
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA thermo-responsive MOF-based H2O2-in-oil “pickering emulsion in situ catalytic system” for highly efficient olefin epoxidation†
Bin
Huang
a,
Wei-Ling
Jiang
b,
Jie
Han
c,
Yan-Fei
Niu
a,
Hai-Hong
Wu
 a and
Xiao-Li
Zhao
*a
a and
Xiao-Li
Zhao
*a
aShanghai Key Laboratory of Green Chemistry and Chemical Processes, Department of Chemistry, East China Normal University, 3663 North Zhongshan Road, Shanghai 200062, P. R. China. E-mail: xlzhao@chem.ecnu.edu.cn; Fax: +86-21-62233179
bKey Laboratory of Synthetic and Natural Functional Molecule of the Ministry of Education, College of Chemistry and Materials Science, Northwest University, Xi’an 710127, P. R. China
cDepartment of Science, School of Science and Technology, Hong Kong Metropolitan University, Hong Kong SAR, P. R. China
First published on 23rd June 2022
Abstract
A newly functionalized MIL-101(Fe) type MOF, namely NaPT@MIL-101-NH-PNIPAM, decorated with a thermo-responsive poly(N-isopropylacrylamide) (PNIPAM) brush outside and loaded with sodium phosphotungstate particles (NaPT) inside has been successfully synthesized. The new thermally responsive amphiphilic NaPT@MIL-101-NH-PNIPAM powder can well serve as a highly efficient Pickering emulsifier to stabilize the H2O2-in-oil emulsion. The resultant H2O2-in-oil Pickering emulsion displayed good catalytic performance in the epoxidation of olefins at room temperature, promoting a greener reaction with good yield and selectivity. Notably, the constitutes (H2O2, olefin) of the Pickering emulsion is also the substrate of the epoxidation of the olefin reaction, which will facilitate further expansion of the reaction scale. Moreover, the catalyst of the Pickering emulsion featured intriguing thermo-responsive properties of swelling/shrinking behavior that can trigger emulsification at room temperature (25 °C) and accomplish demulsification at an elevated temperature (50 °C), mainly benefitting from the thermally responsive amphiphilic nature of the attached PNIPAM polymer. This thermo-responsive feature of the Pickering emulsion catalyst renders the catalyst–product separation and catalyst recycling feasible during the epoxidation of alkenes. This work reveals new insights for the design of a highly efficient heterogeneous catalytic system in H2O2-based epoxidation and may have potential for application in industry in the future.
Introduction
Polyoxometalates (POMs) represent a rich and diverse class of inorganic clusters that exhibit fascinating structural, electrochemical, catalytic, medicinal, and electron-transfer properties.1,2 In view of the controllable redox and acidic properties, numerous POMs have been extensively employed as efficient catalysts in many oxidative reactions, including the epoxidation of olefins to epoxides.3–5 The epoxidation of olefins has drawn much attention since epoxides are indispensable intermediates in both final-chemical and polymer chemistry.6 Green and clean epoxidation of olefins using hydrogen peroxide (H2O2) under mild conditions is regarded as a pivotal reaction due to its intrinsic advantages including low cost, environment amity and high active oxygen content.7 However, POM-based catalysts are much soluble in many polar solvents, leading to the difficulty in separation and recycling of the catalyst. Therefore, great efforts have been devoted to the preparation of heterogeneous POM-based catalytic systems for the highly efficient epoxidation of olefins.8–11 Early methods mainly focused on the immobilization of POMs on various porous supports such as anion-exchange resin, surface-modified silica and layered double hydroxides and so on.12–21 Later, a series of POM-based ionic hybrid nanospheres prepared by protonating and anion-exchanging amino-attached ionic liquid cations with POM-anions have been proved to be a kind of highly efficient heterogeneous catalysis in H2O2-based epoxidation.22 More recently, metal–organic frameworks (MOFs) have been demonstrated to be efficient supports to incorporate POMs for the formation of efficient heterogeneous catalysts.23–26 However, these processes usually suffer from high reaction temperatures and excessive use of catalysts. Most importantly, product separation and catalyst recycling have some difficulties in these systems.23–25In recent years, environmentally responsive Pickering emulsions have attracted great focus as a highly efficient catalytic system due to their excellent catalytic performance and ease of product separation and recyclability.27–33 It has been reported that MOFs can be employed as suitable emulsifiers for Pickering emulsion after post-synthetic modification (PSM) and covalent incorporation with functional polymers.34–39 For example, light-, pH-, and thermo-responsive Pickering emulsions provide efficient solutions for various catalytic reactions.40–45 So far, the reported Pickering emulsions with MOFs as emulsifiers are all oil-in-water systems. However, the water core of the water-in-oil emulsion is considered to be an ideal reactor for the encapsulation of nanoparticles.46 Therefore, the development of water-in-oil Pickering emulsions with MOF emulsifiers is important for improving its catalytic performance. Based on the above-mentioned considerations, using POM-loaded MOFs as emulsifiers to stabilize emulsion catalytic systems can realize the convenient separation and recycling of POM-based catalysts. In addition, the water phase and oil phase in most of the emulsion catalyst systems currently reported only serve as solvents for the substrate and do not participate in the reaction, which greatly limits the scale and efficiency of the reaction. In our opinion, it is highly anticipated to overcome the above shortcomings by using the components involved in the reaction (oxidizing or reducing agent, substrate, etc.) as constituent phases of the emulsion.
Herein, we report the first example of a thermo-responsive MOF-based H2O2-in-oil type Pickering emulsion for improving the catalytic performance toward olefin epoxidation. The target MOF of NaPT@MIL-101-NH-PNIPAM (NaPT: sodium phosphotungstate particles, PNIPAM: poly(N-isopropylacrylamide) as an emulsifier can not only stabilize the Pickering emulsion, but also significantly increase the conversion and selectivity of olefin epoxidation at room temperature. NaPT@MIL-101-NH-PNIPAM bearing PNIPAM chains has a thermally responsive amphiphilic nature and thus can well serve as a highly efficient Pickering emulsifier to stabilize the H2O2-in-oil emulsion. To the best of our knowledge, this is the first time that a H2O2-in-oil system is realized for efficient olefin epoxidation, i.e., H2O2 as an oxidant in this reaction is also used as one of the emulsion phases (aqueous phase), and reactant olefin is used as part of the oil phase, leading to a “Pickering emulsion in situ catalytic system”. In addition, the scale of the olefin epoxidation reaction in this catalytic system is successfully expanded with excellent yield and selectivity compared with other emulsion catalytic systems based on MOFs. Moreover, demulsification can be achieved by adjusting the temperature to separate the oil and aqueous phase, and the aqueous phase with NaPT@MIL-101-NH-PNIPAM can be reused directly in the next catalytic cycle. Therefore, this work has demonstrated a successful example of an MOF-based H2O2-in-oil Pickering emulsion system for the highly efficient olefin epoxidation.
