
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ growth of SeOx films on the surface of Ni–Fe–selenide nanosheets as highly active and stable electrocatalysts for the oxygen evolution reaction†
Xiaonan
Shang
a,
Weiheng
Chen
c,
Zhong-Jie
Jiang
*b,
Changsheng
Song
 *a and
Zhongqing
Jiang
*a
*a and
Zhongqing
Jiang
*a
aKey Laboratory of Optical Field Manipulation of Zhejiang Province, Department of Physics, Zhejiang Sci-Tech University, Hangzhou 310018, P. R. China. E-mail: zhongqingjiang@zstu.edu.cn
bGuangzhou Key Laboratory for Surface Chemistry of Energy Materials & Guangdong Engineering and Technology Research Center for Surface Chemistry of Energy Materials, New Energy Research Institute, College of Environment and Energy, South China University of Technology, Guangzhou 510006, P. R. China. E-mail: eszjiang@scut.edu.cn; zhongjiejiang1978@hotmail.com
cDepartment of Mechanical Engineering, Ningbo University of Technology, Ningbo 315336, P. R. China
First published on 26th January 2022
Abstract
Reasonable design, extremely low-cost, high efficiency and high durability oxygen evolution reaction (OER) catalysts are essentially required to promote the practical large-scale applications of electrocatalytic water splitting for the efficient production of hydrogen. Herein, a rapid, facile, green and controllable electrochemical deposition method is reported for the growth of SeOx films on the surface of Ni–Fe–selenide nanosheets (SeOx/FeNixSe) through the hydrogen evolution reaction (HER) cycle process, which are used as highly active and stable electrocatalysts for the OER. The obtained SeOx/FeNixSe heterojunction nanosheets have interconnected heterostructures, which play a vital role in boosting the catalytic performance by offering rich active sites and convenient pathways for rapid electron transport. Detailed spectroscopic and DFT investigations disclose that the strong electron interaction between FeNixSe and SeOx could reduce the surface charge-transfer-resistance, and improve the electrocatalytic properties and the structural stability of the electrode. The optimized SeOx/FeNixSe (CM7-FeNixSe) catalyst exhibits superior OER activities to the commercial Ir and most of the reported Ni-based selenide catalysts with a small overpotential of 285 mV to achieve a current density of 200 mA cm−2 in 1.0 M KOH. This finding provides a new ultrathin oxide layer preparation strategy to boost the OER catalytic activity.
Introduction
Electrocatalytic water splitting is one of the most promising strategies for the efficient production of hydrogen with environmental benignity and low-cost consumption.1 As a pivotal half reaction in water-splitting, the anodic oxygen evolution reaction (OER) is more complex than the cathodic hydrogen evolution reaction (HER) due to the four electron transfer processes with high overpotential, which greatly hinders its large-scale industrial application.2–5 Although noble metal-based Ir or RuO2 catalysts can significantly reduce the overpotential and accelerate the kinetics of the OER, the high cost impedes their extensive applications.6–10 Thus, over the past few years, many earth-abundant materials with lower cost have been developed to improve the reaction efficiency of the OER, such as transition metal-based selenide,11,12 nitride,13–15 sulfide,16,17 phosphide,18,19 and oxide20–22 supported non-metallic carbon compound electrocatalysts.23,24Among them, transition metal-based selenides have been reported as prospective OER catalysts due to their intrinsically metallic conductive properties which would efficiently enhance their electron transfer capability during the catalytic reaction process.25,26 However, further research in recent years has shown that transition metal-based selenides will transform into metal oxides and/or (oxy)hydroxides during the catalytic process under alkaline conditions, thus showing low stability under alkaline conditions, which negate the catalytic ability of transition metal-based selenides themselves. As a result, the oxide species formed in situ during catalytic reactions are almost invariably identified as the real active species.27–29 Although such a change is conducive to the improvement of catalytic performance, the content of oxides is uncontrollable. On the one hand, too much oxides will reduce the conductivity of the materials; on the other hand, phase changes during the cycling process will affect the stability of the materials, which may lead to structural collapse. It has been demonstrated that the catalytic reaction is dependent on the electrochemically active sites on the surface of materials. Therefore, for the metal oxides converted from selenides in the catalytic reaction process, the surface oxides are key to improving the catalytic performance of the materials, while the internal oxides do not play a major role in the catalytic reaction due to their intrinsic poor conductivity, which is not conducive to electron conduction. Based on the above-mentioned analysis, we propose the formation of a heterostructure by growing an ultrathin metal oxide layer on selenides to improve the catalytic activity without affecting the conductivity of materials. Various different protocols are available for growing oxide films over substrates. The traditional preparation methods, such as the hydrothermal method and high temperature calcination method, have low controllability, which is quite challenging to control the content and thickness of the produced oxide layer. Several recent studies have shown that a thin layer is formed on the surface of the metal phosphates during their HER performance testing.30 Inspired by this, in this work, selenides were used as the substrate and precursor. The electrochemical deposition process was used as a means for the in situ growth of a layer of ultra-thin oxides on the surface of selenides to obtain oxide/selenide heterojunction structures.
With this in mind, herein, three-dimensional (3D) nanosheets with interconnected SeOx/FeNixSe heterostructures are fabricated by the electrochemical deposition process through the hydrogen evolution reaction (HER) cycle process using FeNixSe as the precursor and NiFe foam as the conductive substrate. A single electrochemical deposition process takes only 90 s for completion. Attractively, this electrochemical deposition process effectively avoids the use of any additional solvents and reactants, and a thin layer of SeOx with optimized thickness can be obtained by performing the electrochemical deposition process 7 times. Thus, it is a rapid, facile, green and controllable preparation method. The optimized ultra-thin SeOx film (∼8 nm) heterogenized with FeNixSe nanosheets could considerably enhance the active sites and increase the OER catalytic activity. DFT calculation and spectroscopy analysis have confirmed that the synergistic effect between inner FeNixSe and surface SeOx can reduce the adsorption energy with an oxygenated intermediate and improve the reaction kinetics of the OER. Moreover, the thin layer of SeOx on the surface can prevent continued oxidation of FeNixSe in the subsequent OER test, which improves the structural stability and electrochemical properties. The obtained 3D nanosheets with interconnected heterostructures could offer a large interfacial contact area with electrolytes and a convenient pathway for rapid electron transport. The optimized SeOx/FeNixSe (CM7-FeNixSe) catalyst exhibits superior OER activities to the commercial Ir and most of the reported Ni-based selenide catalysts with a small overpotential of 285 mV to achieve 200 mA cm−2 current density in 1.0 M KOH.
Experimental section
Synthesis of FeNixSe nanosheet arrays with interconnected structures
FeNixSe nanosheet arrays were synthesized by a facile solvothermal reaction and in situ selenization process. A piece of NiFe foam (3 cm × 3 cm) was cleaned by sonicating it in 2.0 M HCl and ethanol for 10 min, respectively. 5 mmol nickel nitrate hexahydrate and 40 mmol urea were added into 70 mL distilled water under stirring for 30 min to form a uniform solution. Then, the above solution and the clean NiFe foam were transferred to a 100 mL Teflon-lined stainless steel autoclave and heated to 135 °C for 12 h. After being cooled to room temperature, FeNi layered double hydroxide (FeNi-LDH) grown on the surface of the NiFe foam was obtained by washing with deionized water and drying naturally. For further selenization, 5 mmol Se powder was added into a 50 mL solution containing 5 mL hydrazine hydrate and 45 mL deionized water under stirring for 60 min. Subsequently, the NiFe foam supported FeNi-LDH was immersed into the mixed solution and transferred into a 100 mL Teflon-lined stainless steel autoclave. After the hydrothermal reaction at 120 °C for 48 h, the FeNixSe nanosheet was formed on the surface of the NiFe foam.Synthesis of SeOx/FeNixSe heterojunctions
Three-dimensional (3D) SeOx/FeNixSe nanosheet heterojunctions with interconnected heterostructures were fabricated by the electrodeposition strategy through the HER cycle process in a standard three-electrode system. Typically, a three-electrode system was connected to an electrochemical workstation using Pt foil as the counter electrode, a saturated calomel electrode (SCE) as the reference electrode, NiFe foam supported FeNixSe (1 cm × 1 cm) as the working electrode, respectively. The electrodeposition strategy was carried out through the HER cycle process in 1 M KOH. The SeOx films with different thicknesses grown on the surface of FeNixSe nanosheets can be obtained by controlling the times of the HER cycle process. The obtained SeOx/FeNixSe nanosheet heterojunction is named CMy-FeNixSe (y represents the HER cycle times). The prepared SeOx/FeNixSe samples do not need any washing and drying processes, and the OER performance can be tested after electrodeposition directly.Characterization
