
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in chemical recycling of polyethylene terephthalate waste into value added products for sustainable coating solutions – hope vs. hype
Krishanu
Ghosal
 * and
Chinmaya
Nayak
* and
Chinmaya
Nayak
Research & Development Laboratory, Shalimar Paints Limited, Nashik, Maharashtra 422403, India. E-mail: krishanughosal01@gmail.com; krishanu.ghosal@shalimarpaints.com; Tel: +91-8240626172
First published on 11th January 2022
Abstract
In the current era of globalization, plastics are an indispensable part of our daily life; from our morning toothbrush to night dinner table, plastics are everywhere in our daily life. In such a polymer governing era, the problems related to their pollution are inevitable as most of the commercially used synthetic plastics are non-biodegradable in nature. Among these non-biodegradable polymers, polyethylene terephthalate (PET) constitutes a significant percentage due to its widespread application in the packaging, food and beverage, and automotive industries. This ever-increasing application of PET generates a large volume of post-consumer waste materials which have to be dumped in most of the cases after a certain time. However, with growing environmental awareness, the recycling of polymeric waste materials into valuable ones has become one of the hot topics in mainstream research. There are already a few research articles where researchers have demonstrated that these polymeric waste material derived products can be used further to synthesize new polymeric materials which have applications in various sectors (such as in the fields of regenerative medicine and organic coatings). From this point of view, here we have summarized the recent progress in recycling of PET waste into value-added products and their subsequent utilization in the field of organic coatings. In addition to that we have reviewed, all the major research studies carried out in chemical recycling of PET waste across the globe which can be later used in the coating industry. The review also highlights the existing challenges in this field involved with upscaling of these processes in industry. Additionally, we have predicted future directions on how these recycled products can be used in a sustainable and economically feasible way, so that this can reduce the carbon footprint significantly and help in achieving a closed-loop circular economy.
 Krishanu Ghosal | Krishanu Ghosal is currently working as an Assistant Technical Manager in Shalimar Paints Limited. Prior to joining Shalimar Paints, he submitted his PhD thesis on “Synthesis of biopolymers from recycled plastic waste for tissue engineering applications” in the Department of Polymer Science and Technology, University of Calcutta, under the supervision of Dr Kishor Sarkar. He received his BSc in Industrial Chemistry and MSc in Applied Chemistry from Ramakrishna Mission Vidyamandira, Howrah, West Bengal, India. During his BSc and MSc, he was actively involved in various research activities as well as industrial training. During his MSc summer internship, he worked under Professor T. P. Radhakrishnan, School of Chemistry, University of Hyderabad. There his research theme was “Synthesis of ultrathin film based on poly-N-octadecylaniline based gold and silver nanoparticles using the air–solution interface”. Additionally, during his master's thesis he worked under the guidance of Dr Atanu Bhattacharya at IISc Bangalore on the topic of “Ground state decomposition mechanism of metal contained nitramine molecule”. His research interests include several diverse fields including synthesis of novel polymers, recycling of polymeric materials into value-added products, synthesis of nanomaterials and their biomedical applications, resins for coating applications, green chemistry, etc. |
 Chinmaya Nayak | Mr Chinmaya Nayak is currently serving as the Head of R&D in Shalimar Paints Limited. Previously he worked as Senior Development Manager in R&D at Berger Paints India Limited, Kolkata. He received his MSc degree in Chemistry from Delhi University and his MTech (Corrosion Science & Technology) from NIT – Durgapur. He has 24 years of experience in the paint industry. As a seasoned R&D professional in the paint industry, he has designed and commercialized more than 2000 formulations. Additionally, he has trained more than 100 professionals in the field of paint technology. He has been a part of numerous technical events and conferences. In addition to that, he has been actively involved in the presentation and publication of technical papers. |
1. Introduction
In the modern era of globalization, plastics are an integral part of our daily life; from our morning toothbrush to night dinner table, plastics are everywhere. The development of plastics has started with the use of natural materials that exhibit intrinsic plastic properties, such as shellac and chewing gum, followed by their chemical modification over time. However, the first discovery of fully synthetic plastics was made in the year 1907 by Leo Baekeland.1 After Baekeland's discovery, several new plastics have been developed by polymer scientists with a different set of characteristics, suitable for a wide range of applications.2 It was also perceived that, with continuous improvement in synthesis processes and manufacturing technology, there is a huge improvement in the properties of plastics compared with those of their predecessors, which leads to replacement of traditionally used materials with plastics. This is the prime reason behind the drastic increase in global plastic consumption from the middle of the 20th century. Recent data show that the global production of plastics has increased by 2.7 times on an average since 1950 with a total production of around 8500 million tons.3,4 Currently plastics are one of the crucial classes of components in various industrial sectors including the packaging, construction, transportation and electronics industries. This ever-increasing production and use of plastics leads to generation of an ever-increasing volume of post-consumer plastic waste as most of these synthetically made plastics are non-biodegradable in nature. In this regard, according to a study by Klankermayer et al., around 150 million tons of plastics are generated each year and the number is continuously increasing.4 However, among this massive amount of manufactured plastics, 79% of the plastics end up as landfill, and as a consequence, 2.41 million tons of these robust waste materials enter into our ecosystem every year. To overcome this problem, researchers now focus on synthesis of novel biodegradable synthetic polymers as well as sustainable strategies for recycling of these plastics so that they can enter into a circular economy. Most of the industrially recognized recycling processes reported until now are not cost competitive and produce plastics with poor physical and chemical properties, which ultimately leads to shrinkage in circular economy. Currently the most used plastics in the world include polyethylene terephthalate (PET), polyethylene (PE), polypropylene (PP), polyvinyl chloride (PVC), polystyrene (PS), polylactic acid (PLA), polycarbonate (PC), polymethyl methacrylate (PMMA), polyoxymethylene (POM) and nylons (PA). Most of these plastics are nonbiodegradable and require recycling processes.In this regard, despite the detrimental effect on the environment, the global consumption of PET is increasing continuously year by year due to its outstanding optical transparency, good mechanical properties and excellent thermoformability. Recent data suggested that the global demand of PET will reach 22.65 million tons by the year 2025 with a current value of USD44.1 billion.5 At present, Asia Pacific is the largest market of PET packaging with a market share of 36.7%, and in the next position North America with a market share of 20.5% followed by Western Europe with 17.9% market share.5 It is also expected that within the time span of 2020–2025 the PET based packaging industry will get a huge boost in the countries of the Middle East and Africa due to their growing economy and emerging socioeconomical status.
These statistics suggest that this huge consumption of PET produces a massive amount of post-consumer plastic waste, which is a serious threat from the environmental point of view. According to a recent report published by Taniguchi et al. in 2019, out of the total PET production, only 28.4% was recycled into sheets, fibers, films, and bottles, and the remaining was discarded into the environment.6 To address this challenge researchers and technologists now focus on recycling of postconsumer PET waste into new value-added products. To enlighten this, in this review we have discussed the current scenario of various recycling processes of PET including physical recycling processes, enzymatic recycling processes and chemical recycling processes. Additionally, we have discussed the advantages of chemical recycling processes over the other recycling processes, along with a brief focus on various chemical recycling routes of PET and use of these chemically recycled products for the development of new value-added polymers for coating applications. We have also highlighted that the existing challenges involved with the upscaling of these processes in industry as well as their probable solutions, so that these recycled products can be used in a sustainable and economically feasible way, which will lead to a closed-loop sustainable ecosystem for a cleaner world.
2. Different recycling routes of plastics
2.1. Primary recycling
The most popular and easy process of recycling is primary recycling because of its easy access and low cost. Primary recycling deals with reuse of materials in their original form. However, the main problem associated with primary recycling is that the constraint in their usability after certain cycles as with time their material properties detoriate.7–92.2. Secondary recycling or mechanical recycling
In the secondary recycling process generally thermoplastic polymers are employed due to their remelting and reprocessibility into end products. Mechanical recycling does not involve any alteration in the polymeric backbone during the processing steps.Secondary recycling or mechanical recycling is also known as physical recycling as it involves cutting, shredding or washing waste polymers into granulates, flakes or pellets of suitable quality based on the requirements, and then melted to make the new product by extrusion. It was observed that, in the case of secondary recycling to obtain better physical and chemical properties of recycled plastics, occasionally virgin plastics are blended in some portions. However, the main disadvantage associated with the secondary recycling process is deterioration of the physicochemical properties of plastics in each cycle which occurs due to the drop in molecular weight in each recycling step and heterogeneity present in the plastic waste.10 The drop in molecular weight of recycled plastics takes place for many reasons such as chain-scission reactions during thermal reprocessing, and the presence of acidic impurities and water molecules. To avoid chain-scission reactions, intensive drying of plastic waste after washing and cleaning and use of chain extenders and reprocessing in a vacuum are recommended.7,8
2.3. Enzymatic recycling
Along with the primary and secondary recycling processes, recently the enzymatic/biological recycling process has also received attention due to its ability to tackle the menace of plastic pollution in an eco-friendly manner.11 Enzymatic recycling of plastics involves secretion of extracellular enzymes by microorganisms (which include bacteria as well as fungi), and subsequent reaction of the excreted enzymes with polymers; this leads to the hydro-peroxidation/hydrolysis of polymers and the formation of monomers as well as short polymer intermediates, which are ultimately assimilated by microbial cells as carbon sources to release CO2.12 In this regard, the enzymatic biodegradation of plastics depends on various critical factors; among them the prime one is what types of bonds are present in the polymeric backbone (since the active sites of related enzymes are individual for any specific bond). Thus, the mechanisms of synthetic plastic degradation are usually distinguished into three categories: (1) polymers with carbon back-bones; (2) polymers with ester-bond back-bones and side-chains; and (3) polymers with hetero-carbon bonds present in the backbone. In addition to this crystallinity of the polymer, the pH of the degrading medium, temperature, enzyme specificity, and substituents/additives present in polymers also influence the degradation process.As of now, most of the synthetic plastics are under investigation for enzymatic degradation. However, some recent studies reported that PE,13,14 PS,15,16 PP,17,18 PVC,19 PET20,21 and PU22 can be recycled enzymatically. In the case of PET, generally four types of enzymes are tested for enzymatic degradation, which include Cutinase, Lipase, PETase and Esterase.11 However, due to several limitations of the enzymatic recycling process, they are not implemented in large scale industry, which include (1) the low catalytic turnover due to inadequate accessibility to active sites in polymer as a result of the high crystallinity of the substrate, (2) inhibition by intermediate metabolites, (3) kinetic instability and loss of enzymatic activity above specific temperature,23 (4) during enzymatic biodegradation/hydrolysis, the reaction medium becoming acidic due to end products, which leads to inactivation of the enzyme, and as a consequence, the reaction proceeds in a sluggish manner with time.
