
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Theoretical insights into molecular design of hot-exciton based thermally activated delayed fluorescence molecules†
Jesni M.
Jacob
and
Mahesh Kumar
Ravva
 *
*
Department of Chemistry, SRM University-AP, Andhra Pradesh, 522240, India. E-mail: mahesh.r@srmap.edu.in
First published on 16th May 2022
Abstract
Despite the recent breakthroughs in the TADF process, more research is needed to understand its mechanism and develop rational molecular designs for structures with higher efficiencies and quantum yield. Hot exciton-based TADF materials, like traditional (cold) TADF, can effectively utilize singlet and triplet excitons, theoretically resulting in 100% IQE. However, in contrast to cold TADF (from low-lying T1 to S1), the RISC process in hot TADF occurs from high-lying triplet to singlet excited states (from Tm(m > 1) to Sn(n > 1)). However, designing materials that satisfy conditions for hot exciton formation, such as large triplet spacing in lower states and a small singlet-triplet gap in higher states, remains a difficult job. In this study, we explore and analyze the fundamental concepts of molecular design and suggest a design strategy by establishing structure-property relationships for hot-TADF molecules using density functional theory methods. This study could lead to new insights into molecular design approaches for organic materials with many hot exciton channels, which could lead to better exciton utilization.
Introduction
For many years, scientists have been working hard to overcome the constraint of spin-statistics in optoelectronic devices.1–3 A lot of research has been done to bring novel strategies to enhance quantum efficiency and methodologies for developing efficient OLED (Organic Light Emitting Diode) devices. Due to the pioneering work of Tang and collaborators in OLED research, a great deal of progress has been made in this domain, and several generations of OLEDs have emerged.4–6 In the traditional fluorescent OLEDs, only 25% of excitons generated in the singlet state are utilized, resulting in the wastage of 75% of the electrons and holes fed into the device. As a result, making better use of passive triplet excitons has become an active and exciting research topic.7–10 To convert non-emissive triplet excitons to emissive singlet excitons, three types of mechanisms are possible based on Reverse Intersystem Crossing (RISC)11 transition channels, namely (i) Thermally Activated Delayed Fluorescence (TADF)7,11–26 (ii) Triplet Triplet Annihilation (TTA)27–31 and (iii) the higher-order RISC (hRISC) based hot-exciton mechanism.32–39 Both TADF and TTA processes can potentially convert excitons from T1 to S1 through RISC and theoretically raise internal quantum efficiency (IQE) to 100% and 62.5%, respectively.12,40,41A breakthrough in this field happened when Ma and coworkers proposed the hot exciton mechanism in 2012.42 The hot exciton mechanism sparked researchers' interest in harvesting triplet excitons via hRISC.43–45 The core notion of the hot-exciton mechanism is hRISC, which is essentially the RISC from high-lying triplet states.32 This process begins with the cross-back of high-lying triplet excitons and ends up as radiative singlet excitons. The hot-exciton property is exhibited by some HLCT (Hybridized Local and Charge Transfer) materials due to its distinct electronic structure and features.46–51 The HLCT state is a hybrid of LE and CT states that bears the benefits of both the LE (Local Excitation) and CT (Charge Transfer) states.35,52 Thus, unlike in the LE and CT states, the hole and electron wavefunctions in HLCT have part of the overlap instead of absolute separation (CT state) or substantial overlap (LE state). The LE state produces a higher radiative rate, and the CT state producing a smaller energy gap results in efficient RISC.33,53
In contrast to the conventional TADF emitters, an ideal hot-exciton material possesses few characteristics. There are a few necessary factors to consider while designing molecules that obey the hot-exciton mechanism. The energy gap between T1 and T2 states is the base for creating hot-exciton channels.33,38,42,54–56 The large gap between T1 and T2 states will undoubtedly obstruct the internal conversion process between these states and prevent long-lived triplet excitons from annihilating.57,58 Another crucial criterion is that the lowest excited state, S1 must have the LE dominating character.53 It is preferable for the hot-exciton mechanism's final emissive state should be a LE excited state.32 This feature will help to achieve a narrower emission spectrum and assure a high chance of S1 radiative transition. This will be beneficial in terms of increasing quantum efficiency. In addition to the aforementioned need, the higher excited states (Sn(n>1), Tm(m>1)) should be charge transfer dominated states. The exciton binding energy will be lowered as a result of exciton delocalization, resulting in a tiny gap (ΔESnTm ∼ 0 eV) between these states. The hRISC process can be accelerated by molecules with higher excited states (Tm → Sn) exhibiting strong CT character and close energy levels.57 This will undoubtedly act as a motivational factor for the hot-exciton channels.
Spin-orbit coupling (SOC) is particularly crucial in determining the pace of RISC, in addition to the negligible singlet-triplet energy splitting in higher excited states.59–62 As per Fermi's Golden rule,63–66 a significantly large SOC between the relevant states can result in a two order of magnitude increase in the RISC rate. Studies on the relationship between the molecular structure and light-emitting characteristics of the hot-exciton emitters are helpful in the development of efficient emitters. Meeting all of the necessary conditions for creating emitters with hRISC based hot-exciton properties is a difficult task. Tremendous attention must be taken while tuning the molecular structures to successfully adjust the excited state of hot-exciton materials. It is hard to obtain organic compounds with considerably large triplet spacing in the lowest states (ΔET2–T1) and higher excited states with CT characteristics.57 In this work, we strive to develop efficient ways for rational molecular designing to get the suitable hot-exciton emitters with desired excited state characteristics.