Results and discussion
Synthesis and characterization of NaPT@MIL-101-NH-PNIPAM
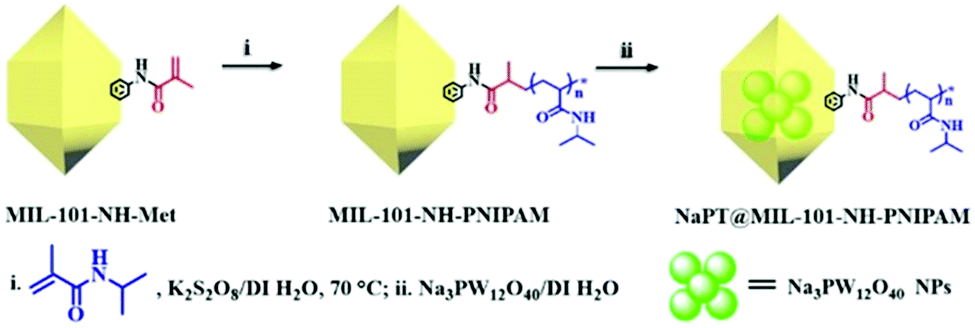
The target composite NaPT@MIL-101-NH-PNIPAM was prepared by post-synthetic modification (PSM) of MIL-101-NH-Met according to a two-step procedure (Scheme 1). First, MIL-101-NH-PNIPAM was obtained as a brown powder via radical polymerization of the N-isopropyl acrylamide monomer (NIPAM) with MIL-101-NH-Met, which was successfully characterized by 1H-NMR spectroscopy (Fig. S1, ESI†). In 1H NMR, the peak ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 (5–6 ppm) of MIL-101-NH-Met almost disappeared (>99%), indicating the successful polymerization of MIL-101-NH-Met. Furthermore, the average molecular weight Mn of PNIPAM from monomer polymerization (Scheme S1 and Fig. S2, ESI†Mn = 5911 Da, PDI = 1.12) and MIL-101-NH-PNIPAM (Mn = 5386 Da, PDI = 1.26) was measured by GPC (Fig. 1a and Table S1, ESI†). This suggested no monomer polymerization species was detected on the synthesis process of MIL-101-NH-PNIPAM.
CH2 (5–6 ppm) of MIL-101-NH-Met almost disappeared (>99%), indicating the successful polymerization of MIL-101-NH-Met. Furthermore, the average molecular weight Mn of PNIPAM from monomer polymerization (Scheme S1 and Fig. S2, ESI†Mn = 5911 Da, PDI = 1.12) and MIL-101-NH-PNIPAM (Mn = 5386 Da, PDI = 1.26) was measured by GPC (Fig. 1a and Table S1, ESI†). This suggested no monomer polymerization species was detected on the synthesis process of MIL-101-NH-PNIPAM.
 | ||
| Scheme 1 Synthetic procedure of NaPT@MIL-101-NH-PNIPAM. | ||
 | ||
| Fig. 1 (a) GPC traces of PNIPAM (pink line) and the free PNIPAM chains obtained by digesting MIL-101-NH-PNIPAM (blue line) in concentrated H2SO4. (b) FT-IR spectra of MIL-101-NH2 (black line), MIL-101-NH-Met (red line), MIL-101-NH-PINIPAM (blue line), NaPT@MIL-101-NH-PINIPAM (green line), PNIPAM (pink line) and NaPT (brown line), respectively. (c) PXRD patterns of a series of MOFs. (d–g) SEM images of MIL-101-NH2 (black line), MIL-101-NH-Met (red line), MIL-101-NH-PINIPAM (blue line) and NaPT@MIL-101-NH-PINIPAM (green line), respectively. | ||
NaPT@MIL-101-NH-PNIPAM was prepared as a red-brown powder (Fig. S3, ESI†) by mixing NaPT (100 mg) and MIL-101-NH-PNIPAM (100 mg) in DI water at room temperature for 72 h (Scheme 1). The loading amount of tungsten (W) was up to 4.63 wt%, which was examined by ICP-MS. In addition, FT-IR spectra and 31P-NMR analysis collectively suggested that NaPT was successfully doped inside the pores of MOFs. Specifically, the bands at 1102 cm−1 were assigned to the stretching frequency of P–O, while peaks at 934 cm−1, 896 cm−1 and 817 cm−1 were assigned to WO, WOb–W and W–Oc–W, respectively. The presence of one peak of 31P NMR spectra suggested NaPT was doped inside the MOFs successfully (Fig. S4, ESI†). Other characterization results including thermogravimetry analysis (TGA) are shown in the ESI† (Fig. 1b and Fig. S5 and S6, ESI†). PXRD and SEM showed that the crystalline nature and structural integrity of MOFs were well maintained after the PSM process and NaPT loading (Fig. 1c–g).
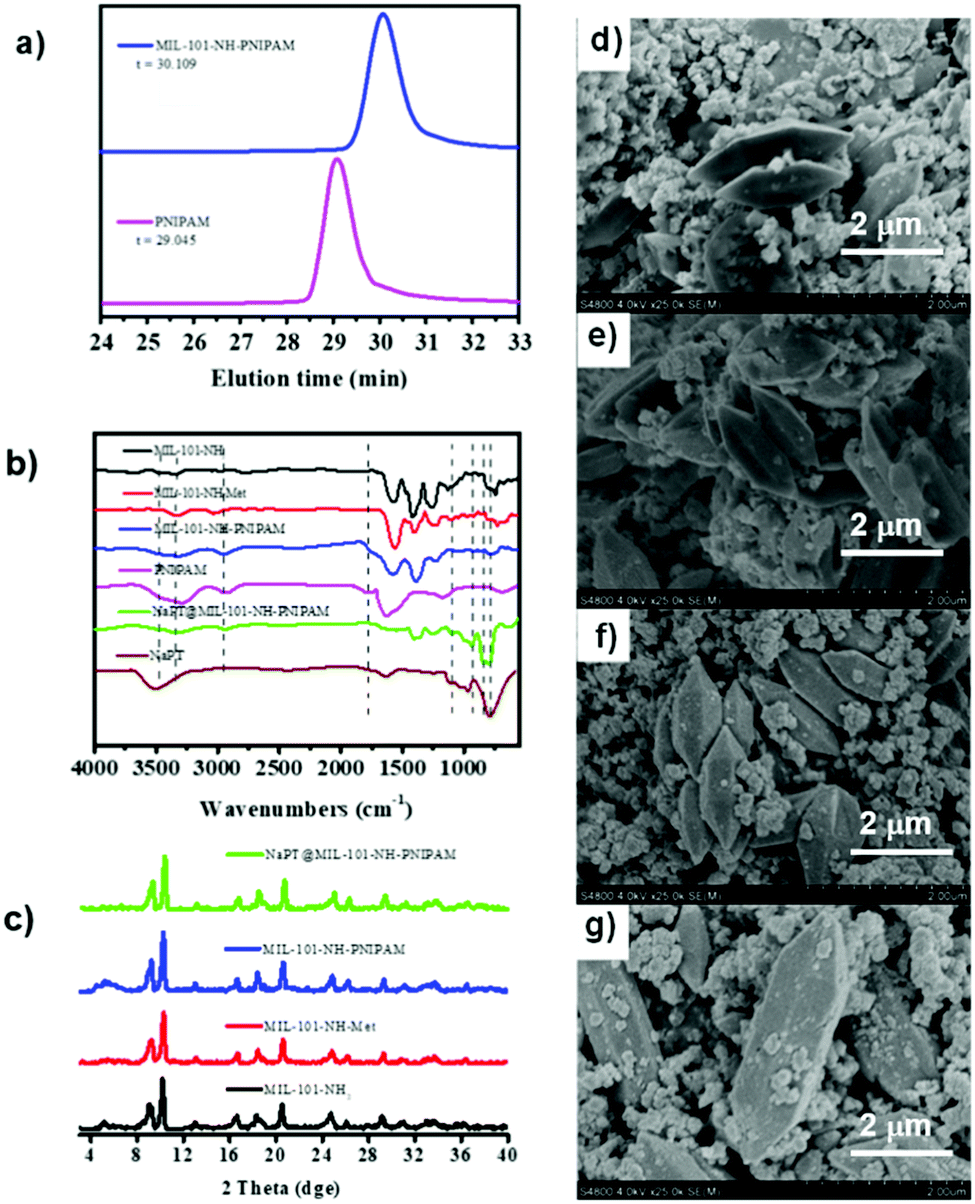
In addition, as MIL-101-NH2 was modified step by step, part of the modified chains grew inside the pores, resulting in a decrease in the total pore volume which was proved by the isotherm study of N2 sorption (Fig. S7, ESI†). The adsorption capabilities of the MOFs declined significantly after PNIPAM grafting and NaPT loading. Moreover, dynamic light scattering (DLS) revealed that the sizes of MOF particles slightly increased after continuous PSM procedures, and centered at ca. 345.2, 348.2, 354.2, and 361.3 nm, for MIL-101-NH2, MIL-101-NH-Met, MIL-101-NH-PINIPAM and NaPT@MIL-101-NH-PINIPAM, respectively (Fig. 2a). It is noteworthy that SEM-EDX elemental mapping confirmed the uniform texture of NaPT@MIL-101-NH-PNIPAM by showing a homogeneous distribution of Fe, W, P, O, N and C elements in the composite material (Fig. 2b).
