
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The formulation of a CMC binder/silicon composite anode for Li-ion batteries: from molecular effects of ball milling on polymer chains to consequences on electrochemical performances†
Mariama
Ndour
ab,
Jean-Pierre
Bonnet
 *a,
Sébastien
Cavalaglio
a,
Tristan
Lombard
a,
Matthieu
Courty
a,
Luc
Aymard
a,
Cédric
Przybylski
cd and
Véronique
Bonnet
*b
*a,
Sébastien
Cavalaglio
a,
Tristan
Lombard
a,
Matthieu
Courty
a,
Luc
Aymard
a,
Cédric
Przybylski
cd and
Véronique
Bonnet
*b
aLaboratoire de Réactivité et Chimie des Solides (CNRS UMR 7314), Université de Picardie Jules Verne, 15 rue Baudelocque, 80039 Amiens Cedex, France. E-mail: jean-pierre.bonnet@u-picardie.fr
bLaboratoire de Glycochimie, des Antimicrobiens et des Agroressources, (CNRS UMR 7378), Université de Picardie Jules Verne, 10 rue Baudelocque, 80039 Amiens Cedex, France. E-mail: veronique.bonnet@u-picardie.fr
cSorbonne Université, CNRS, Institut Parisien de Chimie Moléculaire, 4 Place Jussieu, 75252 Cedex 05 Paris, France
dSorbonne Université, Mass Spectrometry Sciences Sorbonne University, MS3U Platform, UFR 926, UFR 927, 75005, Paris, France
First published on 22nd September 2022
Abstract
The semi-synthetic polysaccharide carboxymethylcellulose (CMC) is one of the most studied and effective polymer binders for silicon-based anodes in Li-ion batteries. The formulation of the corresponding composite negative electrode with an appropriate mixture of electroactive silicon, a CMC binder and a carbon additive is mandatory to ensure a good electrical conductivity. Blending is commonly realized by a highly energetic ball milling treatment of these three aforementioned components. This type of mixing reduces the size of the obtained particles and can also potentially agglomerate them. Morever, it allows the formation of a nanostructured mixture which is essential for both the silicon activation and to achieve good electrochemical performance. However, such strong treatment can also cause a significant degradation of the polymer chains, as we have recently demonstrated for polyacrylic acid (PAA). In the present work, the structural and chemical effects of this mechanical grinding on three commercial CMCs ranging from 90 to 700 kg mol−1 were investigated. All the polymers were characterized using SEC-MALLS, FTIR spectroscopy, MALDI-TOF mass spectrometry and TGA-MS thermal analysis. In all cases, a huge average molecular weight decrease was noticed, leading to the appearance of a bimodal distribution with low (52–72 kg mol−1) to very low molecular weight populations (1–1.8 kg mol−1). From these results, two formulations of a negative electrode were compared, one with ball milling of the three compounds and another one including only ball milling steps for silicon and carbon. After the correlation of the characteristics of this negative electrode composite with the electrochemical results, it was demonstrated that a high number of functions for supramolecular or covalent linkages are keypoints of the herein anode performance. Low molecular weight CMC derivatives (about 64 kg mol−1) obtained by ball milling treatment led to higher stability of the electrode.
Introduction
In the field of lithium-ion batteries (LiBs), silicon is recognized as an anodic material of choice due to its high specific gravimetric capacity of 3579 mA h g−1. Such a capacity is related to the Li15Si4 phase obtained during the lithiation process and is associated with a huge volume expansion of about 280%.1,2 This volume expansion is accompanied by a drastic loss of capacity following three main phenomena which are silicon pulverization, the formation of a continuous unstable and thick layer of a solid electrolyte interphase (SEI), and even the electrical isolation of the battery after delamination of the electrode.3–5 This SEI is a passivation layer on the surface of the electrode following degradation of the electrolyte. It must remain thin and stable to allow the transport of lithium cations. To keep a good reversible capacity of silicon, the composite negative electrode must therefore present certain structural properties,6,7 such as (i) a nanoporous structure to accommodate the variation in volume during electrochemical cycling and facilitate the electrolyte diffusion, (ii) an electrically conductive network percolating through the entire electrode, to counter the low conductivity of silicon and (iii) a stable external surface of the electrode to allow the formation of a layer of the SEI, fine and stable on the surface of the silicon.Consequently, several studies were devoted to improve the electrochemical stability by modifying the morphology of silicon,8–10 using a conductive material,11 tailoring the polymer binder12,13 and also tuning the formulation parameters.14,15 For silicon-based composite negative electrodes, the choice of a polymer binder is here especially critical for the electrochemical stability of the negative electrode and, therefore, the battery.16 Here, polyvinylidene fluoride (PVDF) was one of the first investigated LiB anode synthetic polymer binders.17 However, it was observed that the resulting van der Waals interactions are not sufficient to maintain the cohesion of the electrode.18 Polyelectrolytes such as carboxymethyl cellulose (CMC) and poly acrylic acid (PAA) were then used for their carboxylic acid functions allowing substantial enhancement of electrode performance compared to PVDF. Such a kind of acidic moiety is expected to allow strengthened interactions between Si particles and polymers, by hydrogen bonding or covalent bonding.19–21 CMC (Fig. 1) is now the most widely used polysaccharide in Si-based composite electrode studies. This semi-synthetic polyelectrolyte is constituted of glucose units with β-(1 → 4) linkages. The carboxymethylation is achieved by etherification of hydroxyl groups of cellulose by monochloroacetic acid.
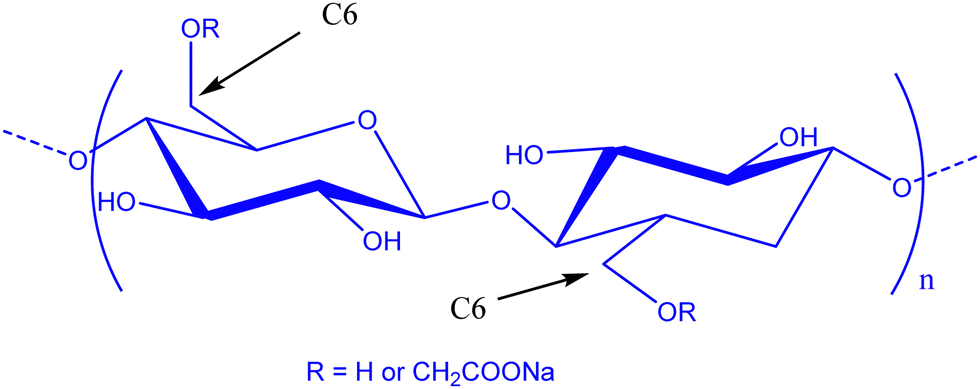 | ||
| Fig. 1 Chemical structure of the carboxymethyl cellulose sodium salt. | ||
In addition to this acidic functionalization of the polysaccharide, several polymer binder parameters can affect the stability of the anode, notably the molecular weight, the degree of substitution (DS), the polymer structure (molecular architecture) and also the type of glycosidic bonds.13,22–25 Most studies in laboratories with a Si/polymer/carbon slurry formulation use a high-energy ball-milling step in the process as “Fritsch Pulverisette” or “SPEX”19,26 for mixing the materials. This milling step is needed to reduce the particle size and eventually decreases the crystallinity of the silicon to facilitate its alloying and the reversibility of the alloying.27 However, on the other hand, the grinding of polymers such as polysaccharides could lead to a strong degradation.28–30 Zhe Ling et al.28 described the effects of ball milling on the structure of cotton cellulose. The authors used an Eberbach E3300 mini cutting Mill, which is considered to be a “vibratory” ball mill. They observed that the molecular weights of the cellulose samples decrease steadily with the increased milling time. Some oxidations, associated with carbonyl group amount, are also reported. Regarding crystallinity, Raman/FTIR spectroscopy studies confirm an increase of the amorphous part with the ball milling process. They suggest that cellulose crystallites are inhomogeneously perturbed during ball-milling steps leading to the so-called “inhomogeneous decrystallization”.
In the present work, we reported efforts to delineate the effect of mechanical grinding on the CMC chemical structure of the various samples and their resulting properties, in particular those involved in the stability of the negative silicon-based electrode. Particular attention was paid to investigate the impact of the most used grinding process, i.e. SPEX ball milling, on the LiB anode formulation and its consequence in the electrochemical performance of the silicon/polymer/carbon slurry composite anode.
