
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHypoxia targeting lutetium-177-labeled nitroimidazole-decorated gold particles as cancer theranostic nanoplatforms†
Sweety
Mittal
 a,
Rohit
Sharma
a,
H. D.
Sarma
b and
Madhava B.
Mallia
*ac
a,
Rohit
Sharma
a,
H. D.
Sarma
b and
Madhava B.
Mallia
*ac
aRadiopharmaceuticals Division, Bhabha Atomic Research Centre, Trombay, Mumbai - 400085, India. E-mail: mallia@barc.gov.in; Fax: +91 22 2550 5151; Tel: +91 22 25590746
bRadiation Biology and Health Science Division, Bhabha Atomic Research Center, Mumbai - 400085, India
cHomi Bhabha National Institute, Anushaktinagar, Mumbai - 400094, India
First published on 27th January 2022
Abstract
Considering the role of hypoxia in cancer progression and poor prognosis, hypoxia-centric theranostic approaches may have immense potential in clinical cancer management. We have successfully developed a hypoxia-selective theranostic nanoplatform, which can be used to deliver a therapeutic dose of radiation to the hypoxic tumor tissue through systemic administration. In addition to delivering a radiation dose to the hypoxic tumor tissue, the versatile nanoplatform developed can also be used to load radiosensitizers or chemotherapeutic drugs to enhance the therapeutic effect.
Tumor hypoxia is a pathological condition arising out of insufficient blood supply in a rapidly growing tumor and poses major challenges to cancer therapy.1 Hypoxic tumors are associated with increased risk of metastasis and poor response to conventional therapeutic modalities. Assessment of the hypoxic status of a cancer can immensely help in modifying the therapeutic strategy for a better clinical outcome to the patient.1 Tumor hypoxia can be assessed by invasive or non-invasive techniques. However, limitations of invasive techniques in clinical settings have directed the focus on non-invasive methods for assessing tumor hypoxia.2–4 Hypoxia-directed external beam radiation therapy of cancer based on the information acquired from hypoxia imaging is a clinically established procedure.5,6 But, studies on hypoxia-directed internal radiotherapy through systemic administration of hypoxia targeting radiopharmaceuticals is limited. A bottleneck to this approach is the non-availability of a suitable platform that could selectively deliver the therapeutic dose of radiation to the hypoxic tumor tissue with a minimal radiation dose to the non-target organs. Escalating the injected dose to deliver the required therapeutic dose to the hypoxic tissue may be limited by unacceptable levels of radiation dose to non-target organs. A possible solution to this problem could be to develop a selective hypoxia-targeting platform that could also carry radiosensitizers or chemotherapeutic drugs along with therapeutic radioisotopes to enhance the therapeutic effect in the hypoxic tissue at a lower radiation dose.
Gold nanoparticles are extensively utilized for targeting cancer, exploiting their passive intra-tumoral accumulation through the enhanced permeation and retention effect (EPR) as well as active uptake achieved via functionalization. The facile surface chemistry, ease of synthesis, tunable size, low cytotoxicity and high adsorption capacity of gold nanoparticles offer effective tools to tailor the pharmacokinetics for targeted applications.7,8 Unique optical properties like surface plasmon resonance (SPR) have made gold nanoparticles the preferred choice for developing agents for photothermal imaging, photothermal therapy and photodynamic therapy for cancer detection and treatment.9–14 Additionally, the large surface-to-volume ratios of nanoparticles open numerous possibilities for drug/radiosensitizer loading, surface modification with target-specific biological vectors, tagging radioisotopes, etc., making them a multipurpose platform for developing targeted systems for drug delivery or radionuclidic therapy, either individually or in combination.15–20
Though systemic administration of radiolabeled nanoplatforms for imaging or therapy of tumors has been reported,21–24 to date, there are limited reports on similar applications targeting hypoxic tissues in vivo.25–27 Nitroimidazoles are extensively studied bioreductive markers for non-invasive detection of hypoxia. They undergo selective, oxygen-dependent, accumulation and retention in hypoxic cells through a series of one-electron reductions mediated by nitroreductase enzymes. There are reports demonstrating the potential of nitroimidazole-based nanoplatforms for hypoxia imaging applications based on photoacoustic, optical and MRI techniques.28,29 However, optical imaging has limitations in visualizing deep-seated hypoxic tumors due to the attenuation of light emitted from the imaging probe by overlying tissues. The MRI approach requires administration of a significant quantity of toxic contrast agents to permit the detection of the oxygen levels in living tissue. In this context, nuclear imaging techniques are much more sensitive than either optical imaging or MRI and radiolabeled nanoparticles decorated with nitroimidazole present a promising approach for real-time imaging and delivery of a therapeutic dose of radiation to the hypoxic tumor site.
In vivo mapping of hypoxia in tissue is carried out by systemic administration of a radiotracer tagged with gamma emitting radioisotopes such as technetium-99m (Eγ = 140 keV; half-life = 6.0 h) or positron emitting radioisotopes such as fluorine-18 (half-life = 109.7 min) or gallium-68 (half-life = 67.7 min). Subsequently, imaging is performed by single photon emission computed tomography (SPECT) or positron emission tomography (PET).3,5,6,30–36 The images showing the presence/absence or distribution of hypoxia in the tissue are then used to select patients who will benefit from hypoxia-directed external beam radiotherapy or other hypoxia-directed treatment strategies. Currently, positron emitting radiopharmaceutical, [18F]-fluoromisonidazole ([18F]-FMISO), is used for clinical imaging of tumor hypoxia.30 Development of a technetium-99m alternative for this purpose is being actively pursued by several research groups including ours.31–35 However, it would be immensely helpful if the vehicle used for imaging hypoxia can also be used for hypoxia-directed internal radiation therapy. This requires a molecular design that will permit radiolabeling with a diagnostic, therapeutic or theranostic radioisotope. The beta–gamma emitting radioisotope lutetium-17737 (Eβmax = 0.49 MeV, 0.18 MeV, Eγ = 208 keV, 112 keV; half-life = 6.7 d) is one such theranostic radioisotope, which can serve the dual purpose of delivering a therapeutic dose of radiation to the target, while at the same time, the associated gamma emission can be used for real-time monitoring of the tracer in vivo.
