
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh affinity zoledronate-based metal complex nanocrystals to potentially treat osteolytic metastases†
Gabriel
Quiñones Vélez
 ab,
Lesly
Carmona-Sarabia
ab,
Alondra A.
Rivera Raíces
bc,
Tony
Hu
d,
Esther A.
Peterson-Peguero
c and
Vilmalí
López-Mejías
*ab
ab,
Lesly
Carmona-Sarabia
ab,
Alondra A.
Rivera Raíces
bc,
Tony
Hu
d,
Esther A.
Peterson-Peguero
c and
Vilmalí
López-Mejías
*ab
aDepartment of Chemistry, University of Puerto Rico, Río Piedras, San Juan, Puerto Rico 00931, USA. E-mail: vilmali.lopez@upr.edu
bCrystallization Design Institute, Molecular Sciences Research Center, University of Puerto Rico, San Juan, Puerto Rico 00926, USA
cDepartment of Biology, University of Puerto Rico, Río Piedras, San Juan, Puerto Rico 00931, USA
dDepartment of Chemistry and the Molecular Design Institute, New York University, 100 Washington Square East, New York, New York 10003-6688, USA
First published on 23rd February 2022
Abstract
Formation of several materials, denoted as bisphosphonate-based coordination complexes (BPCCs), resulted from the reaction between clinically employed bisphosphonate, zoledronate (ZOLE) and bioactive metals (M2+ = Ca2+, Mg2+ and Zn2+). Six ZOLE-based BPCCs were synthesized using different variables (M2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ZOLE molar ratio, temperature, pH, and anion) and their structures were elucidated by single crystal X-ray diffraction (ZOLE–Ca forms I and II, ZOLE–Mg forms I and II, and ZOLE–Zn forms I and II). The dissolution of the ZOLE-based BPCCs was compared to that of ZOLE (Reclast®). Most of the ZOLE-based BPCCs (60–85%, in 18–24 h) present a lower dissolution and equilibrium solubility than ZOLE (∼100%, 30 min) in phosphate buffered saline (PBS), while a significantly higher dissolution is observed in acidic media (88% in 1 h). This suggests the ability to release the ZOLE content in a pH-dependent manner. Moreover, a phase inversion temperature (PIT)-nano-emulsion synthesis was performed, which demonstrated the ability to significantly decrease the crystal size of ZOLE–Ca form II from a micron-range (∼200 μm) to a nano-range (∼150 d nm), resulting in nano-Ca@ZOLE. Furthermore, low aggregation of nano-Ca@ZOLE in 10% fetal bovine serum (FBS):PBS after 0, 24 and 48 h was demonstrated. Additionally, nano-Ca@ZOLE showed an ∼2.5x more binding to hydroxyapatite (HA, 36%) than ZOLE (15%) in 1 d. The cytotoxicity of nano-Ca@ZOLE against MDA-MB-231 (cancer cell model) and hFOB 1.19 (normal osteoblast-like cell model) cell lines was investigated. The results demonstrated significant cell growth inhibition for nano-Ca@ZOLE against MDA-MB-231, specifically at a low concentration of 3.8 μM (%RCL = 55 ± 1%, 72 h). Under the same conditions, the nanocrystals did not present cytotoxicity against hFOB 1.19 (%RCL = 100 ± 2%). These results evidence that nano-ZOLE-based BPCCs possess viable properties in terms of structure, dissolution, stability, binding, and cytotoxicity, which render them suitable for osteolytic metastasis therapy.
:ZOLE molar ratio, temperature, pH, and anion) and their structures were elucidated by single crystal X-ray diffraction (ZOLE–Ca forms I and II, ZOLE–Mg forms I and II, and ZOLE–Zn forms I and II). The dissolution of the ZOLE-based BPCCs was compared to that of ZOLE (Reclast®). Most of the ZOLE-based BPCCs (60–85%, in 18–24 h) present a lower dissolution and equilibrium solubility than ZOLE (∼100%, 30 min) in phosphate buffered saline (PBS), while a significantly higher dissolution is observed in acidic media (88% in 1 h). This suggests the ability to release the ZOLE content in a pH-dependent manner. Moreover, a phase inversion temperature (PIT)-nano-emulsion synthesis was performed, which demonstrated the ability to significantly decrease the crystal size of ZOLE–Ca form II from a micron-range (∼200 μm) to a nano-range (∼150 d nm), resulting in nano-Ca@ZOLE. Furthermore, low aggregation of nano-Ca@ZOLE in 10% fetal bovine serum (FBS):PBS after 0, 24 and 48 h was demonstrated. Additionally, nano-Ca@ZOLE showed an ∼2.5x more binding to hydroxyapatite (HA, 36%) than ZOLE (15%) in 1 d. The cytotoxicity of nano-Ca@ZOLE against MDA-MB-231 (cancer cell model) and hFOB 1.19 (normal osteoblast-like cell model) cell lines was investigated. The results demonstrated significant cell growth inhibition for nano-Ca@ZOLE against MDA-MB-231, specifically at a low concentration of 3.8 μM (%RCL = 55 ± 1%, 72 h). Under the same conditions, the nanocrystals did not present cytotoxicity against hFOB 1.19 (%RCL = 100 ± 2%). These results evidence that nano-ZOLE-based BPCCs possess viable properties in terms of structure, dissolution, stability, binding, and cytotoxicity, which render them suitable for osteolytic metastasis therapy.
Introduction
Cancer remains a major public health concern, being one of the leading causes of death worldwide.1 Among all the new cancer cases recently diagnosed (18.1 M), the three most common cancers in women are breast, lung and colorectal, of which breast cancer alone accounts for 30% of all the estimates.1,2 The highest mortality rate observed in women is primarily due to breast cancer, because of its high potential to metastasize once in an advanced stage.3 Over 80% of patients with advanced breast cancer develop osteolytic metastases (OM), representing a debilitating stage of the disease with very low prognosis.4 Clinically, OM is challenging due to the rapid microarchitectural deterioration of affected sites at the bone marrow, based on the altered coupling between osteoblasts and osteoclasts, all mediated by tumor-driven dysregulation.4 Therefore, progression of the disease is mainly caused by the dysregulation promoted by metastatic cells at the bone microenvironment. As a consequence, these tumor cells are the prominent therapeutic targets when treating OM.Antiresorptive medications, such as bisphosphonates (BPs), are commonly prescribed to treat and delay the progression of breast-cancer-induced OM.4–7 These compounds resist enzymatic hydrolysis due to the presence of a P–C–P bond, in contrast to pyrophosphates (P–O–P bond).7,8 Once the BPs reach their target, their binding to the bone is strong because they exhibit high affinity to calcium ions.9 Preclinical research has demonstrated that BPs promote anti-tumor effects via direct (tumor-cell apoptosis) and indirect (angiogenesis, γδ T cells) mechanisms.10 However, due to the several pharmacological deficiencies presented by BPs (poor bioavailability and low intestinal adsorption <10%), their direct anti-tumor effects remain unclear at the high doses required to provide the desired therapeutic effect.11–13
Among clinically utilized BPs, alendronate, risedronate and ibandronate present a lower therapeutic efficacy compared to zoledronate (ZOLE, Fig. 1), a last generation BP which exhibits the most potent and prolonged osteoclast antiresorptive activity.14,15 At present, an optimal regimen for ZOLE against bone-related diseases and OM is not known.14,16,17 It has been established that most of the drug undergoes renal clearance due to its significantly low bioavailability (<1%) and gastrointestinal permeation.18–20 These attributes result in a maximum plasma concentration of 1 μM for the drug, which represents a 10–100x lower concentration than that needed to kill cancer cells in vitro.21 However, recent studies have demonstrated that ZOLE exhibits direct cytotoxicity effects against human cancer cell models, recommending its use as a chemotherapeutic agent.22–25 Several attempts have been made by employing ZOLE to design effective therapies against bone-related diseases, such as coordination complexes for drug delivery,21,26,27 and radiopharmaceuticals as a bone pain therapy for oncological diseases.15,28–30 However, these approaches focused mainly on the labeling efficiency of ZOLE to beta emitters, adsorption to hydroxyapatite (HA), biodistribution, and cytotoxicity of the proposed systems through in vitro assays employing prostate, lung, and liver cancerous cells.15,21,26–30 Here, we propose to develop novel ZOLE-based coordination complex nanocrystals and assess their potential to treat specifically breast-cancer-induced OM.
 | ||
| Fig. 1 Schematic diagram of the synthesis conditions leading to six crystalline phases of ZOLE-based BPCCs. As products, the coordination of zoledronate (ZOLE) with three different bioactive metals (M2+ = Ca2+, Mg2+, and Zn2+) under several synthesis conditions. Unit cell and polarized optical micrographs of each ZOLE-based BPCC are shown in the product side of each reaction. The variables explored were pH, metal salt anion (NO3−vs. Cl−), and addition of etidronic acid (HEDP) as an auxiliary ligand. | ||
In a previous study, the design of nanocrystals comprising bisphosphonate-based coordination complexes (BPCCs) with the potential to treat breast-cancer-induced OM was described.31 The reported alendronate-based BPCCs presented several promising properties such as high thermal and structural stability, selective degradation under different physiological conditions, and significant cytotoxicity against human breast cancer cells at low concentrations.31 Moreover, metal ions such as Ca2+, Co2+, Mg2+, Mn2+, Ni2+ and Zn2+ have been employed to form coordination complexes (CCs) with ZOLE.32–36 However, these reports focus mainly on the structural properties of these materials.
In this work, ZOLE was employed as a ligand for reaction with three different bioactive metals (M2+ = Ca2+, Mg2+ and Zn2+) to design ZOLE-based BPCCs (Fig. 1) with the potential to treat OM. From the resulting materials, the structural and thermal stability as well as degradation in different physiological media were assessed to provide insights into their ability to be employed for biomedical applications. Furthermore, efficient crystal size reduction through a phase inversion temperature (PIT)-nano-emulsion synthesis allowed the formation of nano-Ca@ZOLE, a ZOLE-based BPCC in the nano-range, for which other biomedical relevant properties were determined. These included the aggregation behavior of the nanomaterial in biologically relevant media, its binding affinity to HA, and cytotoxicity against both triple-negative breast cancer cells that metastasize to the bone (MDA-MB-231) and normal osteoblast cells (hFOB 1.19). This study is intended to assess BPCCs, specifically nano-Ca@ZOLE, and potentiate them as an alternative aimed at treating and preventing breast-cancer-induced OM.