Results and discussion
Silicon ball milling experiments
As a first step, 2witech silicon has been characterized by laser granulometry to determine the particle size distribution and the resulting average diameter of the particles before and after ball milling. The sample is analysed in a liquid aqueous dispersion at 0, 30 and 60 min (Fig. 2a).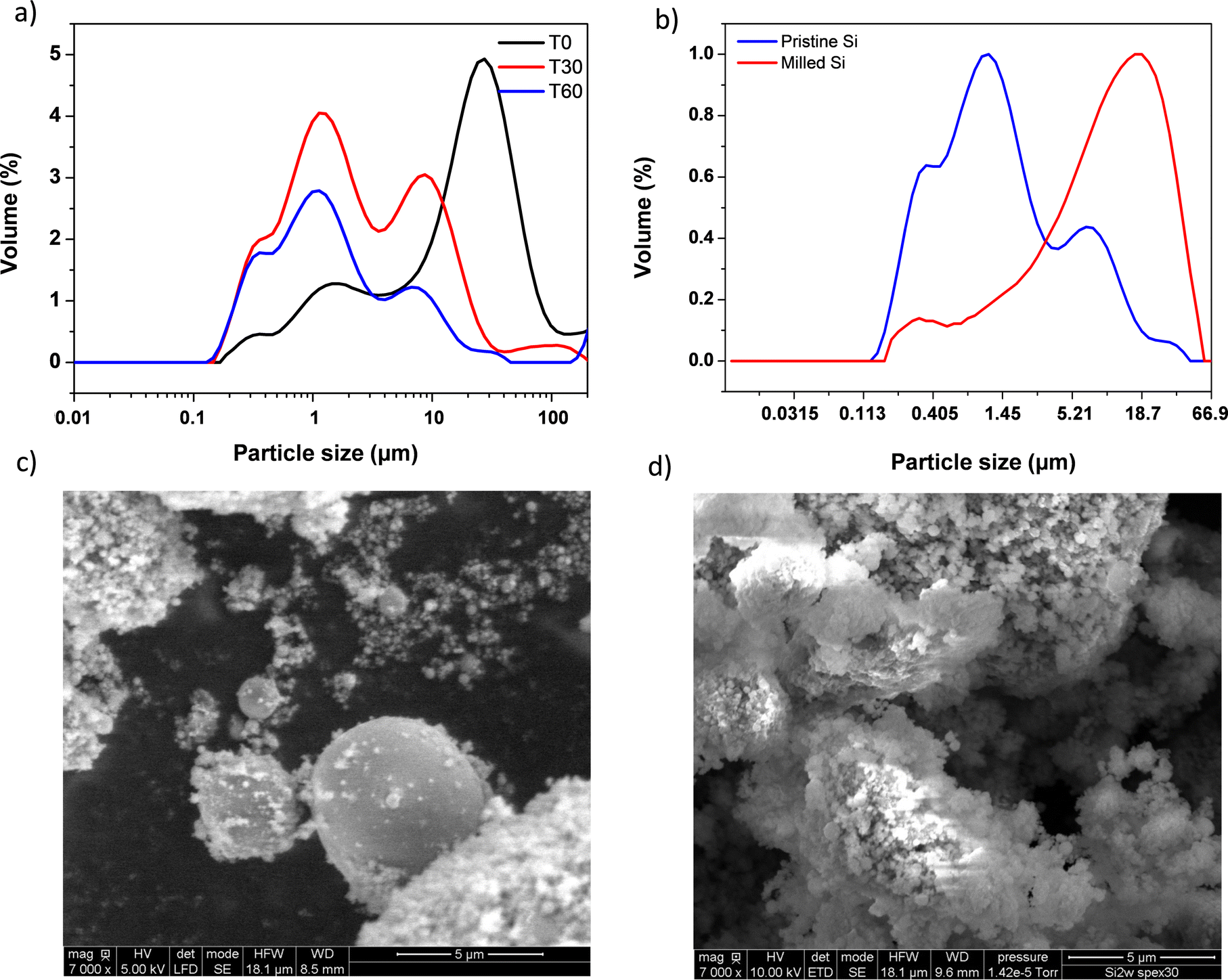 | ||
| Fig. 2 Size distribution curves measured by laser diffraction granulometry of commercial silicon 2witech® in solution at 0 min (black trace), 30 min (red trace) and 60 min (blue trace) (a) and after ball milling SPEX (b). Scanning electron microscopy (SEM) imaging of silicon before and after SPEX grinding (c and d, respectively). | ||
We observed that, from 60 minutes, particle sizes in solution did not change anymore. A multimodal distribution was observed with 4 distinct populations of sizes around 0.3, 1, 10 and 35 μm, respectively (Fig. 2a). After grinding with SPEX for 30 min, 2 populations around 0.3 μm and 19 μm were finally obtained (Fig. 2b). The formation of these two populations confirms some results described elsewhere.26,27 The ball milling created the agglomeration of sub-micrometric particles. This study was carried out with a micrometric silicon of the order of 2 μm undergoing ball milling for 20 h. It is important to note that the ball milling step of Si particles exhibits a beneficial effect which is portrayed by better capacity retention. This enhanced performance is presumably due to nano-structuration thanks to grain boundary creation which leads to faster Li+ diffusion. After just 30 min of SPEX ball milling, we observed similar results to those already described in the literature.26,27
In addition, scanning electron microscopy (SEM) imaging of two 2witech silicon particles (before and after ball milling) allowed us to confirm the formation of agglomerates during ball milling. The images obtained at magnifications of 500 (see Fig. S1, ESI†) and 7000 (Fig. 2) show a clear difference between the pristine silicon (Fig. 2c) and the ball milled silicon (Fig. 2d). In the snapshot of commercial 2witech silicon (Fig. 2c), spherical particles present a very large disparity with an average size of 1–2 μm. After ball milling, we observe even larger aggregates with also a multitude of nanometric particles on the surface. These results are therefore in perfect agreement with previous ones obtained by laser granulometry (Fig. 2a and b).
Study of the binder degradation issued from the ball milling step
Size exclusion chromatography (SEC) was used to determine the average molecular weight![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n, w and the resulting polydispersity index (Đ = w/n) of different CMCs. Three commercial CMCs at 90, 250 and 700 kg mol−1 are picked for a comparative study of the ball milling. The buffer solution (0.173 M citric acid + 0.074 M KOH pH = 3) used during SEC experiments was selected to be the closest from the reference formulation conditions. Fig. 3 shows the SEC profiles issued from the analysis of commercial and post-milled CMCs. The commercial CMC chromatograms showed a trimodal distribution with high polydispersity on different samples (Fig. 3a). This important polydispersity is well known for natural polymers. However, weight average molecular weights (w) are fairly close to the references indicated by the manufacturer.
n, w and the resulting polydispersity index (Đ = w/n) of different CMCs. Three commercial CMCs at 90, 250 and 700 kg mol−1 are picked for a comparative study of the ball milling. The buffer solution (0.173 M citric acid + 0.074 M KOH pH = 3) used during SEC experiments was selected to be the closest from the reference formulation conditions. Fig. 3 shows the SEC profiles issued from the analysis of commercial and post-milled CMCs. The commercial CMC chromatograms showed a trimodal distribution with high polydispersity on different samples (Fig. 3a). This important polydispersity is well known for natural polymers. However, weight average molecular weights (w) are fairly close to the references indicated by the manufacturer.
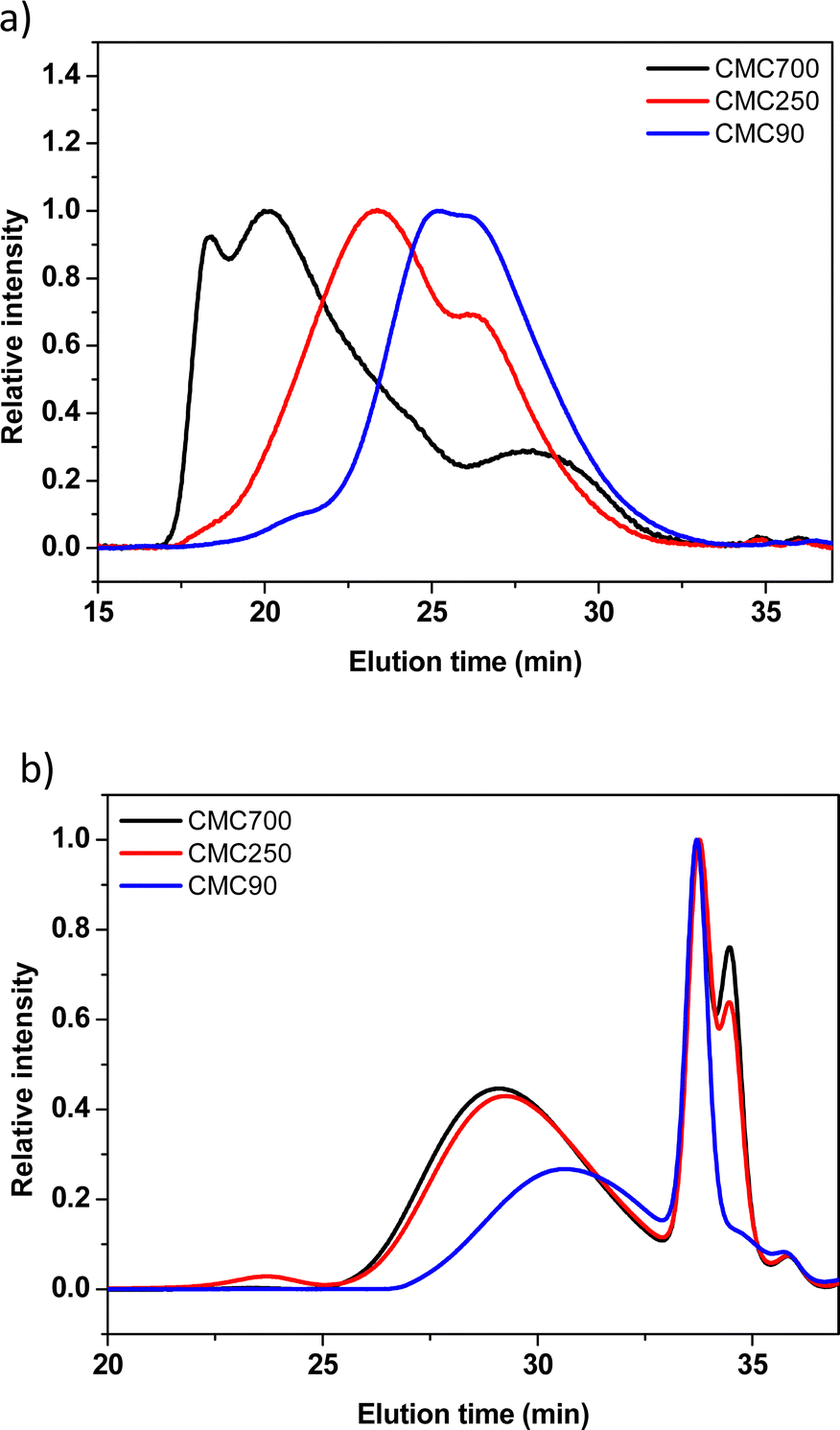 | ||
| Fig. 3 Normalized SEC-RI molar mass distribution profiles of CMC90 (blue trace), CMC250 (red trace) and CMC700 (black trace) before (a) and after ball milling (b). Samples were milled with buffer (citric acid/KOH) pH 3 and eluted in H2O/NaNO3/NaN3. | ||
The comparison of the traces before and after ball milling of the three CMCs shows quasi-similar profiles for the three samples of CMC (Fig. 3b). In detail, two populations are distinguished, the first population eluted between 26 and 32 min is associated with large molecular weights (polymers).
