DOI:
10.1039/D2MA00129B
(Paper)
Mater. Adv., 2022,
3, 5043-5054
Surfactant-free suspension polymerization of hydrophilic monomers with an oil-in-water system for the preparation of microparticles toward the selective isolation of tumor cells†
Received
6th February 2022
, Accepted 6th May 2022
First published on 10th May 2022
Abstract
Circulating tumor cells (CTCs) are derived from a primary tumor or monastic foci, and are found in the bloodstream of patients with tumors. We developed polymer droplets of blood-compatible poly(2-methoxyethyl acrylate) (PMEA) that selectively accumulate in tumor cells. PMEA microparticles, which are larger than the size of cells, have potential as a platform for CTC capture devices without the need to use antibodies. Herein, these microparticles, as well as several, other types of microparticles composed of hydrophilic monomers, were prepared by surfactant-free suspension polymerization, and their selective isolation abilities toward the capture of tumor cells were evaluated via comparative studies. The microparticles possessed smooth and extremely pure surfaces suitable for evaluating the interaction force with tumor cells. The number of human platelets adhered to the PMEA microparticles was clearly lower than the number of platelets adhered to the other polymer microparticles. Interestingly, the PMEA microparticles prepared with a 1% crosslinking ratio showed stronger interactions with tumor cells than the other polymer microparticles. In addition, the PMEA microparticles enabled the efficient recovery of tumor cells from cell suspensions under dynamic conditions in comparison with the other polymer microparticles. These results provide insights into the possible applicability of PMEA microparticles as a platform for CTC capture without using antibodies.
Introduction
Circulating tumor cells (CTCs), which invade the bloodstream from a primary tumor and monastic foci, can act as an effective biomarker to stipulate a reliable method for diagnosing not only tumor progression but also tumor metastasis.1 Some methods for the detection of CTCs have been developed, such as filtration-based assessment, the DEPArray system, and the semi-automated CellSearch system.2–4 Because CTCs express specific membrane antigens, including epithelial cell adhesion molecules (EpCAMs), they have been routinely captured through ligand–receptor interactions and by immunomagnetic separation using microspheres coated with ligands, such as antibodies.5,6 However, the utilization of EpCAM-based approaches is often limited because of the existence of EpCAM-negative CTCs.7 Therefore, there is a need to develop a system for the specific isolation of CTCs that uses technology other than tumor-related antigens and antibodies. We previously reported that poly(2-methoxyethyl acrylate) (PMEA)-coated substrates suppress the adhesion and activation of platelets but enhance the adhesion of integrin-overexpressed cells, such as non-tumorigenic epithelial cells and tumor cells.8–10 PMEA-coated substrates showed no cytotoxicity and an improved viability of tumor cells under serum-free conditions.11 Furthermore, we demonstrated that polymer droplets composed of PMEA selectively accumulate in tumor cells, avoiding normal cells.11–13 Thus, PMEA has the potential to selectively isolate tumor cells from blood samples regardless of their EpCAM expression.
Microparticles have been developed as a valuable platform for microcarrier-based cell culture.14,15 In addition, microparticles conjugated with antibodies or polymers have been readily utilized for the isolation of proteins, immune cells, and stem cells.16–18 Microparticles with high surface area-to-volume ratios can gain an increased contact frequency with cells by using dynamic systems.19,20 Hence, we hypothesize that PMEA-formed microparticles can provide an effective platform for the separation of CTCs from blood.
Suspension polymerization via oil-in-water systems has been used to prepare polymer microparticles.21–23 Conventional suspension polymerization systems are suitable for preparing microparticles composed of hydrophobic monomers (e.g., styrene, methyl methacrylate, and vinyl acetate);21,22 whereas hydrophilic monomers, including 2-methoxyethyl acrylate (MEA), partially or completely dissolve in large amounts of water and are difficult to form oil droplets. The oil droplets are unstable and easily fuse together, resulting in an indefinite aggregation of polymers. Stabilizers, including surfactants, are generally used in suspension polymerization processes to inhibit droplet fusion;23 however, the surfactants often remain on the surface of the resulting microparticles, even if the microparticles are carefully washed with solvents that would dissolve the surfactant. Residual surfactants often affect the function of the particles, including their cell adhesion. For water-soluble hydrophilic monomers, inverse suspension polymerization, which uses water-in-oil systems, is often employed because these monomers dissolve in water.24–26 Inverse suspension polymerization requires not only a surfactant to stabilize the dispersed droplets, but also large amounts of organic solvents.
As such, suspension polymerization with oil-in-water systems is less detrimental to the environment as it uses less organic solvent than inverse suspension polymerization. Okudaira et al. reported a preparation method for hydrophobic polystyrene (PSt) microparticles by a surfactant-free suspension polymerization,27 but there are no such reports on hydrophilic polymer microparticles, owing to the dissolution of hydrophilic monomers in water. Thus, it is challenging to establish a protocol toward hydrophilic polymer microparticles by surfactant-free suspension polymerization with an oil-in-water system.
In the present study, we describe the preparation of PMEA microparticles by a surfactant-free suspension polymerization that enabled the selective adhesion of cells. As controls, hydrophobic poly(2-methoxyethyl methacrylate) (PMEMA) and hydrophilic poly(oligo(ethylene glycol)acrylate) (POEGA), poly(2-hydroxyethyl acrylate) (PHEA), and poly(2-hydroxyethyl methacrylate) (PHEMA) were prepared by the developed protocol, and their affinities for tumor cells were evaluated. Finally, we demonstrated the capturing of tumor cells using the PMEA microparticles under dynamic conditions.
Results and discussion
Preparation of the polymer microparticles by surfactant-free suspension polymerization
We demonstrated the preparation of microparticles composed of PMEA, POEGA, PHEA, PMEMA, and PHEMA by surfactant-free suspension polymerization with an oil-in-water system. To obtain suitable particles for the cell experiments, three difficulties had to be overcome (i.e., removing any remaining surfactant, selecting the appropriate organic solvents, and ensuring low glass transition temperatures (Tg)). Polymers with high Tg (e.g., PSt and PMEMA) form hard particles whose surfaces inhibit their fusion with one another. On the other hand, polymers with low Tg (e.g., PMEA, POEGA, PHEA, and PHEMA) form soft particles with sticky surfaces, resulting in their aggregation. Some research groups have reported the preparation of PHEMA by suspension polymerization;28,29 however, these protocols used an exorbitant amount of crosslinker, which could give resultant particles that do not comprise pure PHEMA. Thus, there is a need to establish a surfactant-free suspension polymerization method that uses a minimal amount of crosslinker to prepare pure microparticles composed of hydrophilic monomers. As such, in this study, we devised a new system for suspension polymerization that combines the salting-out effect of calcium chloride and the stabilization effect of calcium carbonate. Calcium chloride is suitable for this system because it can dissolve in not only water but also in the lower alcohols (e.g., methanol and ethanol), which are used to remove unreacted monomers from the resultant particles; this is notable as general salts tend to be insoluble in such alcohols. Calcium carbonate was employed as the dispersion stabilizer because it can be decomposed by hydrochloric acid into easily removable calcium chloride. Consequently, the particles prepared by our system can be easily purified by washing them with a lower alcohol after treatment with hydrochloric acid. Table 1 summarizes the preparation of the microparticles used in this study. To clarify the effect of the crosslinking ratio (Cr), PMEA microparticles were prepared with Cr values of 1% and 2%, while the other microparticles were prepared using a Cr of 1%. The monomers suspended in a highly concentrated aqueous calcium chloride solution were polymerized in the presence of calcium carbonate with stirring by a propeller agitator. Note that the concentration and volume of calcium chloride depended on the hydrophilicity of the monomers. To obtain sturdy particles from PMEA, PMEMA, or PHEMA, no organic solvent was needed, whereas a minimal amount was required to obtain particles from POEGA and PHEA. In addition, a two-step heating process was necessary to accomplish high monomer conversion and obtain sturdy particles. When these conditions suited very well the properties of the monomers, the microparticles were formed without aggregation (i.e., the combination of the concentration of calcium chloride, the condition of a two-step heating process, and the hydrophilicity of monomers). The resultant microparticles were treated with hydrochloric acid, followed by Soxhlet extraction with methanol, resulting in pure microparticles. The purified microparticles were sorted into two groups based on size via a wet-type classification using a sieve (mesh opening: 300 μm) and methanol; they were then lyophilized after replacement with water to give the pure PMEA microspheres. The smaller microparticles of the two sized-based groups of particles were used for the remainder of the study.
Table 1 Summary of the surfactant-free suspension polymerization via a two-step heating process
| Samples |
Feed composition |
Temp. (°C) |
Polymn. time (h) |
Stirring speed (rpm) |
| Monomer (mol) |
Initiatora (mmol) |
Crosslinkerb (mol%) |
CaCl2 (mol) |
CaCO3 (mol) |
Water (mL) |
Org. solv.c (mL) |
1st |
2nd |
1st |
2nd |
|
2,2-Azobis(isobutyronitrile) (AIBN) was used for PMEA, POEGA, PMEMA, and PHEMA. 2,2-Azobis(4-methoxy-2,4-dimethylvaleronitrile) was used for PHEA.
Ethylene glycol diacrylate was used for PMEA, POEGA, and PHEA. Ethylene glycol dimethacrylate was used for PMEMA and PHEMA.