With these objectives in mind, we envisaged a versatile, hypoxia-targeting platform that could be employed for multifarious applications such as imaging hypoxia, delivery of a therapeutic dose of radiation, delivery of radiosensitizers, delivery of chemotherapeutic drugs or a combination of the above to hypoxic cells through systemic administration. Since gold nanoparticles themselves do not possess hypoxia selectivity, their surface was decorated with a bioreductive marker, 2-nitroimidazole, which could undergo selective oxygen-dependent reduction in hypoxic cells imparting the required selectivity. To achieve the flexibility of radiolabeling with a diagnostic or therapeutic radioisotope, the gold nanoparticle surface was further modified to include a bifunctional chelator, DOTAGA. The DOTAGA chelator permits radiolabeling with a theranostic radioisotope such as lutetium-177, a therapeutic radioisotope such as yttrium-90 or a positron emitting diagnostic radioisotope such as gallium-68.38–40 The various steps involved in the preparation of DOTAGA-conjugated, 2-nitroimidazole-decorated gold nanoparticles are shown in Fig. 1 (also see ESI†). For facile attachment of 2-nitroimidazole to the gold nanoparticles, 2-nitroimidazole (2-NIM) functionalized lipoic acid, NIM–TA (3) was synthesized by coupling 2-nitroimidazole amine (2) with lipoic acid [Fig. 1(a)].
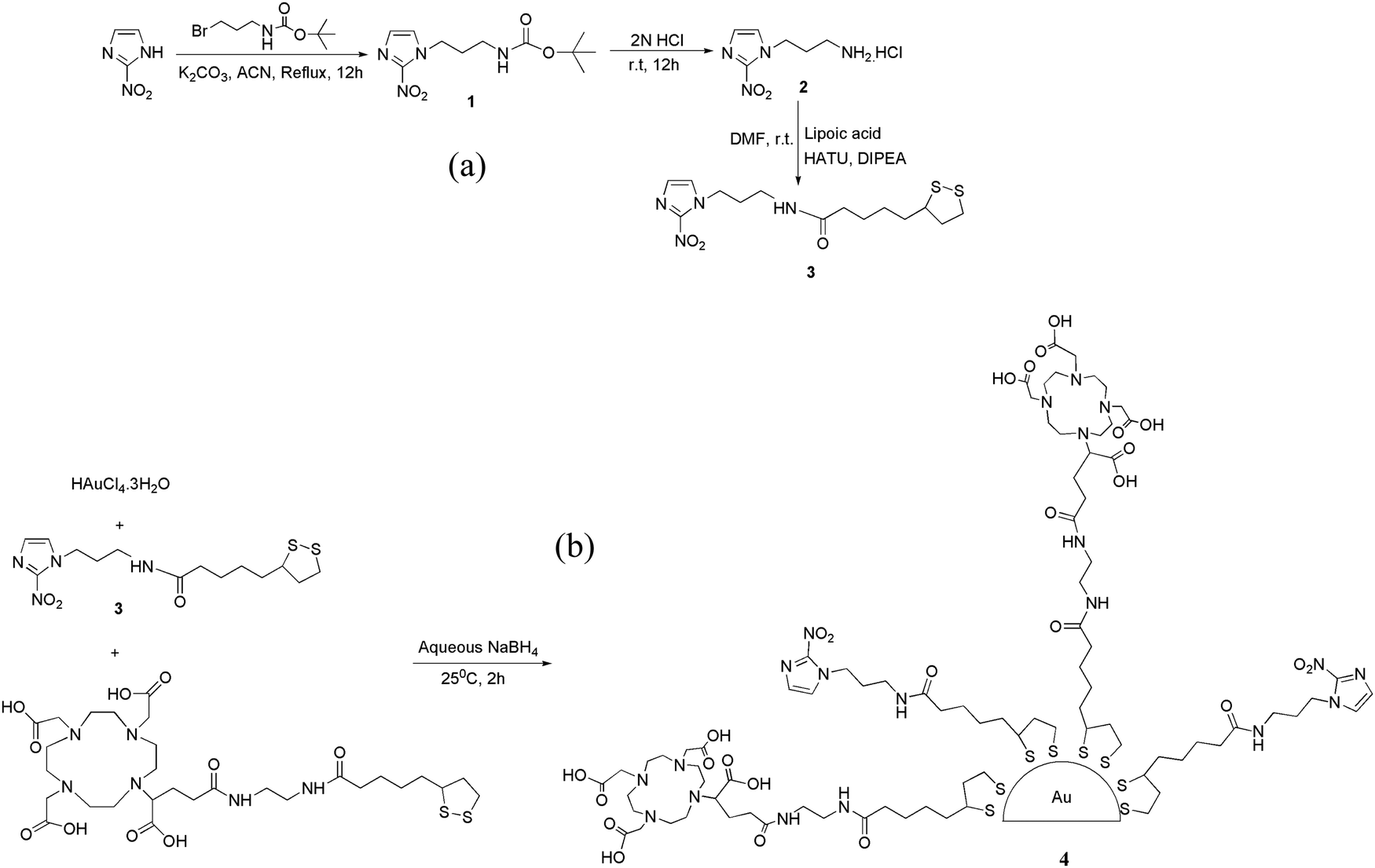 | ||
| Fig. 1 (a) Synthesis of NIM–TA (3). (b) Synthesis of DOTAGA–AuNP–2-NIM (4). | ||
The presence of the characteristic carbonyl amide peak at 1641 cm−1 in the IR spectra confirmed the formation of compound 3 (Fig. S7 in ESI†). The 1H NMR spectra of all the compounds were consistent with the expected structure. A molecular ion peak at m/z 397.1 (M + K)+ in the mass spectra of NIM–TA (3) provided additional evidence for its formation (Fig. S10 in ESI†). The 2-NIM-tagged DOTAGA-conjugated gold nanoparticles (DOTAGA–AuNP–2-NIM) were synthesized by in situ reduction of a gold salt (HAuCl4.3H2O) with sodium borohydride (NaBH4) [Fig. 1(b)] in the presence of excess thiolated ligands, 2-NIM–TA (3) and DOTAGA–TA. The gold nanoparticles were purified by centrifugal filtration and characterized by DLS, UV-Vis spectroscopy, TEM, FT-IR and 1H NMR spectroscopy.