 | ||
| Fig. 2 (a) DLS results of MIL-101-NH2, MIL-101-NH-Met, MIL-101-NH-PINIPAM and NaPT@MIL-101-NH-PINIPAM at room temperature. (b) SEM-EDX elemental mapping of NaPT@MIL-101-NH-PINIPAM. | ||
Thermo-responsivity of MIL-101-NH-PNIPAM and NaPT@MIL-101-NH-PNIPAM
The thermo-responsivity of MIL-101-NH-PNIPAM was characterized by measuring their hydrodynamic diameters in dilute aqueous solution at different temperatures. As shown in Fig. 3a, the diameters of MIL-101-NH-PNIPAM in the swollen state at 25 °C and in the shrunken state at 50 °C were 349.4 and 261.3 nm, respectively, which were in agreement with the temperature-responsive nature of PNIPAM (lower critical solution temperature, LCST = 32 °C). At lower temperature, the PNIPAM chains were hydrophilic and stretched freely in water. When the temperature was higher than the LCST, the PNIPAM chains turned to be hydrophobic and began to compact on the surface of the MOFs, leading to rapid size shrinking. The hydrodynamic diameters of MIL-101-NH-PNIPAM from 25 to 50 °C were measured for five heating–cooling cycles, indicating good thermo-responsive reversibility (Fig. 3b). In contrast, the sizes of MIL-101-NH2 and MIL-101-NH-Met without decorated polymer brushes were almost unchanged at the given temperature (Fig. S8, ESI†).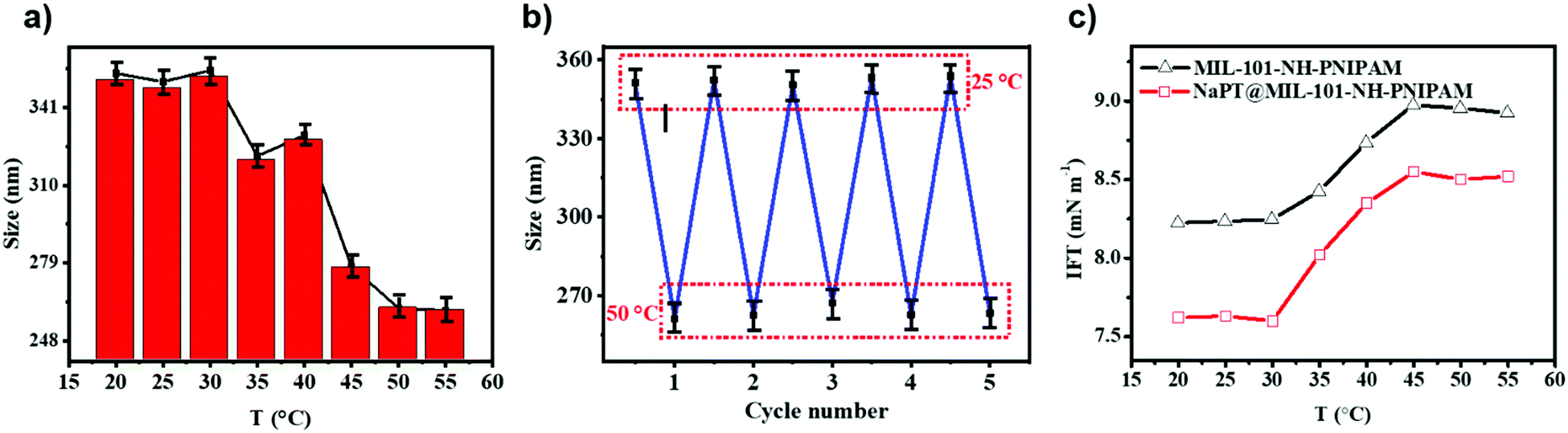 | ||
| Fig. 3 (a) Hydrodynamic diameters of MIL-101-NH-PNIPAM in dilute aqueous solution at different temperatures. (b) Five heating–cooling cycles in the temperature from 25 to 50 °C. (c) IFT of MIL-101-NH-PNIPAM and NaPT@MIL-101-NH-PNIPAM at different temperatures. | ||
In order to evaluate the changes from hydrophilicity to hydrophobicity of the PNIPAM chains below and above the LCST (32 °C), the water contact angle (WCA) of the MOF particles was measured at different temperatures. The photographs of water-drop profiles on the MOF tablets at 25 and 50 °C are shown in Fig. S9 (ESI†). These results exhibited that the surface properties of MIL-101-NH-PNIPAM changed from hydrophilicity with a WCA of 18° ± 2° at 25 °C to hydrophobicity with a WCA of 82° ± 2° at 50 °C. Similarly, the WCA of NaPT@MIL-101-NH-PNIPAM increased from 38 ± 2° to 92° ± 2° upon heating from 25 to 50 °C. This indicated an obvious phase transition of PNIPAM from hydrophilic (25 °C) to hydrophobic (>50 °C). In contrast, the WCAs of MIL-101-NH2 and MIL-101-NH-Met without the PNIPAM decoration were unaffected by temperature.
The co-solution of the two liquids was known to be related to the interfacial tension (IFT). The “barrier” between the two phases gradually disappears to reach the state of co-solution when the interfacial tension decreased. Thus, to further understand the thermo-responsive behaviour of MIL-101-NH-PNIPAM and NaPT@MIL-101-NH-PNIPAM, IFT between cyclohexane and H2O2 was measured using a spinning drop tensiometer from 20 to 55 °C (Fig. 3c). The IFT value for H2O2/cyclohexane had no significant changes with an increase in temperature (Fig. S10, ESI†). Upon addition of MIL-101-NH-PNIPAM, the IFT was down to 8.24 mN m−1 at 20 °C, and then it went through a sharp increase from 8.25 to 8.94 mN m−1 in the range of 30–45 °C, which was similar to the change of H2O2/cyclohexane with PNIPAM. Finally, the IFT decreased to 8.82 mN m−1 when the temperature was up to 55 °C (Fig. 3c and Fig. S10, ESI†). Meanwhile, NaPT@MIL-101-NH-PNIPAM had a similar temperature-dependent change with a lower ITF value of 7.62 mN m−1 at 20 °C, while the IFT data of MIL-101-NH2 and MIL-101-NH-Met had no significant change from 20 °C to 55 °C (Fig. S10, ESI†). Therefore, both MIL-101-NH-PNIPAM and NaPT@MIL-101-NH-PNIPAM could decrease the interfacial tension between two solvents, so that two solvents realized the co-solution state.
