic nitrogen containing group with the potential to bind to the iron of the haem group is included, although indirect water-mediated binding with the haem is occasionally observed and/or binding of the nitrogen containing group with Gln395/Arg386, which would block access of cYY to the active site.6–12 One of the more promising scaffolds is the diarylpyrazole series6,9 with optimal binding observed for imidazole as the nitrogen containing binding group and substitution of one of the aryl rings with lipophilic alkyl groups (Fig. 2).
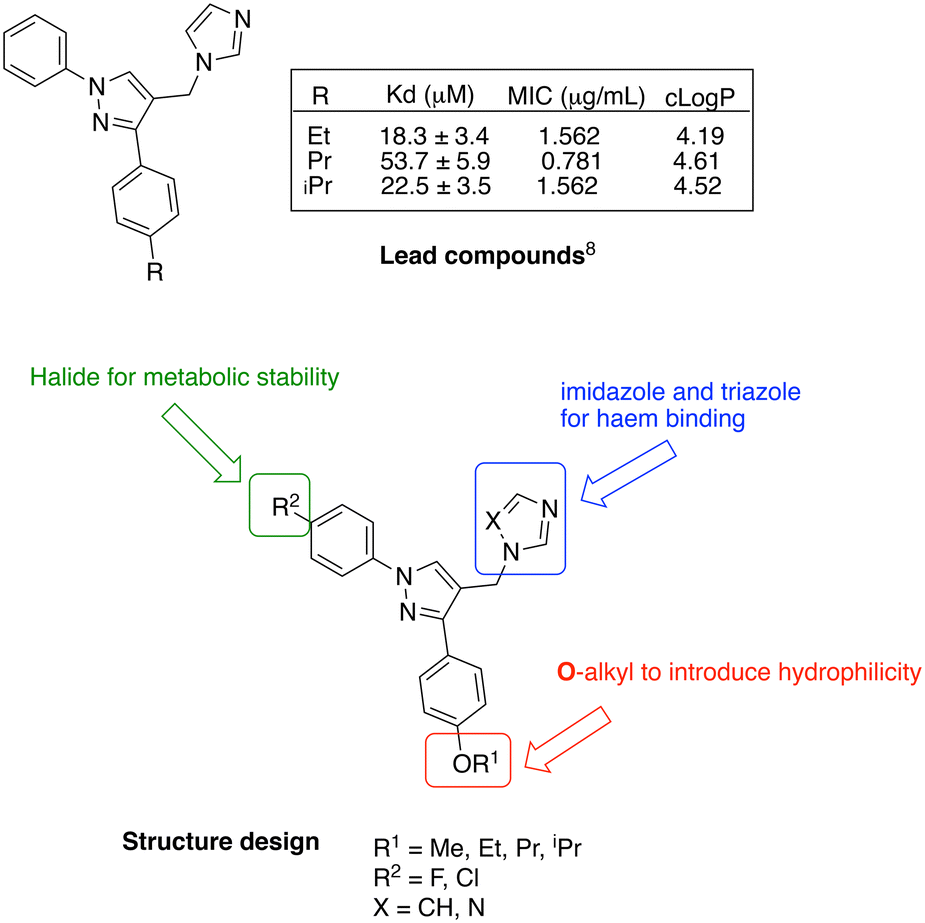 | ||
| Fig. 2 Lead diarylpyrazole derivatives9 and structure design of new compounds to improve drug likeness. | ||
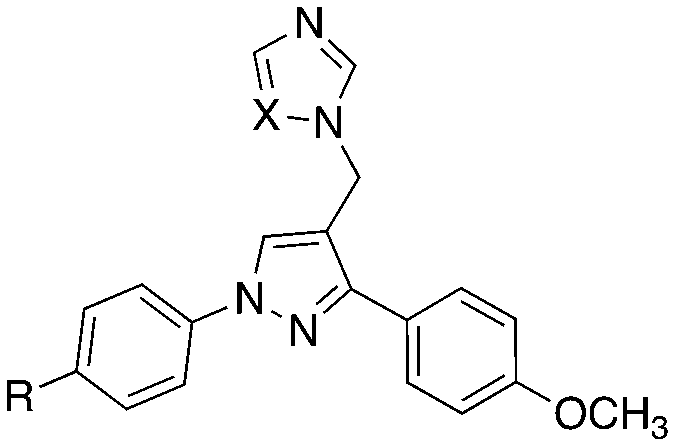
Presented here is a further design of these lead pyrazole derivatives to consider drug likeness, incorporating O-alkyl substitutions in one aryl ring to improve hydrophilic properties compared with the lead compounds, and the addition of a halide (F or Cl) in the second aryl ring for metabolic stability (Fig. 2). Imidazole and 1,2,4-triazole azole moieties are included as the basic nitrogen binding groups to evaluate effect on binding affinity and antimycobacterial activity.
Results and discussion
Chemistry
The hydrazines (7) were prepared following previously described methodology6,9 by reaction of the 4-alkoxy-acetophenones (1–4) with either 4-fluoro or 4-chlorophenylhydrazine hydrochloride (5 or 6) in EtOH with acetic acid as the acid catalyst (Scheme 1). The hydrazines (7) were converted to the aldehydes (8) on reaction with Vilsmeier–Haack reagent, generated in situ from DMF and POCl3, and obtained as solids in good yields (65–100%).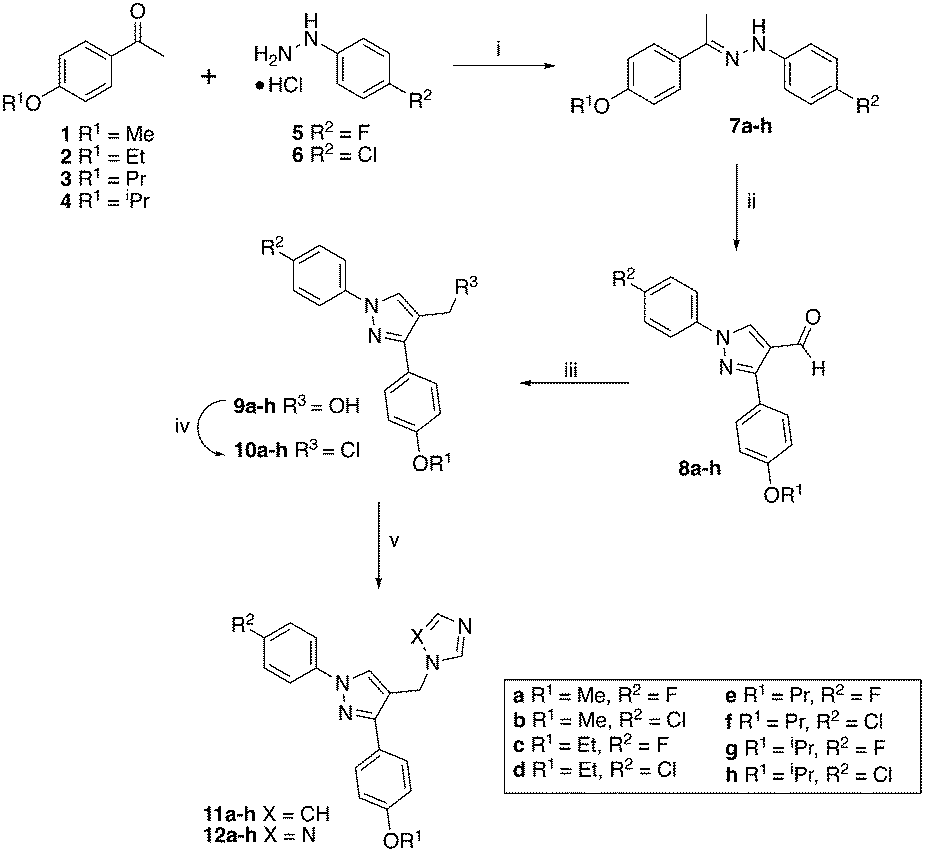 | ||
| Scheme 1 Reagents and conditions: (i) Et3N, AcOH, EtOH, 83 °C, 1 h, 79–100%; (ii) POCl3, DMF, 60 °C, 3 h, 65–100%; (iii) NaBH4, EtOH, r.t, 1 h, 68–98%; (iv) SOCl2, CH2Cl2, 40 °C, 4 h; (v) imidazole or triazole, K2CO3, CH3CN, 45 °C, 1 h then 70 °C, overnight, yields see Table 1. | ||
The aldehydes (8) were reduced with NaBH4 and the alcohols (9), obtained after extraction with EtOAc in yields of 68–98%, were converted to the chlorides (10) on treatment with SOCl2 in CH2Cl2 at 40 °C for 4 h. The crude chlorides (10) were obtained in very high yields (93–100%) except for the isopropyl/fluoride derivative (48%), and were used immediately in the next reaction, which involved the generation of the imidazole or triazole anion by reaction of imidazole or triazole with K2CO3 in CH3CN at 45 °C for 1 h, followed by the addition of the chlorides and heating at 70 °C overnight (Scheme 1). The final imidazole (11) and triazole (12) products were obtained after purification by gradient column chromatography with generally good yields (Table 1).
