
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile room-temperature fabrication of a silver–platinum nanocoral catalyst towards hydrogen evolution and methanol electro-oxidation†
Hau Quoc
Pham
 ab and
Tai Thien
Huynh
*c
ab and
Tai Thien
Huynh
*c
aFuture Materials & Devices Lab., Institute of Fundamental and Applied Sciences, Duy Tan University, Ho Chi Minh City, 700000, Vietnam
bThe Faculty of Environmental and Chemical Engineering, Duy Tan University, Da Nang, 550000, Vietnam
cHo Chi Minh City University of Technology (HCMUT), Ho Chi Minh City, 700000, Vietnam. E-mail: httai@hcmunre.edu.vn
First published on 15th December 2021
Abstract
With rising environmental problems, hydrogen produced from electrolyzing water has emerged as the most sustainable and clean energy source to replace fossil fuels in a variety of applications. Although platinum (Pt) is the most efficient catalyst towards the hydrogen evolution reaction (HER), its high cost is a large limitation. Herein, we report a simple self-growth-assisted reduction route for fabricating a coral-like dendritic AgPt alloy nanocatalyst on a carbon support to lower Pt utilization and enhance the electrocatalytic performance. Due to its unique morphological features and alloy nature, the AgPt nanocoral (NCs)/C catalyst with 50 at% Ag shows excellent HER performance in acidic electrolyte with an extremely low overpotential of 16.61 mV at 10 mA cm−2 and a small Tafel slope of 19.05 mV dec−1, which are improved compared to those of a carbon-supported Pt (E-TEK) catalyst. In addition, the AgPt NCs/C catalyst exhibits enhanced reaction kinetics and superior CO-poisoning resistance for the methanol oxidation process. This study not only describes the design of a cost-effective HER catalyst but also provides a robust catalyst for energy conversion technologies.
Introduction
Due to serious environmental issues related to fossil fuel consumption, hydrogen production from the electrolysis of water has been considered to be one of the most sustainable and economic-efficient energy-generation technologies owing to its advantages of sufficient water sources and unharmful emissions.1,2 Electrocatalysts play an important role in boosting the electrochemical reaction rate by minimizing the overpotential and promoting the reaction kinetics during the HER; therefore, the Pt catalyst is widely used owing to its fast kinetics and low overpotential.1,3 The natural scarcity and high cost of Pt, however, are major limitations for its particular implementation in the HER.4–6 A great number of studies have been performed to fabricate Pt-free catalysts as an approach to reduce the cost of the HER catalyst. For example, Li et al.3 introduced a H-CoP@RGO catalyst that exhibited catalytic performance for the HER with 190 mV overpotential at 10 mA cm−2 in an acidic environment. Sahu et al.7 reported MOF-D CoSe2 for the HER with an overpotential of 195 mV at the current density of 10 mA cm−2 and a Tafel slope of 43 mV dec−1. Nonetheless, low catalytic activity is still a large obstacle to their application; therefore, designing and developing cost-effective and active catalysts is necessary.In the current era, introducing transition metals into the Pt catalyst is considered a robust strategy to boost the electrocatalytic performance and reduce the utilization of Pt by the bifunctional mechanism and the electronic effect.1,8,9 Even though the integration of Pt with inexpensive metals such as Ni, Fe, and Co exhibits an advantage in economic benefit, these nanoalloys are still susceptible to oxidation, resulting in their catalytic instability in long-term operating conditions.10–12 Because its price is equivalent to that of cobalt and less than 2% that of platinum, silver (Ag) has emerged as a favorable alloy component for Pt to obtain excellent electrocatalytic stability owing to its high oxidation resistance.11 In addition, the incorporation of Ag not only modifies the electronic structure but also increases the activation of Pt surface-active sites, which is attributable to the effective removal of adsorbed intermediate species on the Pt surface at a low potential by the formation of Ag(OH)ads.4,13,14 For instance, Huang et al.15 reported PtAg alloy NPs embedded in polyaniline that exhibited higher HER activity than Pt NPs with a 41 mV overpotential at 10 mA cm−2. Nonetheless, zero-dimensional (0D) Pt-based NPs were normally unstable under harsh electrochemical reaction conditions by the aggregation and Oswald ripening process.14,16,17 Adjusting the morphology and structure of Pt-based catalysts has been considered to be a powerful way to enhance the catalytic activity and stability. Song et al.18 prepared a coral-like PdAg catalyst by a polyol route that displayed superior CO-tolerance and great catalytic stability for glycerol oxidation; however, the use of polyvinylpyrrolidone (PVP) as a capping agent for the formation of the coral-like nanostructure can be a limitation because it potentially interferes with the catalyst activities.11,19
Although extensive reports have presented the high catalytic activity of the bimetallic PtAg alloy nanocatalyst for the HER and MOR, either complex processes,4,20 high temperature,14 or surfactant agents15,21 have been required for the fabrication of PtAg alloys. In this work, we introduce a coral-like dendritic AgPt nanoalloy on a carbon support by adjusting the nucleation and growth of Pt and Ag in an aqueous solution at room temperature. With 50 at% Ag, the as-made AgPt NCs/C catalyst showed great electrocatalytic performance for both the HER and MOR in an acidic electrolyte. In terms of the HER, the as-obtained catalyst exhibited an extremely small overpotential (16.61 mV) at 10 mA cm−2 and a low Tafel slope (19.05 mV dec−1) in comparison with the Pt NPs/C (E-TEK) catalyst. In addition, the AgPt NCs/C catalyst demonstrated a negative onset potential (0.32 VRHE), large current density (22.88 mA cm−2), and impressive CO-poisoning resistance toward the MOR. The ADT results also indicated superior catalytic stability of the AgPt NCs/C catalyst for both the HER and MOR. Apart from the synergistic effect of Ag, the unique morphological features with more active sites also contributed significantly to enhancing the catalytic performance of the as-made AgPt NCs/C catalyst.
