DOI:
10.1039/D1MA01154E
(Paper)
Mater. Adv., 2022,
3, 4026-4036
Superatomic salts with controlled ionicity†
Received
6th December 2021
, Accepted 4th April 2022
First published on 5th April 2022
Abstract
We propose a new class of superatomic molecules where the bonding characteristics between two identical superatoms can be modified by using suitable ligands around metal-chalcogen clusters. We demonstrate these intriguing findings by considering the Rh6S8Ln class of superatoms. Our choice of ligands (L) is motivated by the condition that the ligand binds to the metal-chalcogen core via the same atom while the electronegativity of the ligand is altered by attaching various units to this connecting atom. For this purpose, we have chosen the phosphine ligands (PR3, R = functional groups) where the electronegativity of the ligands is changed via the functional (R) group ranging from NMe2 to F. It is also observed that this technique can be further used to induce controllable electronic features in the (PR3)5Rh6S8–Rh6S8(CO)5 series of dimers/salts. The differences in the location of electronic levels on the two sides result in internal charge transfer that not only adds an ionic component to bonding over the covalent component but also leads to dipole moments/internal electric field in an otherwise homo-cluster dimer. Thus, the strength of the dipole moment/internal field of the dimer can be adjusted by changing the electronegativity of the PR3 group offering a unique opportunity of making cluster assembled solids using superatomic molecules with adjustable dipolar strength, internal fields, and location of HOMO/LUMO, useful for various applications including light-harvesting.
1. Introduction
One of the most fundamental questions in physical and chemical sciences is how the forces acting between atoms lead to the formation of aggregates starting from molecules to extended solids. An understanding of the nature of chemical bonds is critical to assess the stability of molecules. Depending on the nature of atoms, there are many types of chemical bonds that characterize molecular bonding; the two most important types of bonds are covalent and ionic bonds.1 The covalent bonds involve sharing of the outer electrons, particularly in atoms with similar electronegativity with no net electron transfer, while the ionic bonds involve an electron donor and an electron acceptor wherein the electron transfer leads to the attractive forces. In particular, for homoatomic molecules, the chemical bonds can not have an ionic component.
In this work, we demonstrate a novel phenomenon whereby a molecule composed of two identical building blocks can simultaneously exhibit covalent and ionic bonding. We demonstrate this intriguing possibility by forming a supermolecule by linking two identical ligated metal-chalcogenide superatoms. This class of clusters is composed of transition metal and chalcogen atoms forming the inner core that is further protected by outer ligands. The units are highly stable, can be formed in solutions, and as first demonstrated by Roy, Nuckolls, and co-workers,2–9 they can form ionic solids when combined with counter-ions maintaining their identity. Unlike metallic clusters where a confined nearly free electron gas10–14 model can identify particularly stable species, the bonding in individual metal-chalcogenide cluster has a covalent component between metal and chalcogens and form a charge–transfer complex between metal and ligands. Consequently, a hybrid model including valence electrons of metals, chalcogens, and the electrons shared with ligands is needed to account for variations in chemical stability as seen through the gap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO).15 What is interesting is that for a given metal-chalcogen core, the redox properties can be controlled by the choice of ligands without altering the effective valence electron count.16–23 Thus, attaching strong σ-donor ligands, e.g., phosphines, to the cluster induces an upward shift of the electronic spectrum, which results in a superatom with enhanced charge donor properties. In contrast, a shift in the opposite direction is induced when strong π-acceptor ligands (e.g., CO) are introduced within the cluster framework. In such a situation, the ligated cluster act as a charge acceptor.
One of the important questions is how the assemblies of superatoms can lead to novel features. It is important to highlight that, unlike atoms, assemblies of superatoms are marked by intra-atomic and inter–atomic interactions. The purpose of the paper is to offer a step in this direction by considering a supermolecule built from the core of identical metal-chalcogen atoms. In this work, we address two questions: (1) How the redox properties of superatomic molecules can be controlled by changing ligands? (2) Is it possible to modify the covalent bonding between superatoms to have an ionic component? We are particularly interested in forming a superatomic salt that does not require distinct metal-chalcogenide units but where the change in ligands induces charge transfer and an internal electric field between otherwise identical cores. The current work, in our view, is a unique example of how intra–cluster and inter–cluster interactions can conspire to lead to novel units that has potential as building blocks for ferroelectric materials or where the individual units could be used for charge separation in photovoltaic applications.24,25
In this work, we focus on Rh6S8Ln (L: Ligand) class of clusters to demonstrate these intriguing findings. Our choice of ligands is motivated by the fact that the ligand atom coordinating to the metal-chalcogen cluster remains the same and that the charge transfer properties are altered by attaching different units to the linking atom. To accomplish this, we focus on tertiary phosphines (PR3, R = functional groups) that represent a unique class of ligands whose electronic and steric properties are directly related to the electronegativity of the attached functional groups (R).26,27 Thus, by using groups of varying electronegativity, one can effectively create a series of PR3 ligands with different donor/acceptor characteristics. For example, although ligands like PEt3 or PMe3 are strong σ donors, PCl3 or PF3 ligands are very strong π- acceptors. The electron-withdrawing capability of PF3 is often comparable to CO. In this present paper, we have used this strategy on the 5 and 6 ligated Rh6S8 metal chalcogenide clusters. It is observed that by changing the attached R groups, it is possible to transform a donor metal chalcogenide cluster to an acceptor without altering the effective valence count or the frontier orbitals. The HOMO−LUMO gaps of the clusters, which are a measure of kinetic stability, have also remained within a similar range for all cases. For a single cluster, in terms of the adiabatic ionization energy (AIE) and electron affinity (AEA), the performance of this technique is observed to be near equivalent to the ligand substitution method,16 which we have proposed earlier for the cobalt telluride clusters (Co6Te8(PEt3)m(CO)n, m + n = 6). Thus, instead of sequentially substituting the donor ligands with acceptors, one can simply use different R groups to attain a similar result. Consequently, this technique can also act as an alternative when the ligand substitution method is not viable or experimentally difficult to achieve. Furthermore, it is noticed that by utilizing a series of PR3 ligands to decorate the donor side of the metal-chalcogenide dimers, one can introduce a similar shift within the electronic spectrum of the dimers as well. Hence, by altering the ligands only on the donor side, a superatomic dimer can also be converted from donor to acceptor or vice versa without significantly changing the characteristics of the frontier orbitals or the HOMO−LUMO gap. Additionally, since the dipole moment of the dimer depends on the donor/acceptor properties of the attached ligands, the same approach also results in simultaneous alteration of the dipole moment of the dimer without changing the direction. The calculated results show that by using a range of PR3 ligands to decorate the donor side, one can create a series of superatomic salts with variable redox properties and dipole moments similar to the series of common ionic molecules (e.g., NaCl, KBr, etc.). The range of the dipole moment is observed to be altered from nearly 1 Debye to as high as 20 Debye and possibly higher for the Rh6S8 based dimer. We have also argued that a similar series can also be constructed simply by using only one type of ligand (i.e., PR3) with differential donor/acceptor characteristics on both sides, which completely eliminates the requirement of a specialized π-acceptor ligand like CO. As shown here, all of the modifications are also achievable by using identical metal chalcogenide cluster on both sides which in this case is Rh6S8. Lastly, since the group electronegativity can be altered simply by chemical design, unlike the ligand substitution method, this approach is not limited to the number of available metal sites on the cluster. Thus, the reported series can further be extended simply by introducing different functional groups within the PR3 series.
2. Results and discussion
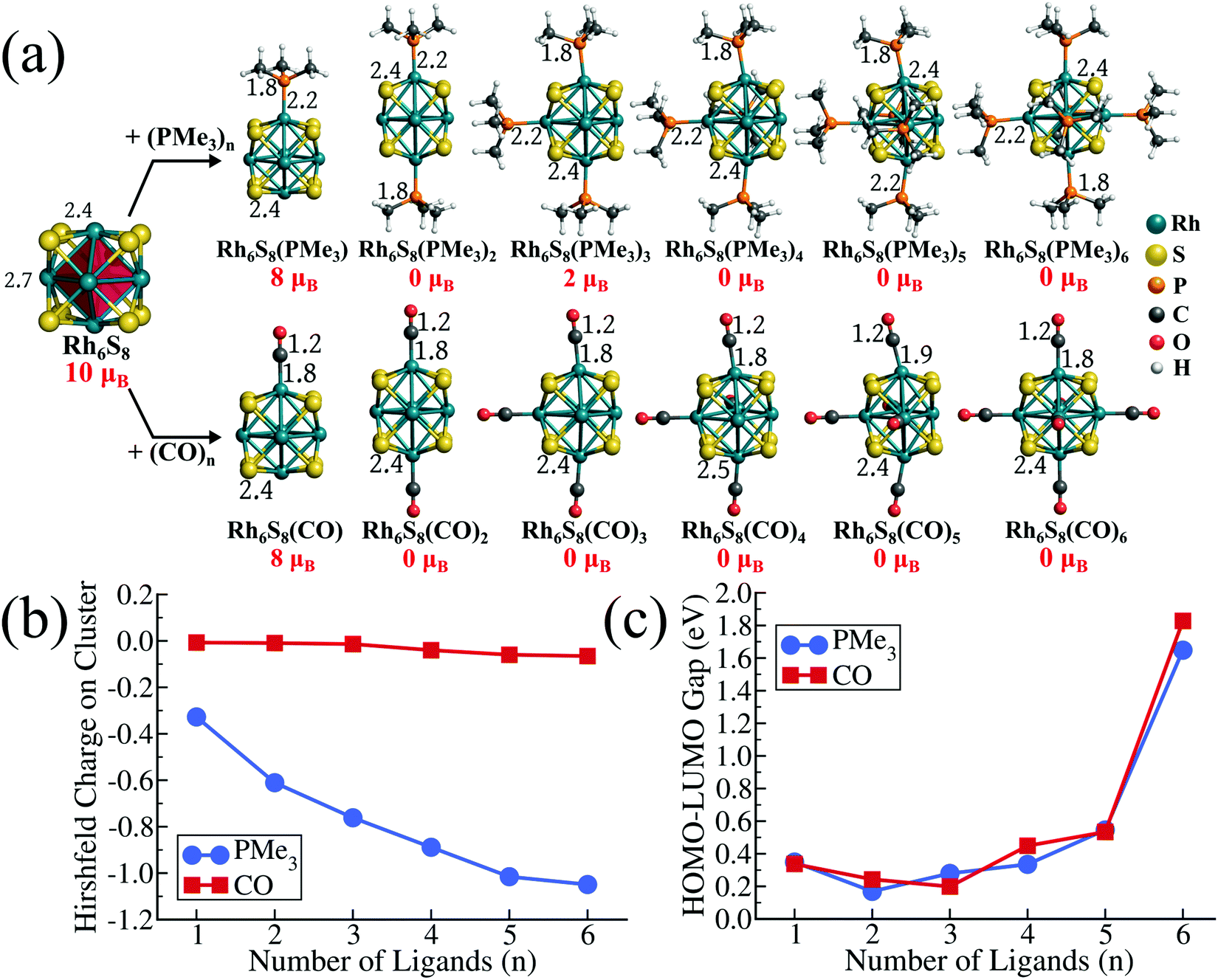
To investigate the effect of different PR3 ligands on the electronic and redox properties of the metal chalcogenide cluster, we have chosen the Rh6S8 cluster as our template system. Our choice of Rh6S8 is motivated by two reasons. Firstly, the fully ligated Rh6S8L6 (L = ligands, e.g., CO or PR3) complexes are stable magic clusters with a large HOMO−LUMO gap; hence such systems can be synthesized by experimentalists.28,29 Secondly, as we will show later, the ground state of 5 and 6 ligated monomers and the composite dimers of Rh6S8 clusters are all found to be closed-shell (0 μB) structures. This makes the energetic comparison of the frontier orbitals and the HOMO−LUMO gaps consistent and relevant. The gradual shifting of the electronic spectrum for such closed-shell systems can also be visualized clearly, as shown in the next sections. The ground-state structure of Rh6S8 is a face-capped octahedron, as shown in Fig. 1a. The structure is composed of a central octahedron of Rh atoms (drawn as a red polyhedron in Fig. 1a) whose triangular faces are capped by the eight sulfur atoms. As a result, the ground state structure belongs to the Oh point group (Fig. S1 of the ESI†). As depicted in Fig. 1a, it is observed that the ground state Rh6S8 cluster has a magnetic moment of 10 μB. The same structure with 8 μB magnetic moment is 0.06 eV higher in energy compared to the global minima (see Table S1 of ESI†). The spin density calculation reveals that the magnetic moments of the ground state cluster are mostly localized on Rh atoms (1.16–1.24 μB) and the sulfur atoms show minor contributions (∼0.35 μB per atom). The cluster has an adiabatic ionization energy (AIE) of 7.56 eV and an adiabatic electron affinity (AEA) of 3.82 eV.