| Compd | R1 | R2 | X | Yield (%) | m.p. (°C) |
|---|---|---|---|---|---|
| 11a | Me | F | CH | 70 | 150–152 |
| 11b | Me | Cl | CH | 61 | 136–138 |
| 11c | Et | F | CH | 45 | 118–120 |
| 11d | Et | Cl | CH | 22 | 124–126 |
| 11e | Pr | F | CH | 66 | 120–122 |
| 11f | Pr | Cl | CH | 63 | 90–92 |
| 11g | iPr | F | CH | 58 | 96–98 |
| 11h | iPr | Cl | CH | 94 | 98–100 |
| 12a | Me | F | N | 58 | 150–152 |
| 12b | Me | Cl | N | 60 | 124–126 |
| 12c | Et | F | N | 52 | 140–142 |
| 12d | Et | Cl | N | 60 | 122–124 |
| 12e | Pr | F | N | 58 | 120–122 |
| 12f | Pr | Cl | N | 72 | 114–116 |
| 12g | iPr | F | N | 53 | 118–120 |
| 12h | iPr | Cl | N | 81 | 98–100 |
Antimycobacterial activity (MIC90)
All compounds were evaluated for MIC90 against Mtb H37Rv using the SPOTi assay as previously described.13 The imidazoles all showed good antimycobacterial activity (Table 2) with optimal activity observed for the propyloxy/Cl (11f, MIC90 4.0 μg mL−1, 10.1 μM) and isopropyloxy/F (11h, MIC90 4.4 μg mL−1, 11.9 μM) derivatives. The activity observed for all the imidazole derivatives (except 11c) and the triazoles 12b, 12d and 12f was comparable with the standard kanamycin (MIC90 7.8 μg mL−1, 16.1 μM), which is an important second line drug for the treatment of MDR-TB.14| Compd | MIC90a (μg mL−1) | MIC90a (μM) | cLogPb |
|---|---|---|---|
| a Results are the average of two independent experiments. b Crippen's fragmentation.15 | |||
| 11a | 4.3 | 12.3 | 3.32 |
| 11b | 6.0 | 16.5 | 3.72 |
| 11c | 12.0 | 33.2 | 3.66 |
| 11d | 5.0 | 13.3 | 4.06 |
| 11e | 6.0 | 15.9 | 4.15 |
| 11f | 4.0 | 10.1 | 4.55 |
| 11g | 4.3 | 11.4 | 3.98 |
| 11h | 6.0 | 15.3 | 4.38 |
| 12a | 12.8 | 36.7 | 2.83 |
| 12b | 4.4 | 11.9 | 3.23 |
| 12c | 25.6 | 70.5 | 3.17 |
| 12d | 6.4 | 16.8 | 3.57 |
| 12e | 20.9 | 55.5 | 3.65 |
| 12f | 6.4 | 16.2 | 4.05 |
| 12g | 20.9 | 55.5 | 3.49 |
| 12h | 25.6 | 65.1 | 3.89 |
| Isoniazid | 0.2 | 1.8 | −0.64 |
| Kanamycin | 7.8 | 16.1 | −7.58 |
| Rifampicin | 0.2 | 0.3 | — |
Mtb CYP121A1 binding affinity
All sixteen final compounds (11a–h and 12a–h) were subject to a preliminary binding affinity screen, which identified seven imidazole (11a, 11c–h) and one triazole derivatives (12b) for further evaluation. Evaluation of inhibitor affinity was carried out in a 96-well plate format. The Kd of cYY in this assay (12 μM) was consistent with its previously reported value.4,16 As a comparison, all inhibitors were titrated manually in a 1 cm quartz cuvette for a single trial. The calculated Kd values closely matched the results from the plate-reader assay.The methoxy triazole derivative with a chloroaryl group (12b) displayed the tightest binding affinity with Kd 5.1 ± 1.5 μM, and in the imidazole series the derivatives with a chloroaryl group were generally more effective in binding to Mtb CYP121A1 compared with the compounds with a fluoroaryl group. The propyloxy derivative (11f, Kd 11.7 ± 5.4 μM) displayed binding affinity comparable with cYY (Kd 12.3 ± 1.1 μM) (Table 3).
| Compd | CYP121A1 Kd (μM) | Soret shift (nm) |
|---|---|---|
| 11a | 65.0 ± 16.6 | 417 to 421 |
| 11c | 54.0 ± 5.2 | 417 to 422 |
| 11d | 18.8 ± 10.2 | 417 to 420 |
| 11e | 22.3 ± 4.2 | 417 to 420 |
| 11f | 11.7 ± 5.4 | 417 to 419 |
| 11g | 38.5 ± 8.7 | 417 to 419 |
| 11h | 17.7 ± 6.9 | 417 to 419 |
| 12b | 5.1 ± 1.5 | 417 to 417 |
| cYY | 12.3 ± 1.1 | 417 to 395 |
As previously stated cYY in the co-crystal structure displays indirect binding with the haem via interstitial water molecules.4,16
However, absorbance changes from cYY titration in solution produce a typical type-I spectral response, indicating displacement of the haem water molecule(s) and induction of a high-spin form of the iron. The pyrazoles described here displayed a type II binding profile suggesting direct binding with the haem (Fig. 3 and S1 and S2†).
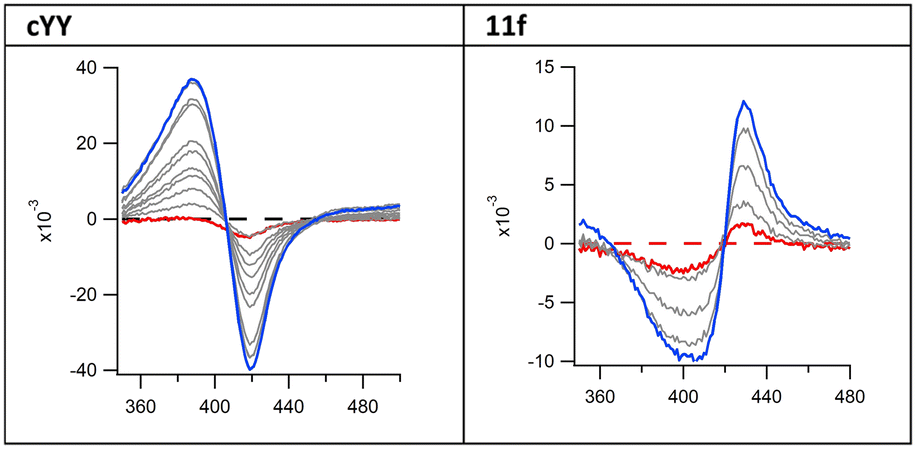 | ||
| Fig. 3 Binding difference spectra of cYY (type I) and pyrazole exemplar 11f (type II). | ||
In the co-crystal structure with cYY, CYP121A1 coordinates the substrate within the active site while also preserving the space and hydrogen-bonding necessary to maintain a two-molecule water bridge to the haem.4 CYP121A1 is also known to coordinate the inhibitor fluconazole concomitantly with the haem water ligand.17 In practice, such coordination is difficult to detect by absorbance difference spectra alone, which rely on ligand-induced displacement of the water molecule followed by direct azole ligation to the haem. In light of this possibility, protein-detected 1D 19F-NMR spectroscopy was utilised as an orthogonal strategy to evaluate ligand binding independent of perturbations at the haem. As described recently,18 BTFA labelling of an S171C mutation located within the CYP121A1 FG-loop serves as a direct reporter of protein–ligand interactions. The FG-loop in CYPs is known to undergo extensive structural remodelling in response to ligand binding.19–21 In ligand-free CYP121A1, the conformational heterogeneity of the loop is indicated by at least three separate resonances (Fig. 4). As reported previously,18 a 3-fold addition of cYY reduces the intensity of the broad down-field resonance at −83.5 ppm while increasing the intensity of the major upfield resonance near −84.5 ppm, thus indicating reduced heterogeneity of the FG-loop conformation and an increase in the ligand-bound state (Fig. 4).
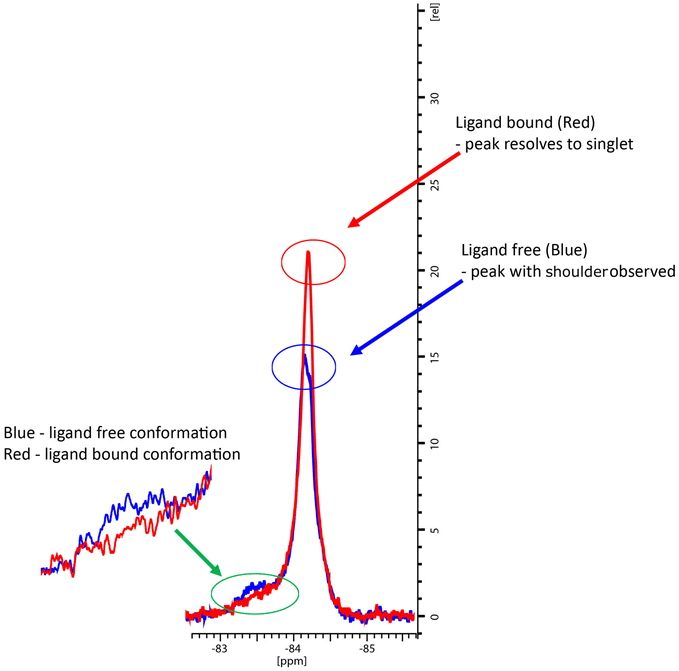 | ||
| Fig. 4 19F NMR spectra of BTFA-labelled CYP121_S171C (blue) alone and with cYY bound (red). | ||
To evaluate the interaction with the compounds, 19F-NMR spectra were acquired for BTFA-labelled CYP121_S171C in the presence of 3-fold excess concentrations of representative imidazole and triazole derivatives. For comparison, spectra were overlayed with ligand-free and cYY-bound spectra recorded from samples from the same batch of labelled protein and at matching protein concentrations.
