
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuppression of phosphine-protected Au9 cluster agglomeration on SrTiO3 particles using a chromium hydroxide layer†
Abdulrahman S.
Alotabi
 abc,
D. J.
Osborn
d,
Shuhei
Ozaki
e,
Yuki
Kataoka
e,
Yuichi
Negishi
e,
Siriluck
Tesana
f,
Gregory F.
Metha
d and
Gunther G.
Andersson
*ac
abc,
D. J.
Osborn
d,
Shuhei
Ozaki
e,
Yuki
Kataoka
e,
Yuichi
Negishi
e,
Siriluck
Tesana
f,
Gregory F.
Metha
d and
Gunther G.
Andersson
*ac
aFlinders Institute for Nanoscale Science and Technology, Flinders University, Adelaide, South Australia 5042, Australia
bDepartment of Physics, Faculty of Science and Arts in Baljurashi, Albaha University, Baljurashi 65655, Saudi Arabia
cFlinders Microscopy and Microanalysis, College of Science and Engineering, Flinders University, 5042 Adelaide, Australia
dDepartment of Chemistry, University of Adelaide, Adelaide, South Australia 5005, Australia
eDepartment of Applied Chemistry, Faculty of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan
fThe MacDiarmid Institute for Advanced Materials and Nanotechnology, School of Physical and Chemical Sciences, University of Canterbury, Christchurch 8141, and National Isotope Centre, GNS Science, Lower Hutt 5010, New Zealand
First published on 14th March 2022
Abstract
Gold clusters have been shown to have great potential for use as co-catalysts in photocatalytic water splitting. Agglomeration of Au clusters deposited onto semiconductor surfaces into larger particles is a major challenge. Metal oxide overlayers can be used to improve the stability of Au clusters on surfaces and avoid their agglomeration. The aim of this work is to investigate the inhibition of phosphine-protected Au9 clusters beneath a Cr(OH)3 overlayer to agglomerate under conditions of photocatalytic water splitting (i.e. UV irradiation). Au9 was deposited on the surface of SrTiO3 using a solution impregnation method followed by photodeposition of a Cr(OH)3 layer. After UV light irradiation for 7 hours for photocatalytic water splitting, uncovered Au clusters on SrTiO3 agglomerated into larger particles. However, agglomeration was inhibited when a thin Cr(OH)3 layer was deposited onto the SrTiO3-Au9 system. From careful XPS measurements, the chemical state of the overlayer is initially determined to be Cr(OH)3 but upon heating at 200 °C for 10 min it converts to Cr2O3. Through photocatalysis experiments it was found that the Cr(OH)3 overlayer blocks the sites for O evolution reaction on the SrTiO3-Au9.
Introduction
Metal clusters are formed by a specific number of metal atoms and are generally less than 2 nm in size. Metal clusters exhibit unique physical and chemical properties which are different to those of nanoparticles formed by the same element and the respective bulk materials.1–4 Clusters can be synthesised with protecting ligands to stabilise the cluster core and prevent agglomeration.5,6 In contrast to gas phase generated clusters, ligand protected clusters can be synthesised on a larger scale. Atomically precise chemically synthesised ligated clusters deposited onto metal oxides are known to be photocatalytically active in water splitting.2,7,8 When using chemically synthesised ligated clusters, the ligands need to be removed to generate naked clusters on a surface as active sites of a photocatalyst.9 Au clusters decorated metal oxide surfaces show enhanced photocatalytic water splitting activity due to the size and dispersibility of ultrasmall metal clusters on surfaces.10–16 However, removal of the ligands of the deposited clusters without causing agglomeration of the clusters is a challenging task.2,7,17 Agglomeration of the clusters render them inefficient as active sites in photocatalysis, particularly when the catalytic performance is based on the size of the clusters. For example, Au25 clusters on BaLa4Ti4O15 surfaces show a strong decrease in photocatalytic activity if the clusters increase in size.13 It is also known that Au clusters on surfaces exposed to constant irradiation can result in agglomeration of the clusters.18,19Inhibiting the agglomeration of Au clusters decorated on metal oxide surfaces can be achieved by using surface modification and coating strategies.20,21 Krishnan et al. have stabilised Au9(PPh3)8(NO3)3 and Au13(dppe)5Cl2Cl3 clusters deposited on ALD TiO2 films by pre-treatment of the surface through heating and sputtering to introduce defects.22,23 These defects sites stabilise the Au clusters against agglomeration on the surface. Recently, Xu et al. reported that the photostability of glutathione-protected Au25 clusters on SiO2 spheres was increased by addition of multifunctional branched poly-ethylenimine (BPEI).24 Subsequent coating with TiO2 shell produced a photocatalyst with improved efficiency and photostability, even after long periods of light irradiation.
It has been shown that deposition of a CrOx overlayer can stabilise nanoparticles and clusters. Domen et al. have developed a method to improve the stability of rhodium nanoparticles using a CrOx overlayer.25–28 CrOx overlayers also can prevent the back reaction H+ and O2 to H2O (oxygen photoreduction reaction) in photocatalysts used for water splitting,29–38 with the effectiveness of the overlayer depending on its thickness.39 CrOx layers have been applied to a range of co-catalysts deposited on other metal oxide substrates, such as platinum nanoparticles,38,40 palladium nanoparticles,41 silver nanoparticles39 and metal oxides (NiOx, RuO2, Rh2O3 and CuOx).28,42 Overlayers of other metal oxides have also been employed to stabilise co-catalysts and to prevent the back reaction in photocatalysis.43–46
Chemically synthesised phosphine-protected Au clusters have attracted much attention because of their fluxional behavior and facile removal of the Au-Ph ligands.2,21,47 The aim of the present study is to investigate the size of Au9(PPh3)8(NO3)3 (hereafter referred to as Au9) deposited onto SrTiO3 nanoparticles before and after the removal of the ligands and after undergoing photocatalysis. This study includes an investigation of the influence of the chromium oxide (CrOx) layer in preventing cluster agglomeration. CrOx overlayers are photodeposited, before the removal of the ligands, to inhibit the Au clusters from agglomeration. The size of Au9 clusters was examined to investigate the effectiveness of the CrOx overlayer to stabilising phosphine-protected Au clusters after heating and UV irradiation. Finally, the influence of CrOx coverage for Au9 deposited onto SrTiO3 on overall photocatalytic water splitting rate is examined.
Experimental methods and techniques
Material and sample preparation
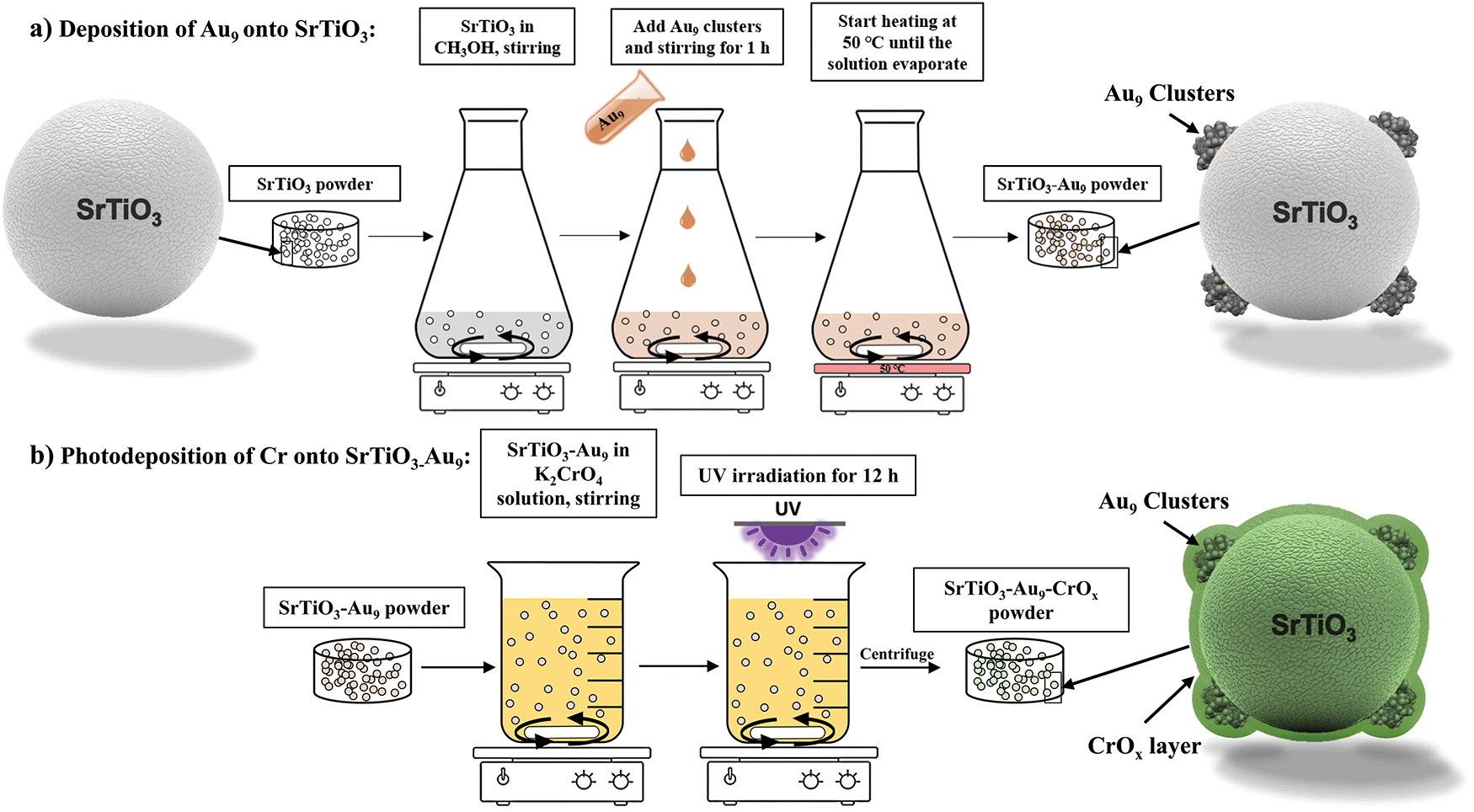 | ||
| Scheme 1 Experimental procedure of (a) Au9 deposition on SrTiO3 particles and (b) photodeposition of CrOx onto SrTiO3-Au9 particles. | ||
(a) Deposition of Au9 onto SrTiO3 (SrTiO3-Au9). SrTiO3-Au9 was prepared using an impregnation method (see Scheme 1a). First, 1 g of SrTiO3 was dispersed in 10 mL of CH3OH. Subsequently, 10 mL of a Au9 cluster solution (0.12 mM) was added to a stirred suspension of SrTiO3 (1 g SrTiO3, 10 mL CH3OH) and left to stir for 1 h at room temperature. The total volume of a CH3OH solution was 20 mL and concentration of Au9 in the solution was 0.06 mM. Assuming complete adhesion of the clusters onto the substrate, the total Au content of the SrTiO3-Au9 sample is 0.2 wt% Au content. The stirred suspension was heated at 50 °C under N2 flow until the CH3OH had evaporated (approximately 8–10 min for evaporating the CH3OH). The SrTiO3-Au9 powder was collected without further treatment.
