
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermomorphological and mechanical properties of vulcanized octenyl succinate/terpenoid-derivatized corn starch composites†
Moira K.
Lauer
a,
Andrew G.
Tennyson
 ab and
Rhett C.
Smith
*a
ab and
Rhett C.
Smith
*a
aDepartment of Chemistry, Clemson University, Clemson, South Carolina 29634, USA. E-mail: rhett@clemson.edu
bDepartment of Materials Science and Engineering, Clemson University, Clemson, South Carolina 29634, USA
First published on 12th April 2022
Abstract
Modification of food starches with octenyl succinic anhydride (OSA) is a well-established protocol to endow the starches with olefin moieties that can subsequently undergo traditional polyolefin crosslinking and polymerization reactions. In the current work, olefin-modified starch derivatives are crosslinked with sulfur, through reactions mechanistically similar to vulcanization of the olefin moieties in rubber, thus yielding the analogous vulcanized starch composites. Given the importance of the olefin moieties in the vulcanization mechanism, OSA-modified corn starch was further derivatized by esterification with geraniol, a terpenol diene. The resulting geraniol-esterified starch (GES) thus contains three olefin moieties per esterified site, affording double the number of crosslinkable-carbon atoms compared to typical OSA-modified starches. GES was reacted with elemental sulfur to generate composites GSSx (where x = wt% sulfur, either 90 or 95 in this work). The thermomorphological and mechanical properties of GSSx were assessed by thermogrametric analysis (TGA), differential scanning calorimetry (DSC), scanning electron microscopy with element mapping by energy-dispersive X-ray analysis (SEM-EDX), mechanical test stand analysis, and dynamic mechanical analysis (DMA) and compared to those of previously-reported composites of OSA-modified starch and elemental sulfur (OSSx). These tests revealed that GSSx underwent significant intramolecular crosslinking to form particles that were well-dispersed in the sulfur-rich networks of the composites. As a result, GSSx materials behaved as particle-reinforced network materials that deformed entirely plastically before breaking under a compressional stress. In contrast, OSSx composites exhibited longer crosslinking chains and more interaction between domains. The result of these morphological differences confers upon GSSx lower stiffness and greater strength relative to their OSSx analogues.
Introduction
Largely unchecked CO2 emissions have been linked to globally increasing temperatures that foreshadow potentially devastating impacts.1 One of the most significant contributors to both anthropogenic CO2 and the destruction of the environment through mining is in the production of Portland cement. It is currently estimated that cement production accounts for 30% of global materials utilization and 8% of anthropogenic CO2 production.2 These statistics can be attributed to current concrete formulations that incorporate massive amounts of cement clinker. Clinker is environmentally deleterious as it is produced by first mining mineral carbonates and then converting them to their respective oxides. This process requires heating the carbonates to more than 1400 °C with concomitant production of stoichiometric CO2. This already gargantuan CO2 output is only forecast to increase in response to current increases in annual cement production3 to meet increasing needs for infrastructure in second and third world countries. These trends coupled with limited ability to recycle end-of-life concrete has only relatively recently led to a significant uptick in research to develop sustainable replacements for cement. Fortunately, this work has borne a variety of innovative approaches and concrete formulations to decrease our reliance on cement clinker. The majority of mineral cements, however, are still inherently acid-sensitive and environmentally vulnerable. One of the primary pitfalls of mineral cements is their high water-uptake and consequent freeze–thaw cycle-induced fissure. This paired with an inability to readily recycle cement at scale renders even these improved formulations as significant waste streams.Sulfur cements were identified over a century ago for their potential to provide the structural integrity of traditional cements but with improved environmental resistance.4–19 Early formulations of sulfur cements were formed by the vulcanization of petrochemical olefins with elemental sulfur to form a hot-set cement to which traditional fines (like sand) and aggregates (like gravel) could be added. Vulcanization – called inverse vulcanization when the reaction involved a majority wt% sulfur (Fig. 1) – involves reaction between thermally-generated oligo/polysulfur radicals with olefins.20–25 Recently, more sustainable high sulfur-content materials (HSMs) have been prepared by vulcanizing bio-derived olefins with elemental sulfur. Such HSMs hold promise for a variety of applications including as adsorbents for environmental remediation,26–32 as IR transparent lenses,33–39 and in lithium sulfur batteries.40–48 The practical application potential for HSMs has also been expanded by advances in novel processing and recycling strategies that have allowed for expanded substrate scope, lower reaction temperatures and solvent- or heat-assisted healable materials.23,49–56 Most pertinent to the current work, however, is that many HSMs possess strength characteristics on par with or better than current cement products, yet the HSMs can be readily reshaped by simple melt-cast processes at moderate temperatures (120–180 °C).57–61 The formulation of HSMs from sulfur – itself a by-product of petroleum refining – and an olefinic by-product of agronomic endeavours makes them attractive candidates for valorization of underutilized good to replace cement. Some of the especially promising bio-derived components of HSMs include derivatives of cellulose,57,62 lignin,58,63–65 mixed lignocellulosic biomass,58,59,66 corn starch,66,67 terpenoids,39,57,61,68–71 triglycerides, and fatty acids.72–74
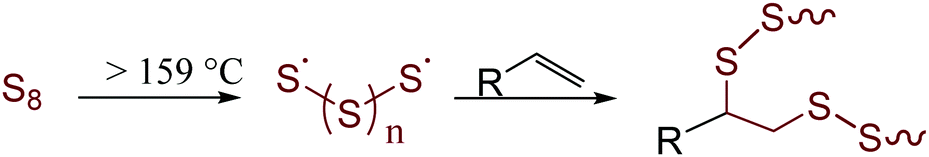 | ||
| Fig. 1 Inverse vulcanization of an olefin with sulfur. | ||
We recently reported materials in which modified corn starch was the organic component of HSMs.66 In the United States alone, corn has been produced in gigaton quantities (∼14–15 Gt) annually since 2013, yet its selling price has fallen by over 50% in the last ten years due to dwindling demand.75,76 Valorisation of corn products therefore presents an exciting economic opportunity. In our previous study, corn starch was modified with octenyl succinic anhydride to prepare OS (degree of substitution = 2.63 ± 0.05%, Fig. 2 and 3) by the established route. Inverse vulcanization of OS with sulfur led to composites OSSx (x = wt% of sulfur in the feed ratio). The OSSx composites exhibited exceptionally long oligosulfur crosslinks between starch chains, endowing the composites with high flexural strength relative to the majority of previously-reported HSMs. Accessing biopolymer-HSMs having good mechanical properties while requiring only a low degree of biopolymer substitution is attractive because it minimizes the non-bio-derived content in the final product. Given the promising properties of OSSx composites, we used a green modification protocol to further derivatize OS and to evaluate the utility of the resultant starch derivative, GES, as a component of starch/sulfur vulcanized composites, GSSx. Starch derivative OS was thus esterified by its reaction with geraniol, a terpenoid alcohol bearing two vulcanizable olefin units, to give GES. This esterification was 100% atom-economical, employing L-leucine as a catalyst and geraniol as the solvent.57GES was then vulcanized with elemental sulfur to generate composite materials GSSx (Fig. 3, x = wt% sulfur in monomer feed). The GSSx composites exhibited significantly different morphological and mechanical characteristics relative to the OSSx analogues.