Cyclohexanol and 2,2,2-trifluoroethanol were used for POEGA and PHEA, respectively.
|
| PMEA_1 |
0.19 |
0.61 |
1 |
0.54 |
0.10 |
200 |
— |
60 |
85 |
0.5 |
1 |
300 |
| PMEA_2 |
0.19 |
0.61 |
2 |
0.54 |
0.10 |
200 |
— |
60 |
85 |
0.5 |
1 |
300 |
| POEGA |
0.052 |
0.61 |
1 |
1.09 |
0.10 |
110 |
2.5 |
60 |
85 |
0.5 |
2 |
300 |
| PHEA |
0.22 |
0.65 |
1 |
1.02 |
0.10 |
150 |
10 |
40 |
60 |
0.5 |
1 |
300 |
| PMEMA |
0.17 |
0.61 |
1 |
0.54 |
0.10 |
220 |
— |
60 |
85 |
0.5 |
1 |
450 |
| PHEMA |
0.19 |
0.61 |
1 |
1.09 |
0.10 |
220 |
— |
60 |
85 |
0.5 |
3 |
300 |
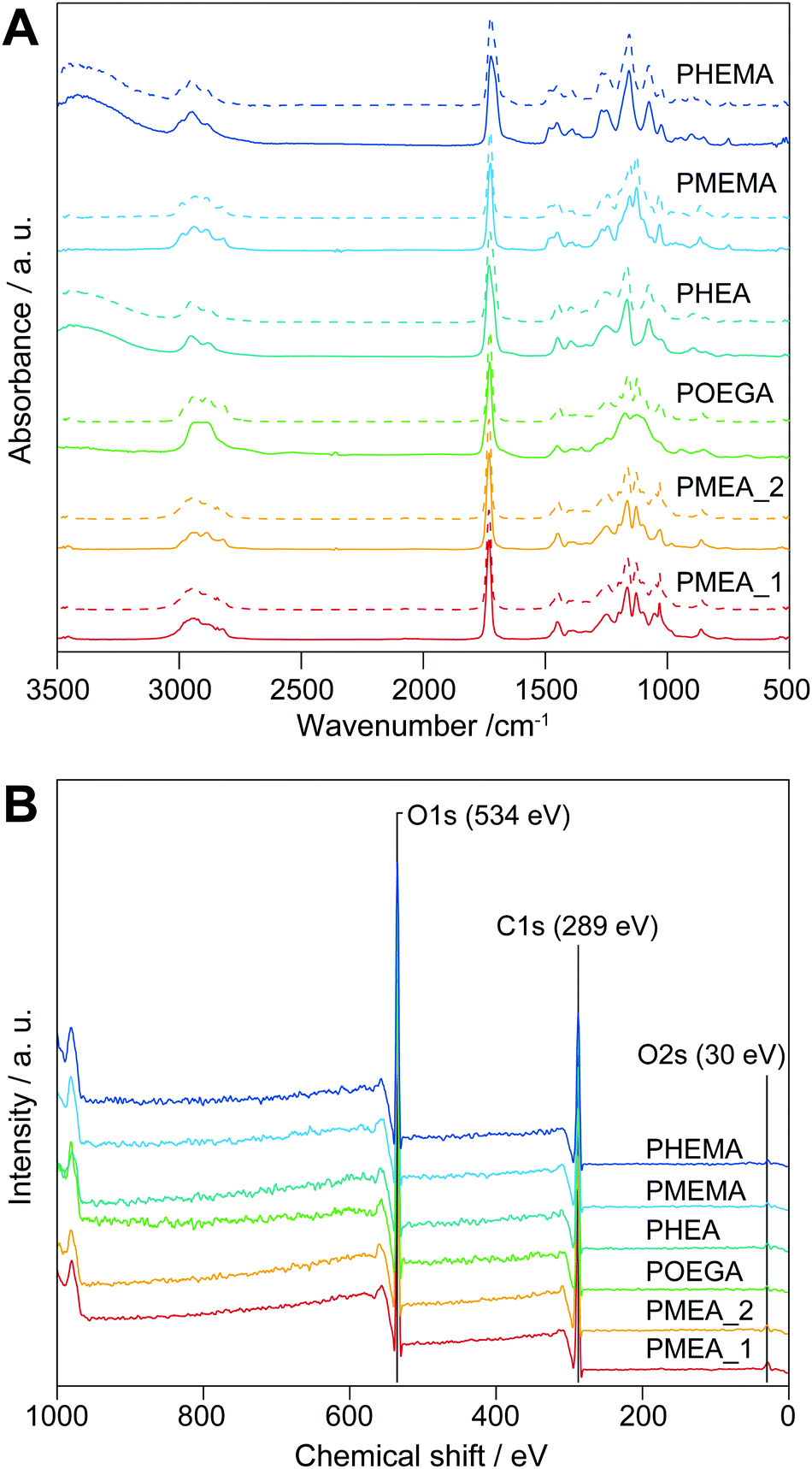
The chemical structures of the microparticles were confirmed by attenuated total reflectance (ATR)-Fourier transform infrared (FTIR) spectroscopy (Fig. 1A, solid lines). The spectra of the PMEA and POEGA microparticles showed bands derived from their respective PMEA and POEGA backbones at 3100–2730 cm−1 (–CH2–, R2CH–, and –O–CH3 C–H stretching), 1731 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of ester for acylate), 1450 cm−1 (C–H bending of –COO–CH2– and –O–CH3), and 1420–800 cm−1 (fingerprint region). As the PHEA microparticles bear hydroxy groups instead of methyl ether groups like PMEA and POEGA, they showed –OH stretching vibrations at 3360 cm−1 (–OH stretching). Moreover, the PMEMA and PHEMA microparticle spectra were almost the same as those of PMEA and PHEA, respectively, except PMEMA and PHEMA exhibited a band at 1728 cm−1 corresponding to the CO stretching of their methacrylate ester moieties. Importantly, all the spectra corresponded to those of the homopolymers synthesized by conventional free-radical polymerization (Fig. 1A, dashed lines). Thus, the surfactant-free suspension polymerizations successfully proceeded to produce objective microparticles composed of PMEA, POEGA, PHEA, PMEMA, and PHEMA. Subsequently, X-ray photoelectron spectroscopy (XPS) measurements of the microparticles were carried out to clarify the state of the outermost surfaces of the microparticles, which significantly influences their cell-attachment functions. Fig. 1B shows the XPS profiles of the microparticles. We did not observe calcium peaks (ca. 347 eV), indicating calcium chloride and calcium carbonate did not remain. In addition, the elemental ratios of carbon and oxygen obtained from the XPS profiles agreed well with the theoretical values (Table S1, ESI†). From these results, we concluded that the obtained microparticles have clean and pure outer surfaces, suggesting that these microparticles are suitable for evaluating the interaction force with tumor cells.
O stretching of ester for acylate), 1450 cm−1 (C–H bending of –COO–CH2– and –O–CH3), and 1420–800 cm−1 (fingerprint region). As the PHEA microparticles bear hydroxy groups instead of methyl ether groups like PMEA and POEGA, they showed –OH stretching vibrations at 3360 cm−1 (–OH stretching). Moreover, the PMEMA and PHEMA microparticle spectra were almost the same as those of PMEA and PHEA, respectively, except PMEMA and PHEMA exhibited a band at 1728 cm−1 corresponding to the CO stretching of their methacrylate ester moieties. Importantly, all the spectra corresponded to those of the homopolymers synthesized by conventional free-radical polymerization (Fig. 1A, dashed lines). Thus, the surfactant-free suspension polymerizations successfully proceeded to produce objective microparticles composed of PMEA, POEGA, PHEA, PMEMA, and PHEMA. Subsequently, X-ray photoelectron spectroscopy (XPS) measurements of the microparticles were carried out to clarify the state of the outermost surfaces of the microparticles, which significantly influences their cell-attachment functions. Fig. 1B shows the XPS profiles of the microparticles. We did not observe calcium peaks (ca. 347 eV), indicating calcium chloride and calcium carbonate did not remain. In addition, the elemental ratios of carbon and oxygen obtained from the XPS profiles agreed well with the theoretical values (Table S1, ESI†). From these results, we concluded that the obtained microparticles have clean and pure outer surfaces, suggesting that these microparticles are suitable for evaluating the interaction force with tumor cells.