Results and discussion
The morphology and particle size of the as-made catalyst were recorded by TEM and HR-TEM analysis. Fig. 1a and b depict a typical coral-like dendritic structure with nanometer size (∼3 nm in diameter and a length of ∼10 nm). Also, the HR-TEM images in Fig. 1c and d indicate that the inter-planar spacing distances of the AgPt NCs/C catalyst were about 0.23 nm, attributed to the indexed (111) facets of the face-cubic-center (fcc) AgPt nanoalloy.4,5 To investigate the elemental distribution of the AgPt NCs, energy-dispersive X-ray spectroscopy (EDX) mapping was performed during STEM. Fig. 1e–g shows the presence of silver and platinum with uniform distribution in the as-obtained catalysts, suggesting the formation of the bimetallic AgPt nanocoral catalyst. In this work, the coral-like AgPt nanoalloy was anchored on the carbon surface by a simple reduction process at room temperature using AgNO3 and H2PtCl6 as metal precursors and HCOOH as a reducing agent, as illustrated in Fig. 1h. For the aqueous formic acid-reducing route, the formate anions produced by the formic acid dehydration were favorable to the development of Pt along the (111) orientation because of the extremely slow reduction reaction and the lowest energy principle.22–24 At low concentrations, the Pt seeds were firstly formed by formic acid due to the slow chemical reduction of Pt ions.25 These seeds can be autocatalytic for the Pt reduction reaction when they obtain a fixed size (about 500 atoms).25,26 Since the standard reduction potential of Ag ions (Ag+) is higher than that of Pt ions (Pt4+ and Pt2+), the reduction reaction kinetics of Pt is slower than that of Ag under the same aqueous conditions. Therefore, the Pt ions must first be reduced to generate crystal seeds for the formation of both Pt and Ag in a particular direction; as a result, the Ag precursor is added to the whole system after the controlled Pt reduction duration.11 In brief, the Pt seeds were first formed and deposited onto the surface of carbon black by the reduction of PtCl62− by formic acid, which acted as sites for the absorption and reduction of Ag+ ions to form clustered particles. Under the extremely slow reduction rate at room temperature, the (111) direction was preferential according to the lowest-energy principle, resulting in the formation of the coral-like dendritic AgPt catalyst. | ||
| Fig. 1 (a and b) TEM images, (c and d) HR-TEM images, (e–g) EDX mapping, and (h) schematic of the preparation process of AgPt NCs/C. | ||
The XRD analysis was performed in the 2θ range from 20° to 80° to investigate the structure information of the AgPt NCs/C catalyst. As can be observed in Fig. 2a, the XRD pattern of the as-made catalyst showed three typical diffraction peaks at the 2θ angles of around 38.82°, 44.79°, and 66.68° of the (111), (200), and (220) facets of the fcc AgPt nanoalloy.27,28 Another peak was considered at a 2θ angle of about 25°, featuring the (002) facet of the hexagonal structure of carbon; this demonstrates that the bimetallic AgPt NCs were successfully anchored onto the carbon surface.4,29 In addition, the diffraction peaks of the as-obtained catalysts lay between those of standard Ag (JCPDS 04-0783) and Pt (JCPDS 04-0802), which were assigned to the lattice distortion by the different atom sizes of Ag and Pt, further confirming the formation of AgPt nanocrystals.27,30 The coherent length (D(khl)) and lattice space (d(hkl)) of the AgPt NCs/C catalyst were calculated by Debye–Scherrer's formula and Bragg's law, respectively. As listed in Table S1 (ESI†), the lattice space values of the AgPt NCs/C and Pt NPs/C catalysts were analogous at elevated order facets, suggesting that the investigated catalysts were developed based on metallic fcc Pt crystal. Additionally, the AgPt NCs/C catalyst showed a lattice expansion of 0.04 Å in both the (111) and (200) facets compared to those of the Pt NPs/C catalyst, implying the favorable local alloying of Ag in these facets; this is appropriately expressed by the increase of the intensity ratio between the (111) facet and other crystal facets (H(111)/H(200) and H(111)/H(220)).31 Furthermore, the conformation of the crystal structure of the catalyst was further certified by the difference of the D(hkl). In the case of the Pt NPs/C (E-TEK) catalyst, the D(111), D(200), and D(220) values were 2.16 nm, 1.89 nm, and 2.11 nm, respectively, showing the unclear discrepancy of the crystallinity; this was further evidenced by the intensity ratio of the facets.31 Meanwhile, the AgPt NCs/C catalyst displayed favorable crystal development along the (111) facet by the largest D(111) among all crystal facets and the highest intensity proportion (7.50) of the crystal facet in the longitude axis (i.e. (111)) and the corresponding lateral facets (i.e. (220)), which agreed generally with the observation from the HR-TEM images. Additionally, the atomic Ag![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pt ratio was close to 1:1 by energy-dispersive X-ray spectroscopy, as shown in Fig. 2b. These results suggested the formation of the AgPt nanoalloy on the carbon surface via a simple reduction process.
:Pt ratio was close to 1:1 by energy-dispersive X-ray spectroscopy, as shown in Fig. 2b. These results suggested the formation of the AgPt nanoalloy on the carbon surface via a simple reduction process.
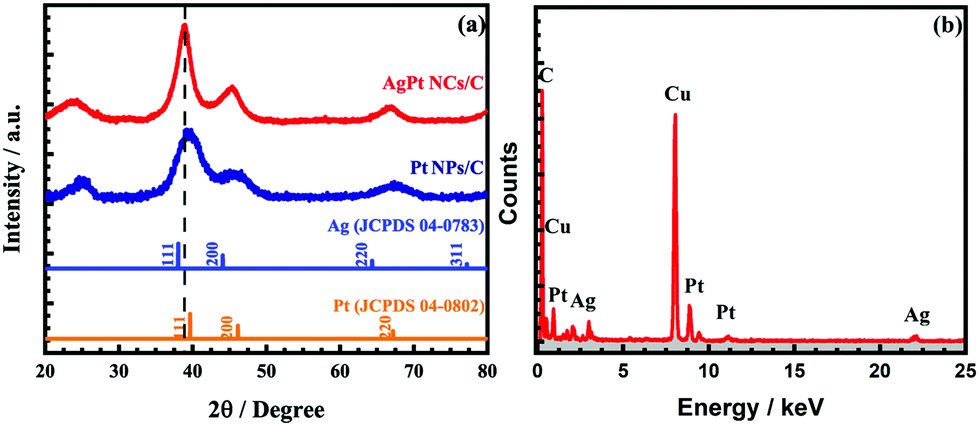 | ||
| Fig. 2 (a) X-ray diffraction patterns in the 2θ range of 20° to 80° at a step size of 0.02°, and (b) energy-dispersive X-ray spectrum of the as-obtained AgPt NCs/C electrocatalyst. | ||
The surface composition of the as-obtained nanocatalyst was investigated by the XPS analysis. The high-resolution spectrum of Pt 4f in Fig. 3a was deconstructed into two pairs of peaks; the strong pair at 70.40 eV (Pt 4f7/2) and 73.72 eV (Pt 4f5/2) was attributed to the zerovalent state (Pt0), whilst another weak couple was observed at 71.46 eV (Pt 4f7/2) and 75.11 eV (Pt 4f5/2), ascribed to the ionic Pt2+ species. The presence of the Pt2+ states in the as-obtained AgPt NCs/C catalysts was attributable to the oxidized surficial Pt in the air or the residual precursors.9,14 The content of Pt0 was calculated as 71.86% by the peak area ratio of all split peaks of Pt0 and Pt2+, indicating the domination of the zerovalent Pt0 state.14 In terms of Ag, the peak pair was considered at 367.94 eV (Ag 3d5/2) and 373.99 eV (Ag 3d3/2), attributed to the zerovalent state (Ag0) in the as-prepared AgPt NCs/C catalysts, as shown in Fig. 3b. More importantly, the binding energy of the Pt 4f orbitals in the AgPt NCs/C catalyst showed a negative shift compared to those of the Pt NPs/C catalyst,8 demonstrating the electronic donation from Ag to Pt owing to their electronegativity difference (Pt of 2.28 and Ag of 1.93).14,32 This electron transfer resulted in a d-band center downshift of Pt, benefiting the removal of oxygen-containing species from the catalyst surface because of the decreased adsorption strength, thereby improving the catalytic performance.9,33 In addition, the binding energy of Ag displayed a adverse trend, with increases of the binding energy from 367.67 to 367.94 eV for Ag0 3d5/2 and from 373.12 to 373.99 eV for Ag0 3d7/2 in comparison with the pure Ag metal,34,35 further confirming the electron transfer from Ag to Pt.