The crystal phase composition of the samples was ascertained by X-ray diffraction (XRD) using Cu Kα radiation with a scan rate of 4° min−1 from 10° to 80°. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) coupled with elemental mapping were used to characterize the morphologies of the products. The micro-nanostructures were recorded using HRTEM. X-ray photoelectron spectroscopy (XPS) was performed on a Kratos Analytical AXIS Supra system. The details of the electrocatalytic measurements and theoretical calculation are shown in the ESI.†Results and discussion
In situ growth of SeOx films on the surface of Ni–Fe–selenide nanosheets and catalyst characterization
The synthetic route of SeOx/FeNixSe (named CMy-FeNixSe, y represents HER cycle times) 3D nanosheets with interconnected heterostructures is illustrated in Scheme 1. Specifically, FeNi-LDH was firstly synthesized via a facile solvothermal reaction. Then, an in situ selenization process was applied to obtain Ni–Fe–selenide (FeNixSe) nanosheets. Finally, CMy-FeNixSe was successfully fabricated by the electrodeposition strategy through different HER cycle times in a standard three-electrode system. SEM images (Fig. S1a, ESI†) showed that ultra-thin FeNi-LDH with a smooth and flat surface was uniformly distributed on the NiFe foam, and interweaved with each other to form a 3D interconnected network structure. This special architecture was conducive to increasing the number of active sites and facilitating electron transport. The TEM images (Fig. S2, ESI†) further demonstrate the 3D interconnected network-like structure of FeNi-LDH, which is consistent with the SEM results. It is worth noting that there is a porous structure inside the nanosheets (Fig. S2, ESI†), which is conducive to the diffusion of the electrolyte. After selenization, the FeNixSe nanosheets maintain their 3D interconnected structure. However, the nanosheets become thicker and coarser, and the secondary structure of the nanosheet is composed of a large number of nanoparticles as shown in Fig. 1a. Elemental mapping (Fig. 1d1), energy dispersive X-ray spectroscopy (EDX) and percentage of elemental weight (Fig. S3, ESI†) show the uniform distribution of Ni, Fe and Se in FeNixSe nanosheets, and the elemental weight ratio is close to 15.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:22.1. The slight excess O may come from the absorbed species on its surface. The uniform distribution of Fe proves that in the hydrothermal process, the Fe element corroded from the NiFe foam substrate is successfully doped into the nickel hydroxide nanosheet and the content is very low (the atomic ratio of Ni and Fe is close to 15:1). It has been reported that the electrocatalytic performance of Ni-based materials can be greatly improved by doping the Fe element.31 TEM images (Fig. 1b1-2) showed that the diameter of most nanoparticles in FeNixSe is distributed within 50 nm, and they have clear edges (Fig. 1c1). Such a unique structure of nanosheets composed of nanoparticles can further increase the specific surface area and active sites, and effectively increase the electrolyte contact in electrochemical applications. The HRTEM image clearly proves the presence of NiSe with interlayer distances of 0.30 nm and 0.24 nm, corresponding to the (101) and (220) facets of NiSe, respectively. The corresponding selected area electron diffraction (SAED) pattern (Fig. 1c2) also shows the highly crystalline nature of NiSe and Ni0.85Se, and the NiSe is the major phase. Due to the low content of Ni0.85Se, no crystalline interplanar spacing of Ni0.85Se was detected in the HRTEM image. The SEM images of CM7-FeNixSe (Fig. 1a3-4) after electrodeposition show no obvious morphological change, however, from the TEM images (Fig. 1b3-4 and c4) we can see that the edge of the particle becomes unclear and thinner, and a mixed thin layer (∼8 nm) composed of crystalline and amorphous phases appeared on the surface of the original particles, which is attributed to the SeOx and Ni(OH)2/NiOOH produced during the HER cycle process,32 indicating the transformation of the structure starting from the surface to forming the transition metal oxides. According to the HRTEM (Fig. 1c6), the lattice fringes corresponding to the (112) and (202) planes of SeO2 are captured, which are generated during the in situ electro-deposition process. The SAED patterns after electrodeposition exhibit clear rings rather than the scattered dots and blurry rings, which are due to the multiple small domains (Fig. 1c5). The elemental mapping (Fig. 1d2) shows that the content of O increased significantly after the HER cycle process, and the EDX (Fig. S3, ESI†) shows that the atomic ratio of O has increased by 24% after the HER, proving that SeOx was indeed generated in situ on the surface of FeNixSe during the electrodeposition process, which was consistent with the TEM results.
:1:22.1. The slight excess O may come from the absorbed species on its surface. The uniform distribution of Fe proves that in the hydrothermal process, the Fe element corroded from the NiFe foam substrate is successfully doped into the nickel hydroxide nanosheet and the content is very low (the atomic ratio of Ni and Fe is close to 15:1). It has been reported that the electrocatalytic performance of Ni-based materials can be greatly improved by doping the Fe element.31 TEM images (Fig. 1b1-2) showed that the diameter of most nanoparticles in FeNixSe is distributed within 50 nm, and they have clear edges (Fig. 1c1). Such a unique structure of nanosheets composed of nanoparticles can further increase the specific surface area and active sites, and effectively increase the electrolyte contact in electrochemical applications. The HRTEM image clearly proves the presence of NiSe with interlayer distances of 0.30 nm and 0.24 nm, corresponding to the (101) and (220) facets of NiSe, respectively. The corresponding selected area electron diffraction (SAED) pattern (Fig. 1c2) also shows the highly crystalline nature of NiSe and Ni0.85Se, and the NiSe is the major phase. Due to the low content of Ni0.85Se, no crystalline interplanar spacing of Ni0.85Se was detected in the HRTEM image. The SEM images of CM7-FeNixSe (Fig. 1a3-4) after electrodeposition show no obvious morphological change, however, from the TEM images (Fig. 1b3-4 and c4) we can see that the edge of the particle becomes unclear and thinner, and a mixed thin layer (∼8 nm) composed of crystalline and amorphous phases appeared on the surface of the original particles, which is attributed to the SeOx and Ni(OH)2/NiOOH produced during the HER cycle process,32 indicating the transformation of the structure starting from the surface to forming the transition metal oxides. According to the HRTEM (Fig. 1c6), the lattice fringes corresponding to the (112) and (202) planes of SeO2 are captured, which are generated during the in situ electro-deposition process. The SAED patterns after electrodeposition exhibit clear rings rather than the scattered dots and blurry rings, which are due to the multiple small domains (Fig. 1c5). The elemental mapping (Fig. 1d2) shows that the content of O increased significantly after the HER cycle process, and the EDX (Fig. S3, ESI†) shows that the atomic ratio of O has increased by 24% after the HER, proving that SeOx was indeed generated in situ on the surface of FeNixSe during the electrodeposition process, which was consistent with the TEM results.
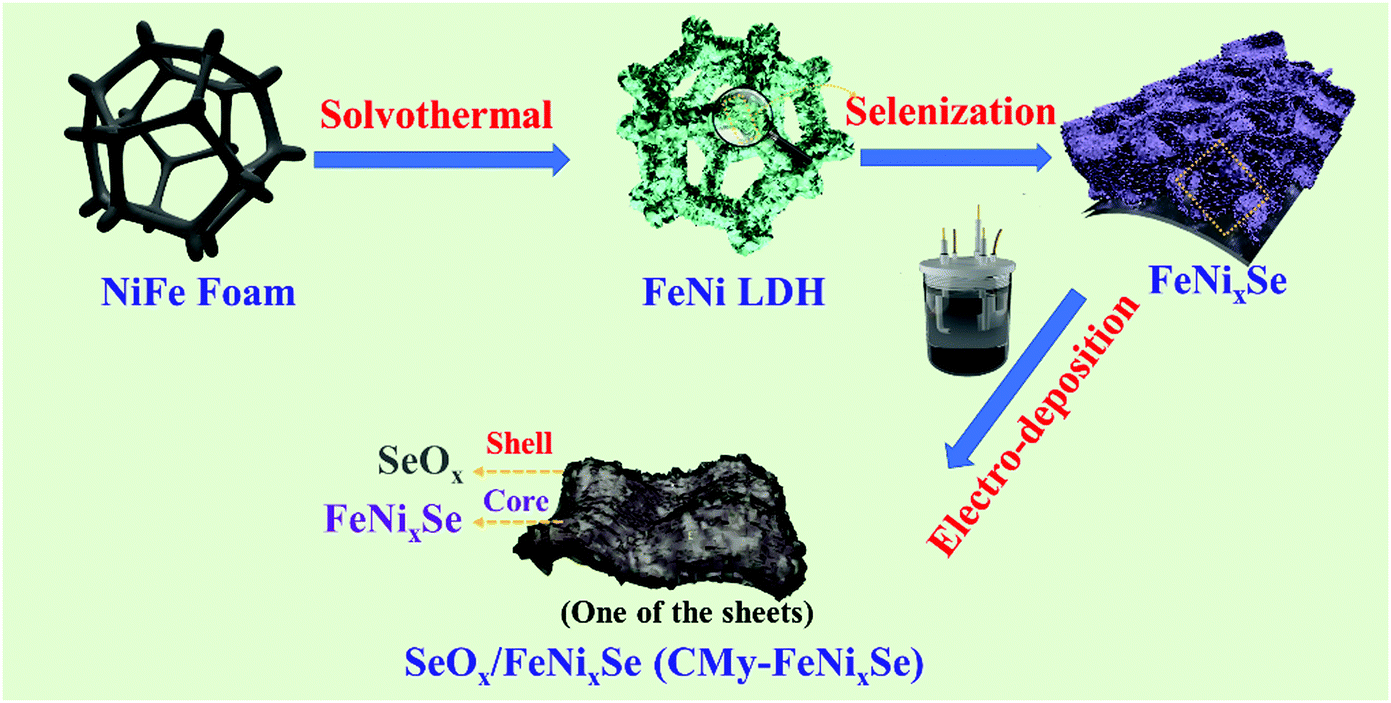 | ||
| Scheme 1 Schematic illustration of the formation process of hierarchical SeOx/FeNixSe (CMy-FeNixSe). | ||
 | ||
| Fig. 1 (a1 and a2) SEM images, (b1 and b2) TEM images, (c1 and c3) HRTEM images, (c2) SAED pattern and (d1) EDX elemental mapping of FeNixSe; (a3 and a4) SEM images, (b3 and b4) TEM images, (c4 and c6) HRTEM images, (c5) SAED pattern and (d2) EDX elemental mapping of CM7-FeNixSe. | ||
Fig. 2a shows the XRD patterns of NiFe Foam, FeNi LDH, FeNixSe and CM7-FeNixSe. The diffraction peaks of NiFe foam match well with those of the FeNi3 alloy (JCPDS no. 38-0419), metallic Ni (JCPDS no. 04-0850) and Fe (JCPDS no. 06-0696). After hydrothermal treatment, the XRD pattern of FeNi LDH shows some new diffraction peaks that appeared at 11.3°, 22.7°, 33.4°, 34.4°, 38.7°, 45.9°, 59.9° and 61.2°, which correspond to the (003), (006), (101), (012), (015), (018), (110) and (113) planes of Ni(OH)2 (PDF 38-0715).33 No diffraction peaks of Fe-related hydroxide can be detected, which may be due to the low content of Fe element, and this result was confirmed by subsequent elemental quantitative analysis of EDX and XPS. Then, an in situ selenization process was applied, and FeNi-LDH was transformed into FeNixSe completely with the appearance of a series of new signal peaks corresponding to the selenides. The characteristic diffraction peaks centered at 2θ = 31.0°, 34.0°, 36.0°, 38.6°, 46.5°, 48.0°, 53.5°, 55.1°, 56.0° and 62.7° in CM7-FeNixSe could be assigned to the (300), (021), (220), (211), (131), (410), (321), (330), (012) and (122) planes of NiSe (JCPDS no. 18-0887),34 and the characteristic diffraction peaks at 2θ = 33.1°, 44.9°, 50.4°, 60.2° and 61.7° can be assigned to the (101), (102), (110), (103) and (201) planes of Ni0.85Se (JCPDS no. 18-0888),35 and both NiSe and Ni0.85Se were found for FeNixSe. We also noticed that no significant changes were found in the diffraction peaks after electro-deposition due to the ultrathin structure and low content of the deposited SeOx layer (Fig. 2a).
