
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced CO2 sorption in a hybrid PEI–Mo oxide film via pulsed electrodeposition†
Mohammad
Tanhaei
 ab,
Ming
Yang
c,
Jayce J. W.
Cheng
a,
Yi
Ren
a,
Arash
Nemati
a,
Jisheng
Pan
a and
Sing Yang
Chiam
*a
ab,
Ming
Yang
c,
Jayce J. W.
Cheng
a,
Yi
Ren
a,
Arash
Nemati
a,
Jisheng
Pan
a and
Sing Yang
Chiam
*a
aInstitute of Materials Research and Engineering, Agency for Science, Technology and Research, 2 Fusionopolis Way, 138634, Singapore. E-mail: chiamsy@imre.a-star.edu.sg; Fax: +65 64168964; Tel: +65 64168964
bDivision of Physics and Applied Physics, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, 637371, Singapore
cDepartment of Applied Physics, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong SAR, China
First published on 28th May 2022
Abstract
Solid sorbents for carbon dioxide (CO2) capture remain both a scientifically challenging and a technologically important area of work. The use of the right material and the understanding of its microstructure are critical for boosting the sorption performance. In this work, we show that the incorporation of PEI in Mo oxide via the introduction of a rejection potential in pulsed electrodeposition can create a hybrid film with an enhanced CO2 sorption performance. By controlling the PEI incorporation and studying the interaction between PEI and Mo oxide, we suggest that the improvements seen can be attributed to the role of PEI as an active surfactant for CO2 sorption, enabling the enhanced microstructure and the resultant CO2 sorption performance.
1. Introduction
One of the most effective and mature CO2 capture techniques at an industrial scale is aqueous amine sorption. However, the technique has its challenges such as high cost (due to equipment corrosion), a high regeneration energy consumption, potential pollution and toxicity.1,2 Alternatively, solid sorbents for CO2 capture that utilize physical sorption via materials such as MOFs, zeolites, activated carbon, and porous silica are potential competing or complementary solutions.3 It has also been reported that their performance at low CO2 partial pressures can be tuned and enhanced via functionalization with active sorbents like amines.4,5 Generally, sorption materials require the combination of high surface area and porosity, coupled with high selectivity and a high regeneration ability for CO2 capture.6–9Compared with more traditional amine-based techniques, solid sorbents are attractive candidates for CO2 sorption since they offer potentially lower energy and regeneration requirements,10 with tunable selectivity,11,12 and can be a retrofittable add-on for CO2 removal.10 They also offer potentially better operational benefits such as lower corrosion, degradation, or detrimental environmental concerns.13,14 However, they do suffer from a lower adsorption capacity, potential pore blockages by water vapor under humid conditions, and poor CO2/N2 selectivity. A hybrid polymer–inorganic sorbent might provide a promising solution to those limitations,15 where surface functionalization with amino groups can yield improved efficiency. With silica-supported polyethyleneimine (PEI) sorbents, increasing the PEI loading has been shown to improve the CO2 capture performance initially, although this results in the possible agglomeration of particles and poorer dispersion that leads to a decrease in the performance with higher loadings.16,17 The molecular weight of PEI also affects the pore sizes, and CO2 sorption rates tend to be lower with any increased molecular weight of PEI used.6 These studies have shown that, besides being an active sorbent, PEI influences the microstructure and plays a critical role in affecting the diffusion of the CO2 gas from the surface to the bulk. This quality remains important and amines can also dynamically change the sorbent viscosity through intermolecular cross-linking between the PEI chains during sorption.6,18
In the silica-supported PEI, the silica primarily provides both an increased surface area and mechanical support for the active sorbent. A recent study in thin-film sorption showed the performance of thin films comprising magnesium and a metal–organic framework (MOF), synthesized via vapor-assisted crystallization, with the film thicknesses ranging from 55 to 1300 nm. CO2 sorption measurements using a QCM device show a similar uptake (∼7.7 mmol g−1) at 1 bar for all the films of different thicknesses.19 High-performing thin films are possible because dead weights are prevented, and our previous studies have demonstrated the high-performing dye-sorption capability of Fe oxide thin films,20 as well as, more recently, a high-performing carbon dioxide (CO2) sorption Mo oxide thin film.21 More importantly, the supported-substrate approach enables detailed studies on the chemical nature of the materials with the sorbents, which is useful for the further design and tuning of materials. Amine is one of the best-performing commercial chemisorption materials. For supported PEI studies, PEI typically yields 7–10 mmol gPEI−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22–24 for the material itself, although the overall performance is lower due to the presence of the support and the increasing gel viscosity upon sorption with a higher PEI loading.23 The use of PEI loading in a coating context has also been demonstrated by Krishnamurthy et al., where silica–PEI and silica–TEPA were incorporated as pigments into a paint formulation for controlling indoor air quality. They reported a promising adsorption capacity of 1 mmol g−1 at 800 ppm of CO2 and 15% RH, even at a significant film thickness of 55 μm.25
22–24 for the material itself, although the overall performance is lower due to the presence of the support and the increasing gel viscosity upon sorption with a higher PEI loading.23 The use of PEI loading in a coating context has also been demonstrated by Krishnamurthy et al., where silica–PEI and silica–TEPA were incorporated as pigments into a paint formulation for controlling indoor air quality. They reported a promising adsorption capacity of 1 mmol g−1 at 800 ppm of CO2 and 15% RH, even at a significant film thickness of 55 μm.25
In this work, we explore the PEI–Mo oxide hybrid film, in particular on how incorporating PEI results in microstructure changes and effects the CO2 sorption performance. The supported-substrate approach enables us to study the chemical nature of the deposited materials and their interaction with the support substrate. We utilize electrodeposition (ED) as it is an excellent coating process that provides control over the thickness and homogeneity of the films. It is simple, economic, versatile and, most importantly, is scalable, even for industrial-type applications.26,27 One persistent challenge of electrodeposition for such a hybrid film is the ability to incorporate non-conducting materials such as PEI, and this greatly restricts its versatility. Here, we introduce a facile protocol and method, to demonstrate how to overcome this hurdle and hence present a route for depositing such non-conducting materials via the electrodeposition process. The protocol opens up the electrodeposition method to the possibilities of varied combinations of both conducting and non-conducting hybrid films. Subsequently, we will also examine the resultant hybrid film properties and examine its CO2 sorption performance and provide an understanding of the hybrid film's evolution and performance through both theoretical calculations and experimental validation.