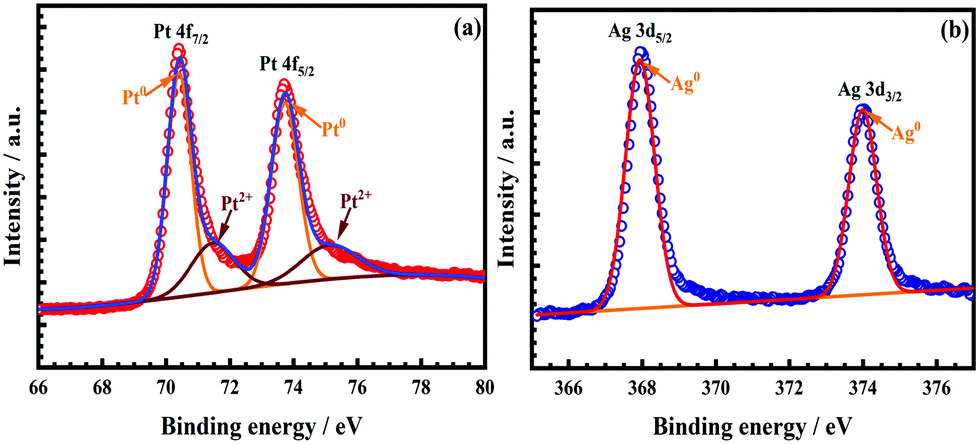 | ||
| Fig. 3 High-resolution spectra of (a) Pt 4f and (b) Ag 3d in the AgPt NCs/C. | ||
The electrocatalytic activity of the as-obtained AgPt NCs/C catalyst was examed by cyclic voltammetry (CV) tests. As illustrated in Fig. 4a, the CV curves of the AgPt NCs/C and Pt NPs/C (E-TEK) catalysts exhibited typical voltammetric features of Pt, with the observed peaks in the potential range of 0.0–0.2 VRHE corresponding to the hydrogen adsorption and desorption regions. Meanwhile, the peaks at 1.0 VRHE in the forward scan and 0.8 VRHE in the backward scan were attributed to the oxidation and reduction of Pt, respectively. The electrochemical surface areas (ECSAs) of the AgPt NCs/C and Pt NPs/C catalysts were 81.36 m2 gMetal−1 and 70.83 m2 mgMetal−1, suggesting that the as-made AgPt NCs/C catalyst contained more active sites and was more accessible for the electroactive species; this was assigned to the unique coral-like dendritic nanostructures.36,37 To investigate the HER performance of the AgPt NCs/C catalyst, linear sweep voltammetry (LSV) was carried out. Fig. 4b demonstrates that the AgPt NCs/C catalyst exhibited an onset potential of 4.78 mV, which was shifted positively by about 1.72 mV compared to the Pt NPs/C (E-TEK) catalyst (6.51 mV). In terms of the HER, the current density at 10 mA cm−2 was similar to that of a solar-to-hydrogen device to obtain about 12% efficiency, which is normally used as a key factor to assess the HER activity of catalysts.38,39 The inset of Fig. 4b illustrates that the overpotential at the current density of 10 mA cm−2 of the AgPt NCs/C catalyst is 16.61 mV, which is 4.69 mV better than that of the C-supported Pt (NPs) catalyst (21.30 mV), suggesting improved catalytic activity of the AgPt NCs/C catalyst for the HER in an acidic environment.
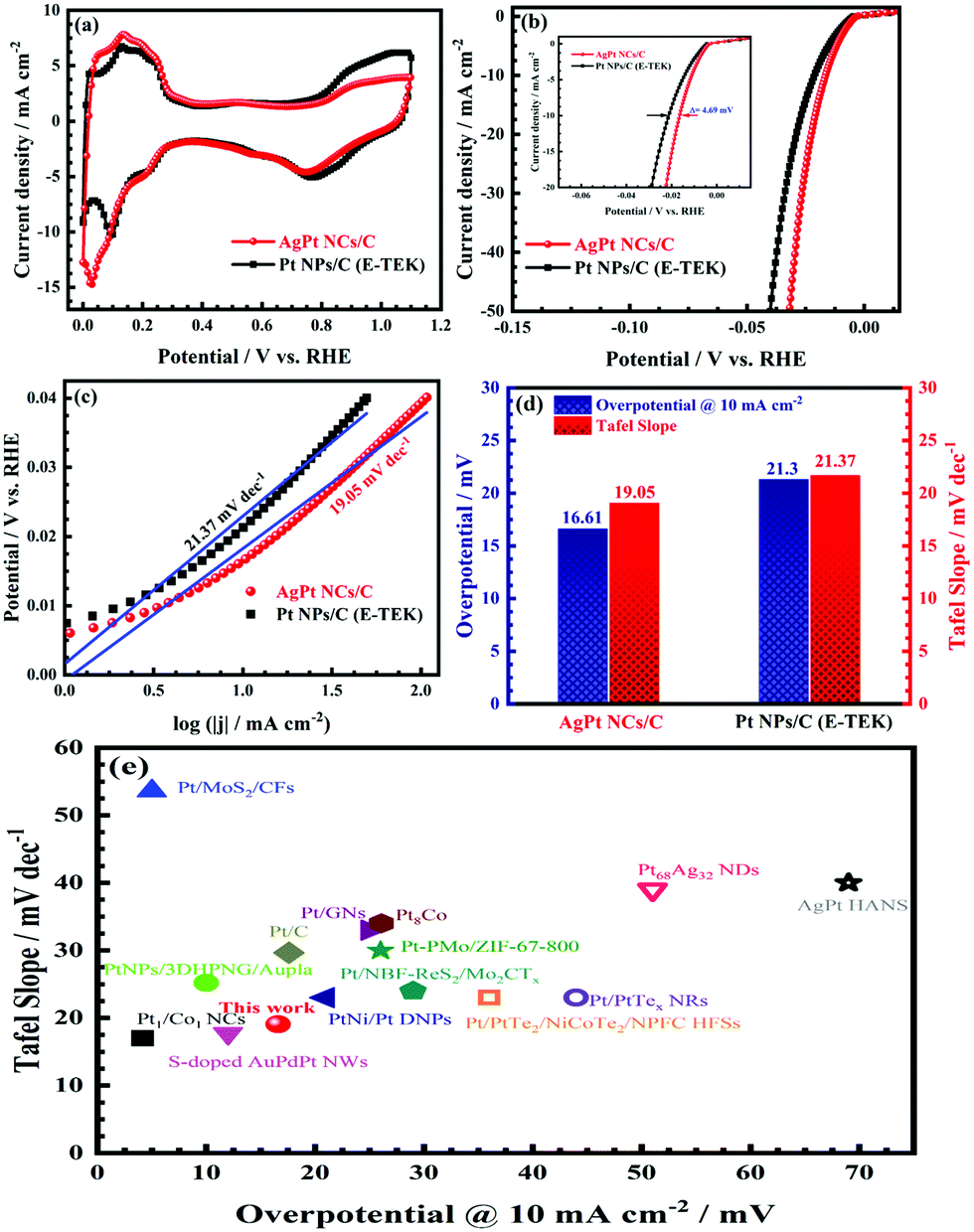 | ||
| Fig. 4 (a) CV curves at a 25 mV s−1 scan rate; (b) LSV curves at a 5 mV s−1 scan rate (inset: overpotentials at 10 mA cm−2 of the catalysts). (c) Tafel plots; (d) overpotentials at 10 mA cm−2 and Tafel slope values; and (e) comparison of the overpotentials at 10 mA cm−2 and Tafel slope values of the catalysts with those of other recently reported HER catalysts in N2-saturated 0.5 M H2SO4 electrolyte. | ||
Furthermore, the Tafel slope was calculated to assess the HER kinetics of the catalysts, as shown in Fig. 4c and d. The as-prepared AgPt NCs/C catalyst had a low Tafel slope (19.05 mV dec−1), which was smaller than that of the C-supported Pt (NPs) catalyst (21.37 mV dec−1), demonstrating the high HER activity of the AgPt NCs/C catalyst because the HER rate increased greatly as the overpotential increased. These results indicated that this AgPt NCs/C catalyst could drastically improve the HER catalytic activity of Pt and decrease the Pt loading, thereby reducing the cost for practical applications. In acidic medium, the HER was carried out according to three pathways, including the Volmer reaction: 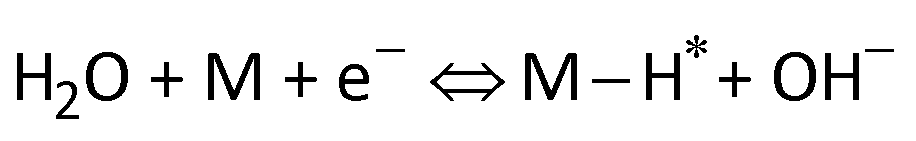 ; the Heyrovsky reaction:
; the Heyrovsky reaction: 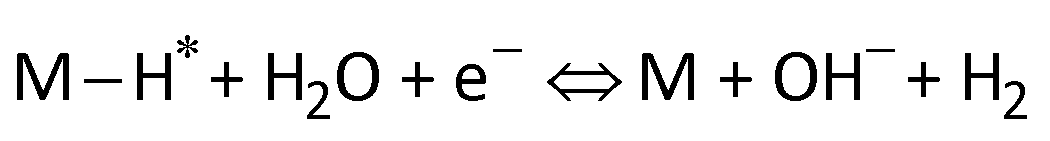 ; and the Tafel reaction:
; and the Tafel reaction: 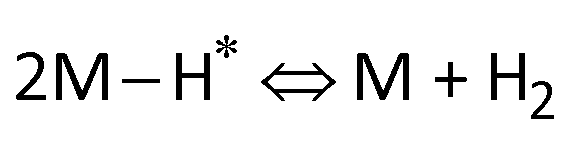 , where M is the active site and Hads denotes the hydrogen atom adsorbed.40,41 The Tafel slope values of the as-obtained AgPt NCs/C and Pt NPs/C catalysts were similar and close to 30 mV dec−1, demonstrating that the HER kinetics of the catalysts was determined by the Tafel step and according to the Volmer–Tafel mechanism.42Fig. 4e and Table S2 (ESI†) show the high HER activity of the AgPt NCs/C catalyst, which was better than that of Pt NPs/C and recently reported Pt-based catalysts in an acidic environment. In addition to the large ECSA value, the enhancement of the AgPt NCs/C catalyst was assigned to the electronic structure modification of the Pt, which accelerated the HER kinetics by decreasing the adsorption strength of Hads on the catalyst surface and facilitated the combination of adjacent Hads and the rapid release of H2.33,43
, where M is the active site and Hads denotes the hydrogen atom adsorbed.40,41 The Tafel slope values of the as-obtained AgPt NCs/C and Pt NPs/C catalysts were similar and close to 30 mV dec−1, demonstrating that the HER kinetics of the catalysts was determined by the Tafel step and according to the Volmer–Tafel mechanism.42Fig. 4e and Table S2 (ESI†) show the high HER activity of the AgPt NCs/C catalyst, which was better than that of Pt NPs/C and recently reported Pt-based catalysts in an acidic environment. In addition to the large ECSA value, the enhancement of the AgPt NCs/C catalyst was assigned to the electronic structure modification of the Pt, which accelerated the HER kinetics by decreasing the adsorption strength of Hads on the catalyst surface and facilitated the combination of adjacent Hads and the rapid release of H2.33,43
To further study the electron transfer kinetics during the HER process of the as-obtained AgPt NCs/C catalysts, electrochemical impedance spectroscopy (EIS) was used. The equivalent circuit diagram of the EIS data in the inset of Fig. 5a exhibit the solution resistance Rs, R1||CPE1 and Rct||CPE2 elements, with R1 and Rct attributed to the electrode surface and electrocatalytic kinetics, respectively.44 According to the Nyquist plots in Fig. 5, the Rct of the as-made AgPt NCs/C catalyst was 18.26 Ω, which was lower than that of Pt NPs/C (26.08 Ω), suggesting a faster electron transfer and preferential kinetics during the HER process on the AgPt NCs/C catalyst. In addition, the excellent HER activity of the as-prepared catalysts was further confirmed by the exchange current density (j0), which was extrapolated from the Tafel plots.33,45 The j0 of the AgPt NCs/C catalyst was 2.68 mA cm−2, which was 3.19-fold greater than that of the Pt NPs/C catalyst (0.84 mA cm−2), demonstrating the high intrinsic activity for the HER of the AgPt NCs/C catalyst (Fig. 5b). Furthermore, the TOF value of AgPt NCs/C was 3.86 H2 s−1, which was superior to that of the commercial Pt NPs/C (1.68 H2 s−1), suggesting the high intrinsical HER activity of the as-prepared catalyst.33,46 Additionally, ADT was performed to record the long-term stability of the HER catalysts, which plays an important role in practical applications. After 1000 cycling tests, the as-prepared AgPt NCs/C catalyst exhibited good catalytic stability, with an overpotential shift at 10 mA cm−2 of 3.5 mV; this was about half the value of 6.6 mV for carbon-supported Pt (NPs) catalyst (Fig. 5c and d), indicating the stability improvement of the AgPt NCs/C catalyst. Table 1 summarizes the HER performance of the AgPt NCs/C catalyst in acidic electrolytes.