As expected, the perturbations induced by the imidazole derivatives 11a and 11g are consistent with an interaction that occurs within the CYP121 active site, since both compounds elicit similar changes in the NMR spectrum of the FG-loop when compared to cYY (Fig. 5A and B). Moreover, the relative extent of these perturbations aligns with the change in the Soret peak of absorbance assays (Table 3). Interestingly, NMR spectra recorded in the presence of the triazole derivatives 12b and 12d were generally not in agreement with the absorbance data. These compounds elicited a weak spectral response in the UV-vis assay when compared with the imidazole derivatives. However, their presence induced all the characteristic changes of ligand-binding in 19F-NMR spectra. For both compounds, perturbations were more intense than that of cYY (Fig. 5C and D). Our interpretation of this apparent disagreement in binding behaviour is that weaker triazole-coordination of the haem may also allow for preservation of the haem-coordinated water ligand, as indicated by weaker absorbance data overall, and similar to the non-haem binding mode observed with fluconazole. Hence, the detection of ligand-binding by 19F-NMR, and thus confirmation that ligand coordination occurs in the substrate-binding pocket despite very modest changes in absorbance, is consistent with the demonstrated growth inhibition potential of these compounds.
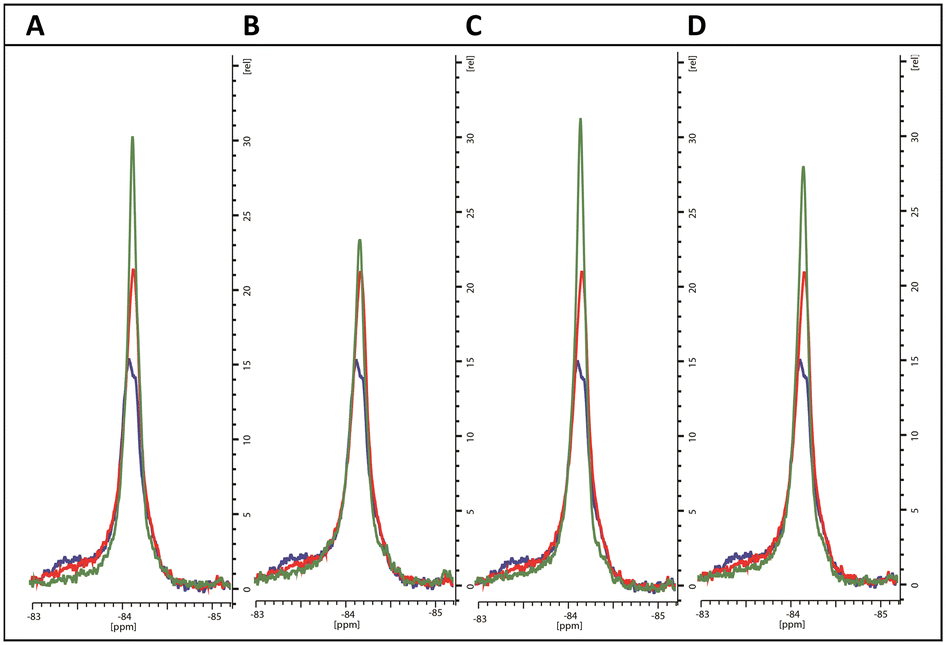 | ||
| Fig. 5 19F NMR spectra of BTFA-labelled CYP121_S171C (blue) alone, with cYY bound (red) and with exemplar pyrazole ligands (green) (A) 11a, (B) 11g, (C) 12b and (D) 12d. | ||
Computational studies
Mtb CYP121A1 protein-ligand complexes were generated through docking of compounds 11 and 12 with the crystal structure of Mtb CYP121A1 co-crystallised with cYY (pdb 3G5H4) using Molecular Operating Environment (MOE) software.22 The protein–ligand complexes were then subject to 150 ns molecular dynamics (MD) simulations using the Desmond programme of Schrödinger software.23cYY sits to one side of the haem and is tethered through a H-bonding interaction between one phenolic OH and Arg386 on one side of the active site and through H-bonding interactions between the diketopiperazine ring C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and NH groups with Val83 and Asn85 on the opposite side of the active site (Fig. 6).
O and NH groups with Val83 and Asn85 on the opposite side of the active site (Fig. 6).
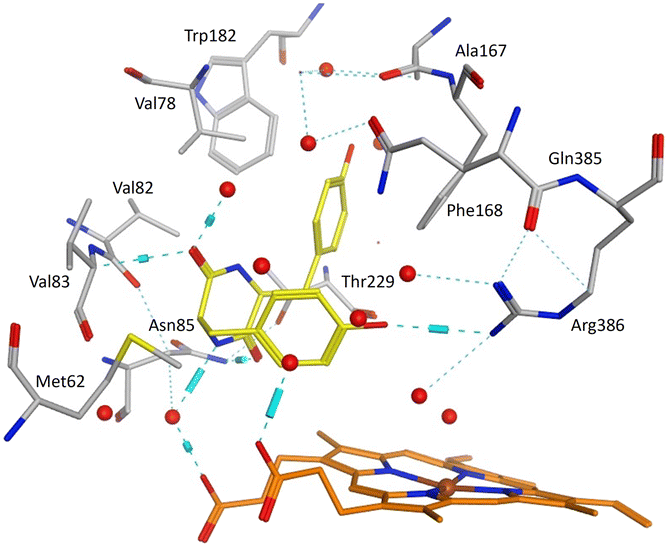 | ||
| Fig. 6 cYY (yellow) binding in Mtb CYP121A1 active site (pdb 3G5H). Haem shown in orange, water molecules as red spheres, H-bonding as blue lines/cylinders. | ||
All the pyrazole derivatives were positioned in a comparable position occupied by cYY, to the side of the haem group (Fig. 7). Water-mediated binding interactions were observed with either the imidazole, triazole or pyrazole ring with Gln385/Arg386 and additional hydrophobic/π–π stacking between one of the benzene rings of the pyrazole and Phe168 (e.g.11b, 12b and 12d) (Fig. 8). A halide interaction was observed for some of the pyrazole derivatives with Leu73 (e.g.11b and 12d) and additional water mediated and multiple hydrophobic interactions (Fig. 8).
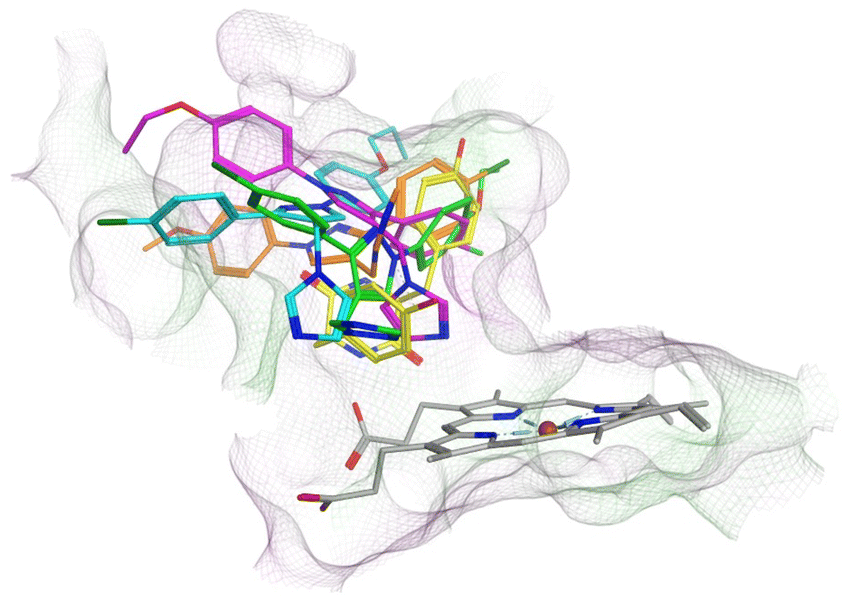 | ||
| Fig. 7 Comparison of binding position of pyrazoles 11c (magenta), 11f (cyan), 12b (orange) and 12d (green) with cYY (yellow). | ||
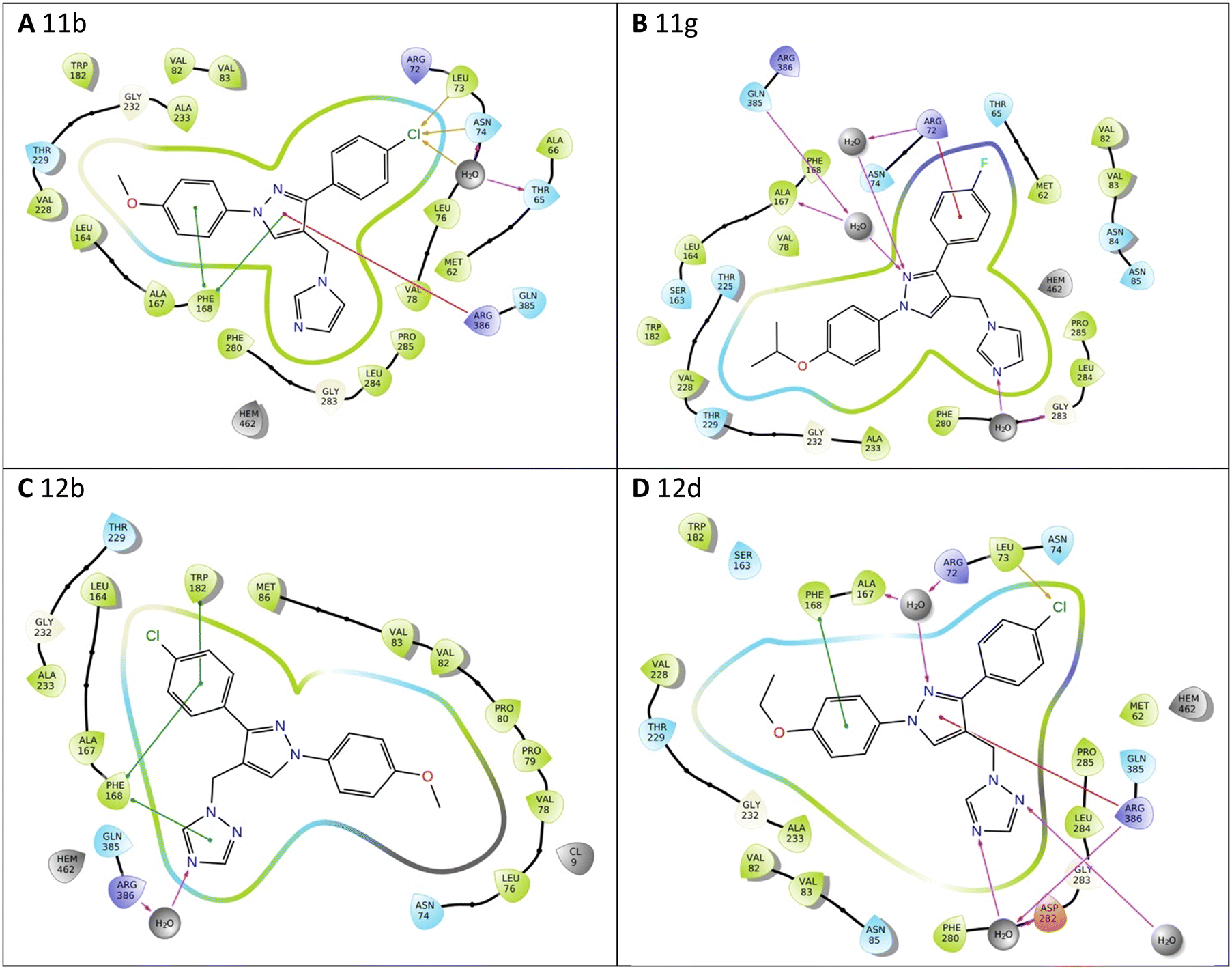 | ||
| Fig. 8 Exemplar 2D ligand interactions with Mtb CYP121A1. (A) 11b, (B) 11g, (C) 12b and (D) 12d. | ||
Conclusions
In this pyrazole series, the imidazole derivatives were generally more active against Mtb with optimal activity observed for the longer/branched chain alkoxy substitutions (e.g.11f and 11g). For the triazole derivatives, the compounds with a chlorobenzene ring were more active against Mtb compared with triazole derivatives containing a fluorobenzene ring. Generally, UV-vis spectral binding assay, which measures binding to the haem, either direct (type II) or indirect (type I through displacement of the 6th axial water ligand of the haem, confirmed type II binding for the imidazole derivatives and reflected Mtb inhibitory (MIC) activity. Compared with lead imidazole compounds (Fig. 2), binding affinity was either comparable or improved but the Mtb inhibitory activity was reduced (e.g.Fig. 2, lead compound R = Pr MIC 0.78 μg mL−1 compared with propyloxy derivative 11f MIC 4.0 μg mL−1). Possibly the reduction in lipophilicity compared with the lead compounds, owing to addition of the oxygen from the alkoxy substitution may be sufficient to reduce uptake across the Mtb cell membrane.Previously we prepared methoxy derivatives with both imidazole and triazole haem binding groups (P1 and P2, Table 4), both of which had MIC90 = 25 μg mL−1.6 Comparison with the methoxy derivatives (11a–b, 12a–b) with the halide substitutions in the second aryl ring showed improved antimycobacterial activity), which may also be related to the increase in lipophilicity and would be consistent with our previous studies.6–9