Experimental section
Materials
Calcium nitrate tetrahydrate [Ca(NO3)2·4H2O, 99% pure], calcium chloride dihydrate [CaCl2·2H2O, USP grade], zinc nitrate hexahydrate [Zn(NO3)2·6H2O, 98% pure], zinc chloride anhydrous [ZnCl2, >98% pure], magnesium nitrate hexahydrate [Mg(NO3)2·6H2O, 99% pure], magnesium chloride anhydrous [MgCl2, ≥98% pure], and etidronic acid 60% aqueous solution (HEDP) were purchased from Sigma-Aldrich (St. Louis, MO). Zoledronic acid monohydrate (C5H10N2O7P2·H2O, >98% pure) was acquired from TCI America (St. Portland, OR). The pH adjustments were made using a stock solution of sodium hydroxide (NaOH, USP grade, 0.3 M). Nanopure water was used as the solvent in all syntheses. Phosphate buffered saline (PBS) tablets, from Sigma Aldrich (St. Louis, MO), were used to prepare PBS solutions (pH = 7.40). Hydrochloric acid (HCl, 37%) and sodium chloride (NaCl, ACS reagent >99.0% pure) from Sigma-Aldrich (St. Louis, MO) were used to prepare fasted-state simulated gastric fluid (FaSSGF) buffered solutions (pH = 1.60). Heptane [CH3(CH2)5CH3, anhydrous 99%] and Brij® L4 [(C20H42O5)n, average Mn ∼ 362 g mol−1] from Sigma-Aldrich (St. Louis, MO) were used to prepare the emulsion for phase inversion temperature (PIT) determination and nano-emulsion synthesis of nano-Ca@ZOLE. Fetal bovine serum (FBS, mammalian and insect cell culture tested) from Sigma-Aldrich (St. Louis, MO) was used for the particle size distribution and aggregation measurements of nano-Ca@ZOLE. Hydroxyapatite (Ca5(OH)(PO4)3, synthetic powder) from Sigma-Aldrich (Milwaukee, WI) was utilized to carry out the binding assays of nano-Ca@ZOLE. Human breast cancer MDA-MB-231 cell line (ATCC® HTB-26™, Manassas, VA), normal osteoblast-like hFOB 1.19 cell line (ATCC® CRL-11372™, Manassas, VA), Dulbecco's Modified Eagle's Medium (DMEM) from Sigma-Aldrich (Milwaukee, WI), 1:1 mixture of Ham's F-12 Medium/Dulbecco's Modified Eagle's Medium (1:1 DMEM:F-12) and geneticin (G418) from Bioanalytical Instruments (San Juan, PR), penicillin–streptomycin (Pen–Strep) from Sigma-Aldrich (St. Louis, MO), and AlamarBlue® from Bio-Rad (Kidlington, Oxford) were employed to investigate the cell proliferation of ZOLE and nano-Ca@ZOLE.
General hydrothermal synthesis of ZOLE-based BPCCs
The hydrothermal syntheses of ZOLE-based BPCCs were carried out by preparing separate solutions of the ligand (ZOLE) and the metal salt in nanopure water at room temperature. If required, 0.3 M NaOH was added to the ligand solution to adjust the pH above several of the ZOLE principal pKa's (pH = 1.23–4.40). HEDP was added in some cases as an auxiliary ligand to decrease the pH below of the pKa's of the ligand (ZOLE). The metal salt solution was added to the ligand solution with a syringe and mixed thoroughly. The formation of metal hydroxides was avoided by adjusting the pH of the resulting solution below the pH that led to M(OH)n precipitation. Heat was applied to the resulting mixture until crystals appeared (between minutes to hours). Once the crystals were visually detected, the vials were removed from heat and left undisturbed to let them grow. The product was collected after vacuum filtration and air-drying. Details of the synthesis conditions leading to each of the BPCCs (ZOLE–Ca forms I and II, ZOLE–Mg forms I and II, and ZOLE–Zn forms I and II) are available in the ESI.†Raman microscopy
Raman spectra were recorded using a Thermo Scientific DXR Raman microscope, equipped with a 780 nm laser, 400 lines per nm grating, and 50 μm slit. The spectra were collected at room temperature over the range of 3400 and 100 cm−1 by averaging 32 scans with exposures of 5 s. The OMNIC for Dispersive Raman software version 9.2.0 was employed for data collection and analysis.Scanning electron microscopy-energy dispersive spectroscopy (SEM-EDS)
X-ray microanalysis was conducted using a JEOL JSM-6480LV scanning electron microscope with an energy dispersive X-ray analysis (EDAX) Genesis 2000 detector. Micrographs were recorded using the same instrument, employing an Evenhart Thomley secondary electron imaging (SEI) detector. SEM samples were coated with a 5–10 nm Au layer with a sputtering target (10 s), employing a PELCO® SC-7 auto sputter coater coupled with a PELCO® FTM-2 film thickness monitor. Images were taken under high vacuum mode with an acceleration voltage of 20 kV, electron beam of 11 mm width, with a spot size value of 36, and SEI signals.Powder X-ray diffraction (PXRD)
Powder diffractograms were collected in transmission mode (100 K) using a Rigaku XtaLAB SuperNova X-ray diffractometer with a micro-focus Cu-Kα radiation (λ = 1.5417 Å) source and equipped with a HyPix3000 X-ray detector (50 kV, 0.8 mA). Powdered samples were mounted in MiTeGen micro loops using paratone oil. Powder diffractograms were collected between 6 and 60° with a step of 0.01° using the Gandalfi move experiment with an exposure time of 300 s. Data were analyzed using the CrystAllisPRO software v. 1.171.3920a.Single crystal X-ray diffraction (SCXRD)
The crystals were observed under polarized light to assess their quality. Optical micrographs were recorded with a Nikon Eclipse Microscope LV100NPOL, equipped with a Nikon DS-Fi2 camera and NIS Elements BR software version 4.30.01. Suitable single crystals were mounted using paraton oil onto MiTeGen micro loops for structure elucidation. Structural elucidation was performed in either of the two instruments.The crystal structure for the ZOLE–Zn form I was determined from the diffraction data collected using a Bruker AXS SMART APEX-II single crystal diffractometer equipped with a Monocap collimator and APEX-II CCD detector with a Mo-Kα (λ = 0.71073 Å) radiation source operating at 50 kV and 40 mA. The data collection was carried out at 100 K using an Oxford Cryosystems Cryostream 700 cooler.
The crystal structures of ZOLE–Ca forms I and II, ZOLE–Mg forms I and II, and ZOLE–Zn form II were determined from the diffraction data collected with a Rigaku XtaLAB SuperNova single micro-focus Cu-Kα radiation (λ = 1.5417 Å) source equipped with a HyPix3000 X-ray detector in transmission mode operating at 50 kV and 1 mA within the CrystAllisPRO software v. 1.171.3920a. Data collection was carried out at 100 K using an Oxford Cryosystems Cryostream 800 cooler. All crystal structures were solved by direct methods. The refinement was performed using full-matrix least squares on F2 within the Olex2 software v1.2. All non-hydrogen atoms were anisotropically refined.
Thermogravimetric analysis (TGA)
TGA patterns of the ligand and all BPCCs were recorded using a Q500 instrument (TA Instruments Inc.). The profile involved the temperature range of 10–700 °C at 5 °C min−1 under a N2 gas purge (60 mL min−1). For all measurements, ∼10 mg of powdered sample was thermally treated. Data were processed with TA Universal Analysis software version 4.3A.Dissolution rate measurements
Dissolution profiles were obtained via direct quantification by measuring absorbance at 208 nm. Rate measurements were recorded for the reagent grade ZOLE, ZOLE–Ca forms I and II, ZOLE–Mg forms I and II, ZOLE–Zn forms I and II in PBS, against a reagent blank. For ZOLE and ZOLE–Ca form II, dissolution measurements were also performed in FaSSGF. Dissolution tests were performed in 100 mL of PBS (pH = 7.40) or FaSSGF (pH = 1.60) buffers at 37 °C under constant stirring at 150 rpm, for 48 h (PBS) or 36 h (FaSSGF). Absorbance measurements were performed using an Agilent Technologies Cary Series UV-Vis Spectrophotometer, Cary 100 UV-Vis model, using the UV Cary Scan software version v.20.0.470. All measurements were performed with a 400–200 nm scan.Determination of the phase inversion temperature (PIT) and PIT-nano-emulsion synthesis of nano-Ca@ZOLE
To reduce the particle size, a PIT-nano-emulsion method was employed during the synthesis of a selected BPCC, specifically ZOLE–Ca form II. The PIT temperature was determined by measuring the conductivity of an aqueous emulsion containing ZOLE in heptane (oil phase) and Brij® L4 (surfactant) in the temperature range of 2–40 °C at 1 °C min−1. After homogenizing the emulsions, conductivity measurements started at 2 °C with an O/W micro-emulsion (∼840 μS). As the temperature is increased (1 °C min−1), a phase inversion (O/W to W/O) occurs. The phase inversion starts at 9 °C and ends at 15 °C, where the conductivity measurements reduce to an average value of ∼8.58 μS. This leads to the conversion of the emulsion into a water-in-oil (W/O) nano-emulsion.The nano-emulsion synthesis of nano-Ca@ZOLE was conducted in a Crystalline™ (Technobis, Crystallization Systems, Alkmaar, Netherlands). The pre-homogenized emulsions (ZOLE, heptane, Brij® L4) prepared for PIT determination were used to perform the nano-Ca@ZOLE synthesis. The emulsion was homogenized before being transferred to the reaction vial and placed in the first reactor at a temperature of 5 °C and 1250 rpm for 30 min. After 30 min, the reaction vial was transferred to a second reactor at 45 °C and 1250 rpm. The emulsion was stirred for 30 min before heating to a reaction temperature of 85 °C. Subsequently, the metal salt solution was added using a syringe and left undisturbed for 30 min. Once completed, the reaction vial was left undisturbed for 30 min before analyzing the supernatant from the aqueous phase using dynamic light scattering (DLS).
Dynamic light scattering (DLS) and aggregation tendency measurements
Samples resulting from the synthesis of nano-Ca@ZOLE were analyzed using a Malvern Panalytical Zetasizer NanoZS equipped with a He–Ne orange laser (633 nm, max 4 mW) (Spectris PLC, Surrey, England). Data were analyzed with Malvern software version 7.12. Aliquots of 50 μL of the supernatant from the aqueous phase were diluted (1:20) with 10% FBS in PBS and transferred to disposable polystyrol/polystyrene cuvettes (REF: 67.754 10 × 10 × 45 mm, Sarsted, Germany). The refractive index of ZOLE in water is 1.333. This value was determined by measuring an aliquot of 2.5 mg mL−1 ZOLE stock solution with a Mettler Toledo Refracto 30GS (Mettler Toledo, Columbus, OH).
For the aggregation measurements, the same procedure was followed for sample preparation. The prepared sample remained undisturbed near the zetasizer for 30 min prior to measurements. Size measurements were performed in the dispersant after 0, 24 and 48 h of sample preparation. Sample equilibration inside the instrument at room temperature (25 °C) was performed for 2 min before measurements.
Hydroxyapatite (HA) binding assay
For the binding assay of nano-Ca@ZOLE, 20 mg of HA was exposed to 3 mL of a nano-Ca@ZOLE in PBS solution (0.5 mg mL−1), for 0–11 d at 37 °C. As control groups, nano-Ca@ZOLE and HA, both in PBS, were employed. As a comparative method, the binding assay for “as received” ZOLE was performed employing the same parameters as for the nanocrystals. For the experimental groups (HA–nano-Ca@ZOLE and HA–ZOLE), collection was performed in duplicate. Supernatants of the control and experimental groups were collected every day for 11 consecutive days. After each time point, the supernatant was collected and centrifuged (1500 rpm, 8 min). Absorbance measurements were performed at 208 nm (λmax) to determine the percentage of ZOLE and nano-Ca@ZOLE bound to HA. Solid samples of HA, HA–ZOLE, and HA–nano-Ca@ZOLE were characterized by EDS.Cell culture methods
The MDA-MB-231 cell line was incubated with DMEM, 1% Pen-Strep, and 10% FBS at 37 °C in 5% CO2. The hFOB 1.19 cell line was incubated with 1:1 DMEM:F-12, 0.3 mg mL−1 G418, and 10% FBS at 34 °C in 5% CO2. Cell passages were observed weekly at 80% of cell confluency, and media were exchanged twice a week.