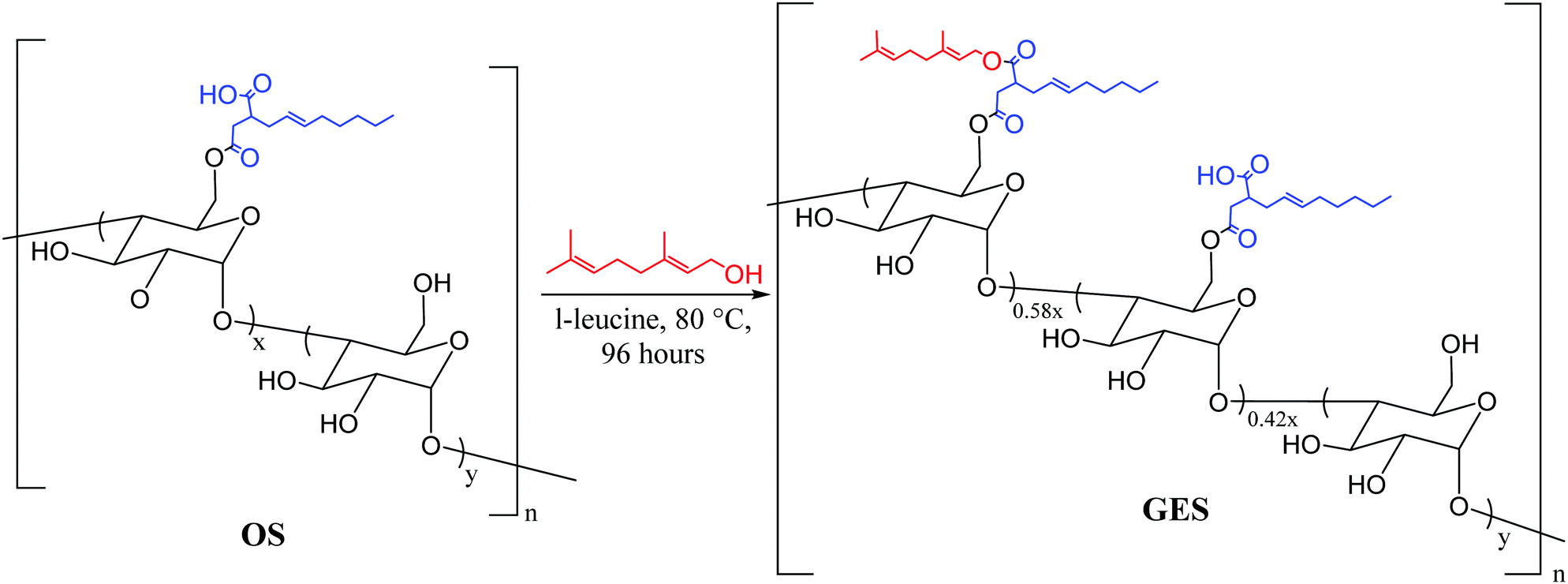 | ||
| Fig. 2 Modification of OS with geraniol to generate GES. The octenyl succinate ester is highlighted in blue and the geraniol portion is in red. The values for x and y are 2.6% and 97.4%, respectively. Upon reaction, 58% of the free carboxylic acid groups are esterified with geraniol. | ||
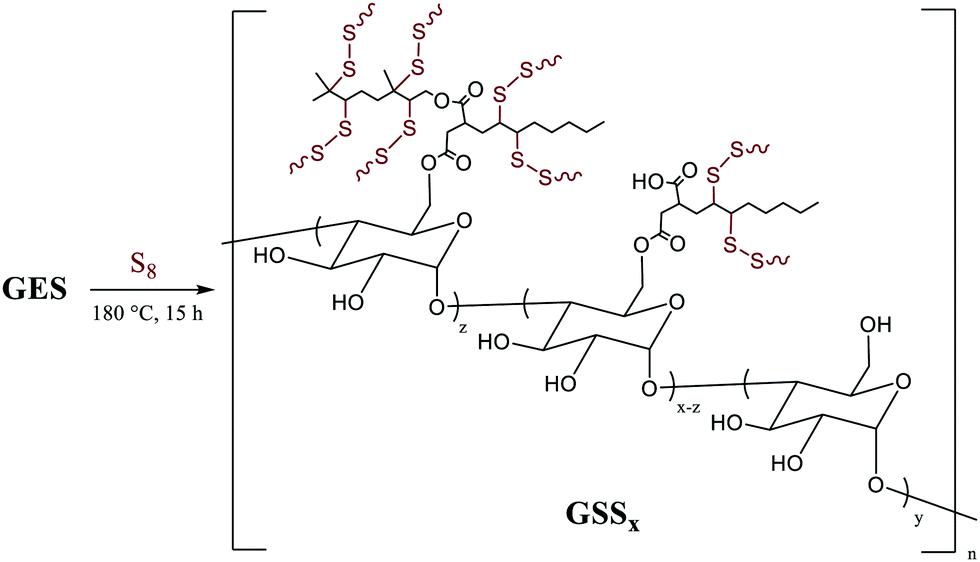 | ||
| Fig. 3 Reaction of GES with sulfur to generate GSSx composites (where x = wt% sulfur in the feed ratio, either 90 or 95). | ||
Results and discussion
Modification of OS
Starch derivative GES was prepared from the same lot of OS (Fig. 2) that was used to prepare previously-prepared composites OSSx to allow direct comparison of composite properties (vide infra).66 In OS, 2.6% of the monomer units have been substituted by their reaction with octenyl succinate to leave a carboxylic acid moiety amenable to esterification. The esterification protocol was based on our previously-reported procedure for the esterification of oxidized cellulose with geraniol.57 Briefly, OS was suspended in an excess of geraniol with catalytic L-leucine. The reaction was carried out in a sealed vessel held at 80 °C for 4 days while stirring. GES was readily isolated by simple filtration and washing with a small amount of ethanol. The purified product was dried in a vacuum oven at 40 °C for 3 days. As in previous studies, excess geraniol was recovered and re-used without changes in its purity (as assessed by GC analysis). The esterification process is thus near 100% atom economy with small amounts of water being produced as a by-product and the green solvent ethanol was the only other compound used in the purification step. Each site esterified by its reaction with geraniol will have three times as many olefin units for crosslinking in the subsequent vulcanization step. The difference in physical properties of the starch derivatives was striking. The difference in bulk starch granule agglomeration following esterification and drying to constant mass was evident to the naked eye. Corn starch presents as clustered granules that are free flowing but can be easily pressed into a thin sheet, whereas OS was a very fine powder and GES looked more similar to corn starch than OS but tended to agglomerate into larger clusters of starch (Fig. 4). | ||
| Fig. 4 Unmodified corn starch (left), OS (center), and GES (right) shown as either a pile of powder (top images) or the top-down view of a vial full of material (bottom images) showing the significant differences in physical properties. | ||
Esterification of starch by this protocol increased its crystallinity to nearly the same crystallinity observed for native corn starch, an important consideration given that biopolymer crystallinity has been shown to affect changes in the mechanical properties of composites made by biopolymer vulcanization and that modification tends to decrease biopolymer crystallinity.57 Powder X-ray diffraction (PXRD) of all three materials – corn starch starting material, OS and GES – exhibit the pattern typical of waxy corn starches that exhibit A-type crystallinity.77 A detailed analysis of structural features contributing to A-type biopolymer crystallinity has been reported previously.78 In the current study, the unmodified corn starch, OS, and GES were found by PXRD to have percent crystallinities of 20.9, 15.8, and 20.2%, respectively (Fig. S1 in the ESI,†). These data reveal a decrease of crystallinity of only 0.7% over the two steps and a small increase of 4.4% upon esterification of OS.