NaPT@MIL-101-NH-PNIPAM stabilized H2O2-in-oil Pickering emulsion
According to the assumption, the hydrophilic PNIPAM chains in NaPT@MIL-101-NH-PNIPAM would point to the H2O2 droplets in the emulsion, while the hydrophobic MOFs skeleton remained in the oil phase, thereby keeping the emulsion stable. Specifically, we needed to consider the ratio of oil to H2O2 and the corresponding content of MOFs to achieve a good emulsification effect. Consequently, the emulsification effect of the obtained Pickering emulsions with a ratio of cyclohexane to H2O2 from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 4:1 (volume) and the mass percentage of MOFs from 0.5 to 3.0 wt% were characterized by observing emulsion in the cuvette and confocal microscopy images. To enhance the visual contrast effect, the fluorescein isothiocyanate (FITC) isomer was added to H2O2 to make it display green fluorescence when excited with a laser at 488 nm. It is noteworthy that the generated emulsion was in the form of H2O2-in-oil, the black continuous cyclohexane phase wrapped the green spherical H2O2 phase (Fig. 4a and b) as evidenced through confocal microscopy, in contrast to the case of the published oil-in-water mode.
:1 to 4:1 (volume) and the mass percentage of MOFs from 0.5 to 3.0 wt% were characterized by observing emulsion in the cuvette and confocal microscopy images. To enhance the visual contrast effect, the fluorescein isothiocyanate (FITC) isomer was added to H2O2 to make it display green fluorescence when excited with a laser at 488 nm. It is noteworthy that the generated emulsion was in the form of H2O2-in-oil, the black continuous cyclohexane phase wrapped the green spherical H2O2 phase (Fig. 4a and b) as evidenced through confocal microscopy, in contrast to the case of the published oil-in-water mode.
 | ||
| Fig. 4 (a) Corresponding images of NaPT@MIL-101-NH-PINIPAM-stabilized Pickering emulsion with different cyclohexane-H2O2 ratios (1:1 to 4:1, 2.0 wt%). (b) Corresponding images with different contents of NaPT@MIL-101-NH-PINIPAM (varying from 0.5 wt% to 3.0 wt%, cyclohexane: H2O2 = 2:1, v/v). For the confocal microscopy images, the H2O2 phase was stained with FITC for the test. | ||
Notably, when the ratio of oil to H2O2 was 1:1, there was a brown solution, not emulsion at the bottom of the cuvette, indicating that H2O2 containing hydrophilic NaPT@MIL-101-NH-PNIPAM was excessive in it. But the transparent oil layer appeared when the ratios of oil and H2O2 were 3:1 and 4:1, which suggested that the oil was excessive at this time, while the 2:1 oil–H2O2 ratio avoided the above two situations (the content of NaPT@MIL-101-NH-PNIPAM was 2.0 wt%). Therefore, we adopted this ratio for establishing the relationship between the mass percentage of MOFs and the effect of the emulsion. It could be found from Fig. 4b that when the content of NaPT@MIL-101-NH-PNIPAM was 0.5 wt%, the emulsification is not uniform and some small bubbles were visible, indicating that the amount of MOFs was insufficient to stabilize the emulsion. The same situation appeared when the ratio was increased to 1.0 wt%. The size of the H2O2 droplets was uniform roughly until the ratio reached 2.0 wt%. When the proportion of MOFs was further increased to 3.0 wt%, although a stable emulsion could be formed, it became too viscous, so that the emulsion still could not flow after the cuvette was turned upside down (Fig. S11, ESI†), which would not be conducive to the subsequent catalytic reaction. In summary, we could conclude the optimal conditions for preparing a stable and roughly homogeneous emulsion: (1) oil:H2O2 ratio = 2:1; (2) 2.0 wt% NaPT@MIL-101-NH-PNIPAM. Furthermore, the resulting emulsion could remain stable for at least a week without conspicuous demulsification (Fig. S12, ESI†).
The thermo-responsivity behaviour of the prepared Pickering emulsion was further investigated. As shown in Fig. 5a, the Pickering emulsion generated from a cyclohexene & cyclohexane–H2O2 mixed solvent system with 2.0 wt% NaPT@MIL-101-NH-PNIPAM was very stable at 25 °C, and it became unstable with a temperature increase under stirring. When the temperature reached 50 °C, the phase separation occurred, i.e., the separated oil and H2O2 phases became clearer and the MOF particles gradually aggregated during the heating process, and finally remained in the oil phase. A reasonable explanation for this phenomenon was that the attached PNIPAM chains compacted on the MOF surface and turned to be hydrophobic above the LCST (32 °C), which led NaPT@MIL-101-NH-PNIPAM to be incompatible with H2O2 (50 °C). When the temperature was cooled down to 25 °C, NaPT@MIL-101-NH-PNIPAM completely moved to the H2O2 phase since PNIPAM chains were outspread and miscible with H2O2 below the LCST. This observation was also supported by WCA measurements at different temperatures (Fig. S9, ESI†). In addition, confocal microscopy images of the upper and lower layers of the system at different temperatures showed that when the temperature exceeded the LCST, demulsification occurred and the H2O2 droplets gradually transferred to the lower layer, while the cyclohexane transferred to the upper layer. Complete demulsification occurred at 50 °C (Fig. S13, ESI†). Upon re-homogenization, a stable emulsion was formed again at 25 °C (Fig. 5b). Most importantly, this thermo-responsivity process was highly reversible. Even after 5 cycles, Pickering emulsions could be formed again steadily. In addition to the cyclohexene & cyclohexane–H2O2 system, NaPT@MIL-101-NH-PNIPAM could also be applied to stabilize the emulsion when other organic solvents were employed as the oil phases, such as n-pentane, n-hexane, benzene, and toluene (Fig. S14, ESI†).