Evidence for the formation of gold nanoparticles could be obtained from the presence of a typical SPR band at ∼520 nm in the UV-Vis spectrum [Fig. 2(a)]. Furthermore, a broad peak seen in the UV-Vis spectrum of DOTAGA–AuNP–2-NIM at 320 nm, which matched with the UV-Vis spectrum of 2-NIM–TA, indicated the presence of 2-NIM–TA on the gold nanoparticle surface [Fig. 2(a)]. The DLS analysis showed that DOTAGA–AuNP–2-NIM has an effective hydrodynamic diameter of 60 ± 1.5 nm [Fig. 2(b)]. The nanoparticles prepared displayed a very low polydispersity index. The zeta potential of the nanoparticles was found to be +20.22 mV at pH 7, indicating good suspension stability. The TEM images showed gold nanoparticles of uniform size, with most of the particles in the range of 10–20 nm [Fig. 2(c)]. The average particle size was 15 ± 1.2 nm. It could be noted that the hydrodynamic size of the nanoparticles determined by DLS is approximately four-times higher than the size of the nanoparticles determined by TEM, which could be attributed to the presence of hydrated layers around the nanoparticles when they are in solution. The amide >C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching (1640–1620 cm−1), C–H stretching (3000–2800 cm−1), amide N–H bending (1589 cm−1) and O–H stretching (3600–2500 cm−1) bands could be seen in the FT-IR spectra of DOTAGA–AuNP–2-NIM [Fig. 2(d)]. For comparison, the FT-IR spectra of 2-NIM–TA and DOTAGA–TA are also included in Fig. 2(d). The presence of ligands (NIM–TA and DOTAGA–TA) on the gold nanoparticle surface was further confirmed by 1H NMR spectroscopy. The peak in the aromatic region (δ 7.52) confirms the presence of 2-nitroimidazole and the peak pattern in the aliphatic region (δ 2.60–3.52) indicates the presence of DOTAGA on the gold surface (Fig. S11 in ESI†). Cyclic voltammetry of DOTAGA–AuNP–2-NIM (see ESI†)41 showed a peak at −1.21 V due to the reduction of the nitro group in 2-nitroimidazole on the gold nanoparticle surface [Fig. 3(a)], which closely matched with the first reduction peak in the cyclic voltammogram of free 2-nitroimidazole [Fig. S12 in ESI†]. It is pertinent to note the absence of any reduction peak in the cyclic voltammogram of DOTAGA–AuNP without 2-NIM decoration. This observation provided additional evidence for the presence of 2-nitroimidazole on the gold nanoparticle surface.