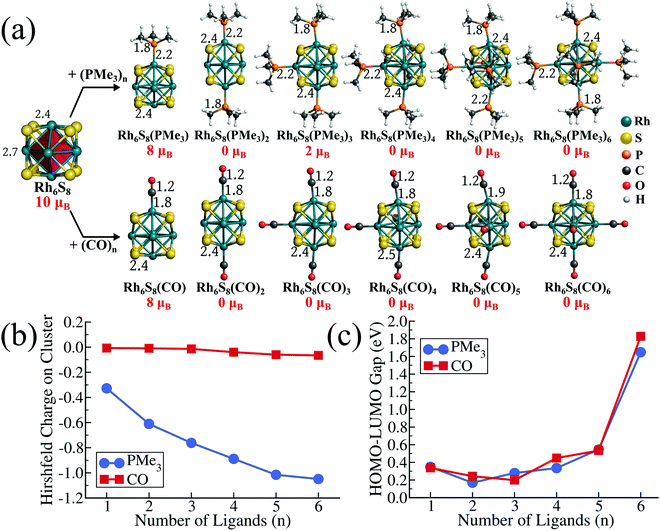 |
| | Fig. 1 (a) The optimized ground state structures and magnetic moments of Rh6S8 and Rh6S8Ln (L = PMe3, CO; n = 1–6) clusters. Bond lengths are in Å. (b) The variation of Hirshfeld charge and HOMO−LUMO gap along the Rh6S8Ln series. | |
To obtain further insight, we have first investigated the effect of sequential ligand attachment on the magnetic and redox properties of the Rh6S8 cluster. For this purpose, we have chosen one donor (i.e., PMe3) and one acceptor (i.e., CO) ligand. The optimized structures of both series are presented in Fig. 1a. It is seen that for both PMe3 and CO ligands, the magnetic moment of the ligated clusters is reduced to 8 μB just after attaching one ligand. Except for Rh6S8(PMe3)3, the inclusion of more than one ligand results in a non-magnetic ligated cluster. Thus, for both PMe3 and CO, all ligated clusters with more than three ligands are found to be non-magnetic. It is noteworthy that even for Rh6S8(PMe3)3, the 2 μB ground-state structure is only marginally (0.03 eV) lower in energy compared to the 0 μB structure. For all the non-magnetic ligated clusters, we have not found any residual moment on any atoms, which proves that the absence of net moment is not due to any antiferromagnetic alignments. Although the effect of both PMe3 and CO ligands on the net moment of ligated clusters seems mostly similar, the electronic features of both series show striking dissimilarities. For example, in Fig. 1b, we have plotted the net Hirshfeld30 charge, which is accumulated on the Rh6S8 cluster as the number of ligands (PMe3 and CO) is sequentially increased. It is evident that being a very strong σ-donor, the negative charge on the cluster has increased consistently as the number of PMe3 ligands changes from 1 to 6. On the other hand, in the case of CO, the net charges on the cluster have remained close to 0 throughout, and accumulation of marginal negative charge (−0.04 e to −0.06 e) is only observed near the end of the series. This is because the CO ligand is not only a strong σ-donor but also a good π-acceptor.27 Both of these effects neutralize each other resulting in the near-zero charge on the cluster core. The striking difference is also reflected in the redox properties of the ligated clusters. Due to the transfer of electrons from ligand to cluster, attaching PMe3 ligands has raised the electronic spectrum of the overall system. Hence, as the number of PMe3 ligands is sequentially increased, the HOMO and LUMO levels are shifted upward (see Table S2 of the ESI†). Consequently, the AIEs of the 5 and 6 PMe3 ligated clusters are calculated as 5.12 and 5.00 eV, respectively, which are much lower compared to the pristine Rh6S8 cluster. In contrast, due to the transfer of electrons to the ligand via back donation, the electronic spectrum of the CO ligated system has lowered. Hence, the AIEs of 5 and 6 CO ligated Rh6S8 cluster has increased to 7.65 and 7.85 eV, respectively. As we sequentially increase the number of the ligands, the HOMO−LUMO gap shows a non-monotonic trend (Fig. 1c) for both ligands; however, a large leap is observed when the last (6th) ligand (PMe3 or CO) is attached, showing the high kinetic stability of the fully ligated Rh6S8 cluster. The HOMO−LUMO gap of Rh6S8(PMe3)6 and Rh6S8(CO)6 clusters are calculated as 1.65 and 1.83 eV, respectively. Our previous investigations15 have shown that the closed-shell metal chalcogenide cluster, i.e., Co6Te8(PEt3)6 with 114 valence electrons, is a closed-shell magic cluster with a large HOMO−LUMO gap. The total valence electron count for Co6Te8(PEt3)6 is obtained by counting 6 electrons per chalcogen, 2 electrons per ligand, and including 9 electrons for each cobalt. Since rhodium shares the same group (Group 9) as cobalt in the periodic table,31 it is expected that the same electron counting rule is equally valid for here as well.
2.1. The electronic properties of Rh6S8(PR3)5 cluster series
From the above discussion, the distinctive electronic features of a σ-donor ligand (e.g., PMe3) and a π-acceptor ligand (e.g., CO) becomes quite evident. So, at this point, it is important to ask if there is any way that such distinctive features can be combined into only one type of ligand? In other words, can we design a single stable class of ligands whose donor–acceptor properties can be controlled by simple chemical modification? The easiest solution to the problem is to consider only one specific type of ligand, i.e., the tertiary phosphines (PR3). It is well known in chemistry26,27 that the tertiary phosphines are a unique class of ligands whose donor–acceptor characteristics can be altered by modifying the functional groups (R). When a PR3 ligand, e.g., PEt3 or PMe3 is attached to a metal atom, they behave as a strong σ-donor since the acceptor σ* (P–R) orbitals reside at higher energy, resulting in a negligible back donation. However, replacing the R group with electronegative atoms/groups stabilizes the vacant σ* orbital, which enables the back donation. Additionally, in such a situation, the contribution of the phosphorous atoms to the σ* orbital also increases,27 resulting in a better overlap with the filled d orbital of the metal atom, which further facilitates the back bonding. Thus, if the R groups are chosen carefully, it is possible to create a sequence of PR3 ligands whose properties can be altered from strong donor to strong acceptor.