The second population, the so-called bimodal, between 32 and 36 min, corresponds to very low molecular weights. Such results clearly demonstrate the detrimental effect of SPEX mechanical milling on the CMC structural integrity, as illustrated by the drastic reduction of apparent molecular weights. In addition to the decrease of the average molecular weight, it was demonstrated in the literature that grinding also induces a crystallinity reduction.28,29
Although, these commercial CMCs are already highly amorphous with a very low crystallinity (CrI: 17%), we observed a significant crystallinity decrease after ball milling for CMC90 and CMC250 (Fig. S2, ESI†). Table 1 shows the values of average molecular weights and the polydispersity of the different CMCs before and after ball milling.
w: weight average molecular weight; n: number average molecular weight; and Đ: polydispersity index (w/n)
| Sample | Commercial | Ball-milled | ||||
|---|---|---|---|---|---|---|
|
n (kg mol−1) |
w (kg mol−1) |
Đ |
n (kg mol−1) |
w (kg mol−1) |
Đ | |
| CMC90 | 9.5 | 124.1 | 13.1 | 2.5 | 6.4 | 2.5 |
| CMC250 | 34.6 | 212.1 | 6.1 | 3.1 | 13.4 | 4.2 |
| CMC700 | 165.6 | 705.5 | 4.2 | 2.9 | 15.0 | 5.0 |
Moreover, it was quoted out that the 2 maxima of the second population (33 and 34 min) presumably ascribed to oligomers are identical for CMC700 and 250. Surprisingly, the CMC90 sample did not exhibit a second peak at around 33–34 min, while the first one presents a similar intensity compared to the two other CMCs.
Another relevant point is thus that regardless of the initial molar mass of CMCs, the molar masses of populations of very low molecular weights are identical. These small molecules could thus correspond to an ultimate stage of degradation. It must be noticed here that a similar behaviour was observed in the case of tribochemical treatments of cellulose (sonochemistry and ball milling), with the statement that degradation proceeded towards a final value, called “limiting molecular weight” (Mwlim), below which no additional chain length decrease took place, even at extended treatment times.31 From this result, the observed significant decrease of the polydispersity index after ball milling for CMC90 and CMC250 (from 13.1/6.1 to 2.5/4.2) and the slight increase, on the contrary, noticed in the case of CMC700 (from 4.2 to 5, respectively) can be mainly understood in terms of the relative weight important of the very narrow distribution of low molecular weight chains in regard with the larger ones. In other terms, the higher the relative percent of these oligomers, the lower the polydispersity index.
The chromatograms obtained using the UV-visible detector (λ = 280 nm) showed the presence of carbonyl functions, for the population of low molecular weights between 32 and 36 min (Fig. S3, ESI†). In addition, no significant UV signal was detected in the large molecules corresponding to the elution time range from the chromatogram. The large molecular weights should be presumably attributed to unmodified cellulose. This mode of degradation suggests that the presence of carboxymethyl and carbonyl moieties is present within small molecular weight CMCs. A similar study carried out by Ling et al.28 on cotton cellulose with different ball milling times revealed that the glycosidic bonds is cleaved by β-elimination on glucose residues. The formation of radicals during tribochemical treatments from milling revealed the increase of the amount of carbonyl groups. According to the authors, the more flexible C-6 groups are not really affected; therefore, this treatment does not change the amount of carboxyl groups.31,32
The SPEX grinding caused an abrasion of the cell which leaves traces of iron in the final products. These traces of iron and carbon pollution avoid any clear interpretation of the NMR and RAMAN spectra, in particular to identify the presence of carbonyl groups on the milled CMC (data not shown).
The FTIR spectra of commercial and milled CMC showed infrared bands commonly observed with oxidized cellulose (Fig. S4, ESI†). Of particular interest, we can note the reduction of the OH stretching band (3600–3000 cm−1) and the OH bending region (1800–1300 cm−1) consecutively to SPEX treatment. The difference can be explained by the β-elimination on glucose residues of the CMC leading to carbonyl groups instead of hydroxyl groups. The drying step can also reduce the OH bending by removing the water molecules. By considering some previously described cellulose based degradation products, possible structures for them can be suggested according to the IR spectroscopy results (Fig. S5, ESI†).
Moreover, the reduction of a bending peak (around 1372 cm−1), corresponding to the proportion of the crystalline part, gives additional evidence of the increase of amorphous fraction previously determined by XRD analysis (Fig. S2, ESI†).
However, it is difficult to access the exact molecular weights of the molecules obtained after grinding using SEC analysis. As aforementioned for NMR and Raman spectroscopies, ball milling is accompanied by the release of iron nanoparticles, leading to a higher chromatogram background with multi-angle light scattering (MALS) detection. Such a phenomenon strongly affects the possibility to obtain the accurate determination of the average molar weights. The two populations were then separated and collected at the SEC outlet for further characterization by mass spectrometry (MS).
The two collected fractions from SEC (large molecular weights/small molecular weights) from CMC90 and CMC700 samples with DS of 0.7 and 0.9, respectively, were further analysed using MALDI-TOF MS (Fig. 4). The MALDI-TOF spectrum of CMC700 in the high mass range showed apparent polymeric chain distributions estimated to be 52.5 and 70.93 kg mol−1, for the number average molecular weight (n) and weight average molecular weight (w), respectively, leading to a calculated dispersity (Đ) of 1.4 (Fig. 4a). As regards the CMC90 sample, n and w are slightly higher with 56.87 and 72.76 kg mol−1, respectively. Nevertheless, polydispersity remains close to that observed for CMC700 with Đ equal to 1.3 (Fig. 4b). Further examination of the spectra with optimized settings for low mass range analysis reveals several peaks which can be ascribed to oligo-glucosides with various degrees of both polymerization (DP) centred at around 3–6 and up to 11, and carboxymethylation for both CMC700 and CMC90 (Fig. 4b and d). It should also be noted that spectra are complicated by the cation exchanges which occurred with acidic moieties, namely H+, Na+, K+ and sometimes traces of Li+.