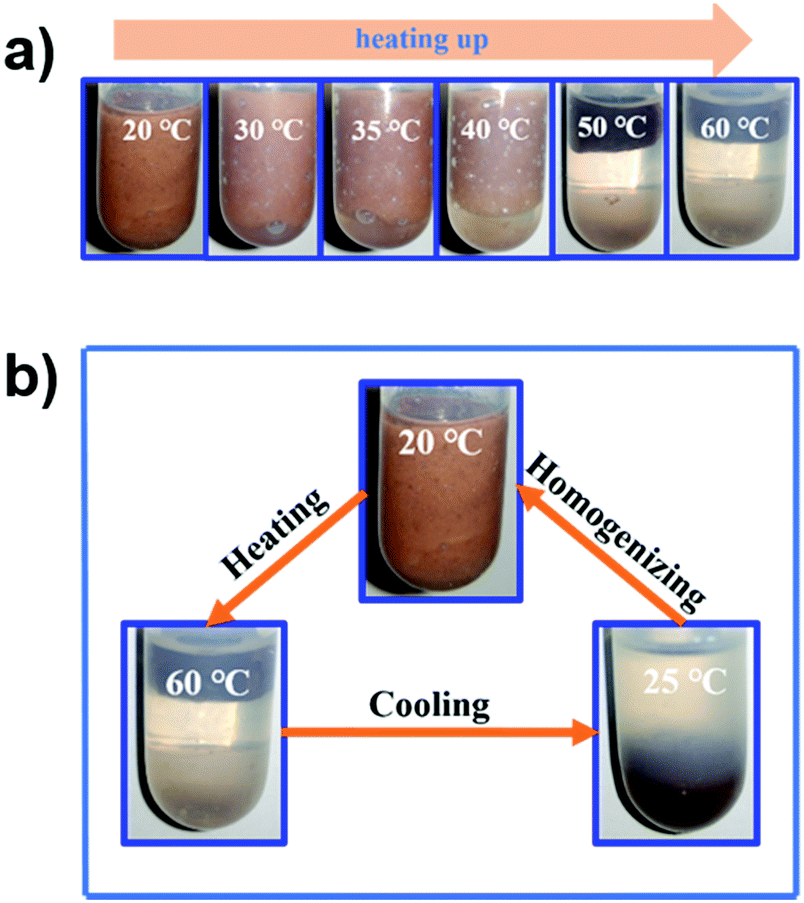 | ||
| Fig. 5 (a) Photographs of the thermo-responsive phenomenon of the Pickering emulsion stabilized by NaPT@MIL-101-NH-PINIPAM (2.0 wt%, cyclohexane-to-H2O2 = 2:1, v/v) in the range of 20 to 60 °C. (b) Photographs of the thermo-responsive cycle of the Pickering emulsion stabilized by NaPT@MIL-101-NH-PINIPAM (2.0 wt%, cyclohexane-to-H2O2 = 2:1, v/v. | ||
Pickering emulsion in situ catalytic systems
Epoxides are useful intermediates in organic synthesis which could be obtained via the epoxidation of olefins.47 The NaPT-loaded Pickering emulsion prepared in this study was an ideal system to promote the epoxidation reaction. First, the epoxidation of cyclohexene was chosen as the model reaction to investigate the catalytic conditions. As shown in Fig. S15, (ESI†) when epoxidation of olefins was performed in the biphasic system without particles of NaPT@MIL-101-NH-PNIPAM, no conversion was observed. Furthermore, epoxidation reactions occurred in low yield when NaPT@MIL-101-NH-PNIPAM was added without emulsification. Upon emulsification, it showed high yield (99%) with excellent selectivity (99%) (Fig. 6a) within 6 hours (Fig. 6b) at room temperature. Moreover, the leaching test confirmed that no further reaction occurred without NaPT@MIL-101-NH-PNIPAM, indicating that the catalyst-encapsulated emulsion system was essential for olefin epoxidation (Fig. 6b). Subsequently, the emulsification effect of the emulsion was investigated under the conditions of oil:H2O2 = 2:1 and n(H2O2): n(cyclohexene) = 5:1. Fortunately, even, the stability of the emulsion prepared was still ensured in the mixed oil phases, and no significant change in the H2O2 droplet size occurred (Fig. S15, ESI†). Therefore, the optimal conditions for the epoxidation of olefins were defined as follows: (1) n(H2O2):n(cyclohexene) = 5:1; (2) ν(oil: solvents and substrates): ν(H2O2) = 2:1; (3) 2.0 wt% of NaPT@MIL-101-NH-PNIPAM; and (4) room temperature.
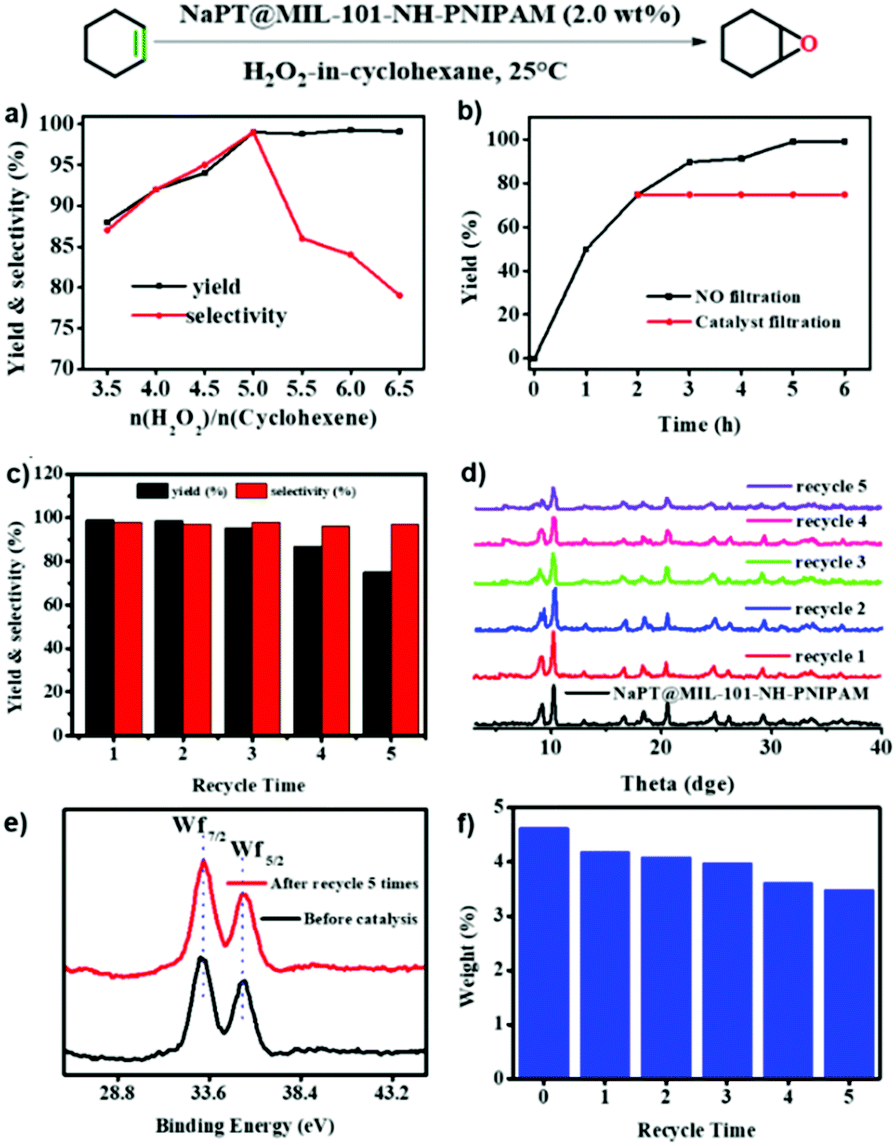 | ||
| Fig. 6 Cyclohexene epoxidation catalyzed by the NaPT@MIL-101-NH-PINIPAM-based Pickering emulsion. (a) Yield and selectivity towards different ratios of H2O2 to cyclohexene. (b) Reaction time examination (black line) and the leaching test (red line). (c) Yield and selectivity after each catalytic cycle. (d) PXRD of NaPT@MIL-101-NH-PINIPAM after each catalytic recycle. (e) Valence state of tungsten (W) before and after the reaction. (f) Tungsten (W) content before and after each catalytic recycle. | ||
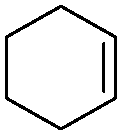
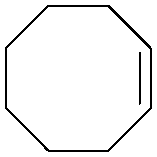
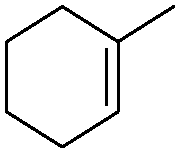
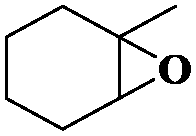
Subsequently, different types of olefins were further explored for epoxidation using H2O2 as an oxidant (Table 1 and Fig. S16–S27, ESI†). On the one hand, it could be found that different ring sizes of cyclopentene, cyclohexene and cyclooctene had little influence on the reaction results (Table 1, entries 1-3). Therefore, the reaction system was suitable for cycloalkenes with different ring tensions. On the other hand, we also investigated aliphatic olefins. Fortunately, the reaction system worked well with aliphatic olefins of different alkyl chain lengths (Table 1, entries 8 and 9). We speculated that the conjugation effect of aromatic substituents would reduce the reactivity of olefins. Surprisingly, the yield and selectivity of aryl substituted olefins were still higher than 95% (Table 1, entries 10–12). To illustrate the effect of the type and position of the substituent on the reaction efficiency, we further explored methylcyclohexene with electron-donating methyl groups at the α-position and γ-position, cyclohexene substituted with the electron-withdrawing carbonyl group at the β-position, and norbornene (Table 1, entries 4–7). Interestingly, the substitution of carbonyl groups significantly reduced the epoxidation yield of olefins, indicating that the reaction system had a negative effect on the epoxidation of electron-deficient olefins (Table 1, entry 6). As a whole, such a “Pickering emulsion in situ catalytic system” exhibits excellent yield and selectivity of versatile olefin epoxidations under mild conditions with excellent regeneration ability over traditional biphasic catalytic systems (Table S2, ESI†). It is noteworthy that the emulsion could be recycled easily after the first cycle