Infrared (IR) spectroscopic data for OS and GES were, unsurprisingly, nearly identical (ESI,† Fig. S2). The presence of geraniol in GES could be qualitatively visualized in its 1H NMR spectrum, where peaks attributable to the methyl groups of geraniol were visible at 1.64 and 1.57 ppm. Most of the other expected resonances attributable to geraniol were obscured by resonances attributable to the starch backbone and the chain of the octenyl succinate functionality (ESI,† Fig. S3). The degree of geraniol-esterification could not be determined with a high degree of certainty from either the 1H NMR spectrum or other solution-based methods due to the limited solubility of GES and the small degree of substitution in the OS precursor. The degree of geraniol esterification was thus determined based on analysis of the thermal decomposition profile of GES from thermogravimetric analysis (TGA) wherein decomposition of geraniol can be observed shortly before the primary decomposition observed for the carbohydrate backbone (see ESI,† Fig. S4, S5, eqn (S1) and Table S1 for full details of the calculation). This analysis revealed that 1.5 ± 0.2% of starch monomers contain the geraniol-esterified unit, while 1.1% ± 0.2% remain in the carboxylic acid form (Fig. 2), equating to successful esterification of 58% of sites available in parent OS. The result is that GES (0.33 mmol olefin g−1) has approximately twice as many olefin units available for vulcanization with sulfur compared to OS (0.16 mmol olefin g−1).
Both starch derivatives OS and GES possessed thermal characteristics quite similar to those of native corn starch. The decomposition of GES proceeded quite similarly to OS with the exception of the decomposition of the geraniol side chain (from ∼150–230 °C), which additionally led to a slightly lower char yield for GES (∼1.2% difference in char at 800 °C, see ESI,† Fig. S4). Differential scanning calorimetry (DSC) data revealed that both derivatives possessed Tg values slightly lower than native corn starch's Tg of 98.4 °C. Interestingly, the Tg for GES (97.5 °C) was slightly increased relative to OS (89.6 °C), indicating that the pendant geraniol chain hindered segmental chain movement relative to the free carboxylic acid group (ESI,† Fig. S6 and Table S2), which is also consistent with the aforementioned trends in crystallinity observed by PXRD. Whereas there was a 9 °C drop in Tg upon modification of native starch to form OS, positive changes in the crystallinity of OS upon esterification actually improved the Tg.
Synthesis and properties of GSSx composites
Composites GSSx (x = wt% sulfur) were readily prepared by simply heating GES with elemental sulfur to initiate the inverse vulcanization reaction (Fig. 1). The reaction of GES with sulfur (Fig. 3) to generate GSS95 and GSS90 required longer heating at 180 °C (∼15 hours) compared to that required to prepare OSSx materials (∼9 hours) due to significant agglomeration of GES powder at the reaction interface. This phenomenon is likely a result of strong intermolecular forces present in the granular structure and higher crystallinity of GES compared to OS. In all cases the materials were heated under nitrogen to minimize the potential for undesired oxidation reactions. Following reaction, the molten composites were poured into cuboid or cylinder-shaped silicone molds to produce samples appropriate for subsequent mechanical analysis. HSMs often contain metastable polysulfur catenates that take time to relax to their ultimate allotropic form. Many HSMs are therefore analyzed after 3–4 days of curing. In the current case, GSSx materials were analyzed after 15 days of curing to match conditions employed for analysis of previously-reported OSSx materials.Thermal analysis of GSSx revealed some interesting results. Both materials possessed two thermal features in the thermogravimetric analysis (TGA) curves. The first feature is attributable to the sublimation of S8 (Tsub), while the second step is attributable to thermal degradation of the organic portion of the composite. In GSS90, Tsub of S8 in GSS90 was slightly depressed relative to GSS95 as the organic portion of the composite essentially acts as an ‘impurity’, disrupting the crystallinity and lowering Tsub (ESI,† Fig. S7). GSS90 also, unsurprisingly, showed a larger step for the decomposition of the organic portion of the material at ∼300 °C. In contrast to most previous HSMs, the char yield of the two GSSx composites did not scale with the amount of organic material in the composite.57,62 Although GSS90 contained less than twice (7.6 and 12.5 wt% organic for GSS95 and GSS90, respectively, ESI,† Table S3) as much organic/crosslinkable material by elemental microanalysis, its char yield was more than three times that observed in GSS95, revealing that a more thermally resistant crosslinked network forms when GSS90 undergoes thermal decomposition. Further evidence for higher-density crosslinking was observed in the heating and cooling curves obtained by DSC for GSSx. Whereas GSS95 exhibited thermomorphological changes quite similar to those observed for pure sulfur (i.e., sulfur melting feature on heating and recrystallization on cooling), GSS90 exhibited a reversible Tg at −40 °C, a feature never before observed for biopolymer–sulfur composites except for sulfur composites prepared from lignin with quite high degrees of crosslinking (Fig. 5).58,63–65 Crystallization on cooling is unsurprisingly delayed (requires cooling to a lower temperature) for GSS90 relative to GSS95 by more than 25 °C and numerous cold-crystallization features are observed upon heating (ESI,† Fig. S8 and S9). These data provide further evidence that the crosslinked portion of GSS90 is more robust and likely contains more highly crosslinked material with shorter sulfur catenates bridging biopolymer crosslinks compared to GSS95.