(b) Photodeposition of Cr onto SrTiO3-Au9. CrOx was photodeposited onto the SrTiO3-Au9 samples to form an overlayer (see Scheme 1b). 500 mg of SrTiO3-Au9 powder was added to a 0.5 mM aqueous K2CrO4 solution (200 mL). The mixture was irradiated for 12 hours using a UV LED source (Vishay, VLMU3510-365-130) while stirring at a speed of 1000 rpm. The radiant power of the UV LED source was 690 mW at a wavelength of 365 nm. After irradiation, the SrTiO3-Au9-CrOx powder was collected by centrifugation and washed with deionised water. ICP-MS analysis determined that approximately 20% of all available Cr was deposited onto the SrTiO3-Au9 particles.
Analytical methods
High-resolution XP spectra were recorded for Au 4f, Sr 3d, Cr 2p, Ti 2p, P 2p, O 1s and C 1s. The XP spectra were calibrated using the main carbon peak at 285 eV to correct for charging effects.51 The peak areas were used to calculate the relative intensities taking into account the transmission function and cross-section.52
Overall water splitting. The photocatalytic activity was tested using a high-pressure Hg lamp (400 W, main wavelength at 365 nm) within a quartz cell (see Scheme S1, ESI†).19 The reaction was carried out in a closed gas flow system with Ar flow of 30 mL min−1 using a solution containing 500 mg of the photocatalyst (i.e. SrTiO3, SrTiO3-Au9 and SrTiO3-Au9-CrOx after heating) with 350 mL of water. Before the photocatalysis experiment, the reaction cell was purged with Ar gas bubbling through the water for 1 h to ensure that air was completely removed from the reaction vessel.
Hydrogen evolution using a sacrificial reagent. This experiment was performed using the same procedure as overall water splitting except that 10% of the water is replaced with methanol.
Oxygen photoreduction reaction. Using the same procedure as hydrogen evolution with the sacrificial reagent, oxygen was introduced into the reaction with a closed gas flow system using a 7
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 mixture of Ar to air at a flow rate of 30 mL min−1.
:3 mixture of Ar to air at a flow rate of 30 mL min−1.
Results and discussion
Before heating
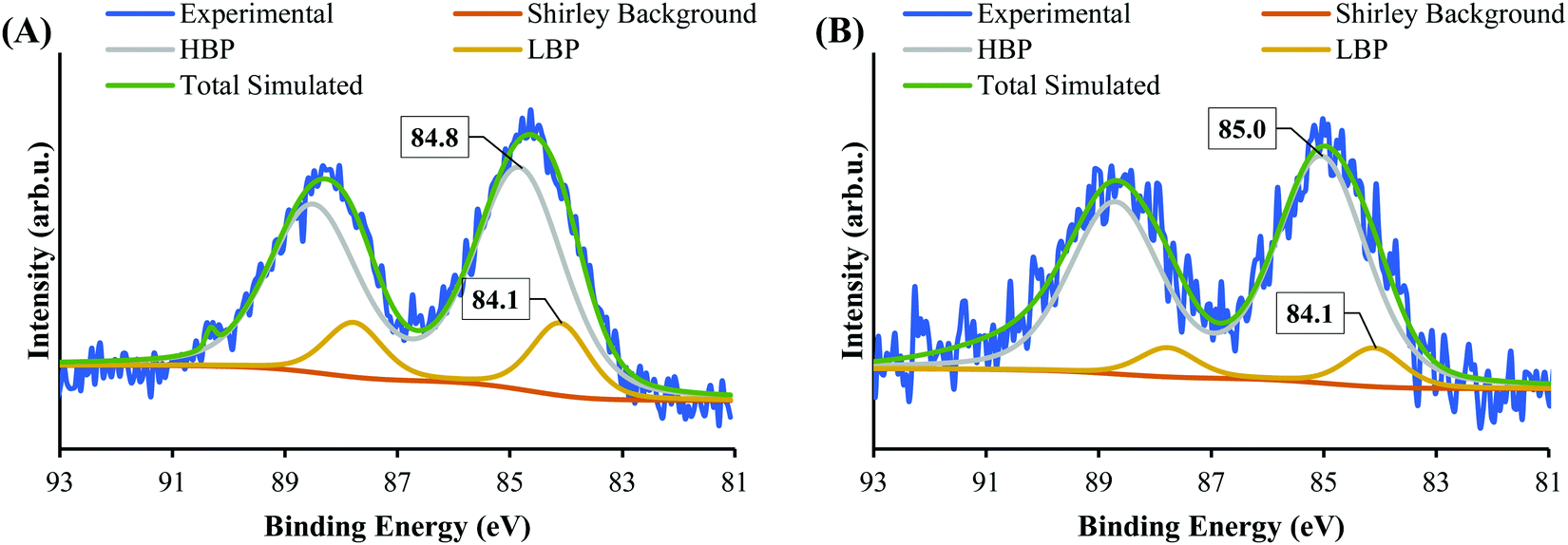 | ||
| Fig. 1 XPS spectra of Au 4f of (A) Au9 deposited on SrTiO3 and (B) SrTiO3-Au9 after photodeposition of a CrOx layer. | ||
The state of Cr photodeposited on SrTiO3-Au9 was investigated using XPS. Fig. 2 shows the Cr 2p spectrum of SrTiO3-Au9-CrOx before heating. The Cr 2p spectrum was fitted with a single doublet peak (2p3/2 and 2p1/2). It was fitted by including the Ti 2s energy loss peak, which occurs in the same energy region. In our previous work, the Cr 2p peak was described and fitted using a fixed relation between the intensity of the Ti 2p3/2 and the Ti 2s loss peak.59 The Cr 2p3/2 peak is found at 577.4 ± 0.2 eV, which can be identified from literature reference data as being Cr(OH)3.60 It should be noted that this is different to the binding energy of Cr2O3 and will be discussed below (vide infra).