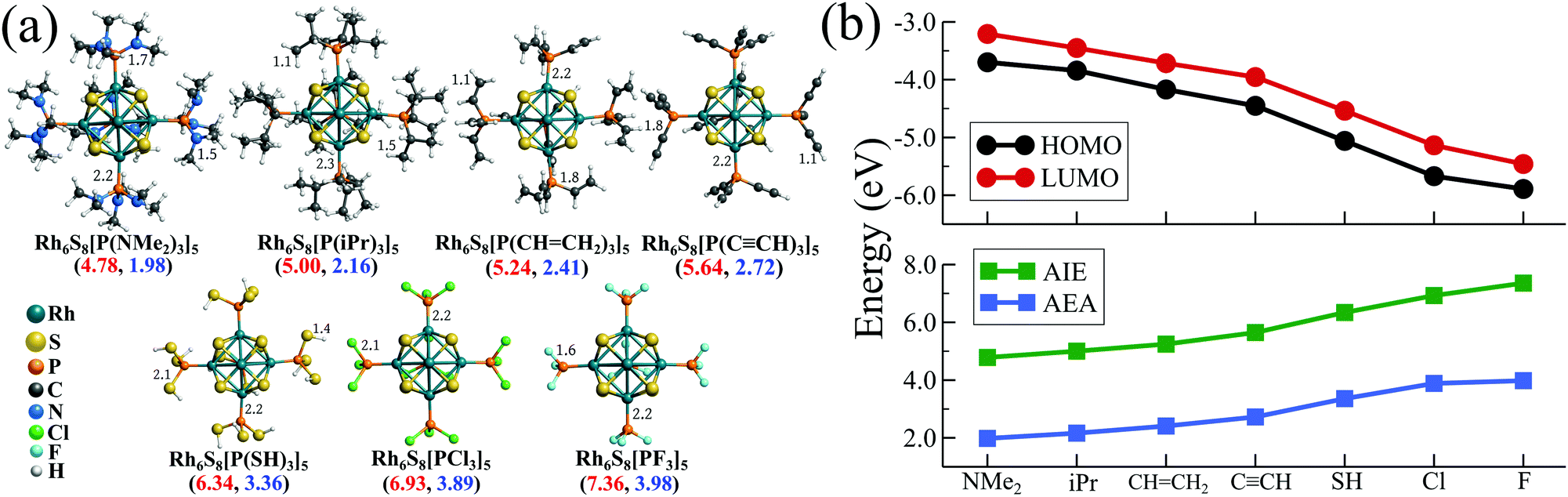
To test this, we have chosen seven such functional groups viz. 1. dimethylamine (–NMe2) 2. isopropyl (–CH(CH3)2 or –iPr) 3. ethenyl or vinyl (–CH = CH2) 4. ethynyl (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH) 5. thiol (–SH) 6. chlorine (–Cl) and 7. fluorine (–F). The groups are chosen in such a way that the group electronegativity increases as we move from 1 to 7. As a result, P(NMe2)3 is expected to act as the strongest σ-donor and PF3 should behave as the best π-acceptor, similar to CO. As a next step, we have decorated the Rh6S8 cluster by 5 of each ligand from the above series, and their optimized ground state structures are shown in Fig. 2a. The electronic structure and associated properties, e.g., HOMO−LUMO gaps, AIEs, and AEAs, are also evaluated. Similar calculations are also performed for the six ligated Rh6S8 clusters. However, the relative trend of the calculated properties for the six ligated series is found to be very similar to the five ligated clusters and hence included in Table S3 of the ESI.†Fig. 2b and Table 1 show the calculated electronic properties of the five ligated Rh6S8 series. It is important to note that upon changing the ligands from P(NMe2)3 to PF3, the energies of both HOMO and LUMO show a near parallel decrease. As a result, the HOMO−LUMO gaps vary within a narrow range (0.39–0.53 eV) throughout the series. This observation is similar to our ligand substitution method,16 which we have proposed before. The energies of HOMO (−3.69 eV) and LUMO (−3.20 eV) are observed to be highest for Rh6S8[P(NMe2)3]5. As we move along the series, both values show a steady decrease and finally reach the lowest for Rh6S8(PF3)5, for which the HOMO and LUMO values are calculated to be −5.89 and −5.46 eV, respectively. As expected, the AIE and AEA show a reverse trend (Fig. 2b), i.e., both the properties show a consistent increase upon moving from left to right. Hence, Rh6S8[P(NMe2)3]5 shows the lowest AIE (4.78 eV) and AEA (1.98 eV), whereas, Rh6S8(PF3)5 shows the highest AIE (7.36 eV) and AEA (3.98 eV) among the series. It is important to note the overall range of AIE and AEA as obtained for both 5 and 6 ligated clusters are similar to our previously reported results16 for the Co6Te8(PEt3)m(CO)n, (m + n = 6) cluster series. In that work, the electron donor PEt3 groups were sequentially substituted by CO. This shows that the current approach can act as an effective alternative to the ligand substitution proposed earlier. If compared with the experimental database,31 then it can be seen that the computed AIE of Rh6S8[P(NMe2)3]5 is between sodium (5.14 eV) and potassium (4.34 eV), and the AEA of Rh6S8(PF3)5 cluster is even higher than the Cl atom (i.e., 3.61 eV), which has the highest electron affinity among all elements. Thus, it is evident that the Rh6S8 cluster can be converted from a strong donor to a strong acceptor simply by altering the nature and the type of the R groups which are attached to the phosphorus atom.
CH) 5. thiol (–SH) 6. chlorine (–Cl) and 7. fluorine (–F). The groups are chosen in such a way that the group electronegativity increases as we move from 1 to 7. As a result, P(NMe2)3 is expected to act as the strongest σ-donor and PF3 should behave as the best π-acceptor, similar to CO. As a next step, we have decorated the Rh6S8 cluster by 5 of each ligand from the above series, and their optimized ground state structures are shown in Fig. 2a. The electronic structure and associated properties, e.g., HOMO−LUMO gaps, AIEs, and AEAs, are also evaluated. Similar calculations are also performed for the six ligated Rh6S8 clusters. However, the relative trend of the calculated properties for the six ligated series is found to be very similar to the five ligated clusters and hence included in Table S3 of the ESI.†Fig. 2b and Table 1 show the calculated electronic properties of the five ligated Rh6S8 series. It is important to note that upon changing the ligands from P(NMe2)3 to PF3, the energies of both HOMO and LUMO show a near parallel decrease. As a result, the HOMO−LUMO gaps vary within a narrow range (0.39–0.53 eV) throughout the series. This observation is similar to our ligand substitution method,16 which we have proposed before. The energies of HOMO (−3.69 eV) and LUMO (−3.20 eV) are observed to be highest for Rh6S8[P(NMe2)3]5. As we move along the series, both values show a steady decrease and finally reach the lowest for Rh6S8(PF3)5, for which the HOMO and LUMO values are calculated