Cell treatments
Both cell lines were treated with ZOLE (control) and nano-Ca@ZOLE (experimental). First, to determine the half-maximal inhibitory concentration (IC50) two-fold serial dilutions of ZOLE (0–200 μM) were prepared. Both cell lines (MDA-MB-231 and hFOB 1.19) were seeded in 96 well plates at 2.5 × 105 cell per mL. The cells were incubated for 24 h at 37 °C (MDA-MB-231) and 34 °C (hFOB 1.19), respectively. After the initial incubation period, both cell lines were treated with 100 μL of the ZOLE solutions previously prepared and incubated for 24, 48, and 72 h at the respective incubation temperatures. For both cell lines, the corresponding culture media (DMEM, Pen-Strep:MDA-MB-231) and (DMEM, F-12, G418:hFOB 1.19) was used as negative control for treatments. AlamarBlue® assay was utilized to determine cell proliferation; for this, 10% of AlamarBlue® solution in PBS was prepared. Finally, the media were removed from the 96 well plates, 100 μL of 10% AlamarBlue® solution was added, and the cells were incubated for 4 h under the same previously mentioned conditions. After the AlamarBlue® assay, the fluorescence (λexc = 560 nm, λem = 590 nm) was evaluated using an Infinite M200 PRO Tecan Microplate Reader. The live cells were assessed comparing the viability of the control group (100%) with the cells treated with the ZOLE solutions. The nonlinear regression method using Graph Pad Prism, version 9.1.2, was applied to fit the dose–response curves (% cell live vs. concentration) to the IC50 values.The percentage of relative cell live (%RCL) for ZOLE (control) and nano-Ca@ZOLE (experimental) was investigated at selected concentrations (1.9, 3.8, 7.5, and 15 μM) in both cell lines. Treatments at these concentrations were carried out at 24, 48, and 72 h for ZOLE and nano-Ca@ZOLE. The cell seeding and AlamarBlue® assay were completed as described above for the IC50 determination in both cell lines. Graph Pad Prism, version 9.1.2, was utilized to plot %RCL found at concentrations of 1.9, 3.8, 7.5, and 15 μM after 24, 48, and 72 h of treatment. All experiments were performed in triplicates and the data were statistically treated using mean, standard deviation, and the coefficient of variation percentage (%CV).
Results and discussion
After exploring the design space for synthesizing ZOLE-based BPCCs, it was found that most of the crystalline materials formed while employing a 1:1 M2+/BP molar ratio at 85 °C and under acidic conditions (pH < 4.40). Concomitant polymorphism was observed in all the hydrothermal reactions between ZOLE and the metals. Phase selection was achieved by varying the anion of the metal salt (NO3−vs. Cl−), adding etidronate (HEDP) as an auxiliary ligand to decrease the pH (pH = 0.93) below the pKa's of the ligand (pH = 1.23–4.40), or applying cooling crystallization once the reaction at 85 °C was complete. A scheme for the hydrothermal syntheses of the ZOLE-based BPCCs is presented in Fig. 1.
Six coordination complexes were obtained with high crystal quality for structural elucidation, as seen under polarized light (Fig. 1). For these ZOLE-based BPCCs, their solid-state characterization, stability in physiological media, particle size and aggregation tendency were assessed to potentiate their biomedical applications as a nanocrystal-based therapy against OM.
Raman spectroscopy analysis
The representative Raman spectra of the isolated ZOLE-based BPCCs were collected from 3400 to 100 cm−1 (Fig. 2). This analysis confirmed that six distinctive crystalline phases were produced by the presence and absence of different Raman shifts among the ligand (ZOLE) and the ZOLE-based BPCCs spectra. Two different intense characteristic signals can be observed in the 3200–2900 cm−1 region of the ZOLE-based BPCCs spectra compared to ZOLE. These bands correspond to the hydrogen phosphate H–OPO2C moieties (3000–2900 cm−1) and the stretching vibrations νO–H/H2O (3200–3100 cm−1) due to coordinated and lattice water molecules in the crystal structure of the resulting complexes. Compared to ZOLE, the increased intensity of the signal at ∼3,000 cm−1 for the coordination complexes corroborates the presence of various strong hydrogen bonds within each lattice. This suggests that extensive hydrogen bonding is present within the crystal structures of the ZOLE-based BPCCs. Moreover, the band at 1290 cm−1 observed in the ZOLE-based BPCC spectra can be attributed to the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O deformation vibration due to coordination with the metal center. The signal at around 1190 cm−1 is characteristic of νPO/δπPOH stretching vibrations. The incorporation of ZOLE in the coordination sphere of the resulting materials is confirmed by two bands at 1100 cm−1 (strong) and 1020 cm−1 (medium), respectively. This strong signal is characteristic of the νasP–O(H) asymmetric stretching vibrations, while the medium signal corresponds to the δPO–H bending of the phosphonate P–O3 groups. These signals are scarcely observed for ZOLE. Similarly to the asymmetric stretching vibration of the P–O bonds, the strong bands at the 950–900 cm−1 region of the ZOLE-based BPCCs spectra are attributed to the symmetric νsP–O(H) stretching vibrations. For ZOLE, these bands are weak in intensity. Different vibrational modes of coordination of the divalent metal ions (M2+) with phosphorus bonded oxygen atoms induce changes in the P–O bond order, generating the differences observed in the symmetric and asymmetric P–O(H) stretching vibrations among the BPCCs and the ligand. Some Raman shifts can be observed at a lower wavenumber (<1000 cm−1), which are assigned to vibrational mode characteristics of the CH2, C–C, C–P, C–OH and M2+–O groups present in the ZOLE-based BPCCs.
O deformation vibration due to coordination with the metal center. The signal at around 1190 cm−1 is characteristic of νPO/δπPOH stretching vibrations. The incorporation of ZOLE in the coordination sphere of the resulting materials is confirmed by two bands at 1100 cm−1 (strong) and 1020 cm−1 (medium), respectively. This strong signal is characteristic of the νasP–O(H) asymmetric stretching vibrations, while the medium signal corresponds to the δPO–H bending of the phosphonate P–O3 groups. These signals are scarcely observed for ZOLE. Similarly to the asymmetric stretching vibration of the P–O bonds, the strong bands at the 950–900 cm−1 region of the ZOLE-based BPCCs spectra are attributed to the symmetric νsP–O(H) stretching vibrations. For ZOLE, these bands are weak in intensity. Different vibrational modes of coordination of the divalent metal ions (M2+) with phosphorus bonded oxygen atoms induce changes in the P–O bond order, generating the differences observed in the symmetric and asymmetric P–O(H) stretching vibrations among the BPCCs and the ligand. Some Raman shifts can be observed at a lower wavenumber (<1000 cm−1), which are assigned to vibrational mode characteristics of the CH2, C–C, C–P, C–OH and M2+–O groups present in the ZOLE-based BPCCs.
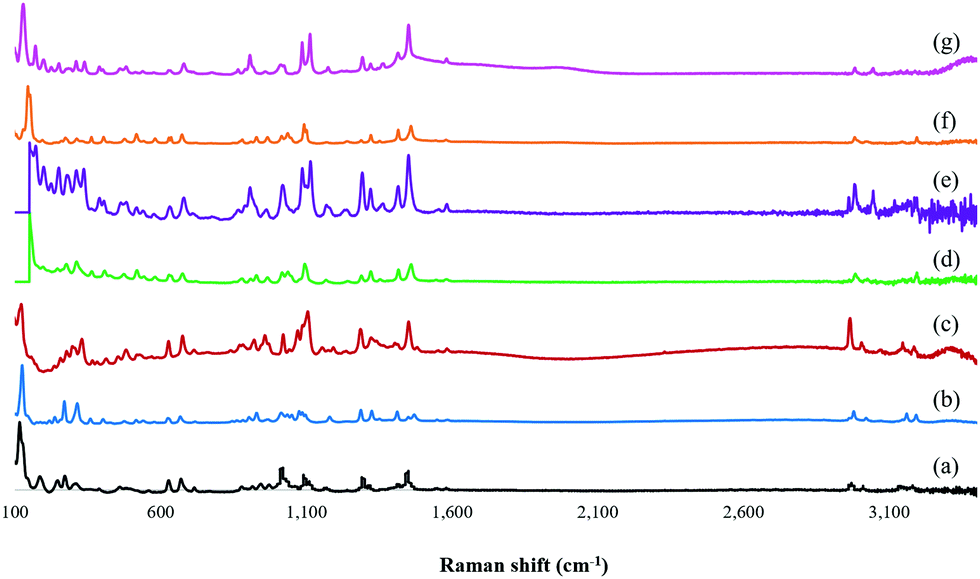 | ||
| Fig. 2 Raman spectra overlay of (a) zoledronate (ZOLE) and ZOLE-based BPCCs; (b) ZOLE–Ca form I, (c) ZOLE–Ca form II, (d) ZOLE–Mg form I, (e) ZOLE–Mg form II, (f) ZOLE–Zn form I, and (g) ZOLE–Zn form II. | ||
Scanning electron microscopy-energy dispersive spectroscopy (SEM-EDS)
The representative SEM images collected for the isolated ZOLE-based BPCCs show crystals with well-defined morphologies (Fig. 3). As observed in Fig. 3, ZOLE-based BPCC form I crystallizes in a prismatic crystal habit, while the ZOLE-based BPCC form II is characterized by the acicular crystal habit. Interestingly, SEM analysis revealed that ZOLE–Zn form II crystallizes in a hollowed acicular crystal habit, which can be observed in Fig. 3f. The diameter of the BPCCs obtained in this study ranges between 10 and 200 μm. | ||
| Fig. 3 Representative scanning electron micrographs and energy dispersive spectra of single crystals of ZOLE-based BPCCs; (a) ZOLE–Ca form I, blue; (b) ZOLE–Ca form II, red; (c) ZOLE–Mg form I, green; (d) ZOLE–Mg form II, purple; (e) ZOLE–Zn form I, orange; and (f) ZOLE–Zn form II, pink. | ||
Representative EDS spectra of these materials present the characteristic signals of the metal and other elements, which are present in the molecular structure of ZOLE (carbon, nitrogen, phosphorous, and oxygen atoms), and had been employed for hydrothermal synthesis (Fig. 3). Thus far, the synthesis of six crystalline phases distinct from the starting materials is supported with these results, along with the Raman spectra analysis.
