
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSystematic photophysical, thermal and electrochemical analysis of a series of phenothiazine cored conjugated aromatic unit appended D–π–A based high-solid state luminescent materials: their applications in reversible mechanofluorochromic and volatile acid sensing†
Bhaswati
Sarkar
,
Edamana
Prasad
 * and
Ramesh L.
Gardas
*
* and
Ramesh L.
Gardas
*
Department of Chemistry, Indian Institute of Technology Madras, Chennai, 600036, India. E-mail: pre@iitm.ac.in; gardas@iitm.ac.in; Fax: +91-44-2257-4202; Tel: +91-44-2257-4232 Tel: +91-44-2257-4248
First published on 4th February 2022
Abstract
The advancement of unique, organic materials possessing exclusive solid-state photoluminescence properties is in high demand due to their noteworthy contribution to materials chemistry and technology. Considering this, a class of novel scissor shaped phenothiazine (PT) derivatives, named PT–Cn–(Ar)2 where n = 2, 12 and Ar = phenyl, naphthyl, anthracenyl, phenanthryl, and pyrenyl units, have been synthesized, which show divergent fluorescence emission characteristics in a variety of solvents by virtue of their twisted intramolecular charge transfer (TICT) state. Among the studied PT derivatives, naphthyl and pyrenyl derivatives generate an unusual blue-shifted aggregation-induced emission (AIE) in the THF–water binary mixture due to the suppression of the TICT state. Interestingly, among all the products, only the phenyl derivative shows reversible mechanofluorochromic (MFC) behavior, where it offers cyan to yellowish-green emission upon mechanical agitation and fumigation concurrently. Mechanistic inspection derived from P-XRD indicates loss in crystallinity, generating redshifted emission upon mechanical pressure. The SEM study shows two distinct morphologies of the MFC material before and after applying the mechanical force. Furthermore, the MFC characteristic has been verified by single-point energy calculations using DFT. Also, a narrow bandgap (Eg) value and lower excited state singlet (S1) and triplet (T1) energy gaps (ΔESTs) derived from DFT calculations for anthracene and pyrene derivatives suggest their potential in organic photovoltaic cells. Furthermore, reversible acid-sensing behavior has been demonstrated by a model PT derivative PT–C2–(Pn)2. Additionally, the high electrochemical stability of the derivatives up to 200 cycles suggests a feasible structural engineering approach for fabricating stable redox-active organic materials for redox flow batteries and OLEDs.
Introduction
Fabrication and synthesis of novel organic molecules having superior solid-state luminescence are highly demanding because of their remarkable contribution in the area of materials science and technology viz., fluorescent probes and switches,1 memory devices,2 data storage devices,3 organic light-emitting diodes (OLEDs),4 and optoelectronic devices.5 They can also be stimulated by mechanical agitation and exhibit mechanofluorochromic (MFC) behavior.6–10 Numerous materials with MFC characteristics such as organometallic compounds and typical organic dyes have been established.11–15 Compared to metal-based MFC materials, which suffer from commercialization challenges owing to the high price and the toxicity of noble metals, organic compounds turn out to be favorable alternatives due to their facile synthetic strategy. These types of materials have attracted special attention from researchers because of their bright potentiality in security inks,16 3D printing,17,18 memory chips,19 and mechanosensors20,21 as they are susceptible to shearing, pressing, ball milling, and grinding, causing a variation in the fluorescence emission color.However, conventional organic luminophores become feebly emissive in their solid-state as they disperse their excited-state energy in a non-radiative pathway because they have a considerable amount of π–π stacking in their aggregated state, termed as aggregation caused quenching (ACQ).22–24 Consequently, ACQ characteristics heavily hinder the practical applicability of MFC materials.25 Tang and co-workers have successfully addressed this shortcoming by inaugurating the concept of aggregation-induced emission (AIE). The non-planar skeleton of AIEgens diminishes the π–π stacking by restricting intramolecular rotation (RIR), resulting in the enhancement of emission in their aggregated state.26,27 Hence, AIE active molecules are highly appreciable for developing MFC materials. Mechanofluorochromic responses usually instigate from the alteration of physical structures. They are primarily reliant on molecular packing arrangement, the conformation of molecules in their solid-state, and the intermolecular interactions in the molecule.28–32 Furthermore, the dependence of MFC properties on the molecular packing arrangement in the solid-state can be finely tuned by several structural changes, like the incorporation of heavy atoms, halogen atoms, and alkyl or aryl units, the inclusion of heteroatoms and positional changes, and use of a donor–acceptor skeleton, which cause discrete intermolecular interactions.1,33,34
Organic frameworks having donor–acceptor (D–A), donor–π–acceptor (D–π–A), or donor–acceptor–donor (D–A–D) are utilized to produce highly emissive solid-state materials which can demonstrate MFC properties. The combination of non-planar twisted AIE active units, attached in an organic donor–acceptor framework, leads to very high fluorescence emission in their aggregated state by diminishing face-to-face interactions between the aromatic rings. Consequently, they can be easily converted into different metastable states upon mechanical stimuli by assuming more planarity.35–37 Phenothiazine (PT) is a well-known core having potentiality in device formation viz. chemosensors,38 solar cells,39 OLEDs,40 and thermally activated delayed fluorescence (TADF) materials.41 The PT-cored organic materials are also one of the highly desirable components as MFC materials resulting from their strong electron-donating character due to the “S” and “N” atoms and an unusual butterfly shape of the middle heterocyclic ring.42
Phenyl, naphthalene, anthracene, phenanthrene, and pyrene units act as electron acceptors when connected with a highly influential electron donor unit, viz. the PT core. In this article, we have designed and synthesized (D–π–A) isomers by appending aromatic chromophores with increasing conjugation to a PT moiety via a vinylidene linkage, to obtain PT–Cn–(Ar)2 where n = 2, 12 and Ar = phenyl (Ph), naphthyl (Nap), anthracenyl (An), phenanthryl (Pn), and pyrenyl (Pr) units. We have hypothesized that the non-planar donor and highly conjugated chromophoric units can act as a D–π–A system, and by virtue of the butterfly conformations of the PT core, it can act as a MFC material. The attachment of the donor PT unit having different chromophores with extended conjugation acting as an acceptor possibly influences the donor–acceptor character and the molecular packing modes in different derivatives, resulting in distinct photophysical and electrochemical characteristics. Also, the size of the alkyl chain length on the N atom of the PT core has been altered to investigate whether there is any effect on their MFC properties.
Photophysical properties have been investigated in detail using UV-vis and fluorescence spectroscopy. AIE phenomena of the PT derivatives also have been investigated. Besides, their thermal and electrochemical properties have been investigated. Furthermore, their MFC properties have been studied by UV-vis, fluorescence, P-XRD, FT-IR, TCSPC, and SEM. Moreover, a DFT study has been performed to understand the electronic structures of the derivatives and to get an in-depth view of the mechanochromism mechanism of the studied PT derivatives. Additionally, reversible volatile acid, trifluoroacetic acid (TFA), sensing has been investigated. This study shows a new possibility for the development of mechanofluorochromic, organoelectric, and solid-state volatile acid sensors.