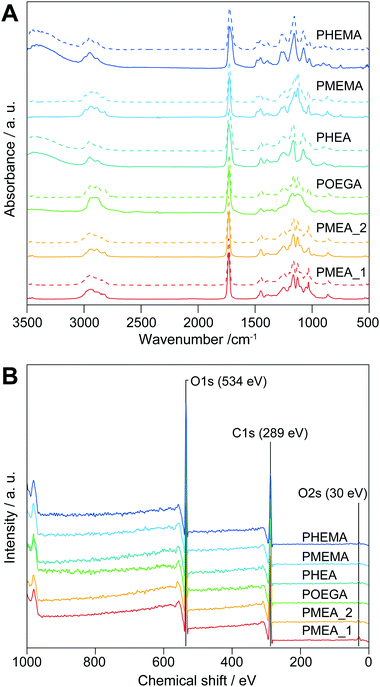 |
| | Fig. 1 (A) ATR-FTIR and (B) XPS profiles of the polymer microparticles synthesized in this study. Dashed lines in A show the spectra of the homopolymers. | |
Characterization of the polymer microparticles
The surface morphology also directly affects cell attachment. We confirmed the shape of the microparticles using scanning electron microscopy (SEM) (Fig. 2); the SEM image of the commercially available PSt microparticles used in this study as a control are shown in the same figure. All the polymer microparticles possessed true, spherical shapes with smooth surfaces, which were appropriate for the cell experiments; although, the diameters of the PHEA and PHEMA microparticles were smaller than those of the others. There was no difference between the sizes of the PMEA_1 and PMEA_2 microparticles, suggesting that the effect of Cr was small for our surfactant-free suspension polymerization system. These microparticles were used for the cell experiments under wet conditions. In the presence of water molecules, the particles swelled and became larger. We evaluated these changes in size using optical microscope observations and image analysis under dry and wet conditions (Fig. S1, ESI†). Note that phosphate buffered saline (PBS (−)) was used as a solvent for the experiments under wet conditions to obtain an environment close to cell culturing conditions, and that the microparticles were immersed for a sufficient amount of time. The results of this analysis are summarized in Table 2. The diameters of the PMEA, POEGA, PHEA, and PHEMA microparticles clearly increased in size under wet conditions. In contrast, the diameters of the PMEMA and PSt microparticles remained nearly unchanged. The POEGA microparticles exhibited the maximum diameter (235.8 ± 33.3 μm), and the PHEA microparticles possessed the minimum diameter (76.2 ± 11.2 μm). Notably, the diameters of all the microparticles were sufficiently larger than the sizes of the cells (>10 μm). The surface areas and volumes of all the microparticles under wet conditions were calculated from their diameters and are shown in Table 2. The swelling ratios of the polymer microparticles were calculated using eqn (6). The calculated swelling ratios of the PMEA microparticles were 125.1% (Cr = 1%) and 114.3% (Cr = 2%), respectively. These results are reasonable because the large amount of crosslinker decreases the extent of the polymer chains and interrupts the diffusion of water molecules into the microparticles. The more hydrophilic POEGA, PHEA, and PHEMA microparticles showed more drastic swelling (340.7% for POEGA, 284.1% for PHEA, and 241.0% for PHEMA) than the PMEA microparticles. Furthermore, hydrophobic PMEMA slightly swelled (102.3%) and PSt shrank (98.9%). Using eqn (8), the water contents of the polymer microparticles were defined. The PMEMA and PSt microparticles hardly contained water, while the POEGA, PHEA, and PHEMA microparticles contained large amounts of water (ca. 60–70 wt%). The PMEA_1 and PMEA_2 microparticles also swelled, and their water contents were 20.1 wt% and 12.5 wt%, respectively. This trend almost agreed with the theoretical values of the octanol–water partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P), which give the hydrophilic indices of the polymers (Table 2). Although the logP value of PHEA (−5.07) was lower than that of POEGA (−3.58), a higher amount of water was captured in the POEGA particles. It seems that the network of PHEA is restricted from expanding due to the crosslinking via hydrogen-bonding formation of hydroxy groups on the side chains. The water content of PMEA_2 was smaller than that of PMEA_1 for the same reason that their swelling ratios were different: the higher amount of crosslinker constrains the polymer chains and interrupts the permeation of water molecules. Interestingly, the water contents of these PMEA microparticles were definitely higher than that of the PMEA homopolymer (9 wt%).9 This indicates that the specific surface area of PMEA, whose outermost surface swells more easily than that of the polymer in the bulk state, increased upon forming microparticles and this promoted hydration.30,31 Subsequently, differential scanning calorimetry (DSC) measurements of the polymer microparticles were performed under wet conditions. We have previously reported that the PMEA homopolymer and its derivatives form cold-crystallizable water, called intermediate water (IW), at approximately −40 °C, which plays a key role in the blood compatibility of polymer surfaces.9,32–35 In the case of the PMEA microparticles synthesized by surfactant-free suspension polymerization, peaks ascribed to the cold crystallization of IW were observed (Fig. S2, ESI†). This result suggests that the PMEA microparticles display excellent blood compatibility similar to that of the PMEA homopolymer.
P), which give the hydrophilic indices of the polymers (Table 2). Although the logP value of PHEA (−5.07) was lower than that of POEGA (−3.58), a higher amount of water was captured in the POEGA particles. It seems that the network of PHEA is restricted from expanding due to the crosslinking via hydrogen-bonding formation of hydroxy groups on the side chains. The water content of PMEA_2 was smaller than that of PMEA_1 for the same reason that their swelling ratios were different: the higher amount of crosslinker constrains the polymer chains and interrupts the permeation of water molecules. Interestingly, the water contents of these PMEA microparticles were definitely higher than that of the PMEA homopolymer (9 wt%).9 This indicates that the specific surface area of PMEA, whose outermost surface swells more easily than that of the polymer in the bulk state, increased upon forming microparticles and this promoted hydration.30,31 Subsequently, differential scanning calorimetry (DSC) measurements of the polymer microparticles were performed under wet conditions. We have previously reported that the PMEA homopolymer and its derivatives form cold-crystallizable water, called intermediate water (IW), at approximately −40 °C, which plays a key role in the blood compatibility of polymer surfaces.9,32–35 In the case of the PMEA microparticles synthesized by surfactant-free suspension polymerization, peaks ascribed to the cold crystallization of IW were observed (Fig. S2, ESI†). This result suggests that the PMEA microparticles display excellent blood compatibility similar to that of the PMEA homopolymer.
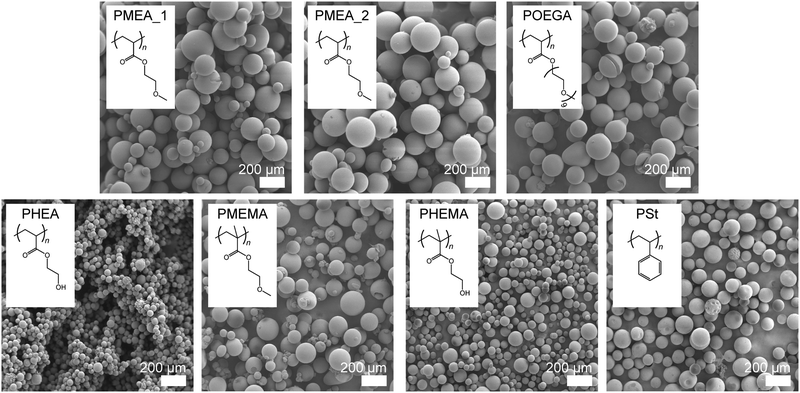 |
| | Fig. 2 SEM images of the polymer microparticles used in this study. | |
Table 2 Summary of the characterization of the microparticles used in this study
| Samples |
Diametera (μm) |
Surface areab (wet) × 10−4 (cm2 particles−1) |
Surface areac (wet) (cm2 g−1) |
Volumed (wet) × 10−6 (cm3 particles−1) |
Swelling ratioe (%) |
Water contentf (wt%) |
LogP valuesg (10−3 Å−2) |
| Dry |
Wet |
|
The diameter of the microparticles was determined as Feret's diameter by analysis of the optical microscope images of the microparticles (n = 200).
The surface area of the microparticles per particle was calculated from eqn (1).
The surface area of the microparticles per gram was calculated from eqn (5).
The volume of the microparticles per particle was calculated from eqn (2).
The swelling ratio of the microparticles was calculated form eqn (6).
The water content of the microparticles was calculated from eqn (8).
The value of the computational octanol–water partition coefficients (logP)/Connolly molecular surface area (SA). LogP and SA were calculated by Chem 3D (ver. 19.1) after MM2 minimalization of the models.13,50
|
| PMEA_1 |
172.6 ± 47.3 |
186.0 ± 40.0 |
10.86 |
32.26 |
33.68 |
125.1 |
20.1 |
−0.76 |
| PMEA_2 |
186.5 ± 38.2 |
195.0 ± 19.5 |
11.94 |
30.77 |
38.80 |
114.3 |
12.5 |
−0.76 |
| POEGA |
156.7 ± 27.5 |
235.8 ± 33.3 |
17.46 |
25.45 |
68.61 |
340.7 |
70.7 |
−3.59 |
| PHEA |
53.8 ± 9.9 |
76.2 ± 11.2 |
1.823 |
78.74 |
2.315 |
284.1 |
64.8 |
−5.07 |
| PMEMA |
117.7 ± 30.6 |
118.6 ± 34.2 |
4.417 |
50.59 |
8.730 |
102.3 |
2.3 |
1.11 |
| PHEMA |
58.1 ± 11.8 |
77.9 ± 14.4 |
1.905 |
77.02 |
2.474 |
241.0 |
58.5 |
−3.05 |
| PSt |
112.2 ± 17.5 |
111.8 ± 14.3 |
3.925 |
53.67 |
7.313 |
98.9 |
— |
24.9 |
Measurement of antithrombogenicity for the polymer microparticles
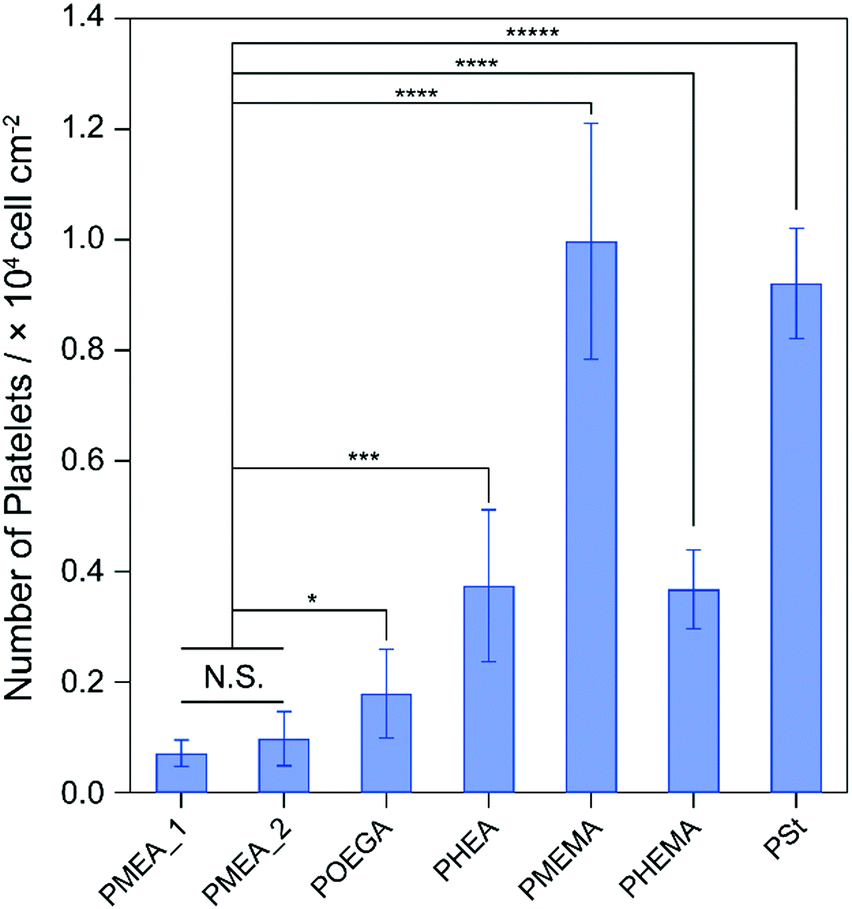
Platelets, as components of blood, play a central role in the formation of clots to maintain hemostasis.36 The adhesion and activation of platelets lead to thrombogenicity on the surface of biomaterials in contact with blood. Because the formation of thrombus on tumor-capturing devices interrupts the adhesion of tumor cells on the device surface, antithrombogenic devices are needed. To clarify the antithrombogenicity of our polymer microparticles, the adhesion of platelets to their surfaces were evaluated by the lactate dehydrogenase (LDH) method (Fig. 3).37 The numbers of platelets adhered to the PMEMA and PSt microparticles were significantly higher than that adhered to the other microparticles. The PMEA_1, PMEA_2, and PHEMA microparticles suppressed platelet adhesion on their surfaces. The number of platelets adhered to PMEA_2 was 0.098 times that of PMEMA, 0.11 times that of PSt, 0.55 times that of POEGA, and 0.26 times that of PHEA. The amount of serum proteins, such as fibronectin, vitronectin, and fibrinogen (in particular), on the surface is related to platelet adhesion and activation.38 Our previous reports demonstrated that PMEA-coated surfaces repelled fibrinogen adsorption and suppressed conformational changes induced by fibrinogen adsorbing to the surface.33,39 Kureha et al. reported that sub-micrometer particles composed of PMEA showed plasma–protein adsorption resistance on their surface.40 Furthermore, fibrinogen adsorption and platelet adhesion on the polymer-coated surface were correlated to the amount of IW in the polymers.41 Specifically, the amount of IW per gram of the polymer microparticles increased in the order of PS ≈ PMEMA ≈ PHEMA ≤ PHEA < POEGA ≤ PMEA_2 < PMEA_1 (Fig. S2, ESI†). This suggests that the PMEA microparticles have antithrombogenic properties.