Powder X-ray diffraction (PXRD) analysis
The representative PXRD diffractograms of the six ZOLE-based BPCCs are shown in Fig. 4. Each X-ray diffractogram reveals a high degree of crystallinity for the isolated materials due to the low amorphous background. Additionally, it was noticed that each phase presents a unique crystal structure compared to the starting materials employed during the syntheses, thus discarding the possibility of concomitant recrystallization of the ligand (ZOLE) and the metal salt. Interestingly, the crystal phases that presented a prismatic crystal habit (all form I of the ZOLE-based BPCCs), resulted in the formation of three isostructural materials, that are distinct to the other polymorphs described within this work. In contrast, the diffractogram of other ZOLE-based BPCCs (forms II) are structurally distinct, despite presenting the same acicular crystal habit. The presence of high angle peaks (>5° in 2θ) suggests that these materials are composed of 2D layers instead of 3D porous networks.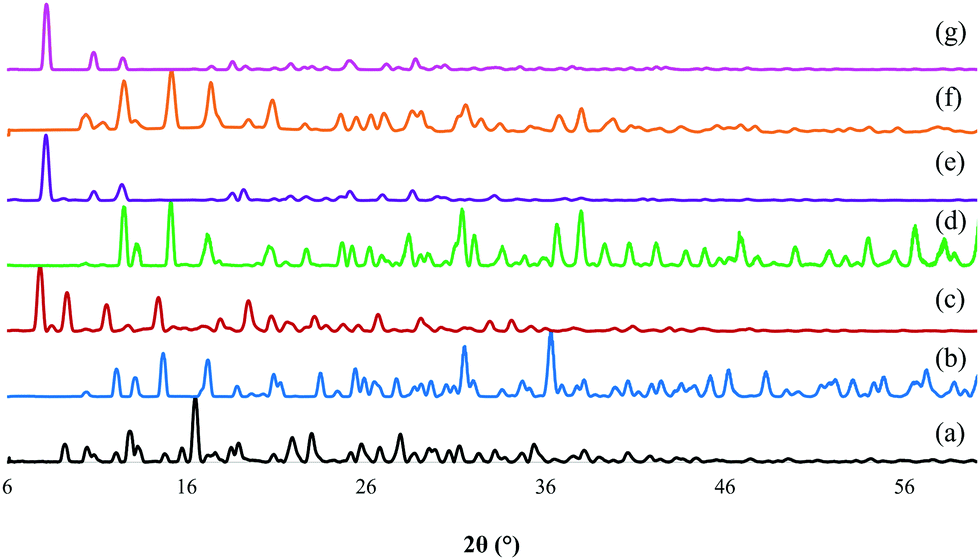 | ||
| Fig. 4 PXRD overlay of (a) zoledronate (ZOLE) and ZOLE-based BPCCs; (b) ZOLE–Ca form I, (c) ZOLE–Ca form II, (d) ZOLE–Mg form I, (e) ZOLE–Mg form II, (f) ZOLE–Zn form I, and (g) ZOLE–Zn form II. | ||
Previously reported structures of coordination complexes containing ZOLE were compared to the ones described within this work.32–36 ZOLE–Ca forms I and II resulted in unique materials compared to a previously reported phase containing the same metal as demonstrated by the overlay of their simulated PXRDs (ESI†).33 Although known, the structures of ZOLE–Mg form I and ZOLE–Zn form I were re-determined to increase the quality of structural refinement (ESI†).33,34 Moreover, both ZOLE–Mg form I and ZOLE–Zn form I resulted in an isostructural to those presented by ZOLE–Co, ZOLE–Mn and ZOLE–Ni which have been reported in literature (ESI†).35,36 In addition to ZOLE–Zn forms I and II reported here, another ZOLE–Zn coordination complex has been previously described.32 Additionally, ZOLE–Zn form II and ZOLE–Mg form II resulted in unique materials that have not been previously described.
Single crystal X-ray diffraction (SCXRD) analysis
Crystal structure elucidation performed by SCXRD provided unambiguous evidence of the formation of the six ZOLE-based BPCCs. Crystal structures were collected at a low temperature (100 K) and solved using direct methods. The crystallographic parameters of the structure refinement for each crystalline phase are summarized in Table 1, and their respective packing motifs are shown in Fig. 5.| Compound | ZOLE–Ca | ZOLE–Mg | ZOLE–Zn | |||
|---|---|---|---|---|---|---|
| Form I | Form II | Form I | Form II | Form I | Form II | |
| Abbreviations: λ (X-ray source wavelength, Å), a/b/c (unit cell lengths, Å), α/β/γ (unit cell angle, °), V (unit cell volume, Å3), Z (number of formula units per unit cell), ρcalc (unit cell calculated density, g cm−3), Rwp (weighted R-factor, %), and Rp (R-factor, %). | ||||||
| Empirical formula | [Ca(C10H18N4O14P4)(H2O)2] | [Ca(C10H18N4O14P4)]·3H2O | [Mg(C10H18N4O14P4)(H2O)2] | [Mg(C10H18N4O14P4)]·4H2O | [Zn(C10H18N4O14P4)(H2O)2] | [Zn(C10H18N4O14P4)]·2H2O |
| FW (g mol−1) | 618.27 | 636.29 | 602.51 | 638.54 | 643.59 | 679.60 |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Pbca |
P |
P21/n |
P |
P21/n |
| Temperature (K) | 100 (10) | 100 (10) | 100 (4) | 100 (10) | 100 (0) | 101 (1) |
| λ (Å) | 1.54184 | 1.54184 | 1.54184 | 1.54184 | 0.71073 | 1.54184 |
| a (Å) | 7.3520(2) | 10.2301(2) | 7.4070(2) | 4.96400(10) | 7.3868(5) | 4.96680(10) |
| b (Å) | 8.9793(2) | 20.8833(5) | 8.40350(10) | 16.3671(3) | 8.4456(5) | 16.4043(3) |
| c (Å) | 9.5484(2) | 22.4723(5) | 9.7306(2) | 14.4253(3) | 9.7191(6) | 14.4177(4) |
| α (°) | 105.677(2) | 90 | 105.249(2) | 90 | 105.3330(10) | 90 |
| β (°) | 108.739(2) | 90 | 111.368(2) | 93.241(2) | 111.2460(10) | 93.452(2) |
| γ (°) | 97.937(2) | 90 | 97.085(2) | 90 | 97.4060(10) | 90 |
| V (Å3) | 556.97(2) | 4801.0(2) | 527.74(2) | 1170.13(4) | 527.70(6) | 1172.58(5) |
| Z | 1 | 8 | 2 | 2 | 2 | 2 |
| ρ calc (g cm−3) | 1.843 | 1.761 | 1.896 | 1.812 | 2.025 | 1.925 |
| R wp | 0.0609 | 0.1211 | 0.0732 | 0.3380 | 0.0787 | 0.1321 |
| R p | 0.0224 | 0.0448 | 0.0272 | 0.1210 | 0.0304 | 0.0511 |
 | ||
| Fig. 5 Packing motifs of (a) ZOLE–Ca form I along the b-axis, (b) ZOLE–Ca form II along the a-axis, (c) ZOLE–Mg form I along the b-axis, (d) ZOLE–Mg form II along the a-axis, (e) ZOLE–Zn form I along the b-axis, and (f) ZOLE–Zn form II along the a-axis. | ||
Additional unit cell packings, asymmetric units and Oak Ridge Thermal Ellipsoid Plots (ORTEPs) can be accessed in the ESI.† PXRD overlays of the simulated and experimental powder patterns for each ZOLE-based BPCC corroborate that a representative solution has been obtained (ESI†). Crystallographic data have been submitted to the Cambridge Crystallographic Data Center (CCDC) for ZOLE–Ca form I (2120919), ZOLE–Ca form II (2120918), ZOLE–Mg form I (2120917), ZOLE–Mg form II (2120916), ZOLE–Zn form I (2120920), and ZOLE–Zn form II (2120915).†
Structural description of ZOLE–Ca form I
The compound [Ca(C10H18N4O14P4)(H2O)2] crystallizes in the space group P, containing a half molecule in the asymmetric unit with a calcium atom located in an inversion center. A distorted CaO6 octahedron (supplementary angles: O1–Ca1–O7, 83.29°, O1–Ca1–O8, 94.50°, and O8–Ca1–O7, 80.71°) is bonded to four phosphonate oxygen atoms from two distinct bidentate ZOLE ligands. Water molecules occupy the remaining two axial sites. The phosphonate oxygen atoms are coordinated to the equatorial positions of the octahedral Ca2+ center forming two identical six-membered chelate rings. The Ca–O bond distances are 2.322 Å (Ca1⋯O1) and 2.286 Å (Ca⋯O7). The conformation of the ligand is reinforced by intramolecular hydrogen bonds (O6⋯O8, 2.899 Å and O2⋯O6, 2.960 Å). The ZOLE molecules are linked into molecular chains that propagate tilted along the b-axis through strong intermolecular hydrogen bonds (O6⋯O5, 2.539 Å) between the oxygen atoms from the phosphonate moieties. The ZOLE molecules are linked into molecular chains that propagate tilted along the a-axis through intermolecular hydrogen bonds (O8⋯O5, 2.677 Å), between the coordinated water molecule and the oxygen atom from the phosphonate moiety. Adjacent chains are linked by a single intermolecular hydrogen bond (N2⋯O1, 2.710 Å) that propagates these chains along the bc-plane. This represents a unique packing mode when compared to the other ZOLE–Ca metal complexes previously reported in the literature (ESI†).33
Structural description of ZOLE–Ca from II
The compound [Ca(C10H18N4O14P4)]·3H2O crystallizes in the Pbca space group, and it is completely distinct from any other ZOLE–Ca metal complexes previously reported.33 The asymmetric unit has two ZOLE molecules coordinated to a Ca2+ center, surrounded by three uncoordinated water molecules. The Ca2+ center is in a distorted octahedral environment (supplementary angles: O1–Ca1–O4, 80.88°, O1–Ca1–O3, 86.29°, and O4–Ca1–O3, 80.42°), with four ZOLE ligands coordinated. Two different binding modes are observed for the ZOLE molecules. One ZOLE ligand is coordinated to the Ca2+ cation in a bidentate mode alternating oxygen from the bisphosphonate group. The Ca–O bond distances range between 2.296 and 2.375 Å. The metal cluster is linked by a single ligand coordinated to form a chain (Ca1–O4–P2–O5–Ca1) that propagates slightly tilted along the a-axis. This chain is reinforced by intermolecular hydrogen bonds (O4⋯O9, 2.586 Å; O5⋯O13, 2.636 Å; and O5⋯O11, 3.020 Å). Adjacent chains are linked by uncoordinated water molecules forming hydrogen bonds along the b-axis (O16⋯O12, 2.643 Å and O17⋯O10, 2.643 Å). An additional intermolecular hydrogen bond is formed through the b-axis, which involves the nitrogen from an imidazole group and the oxygen from an adjacent phosphonate moiety (N1⋯O11, 2.743 Å). An extensive network of intermolecular hydrogen bonds facilitated by the uncoordinated water molecules, serves to propagate the chain along the c-axis (O14⋯O15, 2.890 Å; O15⋯O16, 2.702 Å; O16⋯O7, 2.812 Å; and N3⋯O10, 2.657 Å).Structural description of ZOLE–Mg form I
The structure of the compound [Mg(C10H18N4O14P4)(H2O)2], which crystallizes in the P space group, has been previously reported at 294 K.33 The crystal structure was re-determined at 100 K, and the R-factor was improved (5.23% vs. 2.72%).33 Two bidentate ZOLE ligands and two water molecules are coordinated to a Mg2+ atom in an inversion center. The octahedral Mg2+ center equatorial positions are occupied by two bidentate ZOLE ligands, forming two identical six-membered chelate rings. In the remaining axial positions, water molecules are coordinated. A rather regular octahedral environment is observed for the metal cation (supplementary angles: O1–Mg1–O5, 89.66°, O5–Mg1–O8, 88.21°, and O1–Mg1–O8, 94.14°). The equatorial Mg–O bond distances are 2.033 Å (Mg1–O1) and 2.078 Å (Mg1–O5), while the axial Mg–O bond distance is 2.097 Å (Mg1–O8). The compound is reinforced by a single intramolecular hydrogen bond (O3⋯O7, 2.974 Å). The ZOLE ligands are linked by several intermolecular hydrogen bonds, the strongest of which (O3⋯O2, 2.583 Å) propagates the metal cluster laterally along the b-axis forming chains reinforced by additional hydrogen bonds (O4⋯O6, 2.868 Å; and O7⋯O6, 2.660 Å). A single intermolecular hydrogen bond (N2⋯O5, 2.724 Å) links adjacent chains and propagates these along the c-axis. Additional chains are linked by intermolecular hydrogen bonds along the a-axis (O8⋯O2, 2.726 Å). Interestingly, ZOLE–Mg form I did not incorporate the auxiliary ligand (HEDP) into its crystal lattice. This material is isostructural to other ZOLE metal complexes containing divalent metals such as Co(II),36 Mn(II),35 Ni(II),36 and Zn(II).34
Structural description of ZOLE–Mg form II