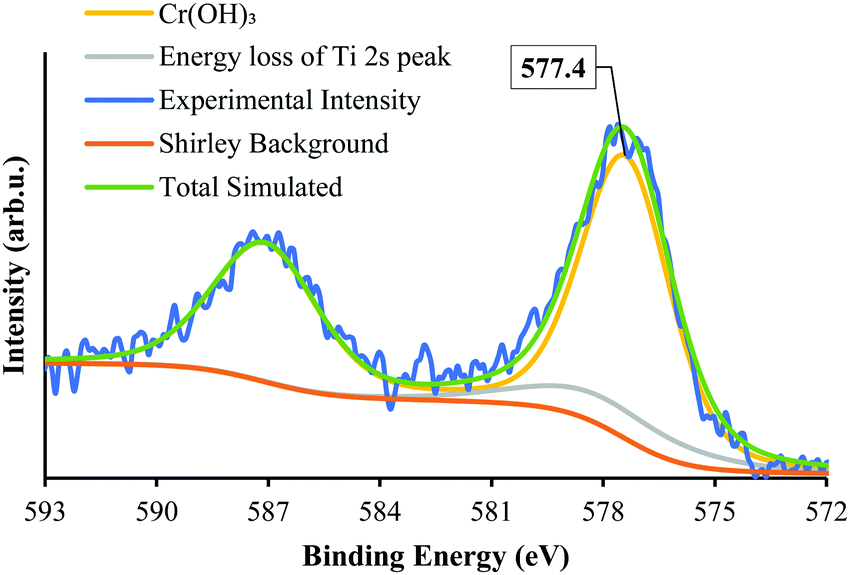 | ||
| Fig. 2 XPS Cr 2p spectrum of SrTiO3-Au9 after photodeposition of a CrOx layer. | ||
Fig. 3 shows the HAADF-STEM image of Au9 deposited onto the surface of SrTiO3. The figure includes EDX elemental mapping of Au and Ti as well as line analysis of Au. It should be noted that P was difficult to analyse using STEM-EDX due to the intensity of P in EDX being significantly lower than that of Au and due to overlapping of the P Kα and Au Mα peaks.61 In Fig. 3A, the HAADF-STEM image shows small bright particles, which can be identified from the EDX mapping as Au features. The line analysis of L1 and L2 confirmed the size of Au features as approximately 5.5 and 1.3 nm, respectively. The HAADF-STEM image shows further small Au clusters, indicated with red circles, that were not detected clearly by EDX mapping (see Fig. 3A). A possible reason for this is that EDX is not suitable for size analysis due to its lower spatial resolution compared with STEM. The size of these Au clusters in the HAADF image is approximately 0.7–0.9 nm. Previous studies have shown that the size of a single Au9 cluster is approximately 0.6 ± 0.2 nm.56 In the present experiment, the small Au clusters (0.7–0.9 nm) in the HAADF-STEM image are of a similar size as Au9. Therefore, we conclude that these are non-agglomerated clusters. The Au feature from the STEM-EDX line analysis (Fig. 3L1), which is 5.5 nm in size, is due to many adjacent Au9 clusters or agglomerations of Au clusters. The Au feature from L2 is 1.3 nm in size, suggesting two adjacent clusters or an agglomeration of two clusters due to the effect of the high-energy STEM electron beam (see Fig. S2, ESI† for more information). This finding is in line with the XPS data that suggested some of the Au clusters had agglomerated on SrTiO3-Au9 STEM shows the Au cluster sizes for only a small selected area of the sample, which could also be subject to electron beam damage of the clusters. Therefore, the XPS data is considered as being more accurate in identifying the fraction of agglomerated and non-agglomerated clusters because XPS averages over a significant larger number of Au cluster compared to STEM and also because XPS does not cause damage to the clusters resulting in their agglomeration.53,54 XPS shows that 83% of Au clusters remain non-agglomerated.
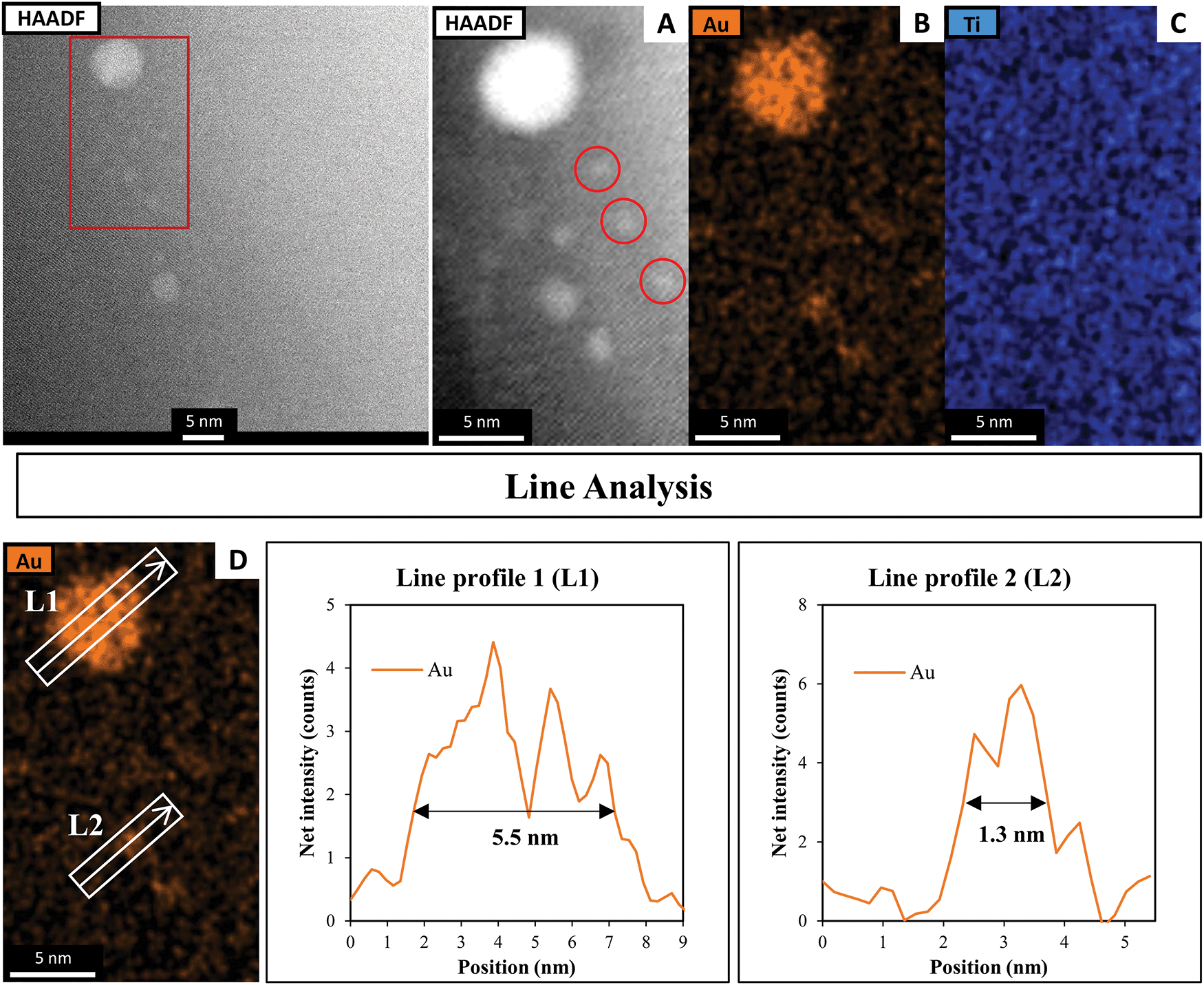 | ||
| Fig. 3 (A) HAADF-STEM image with EDX elemental mapping of (B) Au and (C) Ti in SrTiO3-Au9 before heating and (D) line analyses of the Au signal. | ||
A HAADF-STEM image of SrTiO3-Au9-Cr(OH)3 with EDX elemental mapping and a line analysis of Cr, Au and Ti is shown in Fig. 4. Fig. 4A–C shows the first STEM mapping region which is focused on the edge of SrTiO3-Au9-Cr(OH)3 to determine the thickness and distribution of Cr(OH)3 at the surface of SrTiO3. The STEM-EDX elemental map L1 at the edge of the SrTiO3-Au9-Cr(OH)3 shows that the Cr(OH)3 layer is distributed over the entire SrTiO3 surface (Fig. 4B) and has a thickness of 1.6 nm (Fig. 4L1). The line analysis of Cr, Ti and Au (L2) shows that the presence of Cr(OH)3 around the Au clusters (Fig. 4L2). Using the line analysis of L2 and L3 in Fig. 4, the Au clusters are shown to be approximately 1.3–1.6 nm in diameter. This is similar to the size of the Au clusters determined for SrTiO3-Au9 (i.e. without the Cr(OH)3 covering layer, Fig. 3). Note that the STEM-EDX mapping shows Au features with a larger diameter compared to a single Au9 cluster. Here, the increase in the size of the Au clusters is most likely due to the effect of the high-energy STEM electron beam, (see Fig. S2, ESI† for further information). Again, XPS is considered to be the more accurate method for determining the extent of cluster agglomeration, which was determined to be 90% of Au clusters remain non-agglomerated on SrTiO3 with Cr(OH)3 coverage.