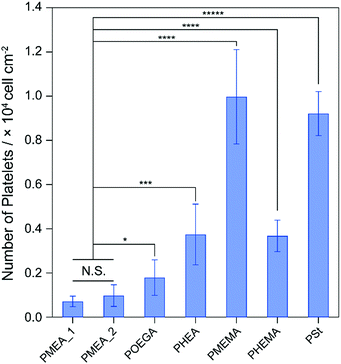 |
| | Fig. 3 Number of human platelets adhered on the polymer microparticles for 1 h of incubation. *p < 0.05; ***p < 0.005; ****p < 0.001; *****p <0.0005; N.S., not significant. | |
Interaction strength between the polymer microparticles and tumor cells
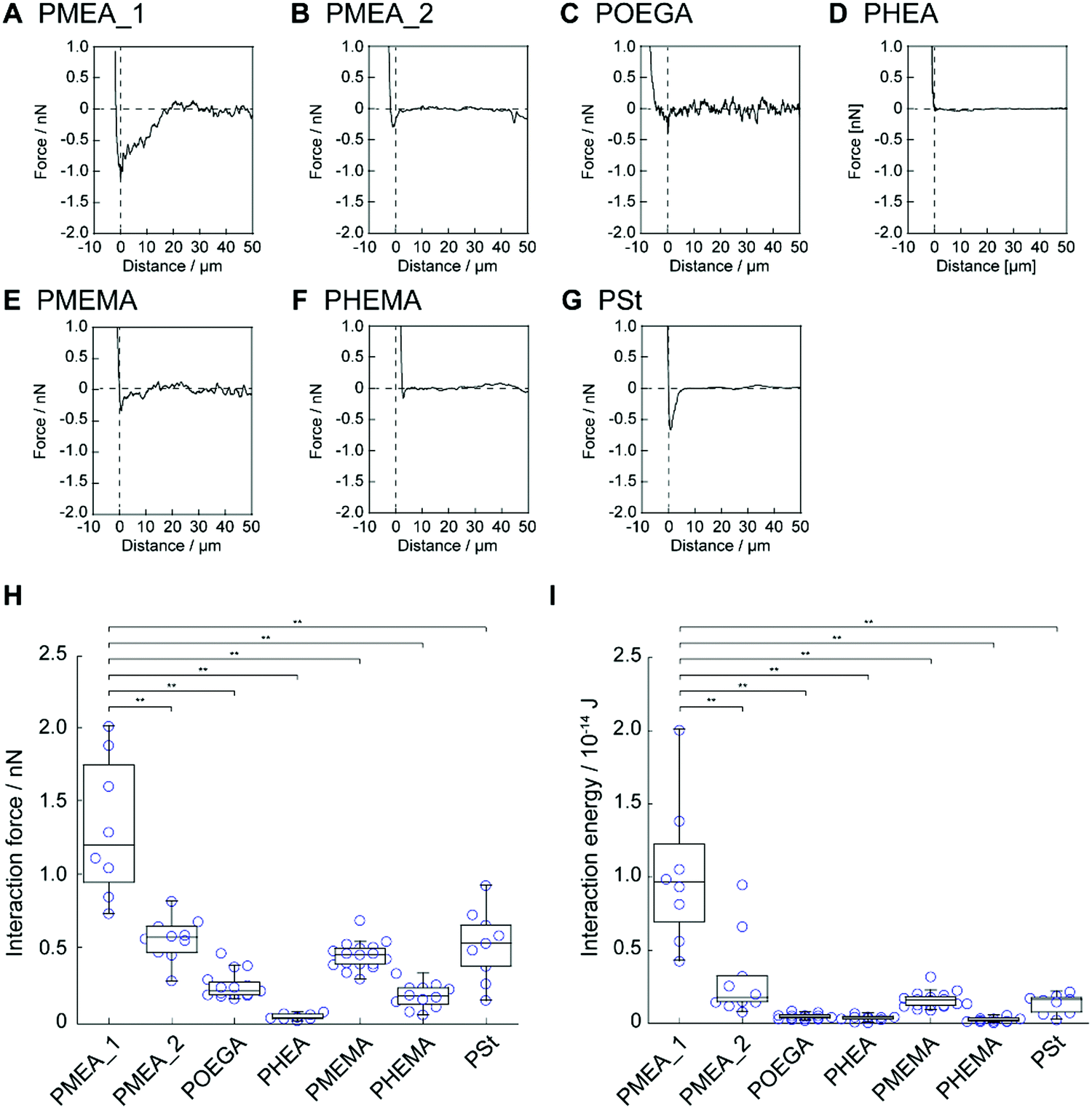
Next, we investigated the interaction strength between the polymer microparticles and tumor cells using AFM. Colloidal cantilevers composed of polymer microparticles were fabricated by immobilizing the polymer microparticles, i.e., PMEA_1, PMEA_2, POEGA, PHEA, PMEMA, PHEMA, and PSt, on a tip-less cantilever (Fig. S3, ESI†). The colloidal cantilevers interacted with human cervical adenocarcinoma HeLa cells for 1 s in the presence of serum proteins (10%). The retraction-force–distance curve was recorded for each colloidal probe (Fig. 4A–G). The force curve of PMEA_1 showed a higher force than those of the other probes; the force curves of POEGA, PHEA, and PHEMA exhibited negligible forces. The value of the minimum force was quantified as the interaction strength between the colloidal cantilevers and the cell surface (Fig. 4H). The interaction strengths of PMEA_2, PMEMA, and PSt were 0.58 ± 0.15, 0.46 ± 0.09, and 0.54 ± 0.24 nN, respectively. Remarkably, PMEA_1 showed an interaction strength of 1.33 ± 0.47 nN, resulting in its interaction with HeLa cells being stronger than those of the other colloidal cantilevers. Furthermore, the interaction energy was calculated as the area enclosed by the force curve and the baseline (Fig. 4I). The interaction energies for colloidal cantilevers other than PMEA_1 were negligibly low. These results suggest that PMEA_1 microparticles strongly interact with tumor cells. Although the adsorption and conformational changes of fibrinogen were suppressed on the PMEA-coated surface, PMEA enhanced the conformational changes induced by fibronectin adsorbing to the PMEA-coated surface.10 Because cell binding motifs, including arginine–glycine–aspartic-acid peptide, were exposed to the conformational change of fibronectin, PMEA could enhance the adhesion of tumor cells on its surface. In this regard, tumor cells with high expression levels of integrin were promoted to adhere to PMEA. The expression level of integrin in tumor cells, including HeLa cells, was relatively higher than that in normal cells.42,43 Therefore, PMEA_1 exhibited a significantly high interaction strength and energy when interacting with tumor cells. Interestingly, PMEA_2 showed a negligible interaction strength with HeLa cells. Because PMEA_2 is composed of a higher crosslinker concentration than PMEA_1, the elasticity of PMEA_2 might be higher than that of PMEA_1. Many researchers have demonstrated that the elasticity of a substrate affects the activation and internalization of integrin in adhering cells, the amount of fibronectin that can adsorb to the cell surfaces, and the conformational changes induced by the adsorbed fibronectin. As such, the elasticity of microparticles may be a key parameter in modulating their cell adhesion. However, the numbers of platelets adhered to PMEA_1 and PMEA_2 were negligibly different. The mechanism by which PMEA_1 interacted with tumor cells with high interaction strength is unclear; hence, further investigation is necessary in terms of its protein adsorption behavior.