Results and discussion
Scheme 1 presents the structures and the synthetic strategy of a set of novel scissor-shaped divinyl MFC materials, for example, PT–C2–(Ph)2 (1a), PT–C12–(Ph)2 (1b), PT–C2–(Nap)2 (2a), PT–C12–(Nap)2 (2b), PT–C2–(An)2 (3), PT–C2–(Pn)2 (4) and PT–C2–(Pr)2 (5) (Chart 1) using Suzuki–Miyaura cross-coupling of commercially available aryl-boronic acid (Ar–B(OH)2) and dibromovinyl-10-alkyl-10H-phenothiazine.43 The PT-derivatives were synthesized using literature procedures (ESI) and characterized using 1H NMR and 13C NMR spectroscopy, and ESI-mass spectrometry (Fig. S1–S36, ESI†). | ||
| Scheme 1 Synthetic pathway of the PT derivatives. | ||
 | ||
| Chart 1 Fluorophores synthesized in the present work. | ||
Photophysical properties
To study the photophysical properties of PT derivatives, UV-Vis and fluorescence measurements were carried out. Fig. 1 shows the normalized electronic absorbance and emission spectra of 10 μM DCM solution of the PT derivatives. Here, Fig. 1(a) shows an absorption band in the range of 250–340 nm of various chromophores having a very high probability of electronic transition, which can be assigned to π–π* (S0–S2) transitions from the various chromophoric components. Additionally, the other absorption bands having a low probability of electronic transition in the range of 340–460 nm could be ascribed to the n–π* (S0–S1) transition.44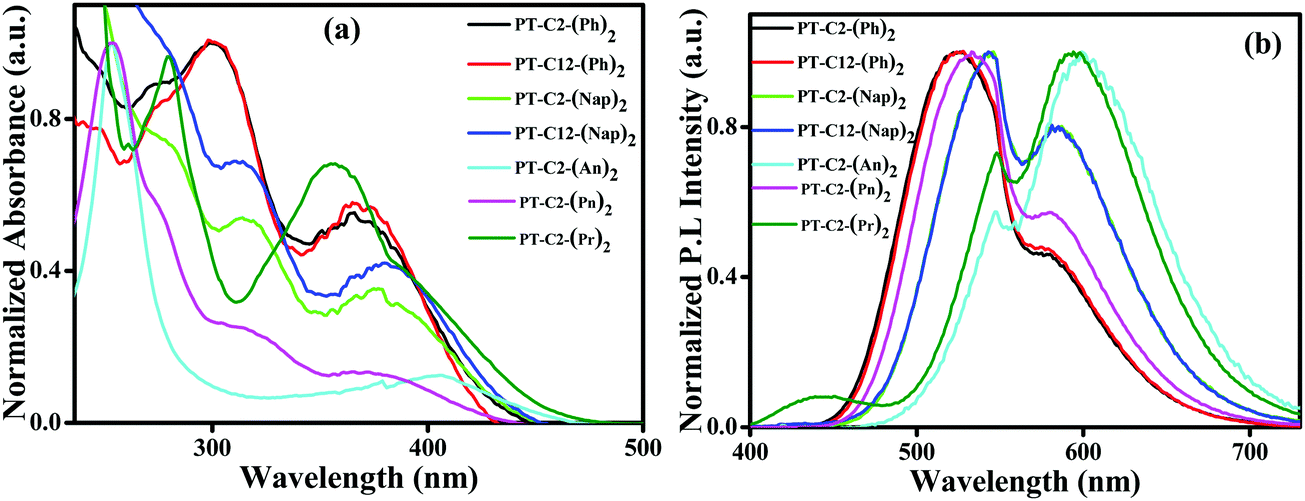 | ||
| Fig. 1 (a) Normalized UV-Vis absorption spectra and (b) emission spectra of PT–C2–(Ph)2 (λEx = 381 nm), PT–C12–(Ph)2 (λEx = 370 nm), PT–C2–(Nap)2 (λEx = 381 nm), PT–C12–(Nap)2 (λEx = 381 nm), PT–C2–(An)2 (λEx = 405 nm), PT–C2–(Pn)2 (λEx = 365 nm) and PT–C2–(Pr)2 (λEx = 372 nm), measured in ca. 10 μM in DCM solution. | ||
This transition arises due to the twisted ground state intermolecular charge transfer taking place between the non-planar donor (D) PT and the acceptor (A) (Ar) (viz. Ph, Nap, An, Pn, and Pr). The phenyl appended PT derivatives, PT–C2–(Ph)2 and PT–C12–(Ph)2, exhibited a maximum absorption wavelength, λmax, of 299 nm, whereas a 14 nm redshift was detected in PT–C2–(Nap)2 and PT–C12–(Nap)2, which could be due to the extended conjugation of the acceptor unit. However, PT–C2–(An)2, PT–C2–(Pn)2 and PT–C2–(Pr)2 demonstrated high energy π–π* (S0–S2) transitions, in the range of 234–290 nm largely as a result of the higher energy necessitated for the electrons to circumnavigate across the larger aromatic rings, i.e., An, Pn and Pr units, and also could be the result of the sterically bulky nature of An, Pn and Pr units.45,46
Sharp CT bands were noticed for PT–C2–(Ph)2, PT–C12–(Ph)2, PT–C2–(Nap)2, PT–C12–(Nap)2, PT–C2–(An)2, and PT–C2–(Pr)2. In contrast, a weak and broad ground state CT band centered at 365 nm was observed in PT–C2–(Pn)2. This can be explained in terms of the dihedral angle between PT and the Ar units. The dihedral angles between the PT unit and Ar units are relatively small in PT–C2–(Ph)2, PT–C12–(Ph)2, PT–C2–(Nap)2, PT–C12–(Nap)2, PT–C2–(An)2, and PT–C2–(Pr)2 (Fig. S37 and Table S1, ESI†), while larger dihedral angles between the PT unit and Pn have been detected in PT–C2–(Pn)2. It is well recognized that the smaller the dihedral angle between the donor and acceptor, the stronger the electronic coupling.46 The CT band in the visible region, i.e., 405 nm, for PT–C2–(An)2, results from the low-lying excited state leading to a low energy band gap (3.03 eV) (Fig. S37 and Table S1, ESI†) as obtained from DFT calculations.
Usually, the functionalized PT core turns into a twisted intramolecular charge transfer state (TICT) from its locally excited state (LE) by virtue of its “butterfly-like” conformation having two terminal planar phenyl rings, and the central ring being in a to some extent flattened boat form in polar solvents. This TICT emission results in more prolonged wavelength emission than LE emission, causing a significant Stokes shift. Fig. 1(b) shows the dual emission from the PT derivatives where the high energy emission corresponds to the emission from the locally excited (LE) state and the low energy emission corresponds to the TICT state. PT–C2–(Ph)2 and PT–C12–(Ph)2 show emission maxima at ca. 524 nm with shoulder peaks at 579 and 581 nm, respectively. Similarly, an emission peak appears for PT–C2–(Nap)2 and PT–C12–(Nap)2 at ca. 547 nm, accompanied by a shoulder peak at ca. 585 nm. Both PT–C2–(An)2 and PT–C2–(Pn)2, containing identical structural isomers (anthracene and phenanthrene), show a different pattern of emission spectra, while PT–C2–(Pr)2 shows similar emission spectra to PT–C2–(An)2. The emission maxima for PT–C2–(Pn)2 were observed at 533 nm containing one shoulder peak at 579 nm, whereas both PT–C2–(An)2 and PT–C2–(Pr)2 exhibited a sharp emission band at 594 and 601 nm, respectively, along with a shoulder peak at 548 nm (Table 1).
| Compd |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Abs λmaxε [104] [nm] (M−1 cm−1) Abs λmaxε [104] [nm] (M−1 cm−1) |
E
m, λmax (nm) |
Onset/S1 [nm eV−1] | Stokes shift (cm−1) | λ onset (nm), Eg (eV) [f] | φ f | FWHM (nm) |
|---|---|---|---|---|---|---|---|
| a Measured in DCM (10−5 M). b Fluorescence quantum yields were recorded using Coumarin 152 as a standard in cyclohexane solution. | |||||||
| PT–C2–(Ph)2 | 299 (2.42), 381 (1.34) | 524, 581 | 453/2.74 | 7163 | 436, 2.84 | 0.41 | 75 |
| PT–C12–(Ph)2 | 290 (1.67), 370 (0.85) | 524, 579 | 422/2.94 | 7943 | 434, 2.86 | 0.43 | 74 |
| PT–C2–(Nap)2 | 279 (4.03), 315 (2.84), 381 (2.0) | 546, 585 | 459/2.70 | 7932 | 446, 2.78 | 0.15 | 114 |
| PT–C12–(Nap)2 | 281 (2.2), 311 (1.4), 381 (1.17) | 547, 583 | 444/2.79 | 7965 | 442, 2.80 | 0.14 | 114 |
| PT–C2–(An)2 | 253 (16.5), 405 (2.23) | 548, 601 | 421/2.94 | 8024 | 465, 2.67 | 0.07 | 121 |
| PT–C2–(Pn)2 | 253 (10.6), 315 (2.8), 365 (1.57) | 533, 579 | 443/2.80 | 8635 | 435, 2.85 | 0.44 | 103 |
| PT–C2–(Pr)2 | 280 (5.80), 359 (4.21) | 548, 594 | 443/2.80 | 11255 |
461, 2.69 | 0.17 | 112 |