to be −5.89 and −5.46 eV, respectively. As expected, the AIE and AEA show a reverse trend (Fig. 2b), i.e., both the properties show a consistent increase upon moving from left to right. Hence, Rh6S8[P(NMe2)3]5 shows the lowest AIE (4.78 eV) and AEA (1.98 eV), whereas, Rh6S8(PF3)5 shows the highest AIE (7.36 eV) and AEA (3.98 eV) among the series. It is important to note the overall range of AIE and AEA as obtained for both 5 and 6 ligated clusters are similar to our previously reported results16 for the Co6Te8(PEt3)m(CO)n, (m + n = 6) cluster series. In that work, the electron donor PEt3 groups were sequentially substituted by CO. This shows that the current approach can act as an effective alternative to the ligand substitution proposed earlier. If compared with the experimental database,31 then it can be seen that the computed AIE of Rh6S8[P(NMe2)3]5 is between sodium (5.14 eV) and potassium (4.34 eV), and the AEA of Rh6S8(PF3)5 cluster is even higher than the Cl atom (i.e., 3.61 eV), which has the highest electron affinity among all elements. Thus, it is evident that the Rh6S8 cluster can be converted from a strong donor to a strong acceptor simply by altering the nature and the type of the R groups which are attached to the phosphorus atom.
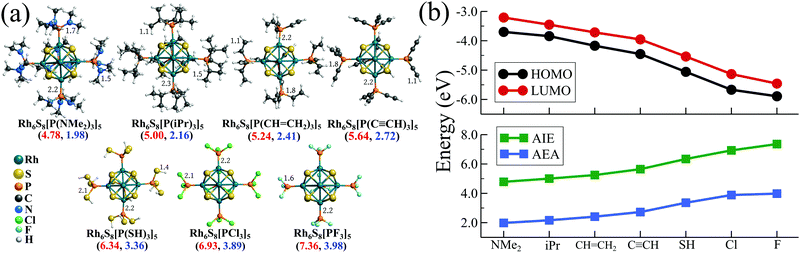 |
| | Fig. 2 (a) The optimized ground state structures of Rh6S8(PR3)5 (R = NMe2 to F). Bond lengths are in Å. The AIE (red) and AEA (blue) values (in eV) are included within parenthesis. (b) The plot of HOMO−LUMO gap, AIE and AEA of Rh6S8(PR3)5 clusters. | |
Table 1 The magnetic moment (in μB) and the calculated electronic properties (in eV) of the Rh6S8(PR3)5 (R = functional groups) cluster series
| Systems |
Mag. moment (μB) |
Electronic properties (eV) |
| HOMO |
LUMO |
HL gap |
AIE |
AEA |
| Rh6S8[P(NMe2)3]5 |
0 |
−3.69 |
−3.20 |
0.49 |
4.78 |
1.98 |
| Rh6S8[P(iPr)3]5 |
0 |
−3.84 |
−3.45 |
0.39 |
5.00 |
2.16 |
Rh6S8[(P(CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)3]5 CH2)3]5 |
0 |
−4.17 |
−3.71 |
0.46 |
5.24 |
2.41 |
| Rh6S8[(P(CCH)3]5 |
0 |
−4.45 |
−3.95 |
0.50 |
5.64 |
2.72 |
| Rh6S8[(P(SH)3]5 |
0 |
−5.06 |
−4.54 |
0.52 |
6.34 |
3.36 |
| Rh6S8(PCl3)5 |
0 |
−5.67 |
−5.14 |
0.53 |
6.93 |
3.89 |
| Rh6S8(PF3)5 |
0 |
−5.89 |
−5.46 |
0.43 |
7.36 |
3.98 |
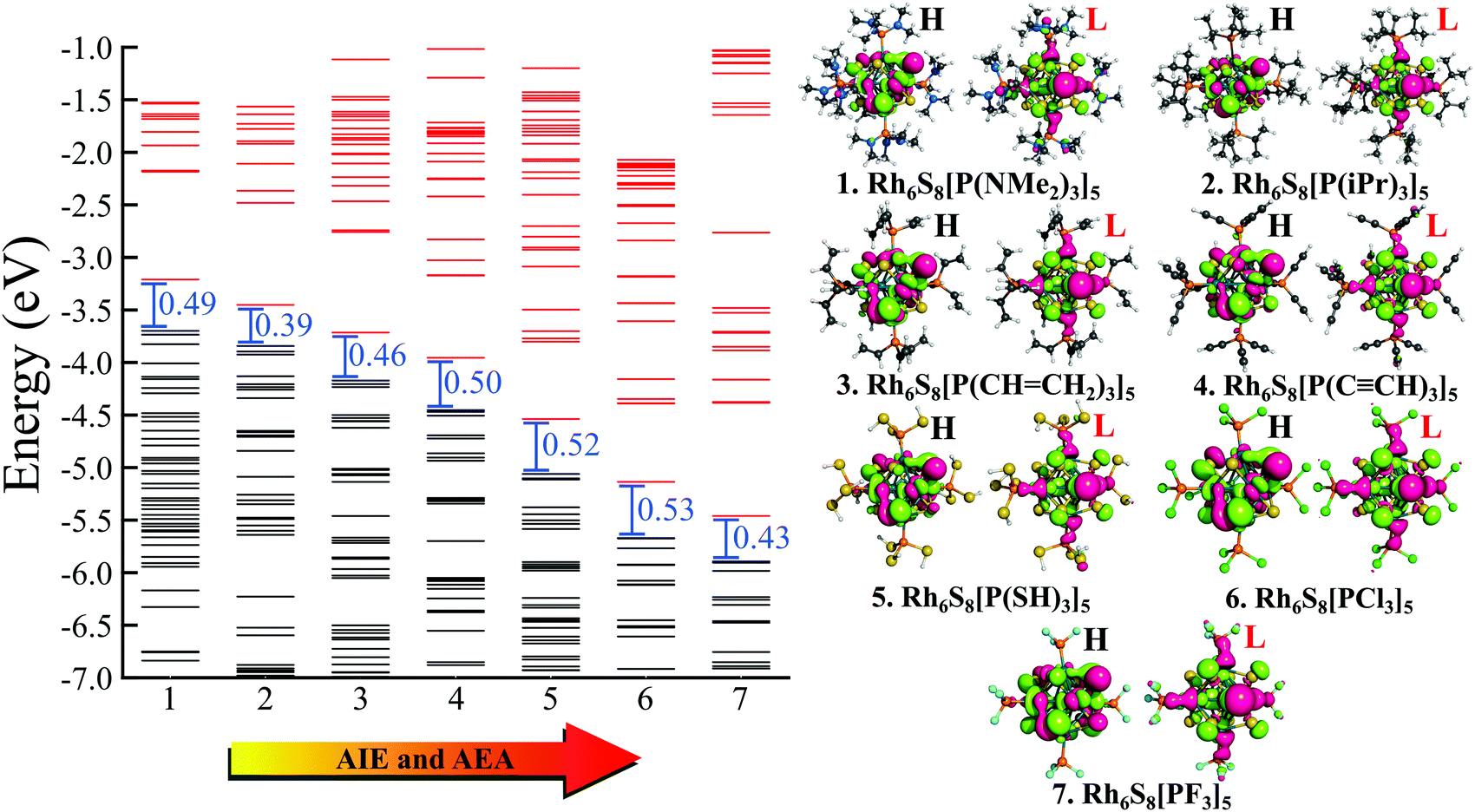
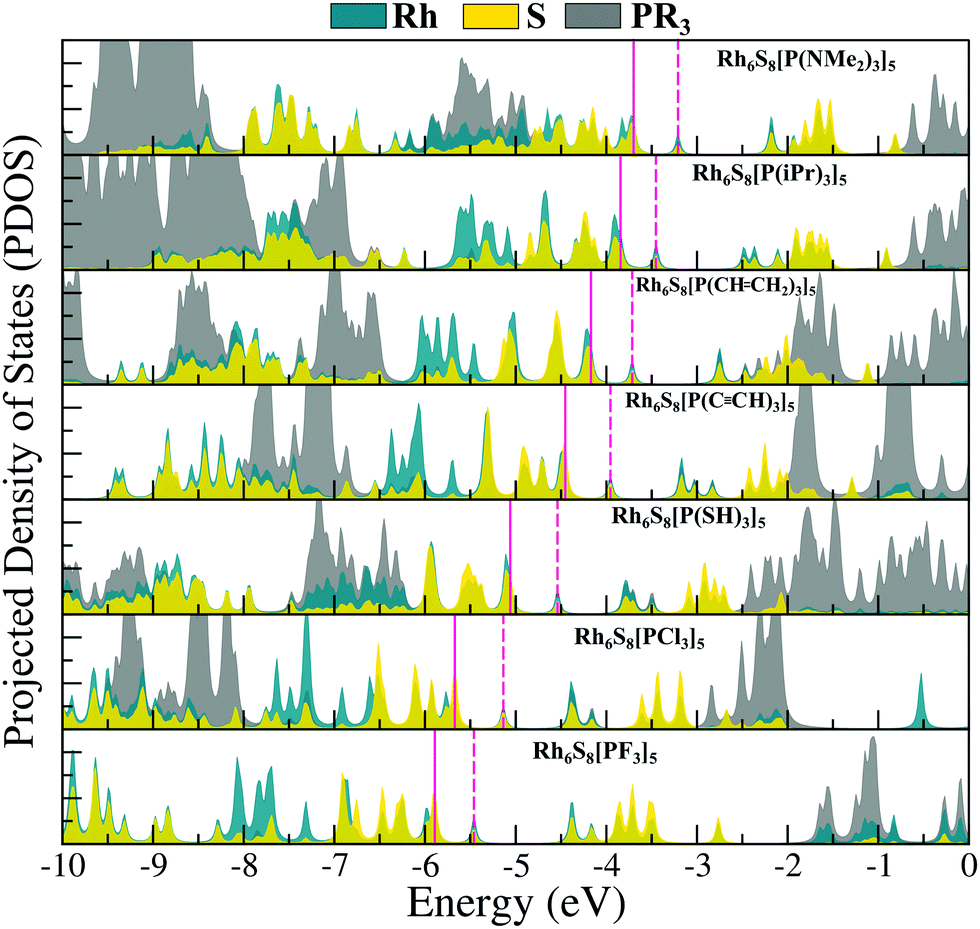
Next, we have plotted (Fig. 3) the molecular orbital (MO) diagrams for all seven clusters, each of which are decorated by five different PR3 ligands. It can be clearly seen from Fig. 3 that as the electronegativity of the R group increases from left to right, the whole electronic spectrum is shifting downward to lower energy, preserving the HOMO−LUMO gaps within a small range of 0.39–0.53 eV. The HOMO and LUMO isosurfaces are also included at the right-hand side of Fig. 3. Despite the change in the attached R groups throughout the series, the isosurfaces of both HOMO and LUMO have remained identical for all cases. This can be confirmed by looking into the salient features (i.e., the location and shape of the positive and negative lobes) of the plotted isosurfaces in Fig. 3. This is direct proof that altering the R groups results in a Coulomb-like shift of the whole electronic spectrum without altering the relative ordering and the occupation numbers of the orbitals. The same conclusion can be reconfirmed from the projected density of states (PDOS) diagram (Fig. 4) as well. Firstly, for all the seven systems, both the HOMO and LUMO show a very similar composition of Rh and S. The attached ligands (PR3) show nearly zero contribution to both HOMO and LUMO in all cases. Secondly, the shift of the whole electronic spectrum can be easily visualized simply by tracking the locations of the PDOS peaks in Fig. 4. As we alter the R groups from NMe2 to F, all the PDOS peaks have simultaneously shifted toward lower energy, clearly showing that it is the overall electronic spectrum that is shifting and not some specific molecular orbitals.
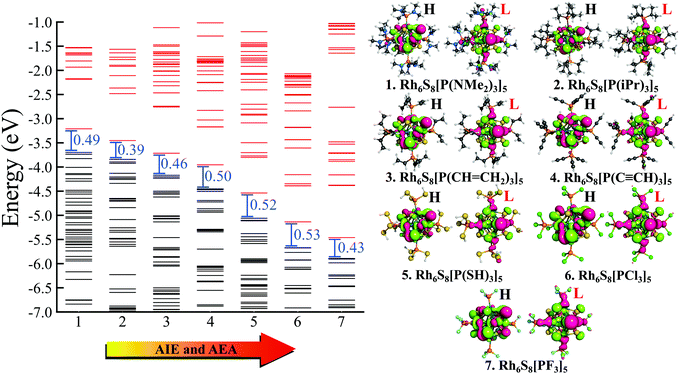 |
| | Fig. 3 The molecular orbital (MO) diagram of Rh6S8(PR3)5, (R = NMe2 to F) clusters. The black and red lines show the occupied and unoccupied MOs, respectively. The HOMO (H) and LUMO (L) isosurfaces for all ligated clusters are shown on right. | |
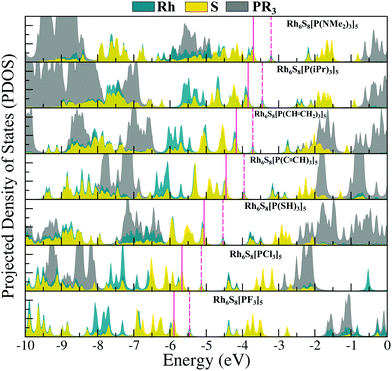 |
| | Fig. 4 The projected density of state (PDOS) diagram of Rh6S8(PR3)5, (R = NMe2 to F) clusters. The location of the HOMO and LUMO levels are marked by solid and dashed lines, respectively. The contributions from Rh, S and the PR3 ligands are shown in different colors. | |