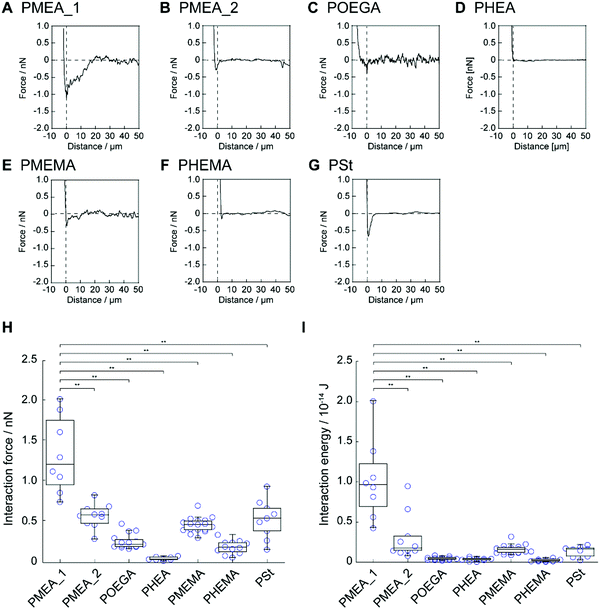 |
| | Fig. 4 Representative force–distance curves of HeLa cells interacting with colloidal cantilevers composed of (A) PMEA_1, (B) PMEA_2, (C) POEGA, (D) PHEA, (E) PMEMA, (F) PHEMA, and (G) PSt; these were recorded with the following parameters: set point = 3 nN, approach rate = 2 μm s−1, retraction rate = 5 μm s−1, and holding time = 1 s. Dot plots of (H) interaction strength and (I) energy of HeLa cells interacting with the same colloidal cantilevers (n = 8–17, **p < 0.01). | |
Capture of tumor cells using PMEA microparticles
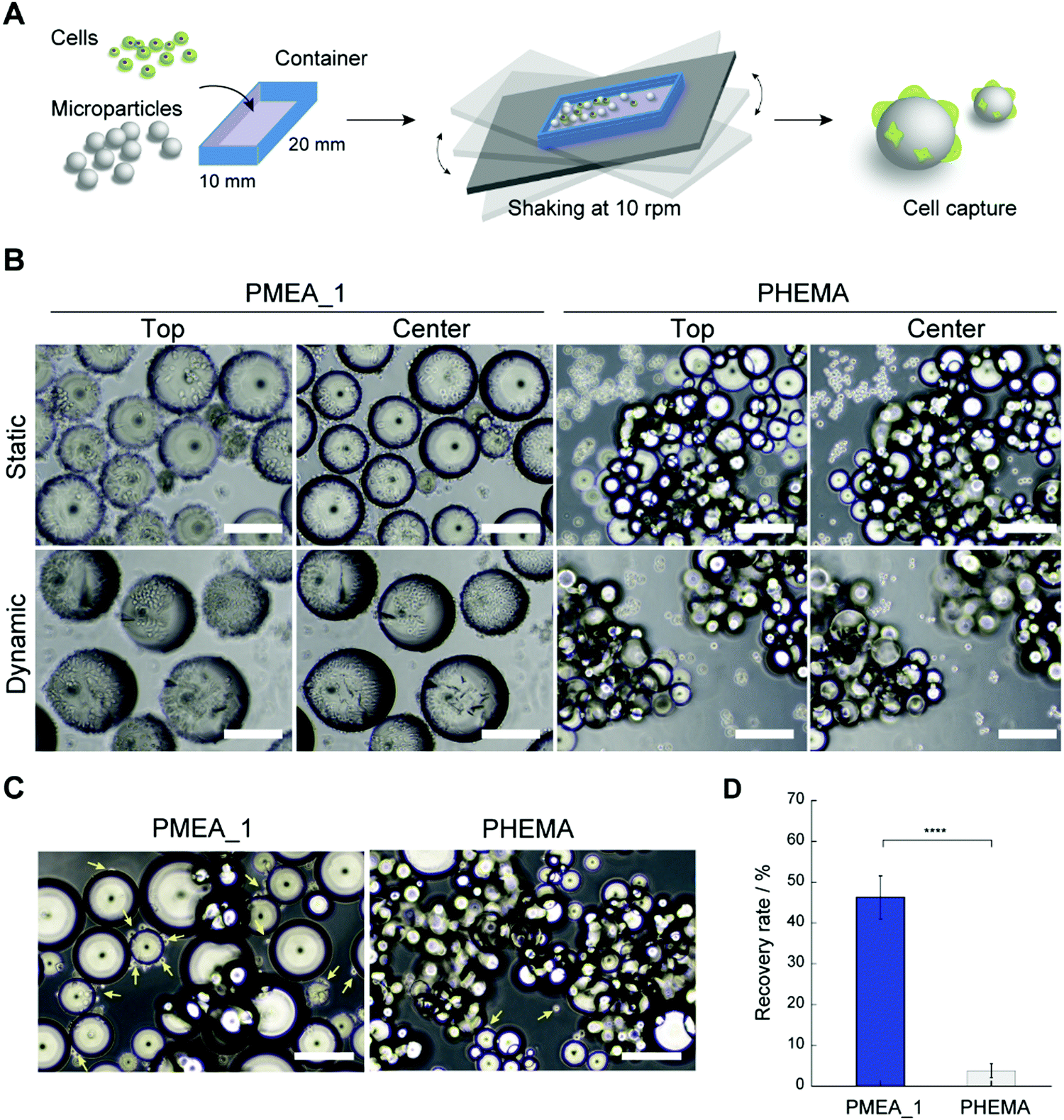
The PMEA microparticles showed low adhesion with platelets and high adhesion with tumor cells, suggesting their potential use as a CTC capture device. Hence, we investigated whether PMEA microparticles could capture tumor cells from cell suspensions. Unfortunately, when capturing CTCs from blood under static conditions, the large amount of red blood cells present obstructs the adhesion of the CTCs to the microparticles due to the precipitation of the red blood cells. However, cell capturing under dynamic conditions has been developed as a methodology to avoid such precipitation of red blood cells.44 To verify the ability of the microparticles to capture tumor cells, PMEA microparticles were shaken in tumor-cell-containing suspensions, and the number of tumor cells adhered to them was calculated (Fig. 5A). The efficacy of tumor-cell capture by the PMEA microparticles was compared to that of the control PHEMA microparticles, which showed a low adhesion of both platelets and tumor cells. The PMEA microparticles exhibited a significant number of adhered HeLa cells (EpCAM negative) on their surfaces in comparison with the other polymer microparticles after 24 h of incubation under static and dynamic conditions (Fig. 5B and Fig. S3, ESI†).45 Additionally, one of our previous reports revealed that MDA-MB-231 cells showed higher expression levels of integrin β1 than normal human fibroblasts;46 therefore, the PMEA microparticles developed here should efficiently recognize and capture MDA-MB-231 cells. In the present study, human breast tumor cells (EpCAM-negative MDA-MB-231 cell line) were used as one of model cells for testing our tumor capture technology.45,47 After incubation for 24 h, the MDA-MB-231 cells adhered to the PMEA microparticles, but only negligibly adhered to the PHEMA microparticles (Fig. 5C). The recovery rate of MDA-MB-231 cells from the cell suspensions was 12.2-fold higher when using the PMEA microparticles than when using the PHEMA microparticles (Fig. 5D).
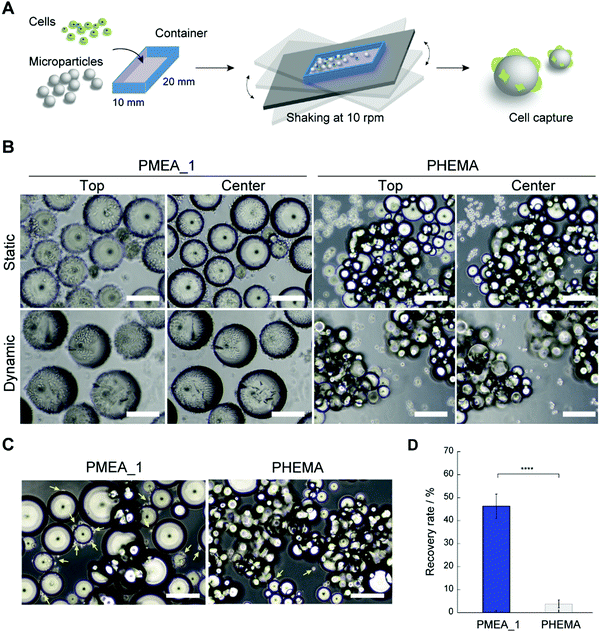 |
| | Fig. 5 (A) Diagram of the methodology for capturing tumor cells with the microparticles under dynamic conditions. (B) Phase contrast images of PMEA_1 and PHEMA microparticles treated with HeLa cells for 24 h under static conditions (Scale bars: 200 μm). (C) Phase contrast images of PMEA_1 and PHEMA microparticles treated with MDA-MB-231 cells for 24 h under static and dynamic conditions (Scale bars: 200 μm). Yellow arrows represent MDA-MB231 cells. (D) Recovery rate of MDA-MB-231 cells using PMEA_1 and PHEMA microparticles (****p < 0.0001). | |
PMEA microparticles captured tumor cells under both static and dynamic condition. Our previous reports demonstrated that PMEA-coated substrates did not cause substantial cytotoxicity under serum-free conditions, but rather improved cell viability.11 PMEA microparticles would not cause insignificant damage to captured tumor cells. On the other hand, the advantage of dynamic conditions using microparticles would be demonstrated in the presence of multiple cells, including tumor cells, platelets, and white and red blood cells. Therefore, the result that tumor cells could adhere on to the microparticles under dynamic as well as static conditions was an important consideration in the present study. The utilization of PMEA microparticles under dynamic conditions shows promise as a methodology for capturing EpCAM-positive and -negative tumor cells from whole blood without using antibodies.