The unmodified PT core exhibits a very poor quantum yield of 0.0016 in cyclohexane because of its forbidden electronic transition and the non-radiative decay resulting from the nonplanarity of the molecule.47 However, it is noteworthy that when the PT core is in conjugation with the simple Ph unit, the emission quantum yield (φf) value increases remarkably. Among all the luminogens, PT–C2–(Ph)2, PT–C12–(Ph)2, and PT–C2–(Pn)2 show higher values of photoluminescence quantum yields (φf) of 0.41, 0.43, and 0.44, respectively, in DCM.48 The combination of the high value of the lifetime component (τ2) and its considerable contribution (B2) shows significant TICT characteristics, resulting in the high fluorescence lifetime (τf) of PT–C2–(Ph)2, PT–C12–(Ph)2 and PT–C2–(Pn)2, whereas the former component (τ1) represents the LE properties (Table S2 and Fig. S38, ESI†). In general, phenanthrene derivatives show shorter fluorescence lifetimes (from 20 ns to 45 ns) and higher φf values (from 0.14 to 0.3) than unsubstituted phenanthrene. This can be explained by the breakage in symmetry in the substituted phenanthrene.49 In contrast, PT–C2–(Nap)2, PT–C12–(Nap)2, and PT–C2–(Pn)2 show relatively lower values of quantum yields (0.15, 0.114, and 0.17).50,51 Also, knr is ca. six times higher than kr in PT–C2–(Nap)2 and PT–C12–(Nap)2 and four times higher in PT–C2–(Pn)2 because of the unrestricted rotation of the diaryl units in their solution state (Fig. S38 and Table S2, ESI†). The negligible φf value of 0.07 of PT–C2–(An)2 could be attributed to the drastic luminescence quenching as a result of the strong π–π stacking by the An units in PT–C2–(An)2, which results in two times more knr and five times less kr values than PT–C2–(Pn)2 (Table S2, ESI†).52 Additionally, substantial Stokes shift values were observed for some of the PT derivatives, making them promising candidates for super-resolution microscopy.53 The lower value of FWHM in PT–C2–(Ph)2 and PT–C12–(Ph)2 indicates that it can produce images generated by highly pure emission spectra.54
Solvatochromism
It is well-recognized that phenothiazine-appended D–π–A based fluorophore molecules impart a significant dipole moment that leads to TICT and consequently could be superior candidates for acquiring MFC organic materials.48 Also, the excited-state dipole moment of D–π–A based chromophores is susceptible to solvent polarity, and hence to gain an insight into the D–π–A conjugated systems of PT derivatives, UV-visible absorption, and fluorescence emission spectra were recorded by varying the polarity from cyclohexane (CHX), benzene (Benz) toluene (Tol), tetrahydrofuran (THF), and dichloromethane (DCM) to dimethyl sulfoxide (DMSO). It is evident from Fig. S39 (ESI†) that, in the cases of both PT–C12–(Ph)2 and PT–C12–(Nap)2, the absorption spectra redshifted with an increase in solvent polarity, whereas minor changes were observed in the cases of PT–C2–(Ph)2 and PT–C2–(Nap)2. This can be ascribed to the effect of the alkyl chain on the donor ‘N’ atom enhancing the electron transfer by its +I effect. PT–C2–(An)2 and PT–C2–(Pn)2 show ca. 16 and 47 nm redshifts, respectively, when the polarity increases from cyclohexane to DMSO, indicating a substantial ground state CT in PT–C2–(Pn)2. In contrast, minor changes were observed in the absorption spectra of PT–C2–(Pr)2 (Fig. S39, ESI†), which is assigned to its insensitivity to the solvent milieu (Table S1, ESI†). Emission maxima (Fig. 2) of the PT derivatives show significant redshifts of 52, 55, 102, 90, 118, 126, and 98 nm for PT–C2–(Ph)2, PT–C12–(Ph)2, PT–C2–(Nap)2, PT–C12–(Nap)2, PT–C2–(An)2, PT–C2–(Pn)2, and PT–C2–(Pr)2, respectively, accompanying broadening in the vibronic spectra with an increase in the solvent polarity from cyclohexane to DMSO, which indicates excited state TICT from the non-planar donor PT to the Ar units.55 The highest redshift in PT–C2–(An)2, PT–C2–(Pn)2, and PT–C2–(Pr)2 in DMSO could be attributed to the high excited-state dipole moment (μe) and comparatively large planarity in the PT core (Fig. 2 and Table S1, ESI†).55 The photographs of the derivatives in the solvents mentioned above are shown in Fig. S40 (ESI†).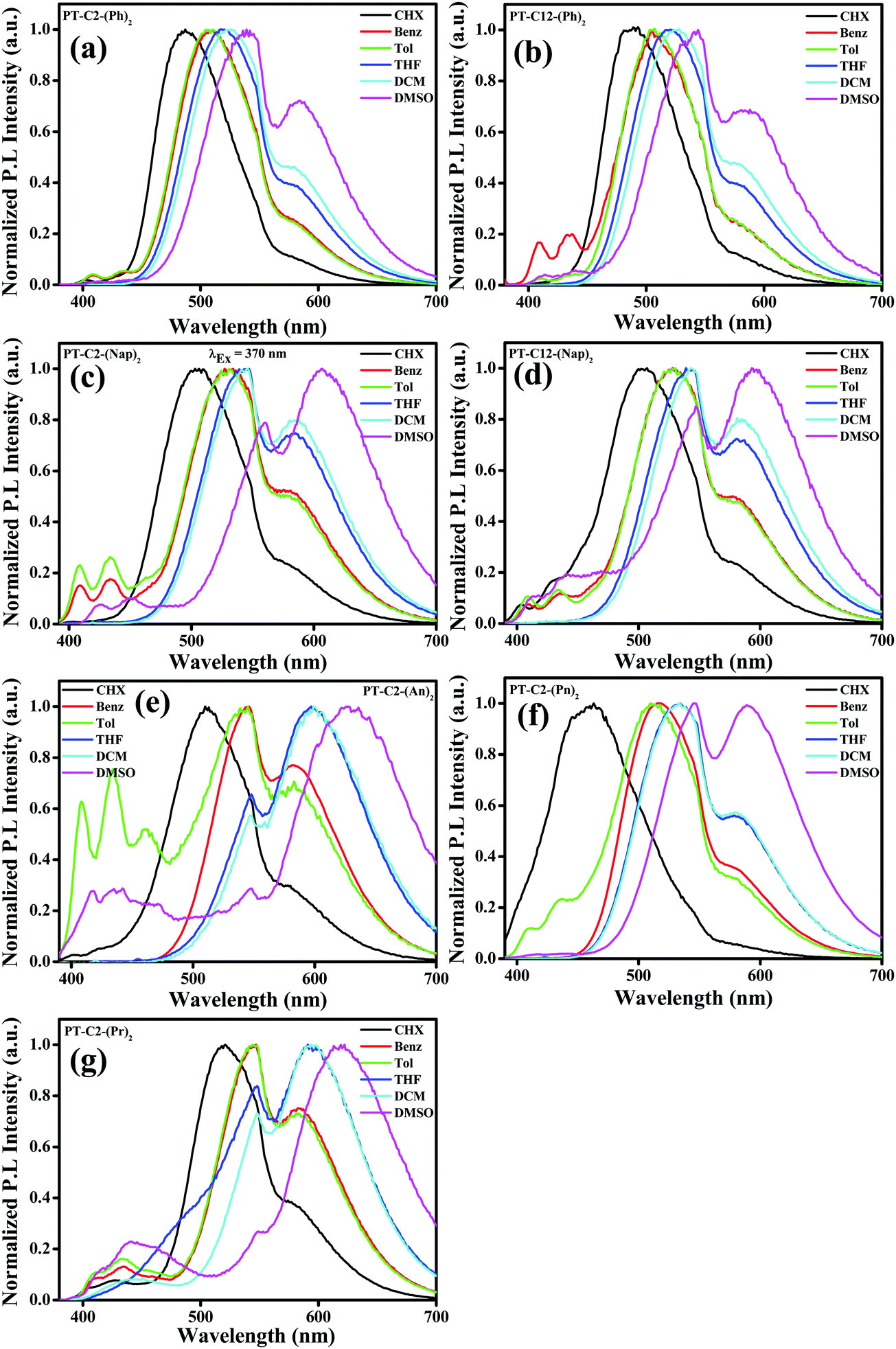 | ||
| Fig. 2 Emission spectra of (a) PT–C2–(Ph)2, (b) PT–C12–(Ph)2, (c) PT–C2–(Nap)2, (d) PT–C12–(Nap)2, (e) PT–C2–(An)2 and (f) PT–C2–(Pn)2 and (g) PT–C2–(Pr)2 in solvents (cyclohexane (CHX), benzene (Benz), toluene (Tol), DCM, THF and DMSO) of varying polarities (λEx = 370 nm). | ||
Aggregation-induced emission
Incorporating non-planar PT units with highly planar conjugated aromatic Ar (Ph, Nap, An, Pn, and Pr) units can facilitate emission in their aggregated state by virtue of their non-planar frameworks. In a polar solvent, a molecule with a twisted conformation results in a TICT emission peak at a longer wavelength when compared to LE emission, producing a significant Stokes shift. Also, the TICT emission peak gets further enhanced due to the AIE effect. By considering this, AIE properties have been explored via absorption (Fig. S41, ESI†) and emission spectroscopy (Fig. 3). Since the PT derivatives are readily soluble in THF but insoluble in water, the successive increase of the water fraction (fw (%)) in the THF solution of PT derivatives triggers the nanoaggregate formation.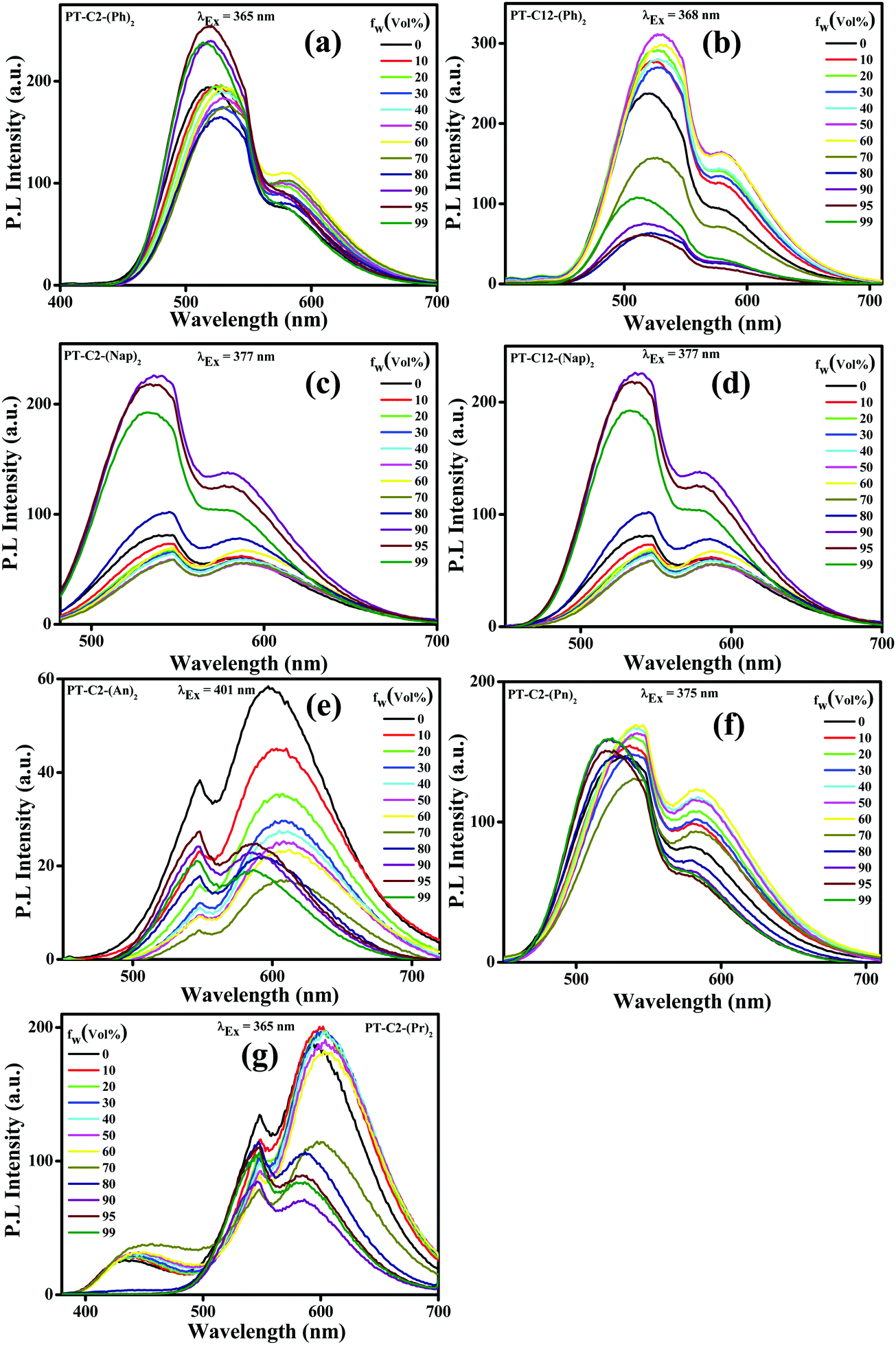 | ||
| Fig. 3 Emission spectra of (a) PT–C2–(Ph)2 (λEx = 365 nm), (b) PT–C12–(Ph)2 (λEx = 368 nm), (c) PT–C2–(Nap)2 (λEx = 377 nm), (d) PT–C12–(Nap)2 (λEx = 377 nm), (e) PT–C2–(An)2 (λEx = 401 nm), (f) PT–C2–(Pn)2 (λEx = 365 nm) and (g and f) PT–C2–(Pr)2 (λEx = 365 nm) in THF–water mixtures with different water fractions (fw (%)) (luminogen conc. 10 μM). | ||
In this study, absorption spectra showed a significant red shift along with the broadening in the spectra beyond fw ≥ 70%, attributed to the poor solubility of the luminogen generated from substantial heterogeneity in its microenvironment, which turned the monomeric form into its aggregated form. Moreover, the tailing observed beyond the fw range of 70–95% in the absorption spectra at the more extended wavelength range suggested the ‘Mie scattering effect,’ signifying the formation of nano-aggregates at higher water fractions.56
Traditionally, AIE-gens are emissive exclusively in their aggregated state, while all the PT derivatives synthesized here demonstrate AIE activities even in their solution state as a result of the combined effect of LE and CT emissions.28 The emission spectra of both the phenyl derivatives were altered in THF with increased fw (%) values (Fig. 3(a) and (b)), resulting in 1.3 times emission enhancement when fw (%) = 95 and 50 for PT–C2–(Ph)2 and PT–C12–(Ph)2, respectively. Furthermore, there is a drop in the emission intensity beyond the fw (%) value, which might be due to the formation of low emissive amorphous aggregates (Fig. S42 and S43, ESI†).38 Similarly, both PT–C2–(Nap)2 and PT–C12–(Nap)2 (Fig. 3(c) and (d)) demonstrate high-intensity fluorescence emission initiating from the LE state of ca. 548 nm in pure THF with slight changes up to 70% water fraction. However, a new emission maximum of ca. 536 nm with a 12 nm hypsochromic shift is generated due to the formation of nanoaggregates for 90% water fraction with 2.79 times emission enhancement as compared to the emission in 100% THF solution. Although above 90% water fraction, a slight drop in intensity was observed due to the development of large aggregates (Fig. 3(c)).57 The observation also shows an aggregation-induced unusual blue-shift in PT–C2–(Nap)2 and PT–C12–(Nap)2, which might be due to the inhibition TICT in the aggregated state of the naphthyl derivatives.58 In general, the AIE effect leading to a blue shift are relatively less compared to those which exhibit a red shift upon aggregation. Among PT–C2–(An)2, PT–C2–(Pn)2 and PT–C2–(Pr)2 (Fig. 3(e), (f) and (g)) both the anthracene and pyrene derivatives show emission from the CT state, whereas phenanthrene shows emission from the LE state. In contrast to other derivatives, PT–C2–(An)2 alone shows ACQ phenomena, upon increasing the water fraction. This can be attributed to the enhanced π–π interaction in their aggregated state, which diminishes the fluorescence emission generating a non-radiative pathway.