2. Experimental section
2.1 Electrochemical deposition
All chemicals used in this work, including sodium molybdate dihydrate (Na2MoO4·2H2O), poly(ethyleneimine) solution (Mw ≈ 2000), and sulfuric acid (H2SO4), acetone and isopropanol, were purchased from Sigma-Aldrich; they are of analytical grade and were used as received without further purification. The two main electrolytes used for electrodeposition in this work are: (1) 20 mM sodium molybdate dihydrate in deionized water, and (2) 20 mM sodium molybdate dihydrate with 0.25 wt% PEI in deionized water. All the solutions are acidified to a pH of 5.5–6 using dilute sulfuric acid. Constant potential (CP) and pulse potential electrodeposition (PPED) methods were used for the deposition of the films on conductive ITO/glass substrates. Cyclic voltammetry (CV) was utilized to study the oxidation and reduction reactions in the respective electrolytes. Substrates were cleaned with acetone, isopropanol (IPA) and deionized water before electrodeposition. The electrodeposition method was performed using a three-electrode potentiostat system (Autolab), with Ag/AgCl (3M KCl) as the reference electrode and platinum as the counter electrode, and the distance between the substrates and the counter electrode was ∼1.5 cm. The pulsed voltage profiles were applied using a Metrohm Autolab (PGSTAT302N) with a high-speed amplifier setup (125 kHz; 8 s ramp time). All the applied potentials reported in this work were referenced against the Ag/AgCl electrode. Samples were rinsed in DI water and dried using a N2 gun before further characterization.2.2 Sample characterizations
A Theta Probe (Thermo Scientific) instrument with a monochromatic Al Kα (1486.6 eV) X-ray source was used for X-ray photoelectron spectroscopy (XPS) analysis. Measurements were recorded using a 400 μm beam spot size, with a pass energy of 40 eV and a step size of 0.1 eV. The adventitious carbon C 1s peak at 284.6 eV was used as the charge-correction reference when appropriate and possible. The recorded spectra were deconvoluted using a least-squares error method in peak fitting using the CasaXPS software. In this work, the following fitting constraints were utilized: (1) spin–orbital splitting of ∼3.13 eV between the Mo 3d5/2 and 3d3/2 core levels, (2) an area ratio of ∼3:2 for Mo 3d5/2:Mo 3d3/2, and (3) consistency across FWHM and line-shapes together with the peak positions recorded. Scanning electron microscopy (SEM) was performed using an FEI Helios 600 instrument and was used to study the surface morphology of the hybrid films.
CO2 sorption isotherms were measured at room temperature (25 °C) under different pressures using ASAP2020 apparatus (Micromeritics). Samples were coated on the appropriate ITO/glass substrates with a surface area of 15 × 8 mm to fit in the tube holder. The samples were degassed at 150 °C for 10 h to prepare all of the porosities prior to analysis. High-purity CO2 gas was employed for sorption analysis. Subsequently, isotherms were modified based on the area and density of the thin films.
2.3. Density-functional theory calculations
The spin-polarized density-functional theory (DFT) calculations were performed using the Vienna ab initio simulation package (VASP.5.4.4.18) with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functionals and the projector augmented wave (PAW) potential. For all the calculations, the cut-off energy, the convergency of the total energy and the force on each atom were set to 400 eV, 10−6 eV and 0.01 eV Å−1, respectively. The van der Waals interaction was considered using the DFT-D3 method. The correlation effect of the Mo d orbital was considered using Hubbard U correction (U = 6.3 eV). For the MoO3 bulk with the orthorhombic structure, the Brillouin zone was sampled using a 12 × 10 × 4 k-point mesh. Based on these settings, the lattice constants of the MoO3 bulk were calculated to be a = 3.74, b = 3.77, and c = 13.71 Å, in line with the experimental values and previous calculations.28–30The adsorption of the PEI molecule on the MoO3 surface was modelled by placing a PEI molecule on a (4 × 4 × 1) MoO3 (001) surface (Mo6+), and on surfaces with one oxygen vacancy at the bridge site (Mo5+) and with one oxygen at the top site (Mo4+), as illustrated and described in Fig. S1 (ESI†). For these slab models, a vacuum layer thicker than 15 Å was applied normal to the MoO3 (001) surface, as well as dipole correction, in which 4 × 4 × 1 k-point meshes were used to sample the Brillouin zones.
3. Results and discussion
3.1. Electrodeposition of Mo oxide
It is important to have a good understanding of the electrodeposition of MoO3 to determine the overpotentials and how the electrodeposition affects the resulting thin films. This is especially crucial for MoO3 as different conditions may yield different stoichiometries that will have an influence on the subsequent co-deposition and the CO2 sorption performance. In general, there are two reduction reactions in molybdate-based electrolytes, and they are associated with the Pourbaix diagram of the Mo–O–H system in acidic pH. The possible reactions are shown via the CV profile in Fig. 1a, and they can be divided into Regions A, B and C. In Region A (−0.2 to −0.7), the observed cathodic current peak at around −0.5 V is related to the reduction of MoO42− ions viaeqn (1):31,32| MoO42− + 2H2O + 2e− → MoO2 + 4OH− | (1) |
| MoO2 + 2OH− + 2H+ → MoO3 + H2O + H2 | (2) |
 | ||
| Fig. 1 (a) Cyclic voltammetry measurement in 20 mM sodium molybdate dihydrate solution across the essential potential range (vs. Ag/AgCl) for Mo oxide thin-film electrodeposition. There are 3 distinct reaction regions indicated as Regions A, B and C. (b) Fitted XPS spectra of the constant-voltage-deposited Mo oxide thin-film across the selected applied potential as indicated. (c) Summary of the fitted XPS spectra from (b), of the representative Mo oxide film across the 3 regions, showing the relative concentrations of the different Mo oxidation states (6+, 5+, 4+). | ||
This reaction shows the conversion to MoO2 that is subsequently oxidized and deposited as MoO3. Based on the enthalpy of formation of MoO3, the conversion of MoO2 to MoO3 in the presence of OH− (eqn (2)) has a large negative formation energy (Section S2 in ESI†) and can potentially proceed if the reactants are available. A deeper understanding of the evolution is investigated in this work through understanding the chemical composition of the resulting film at the respective deposition voltage. The XPS spectra of films derived via constant-voltage deposition are shown in Fig. 1b. In order to achieve a higher consistency over differing deposition rates at the different potentials, all samples from constant-voltage deposition were set to yield the same calculated charge (Q) according to the recorded chronoamperometry, and the resultant defect-concentration-profile distribution is summarized in Fig. 1c.