O stretching (1640–1620 cm−1), C–H stretching (3000–2800 cm−1), amide N–H bending (1589 cm−1) and O–H stretching (3600–2500 cm−1) bands could be seen in the FT-IR spectra of DOTAGA–AuNP–2-NIM [Fig. 2(d)]. For comparison, the FT-IR spectra of 2-NIM–TA and DOTAGA–TA are also included in Fig. 2(d). The presence of ligands (NIM–TA and DOTAGA–TA) on the gold nanoparticle surface was further confirmed by 1H NMR spectroscopy. The peak in the aromatic region (δ 7.52) confirms the presence of 2-nitroimidazole and the peak pattern in the aliphatic region (δ 2.60–3.52) indicates the presence of DOTAGA on the gold surface (Fig. S11 in ESI†). Cyclic voltammetry of DOTAGA–AuNP–2-NIM (see ESI†)41 showed a peak at −1.21 V due to the reduction of the nitro group in 2-nitroimidazole on the gold nanoparticle surface [Fig. 3(a)], which closely matched with the first reduction peak in the cyclic voltammogram of free 2-nitroimidazole [Fig. S12 in ESI†]. It is pertinent to note the absence of any reduction peak in the cyclic voltammogram of DOTAGA–AuNP without 2-NIM decoration. This observation provided additional evidence for the presence of 2-nitroimidazole on the gold nanoparticle surface.
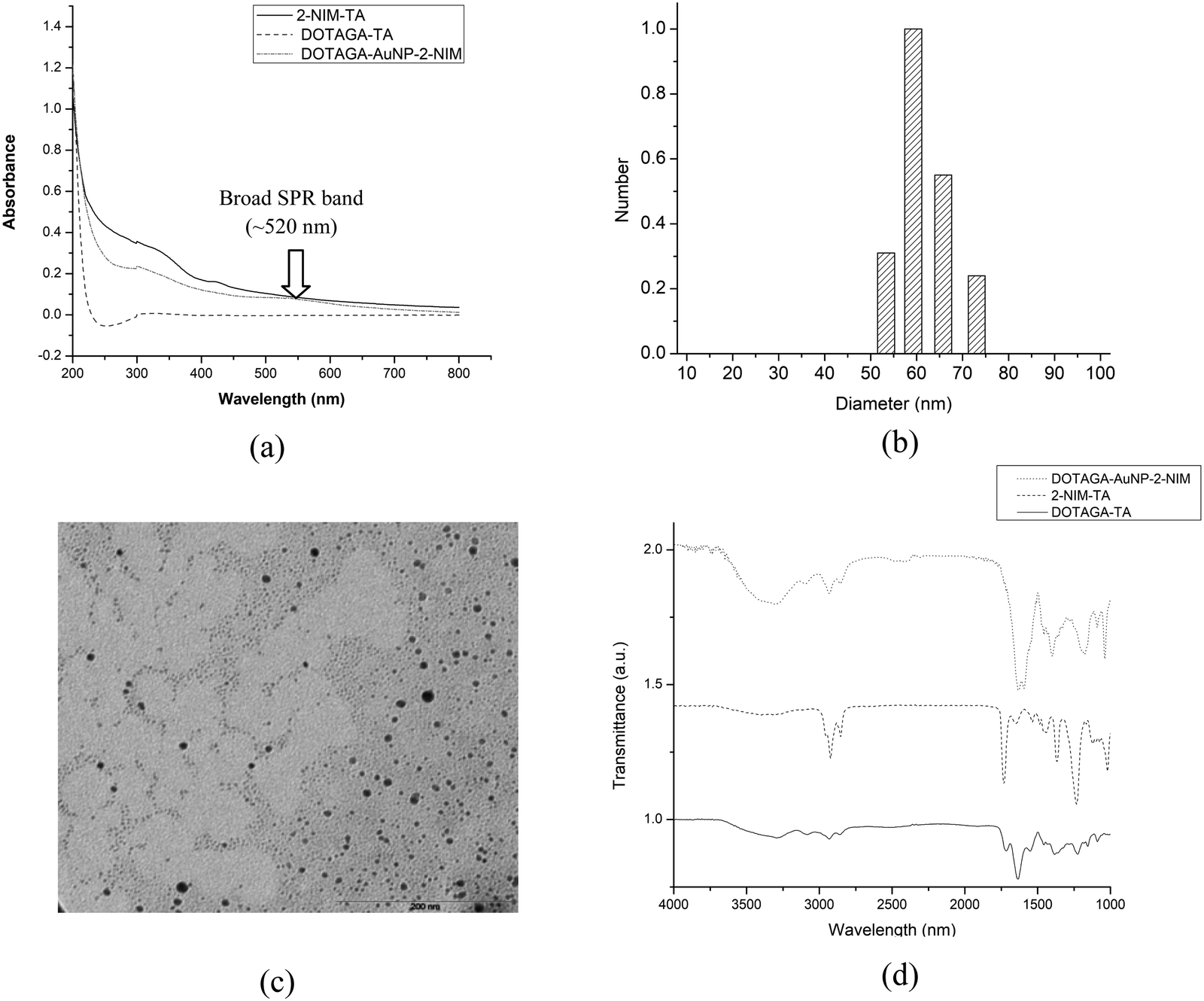 | ||
| Fig. 2 (a) UV-Vis spectra of NIM–TA, DOTAGA–TA and DOTAGA–AuNP–2-NIM. (b) Size distribution data of DOTAGA–AuNP–2-NIM. (c) TEM image of DOTAGA–AuNP–2-NIM. (d) Overlaid FT-IR spectra of NIM–TA, DOTAGA–TA, and DOTAGA–AuNP–2-NIM. | ||
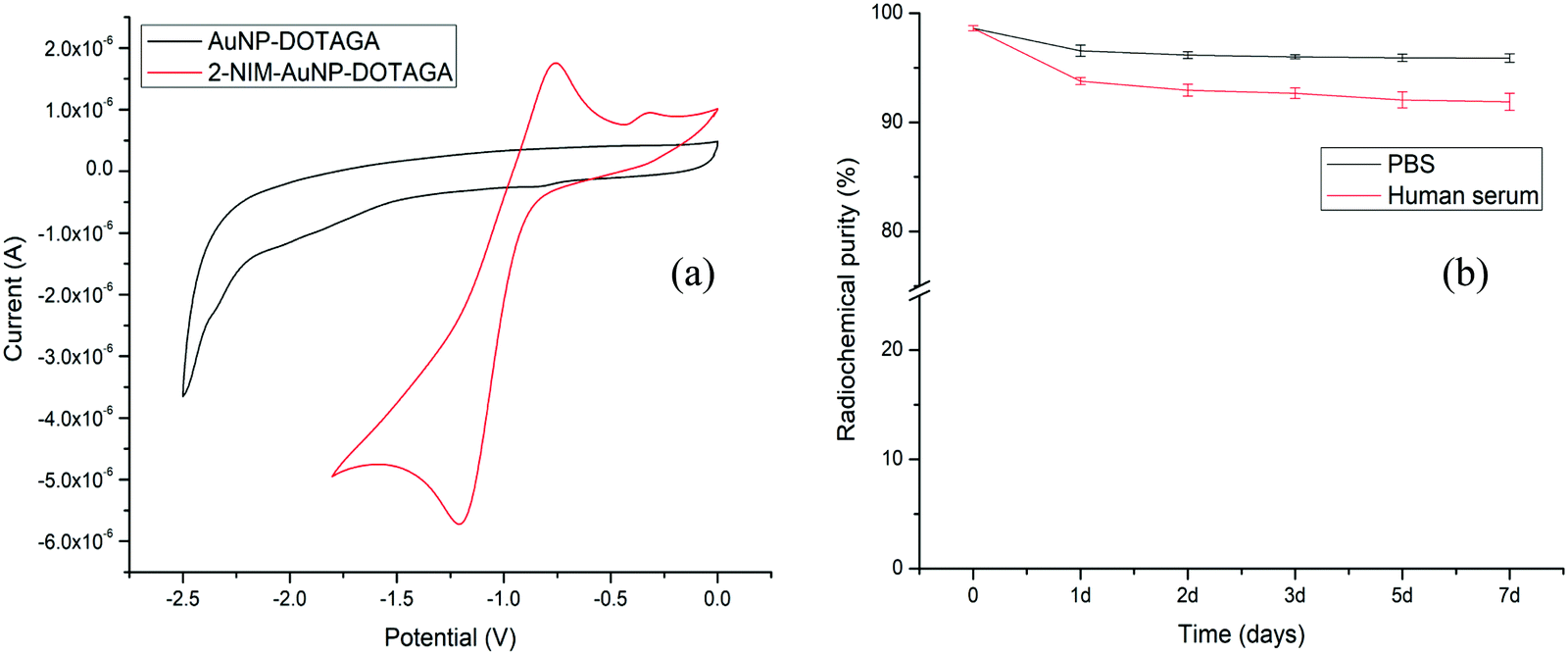 | ||
| Fig. 3 (a) Cyclic voltammograms of AuNP–DOTAGA and DOTAGA–AuNP–2-NIM and (b) variations of RCP of [177Lu]Lu–DOTAGA–AuNP–2-NIM in PBS and human serum over a period of 7 days. | ||
Lutetium-177 in chloride form (177LuCl3) is used for radiolabeling the functionalized gold nanoparticles (see ESI†). The radiochemical yield (RCY) and radiochemical purity (RCP) of [177Lu]Lu–DOTAGA–AuNP–2-NIM were determined by size exclusion HPLC (SE-HPLC) [Fig. S13 in ESI†]. The retention time of [177Lu]Lu–DOTAGA–AuNP–2-NIM in HPLC was found to be 13.45 ± 0.60 min [Fig. S13(a) in ESI†], whereas 177LuCl3 appeared at 22.19 ± 0.40 min [Fig. S13(c) in ESI†]. From peak area measurements, the RCY of [177Lu]Lu–DOTAGA–AuNP–2-NIM was found to be above 90%. The bifunctional chelator used for the surface modification of gold nanoparticles, DOTAGA, was radiolabeled separately for comparison. The peak corresponding to [177Lu]Lu–DOTAGA appeared at 19.89 ± 0.40 min [Fig. S13(b) in ESI†], distinctly different from the peak representing radiolabeled gold nanoparticles. The RCY and RCP determined by PC [Fig. S14 in ESI†] were in concurrence with the values obtained by HPLC. The nanoparticles were further purified by passing through a PD-10 column to improve the RCP to over 95%.