 | ||
| Fig. 2 (a) XRD patterns of NiFe foam, FeNi LDH, CM7-FeNixSe and FeNixSe. (b) XPS survey spectra and high-resolution XPS spectra of (c) Ni 2p, (d) Fe 2p, (e) Se 3d and (f) O 1s of NiFe LDH, CM7-NiFe LDH, CM7-FeNixSe and FeNixSe. | ||
XPS analysis confirms the presence of Ni, Se, Fe, O and C in CM7-FeNixSe and FeNixSe (Fig. 2b). For FeNixSe, the peaks at 852.4 and 870.2 eV belong to metallic Ni 2p from the NiFe foam (Fig. 2c).36,37 The peaks at binding energies of 855.5 and 873.6 eV with two shakeup satellites (861.0 and 879.4 eV) can be attributed to the Ni 2p3/2 and Ni 2p1/2 of Ni2+, respectively, which confirm the Ni2+ state in FeNixSe.38 After electro-deposition, the peak intensity of metallic Ni is reduced and almost disappears. This is because of the significant oxidation that occurred on the surface of the FeNixSe during the electro-deposition process, and a layer of SeOx covered the surface of FeNixSe. Since XPS can only detect the surface state of the samples, the covering of the oxide layer will leave the bottom NiFe foam not easy to be detected. For FeNi LDH, the peak shape of Ni 2p (Fig. 2c) and XRD patterns (Fig. S4b, ESI†) before and after the electrodeposition process did not change significantly, proving that there were no significant valence state changes and crystal phase composition changes on the surface of FeNi LDH during the electrodeposition process. This result could also be proved by the EDX analysis (Fig. S2, ESI†) that there is a minor increased proportion of oxygen for CM7-FeNi LDH, which may be attributed to the surface hydroxylation/oxidation during the HER cycle process. For the Fe 2p spectrum (Fig. 2d) in FeNixSe, the binding energies at 707.2 and 719.94 eV conform to Fe2+, and the peaks at 713.5 eV and 727.2 eV belong to Fe3+.39–41 It is worth noting that new peaks in FeNixSe appear at 711.8 and 725.5 eV, which correspond to FeNixSe, and the absence of satellite peaks of Fe2+ and Fe3+ indicates that Fe2+ and Fe3+ ions were incorporated into NixSe.42 After the electrodeposition process, the binding energies of Fe2+ and Fe3+ shift towards higher binding energies, confirming a higher oxidation state of the Fe element.43 As for the Se 3d spectra of FeNixSe (Fig. 2e), two peaks located at 53.9 and 55.3 eV correspond to Se 3d5/2 and Se 3d3/2, respectively, which shows the presence of metal-selenium bonds of Se2−. The small peak at 58.3 eV is associated with trace amounts of SeOx, which is caused by the FeNixSe exposure to air.44,45 After the electrodeposition process, the intensity of the SeOx peak is significantly enhanced, while the peaks of Se 3d3/2 and Se 3d5/2 decrease and appear at positions with slightly higher binding energies (∼0.3 eV), indicating the leaching and oxidation of Se, and the electrons were transferred from FeNixSe to SeOx.46 Combined with the above results, this strongly suggests that there exists a strong electronic coupling between FeNixSe and SeOx. It is further demonstrated that SeOx is produced on the surface of FeNixSe during the electrodeposition process, which is consistent with the above TEM and SEM results. The phenomenon of the surface oxidation process can also be demonstrated by the increased intensity of the lattice oxygen (O2−) peak at 529.9 eV in FeNixSe after the electro-deposition process (CM7-FeNixSe) (Fig. 2f).47 The increased proportion of lattice oxygen is attributed to the formation of Se-O bonds during the electro-deposition process. For FeNi LDH, the O 1s spectrum showed no obvious change before and after the HER cycle process, although the content of oxygen on the surface increased, which may be due to the increased oxygen from surface hydroxylation/oxidation during the electrodeposition process.
Oxygen evolution reaction performance and stability assessment
The electrocatalytic OER performance was measured in 1 M KOH. Fig. 3a shows that after electrodeposition of SeOx, the resultant CM7-FeNixSe exhibits a great enhancement in OER activity. It exhibits the lowest overpotential (η200) of only 285 mV to achieve a current density of 200 mA cm−2, which is much lower than that of the noble metal Ir (η200 = 326 mV), a benchmark catalyst for the OER. The overpotential is reduced by 55 mV compared with that of FeNixSe (η200 = 340 mV). The small oxidation peak at 1.35–1.45 V can be assigned to the oxidation of Ni2+ to Ni3+ or Ni4+ species.48,49 The lower OER overpotential suggests the superior OER activity, which is compared favorably to many other recently reported selenide based high-performance OER catalysts (Table S1, ESI†). To demonstrate the role of electro-deposited SeOx on OER performance improvement, we also tested the OER performance of FeNi LDH before and after the HER cycle process. It shows that the OER performance of the CM7-FeNi LDH electrode is much worse than that of CM7-FeNixSe, but the η200 of CM7-FeNi LDH (η200 = 310 mV) is also reduced by about 30 mV compared with that of FeNi LDH (η200 = 340 mV), which is attributed to the increased active sites obtained from surface hydroxylation/oxidation during the electro-deposition process. In contrast, the CM7-NiFe foam exhibits similar activity (η200 = 364 mV) to the NiFe foam (η200 = 379 mV), which proves that the HER cycle process has no significant influence on the electrocatalytic activity of NiFe foam. It also indirectly proves that the surface state of NiFe foam is relatively stable, and the HER cycle process does not cause the phase change of the surface composition of NiFe foam, which can be further confirmed by the invariant crystal phase composition in the XRD patterns before and after the HER cycle process (Fig. S4a, ESI†). This result proves that the introduction of both SeOx and surface hydroxylation/oxidation can effectively enhance the OER electrocatalytic activity of the materials, but the promotion effect of the former is even more obvious. Thus, it can be inferred that the SeOx formed on the FeNixSe surface during the electro-deposition process is the real active center.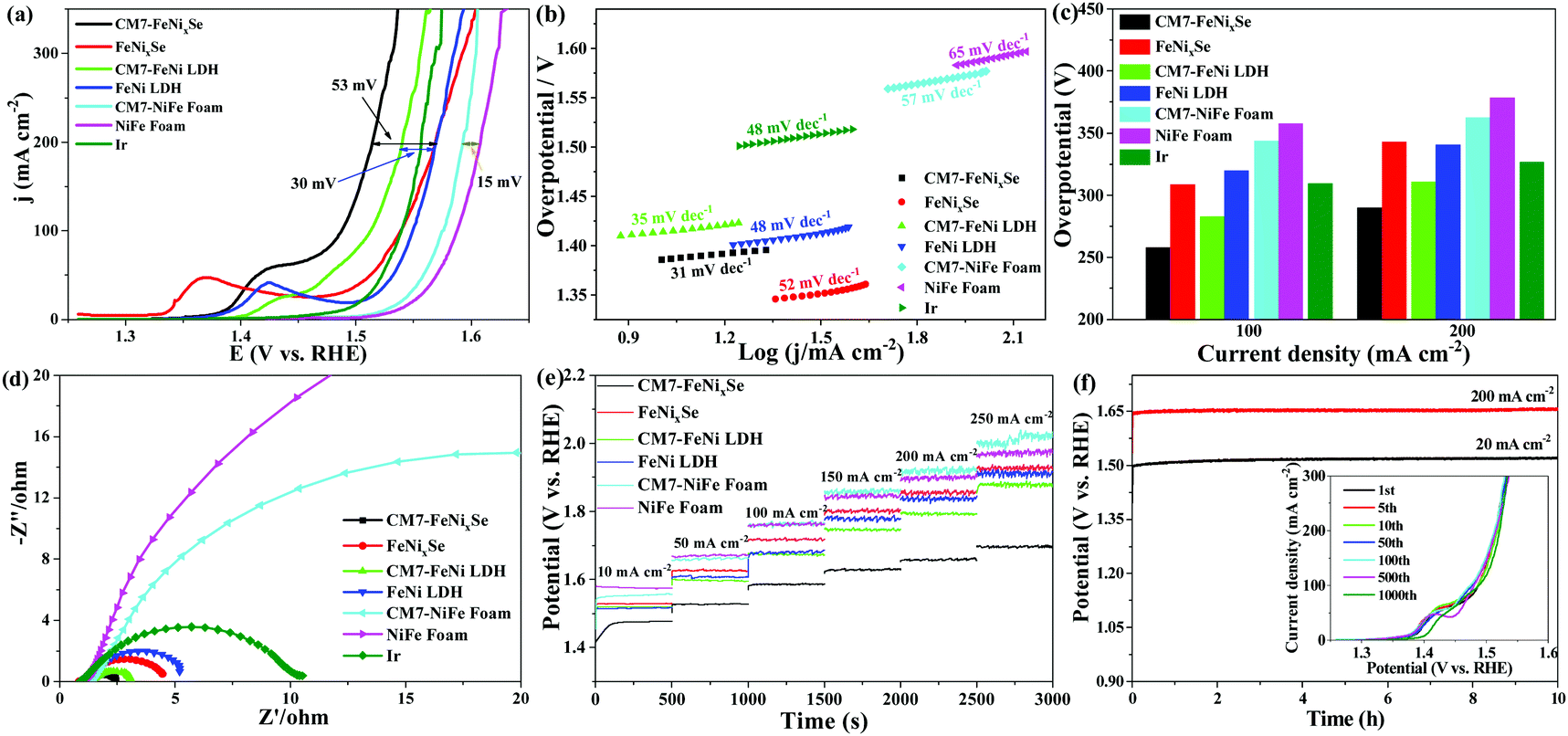 | ||