of the catalytic reaction. When the reaction was complete, the temperature was increased to 50 °C. Then the system was cooled to room temperature after the demulsification was complete. After that, we took out the oil phase and re-added the organic solvent and substrate to start a new cycle of catalysis (Scheme 2). Remarkably, the NaPT@MIL-101-NH-PNIPAM-based Pickering emulsion showed excellent catalytic efficiency and selectivity up to 97% even after five recycles (Fig. 6c). This corresponds to the full contact between the active species [PO4{WO(O2)2}4]3− and the substrate, the former was the active species for H2O2-based epoxidations. When H2O2 completely converted to H2O, [PO4{WO(O2)2}4]3− converted to [PW12O40]3−. After the oxidation was complete, [PO4{WO(O2)2}4]3− regenerated and entered the next catalytic cycle, and FTIR and 31P NMR had proved the mechanism (Fig. S28 and S29, ESI†). With an increase of the recycle times, it was found that the particle size of the water in the emulsion became less uniform, possibly due to the shedding effect of PNIPAM chains (Fig. S30, ESI†). The PXRD results showed that the structural features of the MOF component within the system were well maintained during the first four recycles (Fig. 6d) and only a little bit loss of NaPT particles occurred after the reuse of NaPT@MIL-101-NH-PNIPAM three times (Fig. 6f and Fig. S31, ESI†), leading to the decrease in yields (Fig. 6c). In addition, XPS measurement provided the direct evidence of the remained structural integrity of NaPT. We found that there was no valence change of W species observed after recycling, which may be attributed to the protection of W species by MOF cores (Fig. 6e).
| Entry | Substrate | Product | t (h) | Yieldb (%) | Selectivityb (%) |
|---|---|---|---|---|---|
|
a Reaction conditions: olefins (2.0 mmol), H2O2 (10 mmol), NaPT@MIL-101-NH-PINIPAM-based Pickering emulsion (olefins & cyclohexane: H2O2 = 2:1, v/v, 2.0 wt% NaPT@MIL-101-NH-PINIPAM), r.t., in air.
b Yields were determined by GC-MS.
|
|||||
| 1 |
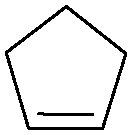
|
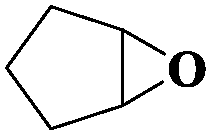
|
6 | 99 | 99 |
| 2 |
|

|
6 | 99 | 99 |
| 3 |
|

|
6 | 96 | 97 |
| 4 |
|
|
8 | 98 | 96 |
| 5 |
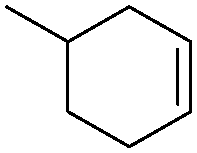
|
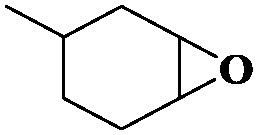
|
6 | 95 | 96 |
| 6 |
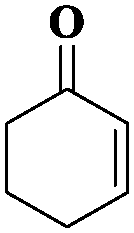
|
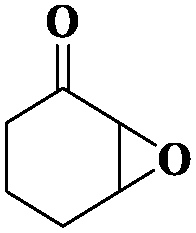
|
6 | 88 | 99 |
| 7 |
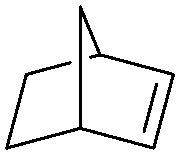
|
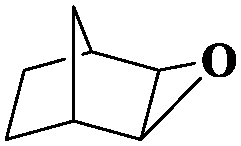
|
8 | 97 | 99 |
| 8 |
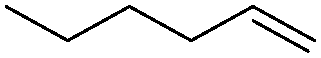
|
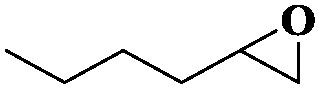
|
8 | 95 | 97 |
| 9 |
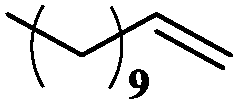
|
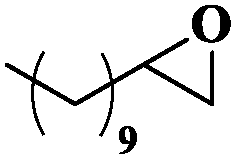
|
10 | 99 | 99 |
| 10 |
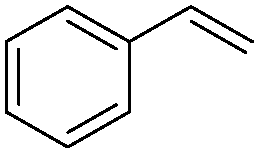
|
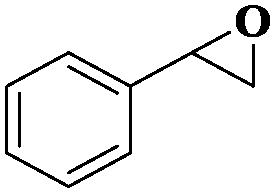
|
8 | 96 | 92 |
| 11 |
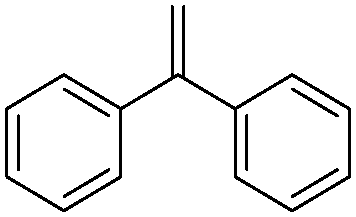
|
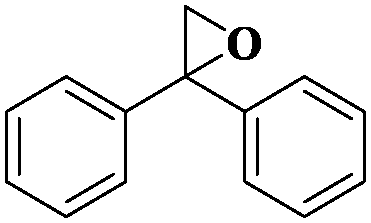
|
10 | 95 | 99 |
| 12 |
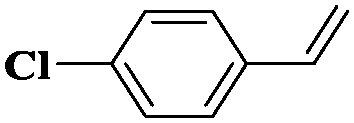
|
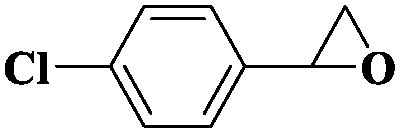
|
8 | 95 | 99 |
 | ||
| Scheme 2 A schematic model for the thermo-responsive NaPT@MIL-101-NH-PINIPAM-based Pickering emulsion catalytic system. | ||
Experimental
Materials and reagents
MIL-101-NH2 and MIL-101-NH-Met were synthesized according to the reported methods.48 The initiator of potassium persulfate (K2O8S2, 99.5%, Energy Chemical) was recrystallized twice from water prior to use. N-Isopropyl acrylamide (NIPAM, ≥99%, Acros Organics) was purified by recrystallization in n-hexane (55 °C). The other reagents of analytical grade were used as received, including Keggin type sodium phosphotungstate (NaPT, Na3O40PW12·12H2O).Preparation synthesis of MIL-101-NH-PNIPAM
A mixture of MIL-101-NH-Met (0.31 g), NIPAM (0.26 g, 2.0 mmol) and potassium persulfate (0.015 g, 0.055 mmol) in DI water (20 mL) was stirred at 70 °C for 24 h under a N2 atmosphere. The unreacted polymer was then removed via centrifugation and the crude product was washed with DI water several times and then dried under vacuum at 80 °C for 4 h to give MIL-101-NH-PNIPAM (472 mg). FT-IR (KBr, cm−1): 3322 (s), 2947 (s), 1750 (m), 1566 (s), 1373 (s), 1210 (m), 991 (s), 766 (m), 724 (m).Synthesis of NaPT@MIL-101-NH-PNIPAM