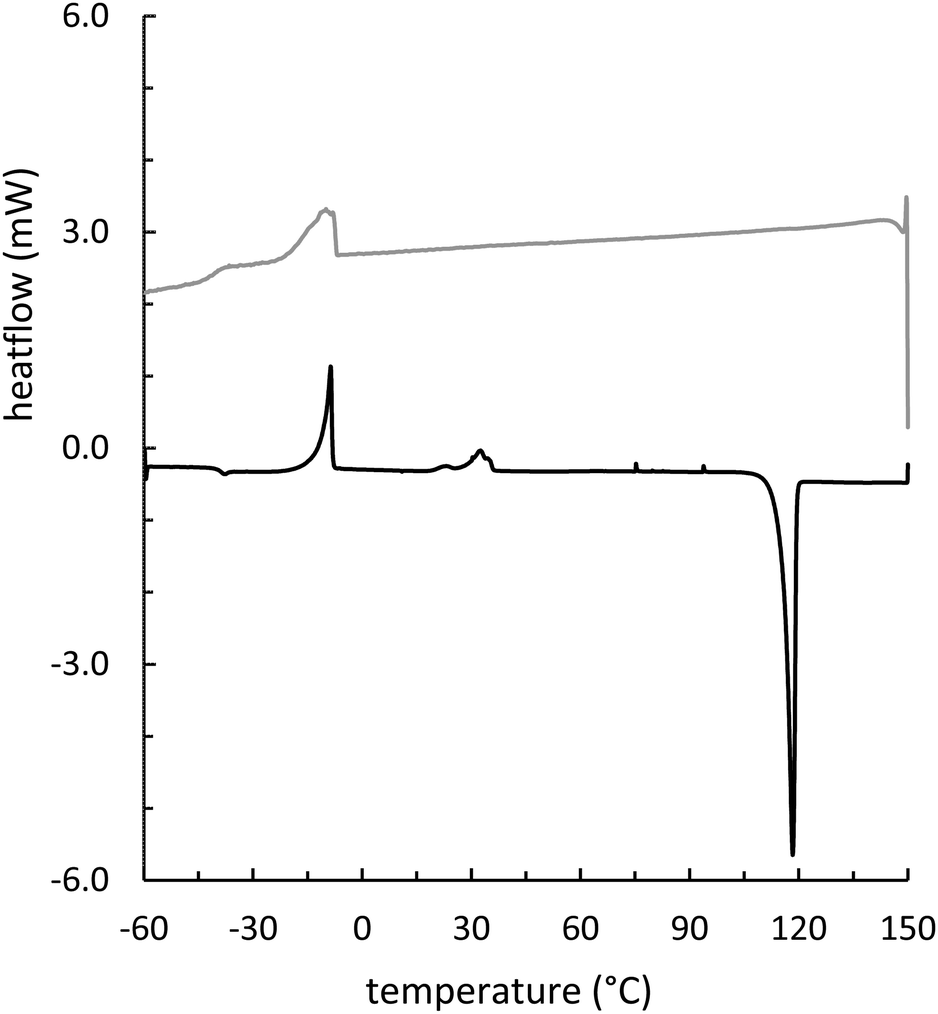 | ||
| Fig. 5 DSC heating (black) and cooling (gray) curves from the third cycle for GSS90. The intensity of the cooling cycle has been multiplied by 4 and shifted by +1.2 mW to make the features appear more prominent. The heating cycle shows a Tg for polymeric sulfur with a midpoint at −39 °C. Two regions of cold crystallization are visible at −24 to −1 °C and from 18 to 39 °C. The melting endotherm for S8(β) appears at 118 °C. In the cooling cycle, a crystallization exotherm occurs at −12 °C and the reversible Tg is apparent with midpoint at −41 °C. | ||
To better understand the differences in crosslinking in the two GSSx materials, each was subjected to fractionation with CS2. This is a common technique for extracting any noncovalently-bound sulfur, which is readily soluble in CS2, from an HSM to leave behind only the organosulfur network in the insoluble fraction. Based on these results, the organic material in GSS95 is able to crosslink 5–6% of the sulfur present in the material whereas the GES in GSS90 crosslinks <1% of the available sulfur. This data is also consistent with the crosslinked portion of the material being much more localized with significantly shorter sulfur catenates. A similar trend was observed in OSSx materials where OSS95 was able to incorporate more sulfur into the crosslinked network but on a much smaller scale (9.5 and 8.3 wt% for OSS95 and OSS90, respectively, ESI,† Table S3). The trend is readily rationalized because although both OS and GES have a low degree of olefin modification, GES has very concentrated densities of olefins due to the necessary proximity of geraniol-derived olefins and the octenyl succinate derived olefin. IR analysis of the CS2 insoluble fractions revealed more sulfur-derived scattering seen as baseline absorbance in GSS95 and a worse signal to noise compared to GSS90 further substantiating this hypothesis (ESI,† Fig. S10).
Although the low degree of substitution did not allow visualization of differences between GSS90 and GSS95 by IR spectroscopy, differences were evident from scanning electron microscopy with elemental mapping by energy dispersive spectroscopy (SEM-EDS, ESI,† Fig. S11) clearly revealing regions of crosslinked organic material. In both GSSx materials the organic fraction of the material was well dispersed within the material but agglomerated into small regions throughout the material. Consistent with what was suggested from thermal and fractionation results, the clusters of organic material were larger and more plentiful in GSS90 than was observed for GSS95.
Mechanical properties of GSSx were assessed by mechanical test stand analysis (compressive strength) and by dynamic mechanical analysis (DMA, flexural strength). Because GSSx materials are morphologically more like a particle-reinforced crystalline material relative to a polymeric material, they deformed entirely plastically before breaking under a compressional stress (Fig. 6A and Table 1). The more uniform dispersion of organic material in the network of GSS90 allowed for better reproducibility but provided less strength than the minimal filling observed in GSS95 (13.4 ± 2.1 MPa and 10.6 ± 0.7 MPa for GSS95 and GSS90, respectively). This trend was nearly identical in the OSSx series. Relative to OSSx materials, however, GSSx materials were less stiff but GSS95 was found to be the strongest material. This fact can similarly be explained by the differences in morphology between the two materials. While OSSx has long crosslinks and more interaction between the domains, GSSx is essentially acting as a particle-reinforced network of sulfur. Recently-reported HSMs comprising variously-sized lignocellulosic particles and sulfur show similar behaviours.79
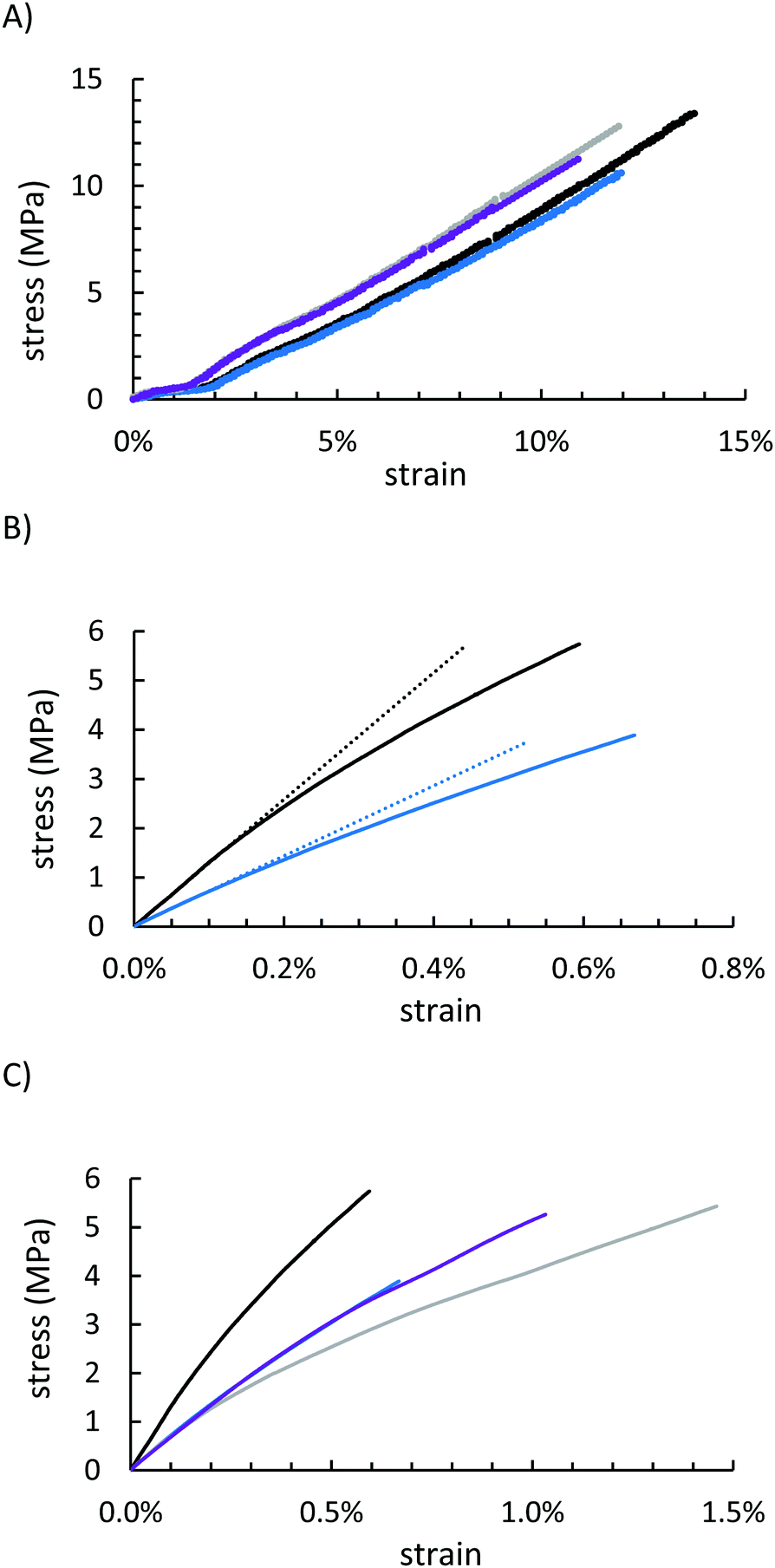 | ||
| Fig. 6 Mechanical data for GSS95 (black), GSS90 (blue), OSS95 (gray), and OSS90 (purple). Compression data for all four materials reveal that each class of materials behaved similarly but with different ultimate strengths where the 95 wt% materials performed better (A). Flexural data for GSS95 and GSS90 (solid lines) plotted with their flexural moduli (dotted lines) (B), revealing significantly higher strength and stiffness of GSS95 relative to GSS90 and all four materials plotted together (C). | ||
| GSS95 | GSS90 | OSS95 | OSS90 | |
|---|---|---|---|---|
| a Defined as a deviation of 0.05 MPa from the linear region. b The area under the linear region up to the limit of linearity (%). c The area under the entire stress–strain curve. | ||||
| Strength (MPa) | 5.7 | 3.9 | 5.4 | 5.3 |
| Limit of linearity (%)a | 0.15 | 0.16 | 0.13 | 0.31 |