The in vitro stability of the [177Lu]Lu–DOTAGA–AuNP–2-NIM nanoparticles was assessed in PBS and human serum (see ESI†) over a period of one week by PC [Fig. 3(b)]. It could be observed that the radiolabeled nanoparticles maintained an RPC of above 90% over the period of study. The size of the radiolabeled nanoparticles in PBS was also evaluated for the same period (1 week). The variation in hydrodynamic size of the radiolabeled nanoparticles with time is shown in Fig. S15 [in the ESI†]. The cytotoxicity of DOTAGA–AuNP–2-NIM to CHO cells was assessed by flow cytometry after incubating them together for 24 h (see ESI†). The typical flow cytometer dot-plot viability data are shown in Fig. S16 (see ESI†). Viable, apoptotic and dead cell populations of CHO cells treated with different concentrations of DOTAGA–AuNP–2-NIM are shown in Fig. 4(a). No significant cell death was observed in the concentration range of 0.004–800 ng mL−1. At a concentration of 1600 ng mL−1, the percentage viability reduced to 74.89 ± 0.05%. In vitro studies were carried out in CHO cells under hypoxic and normoxic conditions to determine the hypoxia selectivity of [177Lu]Lu–DOTAGA–AuNP–2-NIM (see the ESI†). The uptake of radiotracer in the CHO cells under hypoxic and normoxic conditions42 is shown in Fig. 4(b and c) [see ESI†]. The results indicated significant uptake of the radiotracer under hypoxic conditions [Fig. 4(b)] compared to normoxic conditions (P < 0.02). The hypoxic/normoxic ratio for [177Lu]Lu–DOTAGA–AuNP–2-NIM increased from 7.1 at 2 h post-incubation to 9.2 at 4 h PI demonstrating excellent hypoxia selectivity. It is pertinent to note that [177Lu]Lu–DOTAGA–AuNP without 2-NIM decoration showed very low cellular uptake under hypoxic and normoxic conditions [Fig. 4(c)] and the difference in cellular uptake was found to be statistically insignificant (P > 0.05).
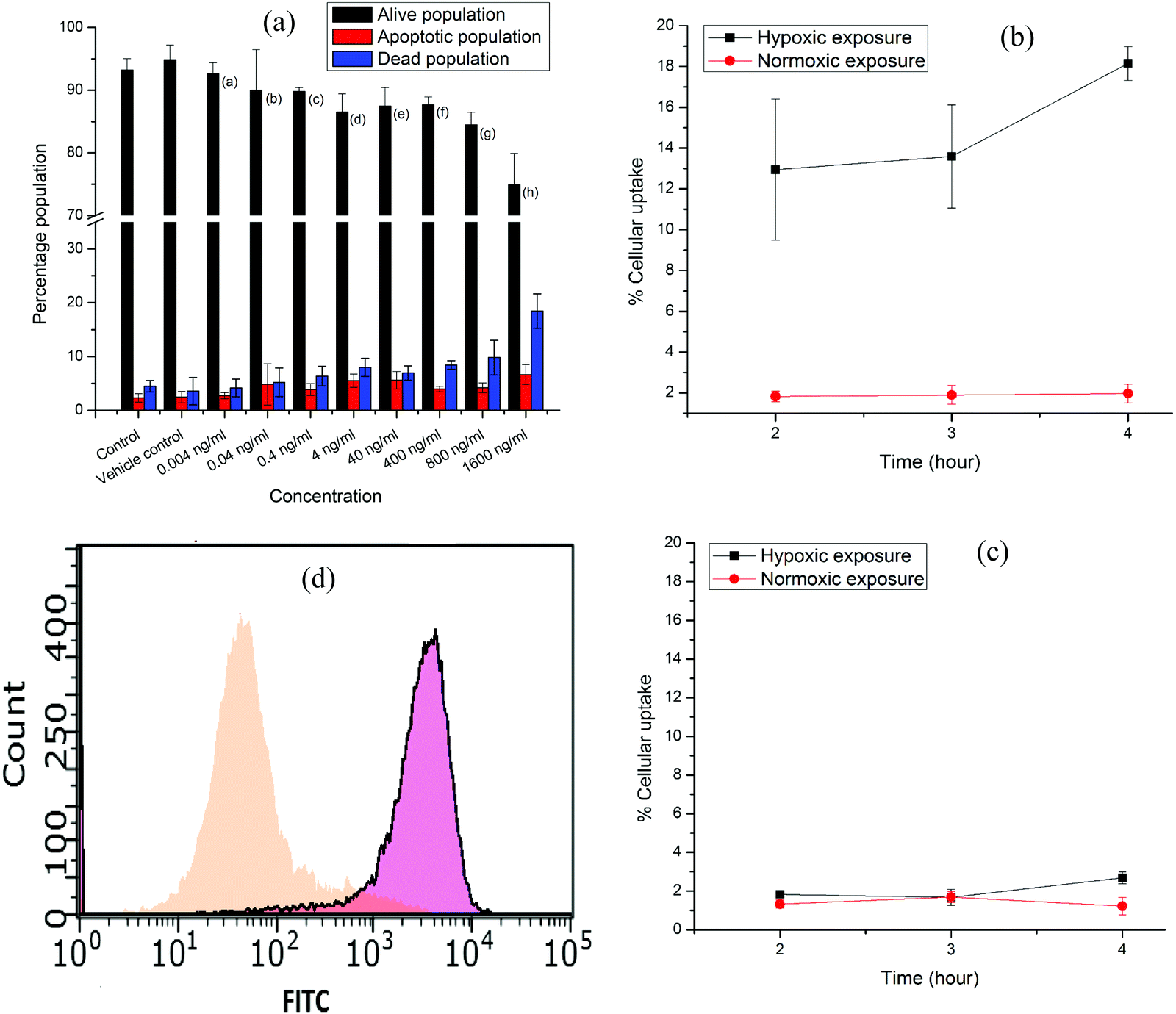 | ||
| Fig. 4 (a) Bar graphs showing viable, apoptotic and dead populations of CHO cells treated with different concentrations (0.004–1600 ng mL−1) of DOTAGA–AuNP–2-NIM. (b) Percentage cellular uptake of [177Lu]Lu–DOTAGA–AuNP–2-NIM, (c) [177Lu]Lu–DOTAGA–AuNP (control) and (d) FITC-conjugated gold nanoparticles, DOTAGA–AuNP(FITC)–2-NIM, in CHO cells under normoxic and (orange) and hypoxic (pink) exposure. | ||
On the other hand, statistical analysis clearly demonstrated significant (P < 0.02) accumulation of 2-nitroimidazole-decorated gold nanoparticles ([177Lu]Lu–DOTAGA–AuNP–2-NIM) in cells under hypoxic conditions at all time points compared to radiolabeled nanoparticles without 2-nitroimidazole ([177Lu]Lu–DOTAGA–AuNP) under similar conditions. Under normoxic conditions, the cellular uptakes of [177Lu]Lu–DOTAGA–AuNP–2-NIM and [177Lu]Lu–DOTAGA–AuNP were similar and statistically insignificant (P > 0.05).