Only one triazole (12b) had a measurable binding affinity using the UV-vis spectral assay, which did not correlate with the Mtb inhibitory activity observed (MIC). The use of the 19F NMR assay to measure changes observed in the FG loop upon binding of ligands using BTFA 19F labelled CYP121_S171C, provided support that for the triazole derivatives (and imidazole derivatives) ligand coordination occurs in the substrate-binding pocket and in all cases with tighter binding than cYY (Fig. 5). Computational studies also support the pyrazole compounds binding in the substrate (cYY) binding pocket (Fig. 7) at the side of the haem rather than perpendicular, which may for some derivatives mean that binding or displacement of the haem water ligand may not occur even though binding in the substrate and access channel is efficient. If no perturbation of the haem/water ligand occurs this reduces the suitability of the UV-vis absorption method and highlights the importance of the results of the 19F NMR assay, which support the use of this method as an additional qualitative assay to determine ligand-CYP121A1 binding. This manuscript describes active triazole derivatives with good MIC values (some comparable with or better than the equivalent imidazole), which was not previously observed6–9 and, having established 1D 19F-NMR spectroscopy as an orthogonal strategy to evaluate ligand binding independent of perturbations at the haem, binding can now be confirmed. Further studies will focus on pyrazole derivative containing a triazole or alternative N-containing heterocycles (e.g. pyridines) as the haem binding moiety, as well as selectivity (human CYP panel) and toxicity assays.
Experimental
General
All chemicals, reagents and solvents were purchased from Sigma-Aldrich, Alfa Aesar, VWR, Acros and Fluka. Solvents were dried prior to use over molecular sieves (4 Å). For column chromatography, a glass column was slurry packed in the appropriate eluent with silica gel (Fluka Kieselgel 60). TLC was performed on pre-coated silica plates (dimension 20 × 20 cm) (ALUGRAM® SIL G/UV254) with visualisation via UV light (254 nm). Melting points were determined on an electrothermal instrument (Gallenkamp) and were uncorrected. 1H, 13C, and 19F NMR spectra were recorded on a Bruker Advance DP500 spectrometer operating at 500, 125 and 470 MHz, respectively. Chemical shifts are given in parts per million (ppm) relative to the internal standard tetramethylsilane (Me4Si). Elemental analysis was performed by MEDAC Ltd (Chobham, Surrey, UK) and HPLC-HRMS was performed at the Department of Pharmacy & Pharmacology, University of Bath, Bath, UK. on a Zorbax Eclipse Plus C18 Rapid Resolution 2.1 × 50 mm, 1.8 μm particle size using a 7.5 minutes gradient method 5:95 v/v water:methanol with 0.1% formic acid as additive. Experimental data for compounds 7, 8, 9 and 10 are provided in ESI.†
Chemistry
:3 v/v and obtained as a light brown solid. Yield 0.39 g (70%), m.p. 150–152 °C, TLC (CH2Cl2–MeOH 95:5 v/v) Rf 0.50. 1H NMR (DMSO-d6): δ 8.49 (s, 1H, pyrazole), 7.90 (m, 2H, Ar), 7.65 (s, 1H, imidazole), 7.56 (d, J = 8.5 Hz, 2H, Ar), 7.37 (m, 2H, Ar), 7.12 (s, 1H, imidazole), 7.02 (d, J = 9.0 Hz, 2H, Ar), 6.89 (s, 1H, imidazole), 5.27 (s, 2H, CH2-imidazole), 3.81 (s, 3H, OCH3). 19F NMR: (DMSO-d6): −116.49 (F–Ar). 13C NMR (DMSO-d6): δ 159.81 and 161.74 (d, 1JCF = 242.7 Hz, C–F, Ar), 159.76 (C, Ar), 150.69 (C, pyrazole), 137.45 (CH, imidazole), 136.41 (d, 4JCF = 2.5 Hz, C, Ar), 130.14 (CH, pyrazole), 129.22 (2 × CH, Ar), 129.08 (CH, imidazole), 125.20 (C, Ar), 120.76 and 120.82 (d, 3JCF = 7.5 Hz, 2 × CH, Ar), 119.61 (CH, imidazole), 116.95 (C, pyrazole), 116.67 and 116.85 (d, 2JCF = 22.6 Hz, 2 × CH, Ar), 114.61 (2 × CH, Ar), 55.64 (OCH3), 40.95 (CH2). Anal. calcd. for C20H17FN4O (348.3814): C 68.59%, H 4.89%, N 15.99%. Found: C 68.54%, H 4.73%, N 16.07%.