Computational methodology
The ground state (S0) geometries were optimized at the DFT/ωB97X/6-31G(d,p) level of theory.67–69 The five lowest excited singlet states (based on S0 structure) and five lowest triplet states (based on optimized T geometry) were calculated by TDA/ωB97X/6-31G(d,p)57,70–73 and the natural transition orbital analysis (NTOs)74 was performed to characterize the nature of excited states. It has been shown previously that the range-separated hybrid functional ωB97X is a better choice for the description of charge-transfer excitations.57,73 In contrast, typical functionals with limited Hartree-Fock exchanges perform poorly in excitation energy estimates and structural optimizations.75 All ground-state and excited-state calculations were performed using Gaussian 16 software.76 Furthermore, the charge transfer ratio was calculated using the Multiwfn code.77 Spin-orbit coupling (SOC) values were estimated by TDA-DFT calculations with the wB97X hybrid functional using Amsterdam Density Functional (ADF) software package.78–83 The Slater-type all-electron basis set (TZP) was used for these computations within the ADF software. The thermal vibration correlation function formalism implemented in MOMAP (Molecular Materials Property Prediction Package) was used to calculate the constants of radiative and non-radiative transition rates.84 One of the most crucial transition processes in TADF is the RISC. The RISC decay rates were calculated using the classical Marcus rate equation.62Molecular design strategy
In this study, we designed a series of novel donor (D)–π–acceptor (A) type molecules that exhibit the hot-exciton mechanism. We explored the newly designed molecules’ electronic properties to contribute to the design of OLED materials using the “hot-exciton” channel. Phenoxazine (PXZ) and carbazole (CZ) were chosen as donor units because of their moderate electron-donating capacity, whereas electron-withdrawing groups including hydrogen (H), fluorine (F), and cyano (CN) groups were substituted on the pyrazine unit as acceptor units. The cyano functionalized naphthothiadiazole (NZ)39,85 and anthracene-thiadiazole (AZ) units were used to connect the donor and acceptor units. Twelve D–π–A framework molecules were designed by joining the aforementioned donor, acceptor, and π-bridge segments (as illustrated in Scheme 1).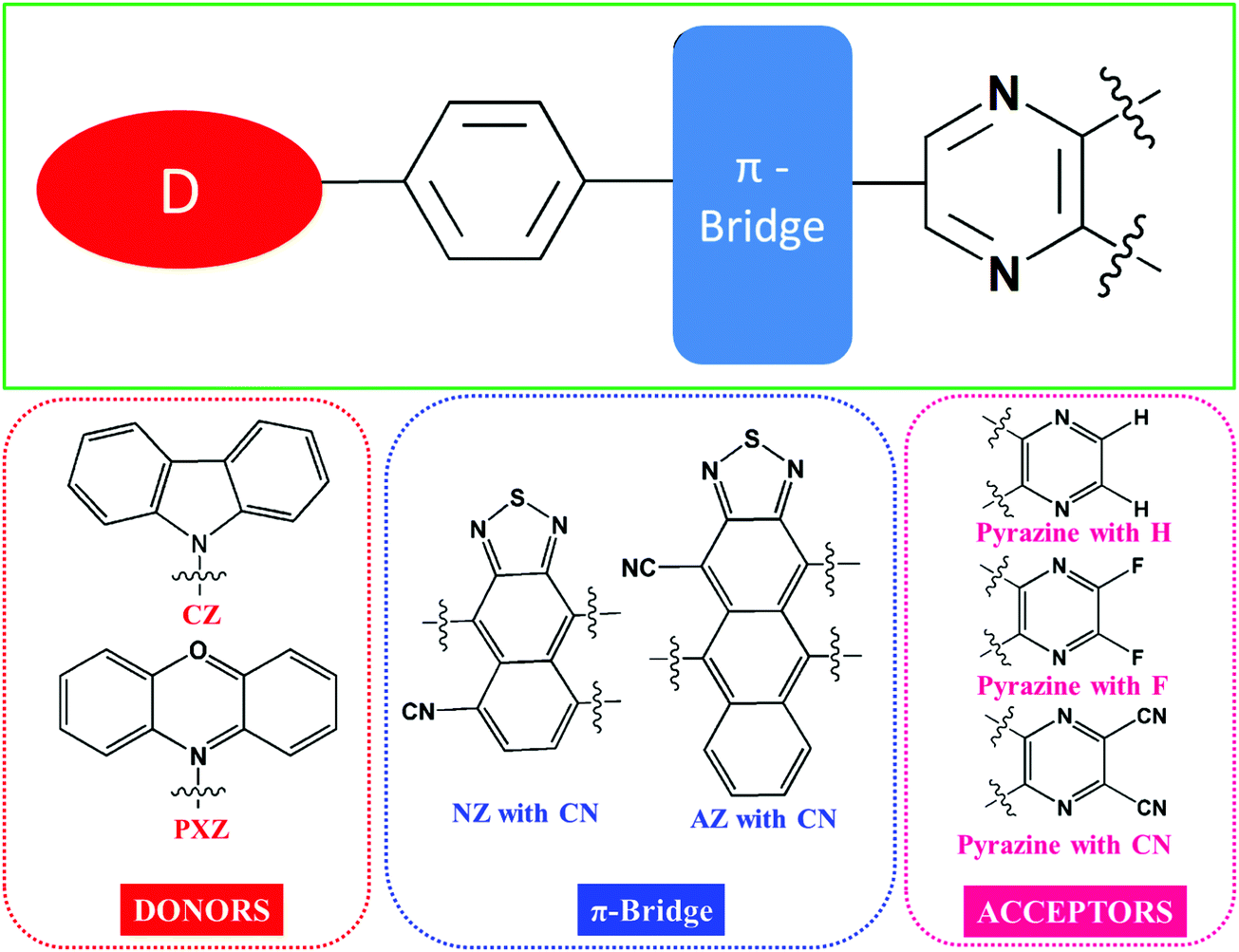 | ||
| Scheme 1 Molecular structures of the different donor, π-bridges and acceptor fragments considered in this study. | ||
Results and discussion
(i) Ground state geometries
Fig. 1 shows the optimized ground state configurations of newly designed molecules along with dihedral angles (ϕ) corresponding to the angle between donor and π-bridge; and π-bridge and acceptor unit. The coupling between donor and acceptor units may be suppressed in a D–π–A type molecule with a large twist angle, resulting in spatially separated HOMO and LUMO orbitals. A lower twist angle may improve π-conjugation and electron delocalization between the donor and acceptor groups. The dihedral angles between the bridging benzene and the donor unit CZ are range from 56° to 60° in the CZ derivatives. The same between the bridging ring and the donor unit PXZ is between 64° and 86° in the PXZ derivatives. PXZ derivatives exhibit higher twist angles than CZ derivatives, which might be related to the difference in donor unit strength and steric interactions between donor unit and π-bridge. Compared to the PXZ moiety, the CZ unit has a weaker electron-donating ability. Also, in the CZ derivatives, hydrogen atoms of the carbazole unit are far away from the π-bridge. As a