| Fig. 3 OER performance of NiFe foam, CM7-NiFe foam, FeNi LDH, CM7-FeNi LDH, CM7-FeNixSe, FeNixSe and Ir conducted in 1 M KOH. (a) Polarization curves and (b) the corresponding Tafel plots. (c) Comparison of overpotential values at a current density of 100 mA cm−2 and 200 mA cm−2. (d) Nyquist plots (overpotential = 250 mV). (e) Multi-step chronopotentiometric curve of samples without iR compensation. (f) Chronopotentiometry curves of CM7-FeNixSe at constant current densities of 20 and 200 mA cm−2 without iR compensation. The inset shows the OER LSV curves of the CM7-FeNixSe catalyst after different CV cycles at a scan rate of 20 mV s−1 in a 1 M KOH electrolyte. | ||
The Tafel plots are investigated to reveal the electrocatalytic kinetics for the OER. As can be seen in Fig. 3b, CM7-FeNixSe possesses a lower Tafel value of 31 mV dec−1, which is much smaller than those of FeNixSe (52 mV dec−1), FeNi LDH (48 mV dec−1), CM7-FeNi LDH (35 mV dec−1), NiFe foam (65 mV dec−1), CM7-NiFe foam (57 mV dec−1), and commercial Ir (48 mV dec−1), suggesting the enhanced OER reaction kinetics with the formation of the SeOx layer. For a further clear comparison of these catalysts, the overpotentials with current densities of 100 and 200 mA cm−2 are plotted and shown in Fig. 3c. The CM7-FeNixSe electrode required overpotentials of 258 mV and 285 mV, respectively, which were much lower than other catalysts. Electrochemical impedance spectroscopy (EIS, Fig. 3d) in the frequency range of 0.01–100 kHz was performed to investigate the OER kinetics of these catalysts. As shown in Fig. 3d, after the electrodeposition process, the electron transfer rate of CM7-FeNixSe becomes faster, which proves that a small amount of the SeOx layer on the surface can provide more electrochemically active sites, which is beneficial for the OER reaction. The result can also be proved by subsequent characterization of the electrochemically active surface areas (ECSAs) of the samples. As shown in Fig. S5 and S6 (ESI†), the electrochemical double-layer capacitances (Cdls) of these samples were measured by the CV method and the corresponding ECSAs were calculated to evaluate the OER activity. Fig. S5 (ESI†) presents the CV results of CM7-FeNixSe, FeNixSe, FeNi LDH, CM7-FeNi LDH, pure NiFe foam, and CM7-NiFe foam at different current scan rates. The calculated Cdl values, shown in Fig. S6 (ESI†), for CM7-FeNixSe, FeNixSe, CM7-FeNi LDH, FeNi LDH, CM7-NiFe foam, and NiFe foam are 26.7, 17.1, 4.76, 3.32, 2.35 and 1.42 mF cm−2, respectively. The ECSAs of the CM7-FeNixSe, FeNixSe, CM7-FeNi LDH, FeNi LDH, CM7-NiFe foam, and NiFe foam were calculated to be 667.7, 427.5, 119.0, 83.0, 58.7 and 35.3 cm2, respectively. The result indicates that CM7-FeNixSe has a much higher ECSA than other samples, and the increased ECSA could increase the reaction sites, hence, more catalytic reactions would occur on the surface, which makes a considerable contribution to its higher OER catalytic activity. The catalytic stability and durability of these electrodes for the OER were also evaluated by multi-step chronopotentiometric tests with the current increasing from 10 to 250 mA cm−2 (Fig. 3e). It can be seen that the potential remains stable in each current density range for all the electrodes, confirming the prominent stability of these samples. Among them, CM7-FeNixSe has the lowest overpotential, which indicated its remarkable conductivity and rapid mass transfer capability for the OER. Furthermore, the long-term stability of the CM7-FeNixSe electrode was evaluated by the chronopotentiometric measurement at a current density of 20 and 200 mA cm−2. The chronopotentiometric curves in Fig. 3f show that the potential remained stable over a period of 10 h. Moreover, the inset in Fig. 3f proves that the LSV polarization curves of CM7-FeNixSe after 1000 CV cycles showed negligible changes, further demonstrating the excellent electrocatalytic stability of CM7-FeNixSe for the OER.
In order to confirm the stability of CM7-FeNixSe in alkaline solution for the OER, we further characterized the structure and morphology of CM7-FeNixSe after a long-term stability test at a current density of 20 mA cm−2 for 10 h. SEM and TEM images show that CM7-FeNixSe after the chronopotentiometry test (CD-CM7-FeNixSe) retains the uniform nanosheet structure (Fig. 4a and b), no obvious morphological change after a long-term stability test, the mixed thin layer composed of crystalline and amorphous phases on the surface remains intact, and the thickness has not changed much (Fig. 4c). Both the XRD pattern and HRTEM image of CD-CM7-FeNixSe are almost the same as those of CM7-FeNixSe. A slight change is that the peak strength of NiSe and Ni0.85Se decreased, which is due to the hydrated oxides and (oxy)hydroxides generated on the surface of CD-CM7-FeNixSe. Elemental mapping (Fig. 4f) shows that after a long period of testing, each element is still evenly distributed on the surface of the nanosheet. This result was confirmed by subsequent elemental quantitative analysis of EDX (Fig. S7, ESI†). Previous reports have shown that in the OER test, the Se inside the selenides will gradually dissolve in the electrolyte during the test, resulting in the decomposition of the electrode materials.27,28,50 However, according to the above characterization, we found that pre-covering with a thin SeOx layer on the surface of FeNixSe can significantly improve the stability of the selenide materials. In the subsequent OER tests, the oxide layer can prevent the internal Se from dissolution, thus showing excellent structural stability and long cycle life. The corresponding high-resolution XPS spectra of CM7-FeNixSe and CD-CM7-FeNixSe also proved the stability of the materials, as shown in Fig. S8 (ESI†), except for the characteristic peak at 532.7 eV, which shows that the adsorbed oxygen species (Fig. S8d, ESI†) has been strengthened, due to the adsorption of water molecules from the alkaline solution on the surface after a long period of testing.51 The other characteristic peaks have not changed from those before the tests. The results further demonstrated that the thin oxide layer deposited on the surface can prevent the internal selenide from directly coming into contact with the electrolyte, thus preventing the loss of Se and maintaining a stable surface state in the OER test.