:3 v/v and obtained as a light orange solid. Yield 0.23 g (61%), m.p. 136–138 °C, TLC (CH2Cl2–MeOH 95:5 v/v) Rf 0.44. 1H NMR (DMSO-d6): δ 8.53 (s, 1H, pyrazole), 7.90 (d, J = 9.0 Hz, 2H, Ar), 7.65 (s, 1H, imidazole), 7.58 (d, J = 9.0 Hz, 2H, Ar), 7.56 (d, J = 9.0 Hz, 2H, Ar), 7.12 (s, 1H, imidazole), 7.02 (d, J = 9.0 Hz, 2H, Ar), 6.88 (s, 1H, imidazole), 5.28 (s, 2H, CH2-imidazole), 3.81 (s, 3H, CH2CH3). 13C NMR (DMSO-d6): δ 159.83 (C, Ar), 150.94 (C, pyrazole), 138.60 (C, Ar), 137.47 (CH, imidazole), 130.86 (C, Ar), 130.10 (CH, imidazole), 129.96 (2 × CH, Ar), 129.25 (2 × CH, Ar), 129.08 (CH, pyrazole), 125.06 (C, pyrazole), 120.33 (2 × CH, Ar) 119.63 (CH, imidazole), 117.32 (C, Ar), 114.63 (2 × CH, Ar), 55.65 (CH3), 40.94 (CH2). Anal. calcd. for C20H17ClN4O (364.8330): C 65.84%, H 4.70%, N 15.35%. Found: C 65.97%, H 4.59%, N 15.38%.
:2 v/v and obtained as a light brown solid. Yield 0.146 g (45%), m.p. 118–120 °C, TLC (CH2Cl2–MeOH 95:5 v/v) Rf 0.47. 1H NMR (DMSO-d6): δ 8.48 (s, 1H, pyrazole), 7.90 (dd, J = 4.5, 9.0 Hz, 2H, Ar), 7.64 (s, 1H, imidazole), 7.55 (d, J = 9.0 Hz, 2H, Ar), 7.37 (t, J = 8.5 Hz, 2H, Ar), 7.12 (s, 1H, imidazole), 7.0 (d, J = 8.5 Hz, 2H, Ar), 6.88 (s, 1H, imidazole), 5.27 (s, 2H, CH2-imidazole), 4.07 (q, J = 7.0 Hz, 2H, CH2CH3), 1.35 (t, J = 7.0 Hz, 3H, CH2CH3). 19F NMR: (DMSO-d6): −116.52 (F–Ar). 13C NMR (DMSO-d6): δ 159.80 and 161.73 (d, 1JCF = 242.7 Hz, C–F, Ar), 159.04 (C, Ar), 150.71 (C, pyrazole), 136.42 (d, 4JCF = 2.5 Hz, C, Ar), 130.10 (CH, pyrazole), 129.22 (2 × CH, Ar), 129.08 (CH, imidazole), 125.06 (C, Ar), 120.74 and 120.81 (d, 3JCF = 8.8 Hz, 2 × CH, Ar), 119.61 (CH, imidazole), 116.96 (C, pyrazole), 116.67 and 116.85 (d, 2JCF = 22.6 Hz, 2 × CH, Ar), 115.04 (2 × CH, Ar), 63.57 (CH2CH3), 40.96 (CH2), 15.12 (CH2CH3). Anal. calcd. for C21H19FN4O (362.4084): C 69.25%, H 5.26%, N 15.38%. Found: C 68.86%, H 5.06%, N 15.16%.
:2.5 v/v and obtained as a white solid. Yield 0.04 g (22%), m.p. 124–126 °C, TLC (CH2Cl2–MeOH 95:5 v/v) Rf 0.55. 1H NMR (DMSO-d6): δ 8.53 (s, 1H, pyrazole), 7.90 (d, J = 9.0 Hz, 2H, Ar), 7.64 (s, 1H, imidazole), 7.58 (d, J = 9.0 Hz, 2H, Ar), 7.56 (d, J = 8.8 Hz, 2H, Ar), 7.12 (s, 1H, imidazole), 7.01 (d, J = 8.8 Hz, 2H, Ar), 6.88 (s, 1H, imidazole), 5.28 (s, 2H, CH2-imidazole), 4.08 (q, J = 7.0 Hz, 2H, CH2CH3), 1.35 (t, J = 7.0 Hz, 3H, CH2CH3). 13C NMR (DMSO-d6): δ 159.11 (C, Ar), 150.95 (C, pyrazole), 138.61 (C, Ar), 137.47 (CH, imidazole), 130.83 (C, Ar), 130.06 (CH, imidazole), 129.96 (2 × CH, Ar), 129.24 (2 × CH, Ar), 129.09 (CH, pyrazole), 124.93 (C, pyrazole), 120.31 (2 × CH, Ar) 119.63 (CH, imidazole), 117.34 (C, Ar), 115.06 (2 × CH, Ar), 63.58 (CH2CH3), 40.96 (CH2), 15.11 (CH2CH3). HPLC: 97.8%, RT = 4.48 min. HRMS (ESI) m/z: calculated, 401.1145 [M + Na]+, Found, 401.1147 [M + Na]+.
:2 v/v and obtained as a light orange solid. Yield 0.20 g (66%), m.p. 120–122 °C, TLC (CH2Cl2–MeOH 95:5 v/v) Rf 0.51. 1H NMR (DMSO-d6): δ 8.48 (s, 1H, pyrazole), 7.90 (m, 2H, Ar), 7.64 (s, 1H, imidazole), 7.56 (d, J = 9.0 Hz, 2H, Ar), 7.36 (t, J = 9.0 Hz, 2H, Ar), 7.12 (s, 1H, imidazole), 7.02 (d, J = 8.50 Hz, 2H, Ar), 6.88 (s, 1H, imidazole), 5.27 (s, 2H, CH2-imidazole), 3.97 (t, J = 6.5 Hz, 2H, OCH2), 1.76 (m, 2H, CH2), 1.0 (t, J = 7.0 Hz, 3H, CH3). 19F NMR: (DMSO-d6): −116.51 (F–Ar). 13C NMR (DMSO-d6): δ 159.80 and 161.73 (d, 1JCF = 242.7 Hz, C–F, Ar), 159.20 (C, Ar), 150.71 (C, pyrazole), 137.45 (CH, imidazole), 136.42 (d, 4JCF = 2.5 Hz, C, Ar), 130.10 (CH, pyrazole), 129.21 (2 × CH, Ar), 129.08 (CH, imidazole), 125.05 (C, Ar), 120.81 and 120.74 (d, 3JCF = 8.8 Hz, 2 × CH, Ar), 119.62 (CH, imidazole), 116.96 (C, pyrazole), 116.67 and 116.85 (d, 2JCF = 22.6 Hz, 2 × CH, Ar), 115.08 (2 × CH, Ar), 69.46 (OCH2), 40.96 (imidazole-CH2), 22.50 (CH2), 10.87 (CH3). Anal. calcd. for C22H21FN4O (376.4354): C 70.20%, H 5.62%, N 14.88%. Found: C 70.00%, H 5.54%, N 14.97%.
:2 v/v and obtained as a light brown solid. Yield 0.20 g (63%), m.p. 90–92 °C, TLC (CH2Cl2–MeOH 95:5 v/v) Rf 0.60. 1H NMR (DMSO-d6): δ 8.53 (s, 1H, pyrazole), 7.91 (d, J = 9.0 Hz, 2H, Ar), 7.65 (s, 1H, imidazole), 7.58 (d, J = 9.0 Hz, 2H, Ar), 7.56 (d, J = 8.50 Hz, 2H, Ar), 7.12 (s, 1H, imidazole), 7.02 (d, J = 9.0 Hz, 2H, Ar), 6.89 (s, 1H, imidazole), 5.28 (s, 2H, CH2-imidazole), 3.98 (t, J = 6.5 Hz, 2H, OCH2), 1.76 (m, 2H, CH2), 1.0 (t, J = 7.0 Hz, 3H, CH3). 13C NMR (DMSO-d6): δ 159.27 (C, Ar), 150.95 (C, pyrazole), 138.61 (C, Ar), 137.48 (CH, imidazole), 130.83 (C, Ar), 130.05 (CH, imidazole), 129.95 (2 × CH, Ar), 129.24 (2 × CH, Ar), 129.10 (CH, pyrazole), 124.93 (C, pyrazole), 120.30 (2 × CH, Ar) 119.63 (CH, imidazole), 117.34 (C, Ar), 115.09 (2 × CH, Ar), 69.46 (OCH2), 40.97 (imidazole-CH2), 22.50 (CH2), 10.87 (2 × CH3). Anal. calcd. for C22H21ClN4O (392.1404): C 67.26%, H 5.39%, N 14.25%. Found: C 67.25%, H 5.19%, N 14.27%.
:1.5 v/v and obtained as a light brown solid. Yield 0.096 g (58%), m.p. 96–98 °C, TLC (CH2Cl2–MeOH 95:5 v/v) Rf 0.44. 1H NMR (DMSO-d6): δ 8.48 (s, 1H, pyrazole), 7.90 (m, 2H, Ar), 7.65 (s, 1H, imidazole), 7.54 (d, J = 9.0 Hz, 2H, Ar), 7.36 (t, J = 9.0 Hz, 2H, Ar), 7.13 (s, 1H, imidazole), 7.0 (d, J = 9.0 Hz, 2H, Ar), 6.89 (s, 1H, imidazole), 5.27 (s, 2H, CH2-imidazole), 4.67 (m, 1H, CH), 1.30 (d, J = 6.0 Hz, 6H, 2CH3). 19F NMR: (DMSO-d6): −116.52 (F–Ar). 13C NMR (DMSO-d6): δ 159.80 and 161.73 (d, 1JCF = 242.7 Hz, C–F, Ar), 157.99 (C, Ar), 150.72 (C, pyrazole), 137.46 (CH, imidazole), 136.43 (d, 4JCF = 2.5 Hz, C, Ar), 130.06 (CH, pyrazole), 129.26 (2 × CH, Ar), 129.08 (CH, imidazole), 124.89 (C, Ar), 120.79 and 120.72 (d, 3JCF = 8.8 Hz, 2 × CH, Ar), 119.63 (CH, imidazole), 116.96 (C, pyrazole), 116.85 and 116.67 (d, 2JCF = 22.6 Hz, 2 × CH, Ar), 116.12 (2 × CH, Ar), 69.67 (CH), 40.97 (CH2), 22.29 (2 × CH3). Anal. calcd. for C22H21FN4O (376.4354): C 70.20%, H 5.62%, N 14.88%. Found: C 69.84%, H 5.48%, N 14.83%.