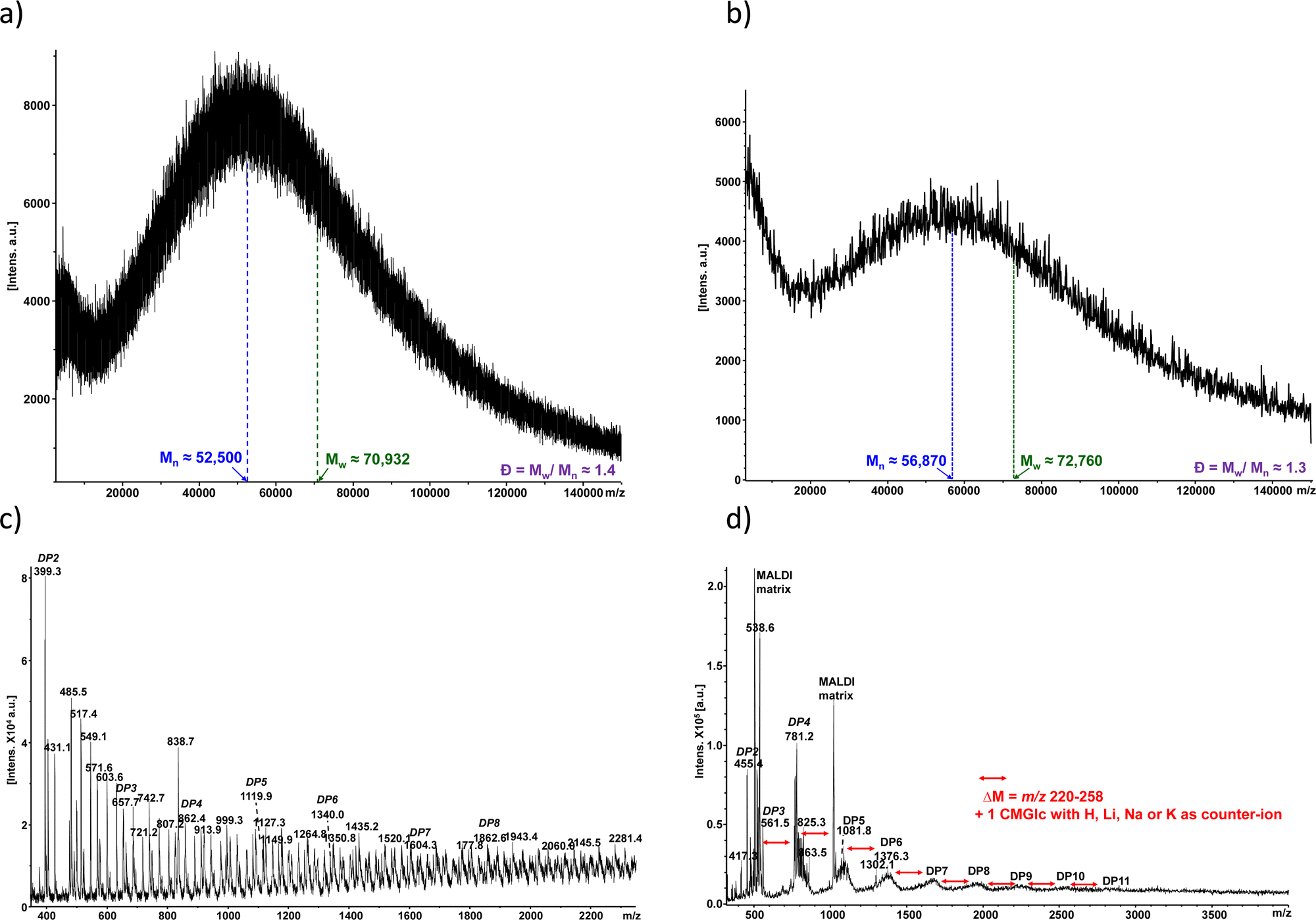 | ||
| Fig. 4 Negative ion linear MALDI-TOF MS spectra of the two collected SEC fractions after ball milling treatment. Detection of either ball-milled CMC 700 in the (a) high mass range and (c) low mass range or CMC 90 (b) in the high mass range (d) and low mass range (see the Experimental section for further details). | ||
Interestingly, these two analyses showed a degradation of the native polysaccharides. Starting from a given degree of substitution (DS = 0.7 or 0.9), ball milling with SPEX of the CMC gives almost the same chromatogram with slightly different size proportions. In addition, the electrode formulated by this method only consists of small or even very small CMCs regardless of the size chosen at the start (700, 250 or 90 kg mol−1). It therefore does not seem very relevant to compare these polysaccharides using this method of preparation. To complete this study, the thermogravimetric analyses of different CMCs were carried out at a heating rate of 5 °C min−1 up to 600 °C before and after ball milling.
The TGA thermograms of both commercial and ball-milled CMC samples are shown in the ESI† (Fig. S5). The three CMCs exhibit a similar three-step degradation process.33–35 The first step (up to approximately 150 °C) corresponds to the loss of bound water molecules (m = 18 g mol−1) associated with the highly hygroscopic nature of this type of polysaccharide. The second part (between 200 and 300 °C) corresponds to a second loss of intramolecular water immediately resulting in a rearrangement of carboxylic acid functions to anhydride with the loss of CO2 (m = 44 g mol−1). The last step corresponds to the degradation of polysaccharide chains with decarboxylation followed by chain cleavage, characterized in particular by the following mass losses (m = 12 C, m = 16 O, m = 17 OH, m = 28 CH2 –CH2, m = 30 g mol−1 CH2O). The comparison of the thermograms of the different CMC samples before ball milling shows a similar degradation temperature (Tonset) at 250 °C (Fig. S5, ESI†). This value at the beginning of the degradation is close to that reported in the literature (280 °C for CMC Na) for this type of cellulose with alkali cations (Li+, Na+, and K+).36 According to the nature of the cellulose and its molecular weight, the Tonset value may present some variations. After ball milling, this temperature is lowered to around 200 °C whatever the CMC used. Such a result gives additional evidences of the presence of smaller macromolecules during grinding and it is in good agreement with the SEC and MALDI-TOF mass spectrometry results.
Electrochemical characterization of the formulated electrode
To better assess the electrochemical performance of silicon following the addition of the polymer binder, we used two formulation methods, with CMC90 and CMC250 as polymer binders: (i) ball milling with SPEX of the Si/C/CMC powder (formulation 1) or (ii) ball milling Si/C powder with SPEX followed by magnetic stirring in a polymer suspension (formulation 2). The porosity and adhesion can give precious pieces of information on the quality of the formulation (Table 2).| Electrode | % porosity | pH of slurry | Adhesion |
|---|---|---|---|
| Formulation 1 CMC90 | 60 | 3.15 | 2B |
| Formulation 2 CMC90 | 56 | 3.15 | 2B |
| Formulation 1 CMC250 | 69 | 3.15 | 2B |
| Formulation 2 CMC250 | 63 | 3.05 | 1B |
The loading is preliminarily set for easier comparison of the electrochemical performances. The porosity of each electrode was calculated using the density of the different components. The porosity of the electrode obtained by formulation 2 is lower than that obtained by formulation 1 (56% versus 60% and 63% versus 69%, for CMC90 and 250, respectively). The SEM images show that such porosity comes from air bubbles due to the formulation technique but also to the presence of small cracks on the electrode (Fig. S6, ESI†). The buffer solution helps promoting adhesion to the current collector (Cu) by inducing the active corrosion of the Cu substrate and the formation of multiple strong Cu(OCO–R)2 anchoring bonds.37 The different films have fairly close adhesion, with a detachment of at least 50% of the grid surface (2B). Such an adhesion feature is suitable to efficiently perform electrochemical tests of the electrode. Only formulation 2 with CMC250 showed a lower adhesion at 1B compare to the other films. High porosity and low adhesion to the copper collector can cause a faster and irreversible loss of electrochemical performances.
The pH of the studied formulations (3.05–3.15) is lower than the pKa of carboxylic acid (4–5), allowing the functions to be kept predominantly under acidic forms to promote the reaction with the oxides of silicon.
Once the electrode film is dry, an infrared analysis is performed to determine the types of interactions involved. The spectrum of 2witech silicon is compared to those of the electrodes obtained after the two formulations used in this study (Fig. 5). In the silicon spectrum, the bands at 975 cm−1 and 1202 cm−1 are associated with Si–O–Si bonds due to the presence of SiO2 on the surface of the silicon particles.
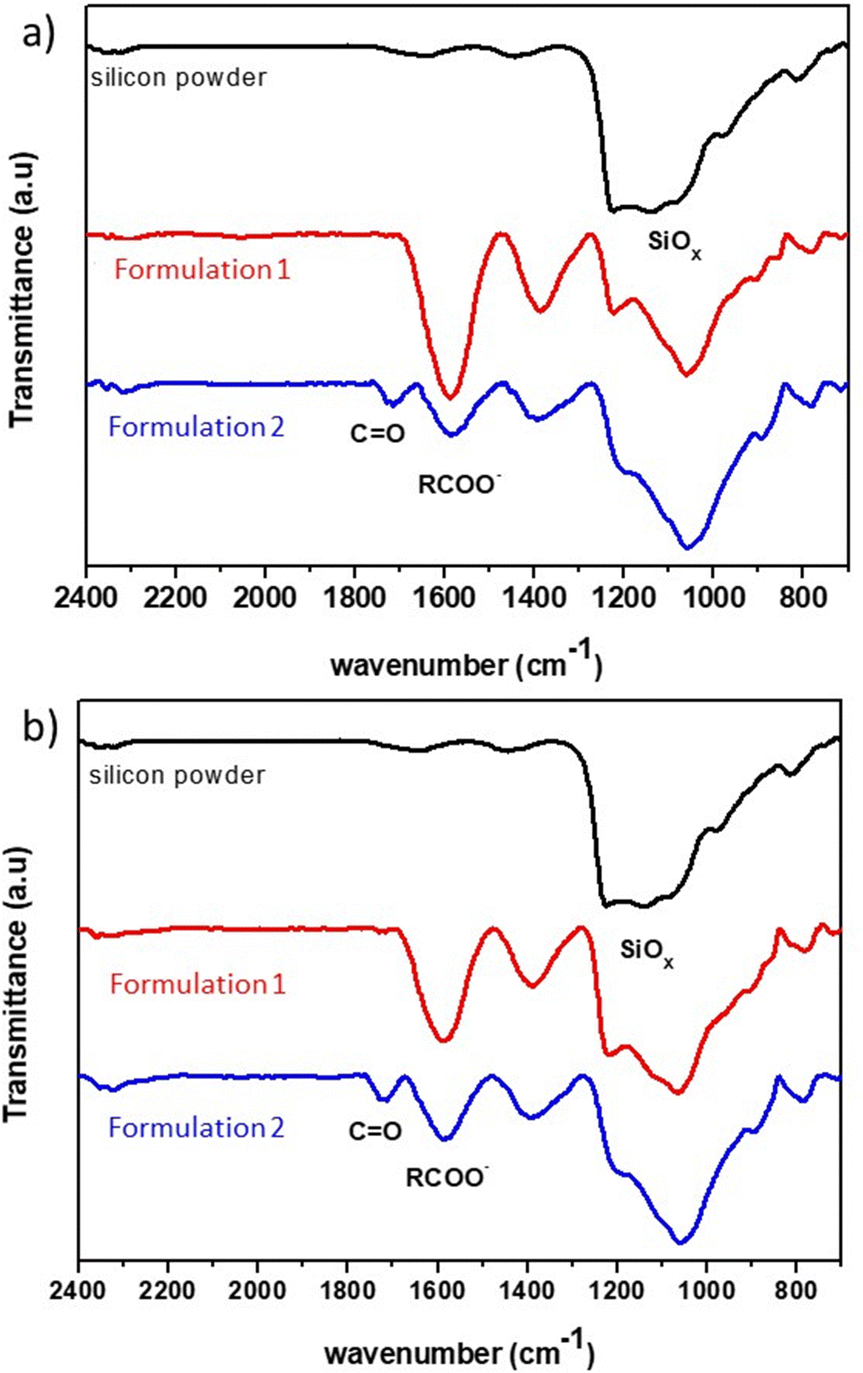 | ||
| Fig. 5 FT-IR spectra of silicon powder (black trace) and mixtures of silicon with a binder (a) CMC90 or (b) CMC250 obtained by formulation 1 (red trace) or 2 (blue trace). | ||
For formulation 1, the electrode analysis shows a strong band at 1604 cm−1 highlighting the presence of carboxylate functions. The decrease of the band at 1202 cm−1 can be associated with the modification of the silicon links, with probably Si–O–C functions. The dehydration of the electrode, after formulation 1, would potentially lead to ionic–dipole interactions.