A mixture of MIL-101-NH-PNIPAM (0.10 g) and NaPT (0.10 g, 0.034 mmol) in DI water (30 mL) was stirred at room temperature for 72 h. The resulting solid was obtained by centrifugation, washed three times with DI water and then dried in a vacuum at 80 °C for 4 h to obtain NaPT@MIL-101-NH-PNIPAM (112 mg) as a reddish-brown powder. ICP measurement indicated that the encapsulated amount of tungsten (W) in MIL-101-NH-PNIPAM was 4.63 wt%. FT-IR (KBr, cm−1): 3315 (s), 2928 (m), 1788 (m), 1613 (s), 1360 (s), 1228 (s), 1102 (m), 934 (m) 896 (s), 817 (s), 615 (m).Emulsification and temperature-triggered demulsification
To prepare the MOF-based Pickering emulsion, cyclohexane and H2O2 were used as the incompatible oil and aqueous phase, respectively. Typically, NaPT@MIL-101-NH-PNIPAM (52 mg) was added into a centrifuge tube containing cyclohexene, cyclohexane and H2O2 (0.2 mL: 1.8 mL: 1.0 mL, 1:9:5 in v/v/v, the volume ratio of the oil phase to the H2O2 phase was 2:1). The mixture was sonicated at 5000 rpm for 300 s to generate a Pickering emulsion with 2.0 wt% of NaPT@MIL-101-NH-PNIPAM. The other types of emulsions with different MOF contents and oil-to-H2O2 ratios were prepared in a similar way. The temperature was controlled using a thermostat oil bath to investigate the temperature-triggered demulsification behaviour of the as-synthesized Pickering emulsions.
Catalytic property
The catalytic performance was based on the “Pickering emulsion in situ catalytic system”. The olefin epoxidation reaction was carried out in the NaPT@MIL-101-NH-PNIPAM-stabilized Pickering emulsion. In a typical procedure, the Pickering emulsion consisting of cyclohexene, cyclohexane and H2O2 (1:9:5 in v/v/v) with NaPT@MIL-101-NH-PNIPAM (52 mg, 2.0 wt%, 2.4 mg of W, 0.65 mol% equiv.) was stirred at room temperature. The reaction process was monitored by GC-MS and the main product was separated and purified, which was further confirmed by 1H NMR spectra. Once the reaction was complete, the temperature of the reaction system was adjusted from 25 to 50 °C under mild stirring. During this process, it was observed that NaPT@MIL-101-NH-PNIPAM gathered together gradually and floated on the oil phase. After complete phase separation, the system was cooled down to 25 °C, accompanied by the migration of MOF nanoparticles from the oil phase to the aqueous phase. The upper oil phase containing the target organic product was isolated, while the bottom aqueous phase containing NaPT@MIL-101-NH-PNIPAM was collected and dried several hours for the next run.
Three control experiments were designed to demonstrate the catalytic effect of the NaPT@MIL-101-NH-PNIPAM-stabilized Pickering emulsion: (1) the reaction was carried out without NaPT@MIL-101-NH-PNIPAM; (2) NaPT@MIL-101-NH-PNIPAM was added into the oil–H2O2 system without emulsification; and (3) the solid catalyst of NaPT@MIL-101-NH-PNIPAM was separated from the catalytic emulsion system after reacting for a given time (2 h). The reaction was continued with the filtrate in the absence of NaPT@MIL-101-NH-PNIPAM. The results of the above experiments were all monitored by 1H NMR and GC-MS.
Conclusions
In summary, the first example of the “Pickering emulsion in situ catalytic system” was successfully developed in this study and it was applied in the epoxidation of olefins efficiently. In this emulsion system, there were two immiscible phases, the green oxidant H2O2 as the aqueous phase, while the olefin and cyclohexane mixture as the oil phase. This system enabled the efficient epoxidation of olefins with excellent yield and selectivity. Furthermore, the resultant new H2O2-in-oil emulsion system was easily separated from the products of the reaction with excellent recycling stability for at least five cycles. Such an emulsion system would compensate the shortcomings of the traditional emulsions, and make the large-scale catalytic reactions possible. Therefore, our study provides a new insight for the application of MOF-based Pickering emulsions for practical organic synthesis.Author contributions
X. L. Zhao designed the research project; B. Huang and W. L. Jiang did the synthetic work and data collection; Characterization studies and the catalytic process were carried out by B. Huang and Y. F. Niu. H. H. Wu and J. Han helped with data analyses and discussions during manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was supported by the National Key Research and Development Program of China (2017YFA0403102).Notes and references
- N. Mizuno, K. Yamaguchi and K. Kamata, Coord. Chem. Rev., 2005, 249, 1944–1956 CrossRef CAS
.