result, less twist angle is observed in CZ derivatives. For the PXZ derivatives, the steric repulsion effect of nearby hydrogen atoms at the 2,6-position of the phenyl ring and AZ unit is responsible for almost orthogonal geometries between AZ moiety and neighboring phenyl ring with dihedral angles over 90°. The stronger repulsions between units lead to a more dihedral angle twist between phenoxazine unit and π-bridge. In CZ derivatives, large dihedral angles are observed in the molecules with AZ bridges than molecules with NZ bridges. The strength of electron-withdrawing units has a marginal impact on dihedral angles (varies from 67° to 64°). The acceptor strength rises in the order H < F < CN, which corresponds to the slight decrease in the dihedral angles between the bridging benzene and acceptor unit in the order as CZ1NPZCN < CZ1NZPZF < CZ1NZPZH. Similar observations are also made in the cases of PXZ derivatives.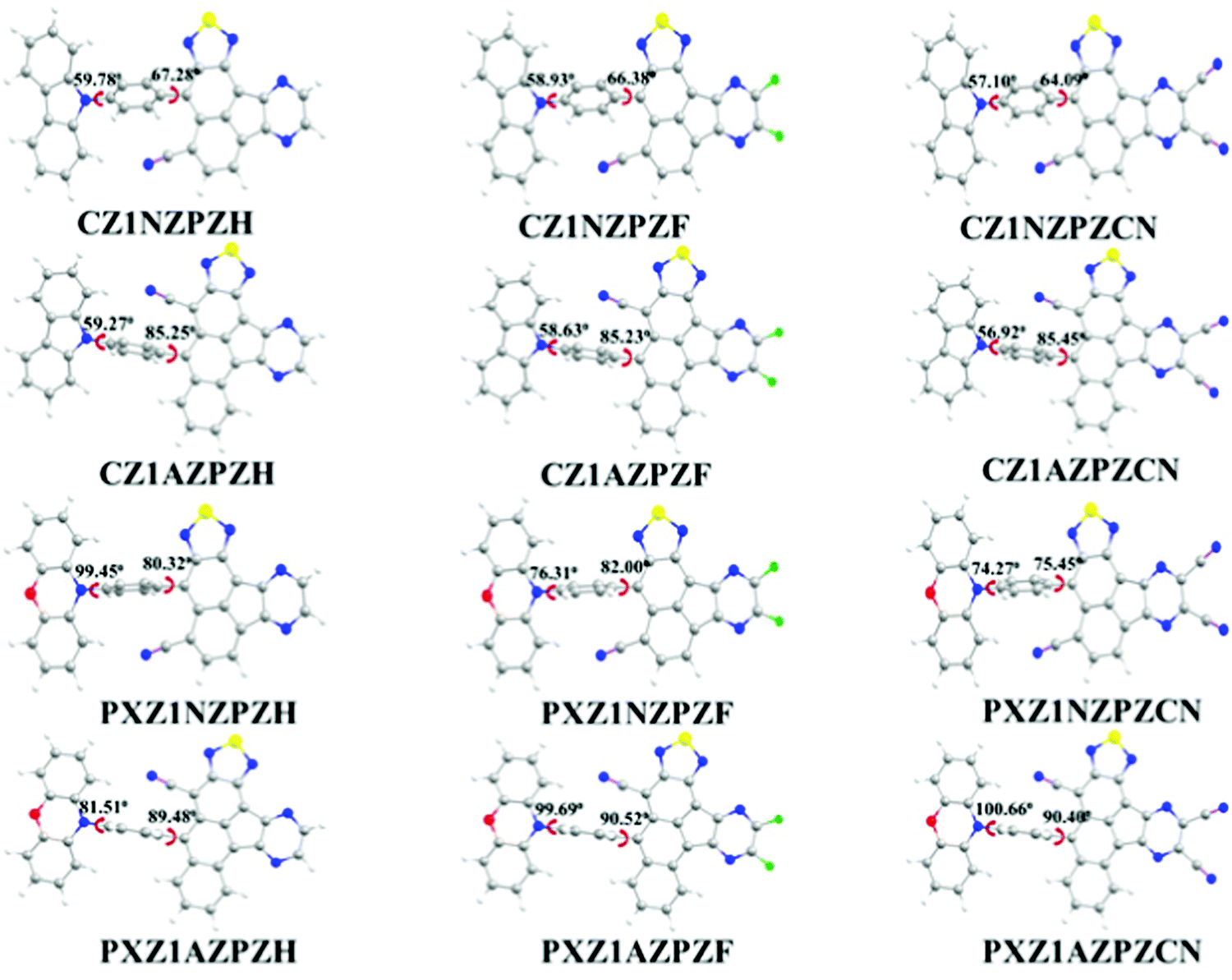 | ||
| Fig. 1 Optimized geometries of newly designed molecules in the ground-state as obtained at ωB97X/6-31G(d,p) level of theory. | ||
The acceptor strength is significantly enhanced with the extended π-conjugation due to the other CN group on the π-bridge in the molecule, which is conjugated through the pyrazine moiety as depicted in Fig. 2. This increased conjugation results in the reduction in ϕ between π-bridge and acceptor unit. However, this does not occur for the CZ derivatives possessing the AZ unit, as the repulsions between the AZ unit and the adjacent H atoms in the bridging benzene ring predominates the conjugation factor within the molecule. When we focus on the molecular skeleton, the strength of donor and acceptor units should be modest.14,57 It should not be too strong or weak. Generally, the hot-exciton emitters are made up of units with moderate electron- donating and electron-withdrawing substituents.
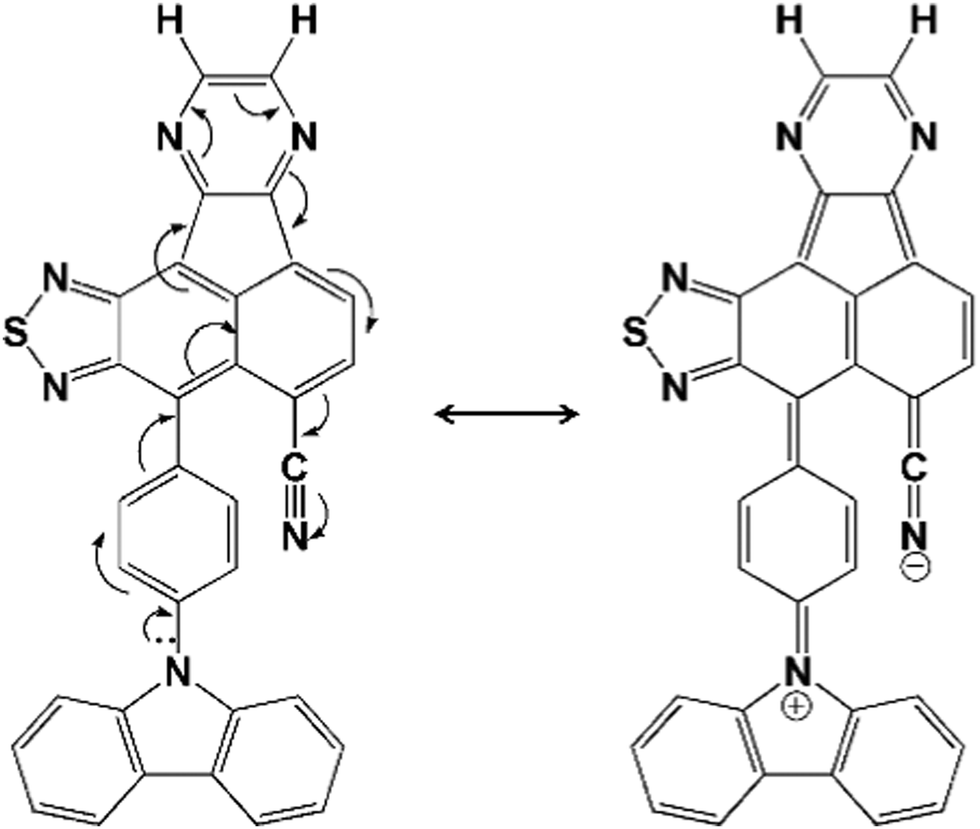 | ||
| Fig. 2 Structures of CZ1NZPZH to show the extended π-conjugation between the donor and cyano group on π-bridge. | ||
(ii) FMO analysis
We then focused on the electronic structure by examining the most important orbitals: highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs). Fig. 3 depicts the electron cloud distribution of HOMO and LUMO orbitals towards the donor, π-bridge, and acceptor units in the molecule. The HOMO is dispersed mainly on the electron-donating CZ unit in CZ derivatives with NZ core and extends to the neighboring phenyl ring. The LUMO is primarily distributed on the electron acceptor moiety and extends slightly to the adjacent benzene ring. The same is true in the case of PXZ derivatives with the NZ unit. However, molecules with AZ core stand different with their HOMO and LUMO localized separately on the donor and acceptor groups. A significant separation of FMO orbitals can be observed in these types of molecules. This reduces the energy gap between the singlet and triplet states to construct RISC channels. Due to this, the excited state properties of the molecules are likely to exhibit strong charge transfer characteristics.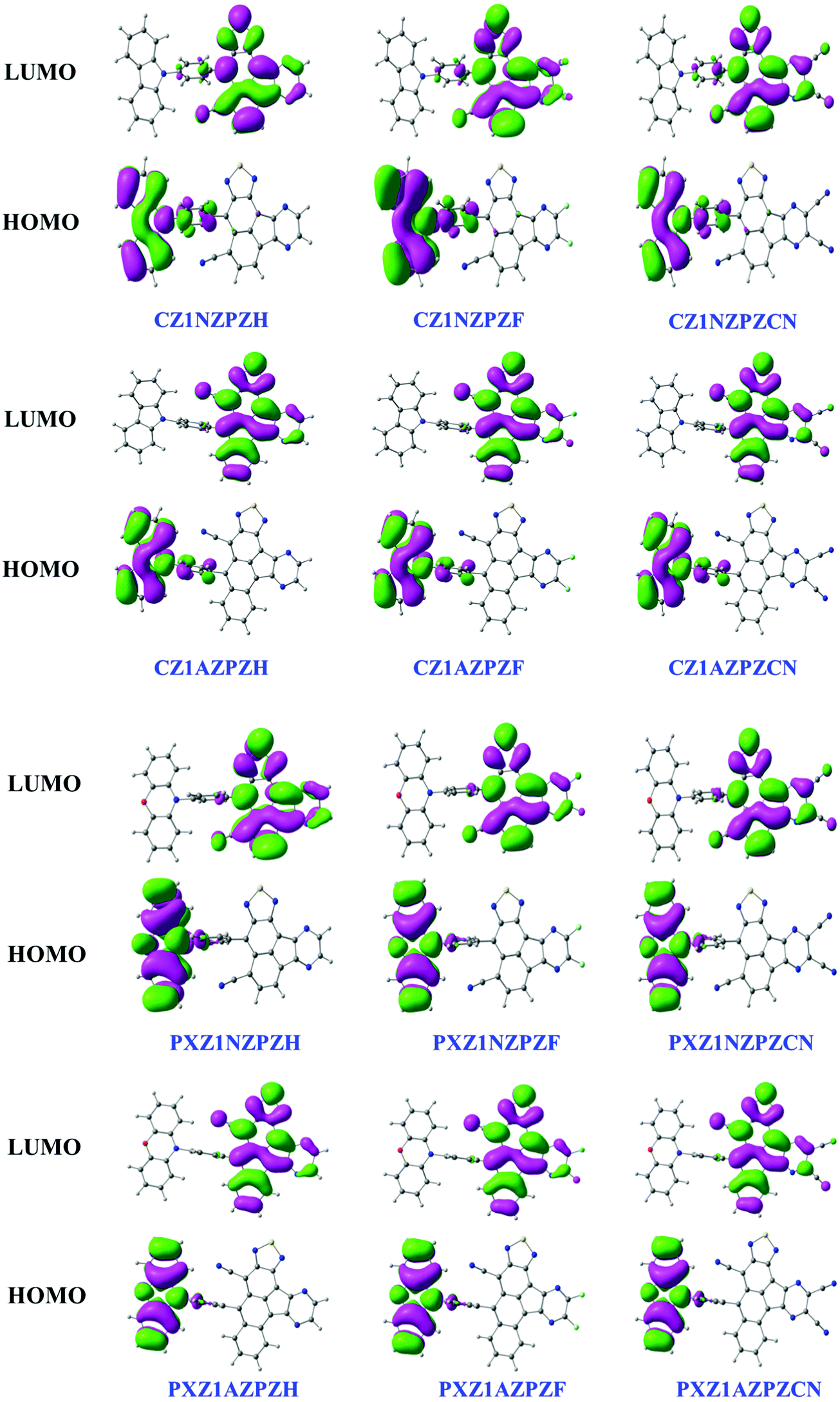 | ||
| Fig. 3 The HOMO–LUMO wavefunctions of the studied molecules at ωB97X/6-31G(d,p) level of theory. | ||
(iii) Excited state characteristics