The Mo 3d spectra for the films deposited in Region A is largely stoichiometric. The film electrodeposited at a high potential (−0.2 V) is generally defect-free as the oxidation of MoO2 appears to be complete. This can be seen via the absence of any Mo5+ or Mo4+ signals that imply the presence of oxygen vacancy defects. A small amount of Mo5+ can be seen as the potential is decreased to −0.6 V, which yields a cathodic peak. The cathodic peak signals a faster deposition rate (eqn (1)), and hence the relative transport and reaction of OH− with MoO2 (eqn (2)) may become the limiting step resulting in the formation of sub-stoichiometric Mo oxides. This further explains and supports the observation that the film deposited at −0.2V is generally defect-free as the lower deposition rates provide a higher chance for the interaction of the −OH reactants with the electrochemically formed MoO2 and explains the observation of the slight Mo5+ presence at the higher deposition potential of −0.6V, as shown in Fig. 1b. In region B (−0.8 to −1.1 V), the increase in the cathodic current can be attributed to the two additional competing reactions shown in eqn (3) and (4):32
| MoO2 + 2H2O + 4e− → Mo + 4OH− | (3) |
| MoO42− + 4H2O + 6e− → Mo + 8OH− | (4) |
The two reactions look to shift the reaction to arrive at more elemental Mo, either through reduction of the previously produced MoO2 layer (eqn (3)) or reduction of MoO42− ions (eqn (4)). Molybdenum metal can still react with the surrounding water or available oxygen to form molybdenum oxide. The availability of oxygen at this stage can potentially favor the formation of more stoichiometric Mo oxide growth.21 Without this, limited oxidative reactants will yield a more defective film, as shown in the XPS plot of the film deposited at −1.0 V, which yields Mo5+ and slight Mo4+ defects. Finally, in Region C (<−1.2 V), the cathodic current rise is accounted for by hydrogen evolution. The XPS spectra of the films deposited at −1.2 V and −1.4 V show a significant level of defect concentration. It is possible that the presence of H+ or hydrogen intercalation has an etching or reducing effect that can potentially increase the Mo5+ defects in the Mo oxide structure.21,33,34
The above relationship between the applied potential and the growth rate and stoichiometry is an important starting step for subsequent co-deposition. This understanding enables us to rationally design the co-deposition recipe, as we will demonstrate via an example with non-conductive PEI, and helps to explain some of the defects evolved in the process.
3.2. Incorporating PEI into thin films via electrodeposition
Non-conductive polymers such as PEI cannot be directly electrodeposited, and to the best of our knowledge we have not come across such reports. One possible way to incorporate them in electrodeposition is through a co-deposition approach. Unfortunately, adding non-conducting elements in the electrodeposition may reduce the ionic and electronic conductivity by increasing the viscosity and reducing the interaction at the electrode surface. Therefore, using PEI as an example, the available literature has only employed it as an additive to suppress dendritic growth35,36 and to provide smaller grain sizes and smooth finishing with lowered growth rates.37,38 Our attempts at the direct electrodeposition of a PEI–Mo oxide hybrid film were also not successful. We employed a simple constant-potential method using an as-prepared solution with 0.25 wt% PEI to incorporate the PEI inside the Mo oxide films. XPS spectra from the surface and depth-profile layers show very little molybdenum signal (Fig. S3a, ESI†) but a strong N 1s peak at ∼398.2 eV (Fig. S3b, ESI†). The peak represents PEI but disappears in the depth-profile scans, showing that it is PEI surface adsorption (Fig. S3b, ESI†). The low growth of Mo oxide films is unexpected given our calibration of the growth rate at the same potential. Therefore, we concluded from the above observations that the PEI adsorbs on the surface and results in strong passivation that prevents the further electrodeposition of Mo oxide. We further investigated the nature of the PEI adsorption via drop-cast PEI on the ITO substrate and immersion of the ITO substrate in PEI solution (Fig. S4, ESI†). The results show the presence of PEI on both surfaces, showing the chemical nature of the surface adsorption of PEI. The strong PEI signal is not surprising since XPS is surface sensitive and usually observed even when there is a small amount of PEI in the electrolyte. Similar results was obtained in a separate study that involves electrodeposition of Au with PEI.38 Based on these observations, constant-potential co-deposition is not only ineffective for incorporating PEI to produce hybrid films, it also significantly constrains the electrodeposition of Mo oxide through the presence of PEI.In order to overcome the strong surface-passivation issue, we introduce a controlled pulsed-electrodeposition (PED) technique to achieve PEI incorporation. The PED technique controls the potential or current swiftly between two different values and it can be used to enhance the passage and distribution of ions,39,40 and also provide removal/etching in the reverse cycle.41,42 PED has demonstrated its ability to tune hydrophilicity/hydrophobicity,43–45 expand the surface area and increase the active sites for electrocatalysis.42,45–47
To utilize PED for the co-deposition of PEI, we need to ascertain the electrochemistry of the PEI solution. Fig. 2a shows the CV profiles of ITO in PEI electrolyte (0.25 wt% PEI in DI water) for different potential ranges. Postulating that there will be an anodic potential whereby PEI may be rejected from the surface, we started with a narrow range of cyclic potential between −1.5 and −1.1 V, and sequentially increased the cyclic window from −1.5 to 0 V. It can be seen that there are no cathodic or anodic peaks at potentials lower than −1.0 V (CV 1 and 2). However, redox peaks appeared as the potential was increased to −0.9 V and remained visible in all cyclic potential windows thereafter. We interpret this as the rejection potential, whereby PEI is rejected from the surface at a potential higher than −0.9 V. After the rejection, PEI can be re-introduced onto the surface and the cyclic-rejection process is repeatable as shown by the redox peaks. This repeatability enables us to introduce PEI into the Mo oxide film via a cyclic process in electrodeposition.
 | ||
| Fig. 2 (a) Cyclic voltammetry measurement of PEI (0.25 wt%)–DI water solution. The potential window (vs. Ag/AgCl) is gradually increased towards higher anodic potentials labelled from (1) to (6), to find the rejection potential of PEI. (b) Cyclic pulses used for the PEI–Mo oxide hybrid film deposition across 3 different pulses with increasing rejection potential moving from Pulse A to Pulse C. | ||
The protocol around designing the PED method based on the CV profiles of PEI and molybdate solutions (Fig. 1a and 2a) is straightforward. Potentials are selected in the range between −0.9 and −1.4 V for three different variations of the pulse recipe (A, B, and C), as shown schematically in Fig. 2b. The holding potential (selected to be the rejection potential) is different for each pulse recipe, while the deposition potential is kept constant.