Thermal properties
To investigate the thermal stability of the PT derivatives, thermogravimetric analysis (TGA) has been carried out under a nitrogen atmosphere (Fig. S44, ESI†). The result shows that PT–C2–(Ph)2, PT–C2–(Nap)2, PT–C2–(An)2, PT–C2–(Pn)2, and PT–C2–(Pr)2 have thermal decomposition temperatures (Tds) of 281, 344, 132, 397 and 264 °C, respectively, at 5% weight loss as shown in Fig. S44 (ESI†). Interestingly, in between two isomeric chromophores PT–C2–(An)2 and PT–C2–(Pn)2, the latter exhibited higher stability than the former one, even the higher among all the PT derivatives. This can be attributed to the result of a complicated interference of numerous (oppositely directed) cyclic conjugation energy effects.59Electrochemical properties
To explore the oxidation and reduction potential and to evaluate the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of the as-synthesized luminogens, cyclic voltammetry (CV) (Fig. S45–S65, ESI†) experiments have been carried out. These compounds were dropcast onto a carbon working electrode, whereas silver and platinum wires were used as the reference and counter electrodes, respectively. The electrochemical characteristics of the luminogens were inspected using 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) solution as an electrolyte consisting of DCM (CH2Cl2) at room temperature under a nitrogen atmosphere. Ferrocene (Fc) dissolved in DCM was taken as an internal standard.The cyclic voltammograms of naphthyl, phenanthrene, and pyrene derivatives have shown two (one-electron) oxidation potentials. In contrast, phenyl and anthracene derivatives have shown three (one-electron) oxidation potentials, which are rational for highly electron-rich organic materials.60 The HOMO energy levels of the PT derivatives have been determined from the onset oxidation potentials (EOX) derived from differential pulse voltammograms (DPVs), using the relation EHOMO = − EOX − 4.8 eV (against Fc/Fc+). Also, the LUMO energy levels have been determined from the optical band gaps (Eg, solution) and the appropriate HOMO energy values (ELUMO = EHOMO + Eg, eV). The oxidation and reduction onsets and the corresponding HOMO and LUMO energy levels are listed in Tables S3 and S4 (ESI†), which lie in the ranges of −5.03 to −5.00 and −2.36 to −2.16 eV, respectively. PT–C2–(An)2, having higher HOMO/LUMO energy levels than the other PT derivatives leading to the low energy gap and the high oxidation potential of 1.57 eV, proves that this compound can behave as a super-oxidant for solar energy purposes as compared to the other derivatives studied here.61–63 Hence, the cyclic voltammograms recognize that all the PT derivatives are comparatively more stable than PT–C2–(An)2 to oxidation.
To further understand the electrochemical stability, all the PT derivatives were treated with 200 charging–discharging cycles at a scan rate of 200 mV s−1 (Fig. 4). It can be seen that they are electronically highly stable up to 200 cycles. However, PT–C2–(Pr)2 has shown moderate stability up to 200 cycles. Hence, by virtue of the high thermal stability up to 264–397 °C and electrochemical stability of the PT derivatives they are potential candidates as organic electrode materials for redox-active organic materials in electrochemical energy devices, redox flow bBatteries, and organic light and organic light-emitting diodes.64–70
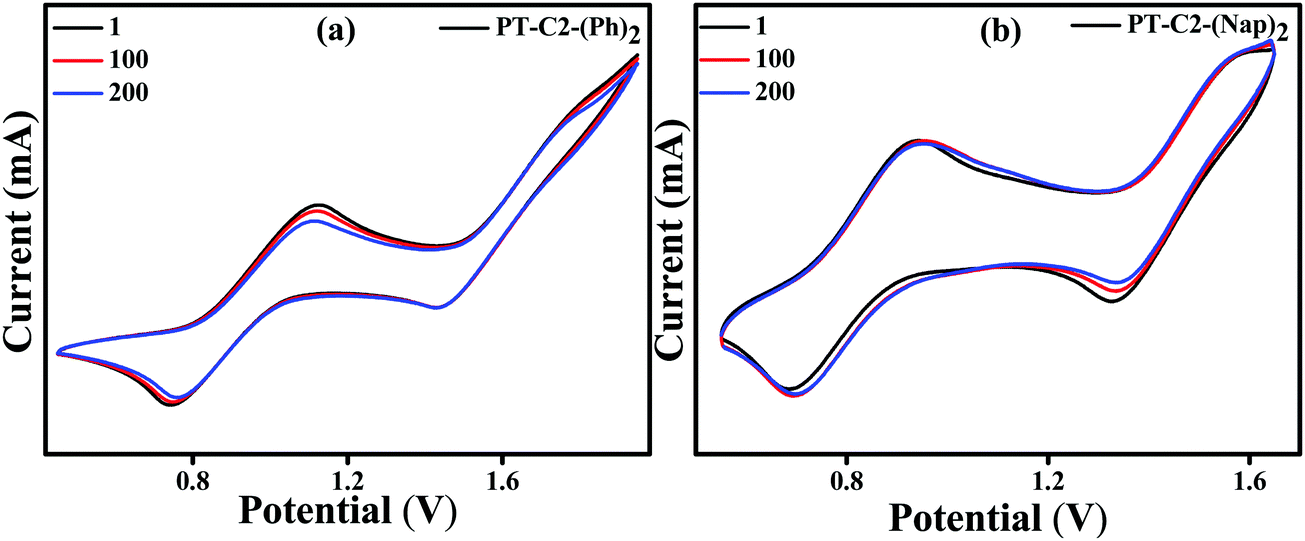 | ||
| Fig. 4 Cyclic voltammograms (200 mV s−1) of PT–C2–(Ph)2 (a) and PT–C2–(Nap)2 (b) (1st, 100th and 200th cycles) in DCM (ca. 1.0 mM) recorded in CH2Cl2 using TBAPF6 (0.1 M) as the supporting electrolyte. | ||
Mechanofluorochromic properties
Combining a highly conjugated planar diaryl unit with a non-planar PT unit accelerated the accomplishment of prominent solid-state emissive derivatives, which might be responsive to external stimuli. In search of the mechanoresponsive properties of the as-synthesized products, the switching of their solid-state emission was examined by employing mechanical force using a mortar and pestle. The mechanofluorochromic properties were explored in detail using UV-vis, fluorescence, lifetime, and powder XRD and SEM measurements in the solid states; however, among the seven PT derivatives (Fig. 5–7 and Fig. S66, S67, ESI†), only PT–C2–(Ph)2 predominantly showed appreciable mechanofluorochromic behaviour. PT–C2–(Ph)2 displayed a grinding-prompted 13 nm bathochromic shift. Consequently, the cyan emission changed to a yellowish-green color with an enhanced emission intensity upon grinding under 365 nm UV light. The changes in absorption in their solid states were recorded for the pristine, ground, and fumed samples (Fig. S67, ESI†).PT–C2–(Ph)2 displayed well-structured absorption maxima at 401 nm in the pristine and fumed states. However, significant broadening was noticed in its absorption spectra for the ground sample compared to its pristine state (Fig. S67, ESI†). To check the reversibility of its mechano-responsive behaviour, when the ground PT–C2–(Ph)2 was kept under DCM vapor, the emission color and the emission maxima reverted to their original position (Fig. 5 and 6). Again, after regrinding, the color changed from cyan emission (491 nm) to yellowish-green (504 nm) (Fig. 6). Additionally, the absorption maxima of ground PT–C2–(Ph)2 returned to its original absorption at 401 nm upon fuming with DCM vapor. Furthermore, it has been seen that upon grinding, the fluorescence lifetime of the ground sample has been enhanced. This can be attributed to the improved interaction between phenothiazine (PT) phenyl (Ph) and PT-long alkyl chain noncovalent interactions58 (viz. π–π, C–H⋯π and C–H⋯H–C) (Fig. 6(c), Table 2 and Table S5, ESI†).