On the other hand, the infrared spectrum (Fig. 5) of the electrode resulting from formulation 2 shows the presence of both carboxylate functions (1604 cm−1), and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (1730 cm−1). This last band can correspond to acid or ester functions. Karkar et al.20 explains the presence of such a band by the formation of Si–OC(O)–R, following the esterification reaction between the SiOH group on the surface of the silicon and the carboxylic acid function of the CMC. This hypothesis suggests an influence of ionic interactions, through the carboxylate functions, but also of covalent bonds in the action of CMC as a polymer binder with this formulation.
O bond (1730 cm−1). This last band can correspond to acid or ester functions. Karkar et al.20 explains the presence of such a band by the formation of Si–OC(O)–R, following the esterification reaction between the SiOH group on the surface of the silicon and the carboxylic acid function of the CMC. This hypothesis suggests an influence of ionic interactions, through the carboxylate functions, but also of covalent bonds in the action of CMC as a polymer binder with this formulation.
SEM images of formulated electrodes exhibit different aspects (Fig. S6, ESI†). The electrodes obtained with formulation 2 have a heterogeneous surface with a compact appearance of the film, whereas with formulation 1, a rather smooth surface and an alveolar film were observed. The porosity is calculated by deducting the void volume after measuring the densities of the various constituents of the electrode. The electrodes obtained by formulation 2 exhibit lower porosity than those obtained by formulation 1. These results are in good agreement with the images obtained by SEM (Fig. S6, ESI†).
The formulated electrodes with CMC90 and 250 are then cycled to compare the effect of the formulation method on the electrochemical performance.
Electrochemical characterization of the Si/C/CMC electrode
The potential versus capacity curves of formulations 1 and 2 with CMC90 and CMC250 are presented in Fig. 6. All the coin cells were cycled under the same conditions for comparison purpose of the electrochemical parameters.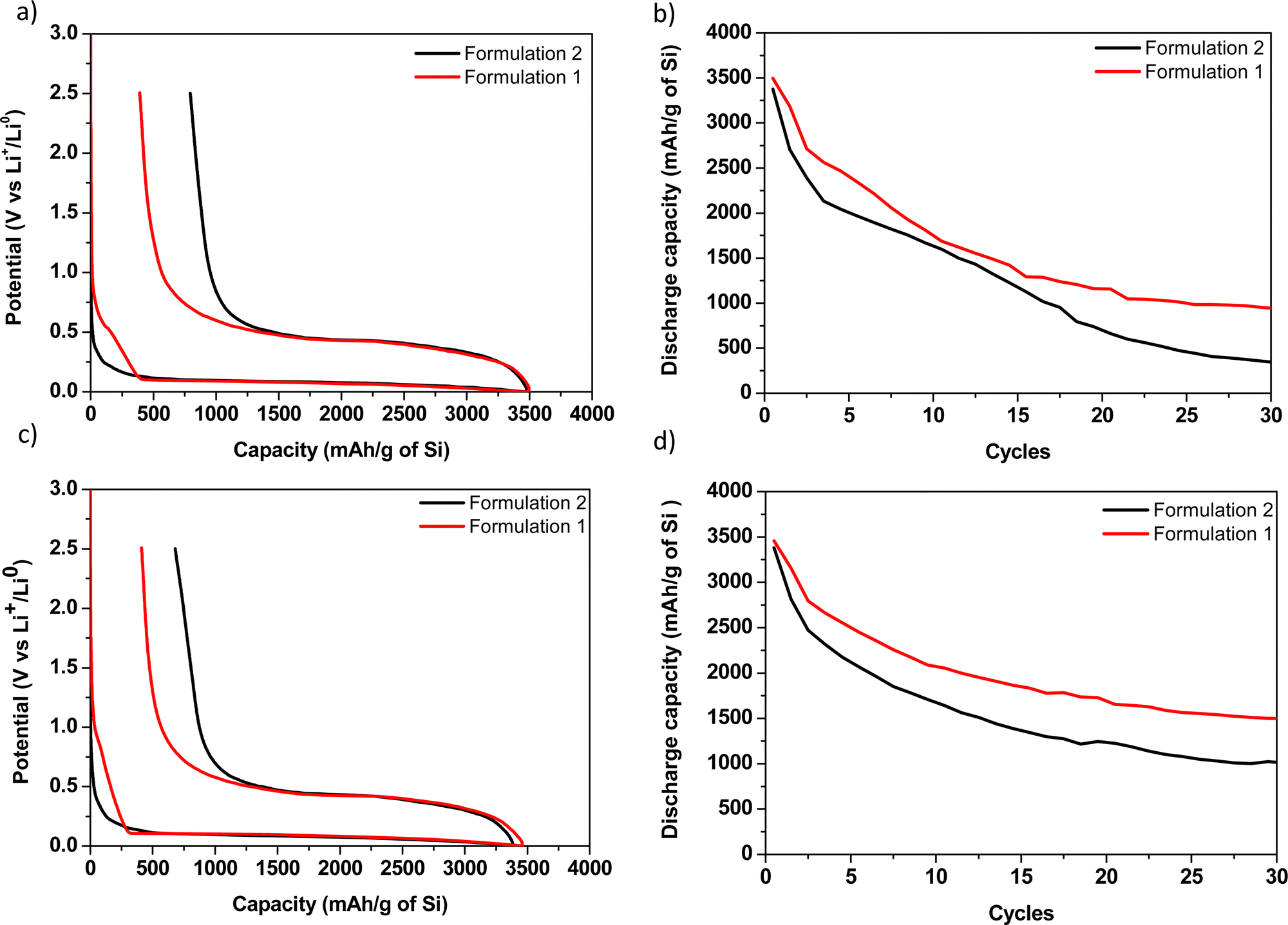 | ||
| Fig. 6 Electrochemical characteristics of silicon electrodes: galvanostatic curves of the first loss of capacity of the Si/Li cell in the presence of CMC90 (a) and CMC250 (c) and silicon capacity retention as a function of the number of cycles in the presence of CMC90 (b) and CMC250 (d) with formulations 1 (red trace) and 2 (black trace). | ||
Here, a significantly lower first loss of capacity is systematically noticed for all the formulations 1 compared with all the formulations 2 (794 and 390 for CMC90 formulations 2 and 1, and 679 and 411 for CMC250 formulations 2 and 1, respectively, Table 3). This result demonstrates the benefit of polymer ball milling with C and Si onto polarization reduction even if a drastic polymer degradation is associated with this ball milling.
| Formulation | CMC90 | CMC250 | ||
|---|---|---|---|---|
| 2 | 1 | 2 | 1 | |
| 1st cycle coulombic efficiency (%) | 77 | 89 | 80 | 88 |
| 1st capacity loss (mA h g−1) | 794 | 390 | 679 | 411 |
| 1st discharge (mA h g−1) | 3477 | 3495 | 3381 | 3459 |
| 30th discharge (mA h g−1) | 400 | 950 | 1021 | 1499 |
| 30th discharge/1st discharge = efficiency (%) | 11 | 27 | 30 | 43 |
Besides, a parasitic reaction beginning at ∼1 V before the starting of the alloying reaction is noticed for both formulations 1. More precisely, a pseudo-plateau at 0.5 V is notified for formulation 1 with CMC90 and absent for formulation 1 with CMC250. This phenomenon is surprisingly not observed for all formulations 2. This is presumably due to the formation of a passivation layer, which can thus be likely related here to the reaction of the species resulting from the CMC degradation in formulations 1. It is followed by the continuous decrease of the potential which is correlated with the reaction with lithium. Bridel et al. suggested that this passivation layer would be formed on the carbon, thus slowing the lithium ionic conductivity and reducing the formation of the LiSi alloy.19
From a chemical point of view, formulation 1 involves the formation of oligosaccharides with low DP, predominantly between 3 and 6. These oligosaccharides could be the origin of the passivation layer on the conductive carbon. On the other hand, the absence of the pseudo-plateau for formulation 2 could be due to the conservation of the length and/or structuration of the original polymeric chains. There, the polymer could initially react with the electrolyte and therefore could prevent the formation of this passivation layer on the carbon.