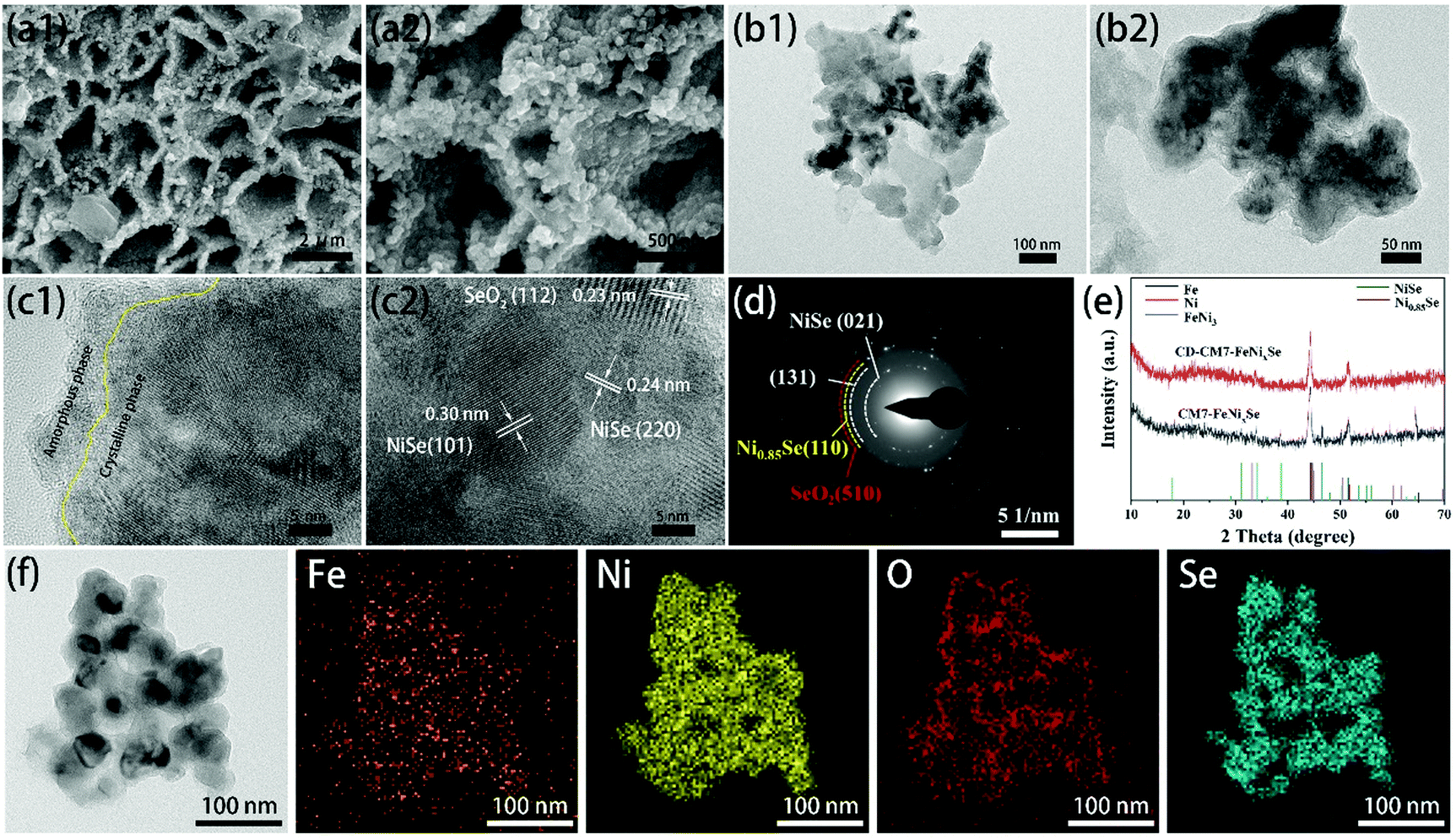 | ||
| Fig. 4 (a1 and a2) SEM images, (b1 and b2) TEM images, (c1 and c2) HRTEM images, (d) SAED patterns, (e) XRD pattern and (f) EDX elemental mapping of CM7-FeNixSe after the chronopotentiometry test (CD-CM7-FeNixSe) at constant current densities of 20 mA cm−2 for 10 h. | ||
The role of the SeOx film thickness
In order to understand the influence of different electro-deposition times on the performance of CMy-FeNixSe, the HER and OER LSV curves and EIS spectra at different electro-deposition times were characterized. As shown in Fig. S9a (ESI†), an obvious cathode peak at ∼−0.13 V (vs. RHE) is observed for CMy-FeNixSe, which may be assigned to the leaching and subsequent oxidation of Se from FeNixSe to selenium oxide, which has been reported in the previous literature.32 Lau et al. found that both the HER and OER processes of selenides in alkaline solution were accompanied by surface oxidation, and the oxidation degree and phase transition rate during the OER is much stronger and faster, while the oxidation rate during the HER was relatively slow.32 They show that in the initial reaction stage of the HER, the outermost layer of selenides (NiSe2) will ionize in strong KOH solution, and the Se22− will leach out and combine with the dissolved oxygen to generate SeOx, accompanied by the generation of Ni(OH)2/NiOOH.52 The specific mechanism is shown as follows:32| Ni2+ + Se22− + 2OH− + xO2 → Ni(OH)2 + 2SeOx + 2e− | (1) |
| Ni(OH)2 + OH− ⇌ NiOOH + H2O + e− | (2) |
We can see that no obvious peak is observed in the first HER, which manifests that there is no obvious decomposition of FeNixSe at this time. With an increase of HER cycle times, the cathode oxidation peak became more and more obvious, which proved that the material had a violent oxidation reaction with the increase of HER cycle times. Until the seventh HER cycle, the curve tends to be stable with little change, indicating that the generated SeOx layer on the surface of FeNixSe has become stable, which can prevent the further decomposition of the internal selenide. The corresponding OER performances with different HER cycle times are displayed in Fig. S9c (ESI†). It shows that the OER activity of CMy-FeNixSe is enhanced with the increase of HER cycle times and tends to stabilise until the seventh cycle. It is proved that the SeOx layer has become stable after seven HER cycles, and the thickness of the SeOx layer is appropriate, which can improve the OER activity of FeNixSe while inhibiting the decomposition of the internal selenide. The OER kinetics with different HER cycle times were further analyzed by EIS (Fig. S9b, ESI†). The charge transfer resistance of CMy-FeNixSe gradually reduced with the increase of HER cycle times and tended to be stable after the seventh HER cycle. The surface SeOx layer brings more active sites for electrochemical reactions, we propose that the superior OER kinetics may be due to the synergistic effect between the core selenides and surface in situ generated SeOx layer.
According to previous reports, the oxides formed on the surface at the beginning stage of electrocatalysis are proposed to be the real active centers of OER catalysts.27,30 However, excessive oxides usually lead to poor electrical conductivity and high charge transfer resistance of the catalysts, which is not conducive to the enhancement of the electrocatalytic performance. In order to investigate the influence of electro-deposition times on the thickness of the oxide layer formed, we selected CM1-FeNixSe, CM7-FeNixSe and CM20-FeNixSe to expound the structure and composition change. Both the SEM and TEM images (Fig. S10a1-c1 and a2-c2, ESI†) show no obvious morphological change of FeNixSe after HER cycling, and it still maintains a three-dimensional nanosheet structure. The high-resolution TEM image indicates that after one HER cycle process, for CM1-FeNixSe, there is no obvious oxide layer formed on the surface of FeNixSe, and only a thin amorphous oxide layer can be observed in the local area (Fig. S10a3, ESI†). Although we have found the corresponding interplanar spacing of SeO2 (Fig. S10a4, ESI†), the diffraction ring of SeO2 was not observed in SAED, due to the low content of SeO2 in CM1-FeNixSe. This result can be confirmed by EDX results (Fig. S10d, ESI†) that the O content of CM1-FeNixSe increased slightly compared with FeNixSe. After 7 HER cycles, the surface of FeNixSe was obviously coated with a thin layer of SeO2. Two new (112) and (202) reflections of SeO2 are captured in Fig. S10b4 (ESI†), which are consistent with the SAED results (Fig. S10b5, ESI†). Additionally, it can be clearly seen from EDX patterns (Fig. S10d and e, ESI†) that the content of O increases from 12.6 to 22.6 wt% upon increasing the HER cycle times. However, with a continuous increase of the HER cycle times, we found that the thickness of the surface oxide layer did not change obviously. According to the EDX results, the O content only increased from 22.6 to 22.9 wt% after 7 to 20 cycles. These results prove that the oxide layer after 7 HER cycles has stabilized, which could prevent the internal selenide from being decomposed continuously during the HER cycle process, and no more new oxides are produced. We noticed that no significant changes were found in the XRD diffraction peaks after different HER cycle times (Fig. S10e, ESI†), which may be due to the oxide layer deposited being very thin and the content being very low, and the SeOx deposited under HER cycles presents a mixed structure of crystalline and amorphous phases.
In order to investigate the elemental composition and surface chemical states, the XPS technique was employed to explain the structure and composition change of FeNixSe after different HER cycle times. As we can see from Fig. S11a (ESI†), with the increase of HER cycle times, the peak intensity of metallic Ni (852.4 and 870.2 eV) is reduced and almost disappears, which is probably due to the covering of the oxide layer leaving the bottom NiFe foam not easy to be detected.53,54 Compared with the Fe 2p spectrum (Fig. S11b, ESI†) of CM1-FeNixSe, a shift to a higher binding energy was observed in the sample of CM7-FeNixSe, indicating a higher oxidation state of the Fe element with an increase of HER cycle times.43 However, CM7-FeNixSe and CM20-FeNixSe showed similar spectra, indicating that the surface state tends to be stable after 7 HER cycles. As for the Se 3d spectra (Fig. S11c, ESI†), the intensity of the SeOx peak (58.3 eV) is significantly enhanced from CM1-FeNixSe to CM7-FeNixSe, and the peaks of Se 3d3/2 and Se 3d5/2 are decreased and appear at positions with slightly higher binding energies (∼0.3 eV), indicating the leaching and oxidation of Se in the FeNixSe with increased HER cycle times.46 It further demonstrated that SeOx is produced on the surface of FeNixSe during the HER cycle process. The increased intensity of the lattice oxygen (O2−) peak at 529.9 eV in CM7-FeNixSe also demonstrate the existence of SeOx (Fig. S11d, ESI†).55 The above results prove that the outermost layer of NiSe will ionize in strong KOH solution during the HER cycle process, and the Se22− will leach out and combine with the dissolved oxygen to form a SeOx covering on the original selenide surface. With the increase of HER cycle times, the oxide layer gradually becomes thicker until the seventh HER cycle. After 7 HER cycles, the oxide layer tends to be stable, and the internal selenide is completely covered by the surface oxide layer and is no longer in contact with the KOH solution. Thus, subsequent decomposition will not continue with the further increase of HER cycle times, showing excellent structural stability.