2.4. Chemical recycling
Chemical recycling of plastics can be defined as the process in which polymers are chemically transformed into monomers or partially depolymerized into oligomers with the help of a chemical reaction/a set of chemical reactions. These monomers which are obtained from the chemical recycling process can be further used to develop new polymers with the desired properties or to resynthesize the mother plastics again. The main advantage of the chemical recycling process over the other recycling processes is that this method is capable of transforming plastic waste into completely new molecules, which are suitable for use as feedstock materials including monomers, oligomers, and mixtures of other hydrocarbon compounds. There are various methods available which can be used for chemical recycling of plastics including hydrolysis, glycolysis, hydrogenation, aminolysis, gasification, ammonolysis, thermal cracking, catalytic cracking, microwave mediated degradation, etc. However, there are only a few companies working on chemical recycling processes of plastics, as these methods need a lot of initial investment, state of the art technology and expert personnel. Other than these processes at present there are a lot of chemical recycling methods that are under investigation for successful implementation at industry, whereas only a few processes are well established on an industrial scale for plastic recycling. For example, currently the gasification process is under extensive research at industrial R&D to optimize the process on an industrial scale, whereas processes such as glycolysis and methanolysis have gained commercial acceptability.8,24–263. Different chemical recycling routes of polyethylene terephthalate (PET)
Polyethylene terephthalate (PET) is a thermoplastic polyester which was first synthesized by DuPont chemists in the year 1941 in search of synthetic fibers; later they branded their PET fibers as “Dacron”. PET is synthesized by either transesterification reaction between dimethyl terephthalate (DMT) and ethylene glycol or condensation reaction between terephthalic acid (TPA) and ethylene glycol. Depending on the chemical recycling process PET totally depolymerizes into monomers or oligomers such as TPA, bis(hydroxylethylene) terephthalate (BHET), DMT, bis(hydroxylethylene) terephthalamide (BHETA) and any other chemical substances. Until now different recycling routes have been reported for depolymerization of PET including hydrolysis, glycolysis, methanolysis, aminolysis, ammonolysis and hydrogenation; in all of these methods different chemical substances are used for PET chain scission.27 Some of these chemical recycling processes of PET are shown in Fig. 1.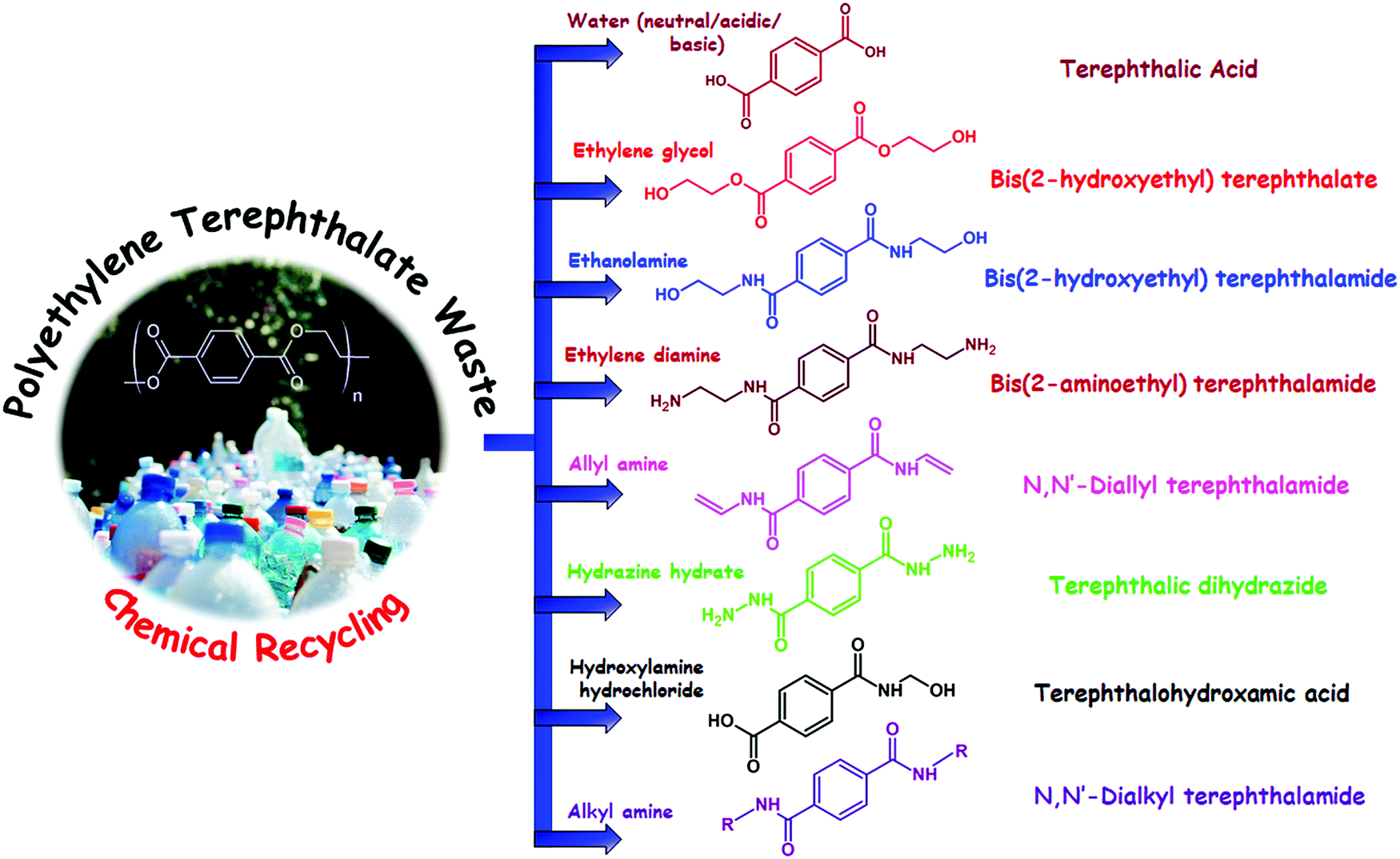 | ||
| Fig. 1 Different materials obtained from chemical recycling of PET waste. From top to bottom: (a) hydrolysis by water, (b) glycolysis by ethylene glycol, (c) aminolysis by ethanolamine, (d) aminolysis by ethylene diamine, (e) aminolysis by ethylene diamine, (f) aminolysis by allyl amines, (g) aminolysis by hydrazine hydrate, (h) aminolysis by hydroxylamine hydrochloride and (i) aminolysis by alkyl amines. | ||
3.1. Hydrolysis
Hydrolysis is one of the well-recognised chemical recycling routes which has been used for several years for depolymerization of PET. In the case of hydrolysis, PET reacts with water molecules under neutral, basic and acidic conditions at high temperature and pressure and produces TPA and ethylene glycol. Alkaline hydrolysis of PET is usually performed in sodium hydroxide solution at concentrations of 4–20%.28,29 In this context, Pitat et al. patented a process of alkaline hydrolysis using 18% sodium hydroxide solution.30 However, studies using other alkaline solutions such as potassium hydroxide, sodium ethoxide, and sodium methoxide have also been performed. For example, Kumar et al. performed extensive research on alkaline hydrolysis of PET waste by aqueous KOH solution under finite solution volume conditions, where they revealed that KOH effectively hydrolyses PET chains and major chain scission takes place at the end chain section.31 On the other hand, in another study, Haith and co-workers investigated the reactivity of NaOH solutions with sodium isopropoxide in isopropyl alcohol, sodium tert-butoxide in tert-butanol, sodium methoxide in methanol, and sodium ethoxide in ethanol for depolymerization of PET.32 They noticed that an ethanolic solution of sodium ethoxide is the most effective for depolymerization of PET, whereas an aqueous solution of NaOH is the least effective for depolymerization. In another study Oku et al. reported that addition of ether along with an alcohol can increase the extent of hydrolysis reaction. They showed that addition of dioxane in methanol can decrease the time of alkaline hydrolysis of PET in the presence of NaOH from 7 h to 0.67 h.33In the case of acid hydrolysis of PET generally concentrated H2SO4 is employed;34,35 however, other concentrated mineral acids such as phosphoric acid36 and nitric acid37–39 are also used for acidic hydrolysis of PET. For example, Mishra and co-workers performed a detailed investigation on the kinetics and thermodynamics during hydrolysis of PET using nitric acid.37,40 However, the major problems associated with acid hydrolysis are severe corrosiveness of inorganic acids and generation of huge quantities of inorganic salts and waste water, whereas neutral hydrolysis of PET is carried out using steam or water in the presence of an alkali metal acetate catalyst.41 Although some recent study also suggests that marine water can also be a potential candidate for neutral hydrolysis of PET,42 the metal ions present in marine water themselves act as a catalyst and no added catalyst is required. Additionally, it has also been observed that under neutral conditions hydrolysis of PET proceeds significantly faster in the molten state than in the solid state, so usually it is performed within a temperature range of 200–300 °C.43 The main advantage of neutral hydrolysis is that it is an eco-friendly process, and thus, no pollution is associated with it; however, the primary drawback related to the neutral hydrolysis is the impurities which are present in PET; in most of the cases they remain in TPA after completion of the recycling process.
3.2. Glycolysis
Glycolysis is the most common and oldest method of PET recycling. The process of glycolysis was first patented in the year 1965. After that, significant efforts have been directed toward improvement and optimization of the process, and currently several industries including DuPont, Shell Polyester, Zimmer, and Goodyear perform recycling of PET using glycolysis. Glycolysis involves transesterification reaction of PET with an excess amount of glycol in a temperature range of 180–240 °C in an inert atmosphere, which leads to the depolymerization of PET and the formation of polyhydric alcohols. Although based on the glycolysis process and glycol used for depolymerization, oligomers with the –OH end group can also be obtained.The structure of polyhydric alcohols depends on the choice of glycol used for depolymerization. However, ethylene glycol is the most well recognised glycol which is used for glycolysis of PET, which generates a diol named bis(2-hydroxyethyl) terephthalate (BHET). Other than ethylene glycol, propylene glycol (PG), polyethylene glycol (PEG), 1,4-butanediol, trimethylolpropane, pentaerythritol and hexylene glycol have also been used for depolymerization of PET using glycolysis.44,45 However, glycolysis by ethylene glycol of PET is considered as a model reaction and investigated in a more detailed fashion. Glycolysis of PET usually involves three steps, the formation of oligomers, dimers and monomers (as shown in Fig. 2). In a typical glycolysis reaction, the glycol molecules diffuse through PET chains, causing them to swell up and subsequently react with ester bonds present in PET chains, leading to degradation of PET into lower fractions.
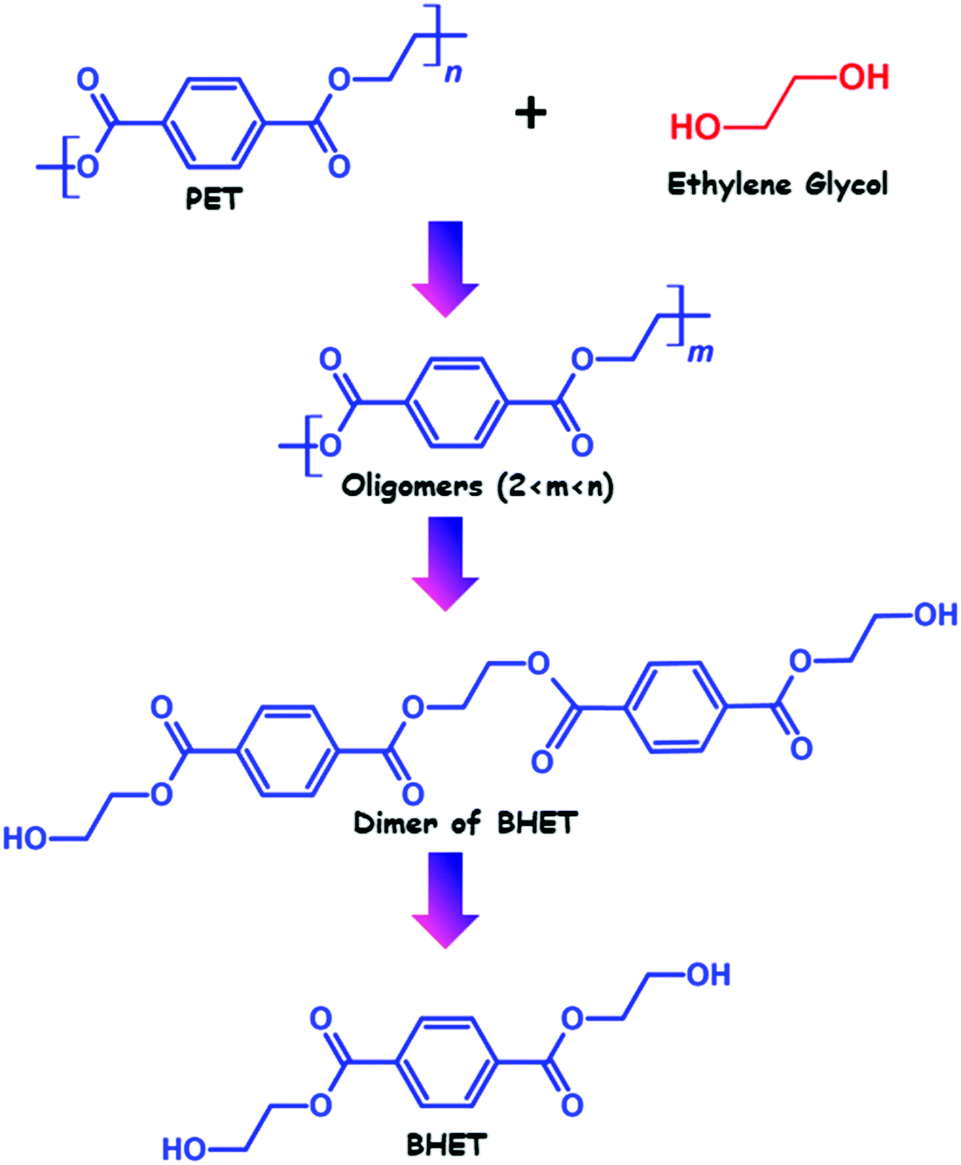 | ||
| Fig. 2 Three different stages of PET depolymerization with ethylene glycol. | ||
During the extensive research on recycling of PET via glycolysis, researchers found that the kinetics of the glycolysis process is very slow and can’t proceed in a faster way without catalyst. It was also observed that without using any catalyst complete depolymerization of PET cannot be achieved and the depolymerization product also contains a mixture of oligomer and monomer in that case, which creates difficulties in obtaining pure monomer. To overcome this problem significant efforts have been directed toward increasing the rate of the reaction and the BHET monomer yield by developing efficient catalysts and optimizing other reaction conditions such as reaction time, temperature, and the PET![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :glycol ratio.
:glycol ratio.
Until now four distinct well-established methods have been used for glycolysis of PET. These are (a) solvent-assisted glycolysis, (b) supercritical glycolysis, (c) microwave-assisted glycolysis and (d) catalyzed glycolysis.
In the case of solvent assisted glycolysis, depolymerization proceeds in the presence of glycol in a reaction medium. For example, Orbay and co-workers used xylene as a solvent for glycolysis of PET, where they observed that glycolysis proceeds in a faster manner in the presence of xylene with an enhanced yield rather than only glycol.46 The main advantage of addition of xylene is that it offers miscibility to the PET–glycol mixture; additionally the glycolysis product is also soluble in xylene, which is an extra advantage. So, as the reaction proceeds, the product moves to the xylene phase, shifting the reaction to the forward direction. However, the main disadvantage associated with this process is that as organic solvents are not eco-friendly the use of this process for PET glycolysis is still highly restricted.
Supercritical glycolysis is carried out at temperature and pressure above the critical point of glycol. Other than glycolysis the applications of supercritical conditions have been employed for other processes such as hydrolysis and methanolysis. The primary advantage of the supercritical process is that there is no need for catalysts for the reaction which are difficult to separate out from the reaction medium. For example, Kim and co-workers studied depolymerization of PET by the supercritical method using ethylene glycol at a temperature of 450 °C and a pressure of 15.3 MPa.47 It was observed that compared to subcritical conditions of 350 °C and 2.49 MPa and 300 °C and 1.1 MPa the reaction under supercritical conditions proceeds in a much faster way. In the case of the supercritical process, a maximum yield of 93.5% BHET was achieved within half an hour of the reaction. The disadvantage of this method is that to carry out the reaction under supercritical conditions very high temperature and pressure are needed.