The compound [Mg(C10H18N4O14P4)]·4H2O crystallizes in the P21/n space group. The asymmetric unit has one ZOLE molecule coordinated to a Mg2+ center, surrounded by two uncoordinated water molecules. A regular octahedral environment is observed for the Mg2+ cation (supplementary angles: O1–Mg1–O5, 90.55°, O1–Mg1–O6, 90.58°, and O5–Mg1–O6, 94.76°), with Mg–O bond distances ranging from 2.037 to 2.122 Å. Intramolecular hydrogen bonds reinforce the metal cluster (O5⋯O1, 2.873 Å; O4⋯O6, 2.634 Å; O1⋯O6, 2.932 Å; and O5⋯O6, 2.577 Å). The ligand is coordinated to the metal center in a bidentate mode (O1–Mg1–O5), forming a six-membered chelate ring. The same ligand is coordinated to the Mg2+ center in a monodentate mode (Mg1–O6). Coordination between the Mg2+ cation and O6 and O5 from the ZOLE molecule results in the formation of an eight-membered chelate ring that fuses adjacent metal centers, resulting in the propagation of molecular chains through the a-axis. An extensive hydrogen bond network reinforces the chains along the b-axis connecting adjacent chains through the imidazole groups of the ligand and the uncoordinated lattice water (N2⋯O5, 2.770 Å; N2⋯O1, 2.985 Å; O2⋯O8, 2.760 Å; O8⋯O4, 2.927 Å; and O8⋯O7, 2.713 Å). Propagation of the metal cluster though the c-axis can be described by the presence of a c glide plane symmetry element, which is perpendicular to b [0, 1, 0] with a glide component [1/2, 0, 1/2]. Intermolecular hydrogen bonds between the two uncoordinated lattice waters, link the chains through the c-axis (O8⋯O9, 2.862 Å and O9⋯O9, 2.810 and 2.845 Å). ZOLE–Mg form II presents a unique packing arrangement when compared to other ZOLE–Mg metal complexes that have been previously described.33Structural description of ZOLE–Zn form I
The structure [Zn(C10H18N4O14P4)(H2O)2] was reported by E. Freire et al. at 293 K, but was re-determined at 100 K with an improvement in the R-factor (5.24% vs. 3.04%).34 The ZOLE–Zn form I BPCC is isostructural to ZOLE–Mg form I33 also re-determined within this work and to other reported ZOLE metal complexes with divalent metals such as Co(II),36 Mn(II),35 and Ni(II).36 The compound crystallizes in the![[P with combining macron]](https://www.rsc.org/images/entities/i_char_0050_0304.gif) space group. The Zn2+ center is coordinated by two bidentate ZOLE ligands and two water molecules in a regular octahedral environment (supplementary angles: O1–Zn1–O5, 90.82°, O5–Zn1–O8, 93.09°, and O1–Zn–O8, 85.45°). There is one half-molecule in the asymmetric unit with the zinc atom located in an inversion center. The phosphonate oxygen atoms are bound to the equatorial positions of the octahedral Zn2+ center forming two identical six-membered chelate rings. Water molecules occupy the remaining two sites of the octahedra (axial positions). The equatorial Zn–O bond distances are 2.042 Å (Zn1–O1) and 2.093 Å (Zn1–O5), while the bond distance of the axial position is 2.130 Å (Zn1–O8). The metal clusters are linked into a chain that propagates laterally through intermolecular hydrogen bonds (O3⋯O2, 2.580 Å; O4⋯O6, 2.866 Å; and O7⋯O6, 2.653 Å) expanding along the b-axis. Adjacent chains are linked by intermolecular hydrogen bonds along the a-axis (O8⋯O2, 2.719 Å). The coordinated water present in ZOLE–Zn form I is responsible for reinforcing this packing through a unique intermolecular hydrogen bond with an adjacent phosphonate moiety along the a-axis (O8⋯O3, 3.009 Å). Adjacent chains are linked by another unique hydrogen bond (N2⋯O5, 2.708 Å) that expands this chain along the c-axis. The packing is reinforced by an intramolecular hydrogen bond between oxygens of different phosphonate moieties of the same ZOLE molecule (O3⋯O7, 2.974 Å) along the c-axis.
space group. The Zn2+ center is coordinated by two bidentate ZOLE ligands and two water molecules in a regular octahedral environment (supplementary angles: O1–Zn1–O5, 90.82°, O5–Zn1–O8, 93.09°, and O1–Zn–O8, 85.45°). There is one half-molecule in the asymmetric unit with the zinc atom located in an inversion center. The phosphonate oxygen atoms are bound to the equatorial positions of the octahedral Zn2+ center forming two identical six-membered chelate rings. Water molecules occupy the remaining two sites of the octahedra (axial positions). The equatorial Zn–O bond distances are 2.042 Å (Zn1–O1) and 2.093 Å (Zn1–O5), while the bond distance of the axial position is 2.130 Å (Zn1–O8). The metal clusters are linked into a chain that propagates laterally through intermolecular hydrogen bonds (O3⋯O2, 2.580 Å; O4⋯O6, 2.866 Å; and O7⋯O6, 2.653 Å) expanding along the b-axis. Adjacent chains are linked by intermolecular hydrogen bonds along the a-axis (O8⋯O2, 2.719 Å). The coordinated water present in ZOLE–Zn form I is responsible for reinforcing this packing through a unique intermolecular hydrogen bond with an adjacent phosphonate moiety along the a-axis (O8⋯O3, 3.009 Å). Adjacent chains are linked by another unique hydrogen bond (N2⋯O5, 2.708 Å) that expands this chain along the c-axis. The packing is reinforced by an intramolecular hydrogen bond between oxygens of different phosphonate moieties of the same ZOLE molecule (O3⋯O7, 2.974 Å) along the c-axis.
Structural description of ZOLE–Zn form II
The compound [Zn(C10H18N4O14P4)]·2H2O crystallizes in the P21/n space group. The asymmetric unit has one ZOLE molecule coordinated to a Zn2+ center, surrounded by two uncoordinated water molecules. The Zn2+ center is in a rather regular octahedral environment (supplementary angles: O1–Zn1–O4, 91.06°, O4–Zn1–O3, 89.40°, and O1–Zn1–O3, 85.72°) with Zn–O bond distances ranging from 2.062 to 2.152 Å. The metal centers coordinate the ligand in a bidentate mode (O1–Zn1–O4) forming a six-membered chelate ring. The same metal center coordinates to the ligand in a monodentate mode (Zn1–O3). The coordination between the Zn2+ cation and O1 and O3 from the ZOLE molecule results in the formation of an eight-membered chelate ring that fuses adjacent metal centers, forming a chain that propagates through the a-axis. This chain is additionally reinforced by intramolecular hydrogen bonds (O1⋯O3, 2.867 Å; and O3⋯O4, 2.966 Å). These chains propagate along the b-axis through an extensive network of hydrogen bonds that form either with the imidazole group or the uncoordinated water molecules (N2⋯O4, 2.972 Å; N2⋯O1, 2.780 Å; O6⋯O8, 2.764 Å; O2⋯O8, 2.721 Å; and O8⋯O7, 2.943 Å). Propagation of the metal cluster though the c-axis can be described by the presence of a c glide plane symmetry element, which is perpendicular to b [0, 1, 0] with a glide component [1/2, 0, 1/2]. Chains that propagate through the c-axis are linked by extensive intermolecular hydrogen bonds between two uncoordinated water molecules (O8⋯O9, 2.868 Å and O9⋯O9, 2.819 and 2.845 Å). Moreover, the metal cluster is reinforced by intramolecular hydrogen bonds (O1⋯O4, 2.890 Å; and O7⋯O3, 2.633 Å) also through the c-axis. ZOLE–Zn form II presents a unique packing arrangement when compared to other ZOLE–Zn metal complexes that have been previously described.32,34Thermogravimetric analysis (TGA)
TGA thermographs of the BPCCs were compared to those of the ligand (ZOLE). Based on the resulting thermographs, all ZOLE-based BPCCs presented higher thermal stability than the ligand (ESI†). ZOLE–Ca form I, ZOLE–Mg form I, and ZOLE–Zn form I present higher thermal stability than ZOLE–Ca form II, ZOLE–Mg form II, and ZOLE–Zn form II maybe due to the presence of coordinated water molecules. In ZOLE–Ca form II, ZOLE–Mg form II, and ZOLE–Zn form II, lattice water molecules require minimal thermal energy for their desolvation to be observed. Most of the ZOLE-based BPCCs are stable up to 100–200 °C, where a minor loss of coordinated and lattice water molecules can be observed. Above 250–300 °C a major decomposition of the ligand was observed for each BPCC and ZOLE. As expected, a minor weight loss was observed at higher temperatures (>400 °C), which accounted for the thermal degradation of the metal/metal oxide.Dissolution profile measurements
The ZOLE (active ingredient of Reclast®) content released from the ZOLE-based BPCCs was quantified in neutral (PBS, pH = 7.40) and acidic (FaSSGF, pH = 1.60) physiological media, via direct UV-Vis spectroscopy quantification (λmax = 208 nm). The dissolution assays employed a maximum concentration of 0.05 mg mL−1 of ZOLE (ESI†), which corresponds to the clinically utilized dosage of this BP.14 Results from dissolution assays in PBS demonstrate that commercial ZOLE has a higher dissolution (100% in 30 min) than the ZOLE-based BPCCs. Most of the ZOLE-based BPCCs presented a lower dissolution and equilibrium solubility (60–85% in 18–24 h) than ZOLE in this media (Fig. 6a). Particularly, ZOLE–Zn form I presented a significant lower dissolution in PBS, reaching the maximum concentration of ZOLE (∼74%) after 6 d (ESI†). | ||
| Fig. 6 (a) Complete dissolution profile for ZOLE (black), ZOLE–Ca form I (blue), ZOLE–Ca form II (red), ZOLE–Mg form I (green), ZOLE–Mg form II (purple), ZOLE–Zn form I (orange), and ZOLE–Zn form II (pink) in PBS for 48 h (dashed line). The extended dissolution profile for ZOLE–Zn form I (orange) up to 192 h, showing the complete release of the ZOLE content. (b) Comparison of the complete dissolution profiles in two different physiological media for ZOLE in PBS (black) and FaSSGF (grey), as well for ZOLE–Ca form II in PBS (dark red) and FaSSGF (bright red), for 36 h (dashed line). | ||
To further investigate if the ZOLE-based BPCCs present a pH-dependent degradation, dissolution of ZOLE–Ca form II in FaSSGF was performed. From the dissolution assays in FaSSGF, the results demonstrate that commercial ZOLE (Reclast®) has a relatively similar dissolution in PBS (100%), but at a lower pH it reaches its maximum equilibrium solubility in 3 h (FaSSGF) rather than in 30 min (PBS). ZOLE–Ca form II presented higher dissolution and equilibrium solubility in acidic media (88% in 1 h), compared to its dissolution in PBS (83% in 24 h, Fig. 6b). The observed pH-dependent dissolution is desirable because it may allow ZOLE–Ca form II nanoparticles to circulate longer allowing them to maintain blood plasma concentrations for ZOLE and reach the target site. Once there, this material might undergo degradation due to the increased acidic microenvironment at the metastatic site.37–39 These results provide evidence of the structural stability of the ZOLE-based BPCCs in physiological media, and hint at their ability to degrade releasing the drug content (ZOLE) in a controlled and pH-dependent manner. Because these materials are not able to encapsulate drugs within their 2D structure, the degradation of the BPCC itself could provide the release of BPs at the metastatic site.