We need to investigate the nature of excited states, energy level alignments, and transition features to gain more insights into the hot-exciton mechanism. The lowest ten singlet and ten triplet excitation energies were calculated to gain an overall picture of the energy landscape. Fig. 4 shows the excited-state energy levels of the studied molecules and the energy values are given in Tables S1 and S2 (ESI†). Furthermore, the Natural Transition Orbitals (NTO) analysis was performed on these states to understand the nature of excited state transition features. NTOs show electronic excitations as concise orbital representations. It portrays the relevant excited states as a single pair of NTO orbitals representing hole and electron wavefunctions.72,86 The calculated NTO maps of studied molecules are provided in Fig. S1 (ESI†).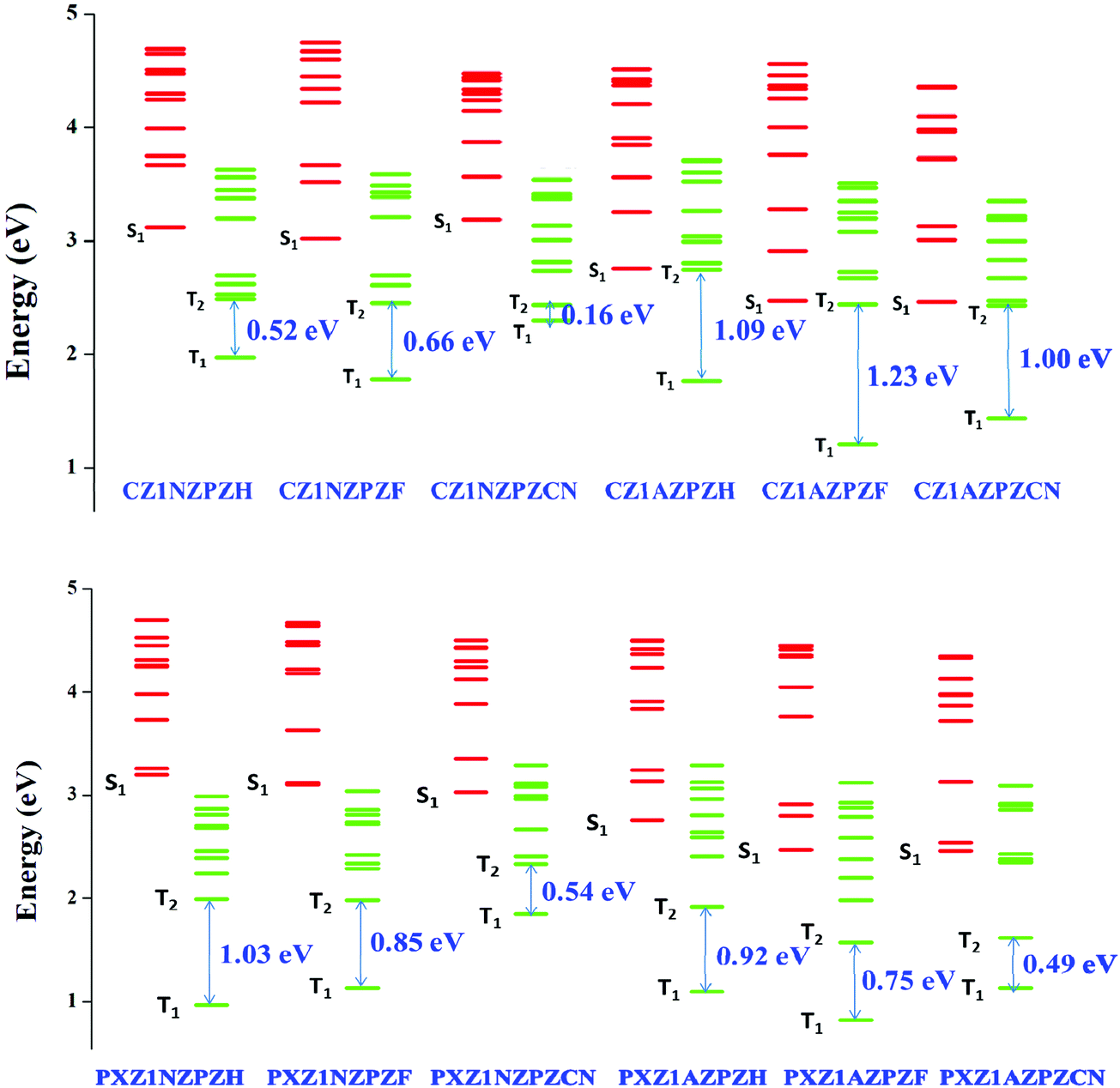 | ||
| Fig. 4 Excited state energy diagrams of molecules, including the lowest ten singlet and ten triplet excited states at TDA/ωB97X/6-31G(d,p) level of theory. | ||
There are three types of excitations in general: Local excitation (LE) occurs when a hole and an electron overlap the same spatial region. Charge-transfer excitation (CT) occurs when the spatial separation between the hole and the electron is significant, resulting in a noticeable shift in charge density. HLCT (Hybridized Local and Charge Transfer) is a type of excited state in which both the LE and CT states exist simultaneously.87–90
The hole and electron wavefunctions (NTO maps) can qualitatively describe the nature of the excited state. Similarly, the Δr index91 (distance between the centroid of the hole and the centroid of the electron) can characterize the nature of excited states quantitatively. We could readily determine whether the excitation is CT/LE/HLCT by inspecting the hole and electron wavefunctions. A LE state is the one in which the hole and electron wavefunctions have a large overlap and smaller Δr value. In a CT state, the hole and electron are spatially separated in various portions of the molecule with a larger Δr value. As described earlier, HLCT is a hybrid state with CT and LE characters. Thus, unlike in the LE and CT states, the hole and electron wavefunctions in HLCT have part of the overlap instead of absolute separation (CT state) or strong overlap (LE state). The calculated Δr indices for the molecules under study are provided in Table S6 (ESI†). The following section describes the necessary conditions to obey hot-exciton TADF properties.
(iv) Evaluation of the conditions for hot-exciton TADF
Upper triplet crossing is a simple way to describe the hot exciton process, i.e., the RISC process occurs between higher triplet and singlet energy levels. The upconversion of triplet excitons through the hot-exciton channel is dependent on precise control of singlet and triplet energy levels and SOC. There must be a small energy difference between the high singlet and triplet states and a substantial energy difference between the T2 and T1 states. As a result, it can open hot-exciton channels between higher Tm and Sn while blocking internal conversion from T2 to T1. For a fast radiative rate and high exciton utilization, the S1 state should have the HLCT character. Excitons in the LE component with large oscillator strength assure good fluorescence radiation efficiency; simultaneously, weakly bounded excitons in the CT component can increase the production of singlet excitons from the high lying excited triplet states in an HLCT excited-state system. As a result, materials having HLCT characteristics can achieve high efficiency above the spin-statistical limit.The most important criteria for the hot exciton process is maintaining a high energy barrier between the lower triplet states, i.e., T2 and T1 levels (to suppress the internal conversion). As clearly shown in Fig. 4, out of the 12 molecules we studied, four molecules (namely CZ1AZPZH, CZ1AZPZF, CZ1AZPZCN, and PXZ1NZPZH) have a reasonably sizeable T2–T1 energy gap (1.0–1.2 eV). Based on the energy gap law, this energy gap will significantly hinder the rapid internal conversion (IC) process. As a result, higher RISC (hRISC) channels are favored, facilitating exciton distribution on S1, compared to the IC channel that consumes T2 energy. Another benefit is that triplet-triplet annihilation (TTA) can be avoided due to the absence of generation or accumulation of triplet excitons.