In case of the microwave assisted recycling process of PET it was perceived that in presence of microwaves the depolymerization reaction time drastically decreased to 35 min from 8 h without any catalyst,48 although it did not affect the product yield. Addition of a suitable catalyst in reaction medium along with microwave irradiation can enhance the reaction efficiency in a synergistic way by increasing the product yield significantly with reduced reaction time. For example, Cho et al. showed that addition of an alkaline catalyst for depolymerization of PET by ethylene glycol in the presence of microwave irradiation can reduce the reaction time drastically with an increased BHET yield.49
Other than these three-methods, glycolysis assisted by catalysts is the most popular and industrially recognised chemical recycling process of PET. It is already well established that glycolysis without catalyst is an extremely slow process and takes a prolonged time to depolymerize the PET with a minimal product yield. Thus, the development of novel catalysts for depolymerization of PET using glycolysis is major research interest among scientists. Until now several types of catalysts have been developed by researchers for glycolysis of PET, which include metal salt-based catalysts, heterogeneous catalysts including zeolites, various nanocatalysts and ionic liquid-based catalysts. Among all these catalysts metal salt-based catalysts are the most prevalent ones due to their easy availability and low cost compared to others. These metal salts include Zn, Mn, Co and Pb salts. In this regard, Baliga et al. investigated the efficiency of four different metal salt (Zn, Mn, Co and Pb) catalysts for depolymerization of PET. They observed that zinc acetate performed best in terms of the extent of depolymerization.50 Later Ghamey and co-workers also discovered that the depolymerization activity of catalysts follows the order of Zn2+ > Mn2+ > Co2+ > Pb2+.51
Meanwhile, Chen et al. found that the best conditions for glycolysis in the presence of manganese acetate are 190 °C with 0.025 mol kg−1 of PET for 90 min,52 whereas Sun and co-workers reported that in the presence of zinc acetate catalyst the optimum temperature for glycolysis of PET using ethylene glycol is 196 °C.53 In that case, the glycol:PET weight ratio should be maintained at 5, and the catalyst:PET weight ratio should be preserved at 0.01. Since zinc and lead are heavy metals which are well known for their detrimental effect on the environment, researchers also investigated mildly alkali metal salt-based catalysts for glycolysis of PET. For example, Shukla and co-workers used sodium carbonate and sodium bicarbonate as catalysts for glycolysis of PET and found that the product yields were almost comparable to those of conventional lead and zinc acetate catalysts.54 In another study they reported that other nonconventional catalysts such as lithium hydroxide, sodium sulfate, potassium sulfate and glacial acetic acid can also deliver similar product yields with traditional heavy metal catalysts for glycolysis of PET.43 The latest catalysts which were developed by Shukla and co-workers are metal chloride salts; these salts are cheap and readily available. However, among these salts, chloride salts especially zinc chloride performed best in terms of production of BHET with a yield of 73.24%.55 Furthermore, it was found that homogeneous catalysts are highly effective for glycolysis of PET. But the main drawback of homogeneous catalyst systems is that it is difficult to separate them out from the reaction medium as they are soluble in ethylene glycol. In most of the cases an additional distillation unit operation is required to separate out these catalysts.
In addition to homogeneous metal salt-based catalysts nowadays heterogeneous catalysts are also gaining popularity for glycolysis of PET due to their outstanding catalysis efficiency, reusability and facile separation techniques. These heterogeneous catalysts include zeolites, spinels and nanomaterial based catalysts. In this regard, Shukla and co-workers reported a novel eco-friendly zeolite catalyst for depolymerization of PET using glycolysis by ethylene glycol.56 Zeolites are well acknowledged for their catalytic activity owing to their microporous and mesoporous structures which act as active sites. The authors found that the yield of BHET is more than 60%, which is analogous with traditionally used heavy metal catalysts such as zinc and lead acetate. In another study, Imran et al. used metal oxide based silica nanoparticles (SNPs)/silica microparticles (SMPs) for catalytic glycolysis of PET at 300 °C temperature, 1.1 MPa pressure with an ethylene glycol-to-PET molar ratio of 11:1 and a catalyst-to-PET-weight ratio of 1.0% for 40–80 min.57 They explored four different catalyst systems for depolymerization of PET including Mn3O4/SNPs, ZnO/SNPs, Mn3O4/SMPs, and ZnO/SMPs. However, among these catalyst systems the Mn3O4/SNP nanocatalyst showed the highest product yield (>90%). This high monomer yield is attributed to its large surface area and porous structures which are present in the nanocatalyst and serve as active sites for the reaction. In their subsequent study they employed novel mesoporous metal oxide and mixed metal oxide based spinel catalysts for glycolysis of PET.58 In this study they used two different metal oxide spinels (Co3O4, Mn3O4) and three different mixed metal oxide based (ZnMn2O4, CoMn2O4, and ZnCo2O4) catalysts; the zinc manganite tetragonal spinel catalyst (ZnMn2O4) performed best among all, with the highest BHET yield (92.2 mol%) at 260 °C temperature and 5 atm pressure. Later in the year 2012 Park and co-workers utilized a graphene oxide–manganese oxide nanocomposite (GO-Mn3O4) for catalytic glycolysis of PET.59 They obtained the highest monomer yield of 96.4% with the nanocomposite containing the lowest amount of Mn3O4, while without GO in the presence of only Mn3O4 they got about 82.7% BHET yield. In another recent study, Guo and co-workers explored the Perkalite F100 nanocatalyst for glycolysis of PET.60 They revealed that at a low nanocatalyst concentration of 0.1 wt% of PET complete depolymerization of PET can take place, and the nanocatalyst is far superior to sodium bicarbonate and zinc acetate metal salt catalysts. In addition to that, the Perkalite F100 nanocatalyst is environmentally benign and economically feasible, which make it one of the suitable catalysts for chemical recycling of PET.
Other than these heterogeneous catalyst systems in recent times ionic liquid-based catalysts have also gained extensive popularity for glycolysis of PET due to their outstanding catalytic ability and easy separability from the solid glycolysis product. Ionic liquids are primarily defined as salts in the liquid state which have melting points below 100 °C. The primary reason behind the low melting point is that ionic liquids are generally composed of large unsymmetrical ions whose charge is disseminated over large volume so the melting points of these ionic liquids can be reach well below 0 °C. Some of the fascinating properties of ionic liquids are good thermal and electrochemical stability, nonvolatility, strong solvent power for both inorganic and organic compounds and low flammability, which make them one of the potential candidates for catalyzed glycolysis of PET.61–63 For example, in a pioneering study, Wang et al. showed that an ionic liquid can catalyze glycolysis of PET with excellent efficiency.64 To carry out the glycolysis they employed 1-butyl-3-methyl-imidazolium bromide ([Bmim][Br]) catalyst within the temperature range of 160 to 195 °C for a time span of 5–10 h in the presence of ethylene glycol. In that case, complete depolymerization of PET was achieved after 8 h of reaction at a temperature of 180 °C. Furthermore, they also investigated the reusability of this catalyst system, where they found that the catalyst can be used multiple times without compromising their catalytic efficacy. Later Yue and co-workers reported that the basic ionic liquid, 1-butyl-3-methylimidazolium hydroxyl ([Bmim][OH]), can exhibit much higher catalytic activity for the glycolysis of PET, compared to 1-butyl-3-methylimidazolium bicarbonate ([Bmim][HCO3]), 1-butyl-3-methylimidazolium chloride ([Bmim][Cl]) and 1-butyl-3-methylimidazolium bromide ([Bmim][Br]).65 Under optimum conditions (a PET:EG mole ratio of 1:10, a dosage of [Bmim][OH] of 0.1 g (5 wt%), a temperature of 190 °C and a time of 2 h), the depolymerization of PET and conversion of the BHET yield were 100% and 71.2% respectively.
Zhang and his group members developed an iron containing ionic liquid, 1-butyl-3-methylimidazolium tetrachloroferrate ([Bmim][FeCl4]), for catalyzed glycolysis of PET.66 This ionic liquid demonstrated much higher catalytic efficiency compared to 1-butyl-3-methylimidazolium chloride ([Bmim][Cl]); however, the main problem associated with this catalytic system is that the monomer product can be easily contaminated (stained) by iron containing ionic liquids and needs to be washed with a large volume of water, which ultimately leads to a loss of product yield. In another very recent study, Yeo and co-workers investigated the mechanistic roles of cations and anions in catalytic glycolysis of PET using ionic liquids.67 They found that cations primarily attack at the carbonyl oxygen of PET to catalyze the reaction, while anions form strong H-bonds with PET and ethylene glycol. They used four different types of ionic liquid catalysts (N1111Ala, N1111Cl, ChCl, MmimCl, BmimCl) for catalyzed glycolysis of BHET, where the cationic part of the ionic liquid coordinates with the carbonyl oxygen of the ester, and the hydroxyl oxygen of ethylene glycol attacks the ester group carbon of PET, with proton transfer to the carbonyl oxygen. Other than these studies, there are many more studies which are focused on catalyzed glycolysis of PET using ionic liquids; some of them are listed in Table 1.
| PET:glycol wt ratio |
Ionic liquid(s) used | Reaction conditions | Product yield | Ref. |
|---|---|---|---|---|
| 1:11 |
[Hmim][ZnCl3], [Hmim][CoCl3], [Hmim][FeCl4] and [Hmim][CuCl3] | 1 atm pressure, 190 °C temperature, 2 h time | 87.1% | 68 |
| 1:20 |
Cu(OAc)2–[Bmim][OAc], Zn(OAc)2–[Bmim][OAc] | 1 atm pressure, 190 °C temperature, 3 h time | 53.95% and 45.6%, respectively | 69 |
| Different amounts of PET:glycol |
[Deim][Zn(OAc)3] | 1 atm pressure, 160–190 °C temperature, 1–4 h time | 70.94% | 70 |
| 3:20 |
[Bmim][OAc] | 1 atm pressure, 190 °C temperature, 3 h time | 58.2% | 71 |
| 1:4 |
[Ch][OAc] | 1 atm pressure, 180 °C temperature, 4 h time | 85.2% | 72 |
| 1:2, 1:6 and 1:10 |
[Bmim][ZnCl3] | 1 atm pressure (constant for all cases); 170 °C temperature, 1 h time;180 °C temperature, 1.5 h time; 190 °C temperature, 2 h time respectively | — | 73 |
3.3. Aminolysis
Aminolysis of PET is also another well-recognised method of chemical recycling which yields the corresponding diamides of terephthalic acid (TPA). However, until now there have been no reports identified which concern with utilization of this process on an industrial scale for chemical recycling of PET. But the aminolysis products of PET have huge potential for further applications in various industrial sectors especially in the coating industry. Aminolysis of PET is usually carried out in the presence of primary amine aqueous solutions such as ethanolamine, ethylamine, and anhydrous n-butylamine within the temperature range of 20–200 °C. Similar to glycolysis, aminolysis is also performed in various methods such as aminolysis without use of any catalyst, catalyzed aminolysis and microwave assisted aminolysis; some of them are summarized below.For example, in a preliminary study in the era of 1960s Zhan et al. reported aminolysis of PET fibers with aqueous solutions of hydrazine, benzylamine, ethylenediamine, hexamethylenediamine, piperidine and aniline, which led to generation of amides of terephthalic acid and oligomeric diacids: terephthalic dihydrazide, terephthalic bis-benzylamide, bis-benzylamides of oligomers and mixtures of oligomeric bis-ethylenediamides, which do not have any potential for further chemical reactions to obtain value added products.74 Later in the year 2006, Shukla and co-workers reported aminolysis of PET by ethanolamine at a molar ratio of 1:6 in the presence of different catalyst systems for different time periods up to a maximum of 8 h.75 These catalysts include glacial acetic acid, sodium acetate and potassium sulfate with PET weights between 0.3 and 1.5%, and produce bis(2-hydroxyethylene) terephthalamide (BHETA) with a maximum yield of 91%. Tawfik and co-workers also reported catalyzed aminolysis of PET using ethanolamine in the presence of dibutyltin oxide (catalyst) at 190 °C temperature and 1 atm pressure.76 The reaction yield varied between 49 and 62% with reaction times of 1 to 4 h and catalyst percentages of 0.5 or 1% of PET weight. In another very recent study Ghosal et al. reported catalytic aminolysis of PET with ethanolamine.77 Briefly they carried out the experiment with a PET:ethanolamine molar ratio of 1:12 at 180 °C for 7 h in the presence of sodium acetate (1% of the total PET weight) catalyst. Spychaj and co-workers performed aminolysis of PET waste using polyamines, such as diethylenetriamine and triethylenetetramine, and their mixtures, as well as mixtures of triethylenetetramine and p-phenylenediamine or triethanolamine.78 Briefly they carried out the reactions at 200–210 °C with a PET:amine ratio of 1:2 and characterized the final products with NMR.
Other than the conventional heating process, microwave irradiation also offers an excellent platform for aminolysis of PET waste without compromising in terms of product yield. In this regard, Shukla and co-workers reported microwave assisted aminolysis of PET in the presence of excess ethanolamine.79 To accelerate the reaction further, they employed low-cost, environment friendly catalysts such as sodium acetate, sodium bicarbonate and sodium sulfate within the system, which led to the formation of BHETA with a yield of up to 90% at a shorter reaction time. In their subsequent study also, they performed aminolysis of PET using hydrazine monohydrate using both microwave irradiation and the conventional heating process in the presence of sodium acetate and sodium sulfate catalysts.80 They observed that in the case of microwave irradiation the yield of terephthalic dihydrazide varied between 74 and 86%, whereas for the conventional heating process it was 40–60%. Additionally, they also discovered that in the presence of microwave irradiation the reaction proceeded much faster (within a couple of minutes), whereas in the case of the conventional heating process it required almost 6 h to get a maximum product yield. In another study, Achilias and co-workers also performed microwave assisted depolymerization of PET waste by ethanolamine; however, in this case they did not utilize any catalyst system for the reaction.81 They observed that in the presence of microwave irradiation (microwave power > 100 W) complete depolymerization of PET took place within 5 minutes with a yield of 100%, which was far superior to thermal depolymerization of PET using aminolysis.