2.2. The properties of the (PR3)5Rh6S8–Rh6S8(CO)5 dimer series
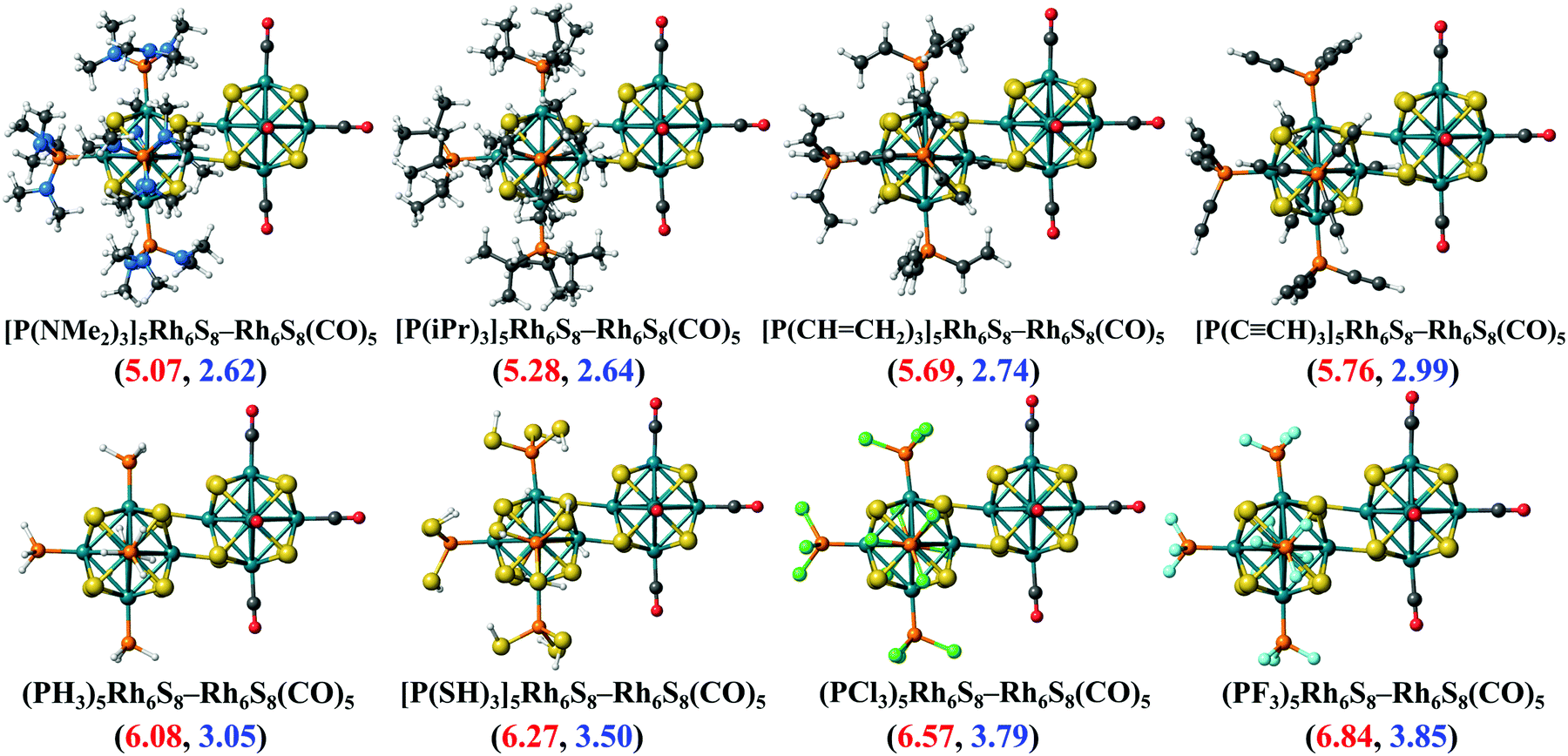
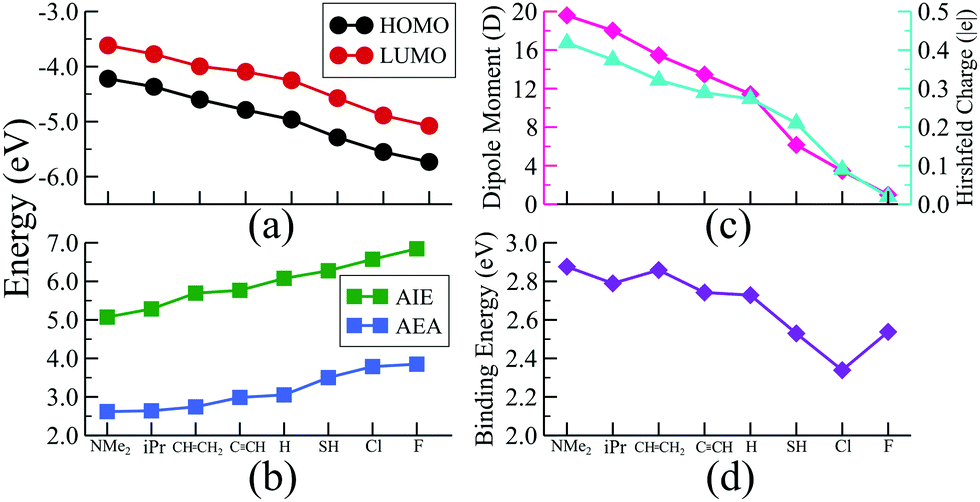
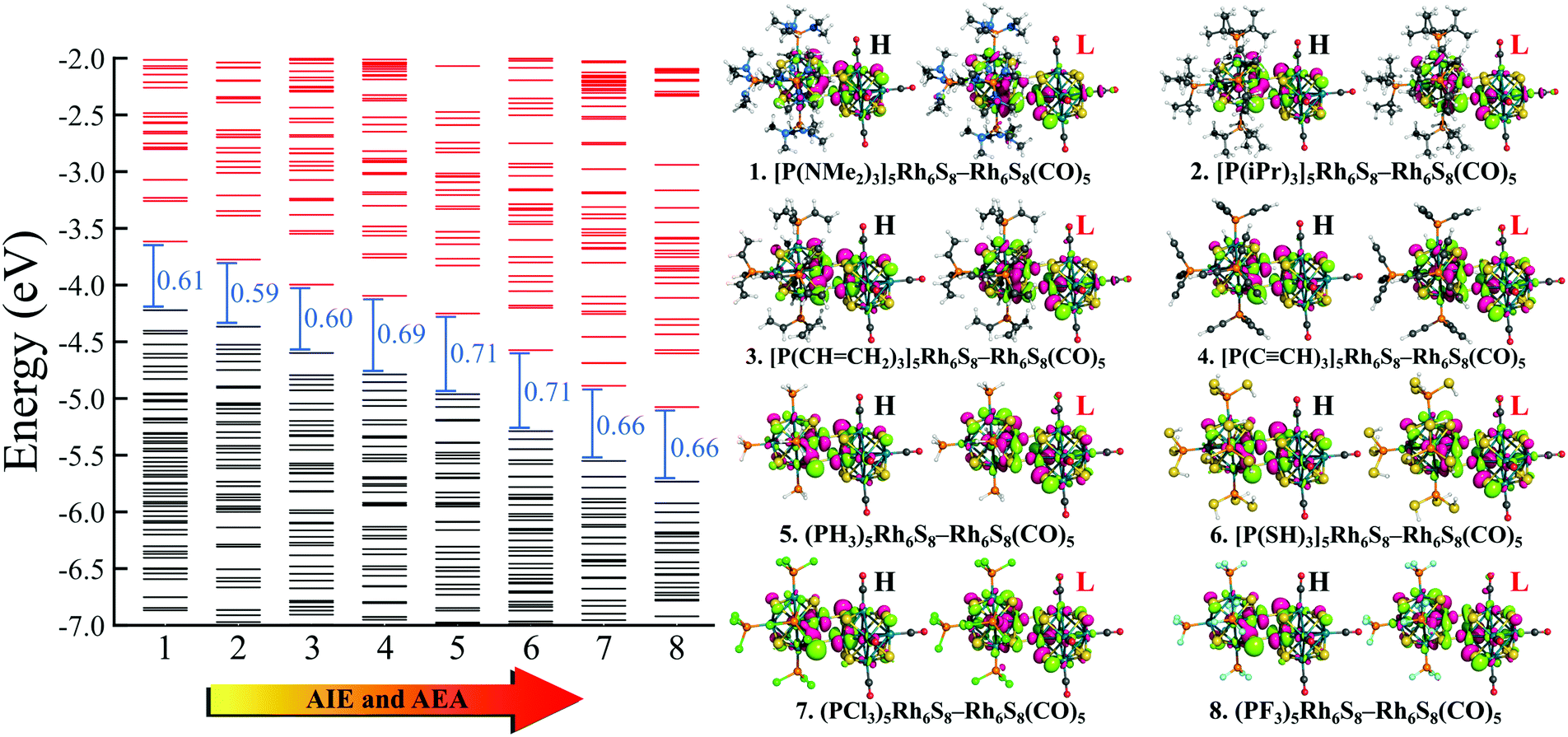
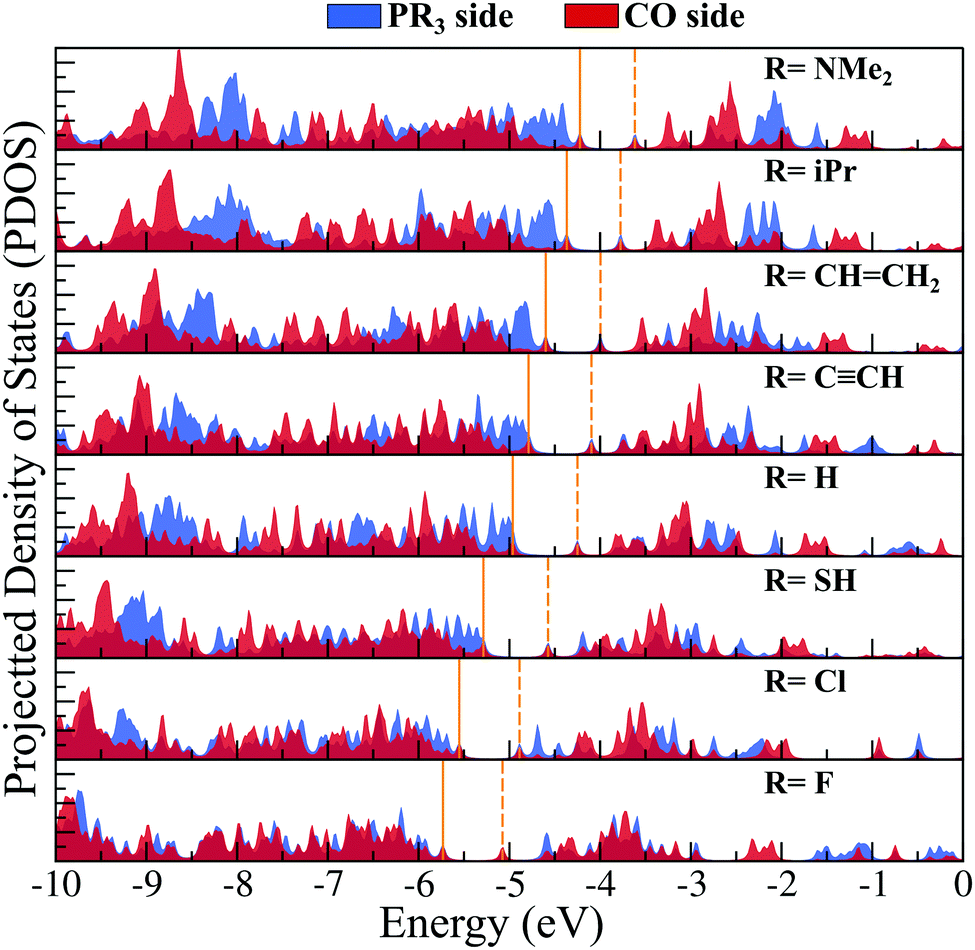
To investigate whether this approach will work beyond the monomeric scale, we have modeled a series of superatomic dimers, in which we have used Rh6S8(CO)5 cluster on one side and the Rh6S8 cluster decorated by 5 PR3 ligands on the other side. For the study of the dimer, we have taken 8 PR3 ligands instead of 7 as before. The new inclusion is the PH3 group which falls near the middle of the series based on the group electronegativity. Fig. 5 shows the optimized structure of all the clusters along with the AIE and AEA values which are mentioned within parenthesis. The plots of the calculated electronic properties are included in Fig. 6, and the numerical values are provided in Table 2. It is noteworthy that for the dimer series, the plotted properties show a similar trend as obtained for the ligated monomers. The HOMO and LUMO energies show a steady downward trend (Fig. 6a) as we change the ligands of the left-hand cluster from P(NMe2)3 to PF3. The energies of HOMO and LUMO also remain near parallel throughout the series, showing that the HOMO−LUMO gaps have not altered significantly. Fig. 6b shows the trend in AIE and AEA for the dimer series. Both parameters show a steady increase as the donor/acceptor characteristic of the PR3 ligands are altered sequentially from left to right. Thus, the lowest AIE (5.07 eV) and AEA (2.62 eV) are obtained for [(P(NMe2)3]5Rh6S8–Rh6S8(CO)5 dimer, which proves that this particular dimer will act as an excellent donor. In contrast, the AIE and AEA of (PF3)5Rh6S8–Rh6S8(CO)5 dimer is calculated as 6.84 and 3.85 eV, respectively, showing the remarkable strong electron acceptor characteristics of this dimer. If compared to the atomic scale,31 the AIE of [(P(NMe2)3]5Rh6S8−Rh6S8(CO)5 is very close to sodium (i.e., 5.14 eV), and the AEA of (PF3)5Rh6S8−Rh6S8(CO)5 is again higher than chlorine (i.e., 3.61 eV). To provide more insight, we have included the MO diagram (Fig. 7) and PDOS plot (Fig. 8) for the dimer series. Observing the MO diagram reveals a similar trend as noticed for the monomers. As the attached R groups of the left-hand Rh6S8 cluster are altered, the electronic spectrum is observed to shift toward lower energies. The HOMO−LUMO gaps again vary within a narrow range of 0.59–0.71 eV throughout the series, and only minor alteration is noticed. It is important to mention that the HOMO−LUMO gap for all the reported dimers is higher by ∼0.1–0.2 eV compared to the Rh6S8(PR3)5 series. The isosurface plots (Fig. 7) reveal that both the HOMO and LUMO have remained identical throughout the series, and no major changes are noticed. The HOMO and LUMO for all dimers are seen to be distributed between both clusters and not localized on one. Similar conclusions can be drawn from the PDOS (Fig. 8) plots as well, where we have plotted the PDOS for the PR3 (left) and CO (right) attached cluster separately. For clarity, in Fig. 8, we have not included the PDOS contribution of either type of ligands. It can be seen from Fig. 8 that the HOMO and LUMO orbitals for all dimers derive contributions from both clusters throughout the series. A more critical examination of PDOS in Fig. 8 reveals that the PDOS from the left cluster (blue regions) is shifting to the left with respect to the PDOS derived from CO side (red regions). As we will show, the apparent shift of the PDOS peaks from the CO side is due to the difference in the amount of charge transferred from the Rh6S8(PR3)5 (left) unit. This suggests that the electronic states on the two sides are undergoing relative displacement as the electronic spectrum in the left side is being lifted/lowered by the electronegativity of the ligand. This difference in the electronic levels leads to a charge transfer from the left to the Rh6S8(CO)5 cluster starting from the case of P[(NMe2)3]5Rh6S8, thus inducing a net dipole moment and an internal electric field. Since the electronegativity of the PR3 group increases as we move from P(NMe2)3 to PF3, it decreases the donor character of the cluster. To show this, we investigated the net Hirshfeld30 charge on each side of the reported dimers. The results are shown in Fig. 6c. Since the PR3 side is acting as the donor for all cases, the charge on