:2.5 v/v and obtained as a beige solid. Yield 0.30 g (94%), m.p. 98–100 °C, TLC (CH2Cl2–MeOH 95:5 v/v) Rf 0.45. 1H NMR (DMSO-d6): δ 8.52 (s, 1H, pyrazole), 7.90 (d, J = 9.0 Hz, 2H, Ar), 7.65 (s, 1H, imidazole), 7.58 (d, J = 9.0 Hz, 2H, Ar), 7.55 (d, J = 9.0 Hz, 2H, Ar), 7.13 (s, 1H, imidazole), 7.0 (d, J = 8.50 Hz, 2H, Ar), 6.89 (s, 1H, imidazole), 5.28 (s, 2H, CH2-imidazole), 4.67 (m, 1H, CH), 1.30 (d, J = 6.0 Hz, 6H, 2 × CH3). 13C NMR (DMSO-d6): δ 158.06 (C, Ar), 150.96 (C, pyrazole), 138.61 (C, Ar), 137.48 (CH, imidazole), 130.82 (C, Ar), 130.01 (CH, imidazole), 129.95 (2 × CH, Ar), 129.29 (2 × CH, Ar), 129.10 (CH, pyrazole), 124.76 (C, pyrazole), 120.29 (2 × CH, Ar) 119.64 (CH, imidazole), 117.34 (C, Ar), 116.12 (2 × CH, Ar), 69.68 (CH), 40.98 (CH2), 22.28 (2 × CH3). Anal. calcd. for C22H21ClN4O (392.1404): C 67.26%, H 5.39%, N 14.26%. Found: C 67.03%, H 5.16%, N 14.21%.
:2.5 v/v and obtained as an orange solid. Yield 0.26 g (58%), m.p. 150–152 °C, TLC (CH2Cl2–MeOH 95:5 v/v) Rf 0.63. 1H NMR (DMSO-d6): δ 8.54 (s, 1H, triazole), 8.54 (s, 1H, pyrazole), 7.99 (s, 1H, triazole), 7.90 (m, 2H, Ar), 7.67 (d, J = 8.50 Hz, 2H, Ar), 7.36 (m, 2H, Ar), 7.04 (d, J = 9.0 Hz, 2H, Ar), 5.48 (s, 2H, CH2-triazole), 3.81 (s, 3H, CH3). 19F NMR: (DMSO-d6): −116.42 (F–Ar). 13C NMR (DMSO-d6): δ 161.77 and 159.83 (d, 1JCF = 242.7 Hz, C–F, Ar), 159.80 (C, Ar), 151.99 (CH, triazole), 150.89 (C, pyrazole), 144.36 (CH, triazole), 136.39 and 136.36 (d, 4JCF = 2.5 Hz, C, Ar), 130.40 (CH, pyrazole), 129.37 (2 × CH, Ar), 125.04 (C, pyrazole), 120.83 and 120.76 (d, 3JCF = 8.7 Hz, 2 × CH, Ar), 116.86 and 116.68 (d, 2JCF = 22.6 Hz, 2 × CH, Ar), 115.69 (C, Ar), 114.59 (2 × CH, Ar), 55.65 (CH3), 43.81 (CH2). Anal. calcd. for C19H16FN5O (349.3694): C 64.99%, H 4.59%, N 19.94%. Found: C 64.67%, H 4.41%, N 19.95%.
:3 v/v and obtained as a light orange solid. Yield 0.32 g (60%), m.p. 124–126 °C, TLC (CH2Cl2–MeOH 95:5 v/v) Rf 0.65. 1H NMR (DMSO-d6): δ 8.59 (s, 1H, triazole), 8.54 (s, 1H, pyrazole), 7.99 (s, 1H, triazole), 7.90 (d, J = 9.0 Hz, 2H, Ar), 7.67 (d, J = 8.5 Hz, 2H, Ar), 7.58 (d, J = 9.0 Hz, 2H, Ar), 7.03 (d, J = 9.0 Hz, 2H, Ar), 5.48 (s, 2H, CH2-triazole), 3.81 (s, 3H, CH3). 13C NMR (DMSO-d6): δ 159.87 (C, Ar), 152.01 (CH, triazole), 151.13 (C, pyrazole), 144.39 (CH, triazole), 138.58 (C, Ar), 130.90 (C, Ar), 130.37 (CH, pyrazole), 129.96 (2 × CH, Ar), 129.39 (2 × CH, Ar), 124.92 (C, pyrazole), 120.32 (2 × CH, Ar), 116.06 (C, Ar), 114.61 (2 × CH, Ar), 55.66 (CH3), 43.79 (CH2). Anal. calcd. for C19H16ClN5O (365.8210): C 62.08%, H 4.39%, N 19.05%. Found: C 61.77%, H 4.14%, N 19.00%.
:1.5 v/v and obtained as a light brown solid. Yield 0.23 g (52%), m.p. 140–142 °C, TLC (CH2Cl2–MeOH 95:5 v/v) Rf 0.56. 1H NMR (DMSO-d6): δ 8.53 (s, 1H, pyrazole), 8.53 (s, 1H, triazole), 7.98 (s, 1H, triazole), 7.90 (m, 2H, Ar), 7.66 (d, J = 9.0 Hz, 2H, Ar), 7.36 (t, J = 8.50 Hz, 2H, Ar), 7.02 (d, J = 9.0 Hz, 2H, Ar), 5.48 (s, 2H, CH2-triazole), 4.08 (q, J = 7.0 Hz, 2H, OCH2), 1.35 (t, J = 7.0 Hz, 3H, CH3). 19F NMR: (DMSO-d6): −116.44 (F–Ar). 13C NMR (DMSO-d6): δ 161.76 and 159.82 (d, 1JCF = 243.9 Hz, C–F, Ar), 159.08 (C, Ar), 151.99 (CH, triazole), 150.91 (C, pyrazole), 144.36 (CH, triazole), 136.39 and 136.37 (d, 4JCF = 2.5 Hz, C, Ar), 130.37 (CH, pyrazole), 129.36 (2 × CH, Ar), 124.90 (C, pyrazole), 120.81 and 120.74 (d, 3JCF = 8.80 Hz, 2 × CH, Ar), 116.85 and 116.67 (d, 2JCF = 22.6 Hz, 2 × CH, Ar), 115.68 (C, Ar), 115.02 (2 × CH, Ar), 63.57 (OCH2), 43.81 (triazole-CH2), 15.11 (CH3). Anal. calcd. for C20H18FN5O (363.3964): C 66.10%, H 4.99%, N 19.26%. Found: C 66.21%, H 4.87%, N 19.17%.
:2 v/v and obtained as a light brown solid. Yield 0.32 g (60%), m.p. 122–124 °C, TLC (CH2Cl2–MeOH 95:5 v/v) Rf 0.49. 1H NMR (DMSO-d6): δ 8.58 (s, 1H, triazole), 8.54 (s, 1H, pyrazole), 7.98 (s, 1H, triazole), 7.90 (d, J = 8.5 Hz, 2H, Ar), 7.66 (d, J = 9.0 Hz, 2H, Ar), 7.57 (d, J = 9.0 Hz, 2H, Ar), 7.01 (d, J = 9.0 Hz, 2H, Ar), 5.48 (s, 2H, CH2-triazole), 4.08 (q, J = 7.0 Hz, 2H, CH2CH3), 1.35 (t, J = 6.5 Hz, 3H, CH2CH3). 13C NMR (DMSO-d6): δ 159.15 (C, Ar), 152.01 (CH, triazole), 151.15 (C, pyrazole), 144.39 (CH, triazole), 138.58 (C, Ar), 130.89 (C, Ar), 130.34 (CH, pyrazole), 129.96 (2 × CH, Ar), 129.39 (2 × CH, Ar), 124.78 (C, pyrazole), 120.31 (2 × CH, Ar), 116.05 (C, Ar), 115.04 (2 × CH, Ar), 63.58 (CH2CH3), 43.80 (CH2), 15.11 (CH2CH3). Anal. calcd. for C20H18ClN5O (379.8480): C 63.24%, H 4.78%, N 18.43%. Found: C 62.91%, H 4.63%, N 18.29%.