Besides, a higher irreversible capacity observed in formulation 2 could be attributed to the lower mechanical cohesion of this electrode, which is in agreement with the more significant fading.
In addition, the studies of Lopez et al. clarify the strained aspect of polyelectrolytes, which is here due to the effect of pH on acidic functions.37,38 When the CMC90 sample contains a w of 124 kg mol−1 (larger chain length), it will tend to cover a maximum of particles to allow binding where the oligomers could only cover a few particles. Improvement in capacity stability is much important with CMC250. Nevertheless, the capacity curves as a function of the number of cycles (Fig. 6b) showed better capacity retention with formulation 1.
The grinding of the composite (Si/C/CMC) by the SPEX first allows the formation of the passivation layer on the carbon. The ball-milling of CMC is at the origin of smaller chains which are more soluble in the medium allowing better formulation of the electrode.39 The slurry is “smoother” allowing the electrode tape to have less visible aggregates.
Moreover, the increase of the carboxylate function content strongly supports the promotion of the capacity retention by providing more interaction sites with the surface of the silicon.28,29,40 The results obtained after cycling of silicon in the presence of CMC90 or CMC250, according to the formulation method, are summarized in Table 3.
In our study, the 1st discharge capacity, less than 3500 mA h g−1, is lower than the typically reported value, more than 4000 mA h g−1. As the theoretical lithiation capacity is 3579 mA h g−1 and that one must add to that the capacity loss in the SEI formation, about 300–400 mA h g−1, one expects more than 4000 mA h g−1 for a well homogenous electrode in which the whole active mass is electronically and ionically wired. Here, the measured 1st discharge capacity values suggest that the electrodes were not well homogeneous or that there is a kinetic (polarization) limitation. The presence of CMC250 leads to the best capacity retention with an efficiency (= 30th discharge/1st discharge) of 43%, whereas a first loss of capacity of 411 mA h g−1 was obtained, due to the formation of the passivation layer on the carbon. The average experimental molecular weight of CMC250 was 64 kg mol−1. It has a slightly smaller average molecular weight in the case of CMC90. Keeping in mind that the starting DS for the two CMCs is 0.7, it would be expected that the electrochemical behaviour will be similar. However, the efficiency is lower in the case of CMC90, with only 27%, much lower that formulation 1.
According to our results, the adequate CMC molecular weight of the electrochemical tests seems to be around 64 kg mol−1. The presence of oligomers tends to improve the formulation and allow better conductivity during cycling. However, this assumption remains to be verified and refined with further studies, especially electrochemical tests.
Experimental
Materials and methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, DS = 0.7) (CMC90), CMC250 (average molecular weight = 250000 g mol−1, DS = 0.7) and CMC 700 (average molecular weight = 700000 g mol−1, DS = 0.9) were purchased from Sigma Aldrich (Saint Quentin Fallavier, France). Silicon nanopowder (Si) was provided by 2witeck (San Diego, California). It consists of four populations of spherical particles, around 300 nm and 1, 10 or 35 μm with major volumetric mass density particles of 1 μm. Silicon has an initial SiO2 content evaluated at 11%. Conducting additive Super C65 carbon (Cc65) was purchased from Timcal (Willebroek, Belgium). The LP30 commercial electrolyte was purchased from Merck (Schnelldorf, Germany). Fluoro ethylene carbonate (FEC), 2-(4-hydroxyphenylazo)benzoic acid (HABA) and 1,1,3,3-tetramethylguanidine (TMG) were purchased from Sigma-Aldrich (Saint Quentin Fallavier, France) and methanol with a purity of ≥99.9% was from VWR (Rosny-sous-Bois, France). Ultrapure water (18.2 MΩ) and distilled water were used.
000 g mol−1, DS = 0.7) (CMC90), CMC250 (average molecular weight = 250000 g mol−1, DS = 0.7) and CMC 700 (average molecular weight = 700000 g mol−1, DS = 0.9) were purchased from Sigma Aldrich (Saint Quentin Fallavier, France). Silicon nanopowder (Si) was provided by 2witeck (San Diego, California). It consists of four populations of spherical particles, around 300 nm and 1, 10 or 35 μm with major volumetric mass density particles of 1 μm. Silicon has an initial SiO2 content evaluated at 11%. Conducting additive Super C65 carbon (Cc65) was purchased from Timcal (Willebroek, Belgium). The LP30 commercial electrolyte was purchased from Merck (Schnelldorf, Germany). Fluoro ethylene carbonate (FEC), 2-(4-hydroxyphenylazo)benzoic acid (HABA) and 1,1,3,3-tetramethylguanidine (TMG) were purchased from Sigma-Aldrich (Saint Quentin Fallavier, France) and methanol with a purity of ≥99.9% was from VWR (Rosny-sous-Bois, France). Ultrapure water (18.2 MΩ) and distilled water were used.
Laser granulometry. Determination of the various populations of spherical particles was performed before and after ball milling experiments. The distribution was estimated using a Mastersizer 3000 laser particle diffraction analyzer equipped with a Hydro EV dispersion system both from Malvern Instruments (Malvern, UK). The particle dispersion was possible with a solution of Triton X-100 (TX-100) as a dispersing agent (500 μL of 1% by weight of TX-100 in 500 ml of distilled water) at a speed of 1600 rpm with 40% ultrasound before performing measurements every 30 min at a speed of 600 rpm.
Scanning electron microscopy (SEM). Imaging was performed using an environmental scanning electron microscope (field effect gun – FEG), model FEI Quanta200F. The samples were placed in a field with a spot at 3.5 V and a voltage of 10 kV.
000 m/z, with a deflection mass of 2000 and a delay of 1500 ns. For low mass range settings, spectra were recorded between 300 and 5000 m/z, with a deflection mass of 300 and a delay of 300 ns. For both settings, the following voltages are applied: 20 kV, 18.8 kV and 6.5 kV for IS1, IS2 and Lens, respectively. Mass spectra were obtained by the accumulation of 2000 laser shots and automatically processed using the Flex Analysis 3.3 software (Bruker Daltonics Inc). The instrument was calibrated using standard peptide and protein mixtures provided by the manufacturer. HABA/TMG2 ionic liquid, used as the matrix, was prepared as described elsewhere.41–44 Briefly, HABA was mixed with TMG at a 1:2 molar ratio in methanol, and the obtained solution was sonicated for 15 min at 40 °C. After removing methanol by centrifugal evaporation, the final solution was then prepared at a concentration of 90 mg mL−1 in methanol, and used as a matrix without further purification. Then, 1.5 μL of the ILM was mixed with an identical volume of the aqueous sample. 1.5 μL of the mixture was spotted onto the MALDI MTP 384 polished steel plate by the dried droplet method and allowed to dry at room temperature and atmospheric pressure for 5 min.
Fourier transformed infrared spectroscopy (FTIR). The analyses were performed in attenuated total reflection (ATR) mode using a Shimadzu IRAffinity-1S spectrophotometer. The IR spectrum was acquired using the manufacturer software LabSolutions IR (MIRacle10 (Ge)) operating in a transmission mode with 32 scans and a resolution of 4 cm−1. The field of study of the wave number σ is between 700 and 4000 cm−1.
Thermogravimetric analyses-mass spectrometry (TGA-MS). The analyses were performed using a STA449C apparatus coupled to a QMS 403 Aeolos Quadrupole-Mass Spectrometer Netzsch (Dardilly, France). Approximately 10 mg of the sample was heated from 25 to 600 °C at 5 °C min−1 under a continuous argon flow (50 mL min−1).
Electrode porosity estimation. The porosity was calculated from theoretical calculations taking into account the electrode thickness and the different materials percentage by weight. Pycnometric densities were calculated using an AccuPyc 1330 gas pycnometer (Micromeritics, Norcross, Georgia, USA) using Helium gas. Runs were performed 5 times using a 3.5 cm3 cell. This analysis allows the determination of the density of each component of the electrode. The knowledge of the mass percentage of each element and the thickness of the electrode allows us to deduce their volume fractions. The porosity is therefore determined by deducting the volume fractions of each component of the electrode (silicon, carbon, binder, citric acid and KOH).
Adhesion test. The adhesion of the dry electrode layers to the copper film is determined using a base cutter (Cross Cut Adhesion Test kit CC3000, TQC). It is thus possible to have comparison values using the ASTM D3359 classification (5B: no peeling up to 0B: complete peeling of the film).