the PR3 side is always positive, and consequently, an equal but negative charge is localized on the CO side. Hence, in Fig. 6c, we have only included the absolute value of the Hirshfeld30 charges accumulated on either side of the dimers. Being the strongest electron donor, the maximum amount of charge (|0.42| e) is observed to be localized on each side of the [(P(NMe2)3]5Rh6S8–Rh6S8(CO)5 dimer. As we move along the series, the amount of the charge accumulated on each side shows a steady decrease and reaches a near-zero value (|0.02| e) for (PF3)5Rh6S8–Rh6S8(CO)5 dimer. Based on the accumulated charge, it can be said that the inter-cluster bonding in [(P(NMe2)3]5Rh6S8–Rh6S8(CO)5 dimer is predominantly ionic. As we move down the series, the ionic nature is observed to reduce and eventually reach a covalent-dominant interaction for (PF3)5Rh6S8–Rh6S8(CO)5 that can be confirmed by the near-zero charge localized on the monomeric fragments. Since the amount of charge accumulated on the donor and acceptor sides determines the magnitude of the net dipole moment, the calculated dipole moments also show a similar trend (Fig. 6c). Thus, the [(P(NMe2)3]5Rh6S8–Rh6S8(CO)5 cluster shows the maximum dipole moment of 19.59 D whereas, the dipole moment of (PF3)5Rh6S8–Rh6S8(CO)5 dimer is the lowest, i.e., only 0.98 D. It is important to mention, that although the value of accumulated charge and the dipole moment is altered, the direction of the dipole moment vector remains the same for all cases, i.e., pointing toward the PR3 side (Fig. 9).
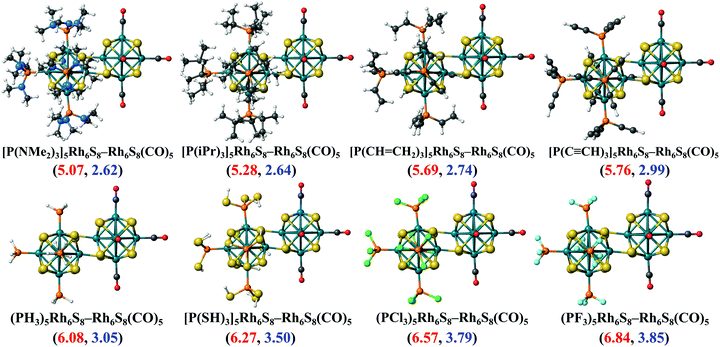 |
| | Fig. 5 The optimized ground state structures of (PR3)5Rh6S8–Rh6S8(CO)5 (R = NMe2 to F) dimers. The AIE (red) and AEA (blue) values (in eV) of each dimer are included within parenthesis. | |
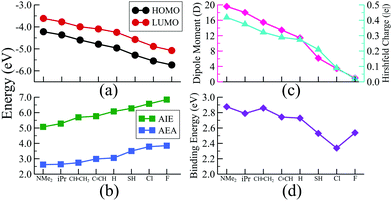 |
| | Fig. 6 The plots of calculated electronic properties a) HOMO−LUMO gap (in eV) b) AIE and AEA (in eV) c) absolute Hirshfeld charge on each side with the dipole moment (in Debye) and d) binding energy (in eV) between the two fragments of (PR3)5Rh6S8–Rh6S8(CO)5 (R = NMe2 to F) series. | |
Table 2 The magnetic moment (in μB), dipole moment (in Debye), and the calculated electronic properties (in eV) of the (PR3)5Rh6S8–Rh6S8(CO)5 (R = functional groups) dimer series
| Dimers |
Mag. moment (μB) |
Dipole moment (Debye) |
Hirshfeld charge |e| |
Electronic properties (eV) |
| HOMO |
LUMO |
HL gap |
AIE |
AEA |
| [(P(NMe2)3]5Rh6S8–Rh6S8(CO)5 |
0 |
19.59 |
0.42 |
−4.22 |
−3.61 |
0.61 |
5.07 |
2.62 |
| [(P(iPr)3]5Rh6S8–Rh6S8(CO)5 |
0 |
18.02 |
0.37 |
−4.36 |
−3.77 |
0.59 |
5.28 |
2.64 |
| [(P(CH = CH2)3]5Rh6S8–Rh6S8(CO)5 |
0 |
15.46 |
0.32 |
−4.59 |
−3.99 |
0.60 |
5.69 |
2.74 |
| [(P(C ≡ CH)3]5Rh6S8–Rh6S8(CO)5 |
0 |
13.45 |
0.29 |
−4.78 |
−4.09 |
0.69 |
5.76 |
2.99 |
| (PH3)5Rh6S8–Rh6S8(CO)5 |
0 |
11.42 |
0.27 |
−4.96 |
−4.25 |
0.71 |
6.08 |
3.05 |
| [(P(SH)3]5Rh6S8–Rh6S8(CO)5 |
0 |
6.15 |
0.21 |
−5.28 |
−4.57 |
0.71 |
6.27 |
3.50 |
| (PCl3)5Rh6S8–Rh6S8(CO)5 |
0 |
3.45 |
0.09 |
−5.55 |
−4.89 |
0.66 |
6.57 |
3.79 |
| (PF3)5Rh6S8–Rh6S8(CO)5 |
0 |
0.98 |
0.02 |
−5.73 |
−5.07 |
0.66 |
6.84 |
3.85 |
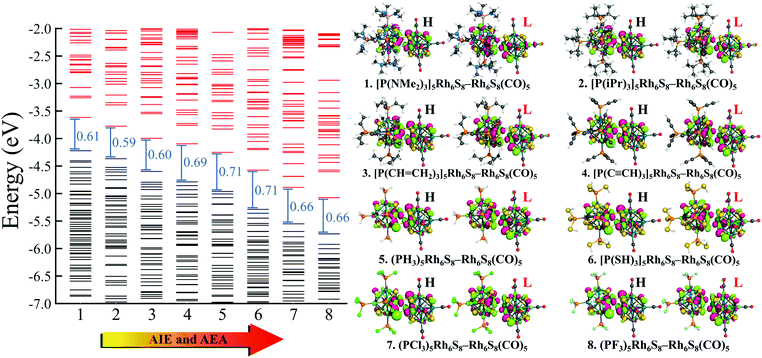 |
| | Fig. 7 The molecular orbital (MO) diagram of (PR3)5Rh6S8–Rh6S8(CO)5 (R = NMe2 to F) dimers. The black and red lines show the occupied and unoccupied MOs, respectively. The HOMO (H) and LUMO (L) isosurfaces for all dimers are shown on right. | |
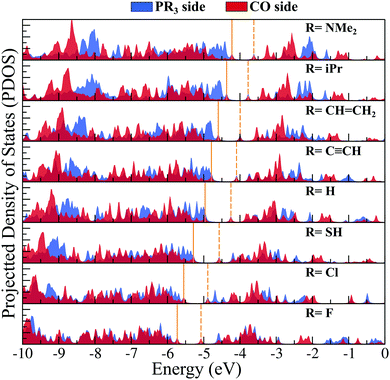 |
| | Fig. 8 The projected density of state (PDOS) diagram of (PR3)5Rh6S8–Rh6S8(CO)5 (R = NMe2 to F) dimers. The location of the HOMO and LUMO levels are marked by solid and dashed lines, respectively. The contributions from PR3 and CO side are shown in blue and red color, respectively. | |
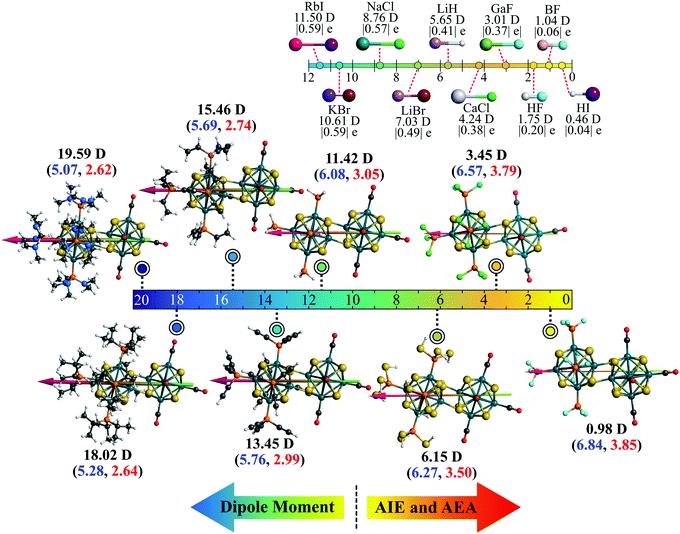 |
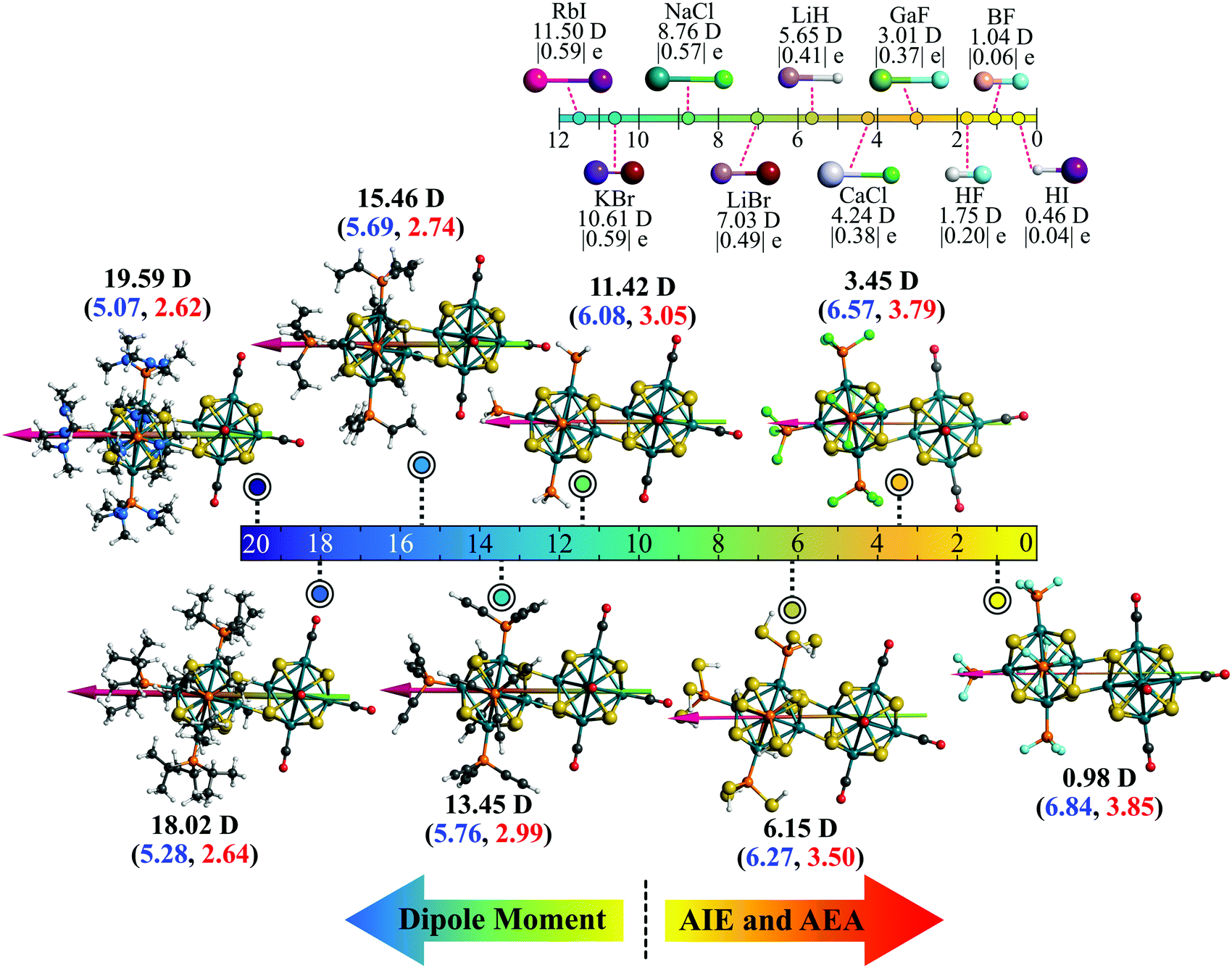
| | Fig. 9 The superionic scale of the (PR3)5Rh6S8–Rh6S8(CO)5 (R = NMe2 to F) dimers. The dipole moment (in Debye) of each is written in black. The AIE (blue) and AEA (red) values (in eV) are included within parenthesis. A similar scale with example ionic molecules alongside the dipole moment (in Debye) and absolute Hirshfeld charge (|e|) is shown on top for comparison. | |