CTCs are detected at a very low level in the blood from cancer patients, and form clusters to suppress anoikis and reduce shear stress in blood.48,49 In addition, blood with a specific viscosity and components affects the recovery rate of tumor cells. PMEA microparticles with a dynamic system may contribute to the capture of CTC clusters in blood with a specific viscosity because of their size (>190 μm), which was sufficiently larger than that of the cells (>10 μm) and the dynamic system was capable of changing the shaking rate. For our tumor capture microparticles to be used in clinical applications for accurately capturing rare CTCs from blood, they require further investigation; specifically, their performance in tumor patient-derived blood should be evaluated, and their tumor-cell capture efficiency should be improved.
Conclusions
In this study, we prepared microparticles composed of hydrophilic monomers via a surfactant-free suspension polymerization with an oil-in-water system, and demonstrated their efficient recovery of tumor cells through comparative studies with several other types of polymer microparticles. All the polymer microparticles types, except for commercially available PSt microparticles, were successfully prepared by the polymerization system. Among them, the PMEA microparticles showed IW formation, which suppressed the adhesion of human platelets. In particular, the PMEA microparticles with a 1% crosslinking ratio strongly interacted with the tumor cells, resulting in a high tumor-cell recovery ratio. Thus, our suspension polymerization system provides an opportunity to design and synthesize various microparticles, composed of hydrophilic monomers, whose surfaces are extremely clean. Conventional methods, such as the filtration-based system, DEPArray system, and semi-automated CellSearch system, rely on the size of the CTCs and the antibody to antigens expressed on the CTCs. Meanwhile, the developed PMEA microparticles with a dynamic system have great potential to selectively capture tumor cells from blood without using antibodies, regardless of their size. We believe that they can be applied in the diagnosis of tumor progression.
Experimental
Materials
Solvents of analytical grade were used unless otherwise stated. Methanol and hydrochloric acid (12 M HCl) were purchased from Kanto Chemicals Co., Ltd, (Japan). Calcium chloride dihydrate, calcium carbonate, and 2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile) (V-70) were purchased from FUJIFILM Wako Pure Chemical Co., Ltd (Japan). Oligo(ethylene glycol) methyl ether acrylate (OEGA) (Mn = 480 g mol−1) was purchased from Merck/Sigma-Aldrich (Germany). Cyclohexanol was purchased from Nacalai Tesque Inc. (Japan). 2-Methoxyethyl acrylate (MEA), 2-hydroxyethyl acylate (HEA), 2-methoxyethyl methacrylate (MEMA), 2-hydroxyethyl methacrylate (HEMA), diethylene glycol diacrylate (DEGDA), diethylene glycol dimethacrylate (DEGDMA), 2,2′-azobis(isobutyronitrile) (AIBN), 2,2,2-tirfluoroethanol (TFE), and polystyrene (PSt) resin (1% divinylbenzene, 100–200 mesh) were purchased from Tokyo Chemical Industry Co., Ltd (Japan). PSt resin was purified by Soxhlet extraction with methanol for 3 days prior to use.
Measurements
The FTIR spectra were acquired by an FT/IR-6600 system (JASCO Co., Japan) using a deuterated l-alanine triglycine sulfate detector fitted with an attenuated total reflection (ATR) accessory that used a germanium internal reflection element (resolution: 4 cm−1, 16 scans). The XPS analyses were carried out with an ULVAC-PHI APEX ESCA system (ULVAC-PHI inc., Japan) at 2.0 × 107 Pa using a monochromatic Al-Kα X-ray source at 150 W. The SEM images were obtained using a Real Surface View VE-7800 system (KEYENCE Co., Japan). The optical microscope images were collected with a VHX-900F system (KEYENCE Co., Japan).
Preparation of poly(2-methoxyethyl acrylate) (PMEA) microparticles
MEA (25 g, 0.19 mol), DEGDA (0.42 g, 1 mol% or 0.84 g, 2 mol%), and AIBN (0.10 g, 0.61 mmol) were mixed. Calcium chloride dihydrate (80 g, 0.54 mol) was dissolved in water (200 mL), and calcium carbonate (10 g, 0.10 mol) was added as a dispersion stabilizer. These solutions were combined and heated to 60 °C with stirring at 300 rpm using a propeller agitator under an argon atmosphere. After 30 min, the suspension solution was allowed to react for 1 h at 85 °C. Hydrochloric acid (12 M, 20 mL) was slowly added to the reaction mixture to decompose calcium carbonate into calcium chloride and carbon dioxide. The resultant PMEA microparticles were purified by Soxhlet extraction with methanol over 2 d. The purified PMEA microparticles were sorted into two size-based groups by wet-type classification using a sieve (mesh opening: 300 μm) with methanol as a solvent, and then lyophilized after replacement with water to give the pure PMEA microparticles. The chemical structures of the particles were evaluated by FTIR and XPS analyses.
[Crosslinking ratio = 1%] yield: 16.4 g (64.5%); 11.2 g (smaller-size group), 5.2 g (larger-size group).
[Crosslinking ratio 2%] yield: 19.1 g (73.9%); 15.6 g (smaller-size group), 3.5 g (larger-size group).
Preparation of poly(oligo(ethylene glycol)methyl ether acrylate) (POEGA) microparticles
OEGA (25 g, 0.052 mol), DEGDA (0.11 g, 1 mol%), and AIBN (0.10 g, 0.61 mmol) were mixed with cyclohexanol (2.5 mL). Calcium chloride dihydrate (190 g, 1.09 mol) was dissolved in water (110 mL), and then calcium carbonate (10 g, 0.10 mol) was added as a dispersion stabilizer. These solutions were combined and heated to 60 °C with stirring at 300 rpm using a propeller agitator under an argon atmosphere. After 30 min, the suspension solution was allowed to react for 2 h at 85 °C. Hydrochloric acid (12 M, 20 mL) was slowly added to the reaction mixture to decompose calcium carbonate into calcium chloride and carbon dioxide. The resultant POEGA microparticles were purified by Soxhlet extraction with methanol for 2 d. The purified POEGA microparticles were sorted into two size-based groups by wet-type classification using a sieve (mesh opening: 300 μm) with methanol as a solvent, and then lyophilized after replacement with water to give the pure POEGA microspheres. The chemical structures of the particles were evaluated by FTIR and XPS analyses.
Yield: 12.6 g (50.2%); 4.3 g (smaller-size group), 8.3 g (larger-size group).
Preparation of poly(2-hydroxyethyl acrylate) (PHEA) microparticles
HEA (25 g, 0.22 mol), DEGDA (0.52 g, 1 mol%), and V-70 (0.20 g, 0.65 mmol) were mixed with TFE (10 mL). Calcium chloride dihydrate (150 g, 1.02 mol) was dissolved in water (150 mL), and then calcium carbonate (10 g, 0.10 mol) was added as a dispersion stabilizer. These solutions were combined and heated to 40 °C with stirring at 300 rpm using a propeller agitator under an argon atmosphere. After 30 min, the suspension solution was allowed to react for 1 h at 60 °C. Hydrochloric acid (12 M, 20 mL) was slowly added to the reaction mixture to decompose calcium carbonate into calcium chloride and carbon dioxide. The resultant PHEA microparticles were purified by Soxhlet extraction with methanol for 2 d. The purified PHEA microparticles were sorted into two size-based groups by wet-type classification using a sieve (mesh opening: 300 μm) with methanol as a solvent, and then lyophilized after replacement with water to give the pure PHEA microspheres. The chemical structures of the particles were evaluated by FTIR and XPS analyses.
Yield: 15.6 g (61.1%); 13.2 g (smaller-size group), 2.4 g (larger-size group).
Preparation of poly(2-methoxyethyl methacrylate) (PMEMA) microparticles
MEMA (25 g, 0.17 mol), DEGDMA (0.42 g, 1 mol%), and AIBN (0.10 g, 0.61 mmol) were mixed. Calcium chloride dihydrate (80 g, 0.54 mol) was dissolved in water (220 mL), and then calcium carbonate (10 g, 0.10 mol) was added as a dispersion stabilizer. These solutions were combined and heated to 60 °C with stirring at 450 rpm using a propeller agitator under an argon atmosphere. After 30 min, the suspension solution was allowed to react for 1 h at 85 °C. Hydrochloric acid (12 M, 20 mL) was slowly added to the reaction mixture to decompose calcium carbonate into calcium chloride and carbon dioxide. The resultant PMEMA microparticles were purified by Soxhlet extraction with methanol for 2 d. The purified PMEMA microparticles were sorted into two size-based groups by wet-type classification using a sieve (mesh opening: 300 μm) with methanol as a solvent, and then lyophilized after replacement with water to give the pure PMEMA microspheres. The chemical structures of the particles were evaluated by FTIR and XPS analyses.
Yield: 46.4 g (61.1%); 10.2 g (smaller-size group), 1.6 g (larger-size group).