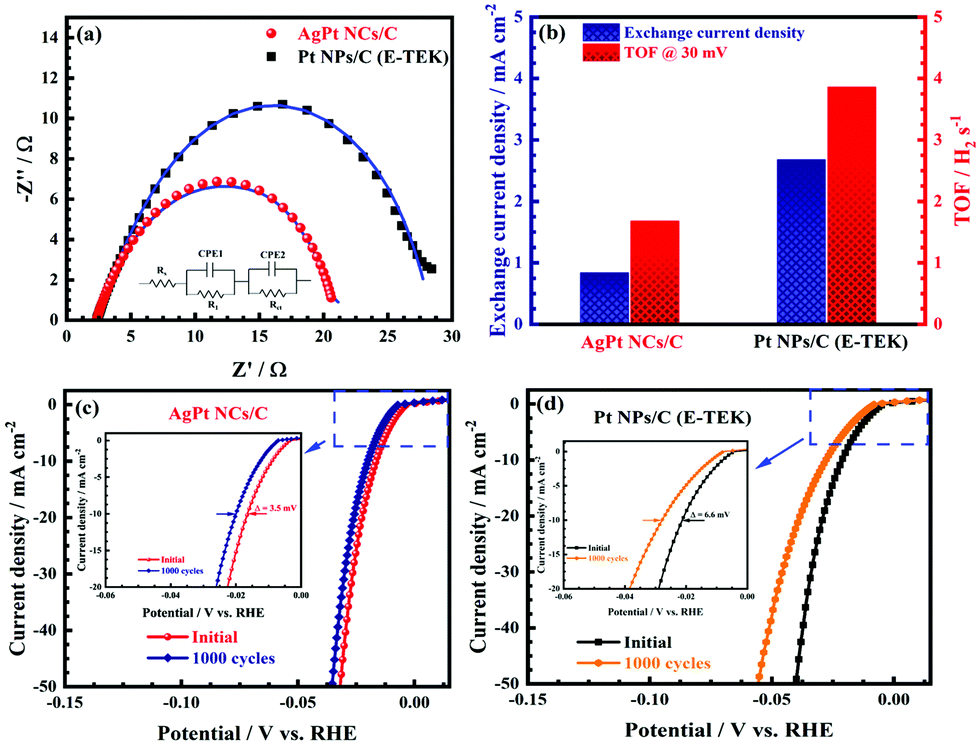 | ||
| Fig. 5 (a) Nyquist plots at an amplitude of 5 mV in the frequency range from 0.1 to 105 Hz; (b) exchange current density and turnover frequency; and (c and d) LSV curves at a 5 mV s−1 scan rate in the first cycle and 1000 cycles of the AgPt NCs/C and Pt NPs/C catalyst in N2-saturated 0.5 M H2SO4 aqueous solution. | ||
| Catalyst | Overpotential at 10 mA cm−2 mV | Tafel slope mV dec−1 | j 0 mA cm−2 | R ct Ω | TOF H2 s−1 |
|---|---|---|---|---|---|
| AgPt NCs/C | 16.61 | 19.05 | 2.68 | 18.26 | 3.86 |
| Pt NPs/C (E-TEK) | 21.30 | 21.37 | 0.84 | 26.08 | 1.68 |
The catalytic performance of the as-made AgPt NCs/C catalyst was further investigated for the methanol electro-oxidation process by CV tests. Fig. 6a depicts that the CV curves exhibited the typical two peaks for the MOR of Pt-based catalysts, including the positive-going scan peak, attributed to the methanol oxidation, and another negative-going scan peak, which was attributed to the oxidation process of the intermediate species produced during the MOR. As expected, the MOR peak of AgPt NCs/C was observed at 0.82 VRHE; this was shifted negatively by 30 mV in comparison with that of the commercial catalyst (0.85 VRHE), suggesting that the methanol oxidation process was easier and faster on the AgPt NCs/C catalyst. In addition, the If/Ib ratio of the AgPt NCs/C catalyst was calculated to be 1.47, which was improved compared to that of the commercial catalyst (0.85), indicating the better CO-like tolerance of the AgPt NCs/C catalyst. Although the Pt loading was reduced significantly, the AgPt NCs/C catalysts still showed a mass activity of 175.25 mA mgMetal−1 and specific activity of 0.35 mA cm−2, which was better than those of the C-supported Pt (NPs) catalyst (161.02 mA mgMetal−1 for mass activity and 0.23 mA cm−2 for a specific activity), as depicted in Fig. 6b and Table S3 (ESI†). The high catalytic activity of the AgPt NCs/C catalyst could be interpreted as due to (i) the unique coral-like dendritic nanostructure of the AgPt NCs, which supplies more active sites and smooth routes for rapid diffusion and transfer of the reactants and products, promoting the oxidation process of small organic molecules;47,48 (ii) the incorporation of Ag into the Pt catalyst, which enhanced the activation of the surface-active sites of the Pt.4,49
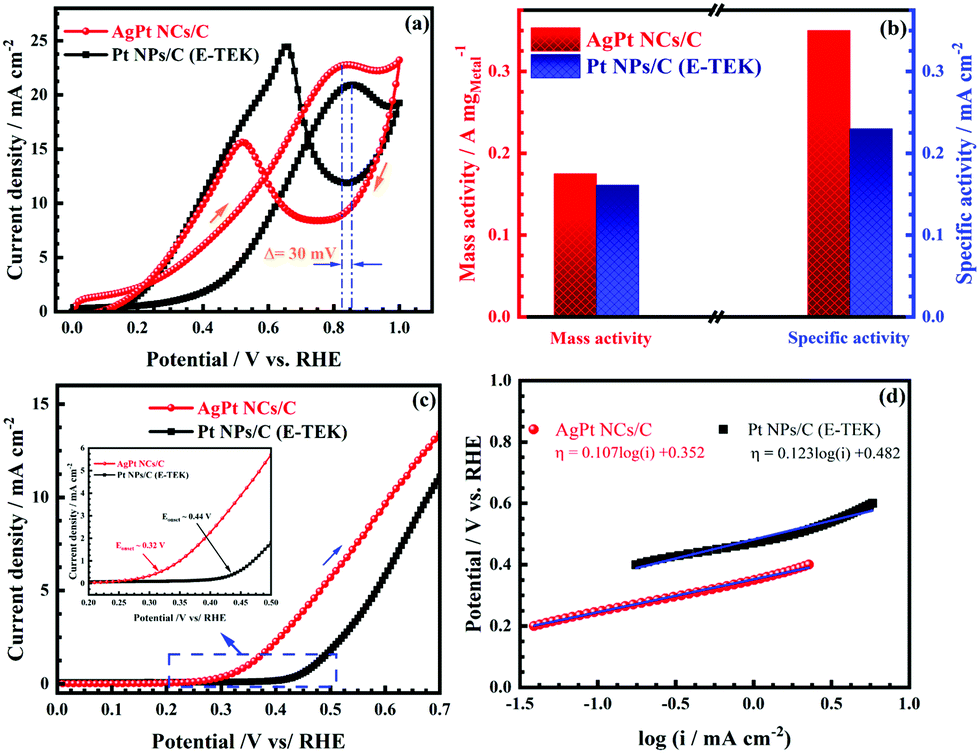 | ||
| Fig. 6 (a) CV curves at a 25 mV s−1 scan rate; (b) mass activity and specific activity; (c) LSV curves at a 1 mV s−1 scan rate; and (d) Tafel plots of the as-made AgPt NCs/C and Pt NPs/C (E-TEK) catalysts in N2-saturated 0.5 M H2SO4 + 1.0 M CH3OH aqueous solution. | ||
The LSV test was performed to further record the electrocatalytic activity of the as-obtained AgPt NCs/C catalyst. As can be seen in Fig. 6c, the AgPt NCs/C electrocatalyst exhibited a MOR onset potential of 0.32 VRHE, which was a much more negative potential than that of 0.44 VRHE for Pt NPs/C. In addition, the Tafel slope of the AgPt NCs/C catalyst was 107.08 mV dec−1, which was smaller than that of the commercial catalyst (123.36 mV dec−1), indicating the enhancement of the MOR kinetics of the AgPt NCs/C catalyst in acidic media (Fig. 6d). By the Tafel equation, the kinetic parameters of the investigated MOR catalysts were also calculated and are listed in Table 2. The exchange current density (j0) of the as-prepared AgPt NCs/C catalyst was 5.13 × 10−4 mA cm−2, which was 4.23-fold greater than that of the C-supported Pt (NPs) catalyst (1.21 × 10−4 mA cm−2). The high j0 of the AgPt NCs/C catalyst implied a faster electrode reaction, contributing significantly to enhancing the MOR activity.50 Also, the charge transfer coefficients (α) of the as-obtained AgPt NCs/C and Pt NPs/C catalysts were 0.56 and 0.48, respectively, demonstrating the improvement of the electron transfer during the MOR process on the AgPt NCs/C catalyst. In brief, the above results suggested high MOR activity of the AgPt NCs/C catalyst.