3.4. Methanolysis
Methanolysis of PET is usually carried out using methanol within a temperature range of 180–280 °C and in a pressure range of 20–40 atm with or without catalyst, which leads to production of dimethyl terephthalate and ethylene glycol.82,83 For example, Gruschke et al. reported depolymerization of PET waste using methanolysis in the absence of catalyst at 210 °C with a depolymerization efficiency of 99%.84 Later Sako et al. proposed a novel process of methanolysis where they used supercritical methanol for depolymerization of PET.85 They performed the process without using any catalyst, above 300 °C at 11 MPa pressure for half an hour, which resulted in complete depolymerization of PET. In the case of catalytic methanolysis typical transesterification catalysts are used such as zinc acetate, cobalt acetate, magnesium acetate and lead dioxide. However, similar to glycolysis, in the case of methanolysis also zinc acetate is the most widely used catalyst. For instance, Mishra and co-workers examined the kinetics and thermodynamics of methanolysis of PET waste in the presence of zinc acetate catalyst.86 They found that the optimum percentage conversion of PET waste into dimethyl terephthalate and ethylene glycol (EG) is 97.8% at 120°C, whereas it is 100% at 130 and 140°C for 2 h of reaction. In another very recent study, Enthaler and co-workers reported depolymerization of end-of-life PET via zinc acetate-catalyzed methanolysis which produced a high dimethyl terephthalate yield of 98%.87 However, the main drawback associated with methanolysis of PET is that, in addition to dimethyl terephthalate, the reaction products of methanolysis also contain glycols, alcohols, and phthalate derivatives. The separation and refinement of each product from others make methanolysis a rather costly process. It was found that the cost of methanolysis-derived dimethyl terephthalate from recycled PET bottles is approximately twice that of virgin dimethyl terephthalate.88 For this reason methanolysis is not that much popular in industry for PET recycling.
3.5. Ammonolysis
Ammonolysis of PET is generally carried out with liquid ammonia, either with catalyst or without catalyst in a temperature range between 70 and 180 °C under pressure.89 In the case of the catalytic ammonolysis process generally zinc acetate is used as the catalyst. The primary product obtained from ammonolysis of PET is 1,4-benzene dicarboxamide, also known as terephthalamide. Unlike the other recycling processes of PET, at present ammonolysis is not that much popular for chemical recycling of PET waste.4. Current developments on sustainable coating solutions based on chemical recycling of PET
In the last couple of years, substantial growth has been noticed in the consumption of chemically recycled PET waste derived products as raw materials for the synthesis of different classes of novel polymers including epoxy resins, alkyd resins, polyester resins, polyurethane resins and vinyl esters. In this section of this review, we have discussed the application of PET waste derived products for synthesis of new polymers for coatings.4.1. Epoxy resins
Epoxy resins are a class of well-recognised reactive intermediates which are used to develop a wide range of thermosetting polymers. They are generally characterized by the presence of a three membered oxirane ring in their structure more commonly known as the 1,2-epoxide ring. Over the last few years, it has been perceived that recycled PET derived products are extensively used as raw materials to produce epoxy resins. For example, Macijauskas and co-workers developed a two-pack epoxy resin system using a polyester polyol as base material and melamine formaldehyde as a hardener, where the polyester polyol produced from transesterification reaction between jatropha oil/castor oil and glycolyzed oligoesters derived from recycled PET waste with a poly(propylene glycol) molecular weight of 2000.90 They noticed that the final coating performance of the resin was significantly influenced by the amount of PET used for glycolysis along with the type of oil used. Atta et al. also synthesized epoxy resins from recycled PET waste derived products for organic coating application.91 To synthesize resins, they chemically recycled PET waste with different ratios of diethanolamine to triethanolamine in the presence of manganese acetate catalyst and reacted the obtained chemolysis products with epichlorohydrin to prepare a series of di- and tetraglycidyl epoxy resins with different molecular weights. To cure the resins, they used p-phenylene diamine and diamino-diphenylether as curing agents. The resins demonstrated outstanding adhesion properties and good thermal stability, which suggests their potential application in organic coatings. In their subsequent work also they prepared epoxy resins from recycled PET waste derived products for coating application.92 Briefly they performed chemical recycling of PET using trimethylol propane (TMP), triethanolamine (TEA), diethylene glycol (DEG) and diethanolamine (DEA) to produce suitable hydroxy-oligomers for epoxy. Subsequently, to prepare the final epoxy resins they reacted these glycolyzed and aminolyzed products of PET with epichlorohydrin, which led to the formation a series of di- and tetraglycidyl epoxy resins with different molecular weights. To cure the epoxy resins, they further reacted the resins with 1-(2-amino ethyl)piperazine at room temperature. The final cured resins demonstrated excellent adhesion properties with steel along with good resistance to alkalis and acids, which is attributed to the high cross-link density of polymer networks due to high epoxy functionalities. In addition to that, the epoxy resins also demonstrated good solvent resistance, salt spray resistance (the salt spray time varied between 300 and 750 h depending on the resin type) and hot water resistance, which indicates their potential application in GI coatings. Bal et al. also synthesized epoxy-based coating systems from the glycolyzed product of PET and evaluated their wet film and dry film properties.93 Briefly to synthesize the epoxy they reacted the glycolyzed products (obtained using ethylene glycol) of PET (i.e. BHET) along with bisphenol-A and epichlorohydrin using toluene as a solvent at 100 °C in the presence of H2SO4 catalyst for 2 h. At the end of the reaction to neutralize the acid present in the reaction medium, they treated the reaction mixture with calcium carbonate. Additionally, to perform the dehydrohalogenation of the epoxy resin they employed thirty percent NaOH solution. Finally, they used the synthesized epoxy resin along with a commercial epoxy resin (solvent-free, two-component epoxy resin) as an epoxy-based paint formulation as a binder. The epoxy resin showed excellent hardness, drying degree, and chemical film forming properties on oven curing; however, certain decreases in abrasion resistance and impact resistance were detected in its film properties. Furthermore, the resin demonstrated outstanding adhesion properties as well as detergent and water resistance, which suggests its potential application as an organic coating. In another similar type of study Czub et al. reported synthesis of epoxy resins from PET waste and investigated their properties for coating application.94 Similar to Bal et al.93 they also reacted the glycolyzed product of PET waste with epichlorohydrin followed by purification stages to obtain the epoxy resin. The block diagram of the synthesis and modification of epoxy resins from recycled PET is shown in Fig. 3.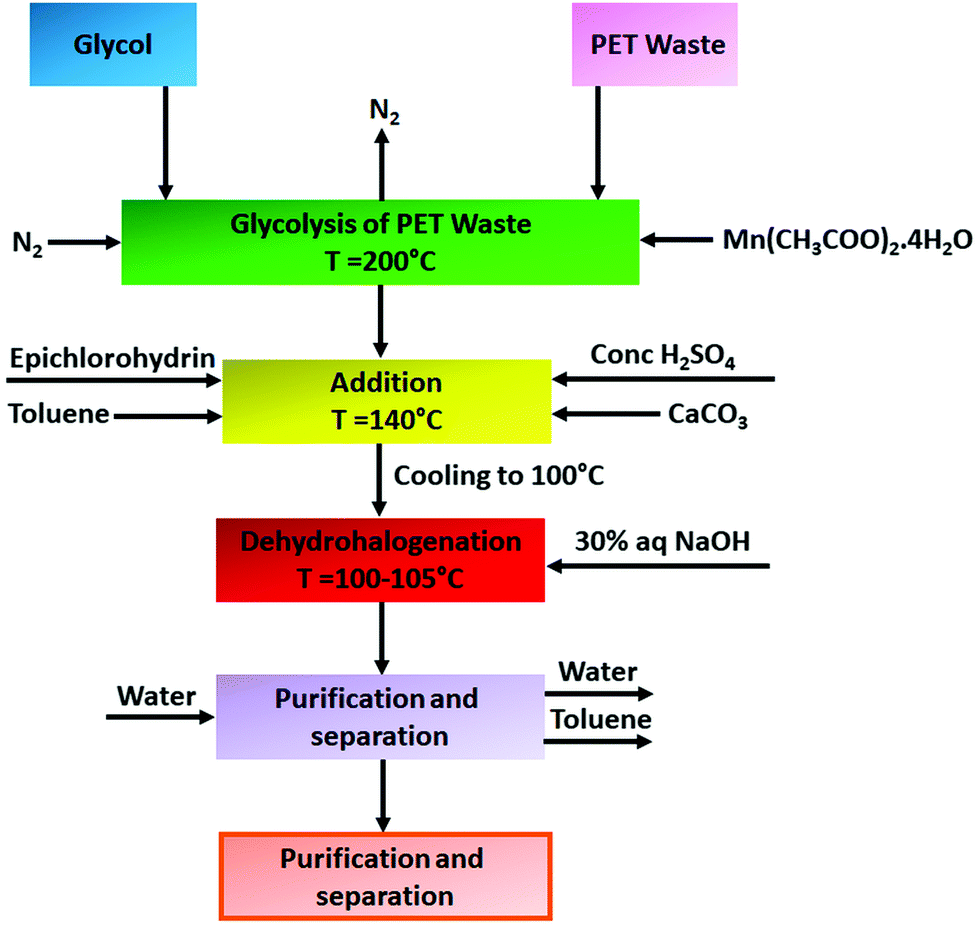 | ||
| Fig. 3 Block diagram of the synthesis of epoxy resins from the glycolyzed product of recycled PET waste and epichlorohydrin. | ||
They also found that incorporation of the glycolyzed product of PET waste led to improvement in the tensile and flexural strengths, hardness, and thermal stability of epoxy resins. Additionally, it was also witnessed that addition of the glycolyzed product of PET improves the overall acid and ethyl acetate resistance performance of the epoxy system. Therefore, from the above studies it can be summarized that incorporation of recycled PET waste derived products (especially the glycolyzed and aminolyzed ones) in the epoxy resin system not only enhances the adhesion properties of the system, but also improves the overall coating performance of the paint system, which include flexibility, water resistance, organic solvent resistance, and acid as well as alkali resistance, which make them promising candidates for coating applications.
4.2. Polyester resins
Similar to epoxy resins, polyesters resins are also an attractive class of candidates for coating application. For example, a recent study predicted that the global market size of polyester resins will grow to USD12.9 billion by the year 2026 from USD9.6 billion in 2021, at a compound annual growth rate of 6.1%, which reflects the massive global consumption of polyester resins in the field of the paint and coating industry.95 Polyester resins are usually prepared by reacting polybasic acid with polyhydric alcohol. Depending on the type of chemical used for depolymerization and the nature of the dicarboxylic acid used for subsequent polycondensation, the synthesized polyester may be saturated or unsaturated. For example, Vaidya et al. in their pioneering study reported synthesis of unsaturated polyester resins from recycled PET waste products for the first time.96 For their resin preparation, at first they depolymerized PET with different amounts of propylene glycol followed by reaction of the depolymerized product with maleic anhydride along with styrene. Furthermore, they compared their developed resins with conventional general-purpose resins and observed that their developed polyester resins are comparable in terms of processibility. However, they did not use them for any end use application purposes. It was noticed that for preparation of unsaturated polyester resins propylene glycol (PG) is favoured over ethylene glycol or diethylene glycol as a glycolytic agent. The reason behind is that PET/PG-derived unsaturated polyester resins are compatible with styrene and increase the activity of the anhydrides before starting the reaction, whereas PET/ethylene glycol- or PET/diethylene glycol-derived resin glycolysis products are not fully compatible with styrene; as a result, the reaction proceeds in a sluggish way. Pimpan et al. synthesized a series of unsaturated polyester resins from postconsumer PET bottles derived from polyhydric alcohol and maleic anhydride.97 Here they depolymerized PET using different glycols such as ethylene glycol (EG), propylene glycol (PG), and diethylene glycol (DEG) in the presence of zinc acetate catalyst and subsequently reacted the depolymerized products with maleic anhydride at 200 °C for 10 h under a nitrogen atmosphere, which resulted in the formation of polyester resins. In another study, Tawfik and co-workers reported synthesis of water-soluble polyester resin based organic coatings using recycled PET waste derived products as starting materials.98 To depolymerize the PET they employed 2,2-dimethyl-1,3-propanediol with different molar ratios in the presence of zinc acetate as a trans-esterification catalyst. Finally, to synthesize the polyester resin, they simply performed melt polycondensation reaction of the glycolyzed product of PET with adipic acid, isophthalic acid, 2,2-dimethyl-1,3-propanediol and trimellitic anhydride in different ratios. FTIR and NMR characterization confirmed that the successful synthesis of the polyester resin took place. For end use coating application, the authors neutralised and cured the polyester resin with different combinations of dimethylethanolamine and triethylamine with methylated high imino melamine formaldehyde. The polyester resin prepared with a glycolyzed product:isophthalic acid:adipic acid:trimellitic anhydride molar ratio of 1:5:1:0.3 and neutralized and cured with dimethylethanolamine and methylated high imino melamine formaldehyde showed the best results for coating application. The final coatings made with this recipe showed excellent mechanical and chemical resistance properties as well as very good hardness along with weatherability, which suggest their application for storing enamels for metal substrates. In addition to enamel-based coatings, recycled PET waste derived polyesters are also used for powder coating application.
For example, Gioa et al. synthesized a polyester by reacting PET derived oligomers with isosorbide and succinic acid for powder coating applications.99 The scheme for synthesis of the polyester from PET, isosorbide and succinic acid is shown in Fig. 4. The polyester coating obtained from this new eco-friendly approach demonstrated very similar applicative properties, such as gloss and direct and reverse impact strength (in some cases superior), to a commercial reference coating obtained from non-renewable resources.