Preparation of poly(2-hydroxyethyl methacrylate) (PHEMA) microparticles
HEMA (25 g, 0.19 mol), DEGDMA (0.47 g, 1 mol%), and AIBN (0.10 g, 0.61 mmol) were mixed. Calcium chloride dihydrate (190 g, 0.109 mol) was dissolved in water (220 mL), and then calcium carbonate (10 g, 0.10 mol) was added as a dispersion stabilizer. These solutions were combined and heated to 60 °C with stirring at 300 rpm using a propeller agitator under an argon atmosphere. After 30 min, the suspension solution was allowed to react for 3 h at 85 °C. Hydrochloric acid (12 M, 20 mL) was slowly added to the reaction mixture to decompose calcium carbonate into calcium chloride and carbon dioxide. The resultant PHEMA microparticles were purified by Soxhlet extraction with methanol for 2 d. The purified PHEMA microparticles were sorted into two size-based groups by wet-type classification using a sieve (mesh opening: 300 μm) with methanol as a solvent, and then lyophilized after replacement with water to give the pure PHEMA microspheres. The chemical structures of the particles were evaluated by FTIR and XPS analyses.
Yield: 13.5 g (53.0%; smaller-size group only).
Calculation of the surface areas, volumes, swelling ratios, and water contents of the polymer microparticles
The surface areas and volumes of the polymer microparticles under wet conditions (per particle) are given by the following formulae:andwhere Swet (cm2 particles−1) is the surface area of the polymer microparticles per particle under wet conditions, Vwet (cm3 particles−1) is the volume of polymer microparticles per particle under wet conditions, and rwet (cm) is the radius of the polymer microparticles estimated from the optical microscope images. Assuming that all the densities are the same (i.e., 1 g cm−3), the relationship between the volume and weight is given by the following formulae:andwhere Vdry (cm3 particles−1) is the volume of the microparticles per particle under dry conditions; Wdry and Wwet (g particles−1) are the weights of the microparticles per particle under dry and wet conditions, respectively. From eqn (1)–(3), the surface area per particle can be translated to the surface area per gram.| | 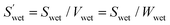 | (5) |
where 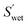 (cm2 g−1) is the surface area of the microparticles per gram under wet conditions. In addition, the swelling ratio of the microparticles is expressed below, derived from eqn (3) and (4).As mentioned above, if the volume is equal to the weight, the amount of water in the microparticles is determined as follows:
(cm2 g−1) is the surface area of the microparticles per gram under wet conditions. In addition, the swelling ratio of the microparticles is expressed below, derived from eqn (3) and (4).As mentioned above, if the volume is equal to the weight, the amount of water in the microparticles is determined as follows:| | | Vwet − Vdry = Vwater = Wwater | (7) |
where Vwater (cm2 particles−1) is the volume of water in the microparticles, and Wwater (g particles−1) is the weight of the water in the microparticles. Therefore, the water content of the microparticles (wt%) can be defined by the following equation:| | | Vwater/Vwet × 100% = Wwater/Vwet × 100% | (8) |
Hydration state analysis for the polymer microparticles using DSC measurements
The hydration state in the hydrated polymer microparticles under wet conditions (in PBS(−)) was analyzed by differential scanning calorimetry (DSC) (X-DSC7000, Seiko Instruments, Japan) with reference to a previously reported method.35 Dry polymer samples were prepared by storing them under vacuum conditions for more than 7 d. Saturated hydration polymer samples were prepared by immersing the dry samples in PBS(−) for more than 3 d. The samples were cooled to −100 °C at a rate of 5 °C min−1, held at −100 °C for 5 min, and then heated to 50 °C at the same rate under a nitrogen purge flow. The amount of IW per gram of the polymer microparticles (WIW (mg g−1)) was calculated using the following equation:| | | WIW = (ΔHcc/ΔHfus)/Wpolymer × 1000, | (9) |
where ΔHcc (J) is the enthalpy change during the cold crystallization of water estimated from the DCS thermogram, and ΔHfus (J g−1) is the heat of fusion of ice (334 J g−1).
Platelet adhesion assay
The polymer microparticles (50 mg) were incubated with PBS for 1 h at 37 °C. Platelets derived from human whole blood (Tennessee Blood Services, USA; density = 4 × 107 cells) were treated with the polymer microparticles for 1 h at 37 °C. The solution was then centrifuged at 1000 rpm for 1 min. The supernatant was removed and the microparticles were washed twice with PBS. The platelet-adhered microparticles were treated with an LDH assay kit (Dojindo, Kumamoto, Japan). After 1 h of incubation, the absorbance of the cells was measured at 490 nm using an Infinite 200PRO M Plex microplate reader (Tecan, Zürich, Switzerland).
Fabrication of the colloidal cantilever
A tip-less cantilever (TL-CONT, spring constant = 0.2 N m−1, NANOSENSORS) was equipped on an atomic force microscope (CellHesion 200, Bruker). A small amount of glue (BOND ULTRA TAYOTO SU Premium SOFT, Konishi Co., Ltd) was placed on the edge of the cantilever. The glue-treated tip-less cantilever interacted with the polymer microparticles, which were spread on a glass slide with cellophane tape. The cantilever approached one microparticle and the particle was pressed for 10 min. The microparticle-attached cantilevers were dried for 1 d at ambient pressure and room temperature to obtain colloidal cantilevers.
Analysis of the interaction strength between the polymer microparticles and cells
The colloidal cantilevers were incubated with DMEM (Wako) containing 10% FBS for 24 h at 37 °C. HeLa cells (1.0 × 104 cells cm−2) (RIKEN BRC, Japan) were seeded on a 35 mm dish and incubated for 24 h. The force curves between the polymer microparticles and cells were recorded using an AFM system (CellHesion 200, JPK) equipped with a colloidal cantilever (set point = 3 nN, approach rate = 2.0 μm s−1, holding time = 1 s, retraction rate = 5 μm s−1). The interaction force was defined as the maximum force for the detachment of the colloidal cantilever from the cell, corresponding to the force at the minimum point of the retraction curve. The interaction energy was estimated as the amount of work required to detach the cells from the substrate, corresponding to the area enclosed by the baseline and retraction curve.
Capture of tumor cells from cell suspensions using the polymer microparticles
The polymer microparticles (50 mg) were incubated with PBS for 1 h at 37 °C. The glass bottom area of the cell culture container (IWAKI) was coated with poly(2-methacryloyoxyethyl phosphorylcholine-co-n-butyl methacrylate) (PMPC; 30:70 mol%, Lipidure-CM5206, NOF Corporation, Tokyo, Japan). HeLa (5.0 × 104 cells) or MDA-MB-231 (ATCC) cells (2.0 × 104 cells) were treated with the polymer microparticles (50 mg) in the container. After 24 h of incubation under shaking conditions (10 rpm), the polymer microparticles were washed with PBS twice and treated with the Cell Courting kit-8 (Dojindo). The absorbance of the cells was measured at 560 nm using an Infinite 200PRO M Plex microplate reader (Tecan, Zürich, Switzerland). The recovery rate of tumor cells was calculated from the ratio of the number of seeded tumor cells to the number of polymer microparticles. HeLa cells were stained with Tracker Green (Thermo Fisher Scientific).
Author contributions
S. N. and K. N. conceived the research and designed the experiments. S. N., S. S., and K. N. performed the experiments. M. T. and K. N., and S. N. obtained founding for the project. M. T. and K. N. oversaw the research. S. N., S. S., K. N., and M. T. co-wrote the manuscript. All authors discussed the results and commented on the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research (KAKENHI) (JP19H05720 to M. T., JP20J00282 and JP20K20201 to S. N., JP19J00686 and 21K18066 to K. N.) from the Japan Society for the Promotion of Science (JSPS), “Dynamic Alliance Open Innovation Bridging Human, Environment and Materials” from the Ministry of Education, Culture, Sports, and Technology of Japan (MEXT). This research was also supported by Grant-in-Aid for the Nakayama Foundation for Human Science (to S. N.), the Japan Prize Foundation (to S. N.) and Fukuoka Public Health Promotion Organization Cancer Research Fund (to K. N.).
References
- P. Paterlini-Brechot and N. L. Benali, Cancer Lett., 2007, 253, 180–204 CrossRef CAS PubMed.
- M. D. Trapani, N. Manaresi and G. Medoro, Cytometry, Part A, 2018, 93, 1260–1266 CrossRef PubMed.
- M. C. Miller, P. S. Robinson, C. Wagner and D. J. O'Shannessy, Cytometry, Part A, 2018, 93, 1234–1239 CrossRef PubMed.
- H. Huebner, P. A. Fasching, W. Gumbrecht, S. Jud, C. Rauh, M. Matzas, P. Paulicka, K. Friedrich, M. P. Lux, B. Volz, P. Gass, L. Haberle, F. Meier-Stiegen, A. Hartkopf, H. Neubauer, K. Almstedt, M. W. Beckmann, T. N. Fehm and M. Ruebner, BMC Cancer, 2018, 18, 204 CrossRef PubMed.
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–1239 CrossRef CAS PubMed.
- F. Farace, C. Massard, N. Vimond, F. Drusch, N. Jacques, F. Billiot, A. Laplanche, A. Chauchereau, L. Lacroix, D. Planchard, S. Le Moulec, F. Andre, K. Fizazi, J. C. Soria and P. Vielh, Br. J. Cancer, 2011, 105, 847–853 CrossRef CAS PubMed.
- T. M. Gorges, I. Tinhofer, M. Drosch, L. Rose, T. M. Zollner, T. Krahn and O. von Ahsen, BMC Cancer, 2012, 12, 178 CrossRef CAS PubMed.