Using the modified PED method, PEI was successfully incorporated into the hybrid films. The films are amorphous in nature as determined via X-ray diffraction measurements (Fig. S5, ESI†). The presence of PEI can be seen from the N 1s peak at 398 ± 0.2 eV.48–50 The depth profile of the pulse-deposited Mo oxide film without PEI is shown in Fig. 3a(i), where only the Mo 3p signal can be observed. The presence of PEI incorporation throughout the XPS depth profiling for the PED of the hybrid film is shown in Fig. 3a(ii). The presence of PEI is also affirmed via energy dispersive X-ray spectroscopy (EDS) mapping (Fig. S6, ESI†), showing a homogenous distribution in the amorphous film. The oxidation state of the electrodeposited Mo oxide and hybrid films were examined through the fitting of the XPS profile for the Mo 3d core-level peaks as shown in Fig. 3b(i–iv). The binding energies of Mo 3d5/2 for Mo4+, Mo5+, and Mo6+ are fitted at 229.7 ± 0.1 eV, 230.6 ± 0.1 eV, and 231.85 ± 0.1 eV, respectively.51–53 The percentage contribution for each fitted chemical oxidation state is shown in Fig. 3c. Firstly, the relative proportion of defective film observed (Mo4+ and Mo5+) is roughly similar between the Mo oxide (70%) and the hybrid films using various pulsed-deposition conditions (72%, 71%, and 66%). However, there is a relative shift towards a larger proportion of Mo4+ defects for the hybrid film. We attribute this to the greater hindrance brought about by PEI in the deposition solution that may impede oxidation of the deposited film (according to eqn (2)). Secondly, when a higher anodic rejection potential is used, there is a slight trend towards reducing Mo4+ and increasing Mo6+. This can be seen more clearly in Fig. 4a, where we compare the surface scans of Mo 3d for the 3 pulses normalized to Mo6+. Relatively speaking, the decrease in the Mo4+ core-level peak is clearly observed as the rejection potential shift towards the anodic range from −1.15 V (Pulse A) to −0.9 V (Pulse C). Fig. 4b shows the fitted ratio between Mo4+ and Mo6+ using the Mo 3d core-level peak and the corresponding N 1s/Mo 3p core-level peak. The ratios are derived from the fitted peak areas, and an example of the fitted spectrum for N 1s/Mo 3p is shown in Fig. S7 (ESI†). The fitted ratios show an increase in the incorporated PEI content, accompanied by a reduction in the Mo4+ concentration.
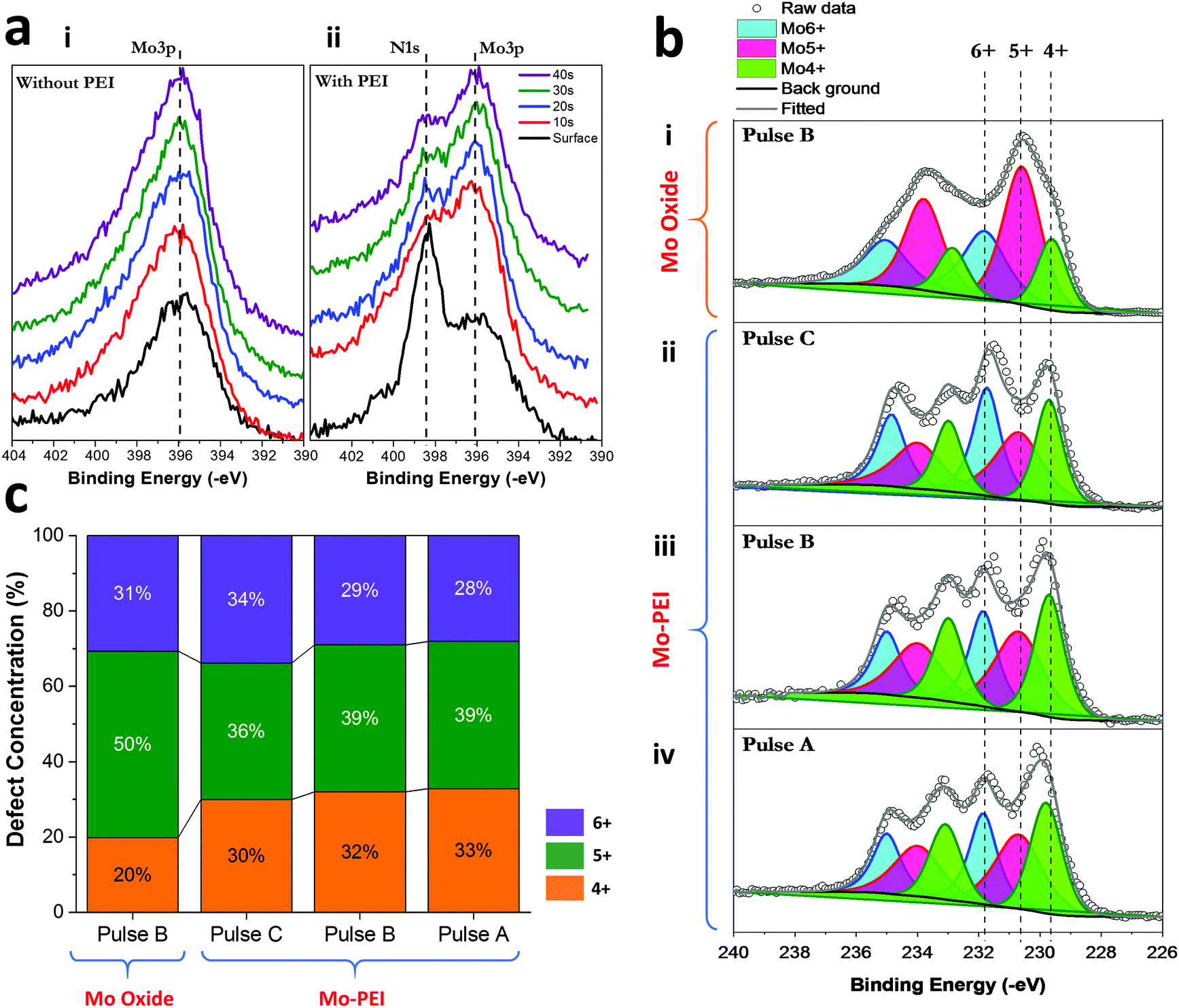 | ||
| Fig. 3 (a) XPS N1s/Mo3p spectra for depth profiling of: (i) the pulsed-deposition Mo oxide film without PEI and (ii) the pulsed-deposition hybrid PEI–Mo oxide film. The sputtering duration is indicated in the legend and represents the chemical profile with increasing depth. (b) XPS spectra for (i) Mo oxide without PEI deposited using Pulse B; (ii) hybrid PEI–Mo oxide film with Pulse C; (iii) hybrid PEI–Mo oxide film with Pulse B; and (iv) hybrid PEI–Mo oxide film with Pulse A. (c) Summary plot showing the different contributions of Mo3d oxidation states for the respective thin films shown in (b). Acquired from representative fitted Mo3d spectra. | ||
 | ||
| Fig. 4 (a) Relative Mo 3d spectra of the hybrid PEI–Mo oxide thin film for 3 different pulse recipes. The spectra are normalized to the Mo6+ peak to show the evolution of the Mo4+ relative contribution. (b) Plot of ratios for Mo4+/Mo6+ and PEI/Mo against the rejection potential (vs. Ag/AgCl). The PEI/Mo ratio is represented by the N 1s and Mo 3p fitted peaks. (c) Film-thickness measurements for samples deposited using electrolytes with different PEI concentrations. The inset on the top left shows the Mo 3d spectrum for PEI concentrations of 0.5, 1 and 3 wt%, while the inset on the bottom right shows the Mo 3d spectrum for the PEI concentration of 0.25 wt%. | ||