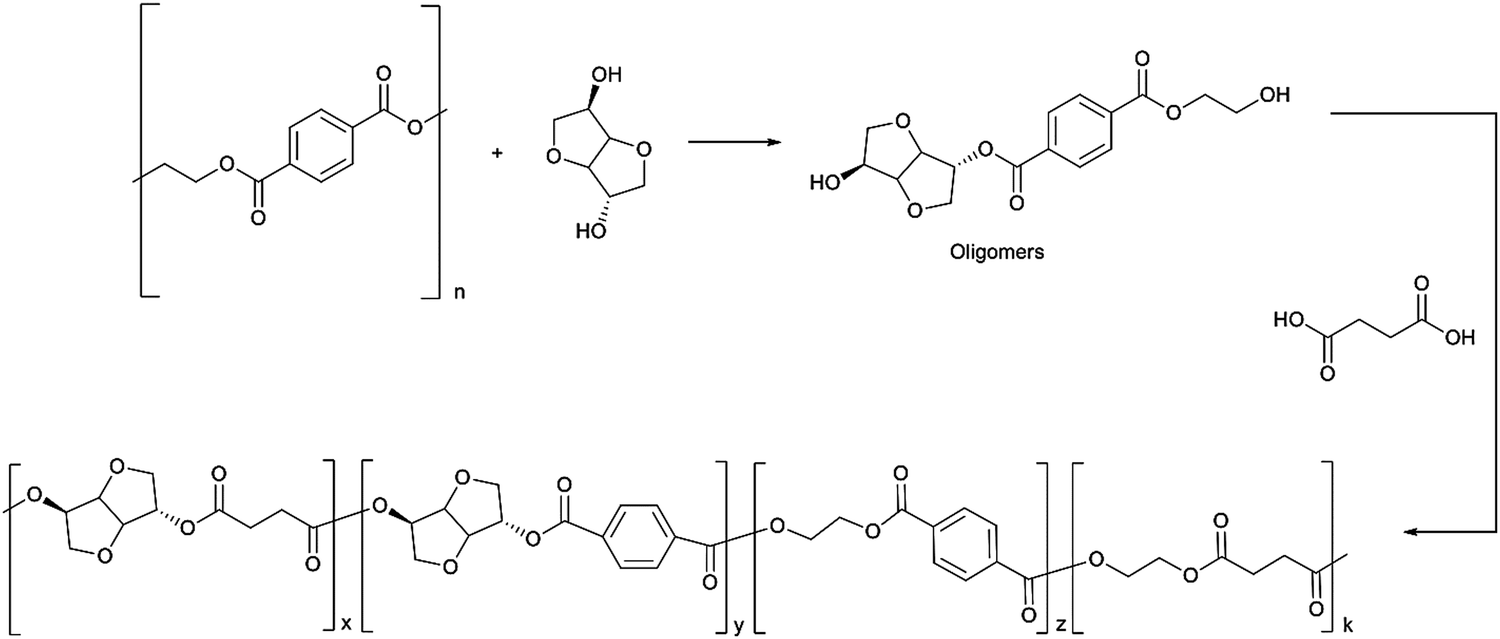 | ||
| Fig. 4 Chemical recycling of PET waste with isosorbide and subsequent polymerization with succinic acid to obtain a polyester for powder coating application. Reproduced with permission from ref. 99. Copyright 2013 Royal Society of Chemistry. | ||
4.3. Alkyd resins
Alkyd resins are oil-based resins which consist of polyols, dibasic acids and triglycerides or fatty acids of oils. They are the most extensively used polymers for a wide range of paint and coating applications due to their low cost, inherent biodegradability, and excellent mechanical and durability characteristics.100–103 In addition to these, their excellent compatibility with a broad range of polymers makes alkyd resins superior to other coating systems in several areas of applications with special demands such as corrosion prevention and hydrophobic antifouling coating.104–106 In this regard, PET based recycled polyols can serve as some of the prime components for preparation of alkyd resins. There are several studies where researchers worked with recycled PET derived polyols for synthesis of alkyd resins for coating application. For example, Atta and co-workers developed alkyd resins derived from recycled PET waste for corrosion resistance protective coating of carbon steel.107 Briefly to prepare the alkyd resins, at first, they glycolyzed PET with pentaerythritol in the presence of manganese acetate catalyst and m-cresol solvent at 220 °C to obtain suitable hydroxyl oligomers. Subsequently, they reacted these PET derived oligomers with sunflower oil/linseed oil, glycerine, and phthalic anhydride, in the presence of butylhydroxytin oxide catalyst to obtain the final alkyd resins. A performance evaluation study revealed that the sunflower oil-based alkyd resin exhibited superior mechanical and adhesion properties with steel. In addition to that, the sunflower oil based alkyd resin exhibited excellent chemical resistance, among the other cured resins. Incorporation of PET derived oligomers also resulted in enhancement of the properties of the alkyd resin. In another recent study, Ma et al. synthesized a waterborne alkyd resin system from recycled PET waste for coating applications.106 For synthesis of alkyd resins similar to Atta et al.,107 they used the glycolyzed product of PET. But, instead of pentaerythritol, they used trimethylolpropane (TMP) for glycolysis of PET. To prepare the final resin formulation along with the glycolyzed product of PET, they used Zanthoxylum bungeanum seed oil (ZSO), phthalic anhydride and 2,2-bis(hydroxymethyl)propionic acid (DMPA) as raw materials. The schematic synthesis procedure of their waterborne alkyd resin is shown in Fig. 5. Compared to the commercially available alkyd resin, the alkyd resin prepared from ZSO and the glycolyzed PET product exhibited enhanced hardness, thermal stability and chemical resistance with a faster drying time, which suggests its potential application in organic coatings. Güclü et al. also formulated an alkyd resin with an oil content of 60% by reacting phthalic anhydride (PA) with simultaneous hydrolysis and glycolysis products derived from PET waste, glycerine (G), sunflower oil fatty acid (SOFA) and ethylene glycol.108 Their developed alkyd resin also showed a better drying time, hardness and abrasion resistance compared to the reference alkyd resin. In addition to that, the PET-based resin showed almost the same water resistance and adhesion strength values as the reference resin, whereas in their subsequent study they reported synthesis of two short oil alkyd resins with high acid values (30–40 mg KOH g−1) utilizing the glycolyzed products of PET waste.109 Phthalic anhydride (PA), coconut oil fatty acids and glycerine were mixed with glycolyzed products of waste PET to obtain the final PET-based alkyd resins.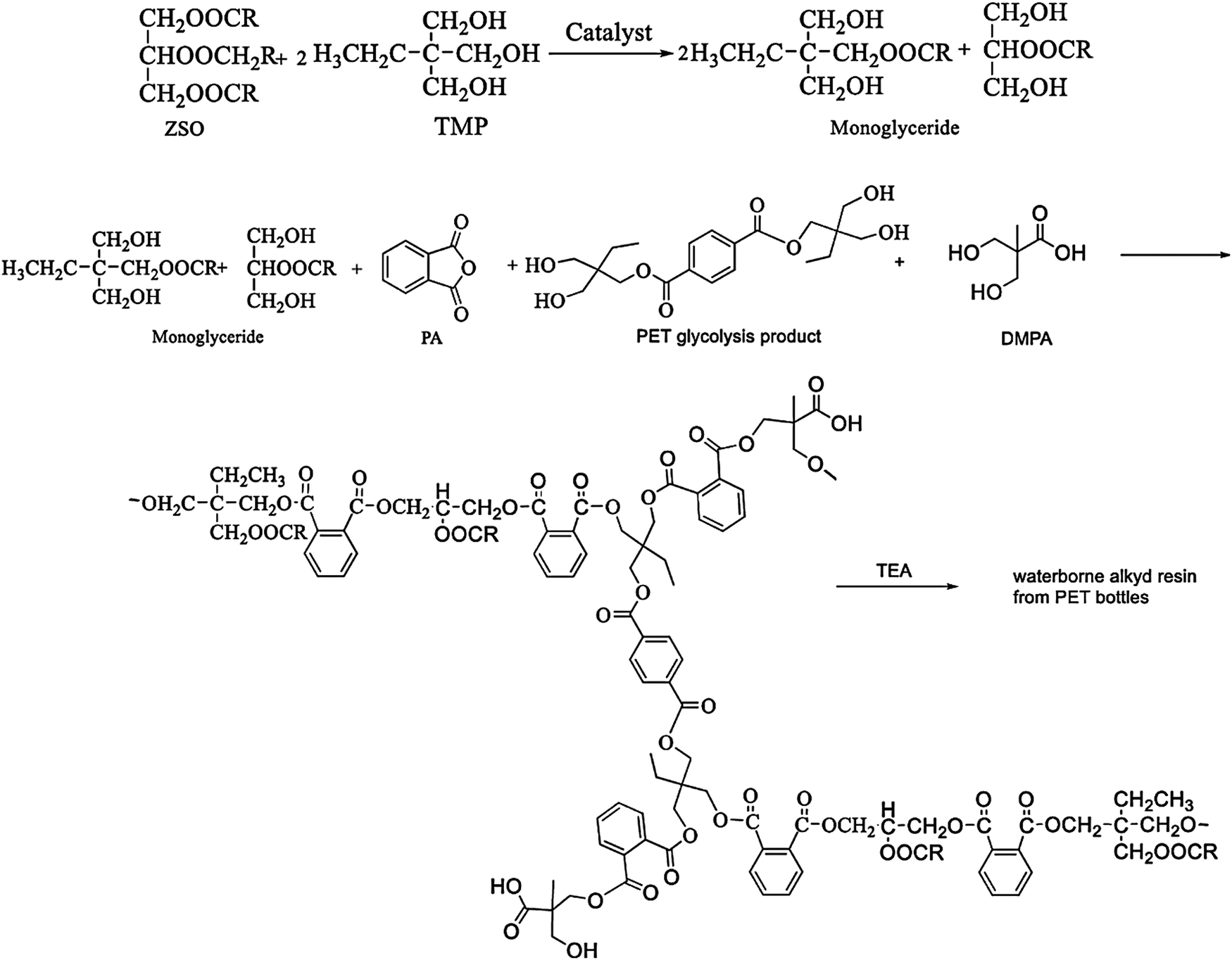 | ||
| Fig. 5 Synthesis of a waterborne alkyd resin from glycolyzed PET and using other monomers. Reproduced with permission from ref. 106. Copyright 2020 Springer. | ||
For final coating applications they blended alkyd resins with 30%, 40%, and 50% commercial urea–formaldehyde, melamine–formaldehyde and urea–formaldehyde/melamine–formaldehyde (1:1 weight ratio) mixtures and heated at 140 °C. The final alkyd-amino resins exhibited excellent thermo-oxidative degradation resistance, hardness and chemical resistance, which indicates that these resins are suitable for manufacturing of industrial baking enamels. Spasojević et al. also reported synthesis of high-performance alkyd resins derived from postconsumer recycled PET waste.110 To obtain the alkyd resins at first they performed glycolysis of PET using a set of difunctional and multifunctional alcohols such as diethylene glycol (DEG), propylene glycol (PG) dipropylene glycol (DPG), glycerol (G), trimethylolethane (TME), trimethylolpropane (TMP) and pentaerythritol (PE), respectively, which led to the formation of di-, tetra- and hexa-functional glycolyzates. In the next step, they carried out a reaction using these glycolyzed products (individually) along with linseed oil fatty acid (LFA), TMP, PE and phthalic anhydride (PA) to get the final set of alkyd resins. In terms of performance, they observed that alkyd resins derived from multifunctional glycolyzates (TME and TMP) showed significantly improved properties in terms of hardness, chemical resistance, drying time and adhesion compared to those synthesized from difunctional glycolyzates and also compared to traditional general-purpose alkyd resins. Other than the glycolyzed product of PET, the aminoglycolyzed product of PET was also employed for synthesis of water reducible alkyd resins. For example, Acar and his team members developed a water reducible alkyd resin from the aminoglycolyzed product of PET waste.111 Their final alkyd formulation was composed of glycerine (G), ethylene glycol (EG), fatty acids (FAs), and phthalic anhydride (PA) or aminoglycolysis depolymerization products (ADPs). From performance test results they found that the properties of the waste PET-based alkyd resin are compatible with the properties of the reference resins. In addition to that, they also revealed that the thermal stability of the water-reducible alkyd resin prepared with the aminoglycolyzed product of PET was far better than that of the commercial reference resin, which makes it an ideal contender for coating application. In another very recent study, Ding and co-workers also reported synthesis of water reducible alkyd resins from postconsumer PET waste derived products.112 For depolymerization of PET, they used trimethylolpropane, which led to terephthalate of trimethylolpropane, EG and unreacted trimethylolpropane. Subsequently, they employed the glycolyzed PET product as a raw material for alkyd synthesis along with tall oil fatty acid (TOFA), PA, PE, EG, benzoic acid (BA) and trimellitic anhydride (TMA) (the detailed synthesis scheme of alkyd resins is shown in Fig. 6). They noticed that the presence of PET enhanced the viscosity of water-reducible alkyd resins compared to water-reducible alkyd resins without PET; the reason behind that is that PET contains harder terephthalic acid monomeric units, whereas the alkyd resins without PET contain more PE with greater steric hindrance, which leads to reduced viscosity.