The influence of the charge accumulation on the monomeric fragments of the dimer also affects the binding energies between the donor and acceptor units. To further illustrate the problem, we show in Fig. 6d the binding energy of the cluster dimer as a function of ligands. The binding energies between the Rh6S8(PR3)5 and Rh6S8(CO)5 monomeric units are higher for the first few members of the series compared to the dimers near the end. Since the charge accumulation on the monomers is significantly higher at the beginning of the series, that results in a strong electrostatic/ionic attraction between the donor and acceptor sides. As we move down the series, the electrostatic attraction between the monomeric units has gotten weaker, and thereby the binding energy as expected for a covalent-dominant interaction. The energy decomposition analysis further proves this point (see Table S4 of ESI†). The electrostatic component (−7.71 eV) between the monomers for [(P(NMe2)3]5Rh6S8–Rh6S8(CO)5 is significantly more negative compared to the values obtained (−6.65 to −7.03 eV) for the dimers near the end. Thus, it can be said with certainty that in addition to the redox properties, the current technique is equally useful to predictably alter the charge accumulation, dipole moments, and binding energies of the superatomic dimers.
In chemistry, it is well known that combining a strong electron donor (e.g., alkali metals) element with a strong electron acceptor (e.g., halogen) results in a salt (e.g., NaCl).32 Arranging such molecules according to their dipole moment produces an ionic series in the molecular domain. Since the dimer series reported herewith are also constructed similarly, i.e., by combining an electron donor superatom with an acceptor, we have asked ourselves if an equivalent series can be constructed in the superatomic domain as well. Fig. 9 shows such a series in which the respective dimers are positioned on a dipole moment scale ranging from 0 to 20 Debye. It is noteworthy that in the superatomic series, although the dipole moment increases from right to left, the AIE and AEA increase in the opposite direction (i.e., from left to right). For qualitative comparison, an equivalent ionic series composed of a few well-known ionic molecules is included on the top-right corner of Fig. 9. The similarity between both series is certainly interesting, especially since we have also observed that such metal-chalcogenide dimers reported herewith show a similar dissociation curve as the ionic molecules showing further resemblance to the molecular scale. An example comparison plot of such is included in the Fig. S2 of ESI†). Moreover, in the series of ionic molecules, the charge on the ions also show a similar trend as the superatomic series (Fig. 9). It also needs to be mentioned that the superionic series presented herewith represents only a fraction of the overall scale. The dipole range can be further extended just by introducing better electron donors than P(NMe2)3 or just by using a different metal chalcogenide cluster. Similarly, attaching stronger electron acceptors (compared to PF3) to the donor side will produce dimers with dipole moments closer to zero. The AIE and AEA range will also extend accordingly. This is an additional advantage compared to our ligand substitution method. In that procedure, for a particular metal chalcogenide dimer and a particular pair of donor–acceptor ligands (e.g., PEt3 and CO), the maximum dipole moment is reached when one side is fully decorated by the donor ligands and the other side is decorated by acceptor ligand. Moreover, precise ligand substitution on specific sites on a small size cluster is always a difficult task. Additionally, not all ligands work in inducing the Coulombic well that is shifting the electronic spectrum. Strong ligands can often perturb the electronic structure of the cluster resulting in undesired results. In the present approach, the functional groups (R) are utilized to alter the donor–acceptor properties of the PR3 group as a whole and are not directly bonded to the cluster. Thus, the chances of perturbing the shell structure of the cluster are largely reduced. It is important to note that although we have used CO ligands on one side of the dimer, it is not necessary. As shown before, since Rh6S8(PF3)5 is also an effective electron acceptor, analogous series can be constructed solely with the PR3 groups without using CO or any different type of π-acceptor ligands. Moreover, if one requires a dimer with some intermediate dipole moment, that is also achievable by using a different combination of the PR3 groups. For example, the dipole moment of (PH3)5Rh6S8–Rh6S8(PCl3)5 dimer is calculated as 9.78 D, which is just between the dipole moment of (PH3)5Rh6S8–Rh6S8(CO)5 and [P(SH)3]5Rh6S8–Rh6S8(CO)5 dimers, respectively (Table 2).