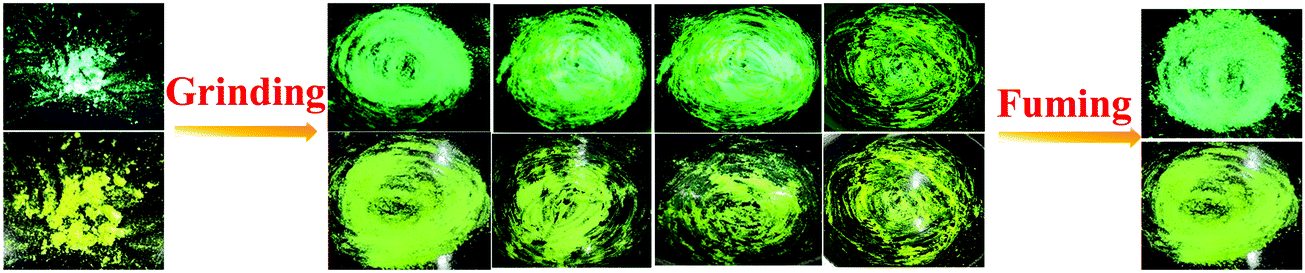 | ||
| Fig. 5 Images of PT–C2–(Ph)2 solids in different states (pristine (left), ground/fumed (right)) under 365 nm light and normal light. | ||
 | ||
| Fig. 6 (a) Fluorescence spectra (λEx = 420 nm) of PT–C2–(Ph)2 upon grinding–fuming cycles, (b) emission color alteration of PT–C2–(Ph)2 upon grinding–fumigation cycles and (c) time-resolved fluorescence decay of PT–C2–(Ph)2 in its pristine and ground state (λEx = 405 nm). | ||
| Compound | Emission | ||||
|---|---|---|---|---|---|
| λ Pristine/λFumed (nm) | λ Grinded (nm) | τ f(Pristine/Fumed) (ns) | τ f(Grinded) (ns) | Δλa (nm) | |
| Where Δλa = λGrinded − λPristine/λFumed. | |||||
| PT–C2–(Ph)2 | 491 | 504 | 4.19 | 6.29 | 13 |
Generally, molecular assemblies play a crucial role in the solid-state emission of organic materials. To achieve an in-depth understanding of the molecular packing arrangement under mechanical force, a diffraction pattern was recorded (Fig. 7(a)) in its different solid states. Pristine PT–C2–(Ph)2 displayed a very intense and sharp diffraction pattern, signifying a well-organized crystalline character. Conversely, ground PT–C2–(Ph)2 demonstrated a substantial decline in the diffraction peaks and the absence of a few peaks compared to its pristine state. This result showed the loss of crystallinity upon grinding.71 The happening can be explained as the mechanical force demolished the ordered stacking in the pristine solid, and a disordered arrangement prevails in the ground state. However, the diffraction peaks recovered when fumigated under DCM vapor, which indicates that the disarrangement of molecular packing returned to the ordered crystalline form. This result indicated that the less crystalline ground state is a metastable phase. Due to increased molecular mobility, a stable crystalline phase will develop upon fuming with solvent vapors.72,73 Furthermore, it can be concluded that the re-emergence of deep green fluorescence emission upon fumigation is mainly due to retrieving its original crystalline state.
 | ||
| Fig. 7 (a) P-XRD patterns of the pristine, ground, and fumed samples of PT–C2–(Ph)2, SEM images of PT–C2–(Ph)2 in its (b) pristine (PT–C2–(Ph)2–P) and (c) ground (PT–C2–(Ph)2–G) states. | ||
Furthermore, to study the effect of mechanical pressure on the morphological structure of PT–C2–(Ph)2, an FE-SEM image was taken before and after grinding of the sample (Fig. 7(b) and (c)). It can been seen that the pristine sample shows a cluster type arrangement. In contrast, there is a breakage in the cluster part upon grinding, and the sheet-type surface has been detected.
Acidochromism
The lone pair electron on the ‘N’ atom that belongs to the phenothiazine core of the PT derivatives is proficient in acid detection due to the protonation response of the derivatives.1 By considering this, as a model acid, highly volatile TFA detection has been carried out using absorbance and fluorescence spectroscopy (Fig. 8).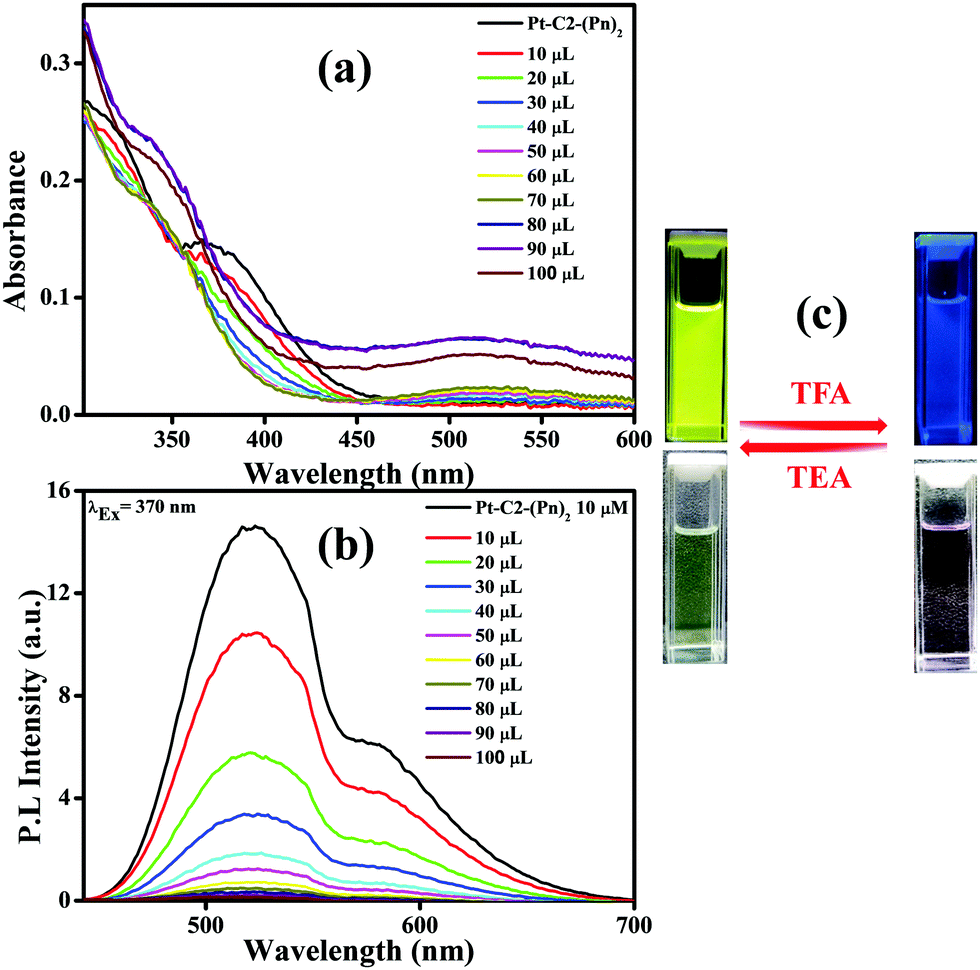 | ||
| Fig. 8 Absorption (a) and emission spectra of (b) PT–C2–(Pn)2 in CHCl3 (conc. 10−5 M) on titration with known amounts of TFA (10–100 mL). (c) Emission photographs of PT–C2–(Pn)2 in reversible response to TFA and TEA under UV light (top) and normal light (bottom). | ||
For demonstrating the sensing response towards TFA, PT–C2–(Pn)2 has been chosen as a sensor due to having a high quantum yield. For understanding the effect of TFA on the electronic spectra of PT–C2–(Pn)2, it has been treated with the commercially available TFA solution in CHCl3 solution (conc. 10−5 M) as shown in Fig. 8.