| Limit of linearity (MPa)a | 1.9 | 1.1 | 0.9 | 2.0 |
| Flexural modulus (MPa) | 1290 | 710 | 690 | 660 |
| Modulus of resilience (kPa)b | 1.5 | 0.9 | 0.6 | 3.2 |
| Toughness (kPa)c | 19.1 | 13.9 | 42.9 | 30.2 |
Under a flexural deformation (Fig. 6B and C), GSS90 and OSS90 behaved very similarly except that GSS90 broke at 1.5 MPa less stress than did OSS90. In general, HSMs having a lower percentage of less CS2-extractable free sulfur are able to withstand flexural stresses-typically leading to a trend where incorporation of a higher feed ratio of the organic component produces a stronger material. This trend was not observed for GSSx materials. Although they contained nearly identical amounts of free-sulfur, GSS95 significantly outperformed GSS90 in the flexural strength test, making it the stiffest and strongest of all of the starch-sulfur vulcanized composites, while also having a high moduli of resilience and reasonable toughness (Table 1 and ESI,† Table S3). This interesting deviation from the general trend observed for HSMs may be attributable to the morphological uniqueness of this series of materials.
Under a flexural deformation (Fig. 6B and C), GSS90 and OSS90 behaved very similarly except that GSS90 broke 1.5 MPa before OSS90. The trend observed in OSSx materials where the material that was less CS2 soluble (i.e. more of the material was part of the crosslinked network), the better the material was able to withstand flexural stresses. Because GSS95 was less CS2 soluble, it outperformed GSS90, and quite significantly, making it the stiffest and strongest material of the series while also having a high moduli of resilience and toughness (Table 1).
Conclusions
Herein is reported a green method to esterify octenyl succinate-modified starch, a commercial food additive, to increase its degree of olefination by 2-fold in new derivative GES. The significant increase in crosslinkable sites paired with the high local density of reactive sites allowed access to a novel morphological regime of biopolymer–sulfur composites produced by inverse vulcanization of the modified starch with elemental sulfur. These vulcanized composites exhibited unprecedentedly low sulfur ranks which ultimately resulted in the materials taking the form of particle-reinforced networks, analogous to common structural materials such as cement. It was found that vulcanization of bulk sulfur with only a 5 wt% feed ratio of modified starch GES, led to materials with excellent stiffness and strength under a flexural and compressional deformation. The trends observed herein between OSSx and GSSx materials seems to suggest that morphology can be controlled depending on the regiochemistry of biopolymer modification prior to its vulcanization. In cases where the crosslinkable sites are necessarily close to one another, short sulfur catenates inevitably form. In this regime, high-strength materials can be produced even with minimal modification, thereby improving the sustainability of the process by decreasing chemical, energy, and waste considerations expenditures while also decreasing the time to material homogenization. These factors ultimately lead to a less expensive and more green cement-surrogate, thus informing future material design.Experimental
General considerations
Fourier transform infrared spectra were obtained using an IR instrument (Shimadzu IRAffinity-1S) with an ATR attachment. Scans were collected over the range 400–4000 cm−1 at ambient temperature with a resolution of 8. TGA was recorded (Mettler Toledo TGA 2 STARe System) over the range 20–800 °C with a heating rate of 10 °C min−1 under a flow of N2 (100 mL min−1). Each measurement was acquired in duplicate and presented results represent an average value. DSC was acquired (Mettler Toledo DSC 3 STARe System) over the range 60 to 150 °C with a heating rate of 5 °C min−1 under a flow of N2 (200 mL min−1). Each DSC measurement was carried out over five heat–cool cycles. Each measurement was acquired in triplicate to ensure consistent results were obtained.DMA was performed (Mettler Toledo DMA 1 STARe System) in single cantilever mode. DMA samples were cast from silicone resin moulds (Smooth-On Oomoo® 30 tin-cure). Samples were manually sanded to ensure uniform dimensions of approximately 15 × 8 × 1.5 mm but due to instrumental limitations (maximum force of 10 N), each sample differed slightly in thickness to obtain a stress at break. Sample dimensions were measured with a digital calliper with 0.01 mm resolution. Clamping was done by hand due to the samples’ brittleness. The force was varied from 0 to 10 N with a ramp rate of 0.2 N min−1 measured isothermally at 25 °C.