| Catalyst | Onset potential VRHE | Tafel slope mV dec−1 | Exchange current density mA cm−2 | Charge transfer coefficient |
|---|---|---|---|---|
| AgPt NCs/C | 0.32 | 107.08 | 5.13 × 10−4 | 0.56 |
| Pt NPs/C (E-TEK) | 0.44 | 123.36 | 1.21 × 10−4 | 0.48 |
The CO-tolerant ability of catalysts plays an important role in boosting their MOR activity; therefore, the CO-tolerance of the AgPt NCs/C catalyst was examed by the CO-stripping test. Fig. 7a depicts the CO-stripping curves of the MOR catalysts, which indicate the impressive CO-tolerance of the as-obtained AgPt NCs/C catalyst. For example, the AgPt NCs/C catalyst exhibited a COads onset potential of 0.63 VRHE, which was 70 mV more negative in comparison with that of the commercial catalyst (0.70 VRHE), suggesting that the COads oxidation occurred more readily on the AgPt NCs/C catalyst. The COads oxidation peak of the AgPt NCs/C catalyst was observed at 0.75 VRHE, which was negatively shifted in comparison with the Pt NPs/C (0.78 VRHE), demonstrating the superior CO anti-poisoning resistance of the AgPt NCs/C catalyst. Additionally, the chronoamperometry (CA) test was used to further evaluate the catalytic activity and possible CO-like poisoning under continuous operating conditions. As illustrated in Fig. 7b, the initial current density of the AgPt NCs/C catalyst was higher than that of Pt NPs/C, indicating the improved MOR activity of the AgPt NCs/C catalyst. Before achieving a pseudo-steady state, deterioration of the current density was detected at the first stage; this was unavoidable because of the methanol concentration difference caused by diffusion on the anode surface and the formation of the Pt oxide.28,51 The current density at the pseudo-steady state of the AgPt NCs/C catalyst was higher than that of Pt NPs/C, confirming the impressive MOR stability of the AgPt NCs/C catalyst. Also, the TEM and HR-TEM images in Fig. S1 (ESI†) indicate that the morphology of the AgPt NCs/C catalyst was almost unchanged after the CA test, indicating the high catalytic stability of the as-made catalyst in long-term duration. Furthermore, the high MOR stability of the as-made catalyst was further exhibited by ADT, as shown in Fig. 7c, d, and Table S4 (ESI†). After the 2000-cycling test, the MOR current density of the AgPt NCs/C catalyst decayed by around 17.52%, against a significant decrease of 28.74% for the Pt NPs/C catalyst. The improvement of the AgPt NCs/C catalyst was assigned to the electronic transfer from Ag to Pt, which was proven by the XPS results, causing the enriched electron density of Pt and weakening the bond strength with the lone electron pair of CO and/or other intermediate species, thereby reducing the binding and poisoning effect.14,52–54 Additionally, the incorporation of Ag into the Pt nanocatalyst can effectively remove the adsorbed carbonaceous intermediates on the surface of the Pt by the formation of OHads on the neighboring Ag surface from water dissociation at a lower potential than Pt.4,13,14 Briefly, the above properties resulted in less accumulation and easier oxidation of CO-like species during the electrochemical oxidation process, leading to improvement of the CO-like tolerance of the AgPt NCs/C catalyst. Meanwhile, the high intrinsic stability of the coral-like nanostructures under electrochemical conditions induced less aggregation and Ostwald ripening compared to the 0D counterparts,18,48 resulting in enhancement of the catalytic stability of the AgPt NCs/C catalyst.
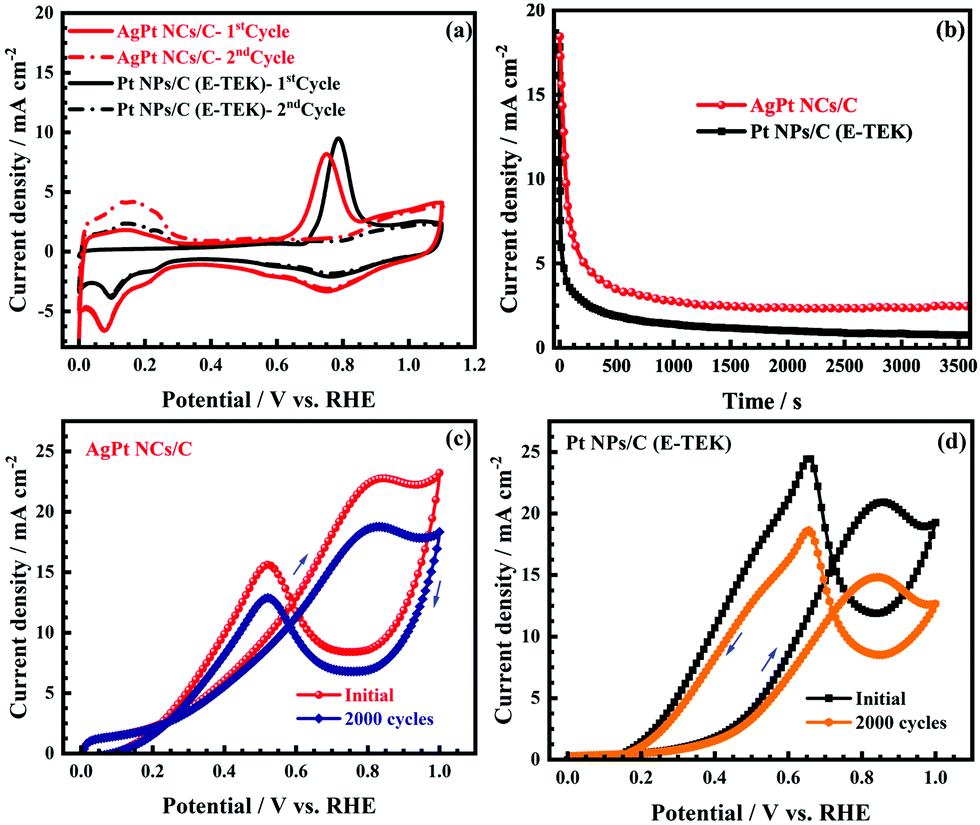 | ||
| Fig. 7 (a) CO-stripping in 0.5 M H2SO4 at a 25 mV s−1 scan rate; (b) CA curves at the fixed potential of 0.7 VRHE for 3600 s; and (c and d) 2000-cycling ADT of the different electrocatalysts in N2-saturated 0.5 M H2SO4 + 1.0 M CH3OH aqueous solution. | ||
These experimental results indicated that the coral-like dendritic AgPt catalyst exhibited high HER and MOR performance in an acidic environment, although its Pt loading was reduced significantly compared to that of the commercial catalyst. The enhancement of the AgPt NCs/C catalyst was assigned to the following reasons: (i) the unique coral-like dendritic nanostructure can not only supply more active sites and greater accessibility to the reactants and products but also promote the charge transfer, which can greatly facilitate the HER and MOR process and thus improve the catalytic performance of the AgPt NCs/C catalyst.4,36,37 Also, the coral-like nanostructures had high intrinsic durability under electrochemical conditions compared to the 0D Pt NPs, thereby enhancing the catalytic stability of the AgPt NCs/C catalyst.18,48 (ii) Alloying of Ag with Pt decreases the d-band center of the Pt by the electronic effect, weakening the affinity of the surface intermediates and/or products to the exposed Pt active sites and eventually endowing the AgPt NCs/C catalyst with enhanced catalytic activity.14,34 In addition, the incorporation of Ag into the Pt-based catalyst can effectively oxidize the COads through the unique ability to produce hydroxyl species from water dissociation at low potential (i.e., bifunctional effect),13,55 contributing to the enhanced CO-tolerant ability of the AgPt NCs/C catalyst.