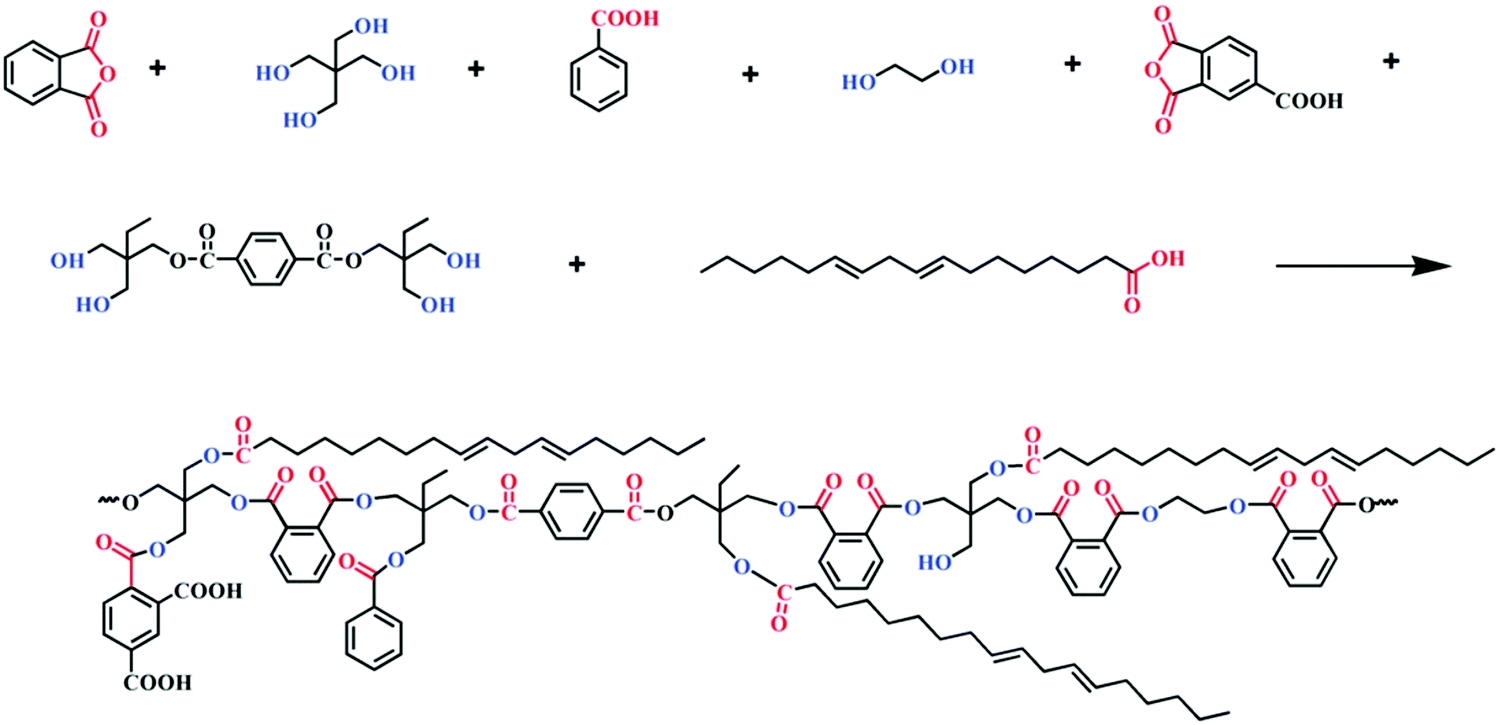 | ||
| Fig. 6 The synthesis reactions of water-reducible alkyd resins from the glycolyzed PET product, TOFA, PA, PE, EG, BA, and TMA. Reproduced with permission from ref. 112. Copyright 2021 Royal Society of Chemistry. | ||
In terms of physical properties (gloss, adhesion, hardness, flexibility and impact resistance) and chemical resistance properties the PET derived water-reducible alkyd coatings demonstrated very similar characteristics to water reducible alkyd coatings prepared without PET waste derived monomers.
4.4. Acrylic resins
Owing to their outstanding durability and weatherability characteristics, acrylic resins are extensively used in architectural, automotive and plastic coatings.113,114 A current report suggests that in 2020 the global market size of acrylic resins was approximately USD16.8 billion, which was projected to reach around USD21.9 billion by the end of 2025 at an annual compound growth rate of 5.5%.115 In this scenario, recycled PET waste derived materials can serve as crucial components for acrylic resin production, especially for preparation of UV curable acrylate crosslinkers. As for example, Karayannidis and co-workers synthesized a methacrylated oligoester crosslinker from glycolyzed PET waste for synthesis of UV curable acrylic resins.116 In their UV curable formulation they employed a homopolymer of methacrylated oligoesters as well as a copolymer of methacrylated oligoesters and styrene along with SiO2 as a reinforcing agent. The oligoesters showed good compatibility with styrene, which suggests that the methacrylated oligoester could be employed as a raw material in the acrylic coating industry. In another similar type of study Soni et al. reported synthesis of a novel acrylic aromatic amide oligomer of aminolysed endproducts of PET for crosslinking of acrylate monomers.117 To obtain the acrylic aromatic amide oligomer, at first they reacted PET waste with hydrazine monohydrate, which resulted in terepthalic dihydrazide (TPD) along with higher analogues of TPD. Subsequently, they further reacted this TPD as well as its higher analogues with acrolyl chloride to get the final products named N,N′-bis-(2-propenamido)benzene 1,4-dicarboxamide (TPDAC) and its higher homologues which were finally used as raw materials for radcure coatings together with other acrylate monomers such as methylmethacrylate, ethylhexylacrylate, acrylic acid, and a photoinitiator. The synthesized resin displayed excellent adhesion in glass as well as metal substrates due to the excellent hydrogen bonding capability of TPDAC and its higher homologues, which indicates that it can be used as a potential alternative for urethane acrylates for radcure coatings. In another study Sabnis and co-workers reported glycolytic depolymerization of PET waste using methylpropanediol and subsequent utilization of the obtained recycled product for acrylic based UV-curable wood coating application.118 Similar to the above discussed studies, here also they employed the PET recycled product for synthesis of crosslinking agents in radcure coatings. To obtain PET derived UV active crosslinking agents, at first, they reacted postconsumer PET waste with methylpropanediol, which led to the formation of oligomers of PET; subsequent reaction of these oligomers with methacrylic acid resulted in UV active crosslinking agents. Their final UV curable formulations consist of a photoinitiator, a commercial epoxy acrylate resin and the synthesized UV active oligomers with different concentrations. The performance report of radcure coating formulations on wood substrates revealed that maximum 50% replacement of epoxy acrylate by the synthesized UV oligomers resulted in very similar coating performance (in terms of hardness, solvent resistance, tensile strength and thermal stability) to the standard formulation using commercial epoxy acrylate. These outcomes indicate that methylpropanediol can be used effectively for depolymerization of PET waste chemically to yield the UV active oligomeric product suitable for radcure wood coatings. In another very recent study, Hakkarainen and co-workers reported synthesis of a series of functional precursors from PET waste which can be used for UV-curable coatings.119 Although they did not use these precursors for direct coating application, they prepared good quality polymer films using these precursors, which suggests their potential application in coatings also. To synthesize the UV active precursors from PET waste, they performed microwave assisted aminolysis of PET with four different amines, which included allylamine, ethanolamine, furfurylamine and hexylamine. Among these amines, only allylamine was used to synthesize the crosslinker named diallylterephthalamide for UV curable resins by reacting it with a thiol through a radical thiol–ene click reaction. Based on the above studies we can say that although there is a huge scope for recycled PET waste derived products for synthesis of UV curable acrylic resins (especially as crosslinkers) a lot of pilot scale studies are still required to commercialize the technology.4.5. Polyurethanes
Since the revolutionary discovery of polyurethanes (PUs) in 1937 by Otto Bayer and co-workers, significant efforts have been directed toward novel PU synthesis, and as a consequence, many ground breaking technologies have been reported in the field of PU synthesis over time. Currently polyurethane technology is so much improved that it has several applications in multiple sectors such as the transportation, adhesive, textile, electronics and building & construction sectors. Other than these applications a significant portion of the total PU consumption is used in the coating industry. A current report revealed that in 2018 the total polyurethane coating market value was ∼USD16 billion with a CAGR of 4% by the end of 2025.120Polyurethanes are usually prepared by reacting polyols with diisocyanates with or without catalyst. For coating application, polyurethanes are one of the prevalent choices especially for high performance coatings due to their outstanding chemical resistance along with good flexibility and impact resistance. As PUs are usually prepared by reacting polyols with diisocyanates, most PU coatings come with two pack systems; the first one contains the polyol part and the second part consists of a hardener (source of isocyanate). However, there are certain recent developments where one pack PU systems are reported for coating applications. In this regard, recycled PET waste derived products can be used as sustainable resources of raw materials for PU production; more specifically, these recycled PET waste derived products can be economical sources of polyols for PU synthesis. For example, the oligoesters obtained by glycolysis of PET waste can be further reacted with aromatic/aliphatic diacids to get polyester polyols, which are some of the prime components for PU coatings. These polyols can be further reacted with diisocyanates to form the urethane linkage within the polymer backbone.
As for example, Akar and co-workers developed novel urethane oil varnishes using glycolyzed PET waste products as raw materials.121 To synthesize the PU oil, at first, they performed transesterification reaction of PET, which led to the formation of oligoesters of PET. In the next step, they conducted a reaction of this glycolyzed PET product with soybean oil and glycerine followed by a reaction with toluene diisocyanate (TDI), which ultimately led to production of urethane varnish oil, which was simultaneously thinned with white spirit to get ∼60% solids. In terms of physical properties such as Koenig hardness, drying time and adhesiveness, they observed that the synthesized urethane oil varnishes have very similar values to commercialized urethane oil. Similar to Akar and co-workers,121 Saravari and his research team also reported synthesis of urethane oils from recycled PET waste.122 Here they employed propylene glycol for glycolysis of PET. The glycolyzed product contained oligomeric diols with number-average molecular weights in the range of 458–844, and was further reacted with soybean oil and toluene diisocyanate to obtain urethane oils at hydroxyl:isocyanate ratios from 1:1 to 1:0.7, with and without methanol, which acts as a blocking agent. They observed that in the case of lower diisocyanate content and in the presence of methanol the urethanes produced exhibited higher viscosity, higher molecular weight, and shorter drying times. Additionally, they noticed that other properties of the PET based urethane oil (in both liquid and dry film forms) were very similar to those of commercial urethane oil. The films of the synthesized urethane oils showed good adhesion, hardness along with excellent water and acid resistance and reasonable alkali resistance. However, in terms of flexibility and wear resistance they did not perform up to the mark compared to commercial urethane oils. In another similar type of study Patel et al. reported synthesis of a two pack PU system from PET waste and biological waste for coating application.123 In their study they used glycol glycosides for depolymerization of PET, which generated oligoesters of PET. Subsequently, they used these oligomers for synthesis of liquid polyester polyols by reacting these oligomers with soya fatty acid (SOFA) for urethane coatings. TDI was used as a source of isocyanate in their preparation. The performance test revealed that the PU coatings took 3.5–4.5 h for drying depending on PET oligomers present in the final coatings, whereas the scratch hardness varied between 2.5 and 3.5 kg. Additionally, the PU coatings also demonstrated good impact hardness, flexibility and chemical resistance, which implies that the urethane coating system can be successfully used in industrial maintenance finishes. In their subsequent study also they got very similar results.124 The only difference was that in this study instead of SOFA they used coco fatty acids to obtain the polyester polyol which was further reacted with TDI to get the final PU product. In terms of performance polyurethane resin coated panels showed good drying times, hardness, adhesion, flexibility and chemical resistance, which suggests their potential for coating application. Sabnis and co-workers reported glycolysis of PET waste using electron beam irradiation and subsequent utilization of the recycled product for PU synthesis which can be further exploited for coating purposes.125 For glycolysis of PET they employed ethylene glycol as a depolymerizing agent, which led to the formation of BHET. Successively the obtained BHET was subjected to esterification reaction with linseed oil fatty acid, followed by epoxidation using formic acid and hydrogen peroxide and hydrolysis to prepare an eco-friendly polyol. To get the final polyurethane coatings they reacted the polyol with hexamethylene diisocyanates and 4,4′-methylene diphenyl diisocyanate (MDI) with 16.5 and 31.5% isocyanate content respectively. All the systems of polyurethane coatings exhibited overall good performance properties including adhesion, hardness, flexibility, and solvent and chemical resistance, which suggest that BHET can be one of the potential raw materials for synthesis of binders to generate value added PUs for coating applications. In another very recent study, Gan and co-workers reported polyurethane coatings derived from recycled PET and palm olein-based polyols.126 To prepare polyols, at first, they prepared alkyds by reacting palm olein, glycerol and phthalic anhydride followed by simultaneous depolymerization of PET by EG and transesterification reactions with the alkyds, which resulted in viscous products (palm olein/PET alkyd polyols). For final PU synthesis, they reacted polyols with methylene diphenyl diisocyanate in the presence of dibutyltin dilaurate catalyst. The PU coatings exhibited significantly improved thermal stability and better anticorrosion properties in the presence of 3.5% NaCl solution as measured by EIS with an enhanced loading of PET into the palm olein alkyd, which indicates their possible application as high-performance PU coatings for industrial or marine applications. In Table 2 different PU materials synthesized from recycled PET waste and their physical properties along with their application as coating systems are given.