Carbon disulfide extractions were performed by suspending 0.3 g of finely ground material (measured to 0.0001 g) in 20 mL of CS2, allowing the solid to settle for 30 minutes, pipetting off the supernatant into a separate vial, and adding another 20 mL of CS2. This process was repeated an additional 3 times so that a total of 5 washes was performed. The residual CS2 was evaporated under a flow of N2, and each vial was weighed to determine the fraction that was soluble (collected as supernatant) or insoluble (remained in the initial vial). This process was performed in duplicate because of unanticipated results yielding identical results both times.
Compressional analysis was performed on a Mark-10 ES30 test stand equipped with a M3-200 force gauge (1 kN maximum force with 1 N resolution) with an applied force rate of 3–4 N s−1. Compression cylinders were cast from silicone resin moulds (Smooth-On Oomoos 30 tin-cure) with diameters of approximately 6 mm and heights of approximately 10 mm. Samples were manually sanded to ensure uniform dimensions and measured with a digital calliper with 0.01 mm resolution. Compressional analysis was performed in triplicate and results were averaged.
Powder X-ray diffraction samples were placed on zero background sample holders and analysed using a Rigaku Ultima IV diffractometer with Cu Kα radiation (l = 1.5406 Å). Data were collected from 5–65 degrees in 2-theta at a rate of 0.5 degree per minute with a sampling interval of 0.02 degrees. Crystallinity calculations were performed by integrating amorphous peaks at 19.01 and 26.43° and crystalline peak integrations at 12.00, 15.35, 17.21, 18.10, and 23.16° as is reported in the literature.80
SEM was acquired on a Schottky Field Emission Scanning Electron Microscope SU5000 operating in variable pressure mode with an accelerating voltage of 15 keV. NMR was conducted based on literature procedures.81 GES or OS was suspended in DMSO-d6 (12.5 mg mL−1) and sonicated for 2 hours at 70 °C. Molecular sieves (4 Å) were added and the samples were left over night. Proton NMR spectra were acquired on a Bruker NEO-300 MHz at room temperature and data was processed with TopSpin 4.0.6 software. All spectra reported were calibrated to the solvent peak at 2.50 ppm.
Author contributions
The author primarily responsible for particular credit roles are provided here. Moira K. Lauer: data curation, formal analysis, investigation, validation, roles/writing – original draft. Andrew G. Tennyson: resources. Rhett C. Smith: conceptualization, funding acquisition, methodology, formal analysis, resources, supervision, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by The National Science Foundation grant number CHE-1708844.Notes and references
- P. A. Arias, N. Bellouin, E. Coppola, R. G. Jones, G. Krinner, J. Marotzke, V. Naik, M. D. Palmer, G.-K. Plattner, J. Rogelj, M. Rojas, J. Sillmann, T. Storelvmo, P. W. Thorne, B. Trewin, K. A. Rao, B. Adhikary, R. P. Allan, K. Armour, G. Bala, R. Barimalala, S. Berger, J. G. Canadell, C. Cassou, A. Cherchi, W. Collins, W. D. Collins, S. L. Connors, S. Corti, F. Cruz, F. J. Dentener, C. Dereczynski, A. D. Luca, A. D. Niang, F. J. Doblas-Reyes, A. Dosio, H. Douville, F. Engelbrecht, V. Eyring, E. Fischer, P. Förster, B. Fox-Kemper, J. S. Fuglestvedt, J. C. Fyfe, N. P. Gillett, L. Goldfarb, I. Gorodetskaya, J. M. Gutierrez, R. Hamdi, E. Hawkins, H. T. Hewitt, P. Hope, A. S. Islam, C. Jones, D. S. Kaufman, R. E. Kopp, Y. Kosaka, J. Kossin, S. Krakovska, J.-Y. Lee, T. M. J. Li, T. K. Maycock, M. Meinshausen, S.-K. Min, P. M. S. Monteiro, T. Ngo-Duc, F. Otto, I. Pinto, A. Pirani, K. Raghavan, R. Ranasinghe, A. C. Ruane, L. Ruiz, J.-B. Sallée, B. H. Samset, S. Sathyendranath, S. I. Seneviratne, A. A. Sörensson, S. Szopa, I. Takayabu, A.-M. Treguier, B. V. D. Hurk, R. Vautard, K. V. Schuckmann, S. Zaehle, X. Zhang and K. Zickfeld, Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, ed. V. Masson-Delmotte, P. Zhai, A. Pirani, S. L. Connors, C. Péan, S. Berger, N. Caud, Y. Chen, L. Goldfarb, M. I. Gomis, M. Huang, K. Leitzell, E. Lonnoy, J. B. R. Matthews, T. K. Maycock, T. Waterfield, O. Yelekçi, R. Yu and B. Zhou, Cambridge University Press, 2021 Search PubMed.
- K. L. Scrivener, V. M. John and E. M. Gartner, Cem. Concr. Res., 2018, 114, 2–26 CrossRef CAS.
- R. M. Andrew, Earth Syst. Sci. Data, 2018, 10, 195–217 CrossRef.
- M. Dehestani, E. Teimortashlu, M. Molaei, M. Ghomian, S. Firoozi and S. Aghili, Data Brief, 2017, 13, 137–144 CrossRef CAS PubMed.
- E. D. Weil, Phosphorus, Sulfur Silicon Relat. Elem., 1991, 59, 325–340 CrossRef CAS.
- U.S. Department of Transportation, Extension and Replacement of Asphalt and Cement with Sulfur (FHWA-RD-78-95), 1978.
- I. Deme, Sulphur Pellet Comprising H2S-Suppressant for Asphalt Paving Mixture, WO2004-EP53357, 2005.
- I. Deme, Adv. Chem. Ser., 1978, 165, 172–189 CrossRef CAS.
- J. E. Gillott, I. J. Jordaan, R. E. Loov, N. G. Shrive and M. A. Ward, Adv. Chem. Ser., 1978, 165, 98–112 CrossRef CAS.
- G. J. Kennepohl and L. J. Miller, Adv. Chem. Ser., 1978, 165, 113–134 CrossRef CAS.