Understanding the high electrocatalytic activity of the SeOx/FeNixSe heterojunction nanosheets with DFT simulations
As demonstrated by the above results, SeOx/FeNixSe exhibits rapid charge transfer capability and high performance and structural stability for the OER. This is mainly due to the thin layer of SeOx electrodeposited on the surface of FeNixSe by performing the HER cycle that built the 3D interconnected heterojunction between the SeOx layer and FeNixSe. Therefore, in order to further understand that the formed heterojunction between the SeOx layer and FeNixSe will reduce the surface charge transfer resistance and increase the catalytic activity of SeOx/FeNixSe, we used density functional theory (DFT) calculations to deeply reveal the mechanism behind the OER process. The optimized geometry models of Ni1−xFexSe and Ni1−xFexSe@SeO2 (Fig. 5a) were used to model FeNixSe before and after the HER cycle. The projected density of states (DOSs) of Ni1−xFexSe and Ni1−xFexSe@SeO2 cross over the Fermi level (Fig. 5b), indicating that both materials show metallic properties.56 In other words, the SeO2 layer with an appropriate thickness grows on the surface of Ni1−xFexSe, which will not change the metallic properties of Ni1−xFexSe. In contrast, the ultrathin SeOx layer on the surface can facilitate the adsorption of intermediates onto Ni1−xFexSe and prevent the decomposition of the internal FeNixSe, thus improving the structural stability of the electrode. In an alkaline electrolyte, the OER proceeds by the adsorption and desorption of a series of *OH, *O and *OOH intermediates formed in the four primary reaction steps with 4 electron transfer (step I: OH− → *OH + e−; step II: *OH + OH− → *O + H2O + e−; step III: *O + OH− → *OOH + e−; step IV:*OOH + OH− → O2(g) + H2O + e−), eventually producing O2 molecules.57 The free energy change (ΔG) values for the four primary steps of the OER in Ni1−xFexSe are −1.28, 0.52, 2.73 and 2.95 eV, respectively (Fig. 5c). Specifically, the first step is exothermic, and the next three steps are endergonic. The step required to absorb most energy to proceed can be considered the rate-determining step (RDS) for the entire OER. It is clear that step IV (*OOH + OH− → O2(g) + H2O + e−) requires the highest free energy change (2.95 eV) for Ni1−xFexSe, indicating that the rate-limiting step is the dehydrogenation of adsorbed *OOH. However, for Ni1−xFexSe@SeO2, the growth of selenium oxide on the catalyst surface changes the surface electronic structure of Ni1−xFexSe and reduces the adsorption energies of the reaction intermediates. The free energy change (ΔG) values for the four primary steps of the OER in Ni1−xFexSe@SeO2 were reduced to more favorable values of −0.71, 0.36, 2.77 and 2.5 eV, respectively (Fig. 5c). It can be found that the RDS for Ni1−xFexSe@SeO2 changes to step III (*O + OH− → *OOH + e−). That is, the RDS in this case (2.77 eV) is the hydrogenation of adsorbed *O, which is significantly smaller than that of Ni1−xFexSe (2.95 eV). The UL(OER) value can be calculated using the equation, UL(OER) = Maxi(ΔGi)/ne − 1.23 eV. The smaller the UL(OER) value, the higher the OER catalytic activity. In particular, the UL(OER) value for Ni1−xFexSe@SeO2 was calculated to be 1.54 V, which is smaller than that of Ni1−xFexSe (UL = 1.72 V). This indicates that Ni1−xFexSe@SeO2 has higher catalytic activity than Ni1−xFexSe. Therefore, DFT calculation is in good agreement with the experimental results, highlighting the synergetic coupling between Ni1−xFexSe and SeOx in producing a favorable surface electronic environment and thereby boosting the OER performance.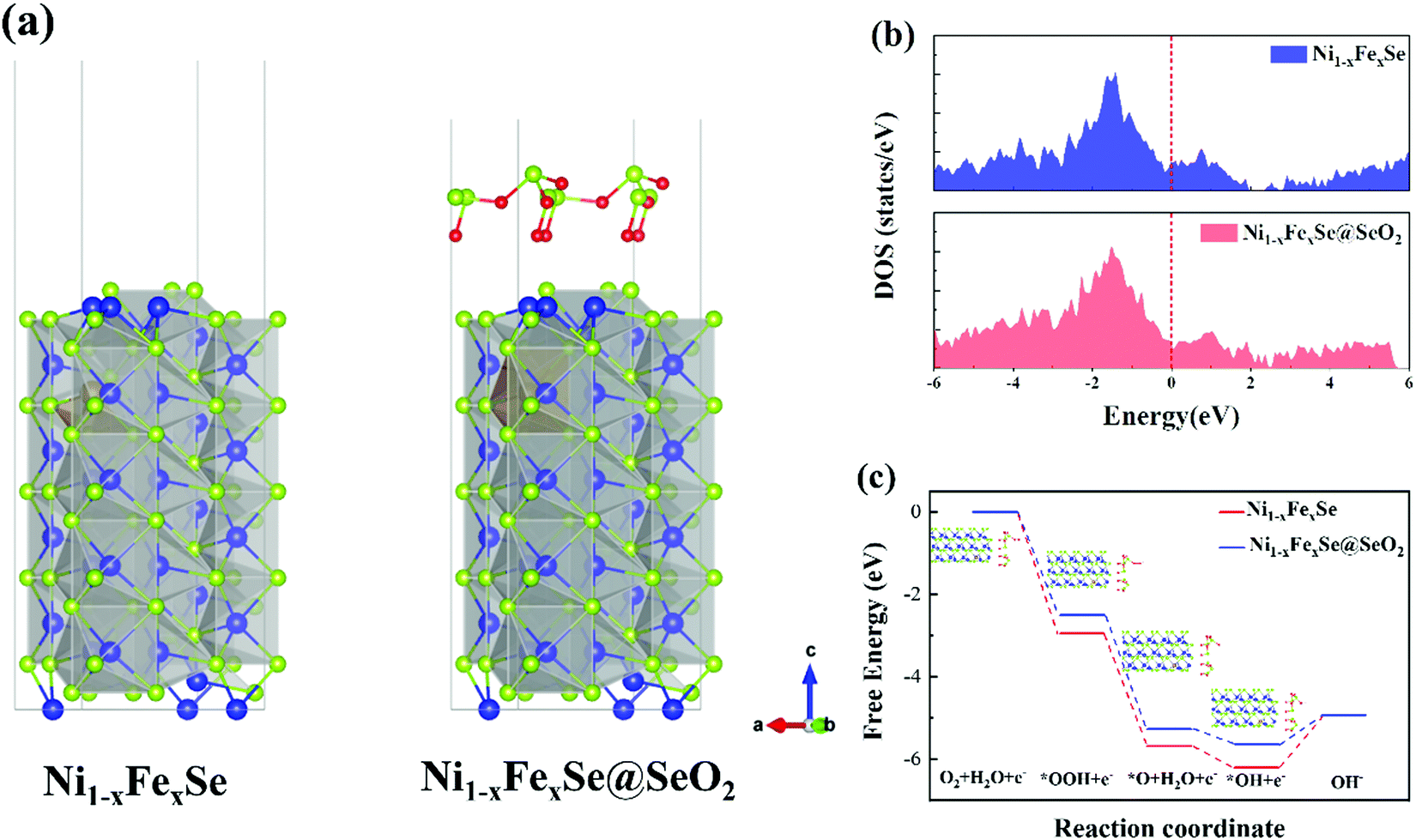 | ||
| Fig. 5 Theoretical calculations of Ni1−xFexSe and Ni1−xFexSe@SeO2 electrocatalysts. (a) Crystal structures, (b) calculated DOS, (c) free energy diagram for the OER over four optimized adsorption geometry of O2, *OOH, *O, and *OH. | ||
Overall water splitting application
Finally, to further evaluate the practical application of CM7-FeNixSe as high efficiency electrochemical catalysts, an electrolyzer was made with a two-electrode configuration using CM7-FeNixSe as the anode and commercial Pt/C as the cathode for overall water splitting in 1.0 M KOH. For comparison, electrolyzer systems using Pt/C‖CM7-FeNi LDH, Pt/C‖CM7-NiFe foam and Pt/C‖Ir were also tested. It can be seen that the electrolyzer Pt/C‖CM7-FeNixSe needs a cell voltage of 1.637 V to achieve a current density of 50 mA cm−2 (Fig. 6a), which is much lower than that of the other electrolyzer systems at the same current density (Pt/C‖CM7-FeNi LDH: 1.672 V; Pt/C‖CM7-NiFe Foam: 1.767 V; Pt/C‖Ir: 1.667 V). The performance is also superior to most previously reported selenide catalysts as shown in Table S2 (ESI†). Attractively, Pt/C‖CM7-FeNixSe presents a faradaic efficiency close to 99% for the OER (Fig. 6b). Furthermore, to evaluate the system stability of overall water splitting, multi-step chronopotentiometric tests (Fig. 6c) were conducted with the current density increasing from 20 mA cm−2 to 100 mA cm−2. The CM7-FeNixSe based electrolyzer system exhibited the most beneficial electrocatalytic activity at different current densities, and upon increasing the current density, the corresponding potential increases and remains stable, which shows the excellent cycling stability of the CM7-FeNixSe electrode.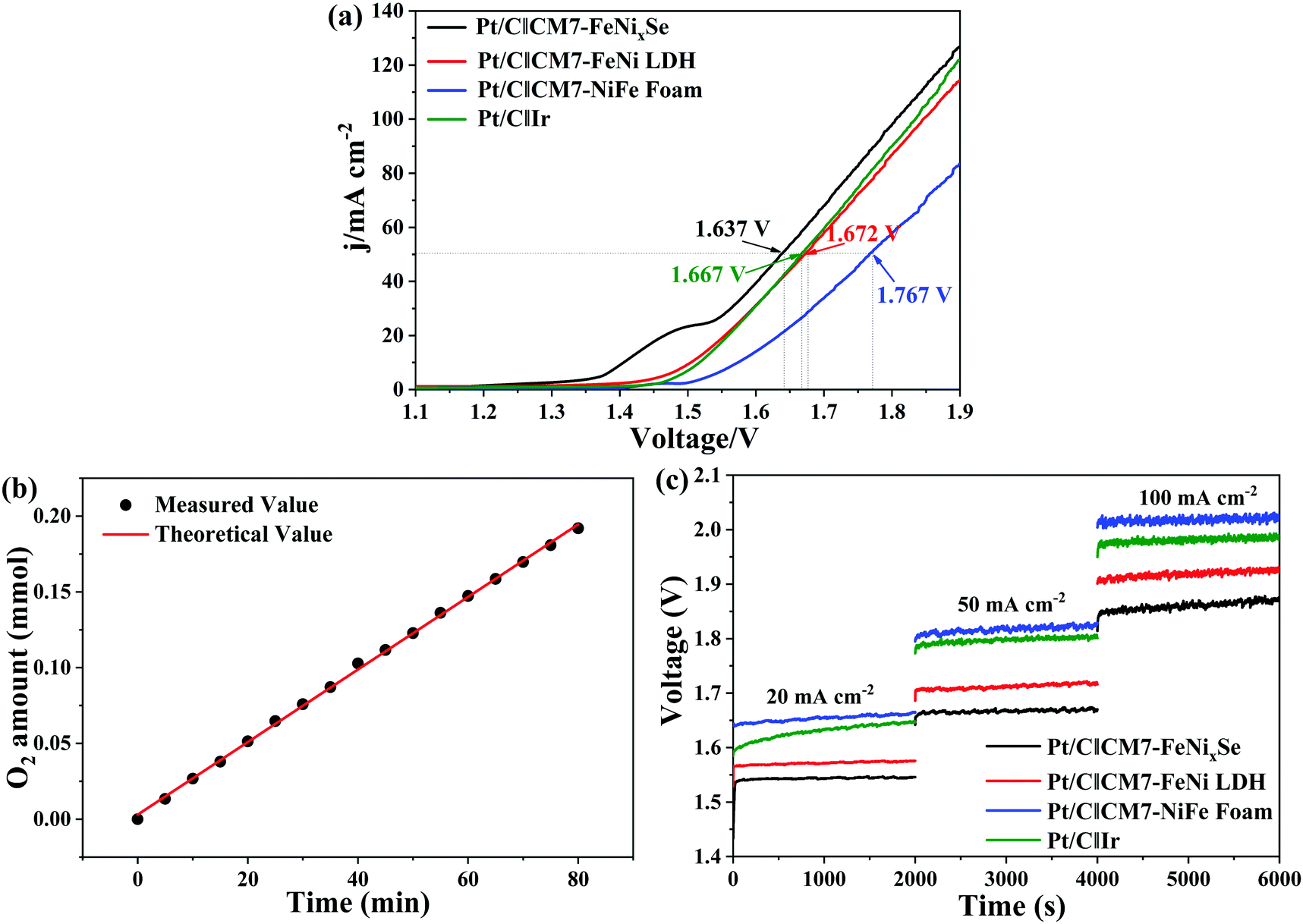 | ||