The energy gap between singlet and triplet excited states (ΔEST) is critical in determining the efficiency of OLED materials as the energy gap between the essential singlet and triplet states impacts the rate of RISC. According to the Marcus rate equation, the rate of RISC is as follows:62
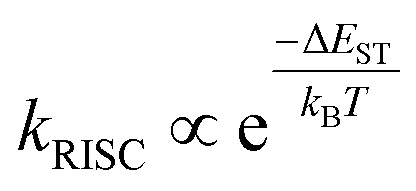 | (1) |
Fig. S1 (ESI†) shows the hole and electron wave functions derived from the NTO analyses of the lowest five singlet excited states and lowest five triplets excited states of the four potential molecules identified based on T1–T2 energy gaps. In all the four potential molecules, the distributions of hole and electron are localized on the acceptor unit in the lower singlet (S1) excited state, which clearly indicates that the S1 state is dominated by a strongly bound localized state (LE) character. Similarly, the T1 state of all molecules exhibits LE character except in the case of PXZ1NZPZH, which is a CT-dominated HLCT state with electron cloud dispersed across the bridging benzene ring. The excited state features indicated by NTO analysis also agree with the calculated charge transfer ratios. Charge transfer ratios are calculated by dividing the molecule into three fragments: donor, acceptor, and π-bridge. The outcomes of the charge transfer ratio analysis are listed in Table S3 (ESI†). The results of both qualitative and quantitative approaches are complementary, thus validating our observations. The TDA-DFT and NTO analysis point to the fact that the three molecules under investigation, CZ1AZPZH, CZ1AZPZF, and CZ1AZPZCN, show considerable overlap between corresponding transition orbitals. As a result of the dominant local character of the lowest singlet and triplet states, a substantial gap between S1 and T1 states emerged, as shown in Fig. 4. The high energy barrier between the lowest singlet and triplet states makes upconversion of triplet excitons to singlet excitons through the thermally activated RISC route impossible. Therefore, the traditional/cold TADF mechanism is no longer feasible. It becomes necessary to envisage a new route for higher RISC (hRISC) to collect the triplet excitons since the chance for the RISC process from the lowest T1 state and S1 state is significantly less due to the high energy barrier between these states.
According to the Fermi's Golden rule63,64 the rate of RISC (kRISC) is directly proportional to the spin orbit coupling and inversely proportional to the singlet-triplet energy splitting (ΔEST).
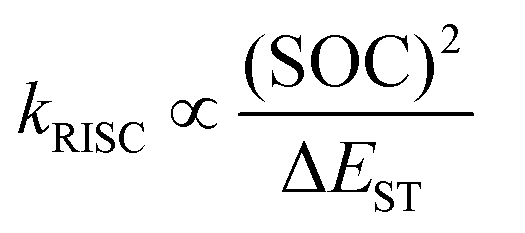 | (2) |
Based on the NTO analysis, Δr index, and charge transfer ratio, several charge transfer dominated states are observed in the high lying excited states. The CT-dominated states create a pathway for several hot exciton-based channels in higher states. In the lowest five excited states, the energy levels of several Sn and Tm levels with dominant CT state features are discovered to be nearly close values for the potential CZ derivatives with the AZ core unit. The ΔESn–Tm is minimal with the sufficient separation of electron and hole wavefunctions. In the lowest five excited states, several possible SOC values between singlet and neighboring triplet states exceeded 0.5 cm−1.