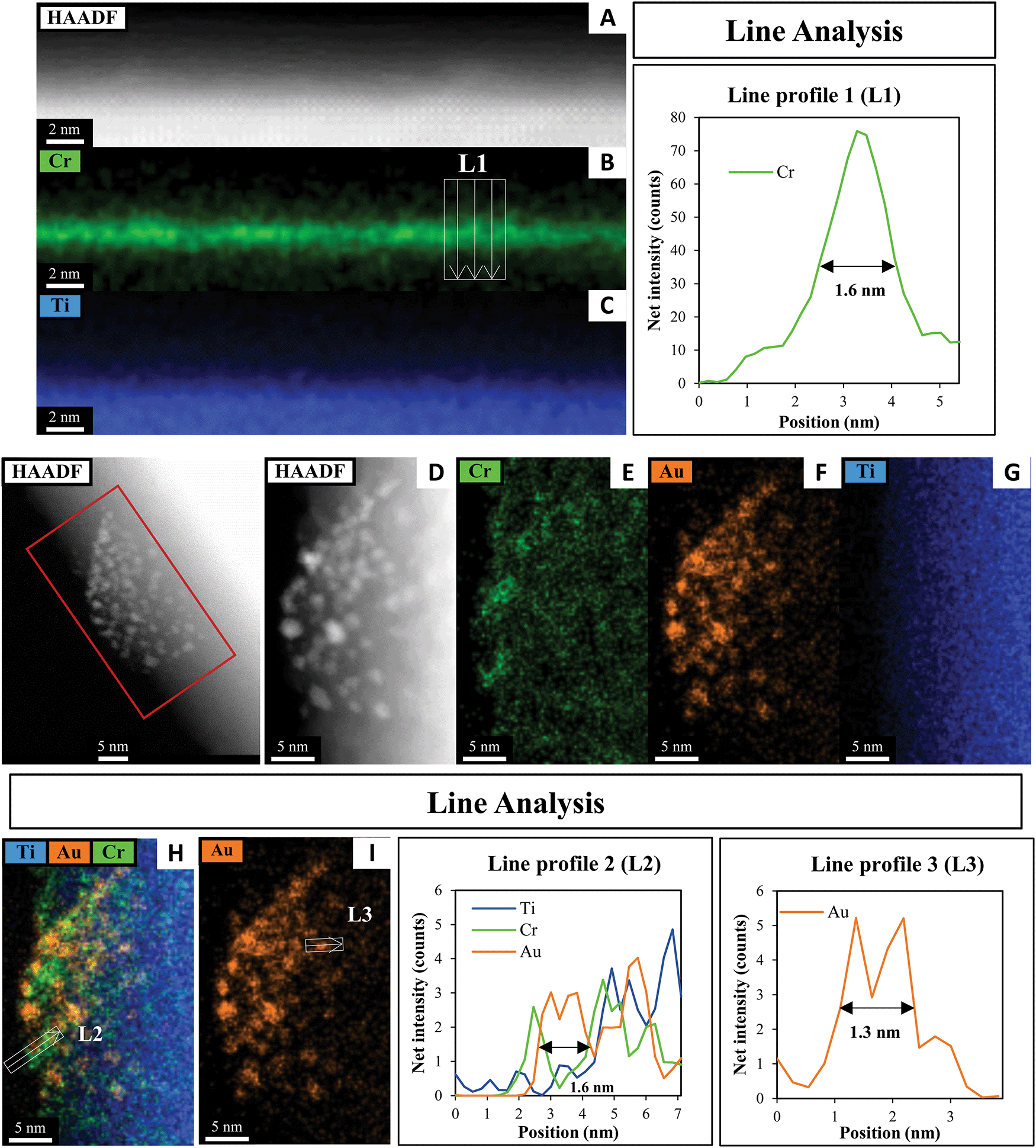 | ||
| Fig. 4 HAADF-STEM image with EDX elemental mapping and line analyses of the Cr, Au and Ti in SrTiO3-Au9-Cr(OH)3. | ||
After heating
Fig. 5 shows the XPS Au 4f spectra of SrTiO3-Au9 and SrTiO3-Au9-CrOx after heating (the CrOx notation is used again here because heating could change the chemical state of the Cr). The Au 4f spectra are again fitted with a HBP and LBP doublet. The peak positions and FWHM are summarised in Table S1 (ESI†). As mentioned above, it is difficult to determine the state of PPh3 ligands on SrTiO3 using XPS, however, previous studies showed that PPh3-ligated Au9 clusters are removed by heating at 200 °C for 10 min.23,49,53–57 As shown in Fig. 5A, the Au 4f spectrum for SrTiO3-Au9 after heating is fitted with 50% Au clusters at HBP (85.4 ± 0.2 eV, FWHM of 2.0 ± 0.2 eV) and 50% agglomerated clusters at LBP (83.7 ± 0.2 eV, FWHM of 1.2 ± 0.2 eV). The feature at 89.9 ± 0.2 eV corresponds to Ba 4d5/2 due to contamination of the commercial SrTiO3 (see Fig. S3, ESI†). After application of the CrOx overlayer and heating, the Au 4f spectrum for SrTiO3-Au9-CrOx is fitted with 65% and 35% of the Au intensity at the HBP (84.9 ± 0.2 eV, FWHM of 2.0 ± 0.2 eV) and LBP (83.8 ± 0.2 eV, FWHM of 1.2 ± 0.2 eV), respectively (see in Fig. 5B). Thus, the comparison of the XPS results between the heated SrTiO3-Au9 and SrTiO3-Au9-CrOx samples indicated that the photodeposition of CrOx improves the stability of the phosphine-protected Au9 clusters, with the majority retaining their size after removal of the ligands through heating to 200 °C.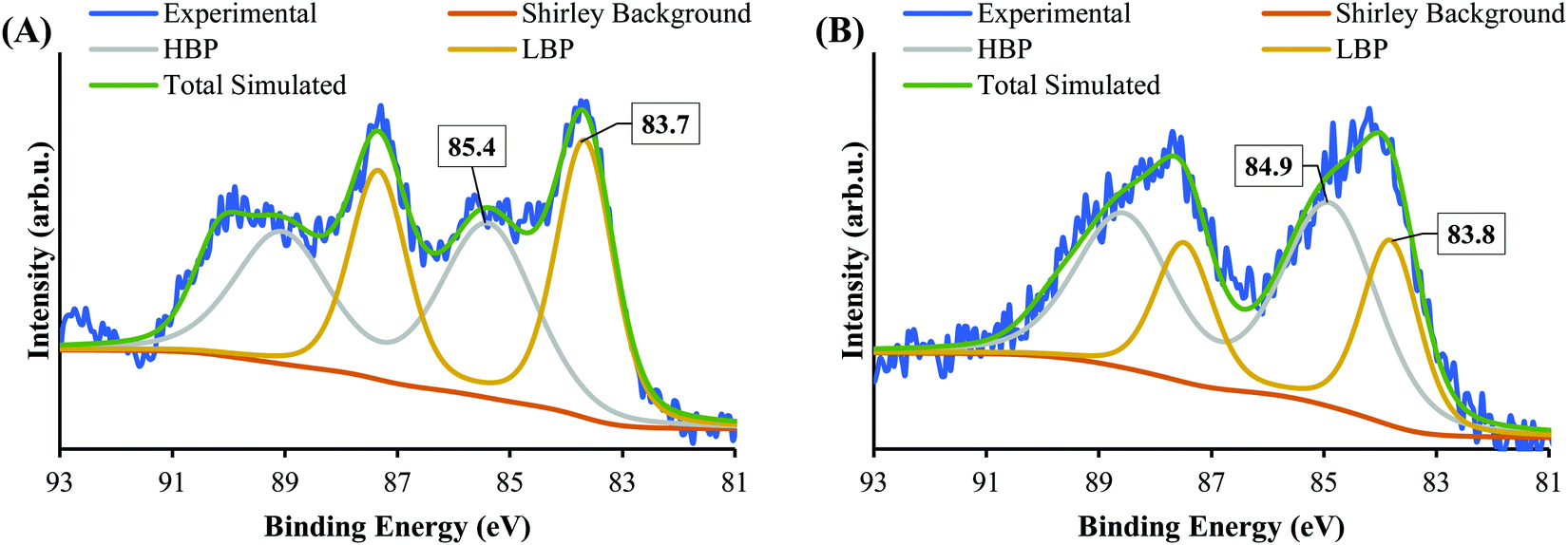 | ||
| Fig. 5 XPS spectra of Au 4f after heating of (A) SrTiO3-Au9 and (B) SrTiO3-Au9-CrOx. | ||
XPS Cr 2p spectra of SrTiO3-Au9-Cr(OH)3 before and SrTiO3-Au9-CrOx after heating are shown in Fig. 6, along with reference spectra for Cr2O3 and Cr(OH)3. A summary of the Cr 2p3/2 peak positions is presented in Table S4 (ESI†). The Cr 2p3/2 peak for SrTiO3-Au9-CrOx after heating shifts slightly to a low binding energy of 577.1 ± 0.2 eV (Fig. S4, ESI†), corresponding to Cr2O3.62 This confirms that a reduction of the Cr(OH)3 layer to Cr2O3 occurs by heating. This finding is in agreement with a previous report that heating reduces photodeposited Cr(OH)3 layer to Cr2O3.16