Nitroimidazole-mediated hypoxia selective internalization and accumulation of gold nanoparticles in CHO cell lines was further demonstrated using fluorescent nanoparticles, DOTAGA–AuNP(FITC)–2-NIM, synthesized following a protocol similar to the preparation of DOTAGA–AuNP–2-NIM (see ESI†). The fluorescent nanoparticles were incubated with CHO cells under both hypoxic and normoxic conditions similar to the study using [177Lu]Lu–DOTAGA–AuNP–2-NIM mentioned earlier. The cells acquired and analyzed using flow cytometry 24 h post-incubation showed a strong fluorescence signal shift in the hypoxic group [∼74.87% of cells with mean fluorescence intensity (MFI) of 3325.83 ± 7.5 (pink)] indicating enhanced retention of DOTAGA–AuNP(FITC)–2-NIM under hypoxic conditions as compared to normoxic conditions [ ∼79.15% of cells with MFI of 45.22 ± 8.56 (orange)] [Fig. 4(d)], further confirming the role of 2-nitroimidazole in hypoxia-specific accumulation. Surface conjugation of FITC also demonstrated the feasibility and the versatility of the nanoplatform to couple other suitable radiosensitizers or chemotherapeutic drugs for possible enhancement of the therapeutic effect.
Animal studies were conducted following the protocols approved by the Institutional Animal Ethics Committee of Bhabha Atomic Research Centre, Trombay, Mumbai. Biodistribution studies of [177Lu]Lu–DOTAGA–AuNP–2-NIM were carried out with Swiss mice bearing fibrosarcoma tumors. The distribution of the radiotracer in different organs/tissue is shown in Table 1. Very fast clearance of the nanoparticles could be observed from the animal body with ∼70% of the injected activity excreted within 3 h of injection. A similar observation was also reported for gallium-67-labeled bombesin-conjugated gold nanoparticles.43 Apparently, fast clearance adversely affected the uptake and retention of the radiotracer in the tumor. Generally nanoparticles reach tumor cells by passive diffusion driven by the concentration gradient developed between the blood pool and the intracellular environment. Quick clearance of the radiotracer from the blood can reverse the concentration gradient resulting in poor distribution of the radiotracer in the tumor and limited time available for the radiotracer to undergo oxygen-dependent reduction in tumor cells. However, it is pertinent to note that the activity observed in the tumor at 3 h post-injection (PI) is retained until 72 h PI, indicating hypoxia-specific reduction of the tracer in hypoxic tumor cells.
| Organs | % Injected dose per gram Avg. (s. d)b | |||
|---|---|---|---|---|
| 3 h | 24 h | 48 h | 72 h | |
| a Excretion is calculated by adding percentage of injected dose in all organs and then subtracting from 100. b s.d. – standard deviation. | ||||
| Liver | 13.57 (1.79) | 12.03 (0.84) | 10.97 (2.58) | 8.62 (3.09) |
| Intestine | 0.30 (0.06) | 0.05 (0.01) | 0.05 (0.01) | 0.04 (0.01) |
| Stomach | 0.14 (0.08) | 0.05 (0.01) | 0.06 (0.03) | 0.07 (0.03) |
| Kidneys | 1.14 (0.21) | 0.73 (0.06) | 0.63 (0.20) | 0.42 (0.06) |
| Heart | 0.14 (0.03) | 0.09 (0.04) | 0.00 | 0.00 |
| Lungs | 0.30 (0.12) | 0.36 (0.14) | 0.23 (0.13) | 0.25 (0.04) |
| Spleen | 1.97 (0.57) | 2.13 (0.13) | 2.33 (0.54) | 1.90 (0.18) |
| Blood | 0.10 (0.02) | 0.06 (0.02) | 0.00 | 0.00 |
| Muscle | 0.04 (0.01) | 0.05 (0) | 0.00 | 0.00 |
| Tumor | 0.20 (0.05) | 0.14 (0.01) | 0.10 (0.03) | 0.16 (0.06) |
| Bone | 2.76 (0.43) | 2.16 (0.75) | 2.40 (0.69) | 3.30 (1.04) |
| Excretiona | 69.91 (6.46) | 77.10 (0.77) | 78.31 (9.00) | 75.27 (6.12) |
| Tumor/blood ratio | 1.91 | 2.56 | Very high | Very high |
| Tumor/muscle ratio | 4.81 | 2.76 | Very high | Very high |
Radiotracer uptake was insignificant in the muscle and blood pool, which reduced the below detection limit two days PI. Significant uptake of the nanoparticle radiotracer was observed in the liver and spleen. A similar observation was reported in studies using lipoic acid-coated silver and gold nanoparticles.44 This observation is attributed to the response of the immune system. Despite having very low uptake in the tumor, the tumor to blood ratio and tumor to muscle ratio progressively increased with time owing to the clearance of the radiotracer from the blood and muscles.