| Depolymerization product of PET | Other raw materials used for PU synthesis | Physical properties of coatings | Coating application(s) | Ref. |
|---|---|---|---|---|
| Glycolyzed oligoester polyols obtained by depolymerization of PET using poly(ethylene glycol) | Castor oil, stannous octoate, poly(caprolactone) diol, isophorone diisocyanate, dibutyltin dilaurate, dimethylol propionic acid, 1,4-butane diol and triethylamine | The hardness varied between 20 and 127, the gloss varied between 45 and 140 @ 60°, cross hatch adhesion pass 5B | High performance coatings and adhesive application | 127 |
| Glycolyzed oligoesters obtained by depolymerization of PET using poly(ethylene glycol) | Castor oil, isophorone diisocyanate, dimethylol propionic acid, and hexamethoxymethyl melamine | Good thermal resistance (degradation started at ∼250 °C), the hardness varied between 38 and 99.4, the gloss varied between 120 and 144 @ 60°, cross hatch adhesion pass 5B | Thermal coating application | 128 |
| Transesterification as well as amidation reaction products of PET with different molecular weights of poly(ethylene glycol) and diethanol amine | Castor oil, adipic acid, toluene diisocyanate | The PU coatings showed dielectric strengths within 28.7–30.5 kV mm−1 and dielectric constants (60 Hz) 3.91–5.14 | High performance metallic electrical insulator coatings | 129 |
| Polyhydroxyl compounds derived from glycolyzed PET using poly(ethylene glycol) | Trimethylol propane, toluene diisocyanate, N-methyl aniline | The tensile properties varied between 19.45 and 46.90 MPa, the pencil hardness varied between 2B and HB | Electrical insulating coatings for metals | 130 |
| Bis(2-hydroxyethyl) terephthalate | Phosphorus oxychloride, triethylamine, phthalic anhydride, ethylene glycol, para-toluenesulfonic acid, toluene, diisocynate and hexamethylene diisocynate | The scratch hardness varied between 1.65 and 1.9 kg, and the impact resistance was between 200 and 400 | Flame retardant coatings | 131 |
| Polyol derived from glycolyzed PET using 1,4-butanediol | Dimethylol propionic acid, dibutyltin dilaurate, isophorone diisocyanate | PU coatings had fair impact hardness, good scratch hardness and good flexibility | Environment friendly low volatile content industrial coating applications | 132 |
| Bis(2-hydroxyethylene) terephthalamide | Ether-type polyol, 1,4-butanediol, dibutyltin dilaurate, hexamethylene diisocyanate | Good adhesion properties, excellent mechanical strength with good thermal stability | General industrial coating application | 133 |
4.6. Other corrosion inhibitors
Other than the above-mentioned recycled PET based resins, PET derived products can also be used as corrosion inhibitors for coating applications. For example, the compatibility of BHETA obtained by the aminolysis of PET was evaluated for anticorrosive paint formulations for the protection of steel structures from rust.134 BHETA itself exhibits high hardness and stiffness, good resistance to weathering, creep strength, and high dimensional stability; therefore, inclusion of BHETA in the final formulations of paint improved the overall coating performance of the paint films without affecting their physical and mechanical properties.Additionally, the platelet-shaped BHETA (shown in Fig. 7) acted as a reinforcing agent and reduced the water and gas permeability, conferring excellent anticorrosive properties and a special appearance to the paint film. The paint formulation with a pigment:binder ratio of 2.233 accommodating 10% BHETA showed the best adherence on a steel substrate along with corrosion resistance, and the results were similar to those of other high-build controls. El-Hameed et al. also examined the performance of BHETA obtained by aminolysis of PET waste as a corrosion inhibitor for carbon steel (in HCl corrosive medium) by inspecting weight loss, open circuit potential, and potentiodynamic polarization.135 The polarization curves suggest that BHETA acts as a mixed type inhibitor. The inhibition efficacy imparted by the adsorbed BHETA on carbon steel enhanced with increase in the concentration of the inhibitor and decreased with increase in temperature. Furthermore, it was also observed that corrosion inhibition took place through adsorption of the inhibitor molecules on the metal surface without modifying the mechanism of the corrosion process, which indicates the application of BHETA as a corrosion inhibitor in paint formulations.
 | ||
| Fig. 7 Platelet shaped SEM micrographs of BHETA at magnifications of 15 (a) and 25 × 102 (b). Reproduced with permission from ref. 134. Copyright 2011 Willey. | ||
5. Current challenges involved with the PET recycling process in industry
Although there is a huge scope for use of PET recycled products as raw materials in the coating industry, there are a few prime challenges involved during the upscaling process at industry.• The first major problem is that either due to the lack of appropriate knowledge or to reduce the cost of the overall PET production different types of plastics are often combined with virgin PET in manufacturing processes of PET based products, which makes recycling of PET products much more difficult. To separate out these plastics from recycled PET products, some added purification stage is always desirable, which ultimately leads to an increase in the overall cost of the recycling process. In addition to that, along with plastics there are some organic dyes often used in PET products to colourize PET products; sometimes these dyes also create problems in purification stages.
• Sometimes it is found that virgin raw materials are cheaper compared to recycled ones, in that case there is no benefit from the economical point of view.
• Plastic is a multifaceted material and each type exhibits exclusive properties which lead to its colour, structure and melting point. Hence, it is very important to sort out PET into different categories so that the recycled products fall into the same category, so that the performance of the final coatings does not alter from batch to batch.
• Another main problem is that initially there is a huge investment cost required to set up a plastic recycling plant under proper supervision of expert personnel which is not possible to accommodate for small industries.
6. Conclusion and future directions
From the above studies and discussion, we can conclude that recycling of postconsumer PET waste not only provides a sustainable solution to the solid waste management problem but also delivers enormous opportunities for the development of new raw materials toward many industries, especially for the paint and coating industry. There are already various well-established methods developed by researchers & scientists to synthesize value added products from PET waste and there is a huge scope for translation of these technologies into industry. To encounter the challenges involved with the recycling processes, especially in separation, purification and processing steps (e.g., separation of pigments and other polymers present in PET waste, use of corrosive chemicals and involvement of high temperature and pressure during the depolymerization reaction) proper expertise and supervision is recommended along with advanced reactor designing, appropriate process control measures and all necessary safety precautionary measures.From the application point of view in the coating industry, the various materials obtained from recycling of PET waste can be used as raw materials for synthesis of various resins and polymers. Not only that most of these resins and polymers perform very similar to and sometimes better than commercialized coating systems in terms of drying time, hardness, flexibility and corrosion resistance. In recent times more efforts have been made by researchers to develop coating systems which are based on renewable resources like natural oils and plant carbohydrates to enhance the properties of the recycled PET based coatings and come up with new economical yet efficient high performance industrial coatings, which ultimately leads to a cleaner, safer and sustainable ecosystem. Finally, we can say that there is a massive potential of recycled PET derived products application as a raw material in coatings industry. However, for commercialization of these coating systems in the market some extra time and effort should be given, especially on large scale trials; so that recovery, recycling and reuse of these waste materials lead to a sustainable and closed loop circular economy which is key to the success of any country.
Authors’ contributions
KG conceptualized the idea and was involved in content writing and overall corrections/modifications. CN helped in writing the manuscript and provided important suggestions for improvement of the quality of the overall manuscript.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
The authors acknowledge Shalimar Paints Limited for financial support.References
- D. Crespy, M. Bozonnet and M. Meier, Angew. Chem., 2008, 47, 3322–3328 CrossRef CAS PubMed.
- D. Crespy, M. Bozonnet and M. Meier, Angew. Chem., Int. Ed., 2008, 47, 3322–3328 CrossRef CAS PubMed.
- R. Geyer, J. R. Jambeck and K. L. J. S. A. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- K. Beydoun and J. Klankermayer, Sci. Adv., 2020, 13, 488–492 CAS.
- A. Vijay, 2020, https://www.printweek.in/whatpackaging/global-pet-packaging-demand-to-reach-usd-441-bn-this-year-53801.
- I. Taniguchi, S. Yoshida, K. Hiraga, K. Miyamoto, Y. Kimura and K. Oda, ACS Catal., 2019, 9, 4089–4105 CrossRef CAS.
- J. Hopewell, R. Dvorak and E. J. P. T. O. T. R. S. B. B. S. Kosior, Philos. Trans. R. Soc., B, 2009, 364, 2115–2126 CrossRef CAS PubMed.
- R. Francis, Recycling of polymers: methods, characterization and applications, John Wiley & Sons, 2016 Search PubMed.
- N. Singh, D. Hui, R. Singh, I. Ahuja, L. Feo and F. J. C. P. B. E. Fraternali, Composites, Part B, 2017, 115, 409–422 CrossRef CAS.
- M. E. J. R. Grigore, Recycling, 2017, 2, 24 CrossRef.
- A. Maurya, A. Bhattacharya and S. K. Khare, Bioprocess Eng., 2020, 8, 602325 Search PubMed.
- N. Mohanan, Z. Montazer, P. K. Sharma and D. B. Levin, Bioprocess Eng., 2020, 11, 580709 Search PubMed.
- L. Yang, J. Gao, Y. Liu, G. Zhuang, X. Peng, W.-M. Wu and X. Zhuang, Chemosphere, 2021, 262, 127818 CrossRef CAS PubMed.
- B. J. Cassone, H. C. Grove, O. Elebute, S. M. P. Villanueva and C. M. R. LeMoine, Proc. R. Soc. B, 2020, 287, 20200112 CrossRef CAS PubMed.
- R. Mor and A. J. B. Sivan, Biodegradation, 2008, 19, 851–858 CrossRef CAS PubMed.
- M. Santo, R. Weitsman and A. Sivan, Int. Biodeterior. Biodegrad., 2013, 84, 204–210 CrossRef CAS.
- I. Cacciari, P. Quatrini, G. Zirletta, E. Mincione, V. Vinciguerra, P. Lupattelli and G. G. Sermanni, Appl. Environ. Microbiol., 1993, 59, 3695–3700 CrossRef CAS PubMed.
- A. Arkatkar, A. A. Juwarkar, S. Bhaduri, P. V. Uppara and M. Doble, Int. Biodeterior. Biodegrad., 2010, 64, 530–536 CrossRef CAS.
- L. Giacomucci, N. Raddadi, M. Soccio, N. Lotti and F. Fava, New Biotechnol., 2019, 52, 35–41 CrossRef CAS PubMed.
- S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara, K. Miyamoto, Y. Kimura and K. Oda, Science, 2016, 351, 1196–1199 CrossRef CAS PubMed.
- P. Liu, T. Zhang, Y. Zheng, Q. Li, T. Su and Q. Qi, Engineering Microbiology, 2021, 1, 100003 CrossRef.
- N. Mahajan and P. J. R. A. Gupta, RSC Adv., 2015, 5, 41839–41854 RSC.
- A. N. Shirke, C. White, J. A. Englaender, A. Zwarycz, G. L. Butterfoss, R. J. Linhardt and R. A. Gross, Biochemistry, 2018, 57, 1190–1200 CrossRef CAS PubMed.
- D. Achilias, C. Roupakias, P. Megalokonomos, A. Lappas and E. J. J. O. H. M. Antonakou, J. Hazard. Mater., 2007, 149, 536–542 CrossRef CAS PubMed.
- S. J. M. B. Matsumura, Macromol. Biosci., 2002, 2, 105–126 CrossRef CAS.
- G. P. Karayannidis and D. S. J. M. M. Achilias, Engineering, 2007, 292, 128–146 CAS.
- D. P. S. Jose Aguado, Feedstock Recycling of Plastic Wastes, 1999 Search PubMed.
- G. Karayannidis, A. Chatziavgoustis and D. J. A. I. P. T. J. O. T. P. P. I. Achilias, Adv. Polym. Technol., 2002, 21, 250–259 CrossRef CAS.
- D. Paszun, T. J. I. Spychaj and E. C. Research, Ind. Eng. Chem. Res., 1997, 36, 1373–1383 CrossRef CAS.
- J. Pitat, V. Holcik and M. J. G. P. Bacak, 1959, 822, 834.
- S. Kumar and C. J. J. O. M. S. Guria, J. Macromol. Sci., Part A: Pure Appl. Chem., 2005, 42, 237–251 CrossRef.
- C. G. G. Namboori and M. S. Haith, J. Appl. Polym. Sci., 1968, 12, 1999–2005 CrossRef CAS.
- A. Oku, L.-C. Hu and E. Yamada, J. Appl. Polym. Sci., 1997, 63, 595–601 CrossRef CAS.
- T. Yoshioka, T. Motoki and A. Okuwaki, Ind. Eng. Chem. Res., 2001, 40, 75–79 CrossRef CAS.
- T. Yoshioka, T. Sato and A. J. J. O. A. P. S. Okuwaki, J. Appl. Polym. Sci., 1994, 52, 1353–1355 CrossRef CAS.
- S. Mishra, A. Goje and V. J. P.-P. T. Zope, Engineering, 2003, 42, 581–603 CAS.
- S. Mishra, A. S. Goje and V. S. Zope, Polym. React. Eng., 2003, 11, 79–99 CrossRef CAS.
- T. Yoshioka, N. Okayama, A. J. I. Okuwaki and E. C. Research, Ind. Eng. Chem. Res., 1998, 37, 336–340 CrossRef CAS.
- S. Mishra, A. Goje and V. J. P. R. E. Zope, Polym. React. Eng., 2003, 11, 79–99 CrossRef CAS.
- T. Yoshioka, N. Okayama and A. Okuwaki, Ind. Eng. Chem. Res., 1998, 37, 336–340 CrossRef CAS.
- D. J. Royall and J. L. Harvie, Canadian Pat., 550979, 1993 Search PubMed.
- D. Stanica-Ezeanu and D. Matei, Sci. Rep., 2021, 11, 4431 CrossRef CAS PubMed.
- A. Al-Sabagh, F. Yehia, G. Eshaq, A. Rabie and A. J. E. J. O. P. ElMetwally, Egypt. J. Pet., 2016, 25, 53–64 CrossRef.
- M. Y. Abdelaal, T. R. Sobahi and M. S. I. Makki, Constr. Build. Mater., 2011, 25, 3267–3271 CrossRef.
- Y. Öztürk and G. Güçlü, Polym. – Plast. Technol. Eng., 2005, 43, 1539–1552 CrossRef.
- G. Güçlü, A. Kaşgöz, S. Özbudak, S. Özgümüş and M. Orbay, J. Appl. Polym. Sci., 1998, 69, 2311–2319 CrossRef.
- M. Imran, B.-K. Kim, M. Han, B. G. Cho and D. H. Kim, Polym. Degrad. Stab., 2010, 95, 1686–1693 CrossRef CAS.
- N. Pingale and S. J. E. P. J. Shukla, Eur. Polym. J., 2008, 44, 4151–4156 CrossRef CAS.
- J. Y. Cho, C.-J. Hong, H.-M. J. I. Choi and E. C. Research, Ind. Eng. Chem. Res., 2013, 52, 2309–2315 CrossRef CAS.
- S. Baliga and W. T. Wong, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 2071–2082 CrossRef CAS.
- M. Ghaemy and K. Mossaddegh, Polym. Degrad. Stab., 2005, 90, 570–576 CrossRef CAS.
- C.-H. Chen, J. Appl. Polym. Sci., 2003, 87, 2004–2010 CrossRef CAS.
- G. Xi, M. Lu and C. Sun, Polym. Degrad. Stab., 2005, 87, 117–120 CrossRef CAS.
- S. R. Shukla and K. S. Kulkarni, J. Appl. Polym. Sci., 2002, 85, 1765–1770 CrossRef CAS.
- N. D. Pingale and S. R. Shukla, Eur. Polym. J., 2008, 44, 4151–4156 CrossRef CAS.