The observation is consistent with our suggestion that the more anodic rejection potential leads to a higher removal of PEI from the surface or near-surface. The relative reduction of Mo4+ when normalized against Mo6+, in the overall film, also points to preferential incorporation around Mo4+ sites (potentially converting to Mo6+). The amount of PEI that can be incorporated with a higher anodic rejection pulse is also observed to not proceed at an equal rate with the reduction of the Mo4+/Mo6+ ratio. The reason for this could be due to the higher deposition rate with the increased rejection of PEI yielding more Mo6+ in film deposition. Overall, we identified Pulse B as the ideal recipe for PEI incorporation for our purpose in balancing the presence of Mo4+, which is useful for CO2 sorption,21 and PEI incorporation. We also examined the effect of PEI concentration using the saturating Pulse B recipe for higher weight percentages of PEI, at 0.5, 1 and 3 wt% PEI solutions. Interestingly, 0.25 wt% remains the highest loading amount possible for film growth, whereby the increase in PEI concentration leads to a drastic reduction in Mo oxide growth, as shown in Fig. 4c. The 0.25 wt% loaded solution produces a film thickness of ∼47 nm (the cross-section SEM result in Fig. S8 in ESI,† shows a thickness of ∼48 nm, while surface profilometry shows ∼46 nm), while using a higher wt% yields little presence of Mo oxide, showing that the PEI impedes the film growth, as supported by the corresponding Mo 3d core-level peak insets in Fig. 4c. The thickness of the films with very low thicknesses is estimated using overlayer attenuation of the substrate (In 3d) signal (Section S9 in ESI†). This finding also supports our earlier hypothesis that PEI in the deposition solution can impede the electrodeposition of Mo oxide. We also report that the same recipe (Pulse B) for a PEI-free solution yielded a film thickness of ∼37nm (as determined using cross-sectional SEM). The larger thicknesses of the hybrid film (∼25%) can be explained through the incorporation of PEI, and this corroborates well with the expected PEI concentration (∼30%) obtained via the fitted atomic concentration percentage of PEI from the XPS plots. This also implies that the rejection potential is adequate to remove the influence of the PEI, meaning that the deposition of Mo oxide can remain similar for both the PEI and PEI-free solutions using the modified pulse-deposition technique.
To further understand the preferential interaction of PEI molecules with molybdenum oxide films, we studied the interaction of the PEI molecule with the surface of a Mo oxide using first-principles calculations. This can be a good representation of the deposition conditions whereby the molecule–surface interaction approximates the reaction propensity when PEI is near the oxide surface, which is a likely case during the pulse deposition. In comparison, we examined 3 different configurations as shown in Fig. 5b–d that represent interactions with the stoichiometric (Mo6+) and defect (Mo5+ and Mo4+) states. The Mo4+ and Mo5+ defect states are represented by the formation of an oxygen vacancy at the top and bridge site, respectively as shown in Fig. S1 in ESI.†Fig. 5a shows the summarized adsorption energies for the representative Mo4+, Mo5+ and Mo6+ surfaces. We observe the highest negative adsorption energy for the PEI interaction with the Mo4+ surface (−4.24 eV), followed by that for the Mo5+ surface (−3.13 eV). By contrast, the PEI–Mo6+ interaction yields the smallest negative adsorption energy (−1.74 eV). The more negative adsorption energy indicates a stronger interaction. As for the Mo4+ surface, we observe the formation of chemical bonds between nitrogen in PEI and the surface Mo atoms near the defect from the optimized atomic structure (Fig. 5d). The stronger interaction between PEI and the Mo oxide with Mo4+ is further confirmed by the more pronounced charge density redistribution, as shown in Fig. 5d, where the charge redistribution extended to neighboring C-atoms. These findings here support Mo4+ as preferential sites for PEI interaction and explain well the relative reduction of the Mo4+ signal when a higher PEI amount is incorporated, which is in agreement with our experimental observations. The results therefore show a facile method for incorporating a non-conductive polymer via a modified pulsed-electrodeposition method. This has been elusive from previous available reports using electrodeposition. Our introduced method only requires pre-deposition test in ascertaining the rejection potential for the polymer before proceeding with the pulse recipe to produce the hybrid film.