Phase inversion temperature (PIT)-nano-emulsion synthesis of nano-Ca@ZOLE
Particle size reduction of ZOLE–Ca form II was performed employing the PIT-nano-emulsion method. Selection of this particular BPCC was based on its favorable conditions for crystallization (i.e., absence of concomitant polymorphism), high thermal stability, and selective degradation in physiological media compared to other ZOLE-based BPCCs. After conductivity measurements were performed in triplicate, the average PIT was observed at ∼12 °C for this system. The hydrothermal synthesis of ZOLE–Ca form II was coupled to the PIT method, to decrease the particle size of this material to the nano-range (Fig. 7a).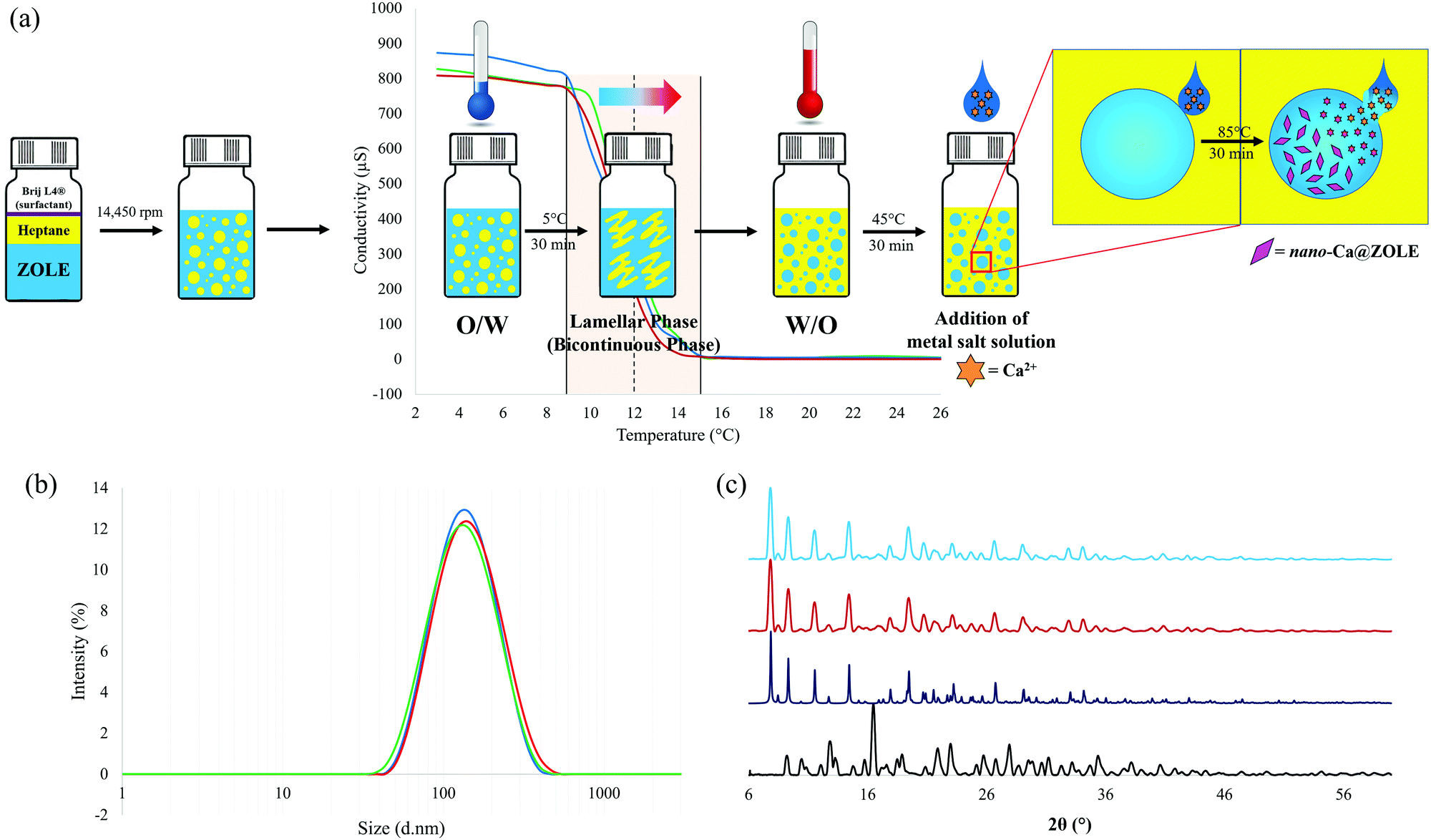 | ||
| Fig. 7 (a) Schematic diagram of the PIT-nano-emulsion synthesis of nano-Ca@ZOLE, showing phase inversion at a temperature of approximately ∼12 °C (dashed line). Phase inversion starts at ∼9 °C and ends at ∼15 °C (light orange region). (b) Dynamic light scattering (DLS) spectra showing average size distribution (∼150 d nm) of nano-Ca@ZOLE particles for the three replicate syntheses, and (c) PXRD overlay of ZOLE (black), ZOLE–Ca form II simulated powder pattern (navy blue), ZOLE–Ca form II bulk crystals (red), and agglomerated nanocrystals of nano-Ca@ZOLE (light blue). | ||
The presumed formation of nano-Ca@ZOLE occurred after performing the PIT-nano-emulsion synthesis. Once the reaction completed, the aqueous supernatant was analyzed by DLS to determine the particle size distribution of the resulting material. The DLS results demonstrate average particle size distribution and polydispersity index (PDI) values of 144.4 d nm (0.202), 146.3 d nm (0.202), and 155.2 d nm (0.206), respectively, for three replicate syntheses (Fig. 7b). PXRD analysis was carried out on a micron-sized agglomerate (ESI†) of the nanocrystals resulting from the PIT-nano-emulsion synthesis, to verify this crystal phase against the bulk material (ZOLE–Ca form II). PXRD analysis confirms that the crystal phase of the nano-Ca@ZOLE nanoparticles is isostructural to that of ZOLE–Ca form II bulk crystals (Fig. 7c). Therefore, the application of the PIT nano-emulsion method successfully decreases the crystal size from a micron-range (∼200 μm, Fig. 3b) to a nano-range (∼150 d nm, Fig. 7b), while maintaining the crystal phase of ZOLE–Ca form II.
Aggregation measurements of nano-Ca@ZOLE in the biorelevant dispersant
Aggregation measurements were performed under biologically relevant conditions (10% FBS:PBS), after 0, 24 and 48 h. This analysis can provide insights into the potential of nano-Ca@ZOLE to maintain its particle size (<500 nm) and the ability to deliver the ZOLE content when suspended in physiological media.40 The results demonstrate a homogeneous particle size distribution in 10% FBS:PBS after 0, 24 and 48 h of being synthesized. After being suspended in the media, nano-Ca@ZOLE presented particle size distribution values of 137.4, 175.5, and 176.9 d nm after 0, 24 and 48 h, respectively (ESI†). Furthermore, nano-Ca@ZOLE particles remained highly monodispersed along the three-time points, showing PDI values of 0.122 (0 h), 0.154 (24 h), and 0.159 (48 h) (ESI†). These results confirmed that nano-Ca@ZOLE has a low aggregation tendency under biologically relevant conditions (10% FBS:PBS), demonstrating that the nanomaterial can maintain its particle size without forming larger aggregates.Binding assays of nano-Ca@ZOLE to HA
The ability of nano-Ca@ZOLE to bind under simulated physiological conditions to the main constituent of the bone microenvironment, HA, was probed. The binding to HA was determined by monitoring the decrease in the ZOLE concentration of the supernatant using absorption measurements (λmax = 208 nm). Binding curves (Fig. 8a) demonstrate that 15% of ZOLE (control) binds to HA in 1 d and a maximum binding of 82% is reached in 8 d under simulated physiological conditions (PBS, pH = 7.40, and 37 °C). The binding curve results of nano-Ca@ZOLE demonstrate that the nanomaterial reaches its maximum binding of 36% to HA in 1 d which remains constant up to 11 d under the same conditions. These results provide evidence of the higher binding (∼2.5x) for nano-Ca@ZOLE compared to ZOLE within a relevant time frame. Here, it is suspected that the uncoordinated phosphate groups at the surface of the nanocrystals are responsible for binding to HA. | ||
| Fig. 8 (a) Binding curves of ZOLE (control, black) and nano-Ca@ZOLE (experimental, light blue) to HA in PBS, showing their maximum binding of 82% (black dashed line) and 36% (light blue dashed line), respectively. Error bars for duplicate measurements fall below a five percent (<5%) error. EDS analysis of (b) HA (control), (c) HA–ZOLE (control), and (d) HA–nano-Ca@ZOLE (experimental) after the binding assay. EDS spectra include the insertion of a schematic for the proposed binding of this material to the HA. | ||
Further characterization was performed to corroborate the binding of nano-Ca@ZOLE to HA. After the binding assay was completed, an elemental analysis performed by EDS confirmed the effective binding of ZOLE and nano-Ca@ZOLE to HA (Fig. 8b–d). The surface composition of HA (control) was compared with that of HA–ZOLE and HA–nano-Ca@ZOLE and contrasted with their respective weight percent (wt%, Table 2).
000x in all surface measurements
EDS analysis of HA [Ca5(OH)(PO4)3, control] corroborated the elemental composition of this mineral (Fig. 8a). To support the effective binding of both ZOLE (C5H10N2O7P2, control) and nano-Ca@ZOLE [Ca(C10H18N4O14P4)]·3H2O, experimental] to HA, it is expected to observe a significant difference in the relative concentration of calcium. For HA–ZOLE (34.17 wt%), the calcium concentration decreased significantly when compared to that for HA (42.73 wt%). This might be because ZOLE (C5H10N2O7P2) has been incorporated as a monolayer onto the surface of HA shielding detection (Fig. 8b). For HA–nano-Ca@ZOLE, the relative concentration of calcium (41.54 wt%) was higher than that for HA–ZOLE (34.17 wt%) but lower than that for HA (42.73 wt%). This can be attributed to the proposed binding of nano-Ca@ZOLE, which suggests the formation of layers of the nanocrystals on the HA surface that shield the detection of calcium ions (Fig. 8c). The difference in the relative concentration of calcium for HA–ZOLE and HA–nano-Ca@ZOLE is based on the molecular proportion of this metal in ZOLE (0 calcium atoms per formula unit) and nano-Ca@ZOLE (1 calcium atom per formula unit), resulting in a slight increase in the detection of calcium for the nanomaterial. A small increase in the phosphorous signals between the controls and the experimental groups was observed as a consequence of the similar composition of this element in these materials. The relative concentration of oxygen increased for both experimental (HA–nano-Ca@ZOLE) and control group (HA–ZOLE) compared to HA, suggesting the presence of ZOLE or nano-Ca@ZOLE bound to the surface of HA. A higher concentration of oxygen was observed for HA–ZOLE (35.11 wt%) when compared to HA–nano-Ca@ZOLE (30.38 wt%), because the ligand (82%) achieves a higher maximum binding when compared to the nanomaterial (36%) after 11 d. HA presents a carbon signal (7.14 wt%) that can be attributed to the conductive tape used for mounting the solid samples. The relative concentration of carbon for HA–ZOLE (11.73 wt%) and HA–nano-Ca@ZOLE (8.37 wt%) slightly increased due to the presence of this element in both materials.
Cytotoxicity assays of nano-Ca@ZOLE.