Conclusions
In brief, a coral-like dendritic AgPt nanocatalyst was successfully fabricated by a facile room-temperature fabrication process without using any structure-directing agent. The unique morphological features endowed the catalyst with geometric and surface electronic merits to enhance the reaction kinetics towards the HER and MOR in an acidic environment. Due to the synergistic and electronic effect between Ag and Pt, improved performance of the AgPt NCs/C catalyst with a low overpotential at the current density of 10 mA cm−2 and a small Tafel slope compared to those of the commercial catalyst for the HER were obtained. In addition, the as-made catalyst demonstrated high HER stability, with an overpotential deterioration of 3.5 mV against a large decay of 6.6 mV for the commercial Pt NPs/C catalyst. Furthermore, the as-prepared AgPt NCs/C catalyst displayed a high current density and catalytic stability as well as superior CO-tolerant ability towards the MOR. This work could provide cost-efficient and robust catalysts for the HER and MOR, which plays an important role in clean energy generation and conversion technologies.Experimental
In a typical fabrication of the AgPt NCs/C catalyst,11 25 mg of carbon black was ultrasonicated in 50 mL of distilled water (DW) for 45 min to generate a homogeneous suspension. Afterward, 1 mL of H2PtCl6 0.01 M in 20 mL of DW was gradually dropped into the obtained suspension; then, 10 mL HCOOH was added. After 6 h, 2 mL of AgNO3 0.05 M in 20 mL DW was added under stirring for 30 min; then, the resulting mixture was stored in the dark at room temperature for 90 hours to finish the reaction. The formed product was rinsed repeatedly with DW and absolute ethanol, followed by drying at 80 °C. Detailed information of the material properties and electrochemical tests are demonstrated in the ESI.†Author contributions
Hau Quoc Pham: conceptualization, writing–original draft preparation, and software. Tai Thien Huynh: methodology, investigation, data curation, reviewing and editing, and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Hau Quoc Pham was funded by Vingroup Joint Stock Company and supported by the Domestic Master/PhD Scholarship Programme of Vingroup Innovation Foundation (VINIF), Vingroup Big Data Institute (VINBIGDATA), VINIF.2020.TS.108.Notes and references
- F. Guo, Z. Zou, Z. Zhang, T. Zeng, Y. Tan, R. Chen, W. Wu, N. Cheng and X. Sun, J. Mater. Chem. A, 2021, 9(9), 5468–5474 RSC.
- L. Y. Zhang, Z. L. Zhao and C. M. Li, Nano Energy, 2015, 11, 71–77 CrossRef CAS.
- Z. Li, D. Wu, Y. Ouyang, H. Wu, M. Jiang, F. Wang and L. Y. Zhang, Appl. Surf. Sci., 2020, 506, 144975 CrossRef CAS.
- H. Xu, B. Yan, S. Li, J. Wang, C. Wang, J. Guo and Y. Du, ACS Sustainable Chem. Eng., 2018, 6(1), 609–617 CrossRef CAS.
- Q. Liu, Y.-M. He, X. Weng, A.-J. Wang, P.-X. Yuan, K.-M. Fang and J.-J. Feng, J. Colloid Interface Sci., 2018, 513, 455 CrossRef CAS PubMed.
- Z. L. Zhao, L. Y. Zhang, S. J. Bao and C. M. Li, Appl. Catal., B, 2015, 174-175, 361–366 CrossRef CAS.
- N. Sahu, J. K. Das and J. N. Behera, Sustainable Energy Fuels, 2021, 5(19), 4992–5000 RSC.
- T. T. Huynh, N. N. Dang and H. Q. Pham, Catal. Sci. Technol., 2021, 11(4), 1571–1579 RSC.
- H. Q. Pham and T. T. Huynh, ACS Appl. Nano Mater., 2021, 4(5), 4983–4993 CrossRef CAS.
- R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, T. Zawodzinski, J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K.-I. Kimijima and N. Iwashita, Chem. Rev., 2007, 107(10), 3904–3951 CrossRef CAS PubMed.
- E. Fidiani, G. Thirunavukkarasu, Y. Li, Y.-L. Chiu and S. Du, J. Mater. Chem. A, 2020, 8(23), 11874–11883 RSC.
- F. Maillard, L. Dubau, J. Durst, M. Chatenet, J. André and E. Rossinot, Electrochem. Commun., 2010, 12(9), 1161–1164 CrossRef CAS.
- G.-T. Fu, B.-Y. Xia, R.-G. Ma, Y. Chen, Y.-W. Tang and J.-M. Lee, Nano Energy, 2015, 12, 824–832 CrossRef CAS.
- X. Fu, C. Wan, A. Zhang, Z. Zhao, H. Huyan, X. Pan, S. Du, X. Duan and Y. Huang, Nano Res., 2020, 13(5), 1472–1478 CrossRef CAS.
- M. Huang, H. Zhang, S. Yin, X. Zhang and J. Wang, ACS Appl. Nano Mater., 2020, 3(4), 3760–3766 CrossRef CAS.
- H. Q. Pham, T. T. Huynh, A. T. N. Mai, T. M. Ngo, L. G. Bach and V. T. T. Ho, Sci. Rep., 2019, 9(1), 14791 CrossRef PubMed.
- Y. Gong, X. Liu, Y. Gong, D. Wu, B. Xu, L. Bi, L. Y. Zhang and X. S. Zhao, J. Colloid Interface Sci., 2018, 530, 189–195 CrossRef CAS PubMed.
- P. Song, J.-J. Feng, F.-Y. Guo and A.-J. Wang, J. Mater. Chem. A, 2015, 3(31), 15920–15926 RSC.
- G. Zhang, Z. Yang, W. Zhang, H. Hu, C. Wang, C. Huang and Y. Wang, Nanoscale, 2016, 8(5), 3075–3084 RSC.
- Y. Kim, H. Kim and W. B. Kim, PtAg nanotubes for electrooxidation of ethylene glycol and glycerol in alkaline media, Electrochem. Commun., 2014, 46, 36–39 CrossRef CAS.
- F. Gao, H. Xu, Y. Zhang, J. Wang, C. Wang and Y. Du, Int. J. Hydrogen Energy, 2018, 43(20), 9644–9651 CrossRef CAS.