- Y. Leng, J.-H. Wu, P.-P. Jiang and J. Wang, Catal. Sci. Technol., 2014, 4, 1293–1300 RSC
- Y. M. A. Yamada, M. Ichinohe, H. Takahashi and S. Ikegami, Org. Lett., 2001, 3, 1837–1840 CrossRef CAS PubMed
- X. Engelmann, D. D. Malik, T. Corona, K. Warm, E. R. Farquhar, M. Swart, W. Nam and K. Ray, Angew. Chem., Int. Ed., 2019, 58, 4012–4016 CrossRef CAS PubMed
- L. Bromberg, Y. Diao, H.-M. Wu and S. A. Speakman, Chem. Mater., 2012, 24, 1664–1675 CrossRef CAS
- Q.-H. Xia, H.-Q. Ge, C.-P. Ye, Z.-M. Liu and K.-X. Su, Chem. Rev., 2005, 105, 1603–1662 CrossRef CAS PubMed
- N. S. Patil, B. S. Uphade, P. Jana, S. K. Bharagava and V. R. Choudhary, J. Catal., 2004, 223, 236–239 CrossRef CAS
- P. J. Cordeiro and T. D. Tilley, ACS Catal., 2011, 1, 455–467 CrossRef CAS
- X. Engelmann, D.-D. Malik, T. Corona, K. Warm, E. R. Farquhar, M. Swart, W. Nam and K. Ray, Angew. Chem., Int. Ed., 2019, 58, 4012–4016 CrossRef CAS PubMed
- W. Lueangchaichaweng, N. R. Brooks, S. Fiorilli, E. Gobechiya, K.-F. Lin, L. Li, S. Parres-Esclapez, E. Javon, S. Bals, G. V. Tendeloo, J. A. Martens, C. E. A. Kirschhock, P. A. Jacobs and P. P. Pescarmona, Angew. Chem., Int. Ed., 2014, 53, 1585–1589 CrossRef CAS PubMed
- M. Feng, M. Halb, M. Feuchter, G. Pacher and G. Pinter, ACS Catal., 2011, 1, 1035–1042 CrossRef
- J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. B. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC
- A. Di, J. Schmitt, M. A. Silva, K. M. Z. Hossain, N. Mahmoudi, R. J. Errington and K. J. Edler, Nanoscale, 2020, 12, 22245–22257 RSC
- F. Marme, G. Coudurier and J. C. Védrine, Microporous Mesoporous Mater., 1998, 22, 151–163 CrossRef CAS
- X. Xue, F. Yu, J.-G. Li, G. Bai, H. Yuan, J. Hou, B. Peng, L. Chen, M.-F. Yue, G. Wang and C. Wang, Int. J. Hydrogen. Energ., 2020, 45, 1802–1809 CrossRef CAS
- K. Liu, Y. Xu, Z. Yao, H. N. Miras and Y.-F. Song, Chem. – Eur. J., 2015, 21, 10812–10820 CrossRef PubMed
- P. Liu, H. Wang, Z. Feng, P. Ying and C. Li, J. Catal., 2008, 256, 345–348 CrossRef CAS
- T. Kwon, G. A. Tsigdinos and T. J. Pinnavaia, J. Am. Chem. Soc., 1988, 110, 3653–3654 CrossRef CAS
- L. Ni, H. Li, H. Xu, C. Shen, R. Liu, J. Xie, F. Zhang, C. Chen, H. Zhao, T. Zuo and G. Diao, ACS Appl. Mater. Interfaces, 2019, 11, 38708–38718 CrossRef CAS PubMed
- Y. Duan, W. Wei, F. Xiao, Y. Xi, S.-L. Chen, J.-L. Wang, Y. Xu and C. Hu, Catal. Sci. Technol., 2016, 6, 8540–8547 RSC
- X. Jing, Z. Li, B. Lu, Y. Han, Y. Chi and C. Hu, Appl. Catal., A, 2020, 598, 117613 CrossRef CAS
- Y. Leng, J. Wang, D. Zhu, M. Zhang, P. Zhao, Z. Long and J. Huang, Green Chem., 2011, 13, 1636–1639 RSC
- Y.-M. Zhang, V. Degirmenci, C. Li and E. J. M. Hensen, ChemSusChem, 2011, 4, 59–64 CrossRef CAS PubMed
- J. Juan-Alcañiz, E. V. Ramos-Fernandez, U. Lafont, J. Gascon and F. Kapteijn, J. Catal., 2010, 269, 229–241 CrossRef
- L. Bromberg, Y. Diao, H.-M. Wu, S. A. Speakman and T. A. Hatton, Chem. Mater., 2012, 24, 1664–1675 CrossRef CAS
- N. V. Maksimchuk, K. A. Kovalenko, S. S. Arzumanov, Y. A. Chesalov, M. S. Melgunov, A. G. Stepanov, V. P. Fedin and O. A. Kholdeeva, Inorg. Chem., 2010, 49, 2920–2930 CrossRef CAS PubMed
- J.-T. Tang, P. J. Quinlan and K. C. Tam, Soft Matter, 2015, 11, 3512–3529 RSC
- J. Wu and G.-H. Ma, Small, 2016, 12, 4633–4648 CrossRef CAS PubMed
- L. Tao, M. Zhong, J. Chen, S. Jayakumar, L. Liu, H. Li and Q. Yang, Green Chem., 2018, 20, 188–196 RSC
- N. Xue, G. Zhang, X. Zhang and H. Yang, Chem. Commun., 2018, 54, 13014–13017 RSC
- B.-H. Wu, C.-J. Yang, Q. Xin, L.-L. Kong, M. Eggersdorfer, J. Ruan, P. Zhao, J.-Z. Shan, K. Liu, D. Chen, D. A. Weitz and X. Gao, Adv. Mater., 2021, 2102362 CrossRef CAS PubMed
- A. M. B. Rodriguez and B. P. Binks, Soft Matter, 2020, 16, 10211–10243 RSC
- Z. Meng, M. Zhang and H. Yang, Green Chem., 2019, 21, 627–633 RSC
- H. Zhu, Q. Zhang and S.-P. Zhu, Chem. – Eur. J., 2016, 22, 8751–8755 CrossRef CAS PubMed
- Y. Shi, D. Xiong, Z. Li, H. Wang, J. Qiu, H. Zhang and J. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 53385–53393 CrossRef CAS PubMed
- Z. Yang, L. Cao, J. Li, J. Lin and J. Wang, Polymer, 2018, 153, 17–23 CrossRef CAS
- Z. Li, J. Zhang, T. Luo, X. Tan, C. Liu, X. Sang, X. Ma, B. Han and G. Yang, Soft Matter, 2016, 12, 8841–8846 RSC
- B. Xiao, Q. Yuan and R. A. Williams, Chem. Commun., 2013, 49, 8208–8210 RSC
- F. Zhang, L. Liu, X. Tan, X. Sang, J. Zhang, C. Liu, B. Zhang, B. Han and G. Yang, Soft Matter, 2017, 13, 7365–7370 RSC
- J. Huo, M. Marcello, A. Garai and D. Bradshaw, Adv. Mater., 2013, 25, 2717–2722 CrossRef CAS PubMed
- W. Xue, H.-Q. Yang and Z.-P. Du, Langmuir, 2017, 33, 10283–10290 CrossRef CAS PubMed
- H.-Q. Yang, T. Zhou and W.-J. Zhang, Angew. Chem., Int. Ed., 2013, 52, 7455–7459 CrossRef CAS PubMed
- J.-P. Huang and H.-Q. Yang, Chem. Commun., 2015, 51, 7333–7336 RSC
- W.-L. Jiang, Q.-J. Fu, B.-J. Yao, L.-G. Ding, C.-X. Liu and Y.-B. Dong, ACS Appl. Mater. Interfaces, 2017, 9, 36438–36446 CrossRef CAS PubMed
- B.-J. Yao, Q.-J. Fu, A.-X. Li, X.-M. Zhang, Y.-A. Li and Y.-B. Dong, Green Chem., 2019, 21, 1625–1634 RSC
- K. Zhang, C. H. Chew, S. Kawi, J. Wang and L. M. Gan, Catal. Lett., 2000, 64, 179–184 CrossRef CAS
- N. Mizuno, K. Yamaguchi and K. Kamata, Coord. Chem. Rev., 2005, 249, 1944–1956 CrossRef CAS
- J.-W. Zhou and B. Wang, Chem. Soc. Rev., 2017, 46, 6927–6945 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00245k |
| This journal is © The Royal Society of Chemistry 2022 |