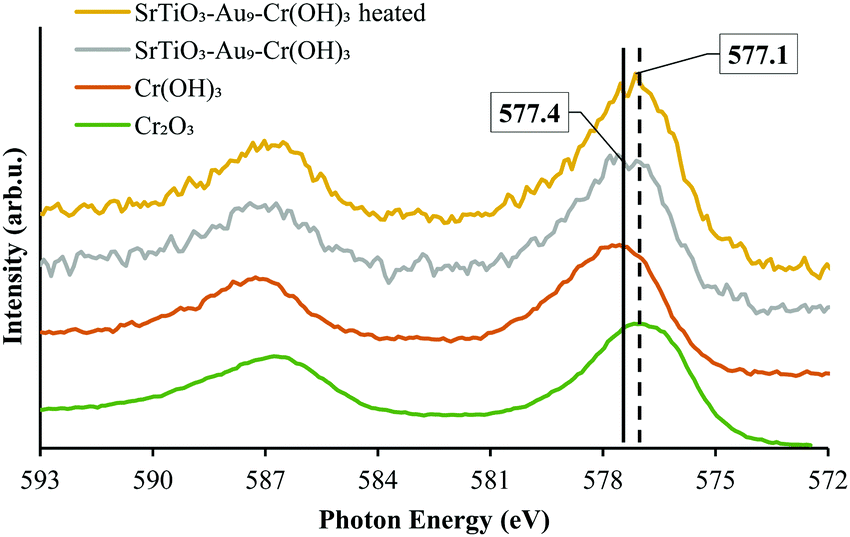 | ||
| Fig. 6 XPS spectra of Cr 2p of SrTiO3-Au9-Cr(OH)3 before heating and SrTiO3-Au9-CrOx after heating with reference spectrum of Cr2O3 and Cr(OH)3 (Fig. S4, ESI† shows complete fitting to all components). | ||
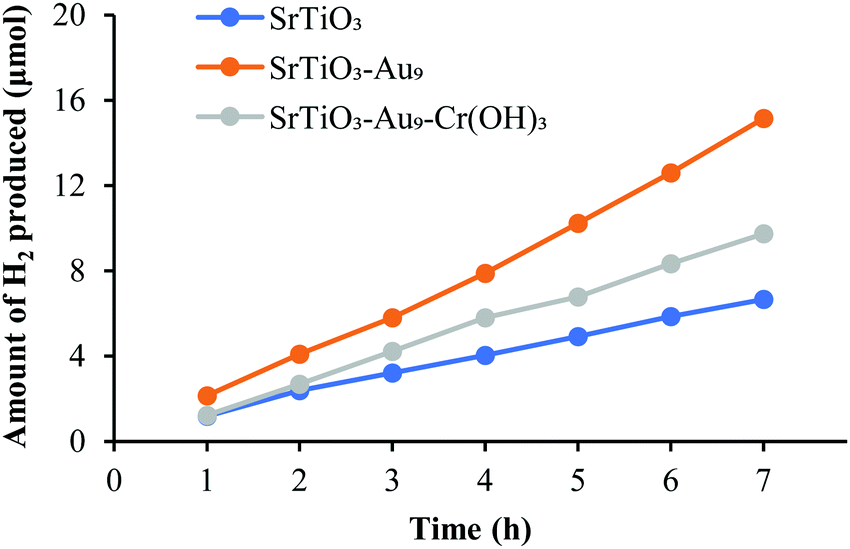 | ||
| Fig. 7 H2 evolution by overall photocatalytic water splitting of SrTiO3, SrTiO3-Au9 and SrTiO3-Au9-Cr2O3 (i.e. after heating to remove the ligands). | ||
While the addition of a CrOx overlayer is expected to increase the H2 production rate for the overall water splitting reaction due to blocking the back reaction,29–38 there is a possibility for the observed decrease in H2 production rate due to an even distribution and a too thick Cr2O3 overlayer on the surface of the photocatalyst (SrTiO3). STEM images shows that Cr2O3 is deposited in a homogeneous layer with a thickness of ca. 1.6 nm across the catalyst (Fig. 4). Having a uniform and too thick Cr2O3 layer covering the entire surface could result in stopping the oxidation reaction to occur through blocking the respective sites on the catalyst surface, which blocks the overall water splitting reaction. Kurashige et al. noted a similar result in their study of an Au25-CrOx-BaLa4Ti4O15 system in which CrOx was deposited at various concentrations (0.1–1.5 wt%).19 The authors found that higher Cr contents led to an increase in the coverage and thickness of the CrOx layer (1.2–2.0 nm), which significantly decreased the water splitting rate. In the above-mentioned studies, this decrease in activity was assumed to be due to a reduction in the numbers of O2 generation sites.19 This shows that the amount of CrOx deposited impacts here the O reduction sites rather the H evolution sites.
After photocatalysis
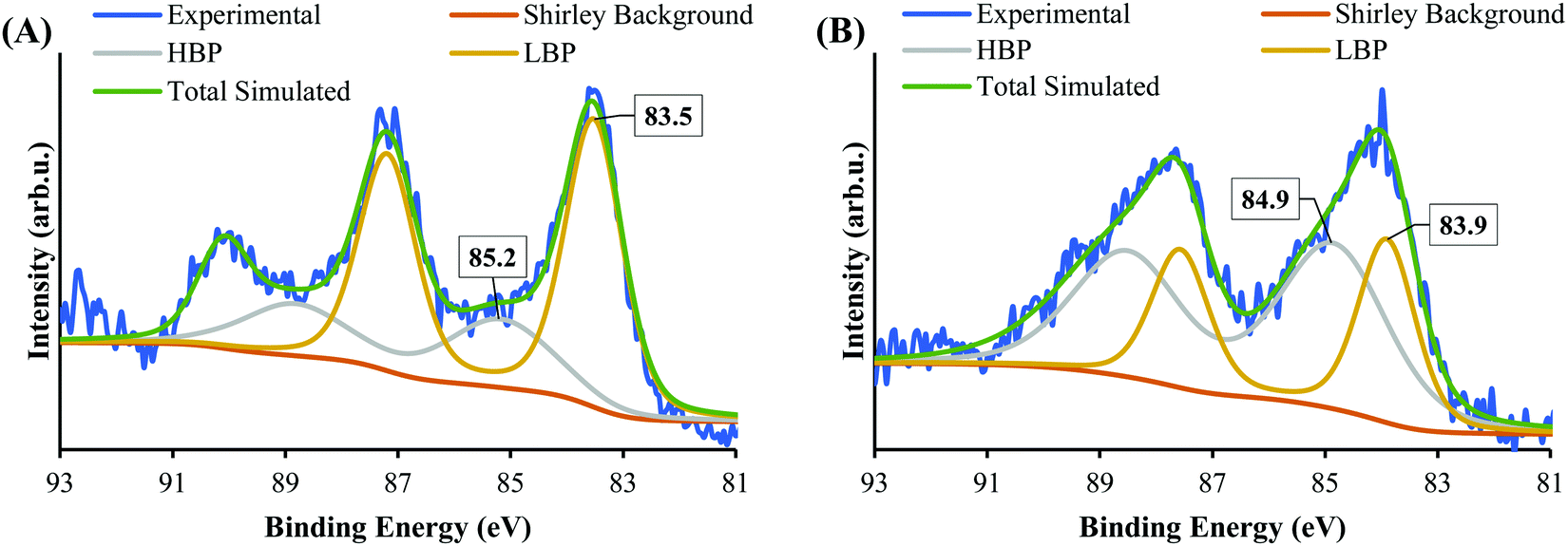 | ||
| Fig. 8 XPS spectra of Au 4f after 7 h irradiation of (A) SrTiO3-Au9 and (B) SrTiO3-Au9-CrOx. | ||
XPS Cr 2p spectra of SrTiO3-Au9-CrOx before and after heating and after 7 h irradiation, with reference spectra of Cr2O3 and Cr(OH)3, are shown in Fig. 9 and summarised in Table S4 (ESI†). After 7 h irradiation, the Cr 2p3/2 peak for SrTiO3-Au9-CrOx appears at 577.4 ± 0.2 eV (Fig. S7, ESI†), corresponding to Cr(OH)3, indicating that the Cr layer converts back to Cr(OH)3 during photocatalysis.16