- C.-W. Liu, Y.-C. Wei, C.-C. Liu and K.-W. Wang, J. Mater. Chem., 2012, 22(11), 4641–4644 RSC.
- S. Sun, F. Jaouen and J.-P. Dodelet, Adv. Mater., 2008, 20(20), 3900–3904 CrossRef CAS.
- S. H. Sun, D. Q. Yang, D. Villers, G. X. Zhang, E. Sacher and J. P. Dodelet, Adv. Mater., 2008, 20(3), 571–574 CrossRef CAS.
- Y. Song, Y. Yang, C. J. Medforth, E. Pereira, A. K. Singh, H. Xu, Y. Jiang, C. J. Brinker, F. van Swol and J. A. Shelnutt, J. Am. Chem. Soc., 2004, 126(2), 635–645 CrossRef CAS PubMed.
- E. Greenbaum, J. Phys. Chem., 1988, 92(16), 4571–4574 CrossRef CAS.
- Z. Peng, H. You and H. Yang, Adv. Funct. Mater., 2010, 20(21), 3734 CrossRef CAS.
- X. Cao, N. Wang, Y. Han, C. Gao, Y. Xu, M. Li and Y. Shao, Nano Energy, 2015, 12, 105–114 CrossRef CAS.
- Y. Zhao, L. Yang, S. Chen, X. Wang, Y. Ma, Q. Wu, Y. Jiang, W. Qian and Z. Hu, J. Am. Chem. Soc., 2013, 135(4), 1201–1204 CrossRef CAS PubMed.
- X. Weng, Q. Liu, A.-J. Wang, J. Yuan and J.-J. Feng, J. Colloid Interface Sci., 2017, 494, 15–21 CrossRef CAS PubMed.
- T.-H. Huang, D. Bhalothia, S. Dai, C. Yan, K.-W. Wang and T.-Y. Chen, Sustainable Energy Fuels, 2021, 5(11), 2960–2971 RSC.
- J.-J. Feng, X.-X. Lin, L.-X. Chen, M.-T. Liu, J. Yuan and A.-J. Wang, J. Colloid Interface Sci., 2017, 498, 22–30 CrossRef CAS PubMed.
- X. Zhang, S. Wang, C. Wu, H. Li, Y. Cao, S. Li and H. Xia, J. Mater. Chem. A, 2020, 8(45), 23906–23918 RSC.
- A. B. Yousaf, M. Imran, A. Zeb, T. Wen, X. Xie, Y.-F. Jiang, C.-Z. Yuan and A.-W. Xu, Electrochim. Acta, 2016, 197, 117–125 CrossRef.
- S.-S. Chen, L.-J. Nan, J.-J. Feng, L. Zhang, K.-M. Fang, X. Luo and A.-J. Wang, Int. J. Hydrogen Energy, 2018, 43(12), 6096–6106 CrossRef CAS.
- H. Huang, L. Ma, C. S. Tiwary, Q. Jiang, K. Yin, W. Zhou and P. M. Ajayan, Small, 2017, 13(10), 1603013 CrossRef PubMed.
- J.-P. Zhong, C. Hou, L. Li, M. Waqas, Y.-J. Fan, X.-C. Shen, W. Chen, L.-Y. Wan, H.-G. Liao and S.-G. Sun, J. Catal., 2020, 381, 275 CrossRef CAS.
- X. Chia, N. A. A. Sutrisnoh and M. Pumera, ACS Appl. Mater. Interfaces, 2018, 10(10), 8702–8711 CrossRef CAS PubMed.
- M. Wei, L. Huang, S. Huang, Z. Chen, D. Lyu, X. Zhang, S. Wang, Z. Q. Tian and P. K. Shen, J. Catal., 2020, 381, 385–394 CrossRef CAS.
- J. Hu, C. Fang, X. Jiang, D. Zhang and Z. Cui, Inorg. Chem. Front., 2020, 7(22), 4377–4386 RSC.
- L. Li, S. Wang, L. Xiong, B. Wang, G. Yang and S. Yang, J. Mater. Chem. A, 2019, 7(20), 12800–12807 RSC.
- Y. Zhang, L. Liu, C. Sun, Y. Du, Y. Zhou and Q. Xie, J. Alloys Compd., 2021, 888, 161564 CrossRef CAS.
- X. Wang, L. Bai, J. Lu, X. Zhang, D. Liu, H. Yang, J. Wang, P. K. Chu, S. Ramakrishna and X.-F. Yu, Angew. Chem., Int. Ed., 2019, 58(52), 19060–19066 CrossRef CAS PubMed.
- B. Jiang, H. Yuan, Q. Dang, T. Wang, T. Pang, Y. Cheng, K. Wu, X. Wu and M. Shao, Int. J. Hydrogen Energy, 2019, 44(59), 31121 CrossRef CAS.
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. L. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Adv. Mater., 2019, 31(31), 1806296 CrossRef PubMed.
- X. Bao, Y. Gong, Y. Chen, H. Zhang, Z. Wang, S. Mao, L. Xie, Z. Jiang and Y. Wang, J. Mater. Chem. A, 2019, 7(25), 15364–15370 RSC.
- L.-Y. Jiang, A.-J. Wang, X.-S. Li, J. Yuan and J.-J. Feng, ChemElectroChem, 2017, 4(11), 2909–2914 CrossRef CAS.
- X. Jiang, Y. Liu, J. Wang, Y. Wang, Y. Xiong, Q. Liu, N. Li, J. Zhou, G. Fu, D. Sun and Y. Tang, Nano Res., 2019, 12(2), 323–329 CrossRef CAS.
- X. ong, C. Tan, J. Liu, J. Yang, X.-J. Wu, Z. Fan, Z. Luo, J. Chen, X. Zhang, B. Chen and H. Zhang, J. Am. Chem. Soc., 2015, 137(4), 1444–1447 CrossRef PubMed.
- S. Lee, H. J. Kim, S. M. Choi, M. H. Seo and W. B. Kim, Appl. Catal., A, 2012, 429–430, 39–47 CrossRef CAS.
- C. Fang, J. Zhao, G. Zhao, L. Kuai and B. Geng, Nanoscale, 2016, 8(32), 14971–14978 RSC.
- L. Y. Zhang, F. Wang, S. Wang, H. Huang, X. Meng, Y. Ouyang, W. Yuan, C. X. Guo and C. M. Li, Adv. Funct. Mater., 2020, 30, 2003933 CrossRef CAS.
- Y. Zhang, F. Gao, P. Song, J. Wang, J. Guo, Y. Shiraishi and Y. Du, ACS Sustainable Chem. Eng., 2019, 7(3), 3176–3184 CrossRef CAS.
- Y. Ouyang, H. Cao, H. Wu, D. Wu, F. Wang, X. Fan, W. Yuan, M. He, L. Y. Zhang and C. M. Li, Appl. Catal., B, 2020, 265, 118606 CrossRef CAS.
- J.-J. Lv, J.-X. Feng, S.-S. Li, Y.-Y. Wang, A.-J. Wang, Q.-L. Zhang, J.-R. Chen and J.-J. Feng, Electrochim. Acta, 2014, 133, 407–413 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma01077h |
| This journal is © The Royal Society of Chemistry 2022 |