| Fig. 6 (a) Polarization curves for overall water splitting. (b) Amounts of gas experimentally measured and theoretically calculated vs. time for the electrolyzer Pt/C‖CM7-FeNixSe at 10 mA cm−2. (c) Chronopotentiometry curves tested at constant current densities of 10, 50 and 100 mA cm−2. | ||
Conclusions
In summary, a rapid, facile, green and controllable electrodeposition method was developed to synthesize three-dimensional (3D) SeOx/FeNixSe heterojunction nanosheets with interconnected heterostructures through the hydrogen evolution reaction (HER) cycle process. It only takes 90 s for one time electrodeposition, and a stable SeOx layer with optimum thickness can be formed on the surface of FeNixSe after seven electrodeposition cycles. Detailed spectroscopic and DFT investigations disclose that the in situ electrodeposited ultrathin SeOx layer could significantly modify the surface electronic structure and provide high electrochemical surface areas and rich active sites, and the electrocatalytic activity of FeNixSe was significantly improved by the synergistic effect between FeNixSe and the SeOx layer. Moreover, the strong electron interaction between FeNixSe and SeOx could reduce the surface charge transfer resistance and speed up the catalytic reaction kinetics in the electrocatalytic reaction. The ultrathin SeOx layer on the surface can prevent the decomposition of the internal FeNixSe and thus improve the structural stability of the electrode. The optimized SeOx/FeNixSe (CM7-FeNixSe) electrode shows excellent OER performance with a current density of 200 mA cm−2 occurring at a small overpotential of 285 mV in 1.0 M KOH. This study offers a new strategy for the construction of a metal selenide heterojunction protected by an ultra-thin selenium oxide coating.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support from the National Natural Science Foundation of China (No. 11975205), the Zhejiang Provincial Natural Science Foundation (No. LR22E070001), and the Guangdong Basic and Applied Basic Research Foundation (No. 2020B1515120048).References
- G. Zhang, J. Zeng, J. Yin, C. Zuo, P. Wen, H. Chen and Y. Qiu, Appl. Catal., B, 2021, 286, 119902 CrossRef CAS.
- B. You, M. T. Tang, C. Tsai, F. Abild-Pedersen, X. Zheng and H. Li, Adv. Mater., 2019, 31, 1807001 CrossRef PubMed.
- J. Zhang, Q. Zhang and X. Feng, Adv. Mater., 2019, 31, 1808167 CrossRef PubMed.
- H. Zhang, M. Zhu, O. G. Schmidt, S. Chen and K. Zhang, Adv. Energy Sustainability Res., 2021, 2, 2000090 CrossRef.
- X. Gu, Z. Liu, M. Li, J. Tian and L. Feng, Appl. Catal., B, 2021, 297, 120462 CrossRef CAS.
- J. Yuan, J. Wu, W. J. Hardy, P. Loya, M. Lou, Y. Yang, S. Najmaei, M. Jiang, F. Qin, K. Keyshar, H. Ji, W. Gao, J. Bao, J. Kono, D. Natelson, P. M. Ajayan and J. Lou, Adv. Mater., 2015, 27, 5605–5609 CrossRef CAS PubMed.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K.-Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- N. Jiang, B. You, M. Sheng and Y. Sun, Angew. Chem., Int. Ed., 2015, 54, 6251–6254 CrossRef CAS PubMed.
- S. Choi, J. Kwon, S. Jo, S. Kim, K. Park, S. Kim, H. Han, U. Paik and T. Song, Appl. Catal., B, 2021, 298, 120530 CrossRef CAS.
- X. Hao, Z. Jiang, B. Zhang, X. Tian, C. Song, L. Wang, T. Maiyalagan, X. Hao and Z.-J. Jiang, Adv. Sci., 2021, 8, 2004572 CrossRef CAS PubMed.
- X. Shi, H. Wang, P. Kannan, J. Ding, S. Ji, F. Liu, H. Gai and R. Wang, J. Mater. Chem. A, 2019, 7, 3344–3352 RSC.
- C. Hu, L. Zhang, Z.-J. Zhao, A. Li, X. Chang and J. Gong, Adv. Mater., 2018, 30, 1705538 CrossRef PubMed.
- M. Chen, D. Liu, B. Zi, Y. Chen, D. Liu, X. Du, F. Li, P. Zhou, Y. Ke, J. Li, K. H. Lo, C. T. Kwok, W. F. Ip, S. Chen, S. Wang, Q. Liu and H. Pan, J. Energy Chem., 2022, 65, 405–414 CrossRef.
- H. Guo, A. Wu, Y. Xie, H. Yan, D. Wang, L. Wang and C. Tian, J. Mater. Chem. A, 2021, 9, 8620–8629 RSC.
- A. K. Tareen, G. S. Priyanga, K. Khan, E. Pervaiz, T. Thomas and M. Yang, ChemSusChem, 2019, 12, 3941–3954 CrossRef CAS PubMed.
- M. Zhao, W. Li, J. Li, W. Hu and C. M. Li, Adv. Sci., 2020, 7, 2001965 CrossRef CAS PubMed.