In the lowest five excited states of CZ1AZPZH, CZ1AZPZF, and CZ1AZPZCN, the potential channels are portrayed in Fig. 5. One could also see the possibility of multiple hot-exciton channels based on the data given in Tables S4 and S5 (ESI†). The transition properties of S2 and T4 in CZ1AZPZH are CT-dominated states, resulting in a ΔEST of 0.28 eV. The SOC values for these states are higher (1.01 cm−1) than those typically seen in organic compounds. The small ΔEST and large SOC values made it possible to achieve hRISC rates of up to 3.8 × 107 s−1. The high-lying excited states pathways, S2–T4 and S3–T5 with small ΔEST and large SOC are probable channels in the case of CZ1AZPZF. The associated RISC rates were found to be extremely high, reaching 4.1 × 108 s−1 for the S2–T4 route and 1.8 × 107 s−1 for the S3–T5 pathway. In higher excited levels, this also ensures the possibility of many hRISC based hot-exciton channels. In the case of CZ1AZPZCN, the S1–T2 channel has a low SOC (0.09 cm−1) but a small ΔEST (0.02 eV) value, and the rate of hRISC from T2–S1 are significantly lowered for this channel, dropping to 2.9 × 106 s−1. However, ΔEST = 0.18 eV and SOC = 0.13 cm−1 in the S2–T5 channel contribute to a high rate of hRISC from T5 to S2, in the order of 1.1 × 107 s−1, which is significantly quicker than the previously reported kRISC range of 102–106 s−1.92 The calculated radiative (kr) and non-radiative (knr) rates are in the range of previously reported values (Table 1).92
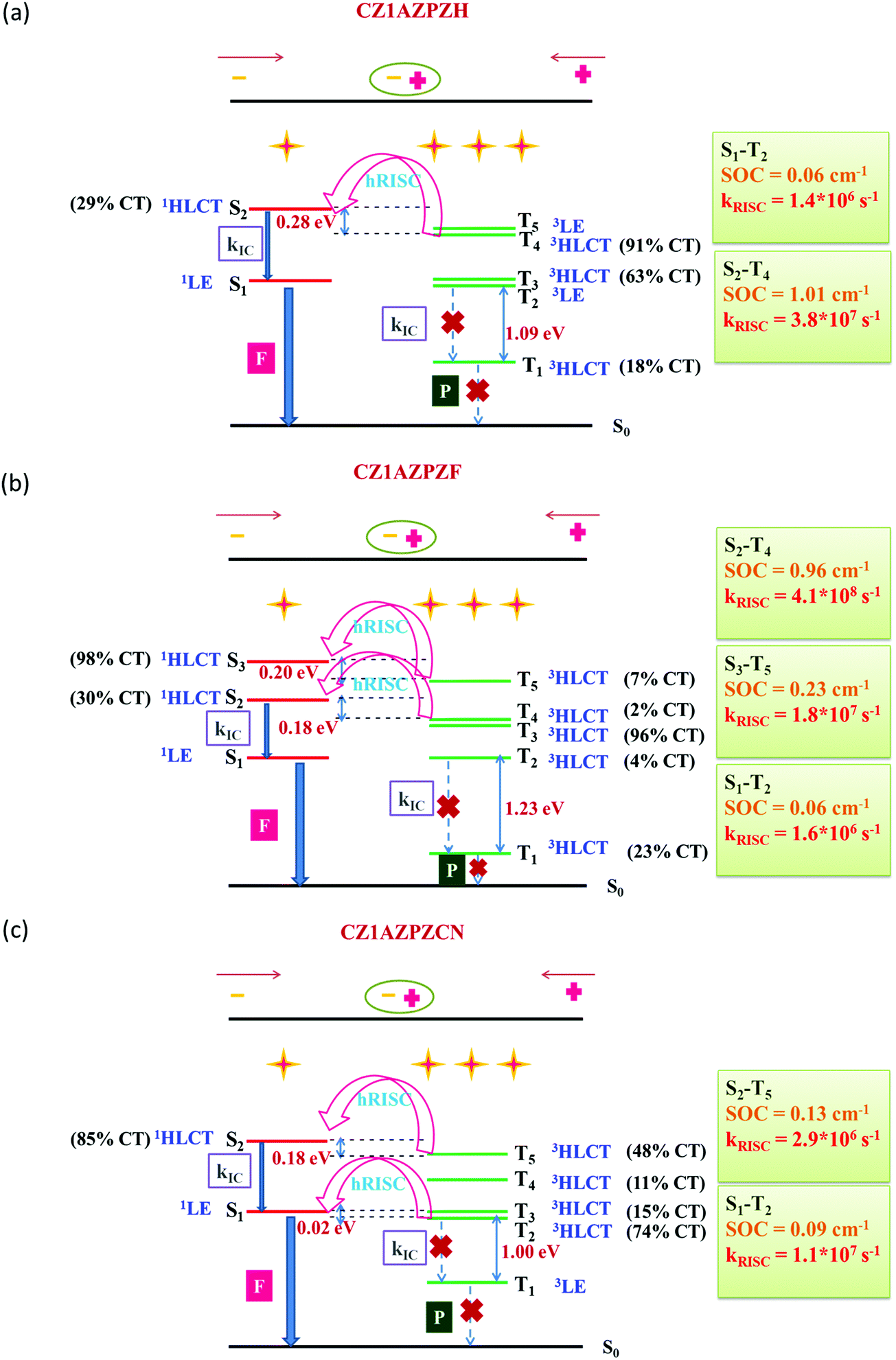 | ||
| Fig. 5 Schematic depiction of a favourable hRISC process in (a) CZ1AZPZH, (b) CZ1AZPZF and (c) CZ1AZPZCN with the calculated energy levels, accompanying SOCs, and associated kRISC. | ||
| Molecule | k nr (s−1) (S1–S0) | k r (s−1) (S1–S0) | k RISC (s−1) (Sn–Tm) |
|---|---|---|---|
| CZ1AZPZH | 1.42 × 1012 | 7.71 × 106 | 3.8 × 107 (S2–T4) |
| CZ1AZPZF | 2.07 × 1012 | 3.76 × 106 | 4.1 × 108 (S2–T4) |
| 1.8 × 107 (S3–T5) |
(v) Emission properties
In order to analyze emission characteristics, we relaxed the molecules in the lowest excited state and determined adiabatic excitation energies. All three potential molecules emit light in the red wavelength region. Because their emission energy values are in the range of 1.65 to 2 eV, implying that their emission wavelengths are greater than 620 nm. Table 2 summarizes the emission energies and the wavelengths associated with them. We compared NTOs obtained from adiabatic and vertical calculations and found that the nature of states in both cases is similar. The LE/HLCT character of these states does not differ.| Molecules | Adiabatic S1 energy (eV) | λ (nm) |
|---|---|---|
| CZ1AZPZH | 1.93 | 642 |
| CZ1AZPZF | 1.72 | 719 |
| CZ1AZPZCN | 1.92 | 644 |
Conclusion
In summary, we designed various D–π–A molecules to investigate the connection between electronic/photophysical properties and the molecular design of OLED emitters that take advantage of the hot-exciton route. Results obtained from ωB97X/6-31g(d,p) calculations reveal that various characteristics (such as the high T2–T1 spacing, singlet-triplet energy gap and SOC between related excited states) significantly impact the rates of RISC (kRISC). Three of the twelve molecules under investigation showed TADF with faster kRISC rates from the higher triplet excited states. The CT/HLCT transition occurred more frequently from the higher excited state in these molecules. This allowed several RISC channels to emerge from high-lying states. We could infer from this study that the combined impact of greater ΔET2–T1, smaller ΔESn–Tm, and larger SOC through systematic molecular structural tuning can activate the hot-exciton process in organic electroluminescent molecules. We expect that our research will serve as inspiration for the development of hot-exciton-based OLED emitters.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. M. J. thanks SRM university for the research fellowship. M. K. R thanks Department of Science and Technology (DST), New Delhi, India (DST/INSPIRE/04/2017/001393), for support of this research under a DST-INSPIRE faculty scheme.References
- J. Wang, A. Chepelianskii, F. Gao and N. C. Greenham, Nat. Commun., 2012, 3, 1 Search PubMed.