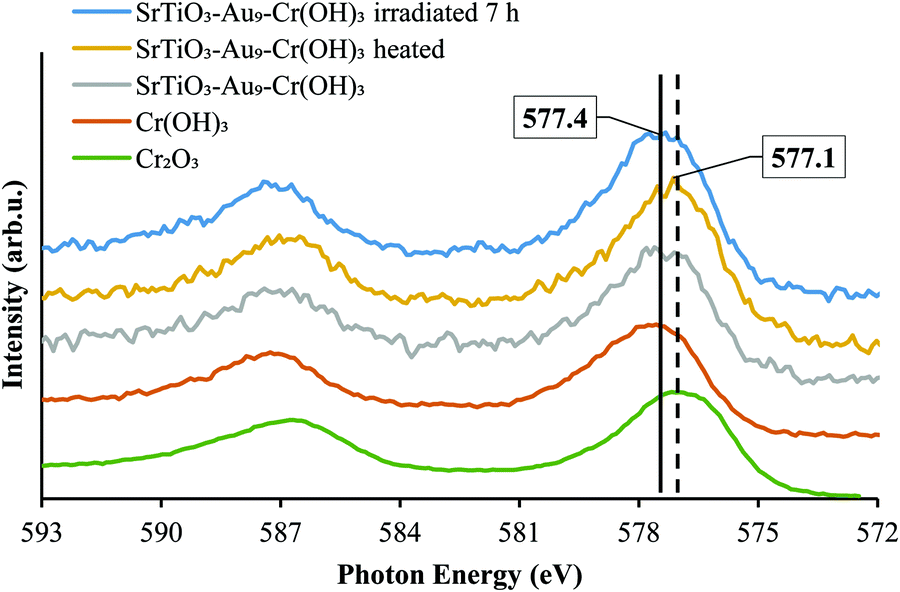 | ||
| Fig. 9 XPS spectra of Cr 2p of SrTiO3-Au9-Cr(OH)3 before heating, SrTiO3-Au9-Cr2O3 after heating and SrTiO3-Au9-CrOx after 7 h irradiation with reference spectrum of Cr2O3 and Cr(OH)3 (Fig. S7, ESI† shows complete fitting to all components). | ||
A HAADF-STEM image of SrTiO3-Au9 after 7 h irradiation with EDX elemental mapping of Au and Ti is shown in Fig. 10 (and summarised in Table S1, ESI†). The image shows that the Au9 clusters have agglomerated into large particles after 7 h irradiation with no Au clusters left on the surface. The line analysis of Au particles (L1 and L2) confirmed the size of Au particles as approximately 3.9–4.6 nm, with some Au particles having a size of 5.0–6.9 nm (as indicated with arrows in Fig. 10D).
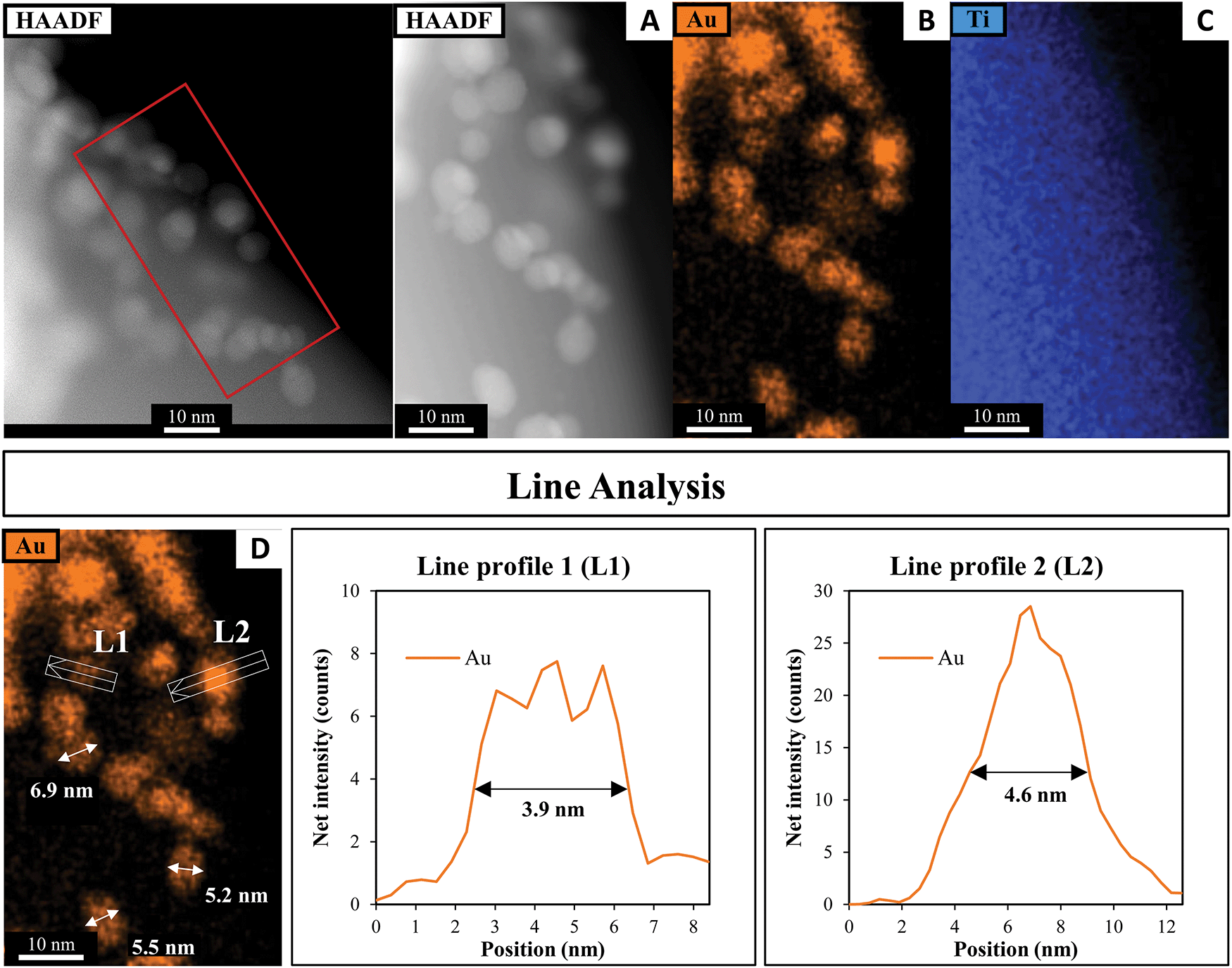 | ||
| Fig. 10 (A) HAADF-STEM image with EDX elemental mapping of (B) Au and (C) Ti in SrTiO3-Au9 after 7 h irradiation and (D) line analyses of the Au signal. | ||
A HAADF-STEM image of SrTiO3-Au9-Cr(OH)3 after 7 h irradiation with EDX elemental mapping of Au, Cr and Ti is presented in Fig. 11. The typical size of the Au clusters as determined using line analysis (L1 and L2) range from 1.4 to 1.6 nm. This size is nearly identical to Au clusters determined before heating and photocatalysis irradiation (Fig. 4 and Fig. S8, ESI†). Note that STEM-EDX elemental mapping shows that some Au features are 2–2.7 nm in size. As discussed above, the slight increase in the size of the Au clusters is assumed to be caused either by adjacent clusters or agglomeration of Au9 clusters due to the effect of the STEM electron beam (this is further discussed around Fig. S2, ESI†). As outlined above, XPS is averaged over a significant larger number of Au clusters and is thus considered as being a more representative analysis whereas STEM is averaged over a small selected area of the sample and also causes beam damage. Through XPS analysis it is shown that 63% of Au clusters remain non-agglomerated (Fig. 8B and Table S1, ESI†).