- M. Al-Ansary, E. Masad and D. Strickland, Adv. Gas Process., 2010, 2, 121–130 CAS.
- A. Taylor, N. Tran, R. May, D. Timm, M. Robbins and B. Powell, J. Assoc. Asphalt Paving Technol., 2010, 79, 403–441 CAS.
- D.-y. Lee, Prod. R&D, 1975, 14, 171–177 Search PubMed.
- S. Gwon, E. Ahn and M. Shin, Composites, Part B, 2019, 162, 469–483 CrossRef CAS.
- S. Gwon, S.-Y. Oh and M. Shin, Constr. Build. Mater., 2018, 181, 276–286 CrossRef CAS.
- S. Gwon and M. Shin, Constr. Build. Mater., 2019, 228, 116784 CrossRef CAS.
- P. Szajerski, A. Bogobowicz and A. Gasiorowski, J. Hazard. Mater., 2020, 381, 121180 CrossRef CAS PubMed.
- M. Lewandowski and R. Kotynia, MATEC Web Conf., 2018, 219, 3006 CrossRef CAS.
- S. Mohammed and V. Poornima, Mater. Today: Proc., 2018, 5, 23888–23897 CAS.
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, A. Somogyi, P. Theato, M. E. Mackay, Y.-E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef CAS PubMed.
- Y. Zhang, R. S. Glass, K. Char and J. Pyun, Polym. Chem., 2019, 10, 4078–4105 RSC.
- T. S. Kleine, R. S. Glass, D. L. Lichtenberger, M. E. MacKay, K. Char, R. A. Norwood and J. Pyun, ACS Macro Lett., 2020, 9, 245–259 CrossRef CAS.
- P. Yan, W. Zhao, B. Zhang, L. Jiang, S. Petcher, J. A. Smith, D. J. Parker, A. I. Cooper, J. Lei and T. Hasell, Angew. Chem., Int. Ed., 2020, 59, 13371–13378 CrossRef CAS PubMed.
- M. J. H. Worthington, R. L. Kucera and J. M. Chalker, Green Chem., 2017, 19, 2748–2761 RSC.
- J. M. Chalker, M. J. H. Worthington, N. A. Lundquist and L. J. Esdaile, Top. Curr. Chem., 2019, 377, 1–27 CrossRef CAS PubMed.
- T. Hasell, D. J. Parker, H. A. Jones, T. McAllister and S. M. Howdle, Chem. Commun., 2016, 52, 5383–5386 RSC.
- M. W. Thielke, L. A. Bultema, D. D. Brauer, P. Theato, B. Richter and M. Fischer, Polymers, 2016, 8, 1–9 CrossRef PubMed.
- D. J. Parker, H. A. Jones, S. Petcher, L. Cervini, J. M. Griffin, R. Akhtar and T. Hasell, J. Mater. Chem. A, 2017, 5, 11682–11692 RSC.
- A. M. Abraham, S. V. Kumar and S. M. Alhassan, Chem. Eng. J., 2018, 332, 1–7 CrossRef CAS.
- H.-K. Lin, Y.-S. Lai and Y.-L. Liu, ACS Sustainable Chem. Eng., 2019, 7, 4515–4522 CrossRef CAS.
- Y. Chen, A. Yasin, Y. Zhang, X. Zan, Y. Liu and L. Zhang, Materials, 2020, 13, 632 CrossRef CAS PubMed.
- A. D. Tikoalu, N. A. Lundquist and J. M. Chalker, Adv. Sustainable Syst., 2020, 4, 1900111 CrossRef CAS.
- J. J. Griebel, S. Namnabat, E. T. Kim, R. Himmelhuber, D. H. Moronta, W. J. Chung, A. G. Simmonds, K.-J. Kim, J. van der Laan, N. A. Nguyen, E. L. Dereniak, M. E. MacKay, K. Char, R. S. Glass, R. A. Norwood and J. Pyun, Adv. Mater., 2014, 26, 3014–3018 CrossRef CAS PubMed.
- S. Namnabat, J. J. Gabriel, J. Pyun and R. A. Norwood, Proc. SPIE, 2014, 8983, 89830D CrossRef.
- S. Namnabat, J. J. Gabriel, J. Pyun, R. A. Norwood, E. L. Dereniak and J. van der Laan, Proc. SPIE, 2014, 9070, 90702H Search PubMed.
- J. J. Griebel, N. A. Nguyen, S. Namnabat, L. E. Anderson, R. S. Glass, R. A. Norwood, M. E. MacKay, K. Char and J. Pyun, ACS Macro Lett., 2015, 4, 862–866 CrossRef CAS.
- L. E. Anderson, T. S. Kleine, Y. Zhang, D. D. Phan, S. Namnabat, E. A. LaVilla, K. M. Konopka, L. Ruiz Diaz, M. S. Manchester, J. Schwiegerling, R. S. Glass, M. E. Mackay, K. Char, R. A. Norwood and J. Pyun, ACS Macro Lett., 2017, 6, 500–504 CrossRef CAS.
- T. S. Kleine, T. Lee, K. J. Carothers, M. O. Hamilton, L. E. Anderson, L. Ruiz Diaz, N. P. Lyons, K. R. Coasey, W. O. Parker, Jr., L. Borghi, M. E. MacKay, K. Char, R. S. Glass, D. L. Lichtenberger, R. A. Norwood and J. Pyun, Angew. Chem., Int. Ed., 2019, 58, 17656–17660 CrossRef CAS PubMed.
- J. Kuwabara, K. Oi, M. M. Watanabe, T. Fukuda and T. Kanbara, ACS Appl. Polym. Mater., 2020, 2, 5173–5178 CrossRef CAS.
- A. G. Simmonds, J. J. Griebel, J. Park, K. R. Kim, W. J. Chung, V. P. Oleshko, J. Kim, E. T. Kim, R. S. Glass, C. L. Soles, Y.-E. Sung, K. Char and J. Pyun, ACS Macro Lett., 2014, 3, 229–232 CrossRef CAS.
- I. Gomez, O. Leonet, J. A. Blazquez and D. Mecerreyes, ChemSusChem, 2016, 9, 3419–3425 CrossRef CAS PubMed.
- A. Hoefling, D. T. Nguyen, Y. J. Lee, S.-W. Song and P. Theato, Mater. Chem. Front., 2017, 1, 1818–1822 RSC.
- S. Zeng, L. Li, L. Xie, D. Zhao, N. Wang and S. Chen, ChemSusChem, 2017, 10, 3378–3386 CrossRef CAS PubMed.
- Y. Zhang, J. J. Griebel, P. T. Dirlam, N. A. Nguyen, R. S. Glass, M. E. MacKay, K. Char and J. Pyun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 107–116 CrossRef CAS.
- P. Dong, K. S. Han, J.-I. Lee, X. Zhang, Y. Cha and M.-K. Song, ACS Appl. Mater. Interfaces, 2018, 10, 29565–29573 CrossRef CAS PubMed.