- Y. Guo, T. Park, J. W. Yi, J. Henzie, J. Kim, Z. Wang, B. Jiang, Y. Bando, Y. Sugahara, J. Tang and Y. Yamauchi, Adv. Mater., 2019, 31, 1807134 CrossRef PubMed.
- S. Yang, J.-Y. Zhu, X.-N. Chen, M.-J. Huang, S.-H. Cai, J.-Y. Han and J.-S. Li, Appl. Catal., B, 2022, 304, 120914 CrossRef CAS.
- D. Lai, Q. Kang, F. Gao and Q. Lu, J. Mater. Chem. A, 2021, 9, 17913–17922 RSC.
- X. Zhang, M. Jin, Q. Lian, O. Peng, S. Niu, Z. Ai, A. Amini, S. Song and C. Cheng, Chem. Eng. J., 2021, 423, 130218 CrossRef CAS.
- R. Zong, Y. Fang, C. Zhu, X. Zhang, L. Wu, X. Hou, Y. Tao and J. Shao, ACS Appl. Mater. Interfaces, 2021, 13, 42852–42860 CrossRef CAS PubMed.
- H. Sancho, Y. Zhang, L. Liu, V. G. Barevadia, S. Wu, Y. Zhang, P.-W. Huang, Y. Zhang, T.-H. Wu, W. You and N. Liu, J. Electrochem. Soc., 2020, 167, 056503 CrossRef CAS.
- C. Hu and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 11736–11758 CrossRef CAS PubMed.
- M. Zhang, Y. Zhang, L. Ye, B. Guo and Y. Gong, Appl. Catal., B, 2021, 298, 120601 CrossRef CAS.
- Y. Huang, L.-W. Jiang, X.-L. Liu, T. Tan, H. Liu and J.-J. Wang, Appl. Catal., B, 2021, 299, 120678 CrossRef CAS.
- L. Tan, J. Yu, H. Wang, H. Gao, X. Liu, L. Wang, X. She and T. Zhan, Appl. Catal., B, 2022, 303, 120915 CrossRef CAS.
- L. Zhai, T. W. Benedict Lo, Z.-L. Xu, J. Potter, J. Mo, X. Guo, C. C. Tang, S. C. Edman Tsang and S. P. Lau, ACS Energy Lett., 2020, 5, 2483–2491 CrossRef CAS.
- L. Zhai, C. H. Mak, J. Qian, S. Lin and S. P. Lau, Electrochim. Acta, 2019, 305, 37–46 CrossRef CAS.
- S. Kim, H. Mizuno, M. Saruyama, M. Sakamoto, M. Haruta, H. Kurata, T. Yamada, K. Domen and T. Teranishi, Chem. Sci., 2020, 11, 1523–1530 RSC.
- Y. Zhang, L. Gao, E. J. M. Hensen and J. P. Hofmann, ACS Energy Lett., 2018, 3, 1360–1365 CrossRef CAS PubMed.
- S. Anantharaj, S. Kundu and S. Noda, Nano Energy, 2021, 80, 105514 CrossRef CAS.
- L. Zhai, C. H. Mak, J. Qian, S. Lin and S. P. Lau, Electrochim. Acta, 2019, 305, 37–46 CrossRef CAS.
- Z.-W. Gao, J.-Y. Liu, X.-M. Chen, X.-L. Zheng, J. Mao, H. Liu, T. Ma, L. Li, W.-C. Wang and X.-W. Du, Adv. Mater., 2019, 31, 1804769 CrossRef PubMed.
- M.-R. Gao, Z.-Y. Lin, T.-T. Zhuang, J. Jiang, Y.-F. Xu, Y.-R. Zheng and S.-H. Yu, J. Mater. Chem., 2012, 22, 13662–13668 RSC.
- H. Li, S. Chen, H. Lin, X. Xu, H. Yang, L. Song and X. Wang, Small, 2017, 13, 1701487 CrossRef PubMed.
- M. Qian, S. Cui, D. Jiang, L. Zhang and P. Du, Adv. Mater., 2017, 29, 1704075 CrossRef PubMed.
- E. Hu, Y. Feng, J. Nai, D. Zhao, Y. Hu and X. W. Lou, Energy Environ. Sci., 2018, 11, 872–880 RSC.
- J. Zhou, L. Yuan, J. Wang, L. Song, Y. You, R. Zhou, J. Zhang and J. Xu, J. Mater. Chem. A, 2020, 8, 8113–8120 RSC.
- Z. Zou, X. Wang, J. Huang, Z. Wu and F. Gao, J. Mater. Chem. A, 2019, 7, 2233–2241 RSC.
- S. Bae, S. Gim, H. Kim and K. Hanna, Appl. Catal., B, 2016, 182, 541–549 CrossRef CAS.
- Y. Song, F. Duan, S. Zhang, J.-Y. Tian, Z. Zhang, Z.-W. Wang, C.-S. Liu, W.-M. Xu and M. Du, J. Mater. Chem. A, 2017, 5, 19378–19389 RSC.
- L. Zhang, P. Yang, W. Chen, L. Cheng, J. Yan and H. Yang, J. Mater. Sci.: Mater. Electron., 2020, 31, 15968–15975 CrossRef CAS.
- J. Kim, J. Lee, C. Liu, S. Pandey, S. Woo Joo, N. Son and M. Kang, Appl. Surf. Sci., 2021, 546, 149124 CrossRef CAS.
- C. Tang, Z. Pu, Q. Liu, A. M. Asiri, X. Sun, Y. Luo and Y. He, ChemElectroChem, 2015, 2, 1903–1907 CrossRef CAS.
- Y. Zhou, H. Xiao, S. Zhang, Y. Li, S. Wang, Z. Wang, C. An and J. Zhang, Electrochim. Acta, 2017, 241, 106–115 CrossRef CAS.
- X. Li, K.-L. Yan, Y. Rao, B. Dong, X. Shang, G.-Q. Han, J.-Q. Chi, W.-H. Hu, Y.-R. Liu, Y.-M. Chai and C.-G. Liu, Electrochim. Acta, 2016, 220, 536–544 CrossRef CAS.
- O. Kasian, S. Geiger, T. Li, J.-P. Grote, K. Schweinar, S. Zhang, C. Scheu, D. Raabe, S. Cherevko, B. Gault and K. J. J. Mayrhofer, Energy Environ. Sci., 2019, 12, 3548–3555 RSC.
- Y. Chen, Z. Ren, H. Fu, X. Zhang, G. Tian and H. Fu, Small, 2018, 14, 1800763 CrossRef PubMed.
- S. Esmailzadeh, T. Shahrabi, G. Barati Darband and Y. Yaghoubinezhad, Electrochim. Acta, 2020, 334, 135549 CrossRef CAS.
- Y. Shi, W. Du, W. Zhou, C. Wang, S. Lu, S. Lu and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 22470–22474 CrossRef CAS PubMed.
- X. Cao, Y. Sang, L. Wang, G. Ding, R. Yu and B. Geng, Nanoscale, 2020, 12, 19404–19412 RSC.
- L. Zhai, T. W. Benedict Lo, Z.-L. Xu, J. Potter, J. Mo, X. Guo, C. C. Tang, S. C. Edman Tsang and S. P. Lau, ACS Energy Lett., 2020, 5, 2483–2491 CrossRef CAS.
- D. Gao, W. Liu, Y. Xu, P. Wang, J. Fan and H. Yu, Appl. Catal., B, 2020, 260, 118190 CrossRef CAS.
- Y. Song, Y. Zhao, G. Nan, W. Chen, Z. Guo, S. Li, Z. Tang, W. Wei and Y. Sun, Appl. Catal., B, 2020, 270, 118888 CrossRef CAS.
- J. Zhao, W. Xi, C. Tu, Q. Dai and X. Wang, Appl. Catal., B, 2020, 263, 118237 CrossRef CAS.
- P. Chen, T. Zhou, M. Zhang, Y. Tong, C. Zhong, N. Zhang, L. Zhang, C. Wu and Y. Xie, Adv. Mater., 2017, 29, 1701584 CrossRef PubMed.
- R. V. Mom, J. Cheng, M. T. M. Koper and M. Sprik, J. Phys. Chem. C, 2014, 118, 4095–4102 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section; SEM, TEM images, EDX, elemental weight percentage and SAED patterns of FeNi LDH and CM7-FeNi LDH; EDX and elemental weight percentage of CM7-FeNixSe and FeNixSe; XRD patterns of NiFe foam, CM7-NiFe foam, FeNi LDH and CM7-FeNi LDH; CVs of CM7-FeNixSe, FeNixSe, CM7-FeNi LDH, FeNi LDH, CM7-NiFe foam and pure NiFe foam; dependence of Δj on the scan rate at different double layer capacitances; elemental weight percentage of CD-CM7-FeNixSe and CM7-FeNixSe; high-resolution XPS spectra of Ni 2p, Fe 2p, Se 3d, and O 1s of CM7-FeNixSe and CD-CM7-FeNixSe; HER polarization curves, Nyquist plots (overpotential = 250 mV), and OER polarization curves of CMy-FeNixSe; SEM, TEM, HRTEM, SAED patterns, EDX, elemental weight percentage, and XRD patterns of CM1-FeNixSe, CM7-FeNixSe, and CM20-FeNixSe; high-resolution XPS spectra of Ni 2p, Fe 2p, Se 3d and O 1s of CM1-FeNixSe, CM7-FeNixSe and CM20-FeNixSe; OER catalytic activity comparison; and overall water splitting catalytic activity comparison. See DOI: 10.1039/d1ma01208h |
| This journal is © The Royal Society of Chemistry 2022 |