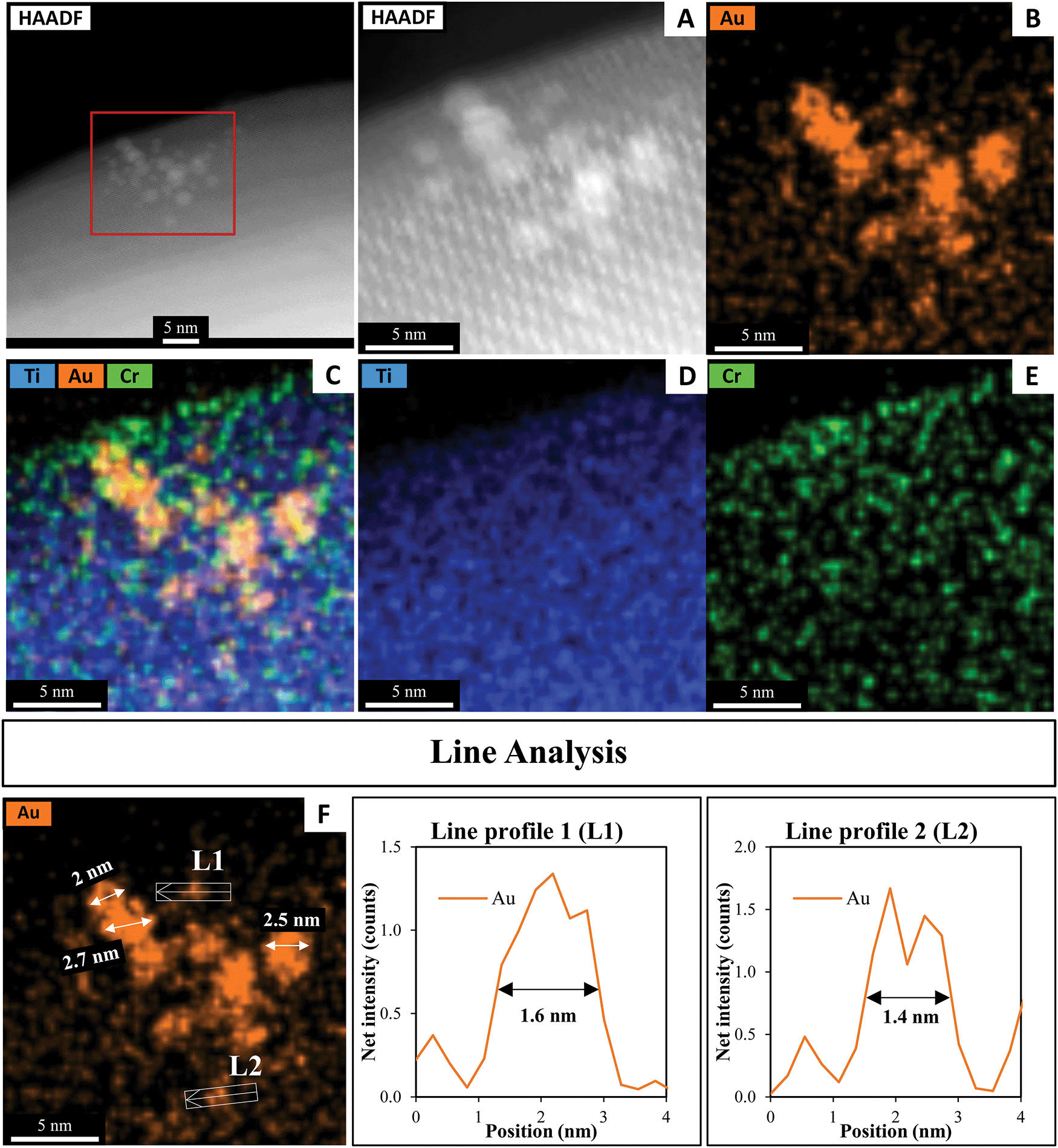 | ||
| Fig. 11 (A) HAADF-STEM image with EDX elemental mapping of (B) Au, (C) Ti. Au and Cr, (D) Ti and (E) Cr in SrTiO3-Au9-Cr(OH)3 after 7 h irradiation and (F) line analyses of the Au signal. | ||
Conclusions
We have demonstrated that application of a Cr(OH)3 overlayer before heating and photocatalytic reaction protects the phosphine-ligated Au9 clusters from agglomeration after the removal of the ligands. The photocatalytic activity of Au clusters deposited on SrTiO3 was investigated as well as the influence of the addition of a Cr2O3 overlayer. For SrTiO3-Au9, the Au clusters agglomerated, forming large particles (up to 8 nm in size) after the overall photocatalytic water splitting reaction. The results show that only 30% of the Au clusters remain non-agglomerated on the SrTiO3 surface after 7 h irradiation. When a Cr(OH)3 overlayer was added, more than 60% Au clusters on the surface of SrTiO3 remained non-agglomerated after heating and with 7 h of photocatalytic water splitting under UV irradiation. The Cr(OH)3 layer was converted to Cr2O3 upon heating and was found to be returned to Cr(OH)3 after photocatalytic water splitting. The H2 production rate reduced after photodeposition of a Cr(OH)3 layer. It is assumed that the decrease of the H2 production is due to the even distribution and thickness of the Cr2O3 layer on the surface of the semiconductor photocatalyst, blocking the O2 generating sites then leading to a decrease of the overall photocatalytic water splitting reaction. Although the deposition of a Cr(OH)3 protective layer has been demonstrated for Au9 clusters deposited onto a SrTiO3 substrate, this approach could be applied to other sized Au clusters on other metal oxide substrates.Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Part of this research was undertaken on the soft X-ray spectroscopy beamline at the Australian Synchrotron, Victoria, Australia (AS1/SXR/15819). We would like to thank Dr Bruce Cowie from the Australian Synchrotron for his assistance. The work is supported by the US army project FA5209-16-R-0017. This research was performed as part of the Australian Solar Thermal Research Institute (ASTRI), a project supported by the Australian Government, through the Australian Renewable Energy Agency (ARENA). Abdulrahman Alotabi acknowledges Albaha University for providing scholarship support. The authors acknowledge the facilities, and the scientific and technical assistance, of Microscopy Australia (formerly known as AMMRF) and the Australian National Fabrication Facility (ANFF) at Flinders University. The authors acknowledge Flinders Microscopy and Microanalysis and their expertise. We thank Dr Ashley Slattery from Adelaide Microscopy for the STEM measurements and A/Prof Vladimir Golovko (Canterbury University) for providing the Au9(PPh3)8(NO3)3 clusters.References
- V. Borman, M. Pushkin, V. Tronin and V. Troyan, J. Exp. Theor. Phys., 2010, 110, 1005–1025 CrossRef CAS.
- V. Sudheeshkumar, K. O. Sulaiman and R. W. J. Scott, Nanoscale Adv., 2020, 2, 55–69 RSC.
- R. Jin, Nanoscale, 2010, 2, 343–362 RSC.
- T. Castro, R. Reifenberger, E. Choi and R. P. Andres, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 8548–8556 CrossRef CAS PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- L. Howard-Fabretto and G. G. Andersson, Adv. Mater., 2020, 32, 1904122 CrossRef CAS PubMed.
- A. Alarawi, V. Ramalingam and J.-H. He, Mater. Today Energy, 2019, 11, 1–23 CrossRef CAS.
- W. Kurashige, Y. Mori, S. Ozaki, M. Kawachi, S. Hossain, T. Kawawaki, C. J. Shearer, A. Iwase, G. F. Metha, S. Yamazoe, A. Kudo and Y. Negishi, Angew. Chem., Int. Ed., 2020, 59, 7076–7082 CrossRef CAS PubMed.
- S. Yang, S. Ding, C. Zhao, S. Huo, F. Yu, J. Fang and Y. Yang, J. Mater. Sci., 2021, 56, 13736–13751 CrossRef CAS.
- C. Wang, P. Lv, D. Xue, Y. Cai, X. Yan, L. Xu, J. Fang and Y. Yang, ACS Sustainable Chem. Eng., 2018, 6, 8447–8457 CrossRef CAS.
- W. Kurashige, Y. Niihori, S. Sharma and Y. Negishi, Coord. Chem. Rev., 2016, 320-321, 238–250 CrossRef CAS.
- Y. Negishi, M. Mizuno, M. Hirayama, M. Omatoi, T. Takayama, A. Iwase and A. Kudo, Nanoscale, 2013, 5, 7188–7192 RSC.
- Y. Negishi, Y. Matsuura, R. Tomizawa, W. Kurashige, Y. Niihori, T. Takayama, A. Iwase and A. Kudo, J. Phys. Chem. C, 2015, 119, 11224–11232 CrossRef CAS.
- W. Kurashige, R. Hayashi, K. Wakamatsu, Y. Kataoka, S. Hossain, A. Iwase, A. Kudo, S. Yamazoe and Y. Negishi, ACS Appl. Energy Mater., 2019, 2, 4175–4187 CrossRef CAS.
- T. Kawawaki, Y. Kataoka, M. Hirata, Y. Akinaga, R. Takahata, K. Wakamatsu, Y. Fujiki, M. Kataoka, S. Kikkawa, A. S. Alotabi, S. Hossain, D. J. Osborn, T. Teranishi, G. G. Andersson, G. F. Metha, S. Yamazoe and Y. Negishi, Angew. Chem., Int. Ed., 2021, 60, 21340–21350 CrossRef CAS PubMed.
- G. G. Andersson, V. B. Golovko, J. F. Alvino, T. Bennett, O. Wrede, S. M. Mejia, H. S. Al Qahtani, R. Adnan, N. Gunby and D. P. Anderson, J. Chem. Phys., 2014, 141, 014702 CrossRef PubMed.
- W. Kurashige, R. Kumazawa, Y. Mori and Y. Negishi, J. Mater. Appl., 2018, 7, 1–11 Search PubMed.