:1.5 v/v and obtained as a light orange solid. Yield 0.185 g (58%), m.p. 120–122 °C, TLC (CH2Cl2–MeOH 95:5 v/v) Rf 0.51. 1H NMR (DMSO-d6): δ 8.53 (s, 1H, pyrazole), 8.53 (s, 1H, triazole), 7.98 (s, 1H, triazole), 7.90 (m, 2H, Ar), 7.66 (d, J = 9.0 Hz, 2H, Ar), 7.36 (t, J = 9.0 Hz, 2H, Ar), 7.03 (d, J = 9.0 Hz, 2H, Ar), 5.48 (s, 2H, CH2-triazole), 3.98 (t, J = 6.5 Hz, 2H, OCH2), 1.76 (m, 2H, CH2), 1.0 (t, J = 7.0 Hz, 3H, CH3). 19F NMR: (DMSO-d6): −116.45 (F–Ar). 13C NMR (DMSO-d6): δ 161.76 and 159.82 (d, 1JCF = 243.9 Hz, C–F, Ar), 159.24 (C, Ar), 151.99 (CH, triazole), 150.91 (C, pyrazole), 144.36 (CH, triazole), 136.39 and 136.37 (d, 4JCF = 2.5 Hz, C, Ar), 130.37 (CH, pyrazole), 129.36 (2 × CH, Ar), 124.90 (C, pyrazole), 120.81 and 120.75 (d, 3JCF = 7.5 Hz, 2 × CH, Ar), 116.86 and 116.67 (d, 2JCF = 23.9 Hz, 2 × CH, Ar), 115.68 (C, Ar), 115.06 (2 × CH, Ar), 69.46 (OCH2), 43.82 (triazole-CH2), 22.50 (CH2), 10.88 (CH3). Anal. calcd. for C21H20FN5O (377.4234): C 66.83%, H 5.34%, N 18.55%. Found: C 66.67%, H 5.20%, N 18.67%.
:1.5 v/v and obtained as a light orange solid. Yield 0.23 g (72%), m.p. 114–116 °C, TLC (CH2Cl2–MeOH 95:5 v/v) Rf 0.69. 1H NMR (DMSO-d6): δ 8.58 (s, 1H, triazole), 8.54 (s, 1H, pyrazole), 7.98 (s, 1H, triazole), 7.91 (d, J = 9.0 Hz, 2H, Ar), 7.67 (d, J = 9.0 Hz, 2H, Ar), 7.58 (d, J = 9.0 Hz, 2H, Ar), 7.03 (d, J = 9.0 Hz, 2H, Ar), 5.48 (s, 2H, CH2-triazole), 3.98 (t, J = 6.5 Hz, 2H, OCH2), 1.76 (m, 2H, CH2), 1.0 (t, J = 7.0 Hz, 3H, CH3). 13C NMR (DMSO-d6): δ 159.31 (C, Ar), 152.01 (CH, triazole), 151.15 (C, pyrazole), 144.40 (CH, triazole), 138.59 (C, Ar), 130.88 (C, Ar), 130.34 (CH, pyrazole), 129.96 (2v × CH, Ar), 129.38 (2 × CH, Ar), 124.78 (C, pyrazole), 120.31 (2 × CH, Ar), 116.06 (C, Ar), 115.08 (2 × CH, Ar), 69.47 (OCH2), 43.81 (triazole-CH2), 22.50 (CH2), 10.88v (CH3). Anal. calcd. for C21H20ClN5O (393.8750): C 64.04%, H 5.12%, N 17.77%. Found: C 63.94%, H 5.02%, N 17.87%.
:1.5 v/v and obtained as a light brown solid. Yield 0.088 g (53%), m.p. 118–120 °C, TLC (CH2Cl2–MeOH 95:5 v/v) Rf 0.52. 1H NMR (DMSO-d6): δ 8.54 (s, 1H, pyrazole), 8.53 (s, 1H, triazole), 7.99 (s, 1H, triazole), 7.90 (m, 2H, Ar), 7.65 (d, J = 9.0 Hz, 2H, Ar), 7.36 (t, J = 9.0 Hz, 2H, Ar), 7.01 (d, J = 9.0 Hz, 2H, Ar), 5.48 (s, 2H, CH2-triazole), 4.67 (m, 1H, CH), 1.30 (d, J = 6.0 Hz, 6H, 2 × CH3). 19F NMR: (DMSO-d6): −116.46 (F–Ar). 13C NMR (DMSO-d6): δ 161.75 and 159.82 (d, 1JCF = 242.7 Hz, C–F, Ar), 158.03 (C, Ar), 152.0 (CH, triazole), 150.93 (C, pyrazole), 144.37 (CH, triazole), 136.39 and 136.37 (d, 4JCF = 3.77 Hz, C, Ar), 130.33 (CH, pyrazole), 129.41 (2 × CH, Ar), 124.74 (C, pyrazole), 120.80 and 120.73 (d, 3JCF = 8.8 Hz, 2 × CH, Ar), 116.86 and 116.67 (d, 2JCF = 23.9 Hz, 2 × CH, Ar), 116.10 (2 × CH, Ar), 115.69 (C, Ar), 69.66 (CH), 43.82 (CH2), 22.29 (CH3). Anal. calcd. for C21H20FN5O (377.4234): C 66.83%, H 5.34%, N 18.55%. Found: C 66.81%, H 5.16%, N 18.68%.
:2.5 v/v and obtained as a brown oil. Yield 0.26 g (81%), TLC (CH2Cl2–MeOH 95:5 v/v) Rf 0.48. 1H NMR (DMSO-d6): δ 8.57 (s, 1H, triazole), 8.54 (s, 1H, pyrazole), 7.99 (s, 1H, triazole), 7.91 (d, J = 9.0 Hz, 2H, Ar), 7.66 (d, J = 9.0 Hz, 2H, Ar), 7.58 (d, J = 9.0 Hz, 2H, Ar), 7.01 (d, J = 9.0 Hz, 2H, Ar), 5.48 (s, 2H, CH2-triazole), 4.68 (m, 1H, CH), 1.30 (d, J = 6.0 Hz, 6H, 2 × CH3). 13C NMR (DMSO-d6): δ 158.10 (C, Ar), 152.02 (CH, triazole), 151.16 (C, pyrazole), 144.40 (CH, triazole), 138.58 (C, Ar), 130.87 (C, Ar), 130.30 (CH, pyrazole), 129.96 (2 × CH, Ar), 129.44 (2 × CH, Ar), 124.61 (C, pyrazole), 120.29 (2 × CH, Ar), 116.10 (2 × CH, Ar), 116.06 (C, Ar), 69.67 (CH), 40.81 (CH2), 22.29 (2 × CH3). HPLC: 97.0%, RT = 4.88 min. HRMS (ESI) m/z: calculated: 394.1435 [M + H]+, found: 394.1443 [M + H]+.
Mtb MIC assay
Spot culture growth inhibition (SPOTi) assay was performed as previously described.13 Briefly, each well of a flat 96-well plate was spotted with 2 μL compound, serially diluted two-fold in DMSO from 150 μg mL−1. 200 μL Middlebrook 7H10 agar supplemented with 0.5% (v/v) glycerol and 10% OADC was dispensed into each well of a flat 96-well plate and the plate shaken to homogenise the compound. The agar was allowed to set before storage at 4 °C and used in the SPOTi assay within 24 h. To inoculate the plate, M. tuberculosis H37Rv was first cultured with shaking at 37 °C in Middlebrook 7H9 media supplemented with 0.5% (v/v) glycerol, 10% OADC and 0.025% Tween 80 up to an OD of 1. The culture was then diluted 1:100 in 7H9 media and 2 μL spotted onto each well of the SPOTi plate. Cultures were incubated at 37 °C for five weeks before MIC90 was recorded for each compound.
CYP121A1 binding affinity assay
Recombinant CYP121A1 protein that was used in spectral binding assays was expressed and purified as described previously.24 Compounds were first screened for binding on a dual-beam Shimadzu 2700 spectrophotometer, in which ligand-free spectra of 1 μM CYP121A1 (50 mM Tris HCl, 300 mM NaCl, pH 7.4) were compared with spectra in the presence of 250 μM of each compound following a 15 min incubation period in a 1 cm quartz cuvette. The shape and intensity of preliminary difference spectra indicated peak maxima between 420–435 nm and minima between 380–415 nm and were consistent with a type-II ligand response.Full titrations were performed in triplicate at ambient temperature using an Agilent BioTek Cytation 5 monochromator-based multimode plate reader. Protein samples at 1 μM concentration and in 200 μL volume were prepared in Corning® 96-well black flat bottom polystyrene 96 well plates. Individual wells were used to acquire 12 intermediate concentration points spanning 0 to 150 μM of compound. Kd values were calculated by plotting the inhibitor concentrations against the blue-shifted type-II response. Data fitting was performed as described previously24 using a single binding mode equation for hyperbolic fitting in Prism GraphPad v7.05. As additional controls, cYY substrate was included in the plate reader assay and all compounds were validated with a single manual titration on the Shimadzu 2700 spectrophotometer.
Protein expression, purification, and sample preparation for NMR
The mutant version of CYP121A1 protein containing C171S was expressed, purified, and labelled for NMR detection as described previously.18 Following purification, the protein was 19F labelled using 3-bromo-1,1,1-trifluoroacetone (BTFA). Briefly, the protein was diluted to 2 μM in 50 mM Tris HCl, pH 7.4, and 300 mM NaCl supplemented with 10 mM BTFA and 5 mM DTT and incubated overnight at 4 °C. Unreacted BTFA and DTT were removed by size exclusion chromatography. The protein was exchanged into the NMR buffer (50 mM potassium phosphate, pH 7.4, 50 mM NaCl, and 10% D2O) using a 10 kDa molecular weight cut-off filter. Protein was aliquoted into separate NMR samples at 100 μM and 450 μL volume. Compounds were added from a DMSO stock to a final concentration of 300 μM and allowed to incubate for 15 min prior to transfer to NMR sample tubes. Spectra were acquired at 25 °C on a Bruker AVANCE III 600 MHz spectrometer for 10000 scans per experiment. The data were processed and analyzed in TopSpin, version 4.1.1.