Gold nanoparticles have been extensively used as a radiosensitizer in the presence of an external radiation source (X-rays) for increased therapeutic effects.45–48 The radio-enhancement effects of gold nanoparticles are due to various interlinked biological, chemical and physical processes.49 However, there are very few reports that utilized the potential of gold nanoparticles as radiosensitizers in the presence of an internal radiation source (radionuclide). Gahlomi et al. demonstrated enhanced radiation damage effects by iron oxide nanoparticles radiolabeled with therapeutic radioisotopes like 213Bi, 223Ra, 90Y, 177Lu, 67Cu, 64Cu and 89Zr in the context of internal radionuclide therapy by computational modeling.50 Only recently, Su et al. demonstrated enhanced efficiency in tumor treatment by using β-emitting radioisotope-tagged tumor targeting gold nanoparticles, 131I–AuNPs–TAT, both by in vitro and in vivo studies.51 A similar enhancement in therapeutic efficacy using [177Lu]Lu–DOTAGA–AuNP–2-NIM is yet to be studied. While gold nanoparticles themselves can act as a radiosensitizer, large surface area permits incorporation of organic radiosensitizers in a way similar to the incorporation of FITC in the present study. That would permit exploring the possibilities of radiosensitizer-enhanced hypoxia-directed internal radiotherapy.
It is a well-known fact that interaction of nanoparticles with complex biological milieu in vivo influences their blood circulation time, biodistribution, cell uptake and host immune response (uptake in the liver and spleen).52 For example, functionalization of nanoparticles with PEG increases their systemic circulation time and decreases their immunogenicity and hence could be a possible strategy to overcome the current problem of low circulation time and high uptake of the radiotracer in the liver and spleen.
Conclusions
The present study describes the preparation and in vitro evaluation of 177Lu-labeled AuNPs decorated with 2-nitroimidazole units for targeting tumor hypoxia. To the best of our knowledge, this is the first report wherein a radiolanthanide (luterium-177)-labeled, nitroimidazole-decorated, and systemically administrable nanoplatform is being evaluated for targeting tumor hypoxia. The in vitro studies demonstrated excellent accumulation of [177Lu]Lu–DOTAGA–AuNP–2-NIM nanoparticles in CHO cells under hypoxic conditions. The DOTAGA–AuNP–2-NIM is a potential hypoxia-targeting platform, which can be loaded with fluorescent markers, diagnostic radioisotopes, radiosensitizers or potent therapeutic radioisotopes for multimodal imaging of hypoxia or radiosensitizer-enhanced hypoxia-directed internal radiotherapy.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors gratefully acknowledge Dr P. K. Pujari, Associate Director, Radiochemistry and Isotope Group, Bhabha Atomic Research Centre (BARC), for his constant encouragement and support. The authors gratefully acknowledge the members of Radiochemicals Section, Radiopharmaceuticals Division, for providing 177Lu for the present study. The authors also thank Dr Anand Ballal, Molecular Biology Division, BARC and Dr Manoj Sharma, Fuel Chemistry Division, BARC for acquiring the TEM images and cyclic voltammograms of the functionalized gold nanoparticles, respectively, presented herein.References
- P. Vaupel and A. Mayer, Cancer Metastasis Rev., 2007, 26, 225–239 CrossRef CAS PubMed.
- O. J. Kelada and D. J. Carlson, Radiat. Res., 2014, 181, 335–349 CrossRef CAS PubMed.
- K. A. Krohn, J. M. Link and R. P. Mason, J. Nucl. Med., 2008, 49, 129S–148S CrossRef CAS PubMed.
- I. Daimiel, J. Breast Cancer, 2019, 22, 155–171 CrossRef PubMed.
- R. L. Wahl, J. M. Herman and E. Ford, Semin. Radiat. Oncol., 2011, 21, 88–100 CrossRef PubMed.
- M. Hodolič, J. Fettich and K. Kairemo, Curr. Radiopharm., 2015, 8, 32–37 CrossRef PubMed.
- Y.-C. Yeh, B. Creran and V. M. Rotello, Nanoscale, 2012, 4, 1871–1880 RSC.
- R. Sardar, A. M. Funston, P. Mulvaney and R. W. Murray, Langmuir, 2009, 25, 13840–13851 CrossRef CAS PubMed.
- M. R. K. Ali, Y. Wu and M. A. El-Sayed, J. Phys. Chem. C, 2019, 123, 15375–15393 CrossRef CAS.
- J. Ribera, C. Vilches, V. Sanz, I. de Miguel, I. Portolés, B. Córdoba-Jover, E. Prat, V. Nunes, W. Jiménez, R. Quidant and M. Morales-Ruiz, ACS Nano, 2021, 15, 7547–7562 CrossRef CAS PubMed.
- P. G. Calavia, G. Bruce, L. Pérez-García and D. A. Russell, Photochem. Photobiol. Sci., 2018, 17, 1534–1552 CrossRef PubMed.
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578–1586 CrossRef CAS PubMed.
- N. Gupta and R. Malviya, Biochim. Biophys. Acta, Rev. Cancer, 2021, 1875, 188532 CrossRef CAS PubMed.
- N. F. Gamaleia and I. O. Shton, Photodiagn. Photodyn. Ther., 2015, 12, 221–231 CrossRef CAS PubMed.
- S. A. Bansal, V. Kumar, J. Karimi, A. P. Singh and S. Kumar, Nanoscale Adv., 2020, 2, 3764–3787 RSC.
- S. Yook, Z. Cai, Y. Lu, M. A. Winnik, J.-P. Pignol and R. M. Reilly, J. Nucl. Med., 2016, 57, 936–942 CrossRef CAS PubMed.
- G. F. Paciotti, L. Myer, D. Weinreich, D. Goia, N. Pavel, R. E. McLaughlin and L. Tamarkin, Drug Delivery, 2004, 11, 169–183 CrossRef CAS PubMed.
- M. U. Farooq, V. Novosad, E. A. Rozhkova, H. Wali, A. Ali, A. A. Fateh, P. B. Neogi, A. Neogi and Z. Wang, Sci. Rep., 2018, 8, 2907 CrossRef PubMed.
- L. Dziawer, P. Koźmiński, S. Męczyńska-Wielgosz, M. Pruszyński, M. Łyczko, B. Wąs, G. Celichowski, J. Grobelny, J. Jastrzębski and A. Bilewicz, RSC Adv., 2017, 7, 41024–41032 RSC.
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC.
- C. Liang, Y. Chao, X. Yi, J. Xu, L. Feng, Q. Zhao, K. Yang and Z. Liu, Biomaterials, 2019, 197, 368–379 CrossRef CAS PubMed.