(1) Usual ball milling formulation (so-called formulation 1): Si, Cc65 and CMC were placed with an adapted quantity of a buffer solution in a stainless-steel ball-miller jar (volume = 50 mL) with 3 stainless steel balls (10 mm of diameter) and subsequently ball milled in a high-energy (SPEX ball-miller) milling for 30 min at a frequency of 50 Hz. The resulting slurry was then coated on a 15 μm thick structured copper foil. The films were dried at room temperature overnight.
(2) Ball milling and magnetic stirring formulation (so-called formulation 2): Si and Cc65 were placed in a stainless-steel ball-miller jar (volume = 50 mL) with 3 stainless steel balls (10 mm of diameter). The jar was shaken in a high-energy SPEX ball-miller for 30 min at a frequency of 50 Hz. The composite powder is added to a solution of the CMC with an adapted quantity of buffer solution and magnetically stirred in a pill for 120 min. The resulting slurry was then coated on a 15 μm thick structured copper foil. The films were dried at room temperature overnight.
No calendaring was performed on these different electrodes.
Conclusions
Here, we demonstrate that ball-milling has a deleterious impact on CMC samples, leading to a drastic and rapid reduction of the average molecular weight of polymers. Interestingly, regardless of the initialw of CMC samples, highly energetic ball milling leads, at glance, to the formation of a bimodal distribution. On the one hand, a large peak ascribed to a polydisperse high molecular weight and on the other hand two narrower ones corresponding to two low molecular weight species can be delineated. An exception is noticed for CMC90, where only one population of low w appears and the reason for this remains, at this stage, unfortunately unclear. These low molecular weight species presumably correspond to the final products from CMC tribochemical degradation, as already observed in the cellulose ball milling, with a “limiting molecular weight” (Mwlim), below which no supplementary polymer chain length decrease occurs.
Regarding C/Si/CMC anode formulation and electrochemical behaviour versus lithium metal, it was noticed that the electrochemical performance is slightly lower for the formulation 2 (ball milling of Si/C followed by magnetic stirring in a polymer suspension) compared with formulation 1 (ball milling of Si/C/CMC). After 30 cycles, electrochemical capacities and efficiencies are thus always superior with formulation 1 compared with formulation 2, whatever the CMC used. Coupled with this result, the 1st cycle coulombic efficiency is always higher for formulation 1 compared to formulation 2. Several assumptions are put forth such as (i) high electrode porosity. The “all SPEX” formulation including the ball milling of the polymer induces a mild porosity growth, compared to formulation 2 (60% versus 56% and 69% versus 63%, for CMC90 and 250, respectively). Grinding of all the electrode constituents, inducing a global average porosity increase, would thus facilitate electrolyte and lithium insertion. (ii) A chemical reaction between CMC and C and/or Si which may occur during ball milling and the presence of new carbonyl functions may increase interactions with Si. It would induce enhanced electrochemical performances. These interactions would not occur if the polymer is simply added by magnetic stirring to the ball milled mixture of C and Si. A passivation layer formation during ball milling, which can likely be related to the reaction of the species resulting from the CMC degradation in formulations 1, can be mentioned. This fact is supported by the parasitic reaction at ∼1 V observed in galvanostatic curves for all formulations 1 and never in formulations 2, before the alloying reaction with Si starts. This chemical reaction of the polymer during ball milling would be consequently significantly more important than the unique consideration of the CMC average molecular weight parameter. Moreover, the oligomers produced by ball milling are more soluble in the medium and, combined with the polymer, would induce a smoother and more homogeneous slurry and therefore a better formulation of the electrode. Finally, more experiments are required to discriminate the most prominent factor affecting the electrochemical performances.
Author contributions
MN: investigation, methodology, and writing – original draft; JPB: conceptualization, investigation, and methodology; SC: investigation, methodology, and validation; TL: investigation, methodology, and validation); MC: investigation, methodology, and validation; LA: supervision electrochemical assays; CP: investigation, methodology, writing – original draft, and writing – review and editing; VB: investigation; methodology; funding acquisition, project administration, resources, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Région Picardie and the FEDER (Fonds Européen de Développement Régional) for the NANOPAM project is gratefully acknowledged.Notes and references
- M. N. Obrovac and L. Christensen, Structural Changes in Silicon Anodes during Lithium Insertion/Extraction, Electrochem. Solid-State Lett., 2004, 7(5), A93, DOI:10.1149/1.1652421.
- M. N. Obrovac and L. J. Krause, Reversible Cycling of Crystalline Silicon Powder, J. Electrochem. Soc., 2007, 154(2), A103, DOI:10.1149/1.2402112.
- J. B. Goodenough and Y. Kim, Challenges for Rechargeable Li Batteries, Chem. Mater., 2010, 22(3), 587–603, DOI:10.1021/cm901452z.
- J. Li and J. R. Dahn, An In Situ X-Ray Diffraction Study of the Reaction of Li with Crystalline Si, J. Electrochem. Soc., 2007, 154(3), A156, DOI:10.1149/1.2409862.
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, Review on Modeling of the Anode Solid Electrolyte Interphase (SEI) for Lithium-Ion Batteries, npj Comput. Mater., 2018, 4(1), 15, DOI:10.1038/s41524-018-0064-0.
- M. Ashuri, Q. He and L. L. Shaw, Silicon as a Potential Anode Material for Li-Ion Batteries: Where Size, Geometry and Structure Matter, Nanoscale, 2016, 8(1), 74–103, 10.1039/C5NR05116A.
- F.-H. Du, K.-X. Wang and J.-S. Chen, Strategies to Succeed in Improving the Lithium-Ion Storage Properties of Silicon Nanomaterials, J. Mater. Chem. A, 2016, 4(1), 32–50, 10.1039/C5TA06962A.
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, High-Performance Lithium Battery Anodes Using Silicon Nanowires, Nat. Nanotechnol., 2008, 3(1), 31–35, DOI:10.1038/nnano.2007.411.
- L. Y. Yang, H. Z. Li, J. Liu, Z. Q. Sun, S. S. Tang and M. Lei, Dual Yolk-Shell Structure of Carbon and Silica-Coated Silicon for High-Performance Lithium-Ion Batteries, Sci. Rep., 2015, 5(1), 10908, DOI:10.1038/srep10908.
- D. Ma, Z. Cao and A. Hu, Si-Based Anode Materials for Li-Ion Batteries: A Mini Review, Nano-Micro Lett., 2014, 6(4), 347–358, DOI:10.1007/s40820-014-0008-2.
- Z. Karkar, D. Mazouzi, C. R. Hernandez, D. Guyomard, L. Roué and B. Lestriez, Threshold-like Dependence of Silicon-Based Electrode Performance on Active Mass Loading and Nature of Carbon Conductive Additive, Electrochim. Acta, 2016, 215, 276–288, DOI:10.1016/j.electacta.2016.08.118.
- N.-S. Choi, S.-Y. Ha, Y. Lee, J. Y. Jang, M.-H. Jeong, W. C. Shin and M. Ue, Recent Progress on Polymeric Binders for Silicon Anodes in Lithium-Ion Batteries, J. Electrochem. Sci. Technol., 2015, 6(2), 35–49, DOI:10.5229/JECST.2015.6.2.35.
- D. Bresser, D. Buchholz, A. Moretti, A. Varzi and S. Passerini, Alternative Binders for Sustainable Electrochemical Energy Storage – the Transition to Aqueous Electrode Processing and Bio-Derived Polymers, Energy Environ. Sci., 2018, 11(11), 3096–3127, 10.1039/C8EE00640G.
- D. Mazouzi, R. Grissa, M. Paris, Z. Karkar, L. Huet, D. Guyomard, L. Roué, T. Devic and B. Lestriez, CMC-Citric Acid Cu(II) Cross-Linked Binder Approach to Improve the Electrochemical Performance of Si-Based Electrodes, Electrochim. Acta, 2019, 304, 495–504, DOI:10.1016/j.electacta.2019.03.026.
- C. Reale Hernandez, Z. Karkar, D. Guyomard, B. Lestriez and L. Roué, A Film Maturation Process for Improving the Cycle Life of Si-Based Anodes for Li-Ion Batteries, Electrochem. Commun., 2015, 61(C), 102–105, DOI:10.1016/j.elecom.2015.10.014.
- Y.-M. Zhao, F.-S. Yue, S.-C. Li, Y. Zhang, Z.-R. Tian, Q. Xu, S. Xin and Y.-G. Guo, Advances of Polymer Binders for Silicon-Based Anodes in High Energy Density Lithium-Ion Batteries, InfoMat, 2021, 3(5), 460–501, DOI:10.1002/inf2.12185.
- S. Jiang, B. Hu, R. Sahore, L. Zhang, H. Liu, L. Zhang, W. Lu, B. Zhao and Z. Zhang, Surface-Functionalized Silicon Nanoparticles as Anode Material for Lithium-Ion Battery, ACS Appl. Mater. Interfaces, 2018, 10(51), 44924–44931, DOI:10.1021/acsami.8b17729.
- X. Zhao, S. Niketic, C.-H. Yim, J. Zhou, J. Wang and Y. Abu-Lebdeh, Revealing the Role of Poly(Vinylidene Fluoride) Binder in Si/Graphite Composite Anode for Li-Ion Batteries, ACS Omega, 2018, 3(9), 11684–11690, DOI:10.1021/acsomega.8b01388.