- T. Hoshiba, M. Nikaido and M. Tanaka, Adv. Healthcare Mater., 2014, 3, 775–784 CrossRef CAS PubMed.
- M. Tanaka, S. Kobayashi, D. Murakami, F. Aratsu, A. Kashiwazaki, T. Hoshiba and K. Fukushima, Bull. Chem. Soc. Jpn., 2019, 92, 2043–2057 CrossRef CAS.
- T. Hoshiba, E. Nemoto, K. Sato, T. Orui, T. Otaki, A. Yoshihiro and M. Tanaka, PLoS One, 2015, 10, e0136066 CrossRef PubMed.
- K. Nishida, S. Sekida, T. Anada and M. Tanaka, ACS Biomater. Sci. Eng., 2022, 8, 672–681, DOI:10.1021/acsbiomaterials.1c01469.
- K. Nishida, S. N. Nishimura and M. Tanaka, Biomacromolecules, 2022, 23(4), 1569–1580, DOI:10.1021/acs.biomac.1c01343.
- K. Nishida, T. Anada, S. Kobayashi, T. Ueda and M. Tanaka, Acta Biomater., 2021, 134, 313–324 CrossRef CAS PubMed.
- S. M. Badenes, T. G. Fernandes, C. A.-V. Rodrigues, M. M. Diogo and J. M.-S. Cabral, J. Biotechnol., 2016, 234, 71–82 CrossRef CAS PubMed.
- S. Derakhti, S. H. Safiabadi-Tali, G. Amoabediny and M. Sheikhpour, Mater. Sci. Eng., C, 2019, 103, 109782 CrossRef CAS PubMed.
- K. Nagase, J. Kobayashi, A. Kikuchi, Y. Akiyama, H. Kanazawa and T. Okano, Biomaterials, 2011, 32, 619–627 CrossRef CAS PubMed.
- I. A. Ly and R. I. Mishell, J. Immunol. Method., 1974, 5, 239–247 CrossRef CAS PubMed.
- A. Palmon, R. David, Y. Neumann, R. Stiubea-Cohen, G. Krief and D. J. Aframian, Methods, 2012, 56, 305–309 CrossRef CAS PubMed.
- A. L. van Wezel, Nature, 1967, 216, 64–65 CrossRef CAS PubMed.
- D. Smith, C. Herman, S. Razdan, M. R. Abedin, W. V. Stoecker and S. Barua, ACS Appl. Bio Mater., 2019, 2, 2791–2801 CrossRef CAS PubMed.
- H. G. Yuan, G. Kalfas and W. H. Ray, J. Macromol. Sci., Part C: Polym. Rev., 1991, 31, 215–299 CrossRef.
- E. Vivaldo-Lima, P. E. Wood and A. E. Hamielec, Ind. Eng. Chem. Res., 1997, 36, 939–965 CrossRef CAS.
-
M. C.-C. Pinto, J. G.-F. Santos, F. Machado and J. C. Pinto, Encyclopedia of Polymer Science and Technology, John Wiley & Sons, Inc., 2013, pp. 1–31 DOI:10.1002/0471440264.pst597.
- M. V. Dimonie, C. M. Boghina, N. N. Marinescu, M. M. Marinescu, C. I. Cincu and C. G. Oprescu, Eur. Polym. J., 1982, 18, 639–645 CrossRef CAS.
- P. J. Dowding, B. Vincent and E. Williams, J. Colloid Interface Sci., 2000, 221, 268–272 CrossRef CAS PubMed.
- G. Wang, M. Li and X. Chen, J. Appl. Polym. Sci., 1997, 65, 789–794 CrossRef CAS.
- G. Okudaira, K. Kamogawa, T. Sakai, H. Sakai and M. Abe, J. Oleo Sci., 2003, 52, 167–170 CrossRef CAS.
- M. Kiremitçi and H. Çukurova, Polymer, 1992, 33, 3257–3261 CrossRef.
- A. Bukowska, W. Bukowski and J. Noworól, J. Appl. Polym. Sci., 2006, 101, 1487–1493 CrossRef CAS.
- T. Hirata, H. Matsuno, D. Kawaguchi, N. L. Yamada, M. Tanaka and K. Tanaka, Phys. Chem. Chem. Phys., 2015, 17, 17399–17405 RSC.
- D. Murakami, K. Yamazoe, S. N. Nishimura, N. Kurahashi, T. Ueda, J. Miyawaki, Y. Ikemoto, M. Tanaka and Y. Harada, Langmuir, 2022, 38, 1090–1098 CrossRef CAS PubMed.
- M. Tanaka, T. Motomura, N. Ishii, K. Shimura, M. Onishi, A. Mochizuki and T. Hatakeyama, Polym. Int., 2000, 49, 1709–1713 CrossRef CAS.
- M. Tanaka, T. Motomura, M. Kawada, T. Anzai, Y. Kasori, T. Shiroya, K. Shimura, M. Onishi and A. Mochizuki, Biomaterials, 2000, 21, 1471–1481 CrossRef CAS PubMed.
- S. Nishimura, T. Ueda, S. Kobayashi and M. Tanaka, ACS Appl. Polym. Mater., 2020, 2, 4790–4801 CrossRef CAS.
- S. Nishimura, T. Ueda, D. Murakami and M. Tanaka, Org. Mater., 2021, 3, 214–220 CrossRef CAS.
- R. K. Andrews and M. C. Berndt, Thrombo. Res., 2004, 114, 447–453 CrossRef CAS PubMed.
- W.-B. Tsai, J. M. Grunkemeier and T. A. Horbett, J. Biomed. Mater. Res., 1999, 44, 130–139 CrossRef CAS PubMed.
- W.-B. Tsai, J. M. Grunkemeier, C. D. McFarland and T. A. Horbett, J. Biomed. Mater. Res., 2002, 60, 348–359 CrossRef CAS PubMed.
- M. Tanaka, A. Mochizuki, T. Motomura, K. Shimura, M. Onishi and Y. Okahata, Colloids Surf., A, 2001, 193, 145–152 CrossRef CAS.
- T. Kureha, S. Hiroshige, S. Matsui and D. Suzuki, Colloids Surf., B, 2017, 155, 166–172 CrossRef CAS PubMed.
- K. Sato, S. Kobayashi, M. Kusakari, S. Watahiki, M. Oikawa, T. Hoshiba and M. Tanaka, Macromol. Biosci., 2015, 15, 1296–1303 CrossRef CAS PubMed.
- T. Riikonen, P. Vihinen, M. Potila, W. Rettig and J. Heino, Biochem. Biophys. Res. Commun., 1995, 209, 205–212 CrossRef CAS PubMed.
- S. Maschler, G. Wirl, H. Spring, D. V. Bredow, I. Sordat, H. Beug and E. Reichmann, Oncogene, 2005, 24, 2032–2041 CrossRef CAS PubMed.
- A. A. Bhagat, H. W. Hou, L. D. Li, C. T. Lim and J. Han, Lab Chip, 2011, 11, 1870–1878 RSC.
- L. Shi, K. Wang and Y. Yang, Colloids Surf., B, 2016, 147, 291–299 CrossRef CAS PubMed.
- S. Nishimura, K. Nishida and M. Tanaka, Chem. Commun., 2022, 58, 505–508 RSC.
- T. Ohnaga, Y. Shimada, K. Takata, T. Obata, T. Okumura, T. Nagata, H. Kishi, A. Muraguchi and K. Tsukada, Mol. Clin. Oncol., 2016, 4, 599–602 CrossRef CAS PubMed.
- N. Aceto, A. Bardia, D. T. Miyamoto, M. C. Donaldson, B. S. Wittner, J. A. Spencer, M. Yu, A. Pely, A. Engstrom, H. Zhu, B. W. Brannigan, R. Kapur, S. L. Stott, T. Shioda, S. Ramaswamy, D. T. Ting, C. P. Lin, M. Toner, D. A. Haber and S. Maheswaran, Cell, 2014, 158, 1110–1122 CrossRef CAS PubMed.
- R. Taftaf, X. Liu, S. Singh, Y. Jia, N. K. Dashzeveg, A. D. Hoffmann, L. El-Shennawy, E. K. Ramos, V. Adorno-Cruz, E. J. Schuster, D. Scholten, D. Patel, Y. Zhang, A. A. Davis, C. Reduzzi, Y. Cao, P. D'Amico, Y. Shen, M. Cristofanilli, W. A. Muller, V. Varadan and H. Liu, Nat. Commun., 2021, 12, 4867 CrossRef CAS PubMed.
- A. J.-D. Magenau, J. A. Richards, M. A. Pasquinelli, D. A. Savin and R. T. Mathers, Macromolecules, 2015, 48, 7230–7236 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00129b |
| ‡ Present address: Faculty of Science and Engineering, Department of Molecular Chemistry and Biochemistry, Doshisha University, Japan. |
| § Present address: Department of Life Science and Technology, School of Life Science and Technology, Tokyo Institute of Technology, Japan. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Kei
Nishida§
,
Kei
Nishida§

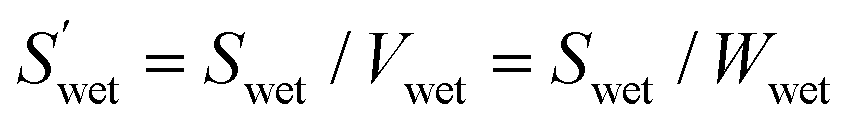
 (cm2 g−1) is the surface area of the microparticles per gram under wet conditions. In addition, the swelling ratio of the microparticles is expressed below, derived from eqn (3) and (4).
(cm2 g−1) is the surface area of the microparticles per gram under wet conditions. In addition, the swelling ratio of the microparticles is expressed below, derived from eqn (3) and (4).