Fig. 8(a) shows that the CT band having absorbance maxima at 366 nm starts diminishing with an increase in the TFA concentration generating a new absorption peak at 518 nm, having an isosbestic point at 456 nm.74 The isosbestic point indicates the formation of the protonated PT–C2–(Pn)2 (PT–C2–(Pn)2H+), which are in reactive equilibrium with the pristine one.
Additionally, the emission intensity of PT–C2–(Pn)2 starts decreasing drastically upon concurrent addition of TFA (10–100 μL) (Fig. 8(b)), showing a blue-shifted emission at the higher concentrations of TFA. The quenching of emission intensity is attributed to the photoinduced electron transfer process.75 The quenching efficiency of the emission maxima for PT–C2–(Pn)2 in its solution state is 99% (Fig. 8(b)). Also, it has been seen that the addition of triethylamine (TEA) into the resulting solution of PT–C2–(Pn)2 and TFA demonstrates reversible color and intensity changes (Fig. 8(c)).
For checking the capability of PT–C2–(Pn)2 as a solid-state acid sensor, the thin film of PT–C2–(Pn)2 was treated with an increased amount of TFA vapor (Fig. 9(a)). Before adding TFA, the film eliminates highly bright greenish-yellow light. However, it was seen that at 0.29 ppm of TFA vapor, the emission intensity was quenched by 67% (Fig. 9(b)). Also, for checking the reusability of the TFA sensor, when it was treated with TEA vapor, the fluorescence intensity reverted to its original color and intensity within a second (Fig. S68, ESI†).
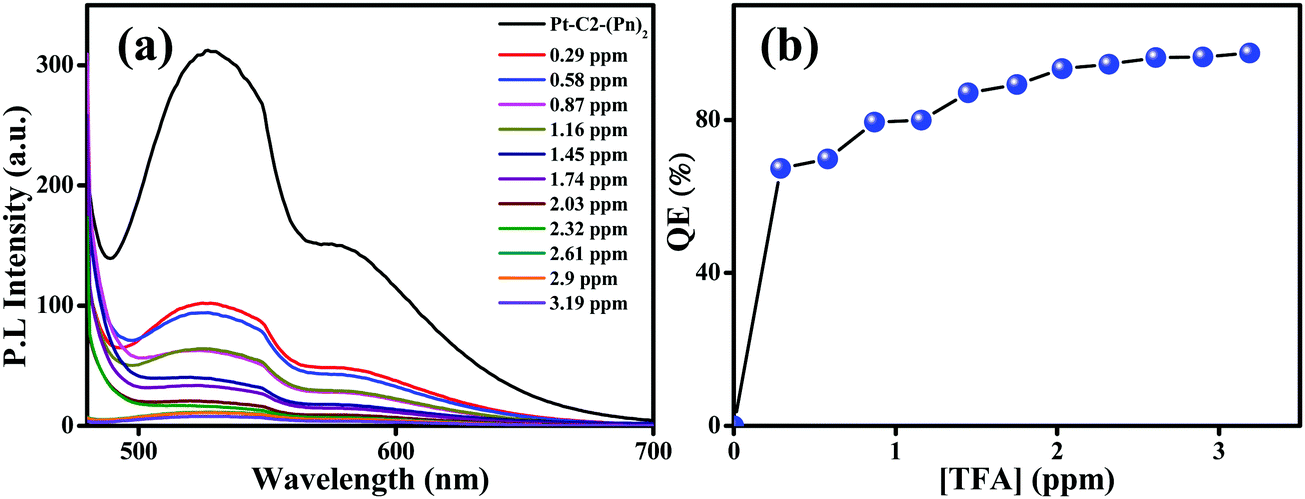 | ||
| Fig. 9 Emission spectra of the (a) PT–C2–(Pn)2 film on titration with known amounts of TFA. (b) Concentration versus quenching efficiency [QE (%)] under increased TFA vapor concentration. | ||
Additionally, for further confirmation about the protonation process, the 1H NMR spectra of PT–C2–(Pn)2 have been recorded before and after the addition of TFA. When the TFA (1.5 equiv.) was added dropwise into a CDCl3 solution of PT–C2–(Pn)2, the aromatic peaks assigned as the PT core peaks vanished immediately accompanied by a drastic color change from bright greenish-yellow to magenta. At the same time, the peaks assigned for the Pn unit become broad, structureless, and downfield shifted. Also, a significant shift of the ethyl proton directly attached with the N atom shows a substantial downfield shift from 3.7 to 5 ppm (Fig. S69, ESI†). However, when the resulting solution was treated with an excess amount of TEA, all the proton peaks of the PT core and the ethyl protons attached with the N atom of the PT core reappeared in their original positions and structures. Hence, the above discussion indicates that the protonation process is the driving force for the proton sensing of the derivative. Therefore, the effective quenching of the emission of PT–C2–(Pn)2 at its solution and solid-state confirms that this can act as a potential solid-state TFA sensor.
Density functional theory
To gain insight regarding the structural aspects by investigating the geometries and the electronic states in the ground-state density functional theory (DFT) has been studied. The ground-state optimization of the PT derivatives was conducted by employing B3LYP as the basis set and 6-311+g(d,p) as the basis function (Fig. S70, ESI†). The value of the highest occupied molecular orbital (HOMO), the lowest unoccupied molecular orbital (LUMO), and the HOMO–LUMO gaps are shown in Table S1 (ESI†). The HOMO for all of the PT derivatives, i.e., PT–C2–(Ph)2, PT–C12–(Ph)2, PT–C2–(Nap)2, PT–C12–(Nap)2, PT–C2–(An)2, and PT–C2–(Pr)2, is mainly localized on the PT unit slightly extending to the terminal Ar units through the vinylic spacer. In contrast, the LUMO is fully localized on the acceptor Ar units. Remarkably, in the case of PT–C2–(An)2 and PT–C2–(Pr)2, the HOMO exists in an end-to-end manner, which reveals the existence of higher electronic communication.28 However, PT–C2–(Pn)2 having well-separated HOMO–LUMO indicates a better donor–acceptor character.28 The HOMO energies obtained from DFT calculation for PT–C2–(Ph)2, PT–C12–(Ph)2, PT–C2–(Nap)2, PT–C12–(Nap)2, PT–C2–(An)2, PT–C2–(Pn)2 and PT–C2–(Pr)2 are −5.35, −5.33, −5.33, −5.32, −5.31, −5.38, and −5.31 eV, respectively. In comparison, the energies for the LUMO of the molecules mentioned above are −1.70, −1.69, −1.85, −1.84, −2.28, −1.85, and −2.21 eV, respectively. Furthermore, the HOMO–LUMO gaps determined for mentioned PT derivatives are 3.65, 3.64, 3.48, 3.48, 3.03, 3.53, and 3.10 eV, respectively. The HOMO level of each chromophore is almost the same irrespective of the extent of π-conjugation. Conversely, the LUMO levels of the compounds slightly differ from each other, where PT–C2–(An)2 shows the lowest LUMO value. Consequently, the lowest LUMO value of this compound results in a decrease of the HOMO–LUMO gap, which is also supported by the experimental results obtained from CV (Tables S3 and S4, ESI†). These results can be attributed to the fact that although anthracene and phenanthrene are geometrical isomers, anthracene having a zigzag conformation shows the lowest optical band gap value compared to phenanthrene because of its lower resonance stabilization values.63Additionally, to institute a proficient structure–property correlation, time-dependent density functional theory (TD-DFT) calculations at the theory level of PBE1PBE/6-311+g(d,p) have been executed on fully optimized structures of the derivatives to investigate the excited-state energy levels and electronic transitions (Fig. S70, ESI†). Theoretical outcomes are in line with the experimentally observed absorption spectra. The high value of oscillator strengths producing the absorption band at a longer wavelength indicates high CT possibility in most derivatives. In contrast, the lower value of oscillator strength noticed in PT–C2–(An)2 shows feeble CT phenomena.
To improve the power consumption efficacy in organic solar cells, increasing the light absorption and decreasing the energy loss by triplet recombination and exciton dissociation are compulsory. Therefore, it is essential to decrease the singlet–triplet energy gap (ΔEST), predominantly for the narrow-bandgap organic compounds that govern the T1 energy of the device, which can efficiently suppress triplet recombination and attain insignificant exciton dissociation driving force.76
Hence, to investigate the potentiality of the present derivatives in organic photovoltaics fabrication, the excited state singlet (S1) and triplet (T1) energy gaps (ΔEST) have been calculated (Table S1, ESI†). The ΔEST values for PT–C2–(Ph)2, PT–C12–(Ph)2, PT–C2–(Nap)2, PT–C12–(Nap)2, PT–C2–(An)2 and PT–C2–(Pn)2 and PT–C2–(Pr)2 were found to be 0.88, 0.87, 0.77, 0.77, 0.59, 0.76, 0.63 eV, respectively. Hence, both PT–C2–(An)2 and PT–C2–(Pr)2 having narrow bands and lower ΔEST values compared to the other derivatives might have better performance in organic photovoltaics.