- J.-S. Bridel, T. Azaïs, M. Morcrette, J.-M. Tarascon and D. Larcher, Key Parameters Governing the Reversibility of Si/Carbon/CMC Electrodes for Li-Ion Batteries, Chem. Mater., 2010, 22(3), 1229–1241, DOI:10.1021/cm902688w.
- Z. Karkar, D. Guyomard, L. Roué and B. Lestriez, A Comparative Study of Polyacrylic Acid (PAA) and Carboxymethyl Cellulose (CMC) Binders for Si-Based Electrodes, Electrochim. Acta, 2017, 258, 453–466, DOI:10.1016/j.electacta.2017.11.082.
- M. Ndour, J.-P. Bonnet, S. Cavalaglio, T. Lombard, C. Przybylski and V. Bonnet, CMC Binding Agent for Silicon Anodes in Li-Ion Batteries: The Impact of the Formulation, ECS Meeting Abstr., 2019, MA2019-04(10), 0471, DOI:10.1149/ma2019-04/10/0471.
- J.-T. Li, Z.-Y. Wu, Y.-Q. Lu, Y. Zhou, Q.-S. Huang, L. Huang and S.-G. Sun, Water Soluble Binder, an Electrochemical Performance Booster for Electrode Materials with High Energy Density, Adv. Energy Mater., 2017, 7(24), 1701185, DOI:10.1002/aenm.201701185.
- B. Hu, I. A. Shkrob, S. Zhang, L. Zhang, J. Zhang, Y. Li, C. Liao, Z. Zhang, W. Lu and L. Zhang, The Existence of Optimal Molecular Weight for Poly(Acrylic Acid) Binders in Silicon/Graphite Composite Anode for Lithium-Ion Batteries, J. Power Sources, 2018, 378, 671–676, DOI:10.1016/j.jpowsour.2017.12.068.
- B.-R. Lee and E.-S. Oh, Effect of Molecular Weight and Degree of Substitution of a Sodium-Carboxymethyl Cellulose Binder on Li4Ti5O12 Anodic Performance, J. Phys. Chem. C, 2013, 117(9), 4404–4409, DOI:10.1021/jp311678p.
- J. Liu, D. G. D. Galpaya, L. Yan, M. Sun, Z. Lin, C. Yan, C. Liang and S. Zhang, Exploiting a Robust Biopolymer Network Binder for an Ultrahigh-Areal-Capacity Li–S Battery, Energy Environ. Sci., 2017, 10(3), 750–755, 10.1039/C6EE03033E.
- M. Gauthier, D. Mazouzi, D. Reyter, B. Lestriez, P. Moreau, D. Guyomard and L. Roué, A Low-Cost and High Performance Ball-Milled Si-Based Negative Electrode for High-Energy Li-Ion Batteries, Energy Environ. Sci., 2013, 6(7), 2145–2155, 10.1039/C3EE41318G.
- M. Gauthier, D. Reyter, D. Mazouzi, P. Moreau, D. Guyomard, B. Lestriez and L. Roué, From Si Wafers to Cheap and Efficient Si Electrodes for Li-Ion Batteries, J. Power Sources, 2014, 256, 32–36, DOI:10.1016/j.jpowsour.2014.01.036.
- Z. Ling, T. Wang, M. Makarem, M. Santiago Cintrón, H. N. Cheng, X. Kang, M. Bacher, A. Potthast, T. Rosenau, H. King, C. D. Delhom, S. Nam, J. Vincent Edwards, S. H. Kim, F. Xu and A. D. French, Effects of Ball Milling on the Structure of Cotton Cellulose, Cellulose, 2019, 26(1), 305–328, DOI:10.1007/s10570-018-02230-x.
- Y. Zheng, Z. Fu, D. Li and M. Wu, Effects of Ball Milling Processes on the Microstructure and Rheological Properties of Microcrystalline Cellulose as a Sustainable Polymer Additive, Materials, 2018, 11(7), 1057, DOI:10.3390/ma11071057.
- H. Liu, Y. Zhang, T. Hou, X. Chen, C. Gao, L. Han and W. Xiao, Mechanical Deconstruction of Corn Stover as an Entry Process to Facilitate the Microwave-Assisted Production of Ethyl Levulinate, Fuel Process. Technol., 2018, 174, 53–60, DOI:10.1016/j.fuproc.2018.02.011.
- B. Stefanovic, K. F. Pirker, T. Rosenau and A. Potthast, Effects of Tribochemical Treatments on the Integrity of Cellulose, Carbohydr. Polym., 2014, 111, 688–699, DOI:10.1016/j.carbpol.2014.05.011.
- I. Solala, U. Henniges, K. F. Pirker, T. Rosenau, A. Potthast and T. Vuorinen, Mechanochemical Reactions of Cellulose and Styrene, Cellulose, 2015, 22(5), 3217–3224, DOI:10.1007/s10570-015-0724-x.
- S. F. El-Kalyoubi and N. A. El-Shinnawy, Thermogravimetric Analysis of Some Chemically Modified Celluloses, J. Appl. Polym. Sci., 1985, 30(12), 4793–4799, DOI:10.1002/app.1985.070301224.
- T. Randriamanantena, F. L. Razafindramisa, G. Ramanantsizehena, A. Bernés and C. Lacabane, Thermal behaviour of three woods of madagascar by thermogravimetric analysis in inert atmosphere, Proc. 4th High-Energy Phys. Int. Conf., Antananarivo, Madagascar, 2009.
- C. Liu, S. Qin, J. Xie, X. Lin, Y. Zheng, J. Yang, H. Kan and Z. Shi, Using Carboxymethyl Cellulose as the Additive With Enzyme-Catalyzed Carboxylated Starch to Prepare the Film With Enhanced Mechanical and Hydrophobic Properties, Front. Bioeng. Biotechnol., 2021, 9 DOI:10.3389/fbioe.2021.638546.
- D. de Britto and O. B. G. Assis, Thermal Degradation of Carboxymethylcellulose in Different Salty Forms, Thermochim. Acta, 2009, 494(1), 115–122, DOI:10.1016/j.tca.2009.04.028.
- C. G. Lopez and W. Richtering, Conformation and Dynamics of Flexible Polyelectrolytes in Semidilute Salt-Free Solutions, J. Chem. Phys., 2018, 148(24), 244902, DOI:10.1063/1.5024242.
- C. G. Lopez, Entanglement of Semiflexible Polyelectrolytes: Crossover Concentrations and Entanglement Density of Sodium Carboxymethyl Cellulose, J. Rheol., 2020, 64(1), 191–204, DOI:10.1122/1.5127015.
- X. Lin, X. Guo, C. Xu and M. Wu, Carboxymethyl Cellulose Assisted Mechanical Preparation of Cellulose Nanocrystals with High Yield, Cellulose, 2019, 26(9), 5227–5236, DOI:10.1007/s10570-019-02452-7.
- Z. S. Wen, The Effect of Ball Milling on the Electrochemical Behavior of Silicon Composite Electrode, Adv. Mater. Res., 2014, 833, 280–285, DOI:10.4028/www.scientific.net/AMR.833.280.
- C. Przybylski, F. Gonnet, D. Bonnaffé, Y. Hersant, H. Lortat-Jacob and R. Daniel, HABA-Based Ionic Liquid Matrices for UV-MALDI-MS Analysis of Heparin and Heparan Sulfate Oligosaccharides, Glycobiology, 2010, 20(2), 224–234, DOI:10.1093/glycob/cwp169.
- D. Ropartz, P.-E. Bodet, C. Przybylski, F. Gonnet, R. Daniel, M. Fer, W. Helbert, D. Bertrand and H. Rogniaux, Performance Evaluation on a Wide Set of Matrix-Assisted Laser Desorption Ionization Matrices for the Detection of Oligosaccharides in a High-Throughput Mass Spectrometric Screening of Carbohydrate Depolymerizing Enzymes, Rapid Commun. Mass Spectrom., 2011, 25(14), 2059–2070, DOI:10.1002/rcm.5060.
- C. Przybylski, F. Blin and N. Jarroux, Toward a More Accurate Structural Determination of High Molecular Weight Polyrotaxanes Based on Cyclodextrins by MALDI−TOF MS, Macromolecules, 2011, 44(7), 1821–1830, DOI:10.1021/ma102641q.
- A. Seffouh, F. Milz, C. Przybylski, C. Laguri, A. Oosterhof, S. Bourcier, R. Sadir, E. Dutkowski, R. Daniel, T. H. van Kuppevelt, T. Dierks, H. Lortat-Jacob and R. R. Vivès, HSulf Sulfatases Catalyze Processive and Oriented 6-O-Desulfation of Heparan Sulfate That Differentially Regulates Fibroblast Growth Factor Activity, FASEB J., 2013, 27(6), 2431–2439, DOI:10.1096/fj.12-226373.
Footnote |
| † Electronic supplementary information (ESI) available: The XRD pattern of CMC, UV traces of SEC, the FTIR spectra of CMC 250, the TGA-MS results and SEM imaging of electrodes. See DOI: https://doi.org/10.1039/d2ma00702a |
| This journal is © The Royal Society of Chemistry 2022 |