Computational studies
Molecular docking and molecular dynamics simulations were performed as previously described.25,26 Briefly docking studies, using the crystal structure of Mtb CYP121A1 co-crystallised with cYY (pdb 3G5H4) were performed to generate PDB Mtb CYP121A1–ligand complexes, using MOE22 until a RMSD gradient of 0.01 kcal mol−1 Å−1 with the MMFF94 forcefield (ligands) and partial charges were automatically calculated. The charge of the haem iron at physiological pH was set to 3+ (geometry d2sp3) through the atom manager in MOE. Docking was performed using the Alpha Triangle placement to determine the poses, refinement of the results was done using the MMFF94 forcefield, and rescoring of the refined results using the London ΔG scoring function was applied. Molecular dynamics simulations were run on the Mtb CYP121A–ligand complexes as previously described26 using the Desmond programme of Schödinger.23 Overlapping water molecules were deleted, and the systems were neutralised with Na+ ions and salt concentration 0.15 M. Force-field parameters for the complexes were assigned using the OPLS_2005 forcefield, that is, a 150 ns molecular dynamic run in the NPT ensemble (T 1/4 300 K) at a constant pressure of 1 bar. Energy and trajectory atomic coordinate data were recorded at each 1.2 ns.Author contributions
Research concept designed by CS, DFE, SJW and SB. LAA performed the chemistry and analysis of all compounds supervised by CS. AK developed the methodology and performed the Mtb CYP121A1 and 19F NMR binding affinity studies supervised by DFE. SJW performed the SPOTi assay, with support and training from GS and SB. Computational studies and visualisation performed by CS. Manuscript reviewed by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. A. A. thanks Princess Nourah bint Abdulrahman University (College of Pharmacy, Department of Pharmaceutical Science) and the Royal Embassy of Saudi Arabia for a full PhD scholarship to complete this research at Cardiff University. Molecular dynamics simulations were undertaken using the supercomputing facilities at Cardiff University operated by Advanced Research Computing at Cardiff (ARCCA) on behalf of the Cardiff Supercomputing Facility and the HPC Wales and Supercomputing Wales (SCW) projects. We acknowledge support of the latter, which is part-funded by the European Regional Development Fund (ERDF) via the Welsh Government.Notes and references
- World Health Organisation, Global tuberculosis report 2021, https://www.who.int/publications/i/item/9789240037021, (accessed 23 March 2022).
- J. L. Khawbung, D. Nath and S. Chakraborty, Drug resistant tuberculosis: a review, Comp. Immunol., Microbiol. Infect. Dis., 2021, 74, 101574 CrossRef CAS.
- K. J. McLean, A. J. Dunford, R. Neeli, M. D. Driscoll and A. W. Munro, Arch. Biochem. Biophys., 2007, 464, 228–240 CrossRef CAS.
- P. Belin, M. H. Le Du, A. Fielding, O. Lequin, M. Jacquet, J. B. Charbonnier, A. Lecoq, R. Thai, M. Courçon, C. Masson, C. Dugave, R. Genet, J. L. Pernodet and M. Gondy, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7426–7431 CrossRef PubMed.
- K. J. Mclean, P. Carroll, D. G. Lewis, A. Dunford, H. E. Seward, R. Neeli, M. R. Cheesman, L. Marsollier, P. Douglas, W. E. Smith, I. Rosenkrands, S. T. Cole, D. Leys, T. Parish and A. W. Munro, J. Biol. Chem., 2008, 283, 33406–33416 CrossRef CAS PubMed.
- I. M. Taban, H. E. A. E. Elshihawy, B. Torun, B. Zucchini, C. J. Williamson, D. Altuwairigi, A. S. T. Ngu, K. J. McLean, C. W. Levy, S. Sood, L. B. Marino, A. W. Munro, L. P. S. de Carvalho and C. Simons, J. Med. Chem., 2017, 60, 10257–10267 CrossRef CAS PubMed.
- H. A. A. Abd El-wahab, M. Accietto, L. B. Marino, K. J. McLean, C. W. Levy, H. M. Abdel-Rahman, M. A. El-Gendy, A. W. Munro, A. S. Aboraia and C. Simons, Bioorg. Med. Chem., 2018, 26, 161–176 CrossRef CAS PubMed.
- S. M. Kishk, K. J. McLean, S. Sood, M. A. Helal, M. S. Gomaa, I. Salama, S. Mostafa, L. P. S. de Carvalho, A. W. Munro and C. Simons, Bioorg. Med. Chem., 2019, 27, 1546–1561 CrossRef CAS PubMed.
- S. M. Kishk, K. J. McLean, S. Sood, D. Smith, J. W. D. Evans, M. A. Helal, M. S. Gomaa, I. Salama, S. Mostafa, C. W. Levy, L. P. S. de Carvalho, A. W. Munro and C. Simons, ChemistryOpen, 2019, 8, 995–1011 CrossRef CAS.
- M. E. Kavanagh, J. L. Gray, S. H. Gilbert, A. G. Coyne, K. J. McLean, H. J. Davis, A. W. Munro and C. Abell, ChemMedChem, 2016, 11, 1924–1935 CrossRef CAS PubMed.
- C. Brengel, A. Thomann, A. Schifrin, G. Allegretta, A. A. M. Kamal, J. Haupenthal, I. Schnorr, S. H. Cho, S. G. Franzblau, M. Empting, J. Eberhard and R. W. Hartmann, ChemMedChem, 2017, 12, 1616–1626 CrossRef CAS PubMed.
- I. Walter, S. Adam, M. V. Gentilini, A. M. Kany, C. Brengel, A. Thomann, T. Sparwasser, J. Köhnke and R. W. Hartmann, ChemMedChem, 2021, 16, 2786–2801 CrossRef CAS PubMed.
- J. D. Guzman, D. Evangelopoulos, A. Gupta, K. Birchall, S. Mwaigwisya, B. Saxty, T. D. McHugh, S. Gibbons, J. Malkinson and S. Bhakta, BMJ Open, 2013, 3, e002672 CrossRef PubMed.
- J. A. Dijkstra, T. van der Laan, O. W. Akkerman, M. S. Bolhuis, W. C. M. de Lange, J. G. W. Kosterink, T. S. van der Werf, J. W. C. Alffenaar and D. van Soolingen, Antimicrob. Agents Chemother., 2018, 62, e01724 CrossRef PubMed.
- A. K. Ghose and G. M. Crippen, J. Chem. Inf. Comput. Sci., 1987, 27, 21–35 CrossRef CAS.
- M. Fonvielle, M.-H. Le Du, O. Lequin, A. Lecoq, M. Jacquet, R. Thai, S. Dubois, G. Grach, M. Gondry and P. Belin, J. Biol. Chem., 2013, 288, 17347–17359 CrossRef CAS.
- H. E. Seward, A. Roujeinikova, K. J. McLean, A. W. Munro and D. Leys, J. Biol. Chem., 2006, 281, 39437–39443 CrossRef CAS.
- C. S. Campomizzi, G. E. Ghanatios and D. F. Estrada, J. Biol. Chem., 2021, 297, 101287 CrossRef CAS.
- P. Urban, T. Lautier, D. Pompon and G. Truan, Int. J. Mol. Sci., 2018, 19, 1617 CrossRef.
- Y.-T. Lee, R. F. Wilson, I. Rupniewski and D. B. Goodin, Biochemistry, 2010, 49, 3412–3419 CrossRef CAS PubMed.
- I. F. Sevrioukova and T. L. Poulos, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 486–491 CrossRef CAS PubMed.
- Molecular Operating Environment (MOE), Chemical Computing Group ULC, 2019, https://www.chemcomp.com/Products.htm.
- Desmond Schrödinger Release 2020-1, Desmond Molecular Dynamics System, D.E. Shaw Research, New York, NY, Maestro Desmond Interoperability Tools, Schrödinger, NewYork, NY, 2020, https://www.schrodinger.com/products/desmond.
- A. Kumar, C. S. Campomizzi, N. Jay, S. Ferguson, E.-J. Scheffler, J. Lioi, C. Tu, J. Qu, C. Simons and D. F. Estrada, Sci. Rep., 2021, 11, 394 CrossRef CAS PubMed.
- N. A. Noureldin, J. Richards, H. Kothayer, M. M. Baraka, S. M. Eladl, M. Wooton and C. Simons, RSC Adv., 2022, 12, 2511–2524 RSC.
- F. A. Binjubair, J. E. Parker, A. G. Warrilow, K. Puri, P. J. Braidley, E. Tatar, S. L. Kelly, D. E. Kelly and C. Simons, ChemMedChem, 2020, 15, 1294–1309 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods and analytical data for compounds 7–10; NMR for compounds 11 and 12. See DOI: https://doi.org/10.1039/d2md00155a |
| This journal is © The Royal Society of Chemistry 2022 |