3. Conclusion
The present paper offers a novel approach of inducing ionic character in an otherwise homo-cluster molecule by attaching ligands that form charge-transfer complexes. For individual Rh6S8 metal-chalcogenide clusters, we have shown that the donors/acceptors character of the ligated clusters can be controlled by using phosphine ligands (PR3, R = functional groups) where the electronegativity of the ligands is altered via the functional (R) group ranging from NMe2 to F. Considering the overall range of AIE/AEA, the approach is observed to show similar performance compared to the ligand substitution method which we have proposed earlier.16 It is also shown that the ligated Rh6S8(PR3)5 clusters can be combined with Rh6S8(CO)5 to generate a series of superatomic salts that are marked by ionic bonding over an otherwise covalent interaction. Further, it is observed that the redox properties and dipole moment of such ionic superatomic molecules can also be predictably altered in a similar way, i.e., by changing the electronegativity of the PR3 group. The possibility of creating superatomic molecules with internal fields and where the location of HOMO and LUMO can be controlled could have important applications for light-harvesting as electron and hole separated by the field could be transferred to the surrounding medium. The superatomic molecules can also be assembled to create superatom-based ferroelectric solids. We are currently collaborating with experimental groups to explore these applications.
4. Materials and methods
4.1. Theoretical techniques
All the reported results in this work were computed using the Amsterdam Density Functional (ADF) software.33 The generalized gradient approximated (GGA) exchange–correlation functional as proposed by Perdew, Burke, and Ernzerhof (PBE) was utilized for all the calculations.34 A Slater type valence triple zeta (ζ) basis set35,36 with two polarization functions (TZ2P) and a large frozen core was chosen for all the elements in the cluster. The geometry optimizations were carried out by the hessian-based quasi-Newton method37,38 without any symmetry restrictions. The default optimization criteria as implemented in ADF software33 is used for all cases. The geometric optimizations for each reported structure were carried out incorporating a wide range of spin multiplicities, and the structure with the lowest energy was chosen as the global minima. Analytical frequencies39,40 for all the optimized structures were also calculated, and it was ensured that all the normal modes were real and positive. This is to verify that the reported optimized structures were true minima and not saddle points. The Hirshfeld30 charges and dipole moments are calculated using the same functional and basis set. The relativistic effects of the elements were accounted for by the scalar relativistic zeroth-order regular approximation (ZORA).41
Data availability
The cartesian coordinates and related data are given in the ESI.† All additional data generated or analyzed during this study are available via request to the corresponding author.
Author contributions
T. S conducted all the calculations. S. N. K and T. S analyzed the results and wrote the manuscript.
Conflicts of interest
The authors declare no competing interests.
Acknowledgements
The authors gratefully acknowledge funding by the US Department of Energy (DOE) under the award DE-SC0006420.
References
-
P. Atkins and T. Overton, Shriver and Atkins’ Inorganic Chemistry, Oxford University Press, USA, 2010 Search PubMed.
- A. M. Champsaur, J. Yu, X. Roy, D. W. Paley, M. L. Steigerwald, C. Nuckolls and C. M. Bejger, ACS Cent. Sci., 2017, 3, 1050–1055 CrossRef CAS PubMed.
- A. Pinkard, A. M. Champsaur and X. Roy, Acc. Chem. Res., 2018, 51, 919–929 CrossRef CAS PubMed.
- X. Roy, C.-H. Lee, A. C. Crowther, C. L. Schenck, T. Besara, R. A. Lalancette, T. Siegrist, P. W. Stephens, L. E. Brus and P. Kim, Science, 2013, 341, 157–160 CrossRef CAS PubMed.
- B. M. Boardman, J. R. Widawsky, Y. S. Park, C. L. Schenck, L. Venkataraman, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2011, 133, 8455–8457 CrossRef CAS PubMed.
- N. A. Gadjieva, A. M. Champsaur, M. L. Steigerwald, X. Roy and C. Nuckolls, Eur. J. Inorg. Chem., 2020, 1245–1254 CrossRef CAS.
- C. H. Lee, L. Liu, C. Bejger, A. Turkiewicz, T. Goko, C. J. Arguello, B. A. Frandsen, S. C. Cheung, T. Medina and T. J. Munsie, J. Am. Chem. Soc., 2014, 136, 16926–16931 CrossRef CAS PubMed.
- E. J. Telford, J. C. Russell, J. R. Swann, B. Fowler, X. Wang, K. Lee, A. Zangiabadi, K. Watanabe, T. Taniguchi and C. Nuckolls, Nano Lett., 2020, 20, 1718–1724 CrossRef CAS PubMed.
- J. Yang, F. Wang, J. C. Russell, T. J. Hochuli, X. Roy, M. L. Steigerwald, X. Zhu, D. W. Paley and C. Nuckolls, J. Am. Chem. Soc., 2020, 142, 11993–11998 CrossRef CAS PubMed.
- W. D. Knight, K. Clemenger, W. A. de Heer, W. A. Saunders, M. Y. Chou and M. L. Cohen, Phys. Rev. Lett., 1984, 52, 2141 CrossRef CAS.
- W. D. Knight, W. A. de Heer, K. Clemenger and W. A. Saunders, Solid State Commun., 1985, 53, 445–446 CrossRef CAS.
- S. N. Khanna and P. Jena, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 13705 CrossRef CAS PubMed.
- D. E. Bergeron, A. W. Castleman, T. Morisato and S. N. Khanna, Science, 2004, 304, 84–87 CrossRef CAS PubMed.
- A. C. Reber and S. N. Khanna, Acc. Chem. Res., 2017, 50, 255–263 CrossRef CAS PubMed.
- S. N. Khanna, A. C. Reber, D. Bista, T. Sengupta and R. Lambert, J. Chem. Phys., 2021, 155, 120901 CrossRef CAS PubMed.
- V. Chauhan, A. C. Reber and S. N. Khanna, J. Am. Chem. Soc., 2017, 139, 1871–1877 CrossRef CAS PubMed.
- V. Chauhan, A. C. Reber and S. N. Khanna, J. Phys. Chem. A, 2016, 120, 6644–6649 CrossRef CAS PubMed.
- V. Chauhan, S. Sahoo and S. N. Khanna, J. Am. Chem. Soc., 2016, 138, 1916–1921 CrossRef CAS PubMed.
- A. C. Reber, V. Chauhan and S. N. Khanna, J. Chem. Phys., 2017, 146, 024302 CrossRef PubMed.
- G. Liu, V. Chauhan, A. P. Aydt, S. M. Ciborowski, A. Pinkard, Z. Zhu, X. Roy, S. N. Khanna and K. H. Bowen, J. Phys. Chem. C, 2019, 123, 25121–25127 CrossRef CAS.
- G. Liu, A. Pinkard, S. M. Ciborowski, V. Chauhan, Z. Zhu, A. P. Aydt, S. N. Khanna, X. Roy and K. H. Bowen, Chem. Sci., 2019, 10, 1760–1766 RSC.
- V. Chauhan, A. C. Reber and S. N. Khanna, Nat. Commun., 2018, 9, 1–7 CAS.
- A. C. Reber, D. Bista, V. Chauhan and S. N. Khanna, J. Phys. Chem. C, 2019, 123, 8983–8989 CrossRef CAS.
- A. C. Reber, V. Chauhan, D. Bista and S. N. Khanna, Nanoscale, 2020, 12, 4736–4742 RSC.
- D. Bista, T. Sengupta, A. C. Reber and S. N. Khanna, Nanoscale, 2021, 13, 15763–15769 RSC.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
-
R. H. Crabtree, The organometallic chemistry of the transition metals, John Wiley & Sons, 2009 Search PubMed.
- G. Thiele, Z. You and S. Dehnen, Inorg. Chem., 2015, 54, 2491–2493 CrossRef CAS PubMed.
- G. Thiele, M. Balmer and S. Dehnen, Z. Naturforsch., B: J. Chem. Sci., 2016, 71, 391–394 CrossRef CAS.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS.
-
D. R. Lide, CRC handbook of chemistry and physics, CRC press, 2004, vol. 85 Search PubMed.
-
A. D. McNaught and A. Wilkinson, Compendium of chemical terminology, Blackwell Science, Oxford, 1997, vol. 1669 Search PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- E. Van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- D. P. Chong, E. Van Lenthe, S. Van Gisbergen and E. J. Baerends, J. Comput. Chem., 2004, 25, 1030–1036 CrossRef CAS PubMed.
- L. Versluis and T. Ziegler, J. Chem. Phys., 1988, 88, 322–328 CrossRef CAS.
- L. Fan and T. Ziegler, J. Chem. Phys., 1991, 95, 7401–7408 CrossRef CAS.
- H. Jacobsen, A. Bérces, D. P. Swerhone and T. Ziegler, Comput. Phys. Commun., 1997, 100, 263–276 CrossRef CAS.
- S. K. Wolff, Int. J. Quantum Chem., 2005, 104, 645–659 CrossRef CAS.
- E. van Lenthe, J. G. Snijders and E. J. Baerends, J. Chem. Phys., 1996, 105, 6505–6516 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  and
Shiv N.
Khanna
and
Shiv N.
Khanna