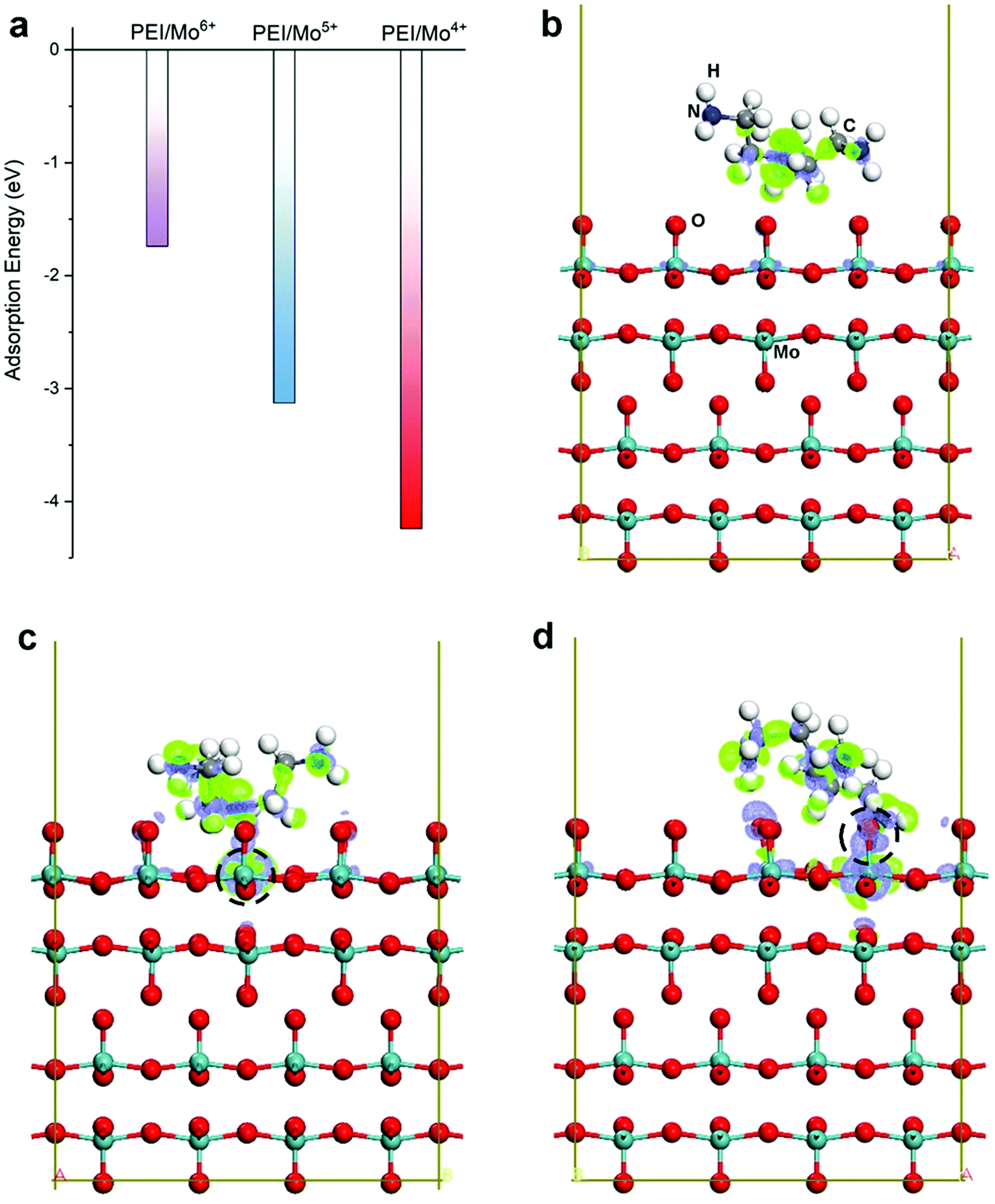 | ||
| Fig. 5 Interaction between the PEI molecule and the MoO3 (001) surface with/without defects. (a) Summary of the adsorption energy for PEI on the MoO3 surface without defects (Mo6+), and with a surface oxygen vacancy at the bridge site (Mo5+) and the top site (Mo4+), respectively. The atomic structure (side view) superimposed with the charge density redistribution for PEI on the MoO3 (001) surface (b) without defects, (c) with a Mo5+ defect, and (d) with a Mo4+ defect, where the location of the oxygen vacancy is indicated by the dashed circle. The charge density redistribution is visualized using an iso-surface value of 1.0 × 10−3 e Å−3 in which the green and blue areas denote charge accumulation and depletion, respectively. | ||
3.3. The effect of PEI on CO2 sorption
PEI is a well-studied material for CO2 capture. The common method for using PEI in sorption is through incorporation into a porous powder, e.g., with silica, to boost its sorption capacity.22 This is attractive as PEI is expected to have strong chemical bonds with CO2 and is a cost-effective polymer. This becomes especially relevant when we are looking at capturing dilute concentrations of CO2, including direct air capture. Here, we examine the performance of the hybrid PEI–Mo oxide film using a room-temperature volumetric CO2 adsorption approach.54–56 The supported thin-film performance is accomplished via the sorption isotherm in a CO2 environment. Fig. 6a shows measurements for the hybrid PEI–Mo oxide film and the pulsed-electrodeposition Mo oxide film (without PEI), after accounting for the sorption on the bare ITO substrate. | ||
| Fig. 6 (a) CO2 sorption isotherms of pulse-deposited Mo oxide and pulse-deposited hybrid PEI–Mo oxide thin film. This absolute sorption quantity is obtained by subtracting the bare ITO substrate that was used as the control sample. (b) SEM micrograph showing the surface morphology of the pulse-deposited Mo oxide thin film. (C) SEM micrograph showing the surface morphology of the pulse-deposited hybrid PEI–Mo oxide film on the ITO substrate. (d) Schematic illustrating the potential effect of PEI on the microstructure of the electrodeposited films. | ||
Sorption quantities are shown in absolute amounts (μmol) at different pressures, and all the isotherms were recorded at ambient temperature. The sorption quantities under ambient conditions (760 mmHg) are an important consideration for direct air capture. The Mo–PEI film shows a good average performance at 1.61 μmol for the hybrid film, compared with 0.57 μmol for Mo oxide under ambient conditions. It is interesting to see that the PEI-incorporated film yields close to a 3 times increase in performance under ambient conditions for the sorption quantity. From a materials perspective (taking into account the thin-film mass only), the Mo oxide yields a capacity of 27.4 mmol g−1. For the hybrid film, an estimation of the mass of the thin film was done by taking the known density values of molybdenum trioxide (4.69 g cm−3) and PEI (1.03 g cm−3). The contributed density is assumed to be proportional to the fitted atomic concentration percentage of PEI in the film (∼30%). Considering this, we achieved a remarkably high result of 79.7 mmol g−1 at 760 mm Hg−1, compared with the range of values reported in the literature (∼1–11 mmol g−1; refer to Table S10 in ESI† for more details). The increase in performance with PEI addition can be attributed to the enhanced morphological structure. Fig. 6b and c show the surface morphology of the pulse-electrodeposited Mo oxide and hybrid PEI–Mo oxide film, respectively. The pulsed addition of PEI resulted in a reduction of the grain sizes of the film, and this can be attributed to the role of PEI acting as a surfactant in the electrodeposition process.37,38 The PEI reduces the surface tension at the interfaces of the Mo oxide films and has the effect of stabilizing smaller particles or grain sizes during the electrodeposition. The difference in microstructure is shown in the cartoon in Fig. 6(d). The smaller grain sizes can provide a higher surface area, and the presence of PEI between the grains can also improve the diffusion and reaction of CO2 with the oxide film, thereby showing enhancement as a hybrid material.