Furthermore, to gain an in-depth insight into the MFC characteristics of PT–C2–(Ph)2, single-point energy calculations were executed by utilizing the B3LYP/6-311+g(d,p) level of theory by fluctuating the dihedral angles (between phenothiazine and phenyl ring (C3–C15–C16–C18, ϕ = −8°)) from −5° to −100° (Fig. 10). According to Lu et al., the increased dihedral angle results in a highly twisted conformation of the molecule.58,77,78
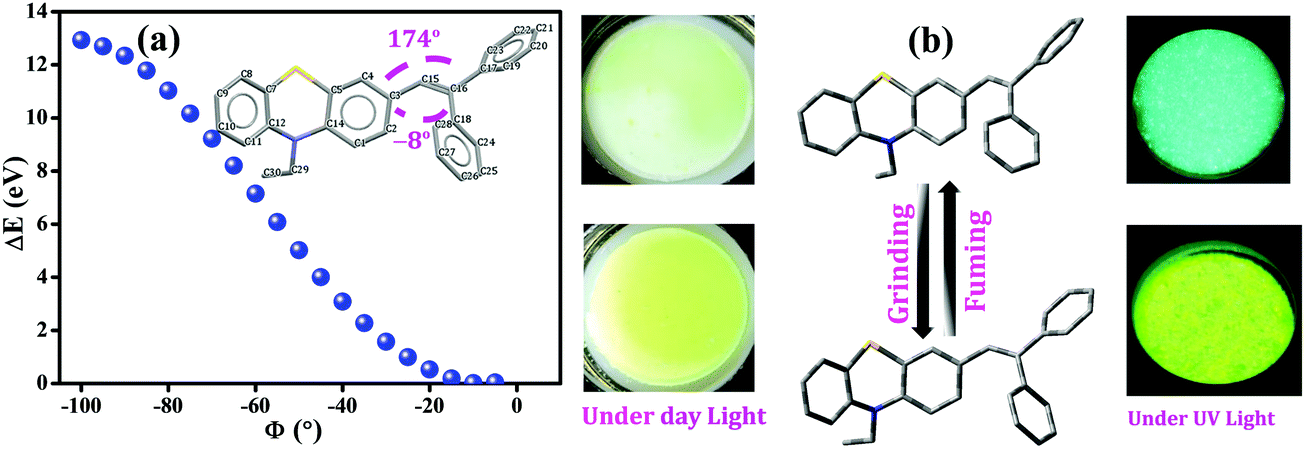 | ||
| Fig. 10 (a) Calculated single point energies vs. varying dihedral angles between phenothiazine and the phenyl ring (C3–C15–C16–C18, ϕ = −8°) from −5° to −100° for PT–C2–(Ph)2 obtained using the B3LYP/6-311+g(d,p) level of theory, (b) schematic representation of the reversible mechanofluorochromic process of PT–C2–(Ph)2 under mechanical force and fumigation under DCM (KBr plate of PT–C2–(Ph)2 has been shown for colour change demonstration). | ||
Fig. 10(a) shows the derived result from where the energy changes (ΔE) have been plotted against dihedral angles (ϕ). The experimentally obtained results show that the fluorescence emission maxima of the compound have been redshifted upon grinding. This observation indicates the lessening of the optical bandgap. The dihedral angles obtained for the fully optimized equilibrium conformers are −8° and 174°, respectively. Fig. 10(a) also shows the reduction in the bandgap when the dihedral angle was <10°; subsequently, it indicates the conversion of a comparatively less twisted conformation upon mechanical agitation on the compound. Hence, insights achieved through theoretical calculations have drawn the triggering mechanism of the experimentally detected fluorescence changes upon mechanical agitation, which drives through conformational changes in the molecules, resulting in red-shifting of the emission maxima (Fig. 10(b)).
Conclusions
A set of novel scissor-shaped divinyl MFC materials has been synthesized. Among all the fluorophores, PT–C2–(Ph)2, PT–C12–(Ph)2, and PT–C2–(Pn)2 appear as highly fluorescent solid-state emitters, wherein in the cases of the former two, the non-fluorescent phenyl in conjugation with the weakly fluorescent PT core, shows the highest emission quantum yields. In contrast to the conventional fluorophore, PT–C2–(Nap)2, PT–C12–(Nap)2, and PT–C2–(Pr)2 demonstrate the blue-shifted AIE phenomenon, which is rare in the literature. All the derivatives show high thermal and electrochemical stability even up to 200 cycles. Surprisingly, only the phenyl derivative displays discrete reversible fluorescence emission color changes from cyan (491 nm) to yellowish-green (504 nm) under mechanical agitation and solvent fumigation, undergoing declination of crystallinity; hence it is a potential candidate for the MFC material. The phenanthrene derivative shows high acid-sensing capability even in its solid-state. Also, the anthracene and the pyrene derivatives having lower ΔEST show potential for possible use in organic photovoltaics. The results taken together showed that phenothiazine with a highly conjugated extended aromatic core can be a potential candidate for security disk application, vapor sensing, acid-sensing, and redox-active organo-electric and organic photovoltaic devices.Conflicts of interest
There are no conflicts of interest.Acknowledgements
RLG acknowledges the financial support from the IIT Madras through the Institute Research & Development Award (IRDA): CY/20-21/069/RFIR/008452. The authors acknowledge the Sophisticated Analytical Instruments Facility (SAIF), IIT Madras, for the SEM facility, and the Indian Institute of Technology of Madras for a fellowship. The authors also acknowledge P. G. Senapathy Centre for computing resources, IIT Madras, for offering us the opportunity to use their supercomputing facilities for executing the DFT and TD-DFT calculations.References
- A. Ekbote, S. M. Mobin and R. Misra, J. Mater. Chem. C, 2020, 8, 3589–3602 RSC.
- C. Zhang, H. Li, Y. Su, Q. Zhang, Y. Li and J. Lu, ACS Appl. Mater. Interfaces, 2020, 12, 15482–15490 CrossRef CAS PubMed.
- Y. Li, Z. Wang, C. Zhang, P. Gu, W. Chen, H. Li, J. Lu and Q. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 15971–15979 CrossRef CAS PubMed.
- X. Nie, T. Wang, W. Huang, H. Su, B. Chen, X. Zhang and G. Zhang, J. Mater. Chem. C, 2021, 9, 15698–15706 RSC.
- Y. Chen, N. Turetta, Y. Yao and P. Samorì, J. Mater. Chem. C, 2022 10.1039/D1TC05055A.
- X. Wang, L. Wang, X. Mao, Q. Wang, Z. Mu, L. An, W. Zhang, X. Feng, C. Redshaw, C. Cao, A. Qin and B. Z. Tang, J. Mater. Chem. C, 2021, 9, 12828–12838 RSC.
- R. Ding, K. Qin, H. Sun, S. Zhou, S. Guo, H. Feng, H. Ma and Z. Qian, J. Mater. Chem. C, 2021, 9, 12177–12183 RSC.
- Y. Chen, X. Li, W. Che, L. Tu, Y. Xie and Z. Li, J. Mater. Chem. C, 2021, 9, 11738–11744 RSC.
- J. Wang, Q. Cheng, S. Feng, L. Zhang and C. Chang, J. Mater. Chem. C, 2021, 9, 6344–6350 RSC.
- W. Yin, Z. Yang, S. Zhang, Y. Yang, L. Zhao, Z. Li, B. Zhang, S. Zhang, B. Han and H. Ma, Mater. Chem. Front., 2021, 5, 2849–2859 RSC.
- S. Yagai, T. Seki, H. Aonuma, K. Kawaguchi, T. Karatsu, T. Okura, A. Sakon, H. Uekusa and H. Ito, Chem. Mater., 2016, 28, 234–241 CrossRef CAS.
- N. M. Wu, M. Ng and V. W. Yam, Angew. Chem., Int. Ed., 2019, 58, 3027–3031 CrossRef CAS PubMed.
- A. E. Norton, M. K. Abdolmaleki, J. Liang, M. Sharma, R. Golsby, A. Zoller, J. A. Krause, W. B. Connick and S. Chatterjee, Chem. Commun., 2020, 56, 10175–10178 RSC.
- S. Jayanty and T. P. Radhakrishnan, Chem. – Eur. J., 2004, 10, 791–797 CrossRef CAS PubMed.
- B. Huitorel, Q. Benito, A. Fargues, A. Garcia, T. Gacoin, J.-P. Boilot, S. Perruchas and F. Camerel, Chem. Mater., 2016, 28, 8190–8200 CrossRef CAS.
- M. Mathivanan, B. Tharmalingam, O. Anitha, C.-H. Lin, V. Thiagarajan and B. Murugesapandian, Mater. Chem. Front., 2021, 5, 8183–8196 RSC.
- Y. Zhang, P. Zhuo, H. Yin, Y. Fan, J. Zhang, X. Liu and Z. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 24395–24403 CrossRef CAS PubMed.
- G. I. Peterson, M. B. Larsen, M. A. Ganter, D. W. Storti and A. J. Boydston, ACS Appl. Mater. Interfaces, 2015, 7, 577–583 CrossRef CAS PubMed.
- Q. Kang, C. Wang, S. Zhou, G. Li, T. Lu, Y. Tian and P. He, ACS Appl. Mater. Interfaces, 2021, 13, 38866–38876 CrossRef CAS PubMed.
- E. Inci, G. Topcu, T. Guner, M. Demirkurt and M. M. Demir, J. Mater. Chem. C, 2020, 8, 12036–12053 RSC.
- B. Poggi, L. Bodelot, M. Louis, R. Métivier and C. Allain, J. Mater. Chem. C, 2021, 9, 12111–12117 RSC.
- G. V. Bünau, Ber. Bunsen. Phys. Chem., 1970, 1294–1295 Search PubMed.
- W. F. Watson and R. Livingston, Nature, 1948, 162, 452–453 CrossRef CAS PubMed.
- J. Weiss, Nature, 1943, 152, 176–178 CrossRef.