- Z. Li, B. Wang, Z. Zhang, B. Wang, Q. Xu, W. Mao, J. Tian, K. Yang and F. Wang, Mol. Ther., 2018, 26, 1385–1393 CrossRef CAS PubMed.
- L. Tian, Q. Chen, X. Yi, J. Chen, C. Liang, Y. Chao, K. Yang and Z. Liu, Small, 2017, 13, 1700640 CrossRef PubMed.
- L. Tian, Q. Chen, X. Yi, G. Wang, J. Chen, P. Ning, K. Yang and Z. Liu, Theranostics, 2017, 7, 614–623 CrossRef CAS PubMed.
- Y. Li, J. Jeon and J. H. Park, Cancer Lett., 2020, 490, 31–43 CrossRef CAS PubMed.
- M. I. Confeld, B. Mamnoon, L. Feng, H. Jensen-Smith, P. Ray, J. Froberg, J. Kim, M. A. Hollingsworth, M. Quadir, Y. Choi and S. Mallik, Mol. Pharmaceutics, 2020, 17, 2849–2863 CrossRef CAS PubMed.
- P. Zhang, H. Yang, W. Shen, W. Liu, L. Chen and C. Xiao, ACS Biomater. Sci. Eng., 2020, 6, 2167–2174 CrossRef CAS PubMed.
- Y. Umehara, T. Kageyama, A. Son, Y. Kimura, T. Kondo and K. Tanabe, RSC Adv., 2019, 9, 16863–16868 RSC.
- M. Filippi, D.-V. Nguyen, F. Garello, F. Perton, S. Bégin-Colin, D. Felder-Flesch, L. Power and A. Scherberich, Nanoscale, 2019, 11, 22559–22574 RSC.
- S. T. Lee and A. M. Scott, Semin. Nucl. Med., 2007, 37, 451–461 CrossRef PubMed.
- K. Vats, M. B. Mallia, A. Mathur, H. D. Sarma and S. Banerjee, ChemistrySelect, 2017, 2, 2910–2916 CrossRef CAS.
- Q. Ruan, X. Zhang, X. Lin, X. Duan and J. Zhang, Med. Chem. Commun., 2018, 9, 988–994 RSC.
- Q. Ruan, X. Zhang and J. Zhang, Appl. Organomet. Chem., 2020, 34, e5798 CrossRef CAS.
- M. B. Mallia, S. Mittal, H. D. Sarma and S. Banerjee, Bioorg. Med. Chem. Lett., 2016, 26, 46–50 CrossRef CAS PubMed.
- M. B. Mallia, A. Mathur, R. Sharma, C. Kumar, H. D. Sarma, S. Banerjee and A. Dash, J. Radioanal. Nucl. Chem., 2018, 317, 1203–1210 CrossRef CAS.
- J. R. Ballinger, Semin. Nucl. Med., 2001, 31, 321–329 CrossRef CAS PubMed.
- S. Banerjee, T. Das, S. Chakraborty and M. Venkatesh, Radiochim. Acta, 2012, 100, 115–126 CrossRef CAS.
- R. Zhao, K. Ploessl, Z. Zha, S. Choi, D. Alexoff, L. Zhu and H. F. Kung, Mol. Pharmaceutics, 2020, 17, 4589–4602 CrossRef CAS PubMed.
- D. Satpati, R. Sharma, H. D. Sarma and A. Dash, Chem. Biol. Drug Des., 2018, 91, 781–788 CrossRef CAS PubMed.
- C. Bernhard, M. Moreau, D. Lhenry, C. Goze, F. Boschetti, Y. Rousselin, F. Brunotte and F. Denat, Chem. – Eur. J., 2012, 18, 7834–7841 CrossRef CAS PubMed.
- L. Mei, Y. Wang and T. Chu, Eur. J. Med. Chem., 2012, 58, 50–63 CrossRef CAS PubMed.
- S. Mittal, R. Sharma, M. B. Mallia and H. D. Sarma, Nucl. Med. Biol., 2021, 96–97, 61–67 CrossRef CAS PubMed.
- F. Silva, A. Zambre, M. P. C. Campello, L. Gano, I. Santos, A. M. Ferraria, M. J. Ferreira, A. Singh, A. Upendran, A. Paulo and R. Kannan, Bioconjugate Chem., 2016, 27, 1153–1164 CrossRef CAS PubMed.
- C. L. D. Lee, S. B. Fashir, M. L. Castilho, M. A. Hupman, L. J. Raniero, I. Alwayn and K. C. Hewitt, J. Pharm. Sci., 2016, 105, 25–30 CrossRef CAS PubMed.
- Y. Zhang, F. Huang, C. Ren, J. Liu, L. Yang, S. Chen, J. Chang, C. Yang, W. Wang, C. Zhang, Q. Liu, X.-J. Liang and J. Liu, Adv. Sci., 2019, 6, 1801806 CrossRef PubMed.
- T. Kong, J. Zeng, X. Wang, X. Yang, J. Yang, S. McQuarrie, A. McEwan, W. Roa, J. Chen and J. Z. Xing, Small, 2008, 4, 1537–1543 CrossRef CAS PubMed.
- Y. Chen, J. Yang, S. Fu and J. Wu, Int. J. Nanomed., 2020, 15, 9407–9430 CrossRef CAS PubMed.
- A. Popovtzer, A. Mizrachi, M. Motiei, D. Bragilovski, L. Lubimov, M. Levi, O. Hilly, I. Ben-Aharon and R. Popovtzer, Nanoscale, 2016, 8, 2678–2685 RSC.
- S. Penninckx, A.-C. Heuskin, C. Michiels and S. Lucas, Cancers, 2020, 12, 2021 CrossRef CAS PubMed.
- Y. H. Gholami, R. Maschmeyer and Z. Kuncic, Sci. Rep., 2019, 9, 14346 CrossRef PubMed.
- W. Su, C. Chen, T. Wang, X. Li, Y. Liu, H. Wang, S. Zhao, C. Zuo, G. Sun and W. Bu, Mater. Horiz., 2020, 7, 1115–1125 RSC.
- T. Lima, K. Bernfur, M. Vilanova and T. Cedervall, Sci. Rep., 2020, 10, 1129 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma01123e |
| This journal is © The Royal Society of Chemistry 2022 |