We believe that the results reported here have some implications for understanding the sorption of CO2 for solid sorbents. If we take every one amino group in PEI to react with one CO2 molecule,16,57 this yields a theoretical maximum efficacy of 23 mmol g−1 for PEI. As it is not expected that PEI will have a fundamentally different sorption potential in a thin film structure (refer to discussion in section S11 in ESI†), this shows that the bigger role played by PEI may be in influencing the microstructure of the thin film, as we have mentioned, via acting as a surfactant. What is useful is that apart from having a purely surfactant role, PEI itself has a relatively high sorption potential and this makes the combination particularly attractive. The second inference is that PEI acting as a surfactant will tend to reside between the grains. Such a hybrid configuration is useful. Not only does it increase the surface area of Mo oxide as a sorbent material but it also potentially provides pathways for CO2 diffusion into the film, contributing to the higher performance recorded. This shows that, for solid sorbents, the gas-diffusion pathways and accessibility are critically important and may be the limiting step in the sorption efficacy of solid sorbents. This further strengthens the role of nanomaterials as solid sorbents and potentially the role of thin-film coatings in reducing the physical pathways for the gas diffusion.
In summary, we have shown for the first time in this work that it is possible to electrodeposit a hybrid film that involves a non-conducting polymer. We demonstrated this using modified PED to yield a hybrid PEI–Mo oxide film. The thin-film approach used in this work enables us to investigate the control of PEI incorporation and showed the relative propensity of PEI to be incorporated at Mo4+ defect sites, which is concurrently supported by first-principles calculations. Overall, we find that incorporating PEI in Mo oxide as a hybrid enables the use of PEI as an effective surfactant that enhances the microstructure of the film, and potentially provides better diffusion pathways. In addition, the PEI surfactant also provides an active sorption capacity, making it ideal in this hybrid approach. This results in an almost 3-fold improvement in the sorption capacity observed, yielding an excellent value of ∼79.7 mmol g−1. We believe this work opens up further exploration of the use of non-conducting polymers via electrodeposition and the vast potential for the synergistic use of PEI as a hybrid material for solid sorbents.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Mohammad Tanhaei would like to thank the Agency for Science, Technology, and Research (A*STAR) for providing its postgraduate scholarship (SINGA). The authors also gratefully acknowledge support from the Institute of Materials Research and Engineering (IMRE) and Nanyang Technological University (NTU).References
- S. Jeon, H. Jung, S. H. Kim and K. B. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 21213–21223 CrossRef CAS PubMed.
- P. Li, R. Yang, Y. Zheng, P. Qu and L. Chen, Carbon, 2015, 95, 408–418 CrossRef CAS.
- J. Qian, F. Jiang, D. Yuan, M. Wu, S. Zhang, L. Zhang and M. Hong, Chem. Commun., 2012, 48, 9696–9698 RSC.
- L. A. Darunte, Y. Terada, C. R. Murdock, K. S. Walton, D. S. Sholl and C. W. Jones, ACS Appl. Mater. Interfaces, 2017, 9, 17042–17050 CrossRef CAS PubMed.
- M. E. Potter, S. H. Pang and C. W. Jones, Langmuir, 2017, 33, 117–124 CrossRef CAS PubMed.
- Y. Zhao, J. Zhou, L. Fan, L. Chen, L. Li, Z. P. Xu and G. Qian, Ind. Eng. Chem. Res., 2019, 58, 19465–19474 CrossRef CAS.
- F. Valdebenito, R. García, K. Cruces, G. Ciudad, G. Chinga-Carrasco and Y. Habibi, ACS Sustainable Chem. Eng., 2018, 6, 12603–12612 CrossRef CAS.
- J. Yu, Y. Zhai and S. S.-C. Chuang, Ind. Eng. Chem. Res., 2018, 57, 4052–4062 CrossRef CAS.
- F. Lou, A. Zhang, G. Zhang, L. Ren, X. Guo and C. Song, Appl. Energy, 2020, 264, 114637 CrossRef CAS.
- M. Khraisheh, F. Almomani and G. Walker, Sci. Rep., 2020, 10, 269 CrossRef CAS PubMed.
- Y. Belmabkhout, Z. Zhang, K. Adil, P. M. Bhatt, A. Cadiau, V. Solovyeva, H. Xing and M. Eddaoudi, Chem. Eng. J., 2019, 359, 32–36 CrossRef CAS.
- J. Duan, Y. Pan, G. Liu, W. Jin, W. Zhang, W. S. Ho and K. Li, Curr. Opin. Chem. Eng., 2018, 20, 122–131 CrossRef.
- R. Chang, X. Wu, O. Cheung and W. Liu, J. Mater. Chem. A, 2022, 10, 1682–1705 RSC.
- J. Wang, L. Huang, R. Yang, Z. Zhang, J. Wu, Y. Gao, Q. Wang, D. O’Hare and Z. Zhong, Energy Environ. Sci., 2014, 7, 3478–3518 RSC.
- H. Li, W. Xu, J. Qian and T. T. Li, Chem. Commun., 2021, 57, 6987–6990 RSC.
- A. Goeppert, H. Zhang, M. Czaun, R. B. May, G. K.-S. Prakash, G. A. Olah and S. R. Narayanan, ChemSusChem, 2014, 7, 1386–1397 CrossRef CAS PubMed.
- A. Goeppert, M. Czaun, R. B. May, G. K.-S. Prakash, G. A. Olah and S. R. Narayanan, J. Am. Chem. Soc., 2011, 133, 20164–20167 CrossRef CAS PubMed.
- L. Wang, M. Al-Aufi, C. N. Pacheco, L. Xie and R. M. Rioux, ACS Sustainable Chem. Eng., 2019, 7, 14785–14795 CrossRef CAS.
- K. J. Kim, J. T. Culp, P. R. Ohodnicki, P. K. Thallapally and J. Tao, ACS Appl. Mater. Interfaces, 2021, 13, 35223–35231 CrossRef CAS PubMed.
- J. Liu, L. M. Wong, L. H. Wong, S. Y. Chiam, S. F.-Y. Li and Y. Ren, J. Mater. Chem. A, 2016, 4, 13280–13288 RSC.
- M. Tanhaei, Y. Ren, M. Yang, F. Bussolotti, J. J.-W. Cheng, J. Pan and S. Y. Chiam, J. Mater. Chem. A, 2020, 8, 12576–12585 RSC.
- A. Sayari, Q. Liu and P. Mishra, ChemSusChem, 2016, 9, 2796–2803 CrossRef CAS PubMed.
- X. Xu, B. Pejcic, C. Heath, M. B. Myers, C. Doherty, Y. Gozukara and C. D. Wood, ACS Appl. Mater. Interfaces, 2019, 11, 26770–26780 CrossRef CAS PubMed.