Here, human breast cancer MDA-MB-231 and the osteoblast-like hFOB 1.19 cell lines were selected to assess the cytotoxicity of nano-Ca@ZOLE. The MDA-MB-231 cell line represents a model of breast-cancer-induced OM as it possess micro-RNAs involved in the development of bone metastasis.3,41 While, the immortalized human fetal hFOB 1.19 cell line is a homogeneous model that allows the study of osteoblast differentiation; in this study, these cells were employed to imitate the normal human bone microenvironment.42 To determine the IC50 values against MDA-MB-231 and hFOB 1.19 cell lines, concentrations of 0–200 μM of ZOLE were employed. While the IC50 for the MDA-MB-231 cell line treated with ZOLE for 72 h was found to be 35 ± 4 μM, treatments at 24 and 48 h produced IC50 values > 200 μM (ESI†). These results demonstrate that ZOLE (0–200 μM) shows cytotoxicity after 72 h of treatment against the MDA-MB-231 cell line. The IC50 for the hFOB 1.19 cell line was >200 μM at 24 h. For treatments at 48 and 72 h, the IC50 values were determined to be 86 ± 3 and 49 ± 4 μM, respectively, indicating that ZOLE (0–200 μM) can cause cell death after 48 h of treatment in the osteoblast cells (ESI†).Furthermore, for both cell lines, the %RCL was investigated at concentrations of 1.9, 3.8, 7.5, and 15 μM for ZOLE (control) and nano-Ca@ZOLE (experimental) at 24, 48, and 72 h.
At a concentration of 1.9 μM, the cell viability decreased minimally for the MDA-MB-231 cell line when treated with the nanocrystals, in contrast to ZOLE where the cell viability was ∼100% (Fig. 9a). The %RCL values for MDA-MB-231 treated with nano-Ca@ZOLE at 3.8 μM decreased significantly to 83 ± 5% and 55 ± 1%, after 48 and 72 h, respectively (Fig. 9b). At this concentration, ZOLE did not cause cell death (%RCL ∼ 100%) against MDA-MB-231. In addition, higher cell growth inhibition of the cancerous model was observed with nano-Ca@ZOLE treatment at 7.5 μM, resulting in %RCL values of 57 ± 1 and 24 ± 2% after 48 and 72 h, respectively (Fig. 9c). Moreover, at 15 μM, nano-Ca@ZOLE presented a much higher cytotoxicity effect against MDA-MB-231 [%RCL: 50 ± 1 (48 h) and 18 ± 2% (72 h)], compared to the one observed for ZOLE at this concentration [%RCL: 80 ± 2 (48 h) and 58 ± 2% (72 h)] (Fig. 9d). These results demonstrate the potential of nano-Ca@ZOLE to induce significant cytotoxicity even at low concentrations (3.8–7.5 μM) against cells that are prone to metastasize to the bone. Surprisingly, nano-Ca@ZOLE is able to achieve significant cytotoxicity effects in vitro against the MDA-MB-231 cell line at concentrations ∼10–100x lower to the ones reported employing ZOLE.21 Additionally, compared to the commercial dosage of ZOLE (Reclast®, 0.05 mg mL−1), the concentrations employed for the nanocrystals are significantly lower.14
 | ||
| Fig. 9 Percentage of relative cell live (%RCL) for the human breast cancer MDA-MB-231 and normal osteoblast-like hFOB 1.19 cell lines, in green controls, ZOLE (black), and nano-Ca@ZOLE (red) at concentrations of (a and e) 1.9, (b and f) 3.8, (c and g) 7.5, and (d and h) 15 μM after 24, 48, and 72 h of treatment. | ||
Treatments were conducted with nano-Ca@ZOLE (experimental) and ZOLE (control) employing the hFOB 1.19 cell line, at the same concentrations used for the MDA-MB-231 assays. The cell viability results demonstrate that no significant cell death was observed after both ZOLE and nano-Ca@ZOLE treatments were performed in the concentration range of 3.8–15 μM after 24, 48, and 72 h. After 72 h of treating the osteoblast-like cells with the nanocrystals, the resulting %RCL values were 97 ± 2% at 1.9 μM (Fig. 9e), 100 ± 2% at 3.8 μM (Fig. 9f), 99 ± 3% at 7.5 μM (Fig. 9g), and 97 ± 2% at 15 μM (Fig. 9h). These results demonstrate that nano-Ca@ZOLE presents significant cytotoxicity against triple-negative breast cancer cells that metastasize to the bone (MDA-MB-231), without affecting negatively normal osteoblast cells (hFOB 1.19) at the metastatic site.
Conclusions
Herein, the hydrothermal syntheses of a series of ZOLE-based BPCCs were reported. These crystalline materials were synthesized by employing clinically utilized BP, ZOLE, as the ligand and three biologically relevant metals (Ca2+, Mg2+, and Zn2+). Six different ZOLE-based BPCCs were obtained as single phases and structurally characterized to provide further insights into the structural motifs observed in these types of materials. All ZOLE-based BPCCs presented higher thermal stability compared to ZOLE, as a result of the presence of coordination bonds and extensive intermolecular hydrogen bonding within their crystal lattices. The dissolution of the ZOLE-based BPCCs was compared to that of ZOLE, to assess the structural stability of these materials in two different simulated physiological media (PBS and FaSSGF). All ZOLE-based BPCCs presented lower dissolution and equilibrium solubility than ZOLE (60–85%, in 18–24 h) in PBS, thus remaining coordinated for a longer period under neutral physiological conditions. Meanwhile, the dissolution profile of the selected BPCC model ZOLE–Ca form II in FaSSGF revealed a higher dissolution and equilibrium solubility (88% in 1 h) in acidic physiological media when compared to PBS (83% in 24 h). These results suggest the ability of this material to release the drug content (ZOLE) in a controlled and pH-dependent manner. The PIT-nano-emulsion method decreased the crystal size of ZOLE–Ca form II significantly, from a micron-range (∼200 μm) to a nano-range (∼150 d nm), thus resulting in nano-Ca@ZOLE. The particle size decrease of nano-Ca@ZOLE presents several advantages towards the therapeutic applications of this BPCC, potentiating its use as a nanocrystal-based therapy. Furthermore, the aggregation behavior of nano-Ca@ZOLE in 10% FBS:PBS was investigated, which provided a further assessment of the potential of this nanomaterial to be employed for drug delivery. Nano-Ca@ZOLE presents a low aggregation behavior under biologically relevant conditions, after 0, 24 and 48 h. These results provide insights into the potential of the nanocrystals to maintain their particle size when in contact with different biological serum-like components without forming larger aggregates, possibly avoiding excretion through phagocytosis mechanisms during cellular uptake. Moreover, the binding affinity of this nanomaterial to HA was addressed to provide insights into its potential to bind at the bone, thus possibly enabling localized therapeutic effects at the metastatic site. The results showed that nano-Ca@ZOLE binds ∼2.5x more (36%) to HA than ZOLE (15%) in 1 d, demonstrating that it could bind to the main constituent of the bone microenvironment with higher affinity. This, along with the dissolution results, could suggest the possibility of the nanocrystals to maintain higher blood plasma concentrations of ZOLE and to degrade selectively at the metastatic site. Furthermore, the cytotoxicity of nano-Ca@ZOLE was compared to that of ZOLE against the human breast cancer MDA-MB-231 and normal osteoblast-like hFOB 1.19 cell lines. The results demonstrated significant cell growth inhibition for nano-Ca@ZOLE against the cancerous model after 72 h of treatment, specifically at a concentration of 3.8 μM (%RCL = 55 ± 1%). At this concentration, the nanocrystals did not present cytotoxicity against the normal osteoblastic cells (%RCL = 100 ± 2%). These results suggest the potential of this nanomaterial to treat cancerous cells that are prone to metastasize with minimal cell death in a model representing a healthy bone microenvironment. These important outcomes provide evidence that nano-ZOLE-based BPCCs possess viable characteristics in terms of structure, dissolution, stability, binding, and cytotoxicity, that render them suitable for OM therapy.Author contributions
Gabriel Quiñones Vélez: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing-original draft, and writing-review & editing. Lesly Carmona-Sarabia: data curation, formal analysis, investigation, methodology, and writing-original draft. Alondra A. Rivera Raíces: data curation, investigation, and methodology. Tony Hu: formal analysis. Esther Peterson: formal analysis and validation. Vilmalí López-Mejías: conceptualization, funding acquisition, investigation, methodology, project administration, resources, supervision, validation, visualization, and writing-review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was initially supported by the NIH–Research Initiative for Scientific Enhancement Grant No. 5R25GM061151-15. The authors thank the New York University's Materials Research Science and Engineering Center (NYU-MRSEC, DMR-1420073) for the opportunity to participate in the Student-Faculty Program during the summer of 2016 and their support in providing the infrastructure to complement these results. The crystal structure of ZOLE–Zn form I was elucidated in the X-ray diffraction facility at NYU (CRIF/CHE-0840277). Additionally, the authors gratefully acknowledge the Institutional Research Funds (FIPI Funds 2017–2019 and 2019–2021) of the University of Puerto Rico, Río Piedras Campus for financial support. The Rigaku XtaLAB SuperNova single crystal X-ray micro diffractometer was acquired through the support of the National Science Foundation under the Major Research Instrumentation Program (CHE-1626103). The present work was additionally funded by the National Institutes of Health, under the award 1SC2GM127223-01A1.The authors would also thank Ms. Sharon Fonseca Williams, cell and tissue culture specialist, for her support and insights regarding the cytotoxicity assays. Furthermore, special thanks to the personnel at the Materials Characterization Center (MCC) of the UPR-Molecular Sciences Research Center (MSRC) for their technical support allowing the collection of SEM-EDS data in this facility.References
- J. Ferlay, M. Colombet, I. Soerjomataram, C. Mathers, D. M. Parkin, M. Piñeros, A. Znaor and F. Bray, Estimating the Global Cancer Incidence and Mortality in 2018: GLOBOCAN Sources and Methods, Int. J. Cancer, 2019, 1941–1953, DOI:10.1002/ijc.31937.
- R. L. Siegel, K. D. Miller and A. Jemal, Cancer Statistics, Ca-Cancer J. Clin., 2019, 69(1), 7–34, DOI:10.3322/caac.21551.
- J. E. Welsh, Animal Models for Studying Prevention and Treatment of Breast Cancer, Animal Models for the Study of Human Disease, Elsevier Inc., 2013, pp. 997–1018 DOI:10.1016/B978-0-12-415894-8.00040-3.
- N. H. Hart, D. A. Galvão, C. Saunders, D. R. Taaffe, K. T. Feeney, N. A. Spry, D. Tsoi, H. Martin, R. Chee and T. Clay, et al., Mechanical Suppression of Osteolytic Bone Metastases in Advanced Breast Cancer Patients: A Randomised Controlled Study Protocol Evaluating Safety, Feasibility and Preliminary Efficacy of Exercise as a Targeted Medicine, Trials, 2018, 19(1), 1–15, DOI:10.1186/s13063-018-3091-8.
- G. R. Mundy, Metastasis to Bone: Causes, Consequences and Therapeutic Opportunities, Nat. Rev. Cancer, 2002, 584–593, DOI:10.1038/nrc867.
- M. Diba, W. A. Camargo, T. Zinkevich, A. Grünewald, R. Detsch, Y. Kabiri, A. P. M. Kentgens, A. R. Boccaccini, J. J. J. P. Van Den Beucken and S. C. G. Leeuwenburgh, Hybrid Particles Derived from Alendronate and Bioactive Glass for Treatment of Osteoporotic Bone Defects, J. Mater. Chem. B, 2019, 7(5), 796–808, 10.1039/c8tb03062f.
- D. Paolino, M. Licciardi, C. Celia, G. Giammona, M. Fresta and G. Cavallaro, Bisphosphonate-Polyaspartamide Conjugates as Bone Targeted Drug Delivery Systems, J. Mater. Chem. B, 2015, 3(2), 250–259, 10.1039/c4tb00955j.