- W. Kurashige, R. Kumazawa, D. Ishii, R. Hayashi, Y. Niihori, S. Hossain, L. V. Nair, T. Takayama, A. Iwase, S. Yamazoe, T. Tsukuda, A. Kudo and Y. Negishi, J. Phys. Chem. C, 2018, 122, 13669–13681 CrossRef CAS.
- C.-L. Tan, F. Zhang, Y.-H. Li, Z.-R. Tang and Y.-J. Xu, Res. Chem. Intermed., 2021, 47, 29–50 CrossRef CAS.
- R. M. Adnan, M. Jenica, A. S. Alotabi, G. F. Metha and G. G. Andersson, Adv. Sci., 2022 DOI:10.1002/advs.202105692.
- G. Krishnan, N. Eom, R. M. Kirk, V. B. Golovko, G. F. Metha and G. G. Andersson, J. Phys. Chem. C, 2019, 123, 6642–6649 CrossRef CAS.
- G. Krishnan, H. S. Al Qahtani, J. Li, Y. Yin, N. Eom, V. B. Golovko, G. F. Metha and G. G. Andersson, J. Phys. Chem. C, 2017, 121, 28007–28016 CrossRef CAS.
- B. Weng, K.-Q. Lu, Z. Tang, H. M. Chen and Y.-J. Xu, Nat. Commun., 2018, 9, 1543 CrossRef PubMed.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. C, 2007, 111, 7554–7560 CrossRef CAS.
- N. Sakamoto, H. Ohtsuka, T. Ikeda, K. Maeda, D. Lu, M. Kanehara, K. Teramura, T. Teranishi and K. Domen, Nanoscale, 2009, 1, 106–109 RSC.
- M. Yoshida, K. Takanabe, K. Maeda, A. Ishikawa, J. Kubota, Y. Sakata, Y. Ikezawa and K. Domen, J. Phys. Chem. C, 2009, 113, 10151–10157 CrossRef CAS.
- K. Maeda, N. Sakamoto, T. Ikeda, H. Ohtsuka, A. Xiong, D. Lu, M. Kanehara, T. Teranishi and K. Domen, Chem. – Eur. J., 2010, 16, 7750–7759 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, H. Masuda, T. Takata, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. B, 2006, 110, 13107–13112 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. B, 2006, 110, 13753–13758 CrossRef CAS PubMed.
- K. Maeda, A. Xiong, T. Yoshinaga, T. Ikeda, N. Sakamoto, T. Hisatomi, M. Takashima, D. Lu, M. Kanehara, T. Setoyama, T. Teranishi and K. Domen, Angew. Chem., Int. Ed., 2010, 49, 4096–4099 CrossRef CAS PubMed.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- K. Maeda, D. Lu and K. Domen, Chem. – Eur. J., 2013, 19, 4986–4991 CrossRef CAS PubMed.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- J. Soldat, G. W. Busser, M. Muhler and M. Wark, ChemCatChem, 2016, 8, 153–156 CrossRef CAS.
- K. E. Sanwald, T. F. Berto, A. Jentys, D. M. Camaioni, O. Y. Gutiérrez and J. A. Lercher, ACS Catal., 2018, 8, 2902–2913 CrossRef CAS.
- M. Qureshi, T. Shinagawa, N. Tsiapis and K. Takanabe, ACS Sustainable Chem. Eng., 2017, 5, 8079–8088 CrossRef CAS.
- R. Pang, K. Teramura, H. Tatsumi, H. Asakura, S. Hosokawa and T. Tanaka, Chem. Commun., 2018, 54, 1053–1056 RSC.
- B. Tian, W. Gao, X. Zhang, Y. Wu and G. Lu, Appl. Catal., B, 2018, 221, 618–625 CrossRef CAS.
- Z. Li, F. Zhang, J. Han, J. Zhu, M. Li, B. Zhang, W. Fan, J. Lu and C. Li, Catal. Lett., 2018, 148, 933–939 CrossRef CAS.
- G. W. Busser, B. Mei, P. Weide, P. C. K. Vesborg, K. Stührenberg, M. Bauer, X. Huang, M.-G. Willinger, I. Chorkendorff, R. Schlögl and M. Muhler, ACS Catal., 2015, 5, 5530–5539 CrossRef CAS.
- J. A. Bau and K. Takanabe, ACS Catal., 2017, 7, 7931–7940 CrossRef CAS.
- A. A.-O. X. Garcia-Esparza, T. A.-O. Shinagawa, S. A.-O. Ould-Chikh, M. Qureshi, X. Peng, N. Wei, D. H. Anjum, A. Clo, T. C. Weng, D. Nordlund, D. Sokaras, J. Kubota, K. Domen and K. A.-O. Takanabe, Angew. Chem., Int. Ed., 2017, 56(21), 5780–5784, DOI:10.1002/anie.201701861.
- T. Takata, C. Pan, M. Nakabayashi, N. Shibata and K. Domen, J. Am. Chem. Soc., 2015, 137, 9627–9634 CrossRef CAS PubMed.
- M. Yoshida, K. Maeda, D. Lu, J. Kubota and K. Domen, J. Phys. Chem. C, 2013, 117, 14000–14006 CrossRef CAS.
- K. Konishi, in Gold Clusters, Colloids and Nanoparticles I, ed. D. M. P. Mingos, Springer International Publishing, Cham, 2014 DOI:10.1007/430_2014_143, pp. 49–86.
- F. Wen, U. Englert, B. Gutrath and U. Simon, Eur. J. Inorg. Chem., 2008, 106–111 CrossRef CAS.
- A. S. Alotabi, Y. Yin, A. Redaa, S. Tesana, G. F. Metha and G. G. Andersson, J. Chem. Phys., 2021, 155, 164702 CrossRef CAS PubMed.
- R. G. Acres, A. V. Ellis, J. Alvino, C. E. Lenahan, D. A. Khodakov, G. F. Metha and G. G. Andersson, J. Phys. Chem. C, 2012, 116, 6289–6297 CrossRef CAS.
- D. Briggs, Auger and X-Ray Photoelecton Spectroscory, 1990, 1, 151–152 Search PubMed.
- I. M. Band, Y. I. Kharitonov and M. B. Trzhaskovskaya, At. Data Nucl. Data Tables, 1979, 23, 443–505 CrossRef CAS.
- D. P. Anderson, J. F. Alvino, A. Gentleman, H. Al Qahtani, L. Thomsen, M. I. Polson, G. F. Metha, V. B. Golovko and G. G. Andersson, Phys. Chem. Chem. Phys., 2013, 15, 3917–3929 RSC.
- D. P. Anderson, R. H. Adnan, J. F. Alvino, O. Shipper, B. Donoeva, J.-Y. Ruzicka, H. Al Qahtani, H. H. Harris, B. Cowie and J. B. Aitken, Phys. Chem. Chem. Phys., 2013, 15, 14806–14813 RSC.
- J.-Y. Ruzicka, F. Abu Bakar, C. Hoeck, R. Adnan, C. McNicoll, T. Kemmitt, B. C. Cowie, G. F. Metha, G. G. Andersson and V. B. Golovko, J. Phys. Chem. C, 2015, 119, 24465–24474 CrossRef CAS.
- H. S. Al Qahtani, K. Kimoto, T. Bennett, J. F. Alvino, G. G. Andersson, G. F. Metha, V. B. Golovko, T. Sasaki and T. Nakayama, J. Chem. Phys., 2016, 144, 114703 CrossRef PubMed.
- H. S. Al Qahtani, G. F. Metha, R. B. Walsh, V. B. Golovko, G. G. Andersson and T. Nakayama, J. Phys. Chem. C, 2017, 121, 10781–10789 CrossRef CAS.
- S. Zong, L. Tian, X. Guan, C. Cheng, J. Shi and L. Guo, J. Colloid Interface Sci., 2022, 606, 491–499 CrossRef CAS PubMed.
- A. S. Alotabi, C. T. Gibson, G. F. Metha and G. G. Andersson, ACS Appl. Energy Mater., 2021, 4, 322–330 CrossRef CAS.
- J. F. Moulder, Handbook of X-ray photoelectron spectroscopy, 1995 Search PubMed.
- H. Mousavi, Y. Yin, L. Howard-Fabretto, S. K. Sharma, V. Golovko, G. G. Andersson, C. J. Shearer and G. F. Metha, Nanoscale Adv., 2021, 3, 1422–1430 RSC.
- W. Jianjun and X. Qunji, Wear, 1994, 176, 213–216 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: The HAADF-STEM images of in situ STEM measurements of the Au9 clusters, the details of the XPS 4f Au spectra, their fitting and quantification and results from the water splitting reaction with and without addition of air (oxygen). See DOI: 10.1039/d1ma01226f |
| This journal is © The Royal Society of Chemistry 2022 |