- S. R. Shukla, V. Palekar and N. Pingale, J. Appl. Polym. Sci., 2008, 110, 501–506 CrossRef CAS.
- M. Imran, K. G. Lee, Q. Imtiaz, B.-K. Kim, M. Han, B. G. Cho and D. H. J. J. O. N. Kim, Nanotechnology, 2011, 11, 824–828 CAS.
- M. Imran, W. A. Al-Masry, A. Mahmood, A. Hassan, S. Haider and S. M. J. P. D. Ramay, Polym. Degrad. Stabil., 2013, 98, 904–915 CrossRef CAS.
- G. Park, L. Bartolome, K. G. Lee, S. J. Lee, D. H. Kim and T. J. Park, Nanoscale, 2012, 4, 3879–3885 RSC.
- Z. Guo, K. Lindqvist and H. de la Motte, J. Appl. Polym. Sci., 2018, 135, 46285 CrossRef.
- J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667–3692 CrossRef CAS PubMed.
- M. J. T. A. Kosmulski, Thermochim. Acta, 2004, 412, 47–53 CrossRef CAS.
- H. Matsumoto, M. Yanagida, K. Tanimoto, M. Nomura, Y. Kitagawa and Y. J. C. L. Miyazaki, Chem. Lett., 2000, 922–923 CrossRef CAS.
- H. Wang, Y. Liu, Z. Li, X. Zhang, S. Zhang and Y. J. E. P. J. Zhang, Eur. Polym. J., 2009, 45, 1535–1544 CrossRef CAS.
- Q. F. Yue, C. X. Wang, L. N. Zhang, Y. Ni and Y. X. Jin, Polym. Degrad. Stab., 2011, 96, 399–403 CrossRef CAS.
- H. Wang, R. Yan, Z. Li, X. Zhang and S. Zhang, Catal. Commun., 2010, 11, 763–767 CrossRef CAS.
- Z. Ju, L. Zhou, X. Lu, Y. Li, X. Yao, S. Cheng, G. Chen and C. Ge, Phys. Chem. Chem. Phys., 2021, 23, 18659–18668 RSC.
- C. Shuangjun, S. Weihe, C. Haidong, Z. Hao, Z. Zhenwei and F. Chaonan, J. Therm. Anal. Calorim., 2021, 143, 3489–3497 CrossRef.
- A. M. Al-Sabagh, F. Z. Yehia, A. M. F. Eissa, M. E. Moustafa, G. Eshaq, A. M. Rabie and A. E. ElMetwally, Polym. Degrad. Stab., 2014, 110, 364–377 CrossRef CAS.
- X. Zhou, X. Lu, Q. Wang, M. Zhu and Z. Li, Pure Appl. Chem., 2012, 84, 789–801 CAS.
- A. M. Al-Sabagh, F. Z. Yehia, A.-M. M. F. Eissa, M. E. Moustafa, G. Eshaq, A.-R. M. Rabie and A. E. ElMetwally, Ind. Eng. Chem. Res., 2014, 53, 18443–18451 CrossRef CAS.
- Y. Liu, X. Yao, H. Yao, Q. Zhou, J. Xin, X. Lu and S. Zhang, Green Chem., 2020, 22, 3122–3131 RSC.
- C. V. G. Silva, E. A. d. Silva, F. Uliana, L. F. R. d. Jesus, C. V. P. d. Melo, R. C. Barthus, J. G. A. Rodrigues and G. J. P. Vanini, Polim.: Cienc. Tecnol., 2018, 28, 450–459 CrossRef.
- H. Zahn and H. Pfeifer, Polymer, 1963, 4, 429–432 CrossRef CAS.
- S. R. Shukla and A. M. Harad, Polym. Degrad. Stab., 2006, 91, 1850–1854 CrossRef CAS.
- M. E. Tawfik and S. B. Eskander, Polym. Degrad. Stab., 2010, 95, 187–194 CrossRef CAS.
- K. Ghosal and K. Sarkar, New J. Chem., 2019, 43, 14166–14178 RSC.
- T. Spychaj, E. Fabrycy, S. Spychaj and M. Kacperski, J. Mater. Cycles Waste Manage., 2001, 3, 24–31 CAS.
- N. D. Pingale and S. R. Shukla, Eur. Polym. J., 2009, 45, 2695–2700 CrossRef CAS.
- Y. S. Parab, N. D. Pingale and S. R. Shukla, J. Appl. Polym. Sci., 2012, 125, 1103–1107 CrossRef CAS.
- D. S. Achilias, G. P. Tsintzou, A. K. Nikolaidis, D. N. Bikiaris and G. P. Karayannidis, Polym. Int., 2011, 60, 500–506 CrossRef CAS.
- M. Goto, H. Koyamoto, A. Kodama, T. Hirose, S. Nagaoka and B. J. McCoy, AIChE. J., 2002, 48, 136–144 CrossRef CAS.
- J. J. J. W. Scheirs, Polymer recycling: science, technology and applications, J. Sons Ltd, Baffins Lane, Chichester, Sussex, PO 19 1 UD, UK, 1998, p. 591 Search PubMed.
- G. Hans, H. Walter and M. Heinz, Process for depolymerizing polyethylene-terephthalate to terephthalic acid dimethyl ester, Google Patents, 1968.
- T. Sako, T. Sugeta, K. Otake, Y. Takebayashi, C. Kamizawa, M. Tsugumi and M. J. K. R. Hongo, J. Mol. Biol., 1998, 55, 685–690 CAS.
- S. Mishra and A. S. Goje, Polym. Int., 2003, 52, 337–342 CrossRef CAS.
- M. Hofmann, J. Sundermeier, C. Alberti and S. J. C. Enthaler, ChemistrySelect, 2020, 5, 10010–10014 CrossRef CAS.
- D. Cornell, Depolymerization of PET for food packaging, Proc. Conf. Recycling RETEC, Society Plastic Engineers, 1993 Search PubMed.
- J. H. Clark, J. A. Alonso, J. A. Villalba, J. Aguado, D. P. Serrano and D. Serrano, Feedstock recycling of plastic wastes, Royal society of chemistry, 1999 Search PubMed.
- G. Macijauskas and V. J. M. S. Jankauskait, Mater. Sci., 2013, 19, 283–290 CrossRef.
- A. M. Atta, Prog. Rubber, Plast. Recycl. Technol., 2003, 19, 17–40 CrossRef CAS.
- A. M. Atta, A. F. El-Kafrawy, M. H. Aly and A.-A. A. Abdel-Azim, Prog. Org. Coat., 2007, 58, 13–22 CrossRef CAS.
- K. Bal, K. C. Ünlü, I. Acar and G. Güçlü, J. Coat. Technol. Res., 2017, 14, 747–753 CrossRef CAS.
- P. Czub, Polym. Adv. Technol., 2009, 20, 183–193 CrossRef CAS.
- I. D. Unsaturated, Polyester Resins Market by Type (Ortho phthalic, End-Use Industry (Building & Construction, Marine, Transportation, Pipes & Tanks, Artificial Stone, Wind Energy, Electrical & Electronics) & Region - Global Forecast to 2026.
- U. R. Vaidya and V. M. Nadkarni, Ind. Eng. Chem. Res., 1987, 26, 194–198 CrossRef CAS.
- V. Pimpan, R. Sirisook and S. Chuayjuljit, J. Appl. Polym. Sci., 2003, 88, 788–792 CrossRef CAS.
- S. Y. Tawfik, M. W. Sabaa, R. T. J. P. Botros and R. Technology, Pigm. Resin Technol., 2017, 46(5), 408–422 CrossRef CAS.
- C. Gioia, M. Vannini, P. Marchese, A. Minesso, R. Cavalieri, M. Colonna and A. Celli, Green Chem., 2014, 16, 1807–1815 RSC.
- P. P. Chiplunkar and A. P. Pratap, Prog. Org. Coat., 2016, 93, 61–67 CrossRef CAS.
- D. İşeri-Çağlar, E. Baştürk, B. Oktay and M. V. Kahraman, Prog. Org. Coat., 2014, 77, 81–86 CrossRef.
- M. S. Selim, M. A. Shenashen, A. Elmarakbi, A. M. El-Saeed, M. M. Selim and S. A. El-Safty, RSC Adv., 2017, 7, 21796–21808 RSC.
- H. R. Ong, R. Ramli, M. M. R. Khan and R. M. Yunus, Prog. Org. Coat., 2016, 101, 245–252 CrossRef CAS.
- M. S. Selim, S. A. El-Safty, M. A. Shenashen, M. A. El-Sockary, O. M. A. Elenien and A. M. El-Saeed, Prog. Org. Coat., 2019, 126, 106–118 CrossRef CAS.
- M. S. Selim, A. Elmarakbi, A. M. Azzam, M. A. Shenashen, A. M. El-Saeed and S. A. El-Safty, Prog. Org. Coat., 2018, 116, 21–34 CrossRef CAS.
- Y. Ma, R. Lei, X. Yang and F. Yang, J. Polym. Environ., 2020, 28, 1083–1094 CrossRef CAS.
- A. M. Atta, R. A. El-Ghazawy and A. M. J. I. J. E. S. El-Saeed, Int. J. Electrochem. Sci., 2013, 8, 5136–5152 CAS.
- G. Güçlü and M. Orbay, Prog. Org. Coat., 2009, 65, 362–365 CrossRef.
- A. Torlakoğlu and G. Güçlü, Waste Manage., 2009, 29, 350–354 CrossRef PubMed.
- P. M. Spasojević, V. V. Panić, J. V. Džunuzović, A. D. Marinković, A. J. J. Woortman, K. Loos and I. G. Popović, RSC Adv., 2015, 5, 62273–62283 RSC.
- I. Acar, A. Bal and G. Guclu, Can. J. Chem., 2013, 91(5), 357–363 CrossRef CAS.
- S. Ouyang, Y. Xie, W. Fu, Y. Ding and L. Shen, R. Soc. Open Sci., 2021, 8, 202375 CrossRef CAS PubMed.
- C. Jiao, L. Sun, Q. Shao, J. Song, Q. Hu, N. Naik and Z. Guo, ACS Omega, 2021, 6, 2443–2449 CrossRef CAS PubMed.
- H. Petit, N. Henry, A. Krebs, G. Uytterhoeven and F. D. Jong, Prog. Org. Coat., 2001, 43, 41–49 CrossRef CAS.
- M. R. Report, Acrylic Resins Market by Solvency (Water-based, solvent-based, and others), Chemistry, Application (Paints & coatings, adhesives & sealants, DIY coatings, elastomers, and others), End-use Industry, and Region – Global Forecast to 2025, https://www.marketsandmarkets.com/Market-Reports/acrylic-resin-market-246195771.html.
- G. P. Karayannidis, A. K. Nikolaidis, I. D. Sideridou, D. N. Bikiaris and D. S. Achilias, Macromol. Mater. Eng., 2006, 291, 1338–1347 CrossRef CAS.
- R. K. Soni, M. Teotia and K. Dutt, J. Appl. Polym. Sci., 2010, 118, 638–645 CAS.
- V. Jamdar, M. Kathalewar and A. Sabnis, J. Coat. Technol. Res., 2018, 15, 259–270 CrossRef CAS.
- E. Bäckström, K. Odelius and M. Hakkarainen, Eur. Polym. J., 2021, 151, 110441 CrossRef.
- M. S. Polyurethane Coating Market – Industry Analysis, Share, Trends, Application Analysis, Growth and Forecast 2021–2026.
- O. Mecit and A. Akar, Macromol. Mater. Eng., 2001, 286, 513–515 CrossRef CAS.
- O. Saravari, B. Vessabutr and V. Pimpan, Energy Procedia, 2004, 92, 3040–3045 CAS.
- J. V. Patel, P. K. Soni and V. Sinha, Int. J. Polym. Mater. Polym. Biomater., 2001, 49, 205–215 CrossRef CAS.
- M. R. Patel, J. V. Patel and V. K. Sinha, Polym. Degrad. Stab., 2005, 90, 111–115 CrossRef CAS.
- V. Jamdar, M. Kathalewar, K. A. Dubey and A. Sabnis, Prog. Org. Coat., 2017, 107, 54–63 CrossRef CAS.
- A. A. Adamu, N. Muhamad Sarih and S. N. Gan, R. Soc. Open Sci., 2021, 8, 201087 CrossRef CAS PubMed.
- S. M. Cakić, I. S. Ristić, M. M. Cincović, N. Č. Nikolić, L. B. Nikolić and M. J. Cvetinov, Prog. Org. Coat., 2017, 105, 111–122 CrossRef.
- S. M. Cakić, I. S. Ristić, M. M. Cincović, D. T. Stojiljković, C. J. János, C. J. Miroslav and J. V. Stamenković, Prog. Org. Coat., 2015, 78, 357–368 CrossRef.
- H. Yeganeh and H. R. Moeini, High Perform. Polym., 2006, 19, 113–126 CrossRef.
- H. Yeganeh and M. A. Shamekhi, J. Appl. Polym. Sci., 2006, 99, 1222–1233 CrossRef CAS.
- A. A. Barot, C. M. Patel and V. K. J. A. O. A. S. R. Sinha, Arch. Appl. Sci. Res., 2016, 8, 12–22 CAS.
- M. R. Patel, J. V. Patel, D. Mishra and V. K. Sinha, J. Polym. Environ., 2007, 15, 97–105 CrossRef CAS.
- R. Shamsi, M. Abdouss, G. M. M. Sadeghi and F. A. Taromi, Polym. Int., 2009, 58, 22–30 CrossRef CAS.
- M. E. Tawfik, N. M. Ahmed and S. B. Eskander, J. Appl. Polym. Sci., 2011, 120, 2842–2855 CrossRef CAS.
- R. A. J. A. A. S. R. Hameed, Adv. Appl. Sci. Res., 2011, 2, 483–499 Search PubMed.
| This journal is © The Royal Society of Chemistry 2022 |