- E. Guenin, D. Ledoux, O. Oudar, M. Lecouvey and M. Kraemer, Structure-Activity Relationships of a New Class of Aromatic Bisphosphonates That Inhibit Tumor Cell Proliferation in Vitro, Anticancer Res., 2005, 25(2A), 1139–1145 CAS.
- E. V. Giger, B. Castagner and J. C. Leroux, Biomedical Applications of Bisphosphonates, J. Control. Release, 2013, 167(2), 175–188, DOI:10.1016/j.jconrel.2013.01.032.
- P. Clezardin, F. H. Ebetino and P. G. Fournier, Bisphosphonates and Cancer-Induced Bone Disease: Beyond Their Antiresorptive Activity, J. Cancer Res., 2005, 65(12), 4971–4974 CrossRef CAS PubMed.
- A. Alasmari, S. C. Lin, S. Dibart and E. Salih, Bone Microenvironment-Mediated Resistance of Cancer Cells to Bisphosphonates and Impact on Bone Osteocytes/Stem Cells, Clin. Exp. Metastasis, 2016, 33(6), 563–588, DOI:10.1007/s10585-016-9798-6.
- J. H. Lin, Bisphosphonates: A Review of Their Pharmacokinetic Properties, Bone, 1996, 75–85, DOI:10.1016/8756-3282(95)00445-9.
- S. Khosla, J. P. Bilezikian, D. W. Dempster, E. M. Lewiecki, P. D. Miller, R. M. Neer, R. R. Recker, E. Shane, D. Shoback and J. T. Potts, Benefits and Risks of Bisphosphonate Therapy for Osteoporosis, J. Clin. Endocrinol. Metab., 2012, 2272–2282, DOI:10.1210/jc.2012-1027.
- A. Grey, Intravenous Zoledronate for Osteoporosis: Less Might Be More, Ther. Adv. Musculoskeletal Dis., 2016, 1, 119–123, DOI:10.1177/1759720X16650866.
- A. Majkowska, M. Neves, I. Antunes and A. Bilewicz, Complexes of Low Energy Beta Emitters 47Sc and 177Lu with Zoledronic Acid for Bone Pain Therapy, Appl. Radiat. Isot., 2009, 67(1), 11–13, DOI:10.1016/j.apradiso.2008.08.014.
- A. Grey, M. Bolland, B. Mihov, S. Wong, A. Horne, G. Gamble and I. R. Reid, Duration of Antiresorptive Effects of Low-Dose Zoledronate in Osteopenic Postmenopausal Women: A Randomized, Placebo-Controlled Trial, J. Bone Miner. Res., 2014, 29(1), 166–172, DOI:10.1002/jbmr.2009.
- A. Grey, M. Bolland, S. Wong, A. Horne, G. Gamble and I. R. Reid, Low-Dose Zoledronate in Osteopenic Postmenopausal Women: A Randomized Controlled Trial, J. Clin. Endocrinol. Metab., 2012, 97(1), 286–292, DOI:10.1210/jc.2011-2081.
- H. Katsumi, T. Mozume, S. I. Yanagi, T. Hasei, T. Watanabe, T. Sakane and A. Yamamoto, Pharmacokinetic and Therapeutic Efficacy of Intrapulmonary Administration of Zoledronate for the Prevention of Bone Destruction in Rheumatoid Arthritis, J. Drug Targeting, 2016, 24(6), 530–536, DOI:10.3109/1061186X.2015.1101768.
- A. De Luca, L. Lamura, M. Gallo, G. Daniele, A. D’Alessio, P. Giordano, M. R. Maiello, M. Pergameno, F. Perrone and N. Normanno, Pharmacokinetic Evaluation of Zoledronic Acid, Expert Opin. Drug Metab. Toxicol., 2011, 7(7), 911–918, DOI:10.1517/17425255.2011.585156.
- A. Varma, P. Laxmi, A. Pai, G. Pai, S. G. Vasantharaju and M. B. Sathyanarayana, Designing of Stable Co-Crystals of Zoledronic Acid Using Suitable Coformers, Chem. Pharm. Bull., 2019, 67(8), 816–823, DOI:10.1248/cpb.c19-00202.
- K. M. Au, A. Satterlee, Y. Min, X. Tian, Y. S. Kim, J. M. Caster, L. Zhang, T. Zhang, L. Huang and A. Z. Wang, Folate-Targeted PH-Responsive Calcium Zoledronate Nanoscale Metal-Organic Frameworks: Turning a Bone Antiresorptive Agent into an Anticancer Therapeutic, Biomaterials, 2016, 82, 178–193, DOI:10.1016/j.biomaterials.2015.12.018.
- J. Zekria, M. Mansour and S. M. Karim, The Anti-Tumour Effects of Zoledronic Acid, J. Bone Oncol., 2014, 3(1), 25–35, DOI:10.1016/j.jbo.2013.12.001.
- U. Varol, M. Degirmenci, B. Karaca, H. Atmaca, A. Kisim, S. Uzunoglu, C. Sezgin, U. A. Sanli and R. Uslu, Zoledronic Acid Increases Cytotoxicity by Inducing Apoptosis in Hormone and Docetaxel-Resistant Prostate Cancer Cell Lines, Tumor Biol., 2015, 36(2), 779–786, DOI:10.1007/s13277-014-2682-6.
- E. Algur, R. M. MacKlis and U. O. Häfeli, Synergistic Cytotoxic Effects of Zoledronic Acid and Radiation in Human Prostate Cancer and Myeloma Cell Lines, Int. J. Radiat. Oncol., Biol., Phys., 2005, 61(2), 535–542, DOI:10.1016/j.ijrobp.2004.09.065.
- C. Gokalp, Cytotoxic and Anti-Angiogenic Effects of Zoledronic Acid in DU-145 and PC-3 Prostate Cancer Cell Lines, Mol. Biol. Rep., 2020, 47(10), 7675–7683, DOI:10.1007/s11033-020-05840-6.
- W. Li, X. Xin, S. Jing, X. Zhang, K. Chen, D. Chen and H. Hu, Organic Metal Complexes Based on Zoledronate-Calcium: A Potential PDNA Delivery System, J. Mater. Chem. B, 2017, 5(8), 1601–1610, 10.1039/c6tb03041f.
- D. Liu, S. A. Kramer, R. C. Huxford-phillips, S. Wang, J. D. Rocca and W. Lin, Coercing Bisphosphonates to Kill Cancer Cells with Nanoscale Coordination Polymers, Chem Commun., 2012, 48, 2668–2670, 10.1039/c2cc17635a.
- M. Nikzad, A. R. Jalilian, S. Shirvani-Arani, M. Arabieh, S. Shanesazzadeh and H. Golchoobian, Development and Assessment of 153Sm-Zoledronate Complex As a Possible Bone Pain Palliative Agent, Iran. J. Nucl. Med., 2017, 25(249), 81–91 CAS.
- M. Nikzad, A. R. Jalilian, S. Shirvani-Arani, A. Bahrami Samani and H. Golchobian, Development of 166Ho-Zoledronate as a Bone Marrow Ablative Agent, Pharm. Biomed. Res., 2016, 2(1), 14–22, DOI:10.18869/acadpub.pbr.2.1.14.
- M. Nikzad, A. R. Jalilian, S. Shirvani-Arani, A. Bahrami-Samani and H. Golchoubian, Production, Quality Control and Pharmacokinetic Studies of 177Lu-Zoledronate for Bone Pain Palliation Therapy, J. Radioanal. Nucl. Chem., 2013, 298(2), 1273–1281, DOI:10.1007/s10967-013-2490-2.
- G. Quiñones Vélez, L. Carmona-Sarabia, W. A. Rodríguez-Silva, A. A. Rivera Raíces, L. Feliciano-Cruz, C. T. Hu, E. A. Peterson and V. Lopez-Mejias, Potentiating Bisphosphonate-Based Coordination Complexes to Treat Osteolytic Metastases, J. Mater. Chem. B, 2020, 8(10), 2155–2168 RSC.
- E. Freire and D. R. Vega, Aquabis[1-Hydroxy-2-(Imidazol-3-Ium-1-Yl)-1,1′-Ethylidenediphophonato-K2 O,O′]Zinc(II) Dihydrate, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65(11), 1430–1431, DOI:10.1107/S160053680904286X.
- E. Freire, D. R. Vega and R. Baggio, Zoledronate Complexes. III. Two Zoledronate Complexes with Alkaline Earth Metals: [Mg(C5H9N2O7P2)2(H2O)2] and [Ca(C5H8N2O7P2)(H2O)]N, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2010, 66(6), 166–170, DOI:10.1107/S0108270110017634.
- E. Freire and D. R. Vega, Diaqua-Bis[1-Hydroxy-2-(Imidazol-3-Ium-1-Yl)-1,1′-Ethyl-Idenediphophonato-K2 O,O′]Zinc(II), Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65(11), 428–429, DOI:10.1107/S1600536809042858.
- Z. C. Zhang, R. Q. Li and Y. Zhang, Diaquabis{[1-Hydr-Oxy-2-(1H-Imidazol-3-Ium-1-Yl)Ethane-1,1-Di-Yl] Bis-(Hydrogen Phospho-Nato)}manganese(II), Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65(12), 1701–1702, DOI:10.1107/S160053680905065X.
- D. K. Cao, Y. Z. Li and L. M. Zheng, Layered Cobalt(II) and Nickel(II) Diphosphonates Showing Canted Antiferromagnetism and Slow Relaxation Behavior, Inorg. Chem., 2007, 46(18), 7571–7578, DOI:10.1021/ic701098t.
- Y. Kato, S. Ozawa, C. Miyamoto, Y. Maehata, A. Suzuki, T. Maeda and Y. Baba, Acidic Extracellular Microenvironment and Cancer, Cancer Cell Int., 2013, 13(1), 1–8, DOI:10.1186/1475-2867-13-89.
- R. Coleman, J. J. Body, M. Aapro, P. Hadji and J. Herrstedt, Bone Health in Cancer Patients: ESMO Clinical Practice Guidelines, Ann. Oncol., 2014, 25, 124–137, DOI:10.1093/annonc/mdu103.
- S. Tsuzuki, S. H. Park, M. R. Eber, C. M. Peters and Y. Shiozawa, Skeletal Complications in Cancer Patients with Bone Metastases, Int. J. Urol., 2016, 1, 825–832, DOI:10.1111/iju.13170.
- A. Pugazhendhi, T. N. J. I. Edison, I. Karuppusamy and B. Kathirvel, Inorganic Nanoparticles: A Potential Cancer Therapy for Human Welfare, Int. J. Pharm., 2018, 104–111, DOI:10.1016/j.ijpharm.2018.01.034.
- M. Croset and P. Clézardin, MicroRNA-Mediated Regulation of Bone Metastasis Formation: From Primary Tumors to Skeleton, Bone Cancer: Primary Bone Cancers and Bone Metastases, Elsevier Inc., 2nd edn, 2014, pp. 479–489 DOI:10.1016/B978-0-12-416721-6.00041-8.
- S. A. Harris, R. J. Enger, L. B. Riggs and T. C. Spelsberg, Development and Characterization of a Conditionally Immortalized Human Fetal Osteoblastic Cell Line, J. Bone Miner. Res., 1995, 10(2), 178–186 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental methodologies for hydrothermal syntheses, additional Raman spectra, PXRD patterns, SEM images, EDS spectra, TGA thermographs, CIF files, ORTEPs, UV-Vis absorption spectra, calibration curves, dissolution profiles, DLS spectra, binding curves, IC50 curves and %RCL tables. CCDC 2120915–2120920. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ma01127h |
| This journal is © The Royal Society of Chemistry 2022 |