- H. Wu, L. Zhang, J. Yang, R. Bo, H. Du, K. Lin, D. Zhang, M. Ramachandran, Y. Shen, Y. Xu, X. Xue, Z. Ma, A. R. Lindstrom, R. Carney, T. Lin and Y. Li, Adv. Funct. Mater., 2020, 30, 1910348 CrossRef CAS.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, B. Z. Tang, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu and D. Zhu, Chem. Commun., 2001, 1740–1741 RSC.
- F. Khan, A. Ekbote, S. M. Mobin and R. Misra, J. Org. Chem., 2021, 86, 1560–1574 CrossRef CAS PubMed.
- S. Sekiguchi, K. Kondo, Y. Sei, M. Akita and M. Yoshizawa, Angew. Chem., Int. Ed., 2016, 55, 6906–6910 CrossRef CAS PubMed.
- X. Meng, G. Qi, C. Zhang, K. Wang, B. Zou and Y. Ma, Chem. Commun., 2015, 51, 9320–9323 RSC.
- S. Xue, W. Liu, X. Qiu, Y. Gao and W. Yang, J. Phys. Chem. C, 2014, 118, 18668–18675 CrossRef CAS.
- N. Jiang, S.-H. Ruan, X.-M. Liu, D. Zhu, B. Li and M. R. Bryce, Chem. Mater., 2020, 32, 5776–5784 CrossRef CAS PubMed.
- H. Gao, D. Xu, Y. Wang, Y. Wang, X. Liu, A. Han and C. Zhang, Dyes Pigm., 2018, 150, 59–66 CrossRef CAS.
- P. Xu, Q. Qiu, X. Ye, M. Wei, W. Xi, H. Feng and Z. Qian, Chem. Commun., 2019, 55, 14938–14941 RSC.
- Y. Yin, F. Zhao, Z. Chen, G. Liu and S. Pu, Tetrahedron Lett., 2018, 59, 4416–4419 CrossRef CAS.
- M. S. Kwon, J. Gierschner, S.-J. Yoon and S. Y. Park, Adv. Mater., 2012, 24, 5487–5492 CrossRef CAS PubMed.
- M. Martínez-Abadía, R. Giménez and M. B. Ros, Adv. Mater., 2018, 30, 1704161 CrossRef PubMed.
- S. Wang, B. Zhu, B. Wang, P. Fan, Y. Jiu, M. Zhang, L. Jiang and J.-T. Hou, Dyes Pigm., 2020, 173, 107933 CrossRef CAS.
- P. Huang, Manju, S. Kazim, L. Lezama, R. Misra and S. Ahmad, ACS Appl. Mater. Interfaces, 2021, 13, 33311–33320 CrossRef CAS PubMed.
- S. Kumar, P. Singh, P. Kumar, R. Srivastava, S. K. Pal and S. Ghosh, J. Phys. Chem. C, 2016, 120, 12723–12733 CrossRef CAS.
- M. Okazaki, Y. Takeda, P. Data, P. Pander, H. Higginbotham, A. P. Monkman and S. Minakata, Chem. Sci., 2017, 8, 2677–2686 RSC.
- M. Fan, Y. Cheng, B. Fang, L. Lai and M. Yin, Dyes Pigm., 2021, 190, 109311 CrossRef CAS.
- A. M. Hamdy, N. Eleya, H. H. Mohammed, Z. Khaddour, T. Patonay, A. Villinger and P. Langer, Tetrahedron Lett., 2013, 54, 3568–3571 CrossRef CAS.
- H.-H. Perkampus, UV-VIS Spectroscopy and Its Applications, Springer Berlin Heidelberg, Berlin, Heidelberg, 1992 Search PubMed.
- S. Kothavale and N. Sekar, Dyes Pigm., 2017, 136, 31–45 CrossRef CAS.
- T. Sachdeva and M. D. Milton, J. Photochem. Photobiol., A, 2020, 402, 112804 CrossRef CAS.
- M. Chen, Y. Lee, Z. Huang, D. Chen and P. Chou, Chem. – Eur. J., 2020, 26, 7124–7130 CrossRef CAS PubMed.
- Y. Ooyama and Y. Harima, J. Mater. Chem., 2011, 21, 8372 RSC.
- P. Zhang, M. A. Winnik and Z. Y. Wang, J. Photochem. Photobiol., A, 1995, 89, 13–17 CrossRef CAS.
- K. Jongseong, K. Shanmugasundaram, R. K. Chitumalla, J. Jang and Y. Choe, J. Lumin., 2018, 197, 383–388 CrossRef CAS.
- S. S. Deshpande, H. S. Kumbhar and G. S. Shankarling, Spectrochim. Acta, Part A, 2017, 174, 154–163 CrossRef CAS PubMed.
- L. Marin, A. Bejan and S. Shova, Dyes Pigm., 2020, 175, 108164 CrossRef CAS.
- B. Huang, Curr. Opin. Chem. Biol., 2010, 14, 10–14 CrossRef CAS PubMed.
- W. H. Jung, J.-Y. Yoo, H. Jun Kim, J.-G. Kim, B. D. Chin and J. S. Kim, Opt. Mater. Express, 2020, 10, 3372 CrossRef CAS.
- P. Xue, B. Yao, J. Sun, Q. Xu, P. Chen, Z. Zhang and R. Lu, J. Mater. Chem. C, 2014, 2, 3942–3950 RSC.
- L. Zhang, Y. Chen and J. Jiang, Dyes Pigm., 2016, 124, 110–119 CrossRef CAS.
- L. Zhu, C. Yang and J. Qin, Chem. Commun., 2008, 6303 RSC.
- S. Suganya, K. Debsharma, E. Ravindran, M. K. Mahato and E. Prasad, ACS Appl. Polym. Mater., 2020, 2, 1222–1233 CrossRef CAS.
- I. Gutman, Maced. J. Chem. Chem. Eng., 2007, 26, 111–114 CAS.
- B. Dandrade, S. Datta, S. Forrest, P. Djurovich, E. Polikarpov and M. Thompson, Org. Electron., 2005, 6, 11–20 CrossRef CAS.
- T. Jafari, E. Moharreri, A. Amin, R. Miao, W. Song and S. Suib, Molecules, 2016, 21, 900 CrossRef PubMed.
- J. A. Christensen, B. T. Phelan, S. Chaudhuri, A. Acharya, V. S. Batista and M. R. Wasielewski, J. Am. Chem. Soc., 2018, 140, 5290–5299 CrossRef CAS PubMed.
- R. Rieger and K. Müllen, J. Phys. Org. Chem., 2010, 23, 315–325 CAS.
- J. Huang, Z. Yang, V. Murugesan, E. Walter, A. Hollas, B. Pan, R. S. Assary, I. A. Shkrob, X. Wei and Z. Zhang, ACS Energy Lett., 2018, 3, 2533–2538 CrossRef CAS.
- F. Otteny, G. Studer, M. Kolek, P. Bieker, M. Winter and B. Esser, ChemSusChem, 2020, 13, 2232–2238 CrossRef CAS PubMed.
- A. Chaudhary, M. Poddar, D. K. Pathak, R. Misra and R. Kumar, ACS Appl. Electron. Mater., 2020, 2, 2994–3000 CrossRef CAS.
- K. S. Weeraratne, A. A. Alzharani and H. M. El-Kaderi, ACS Appl. Mater. Interfaces, 2019, 11, 23520–23526 CrossRef CAS PubMed.
- J. H. Jang, S. Ahn, S. E. Park, S. Kim, H. R. Byon and J. M. Joo, Org. Lett., 2020, 22, 1280–1285 CrossRef CAS PubMed.
- P. Venkatakrishnan, S. Maddala and A. Panua, Chem. – Eur. J., 2021, 27, 16013–16020 CrossRef PubMed.
- C. Zhang, Z. Niu, S. Peng, Y. Ding, L. Zhang, X. Guo, Y. Zhao and G. Yu, Adv. Mater., 2019, 31, 1901052 CrossRef PubMed.
- K. Ishizuki, H. Oka, D. Aoki, R. Goseki and H. Otsuka, Chem. – Eur. J., 2018, 24, 3170–3173 CrossRef CAS PubMed.
- E. Horak, M. Robić, A. Šimanović, V. Mandić, R. Vianello, M. Hranjec and I. M. Steinberg, Dyes Pigm., 2019, 162, 688–696 CrossRef CAS.
- B. Yu, D. Liu, Y. Wang, T. Zhang, Y.-M. Zhang, M. Li and S. X.-A. Zhang, Dyes Pigm., 2019, 163, 412–419 CrossRef CAS.
- B. Sarkar, P. Prabakaran, E. Prasad and R. L. Gardas, ACS Sustainable Chem. Eng., 2020, 8, 8327–8337 CrossRef CAS.
- T. Sachdeva and M. D. Milton, J. Mol. Struct., 2021, 1243, 130768 CrossRef CAS.
- G. Han, T. Hu and Y. Yi, Adv. Mater., 2020, 32, 2000975 CrossRef CAS PubMed.
- T. Virgili, A. Forni, E. Cariati, D. Pasini and C. Botta, J. Phys. Chem. C, 2013, 117, 27161–27166 CrossRef CAS.
- G. Zhang, J. Sun, P. Xue, Z. Zhang, P. Gong, J. Peng and R. Lu, J. Mater. Chem. C, 2015, 3, 2925–2932 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedure and 1H NMR and 13C NMR spectra of PT derivatives; photographs of PT derivatives in different solvents; detailed electrochemical plots and calculations, and DFT and TD-DFT results. See DOI: 10.1039/d1ma01162f |
| This journal is © The Royal Society of Chemistry 2022 |