- M. Segal, A. Baldo, J. Holmes, R. Forrest and G. Soos, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 075211 CrossRef.
- M. A. Baldo, D. F. O’Brien, M. E. Thompson and S. R. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 14422 CrossRef CAS.
- C. W. Tang and S. A. Vanslyke, Appl. Phys. Lett., 1998, 51, 913 CrossRef.
- J. Kido, M. Kimura and K. Nagai, Science, 1995, 267, 1332 CrossRef CAS PubMed.
- Z. Wu, Y. Liu, L. Yu, C. Zhao, D. Yang, X. Qiao, J. Chen, C. Yang, H. Kleemann, K. Leo and D. Ma, Nat. Commun., 2019, 10, 1 CrossRef CAS.
- H. Tanaka, K. Shizu, H. Miyazaki and C. Adachi, Chem. Commun., 2012, 48, 11392 RSC.
- C. Duan, J. Li, C. Han, D. Ding, H. Yang, Y. Wei and H. Xu, Chem. Mater., 2016, 28, 5667 CrossRef CAS.
- X. Lv, W. Zhang, D. Ding, C. Han, Z. Huang, S. Xiang, Q. Zhang, H. Xu and L. Wang, Adv. Opt. Mater., 2018, 6, 1800165 CrossRef.
- Q. Liang, C. Han, C. Duan and H. Xu, Adv. Opt. Mater., 2018, 6, 1800020 CrossRef.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253 CrossRef CAS.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234 CrossRef CAS PubMed.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931 CrossRef CAS.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915 RSC.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 1 CrossRef.
- M. Y. Wong, E. Zysman-Colman, M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326 CrossRef CAS.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707 CrossRef CAS PubMed.
- S. Youn Lee, T. Yasuda, H. Nomura and C. Adachi, Appl. Phys. Lett., 2012, 101, 093306 CrossRef.
- G. Méhes, H. Nomura, Q. Zhang, T. Nakagawa and C. Adachi, Angew. Chem., Int. Ed., 2012, 51, 11311 CrossRef PubMed.
- T. Nakagawa, S. Y. Ku, K. T. Wong and C. Adachi, Chem. Commun., 2012, 48, 9580 RSC.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706 CrossRef CAS PubMed.
- J. Lee, K. Shizu, H. Tanaka, H. Nomura, T. Yasuda and C. Adachi, J. Mater. Chem. C, 2013, 1, 4599 RSC.
- H. Nakanotani, K. Masui, J. Nishide, T. Shibata and C. Adachi, Sci. Rep., 2013, 3, 1 Search PubMed.
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, Chem. Mater., 2013, 25, 3766 CrossRef CAS.
- S. Wu, M. Aonuma, Q. Zhang, S. Huang, T. Nakagawa, K. Kuwabara and C. Adachi, J. Mater. Chem. C, 2013, 2, 421 RSC.
- D. Y. Kondakov, T. D. Pawlik, T. K. Hatwar and J. P. Spindler, J. Appl. Phys., 2009, 106, 124510 CrossRef.
- N. A. Kukhta, T. Matulaitis, D. Volyniuk, K. Ivaniuk, P. Turyk, P. Stakhira, J. V. Grazulevicius and A. P. Monkman, J. Phys. Chem. Lett., 2017, 8, 6199 CrossRef CAS PubMed.
- Y. Luo and H. Aziz, Adv. Funct. Mater., 2010, 20, 1285 CrossRef CAS.
- Y. J. Luo, Z. Y. Lu and Y. Huang, Chin. Chem. Lett., 2016, 27, 1223 CrossRef CAS.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560 CrossRef CAS.
- W. Li, Y. Pan, R. Xiao, Q. Peng, S. Zhang, D. Ma, F. Li, F. Shen, Y. Wang, B. Yang and Y. Ma, Adv. Funct. Mater., 2014, 24, 1609 CrossRef CAS.
- Y. Pan, W. Li, S. Zhang, L. Yao, C. Gu, H. Xu, B. Yang and Y. Ma, Adv. Opt. Mater., 2014, 2, 510 CrossRef CAS.
- L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang and Y. Ma, Angew. Chem., Int. Ed., 2014, 53, 2119 CrossRef CAS.
- L. Yao, B. Yang and Y. Ma, Sci. China: Chem., 2014, 57, 335 CrossRef CAS.
- C. Lin, P. Han, S. Xiao, F. Qu, J. Yao, X. Qiao, D. Yang, Y. Dai, Q. Sun, D. Hu, A. Qin, Y. Ma, B. Z. Tang and D. Ma, Adv. Funct. Mater., 2021, 2106912, 1 Search PubMed.
- Y. Xu, X. Liang, X. Zhou, P. Yuan, J. Zhou, C. Wang, B. Li, D. Hu, X. Qiao, X. Jiang, L. Liu, S. J. Su, D. Ma and Y. Ma, Adv. Mater., 2019, 31, 1 Search PubMed.
- D. Hu, L. Yao, B. Yang and Y. Ma, Philos. Trans. R. Soc., A, 2015, 373, 20140318 CrossRef PubMed.
- Q. Wan, J. Tong, B. Zhang, Y. Li, Z. Wang and B. Z. Tang, Adv. Opt. Mater., 2020, 8, 1901520 CrossRef CAS.
- C. J. Chiang, A. Kimyonok, M. K. Etherington, G. C. Griffiths, V. Jankus, F. Turksoy and A. P. Monkman, Adv. Funct. Mater., 2013, 23, 739 CrossRef CAS.
- S. Xu, R. Chen, C. Zheng and W. Huang, Adv. Mater., 2016, 28, 9920 CrossRef CAS PubMed.
- W. Li, D. Liu, F. Shen, D. Ma, Z. Wang, T. Feng, Y. Xu, B. Yang and Y. Ma, Adv. Funct. Mater., 2012, 22, 2797 CrossRef CAS.
- S. Reindl and A. Penzkofer, Chem. Phys., 1996, 211, 431 CrossRef CAS.
- R. W. Redmond, I. E. Kochevar, M. Krieg, G. Smith and W. G. McGimpsey, J. Phys. Chem. A, 1997, 101, 2773 CrossRef CAS.