- Q. Jiang, Y. Li, X. Zhao, P. Xiong, X. Yu, Y. Xu and L. Chen, J. Mater. Chem. A, 2018, 6, 17977–17981 RSC.
- F. Zhao, Y. Li and W. Feng, Small Methods, 2018, 2, 1–34 CAS.
- T. S. Sahu, S. Choi, P. Jaumaux, J. Zhang, C. Wang, D. Zhou and G. Wang, Polyhedron, 2019, 162, 147–154 CrossRef CAS.
- C. R. Westerman and C. L. Jenkins, Macromolecules, 2018, 51, 7233–7238 CrossRef CAS.
- C. Herrera, K. J. Ysinga and C. L. Jenkins, ACS Appl. Mater. Interfaces, 2019, 11, 35312–35318 CrossRef CAS PubMed.
- K. Orme, A. H. Fistrovich and C. L. Jenkins, Macromolecules, 2020, 53, 9353–9361 CrossRef CAS.
- X. Wu, J. A. Smith, S. Petcher, B. Zhang, D. J. Parker, J. M. Griffin and T. Hasell, Nat. Commun., 2019, 10, 10035–10044 Search PubMed.
- B. Zhang, S. Petcher and T. Hasell, Chem. Commun., 2019, 55, 10681–10684 RSC.
- B. Zhang, H. Gao, P. Yan, S. Petcher and T. Hasell, Mater. Chem. Front., 2020, 4, 669–675 RSC.
- N. A. Lundquist, A. D. Tikoalu, M. J. H. Worthington, R. Shapter, S. J. Tonkin, F. Stojcevski, M. Mann, C. T. Gibson, J. R. Gascooke, A. Karton, L. C. Henderson, L. J. Esdaile and J. M. Chalker, Chem. – Eur. J., 2020, 26, 10035–10044 CrossRef CAS PubMed.
- S. J. Tonkin, C. T. Gibson, J. A. Campbell, D. A. Lewis, A. Karton, T. Hasell and J. M. Chalker, Chem. Sci., 2020, 11, 5537–5546 RSC.
- M. K. Lauer, A. G. Tennyson and R. C. Smith, ACS Appl. Polym. Mater., 2020, 2, 3761–3765 CrossRef CAS.
- M. K. Lauer, M. S. Karunarathna, A. G. Tennyson and R. C. Smith, Mater. Adv., 2020, 1, 2271–2278 RSC.
- M. K. Lauer, M. S. Karunarathna, A. Tennyson and G. R. C. Smith, Mater. Adv., 2020, 1, 590–594 RSC.
- C. V. Lopez, M. S. Karunarathna, M. K. Lauer, C. P. Maladeniya, T. Thiounn, E. D. Ackley and R. C. Smith, J. Poly. Sci., 2020, 58, 2259–2266 CrossRef CAS.
- C. P. Maladeniya, M. S. Karunarathna, M. K. Lauer, C. V. Lopez, T. Thiounn and R. C. Smith, Mater. Adv., 2020, 1, 1665–1674 RSC.
- M. K. Lauer, T. A. Estrada-Mendoza, C. D. McMillen, G. Chumanov, A. G. Tennyson and R. C. Smith, Adv. Sustainable Syst., 2019, 3, 1900062 CrossRef CAS.
- M. S. Karunarathna, M. K. Lauer, T. Thiounn, R. C. Smith and A. G. Tennyson, J. Mater. Chem. A, 2019, 7, 15683–15690 RSC.
- M. S. Karunarathna and R. C. Smith, Sustainability, 2020, 12, 734–748 CrossRef CAS.
- M. S. Karunarathna, A. G. Tennyson and R. C. Smith, J. Mater. Chem. A, 2020, 8, 548–553 RSC.
- M. K. Lauer, A. G. Tennyson and R. C. Smith, Mater. Adv., 2021, 2, 2391–2397 RSC.
- M. K. Lauer and R. C. Smith, Compr. Rev. Food Sci. Food Saf., 2020, 1–53, DOI:10.1111/1541-4337.12627.
- C. P. Maladeniya and R. C. Smith, J. Compos. Sci., 2021, 5, 257 CrossRef CAS.
- M. P. Crockett, A. M. Evans, M. J. H. Worthington, I. S. Albuquerque, A. D. Slattery, C. T. Gibson, J. A. Campbell, D. A. Lewis, G. J. L. Bernardes and J. M. Chalker, Angew. Chem., Int. Ed., 2016, 55, 1714–1718 CrossRef CAS PubMed.
- F. Wu, S. Chen, V. Srot, Y. Huang, S. K. Sinha, P. A. van Aken, J. Maier and Y. Yu, Adv. Mater., 2018, 30, 1706643 CrossRef PubMed.
- J. A. Smith, S. J. Green, S. Petcher, D. J. Parker, B. Zhang, M. J. H. Worthington, X. Wu, C. A. Kelly, T. Baker, C. T. Gibson, J. A. Campbell, D. A. Lewis, M. J. Jenkins, H. Willcock, J. M. Chalker and T. Hasell, Chem. – Eur. J., 2019, 25, 10433–10440 CrossRef CAS PubMed.
- A. D. Smith, T. Thiounn, E. W. Lyles, E. K. Kibler, R. C. Smith and A. G. Tennyson, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1704–1710 CrossRef CAS.
- A. D. Smith, C. D. McMillin, R. C. Smith and A. G. Tennyson, J. Poly. Sci., 2020, 58, 438–445 CrossRef CAS.
- A. D. Smith, R. C. Smith and A. G. Tennyson, Sustainable Chem., 2020, 1, 209–237 CrossRef.
- L. Dai, J. Zhang and F. Cheng, Int. J. Biol. Macromol., 2019, 132, 897–905 CrossRef CAS PubMed.
- P. Paronen and M. Juslin, J. Pharm. Pharmacol., 1983, 35, 627–635 CrossRef CAS PubMed.
- R. Bodîrlău, C.-A. Teacă, I. Spiridon and N. Tudorachi, Monatsh. Chem., 2012, 143, 335–343 CrossRef.
- J. J. G. van Soest, S. H. D. Hulleman, D. de Wit and J. F. G. Vliegenthart, Ind. Crops Prod., 1996, 5, 11–22 CrossRef CAS.
- M. K. Lauer, Z. E. Sanders, A. D. Smith and R. C. Smith, Mater. Adv., 2021, 2, 7413–7422, 10.1039/D1MA00689D.
- Z. Zhang, S. Zhao and S. Xiong, Starch/Staerke, 2010, 62, 78–85 CrossRef CAS.
- S. Schmitz, A. C. Dona, P. Castignolles, R. G. Gilbert and M. Gaborieau, Macromol. Biosci., 2009, 9, 506–514 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional synthetic and characterization including PXRD data, IR spectra, NMR spectra, TGA and DSC data, details on calculations and mechanical testing. See DOI: https://doi.org/10.1039/d1ma01202a |
| This journal is © The Royal Society of Chemistry 2022 |