- X. Xu, M. B. Myers, F. G. Versteeg, B. Pejcic, C. Heath and C. D. Wood, Chem. Commun., 2020, 56, 7151–7154 RSC.
- A. Krishnamurthy, B. Salunkhe, A. Zore, A. Rownaghi, T. Schuman and F. Rezaei, ACS Appl. Mater. Interfaces, 2019, 11, 16594–16604 CrossRef CAS PubMed.
- W. Zhang, H. Li, C. J. Firby, M. Al-Hussein and A. Y. Elezzabi, ACS Appl. Mater. Interfaces, 2019, 11, 20378–20385 CrossRef CAS PubMed.
- Z. Yan, H. Liu, Z. Hao, M. Yu, X. Chen and J. Chen, Chem. Sci., 2020, 11, 10614–10625 RSC.
- H. Negishi, S. Negishi, Y. Kuroiwa, N. Sato and S. Aoyagi, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 64111 CrossRef.
- A. R. Head, R. Tsyshevsky, L. Trotochaud, Y. Yu, L. Kyhl, O. Karslıoģlu, M. M. Kuklja and H. Bluhm, J. Phys. Chem. C, 2016, 120, 29077–29088 CrossRef CAS.
- Y.-H. Lei and Z.-X. Chen, J. Phys. Chem. C, 2012, 116, 25757–25764 CrossRef CAS.
- A. C. Pereira, T. L. Ferreira, L. Kosminsky, R. C. Matos, M. Bertotti, M. H. Tabacniks, P. K. Kiyohara and M. C.-A. Fantini, Chem. Mater., 2004, 16(13), 2662–2668 CrossRef CAS.
- D. Sinkeviciute, J. Baltrusaitis and N. Dukstiene, J. Solid State Electrochem., 2011, 15, 711–723 CrossRef CAS.
- W. J. Dong, J. Ham, G. H. Jung, J. H. Son and J. L. Lee, J. Mater. Chem. A, 2016, 4, 4755–4762 RSC.
- A. Borgschulte, O. Sambalova, R. Delmelle, S. Jenatsch, R. Hany and F. Nüesch, Sci. Rep., 2017, 7, 40761 CrossRef CAS PubMed.
- S. J. Banik and R. Akolkar, Electrochim. Acta, 2015, 179, 475–481 CrossRef CAS.
- M. H. Lin, C. J. Huang, P. H. Cheng, J. H. Cheng and C. C. Wang, J. Mater. Chem. A, 2020, 8, 20637–20649 RSC.
- B.-G. Xie, J.-J. Sun, Z.-B. Lin and G.-N. Chen, J. Electrochem. Soc., 2009, 156, D79 CrossRef CAS.
- X. Ren, Y. Song, A. Liu, J. Zhang, P. Yang, J. Zhang, G. Yuan, M. An, H. Osgood and G. Wu, RSC Adv., 2015, 5, 64806–64813 RSC.
- M. S. Chandrasekar and M. Pushpavanam, Electrochim. Acta, 2008, 53, 3313–3322 CrossRef CAS.
- S. N. Hasan, M. Xu and E. Asselin, Can. Metall. Q., 2019, 58, 1–18 CrossRef CAS.
- J. J. W. Cheng, F. Wei and S. Y. Chiam, ACS Appl. Electron. Mater., 2020, 2, 1041–1047 CrossRef CAS.
- Y. L. Qiu, H. X. Zhong, T. T. Zhang, W. B. Xu, X. F. Li and H. M. Zhang, ACS Catal., 2017, 7, 6302–6310 CrossRef CAS.
- D. K. Kim, H. Kim, H. Park, S. H. Oh, S. H. Ahn, H. J. Kim and S. K. Kim, J. Power Sources, 2019, 438, 227022 CrossRef CAS.
- S. Jiang, Z. Guo, Y. Deng, H. Dong, X. Li and J. Liu, Appl. Surf. Sci., 2018, 458, 603–611 CrossRef CAS.
- G. Barati Darband, M. Aliofkhazraei, S. Hyun and S. Shanmugam, ACS Appl. Mater. Interfaces, 2020, 12, 53719–53730 CrossRef CAS PubMed.
- A. M.-P. Sakita, R. Della Noce, E. Vallés and A. V. Benedetti, Appl. Surf. Sci., 2018, 434, 1153–1160 CrossRef CAS.
- S. Esmailzadeh, T. Shahrabi, G. Barati Darband and Y. Yaghoubinezhad, Electrochim. Acta, 2020, 20, 135549 CrossRef.
- J. Qiu, Z. Yang, Q. Li, Y. Li, X. Wu, C. Qi and Q. Qiao, J. Mater. Chem. A, 2016, 4, 13296–13306 RSC.
- X. Liu, I. S. Amiinu, S. Liu, Z. Pu, W. Li, B. Ye, D. Tan and S. Mu, Adv. Mater. Interfaces, 2017, 4(11), 1601227 CrossRef.
- T. Hudec, V. Izai, L. Satrapinskyy, T. Huminiuc, T. Roch, M. Gregor, B. Grančič, M. Mikula and T. Polcar, Surf. Coat. Technol., 2021, 405, 126536 CrossRef CAS.
- K. Murugappan, E. M. Anderson, D. Teschner, T. E. Jones, K. Skorupska and Y. Román-Leshkov, Nat. Catal., 2018, 1, 960–967 CrossRef CAS.
- M. T. Greiner, L. Chai, M. G. Helander, W. M. Tang and Z. H. Lu, Adv. Funct. Mater., 2013, 23, 215–226 CrossRef CAS.
- J. Światowska-Mrowiecka, S. De Diesbach, V. Maurice, S. Zanna, L. Klein, E. Briand, I. Vickridge and P. Marcus, J. Phys. Chem. C, 2008, 112, 11050–11058 CrossRef.
- C. Gunathilake and M. Jaroniec, J. Mater. Chem. A, 2016, 4, 10914–10924 RSC.
- G. A. Mutch, S. Shulda, A. J. McCue, M. J. Menart, C. V. Ciobanu, C. Ngo, J. A. Anderson, R. M. Richards and D. Vega-Maza, J. Am. Chem. Soc., 2018, 140, 4736–4742 CrossRef CAS PubMed.
- Á. Sánchez-Sánchez, F. Suárez-García, A. Martínez-Alonso and J. M.-D. Tascón, ACS Appl. Mater. Interfaces, 2014, 6, 21237–21247 CrossRef PubMed.
- L. A. Darunte, K. S. Walton, D. S. Sholl and C. W. Jones, Curr. Opin. Chem. Eng., 2016, 12, 82–90 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00105e |
| This journal is © The Royal Society of Chemistry 2022 |