- H. Fukumura, K. Kikuchi, K. Koike and H. Kokubun, J. Photochem. Photobiol., A, 1988, 42, 283 CrossRef CAS.
- Y. Gao, S. Zhang, Y. Pan, L. Yao, H. Liu, Y. Guo, Q. Gu, B. Yang and Y. Ma, Phys. Chem. Chem. Phys., 2016, 18, 24176 RSC.
- Y. Pan, J. Huang, W. Li, Y. Gao, Z. Wang, D. Yu, B. Yang and Y. Ma, RSC Adv., 2017, 7, 19576–19583 RSC.
- S. Zhang, W. Li, L. Yao, Y. Pan, F. Shen, R. Xiao, B. Yang and Y. Ma, Chem. Commun., 2013, 49, 11302 RSC.
- J. Yang, Q. Guo, J. Wang, Z. Ren, J. Chen, Q. Peng, D. Ma and Z. Li, Adv. Opt. Mater., 2018, 6, 1800342 CrossRef.
- J. Jayabharathi, J. Anudeebhana, V. Thanikachalam and S. Sivaraj, RSC Adv., 2020, 10, 8866 RSC.
- B. Liu, Z. W. Yu, D. He, Z. L. Zhu, J. Zheng, Y. D. Yu, W. F. Xie, Q. X. Tong and C. S. Lee, J. Mater. Chem. C, 2017, 5, 5402 RSC.
- X. Tang, Q. Bai, Q. Peng, Y. Gao, J. Li, Y. Liu, L. Yao, P. Lu, B. Yang and Y. Ma, Chem. Mater., 2015, 27, 7050 CrossRef CAS.
- W. Li, Y. Pan, L. Yao, H. Liu, S. Zhang, C. Wang, F. Shen, P. Lu, B. Yang and Y. Ma, Adv. Opt. Mater., 2014, 2, 892 CrossRef CAS.
- C. Wang, X. L. Li, Y. Gao, L. Wang, S. Zhang, L. Zhao, P. Lu, B. Yang, S. J. Su and Y. Ma, Adv. Opt. Mater., 2017, 5, 1700441 CrossRef.
- L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang and Y. Ma, Angew. Chemie, 2014, 126, 2151 CrossRef.
- S. Zhang, W. Li, L. Yao, Y. Pan, F. Shen, R. Xiao, B. Yang and Y. Ma, Chem. Commun., 2013, 49, 11302 RSC.
- Y. Pan, Y. Guo, M. Zhao, C. Li and B. Yang, Mater. Adv., 2021, 2, 1351 RSC.
- J. Fan, L. Cai, L. Lin and C. K. Wang, Phys. Chem. Chem. Phys., 2017, 19, 29872 RSC.
- X. K. Chen, S. F. Zhang, J. X. Fan and A. M. Ren, J. Phys. Chem. C, 2015, 119, 9728 CrossRef CAS.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956 CrossRef CAS PubMed.
- C. M. Marian, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 187 CAS.
- P. K. Samanta, D. Kim, V. Coropceanu and J. L. Brédas, J. Am. Chem. Soc., 2017, 139, 4042 CrossRef CAS PubMed.
- J. L. Brédas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971 CrossRef PubMed.
- K. Schmidt, S. Brovelli, V. Coropceanu, D. Beljonne, J. Cornil, C. Bazzini, T. Caronna, R. Tubino, F. Meinardi, Z. Shuai and J. L. Brédas, J. Phys. Chem. A, 2007, 111, 10490 CrossRef CAS PubMed.
- G. C. Abell and A. Mozumder, J. Chem. Phys., 2003, 56, 4058 Search PubMed.
- S. Xu, Q. Yang, Y. Wan, R. Chen, S. Wang, Y. Si, B. Yang, D. Liu, C. Zheng and W. Huang, J. Mater. Chem. C, 2019, 7, 9523 RSC.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- J. Da Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- J. Da Chai and M. Head-Gordon, Chem. Phys. Lett., 2008, 467, 176 CrossRef.
- T. J. Penfold, J. Phys. Chem. C, 2015, 119, 13535 CrossRef CAS.
- H. Sun, C. Zhong and J. L. Brédas, J. Chem. Theory Comput., 2015, 11, 3851 CrossRef CAS PubMed.
- K. Lee and D. Kim, J. Phys. Chem. C, 2016, 120, 28330 CrossRef CAS.
- Y. Y. Pan, J. Huang, Z. M. Wang, D. W. Yu, B. Yang and Y. G. Ma, RSC Adv., 2017, 7, 26697 RSC.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775 CrossRef CAS.
- Y. Dawei, Z. Xiaojuan, W. Zhiming, Y. Bing, M. Yuguang and P. Yuyu, RSC Adv., 2018, 8, 27979 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J.-A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931 CrossRef CAS.
- T. T. Huang and E. Y. Li, Org. Electron., 2016, 39, 311 CrossRef CAS.
- E. E. Bas, P. Ulukan, A. Monari, V. Aviyente and S. Catak, ChemRxiv, 2021, 1, DOI:10.33774/chemrxiv-2021-vq9dd.
- Y. Gao, X. D. Yang, S. X. Wu and Y. Geng, Adv. Theory Simulations, 2019, 2, 1900076 CrossRef.
- S. Kang, S. H. Jeon, Y. M. Cho, Y. J. Kim, T. Kim and J. Y. Lee, Org. Electron., 2020, 78, 105595 CrossRef CAS.
- Y. Gao, Y. Geng, Y. Wu, M. Zhang and Z. M. Su, Dyes Pigm., 2017, 145, 277 CrossRef CAS.
- Y. Niu, W. Li, Q. Peng, H. Geng, Y. Yi, L. Wang, G. Nan, D. Wang and Z. Shuai, Mol. Phys., 2018, 116, 1078 CrossRef CAS.
- Y. Li, J. Yao, C. Wang, X. Zhou, Y. Xu, M. Hanif, X. Qiu, D. Hu, D. Ma and Y. Ma, Dyes Pigm., 2020, 173, 107960 CrossRef.
- X. K. Chen, D. Kim and J. L. Brédas, Acc. Chem. Res., 2018, 51, 2215 CrossRef CAS PubMed.
- G. Sun, X. H. Wang, J. Li, B. T. Yang, Y. Gao and Y. Geng, Sci. Rep., 2021, 11, 1 CrossRef.
- T. Liu, X. Chen, J. Zhao, W. Wei, Z. Mao, W. Wu, S. Jiao, Y. Liu, Z. Yang and Z. Chi, Chem. Sci., 2021, 12, 5171 RSC.
- X. Chen, D. Ma, T. Liu, Z. Chen, Z. Yang, J. Zhao, Z. Yang, Y. Zhang and Z. Chi, CCS Chem., 2021, 1285 Search PubMed.
- C. Zhou, S. Xiao, M. Wang, W. Jiang, H. Liu, S. Zhang and B. Yang, Front. Chem., 2019, 7, 141 CrossRef CAS PubMed.
- C. A. Guido, P. Cortona, B. Mennucci and C. Adamo, J. Chem. Theory Comput., 2013, 9, 3118 CrossRef CAS PubMed.
- Y. Liu, Y. Yuan, X. Tian and J. Sun, Int. J. Quantum Chem., 2020, 120, 1 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00039c |
| This journal is © The Royal Society of Chemistry 2022 |